Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(12):2068-2083. doi:10.7150/thno.15007 This issue Cite
Research Paper
Crucial Role of miR-433 in Regulating Cardiac Fibrosis
Lichan Tao1*, Yihua Bei2*, Ping Chen2, Zhiyong Lei3, Siyi Fu2, Haifeng Zhang1, Jiahong Xu4, Lin Che4, Xiongwen Chen5, Joost PG Sluijter3, Saumya Das6, Dragos Cretoiu7,8, Bin Xu9, Jiuchang Zhong10, Junjie Xiao2 ![]() , Xinli Li1
, Xinli Li1 ![]()
1. Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China.
2. Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, Shanghai 200444, China.
3. Laboratory of Experimental Cardiology, University Medical Centre Utrecht, Utrecht 3508GA, The Netherlands.
4. Department of Cardiology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China.
5. Cardiovascular Research Center and Department of Physiology, Temple University School of Medicine, Philadelphia, PA 19140, USA.
6. Cardiovascular Division of the Massachusetts General Hospital and Harvard Medical School, Boston, MA 02215, USA.
7. Victor Babes National Institute of Pathology, Bucharest 050096, Romania.
8. Division of Cellular and Molecular Biology and Histology, Carol Davila University of Medicine and Pharmacy, Bucharest 050474, Romania.
9. Innovative Drug Research Center of Shanghai University, Shanghai 200444, China.
10. State Key Laboratory of Medical Genomics & Shanghai Institute of Hypertension, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China.
* These two authors contributed equally to this work.
Received 2016-1-18; Accepted 2016-8-6; Published 2016-9-10
Abstract

Dysregulation of microRNAs has been implicated in many cardiovascular diseases including fibrosis. Here we report that miR-433 was consistently elevated in three models of heart disease with prominent cardiac fibrosis, and was enriched in fibroblasts compared to cardiomyocytes. Forced expression of miR-433 in neonatal rat cardiac fibroblasts increased proliferation and their differentiation into myofibroblasts as determined by EdU incorporation, α-SMA staining, and expression levels of fibrosis-associated genes. Conversely, inhibition of miR-433 exhibited opposite results. AZIN1 and JNK1 were identified as two target genes of miR-433. Decreased level of AZIN1 activated TGF-β1 while down-regulation of JNK1 resulted in activation of ERK and p38 kinase leading to Smad3 activation and ultimately cardiac fibrosis. Importantly, systemic neutralization of miR-433 or adeno-associated virus 9 (AAV9)-mediated cardiac transfer of a miR-433 sponge attenuated cardiac fibrosis and ventricular dysfunction following myocardial infarction. Thus, our work suggests that miR-433 is a potential target for amelioration of cardiac fibrosis.
Keywords: cardiac fibrosis, miR-433, AZIN1, JNK1.
Introduction
Cardiac fibrosis, a hallmark of most cardiomyopathies, is characterized by excessive extracellular matrix accumulation contributing to the destruction of normal tissue architecture and progressive organ dysfunction [1, 2]. Cardiac fibrosis is a strong driver of adverse ventricular remodeling and heart failure that occurs after a variety of different cardiac injuries, such as myocardial infarction (MI) and hemodynamic stress as seen in hypertrophic and dilated cardiomyopathies [3, 4]. Although acetyl choline esterase (ACE) inhibition, angiotensin II receptor antagonists, and recently LCZ696 (an angiotensin II type 1 receptor-neprilysin inhibitor) can partially reverse remodeling, no effective anti-fibrotic therapeutic strategies are currently available [1, 5, 6]. The lack of an effective therapy for cardiac fibrosis and cardiac remodeling is in part responsible for the morbidity, mortality, and healthcare expenditure attributable to heart failure [2, 5]. Therefore, novel anti-fibrotic strategies represent a critical unmet clinical need [2, 5].
MicroRNAs (miRNAs, miRs) are small noncoding RNAs, which repress gene expression by degradation or translational inhibition of target mRNAs [7]. A single mRNA can be regulated by multiple miRNAs, while individual miRNAs are capable of regulating tens to hundreds of distinct target genes [7, 8]. As approximately 60% of protein-coding genes are regulated by miRNAs, they have emerged as powerful regulators for almost all essential biological processes including cellular proliferation, differentiation, apoptosis, development, and metabolism [9, 10]. Emerging data have suggested that aberrant expression of miRNAs could lead to a profound disturbance of target gene network and signaling cascades that participate in many pathological phenotypes. One such example is of adverse cardiac remodeling and fibrosis [1, 11, 12]. Increased pro-fibrotic miRNAs such as miR-21, 22, and 34a and decreased anti-fibrotic miRNAs such as miR-24, 15 family, 26a, and 29b have been reported to contribute to cardiac fibrosis [13-20]. These observations indicate that manipulation of miRNAs may serve as a novel potential therapeutic approach to combat cardiac fibrosis. An unexplored candidate located on chromosome 12, miR-433, has been reported to be up-regulated in renal fibrosis and liver fibrosis [21, 22]. However, the role of miR-433 in the heart and especially in cardiac fibrosis is unclear.
In the present study, based on miRNA arrays, we noted that miR-433 was significantly increased in ventricle samples at 21-days following MI in mice. We further validated up-regulation of miR-433 in a rodent model of doxorubicin-induced cardiomyopathy and human dilated cardiomyopathy (DCM). Also, over-expression of miR-433 increased the proliferation of cardiac fibroblasts and promoted their differentiation into myofibroblasts, whereas knockdown of miR-433 suppressed these responses upon transforming growth factor-β (TGF-β) or Angiotensin II (Ang II) stimulation. Our work further identified AZIN1 and JNK1 as two target genes of miR-433. Importantly, treatment with miR-433 antagomir or adeno-associated virus 9 (AAV9)-mediated cardiac transfer of a miR-433 sponge improved post-MI cardiac function and attenuated cardiac fibrosis in adult mice. Collectively, our findings indicate that miR-433 promotes cardiac fibrosis and therefore inhibition of miR-433 might be useful for the treatment of cardiac fibrosis.
Materials and Methods
Ethics Statement
All animals were raised at the Experimental Animal Center of Nanjing Medical University (Nanjing, China) or Shanghai University (Shanghai, China). All procedures with animals were in accordance with the guidelines on the use and care of laboratory animals for biomedical research published by National Institutes of Health (No. 85-23, revised 1996). The experimental protocol was reviewed and approved by the ethical committees of Nanjing Medical University and Shanghai University. All human investigations conformed to the principles outlined in the Declaration of Helsinki and was approved by the institutional review committees of Nanjing Medical University. All participants gave written informed consent before enrollment in the study. Human left ventricular tissue samples were obtained from 4 patients with dilated cardiomyopathy (DCM) undergoing cardiac transplantation and 4 healthy donors (The First Affiliated Hospital of Nanjing Medical University).
Isolation of Cardiac Fibroblasts, Culture, and Transfection
Cardiac fibroblasts were isolated from 1 to 3-day-old SD rats. Ventricles were finely minced and digested in trypsin buffer (60% trypsin and 40% collagenase). Cell suspensions were centrifuged, resuspended in DMEM (Gibco, Grand Island, CA, USA) with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin, and plated for 2 h under standard culture conditions (37°C in 5% CO2 and 95% O2) which allowed fibroblast attachment to the culture plates.
All transfections and assays on cardiac fibroblasts were conducted in low serum medium (1% FBS). Cardiac fibroblasts at passage 2 were exposed to either miRNA agomir versus negative control (100 nM), or antagomir versus negative control (200 nM) (RiboBio, Guangzhou, China) for 48 h, and treated with 10 ng/ml recombinant human TGF-β1 for 24 h (Peprotech, Rocky Hill, NJ, USA) or 100 nM Ang II for 48 h (Sigma, St. Louis, MO, USA), respectively. siRNAs for AZIN1, JNK1, and negative controls were purchased from Invitrogen Carlsbad, CA. Plasmids over-expressing AZIN1 or JNK1 were purchased from Sangon Biotech, Shanghai, China. Transfections with siRNAs (50 nM) or plasmids (50 nM) for 48 h were carried out using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen). p38 MAP kinase inhibitor SB202190 (Sigma, 10 μM, 1 h), ERK inhibitor U0126 (Sigma, 10 μM, 1 h), and Smad3 inhibitor SIS3 (Millipore, 1 μM, 48 h) were used to treat cells in the presence or absence of miR-433 agomir.
Animal Models
Eight-week-old male C57BL/6 mice were used in this study. MI was generated by ligating the left anterior descending coronary artery (LAD) using a 7/0 silk thread while sham was created by the same process but without LAD ligation. Doxorubicin-induced cardiomyopathy mouse model was induced by chronically treating mice with either doxorubicin or phosphate-buffered saline (PBS) by four intraperitoneal (i.p) injections (day 0, 2, 4 and 6) at a dose of 4 mg/kg. All mice were sacrificed after 4 weeks.
To determine if inhibition of miR-433 is sufficient to prevent cardiac fibrosis in vivo, mice were injected via tail vein with 80 mg/kg antagomir (a 2'OME+ 5'chol modified miR-433 inhibitor) or the scramble control (Ribobio, Guangzhou, China) for 3 consecutive days and subjected to LAD ligation. AAV represents an efficient and safe vector for in vivo gene transfer and serotype 9 is significantly cardiotropic [23-26]. Thus, besides miR-433 antagomir, the cardiotropic miR-433 sponge AAV9 was used to determine further if cardiac inhibition of miR-433 is sufficient to prevent fibrosis in vivo. In brief, mice were randomly chosen to receive a single-bolus tail vein injection of either miR-433 sponge AAV9 or miR-scramble (Hanheng Biotechnology, Shanghai, China) at 1*1011 vg (viral genomes) per animal. After 1 week, mice were subjected to LAD ligation and finally sacrificed at 3 weeks post-MI.
miRNA Array and Gene-Chip Analysis
Total RNA extracted from ventricular tissues 21 days post-MI or sham control was used for miRNA arrays based on Affymetrix 4.0 (OE Biotech's, Shanghai, China). Additionally, total RNA extracted from ventricular tissues 21 days post-MI injected with miR-433 antagomir or scramble control was used for gene-chip analysis based on Agilent SurePrint G3 Mouse GE (8*60K, Design ID: 028005) Microarray (OE Biotech's, Shanghai, China). The MIAME compliant data have been submitted to Gene Expression Omnibus (GEO, platform ID: GSE74135 for miRNA array and GSE74206 for gene-chip analysis, respectively).
Quantitative Real-time Polymerase Chain Reactions (qRT-PCRs)
Total RNAs were extracted from cardiac fibroblasts and heart samples by using miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. Total RNAs (400 ng) were reverse transcribed using Bio-Rad iScriptTM cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) to obtain cDNAs. The expression levels of TGF-β, α-SMA, Col1a1, and Col3a1 were analyzed by using Bio-Rad SYBR qPCR (Bio-Rad, Hercules, CA, USA) on ABI-7900 Real-Time PCR Detection System (7900HT, Applied Biosystems, CA, USA). 18S RNA was used as an internal control for gene expressions. Primer sequences used in the study are listed in Supplemental Table 1. For quantitative miRNA analysis, the Bulge-LoopTM miRNA qPCR Primer Set (RiboBio) was used to determine the expression levels of miRNAs with Takara SYBR Premix Ex TaqTM (Tli RNaseH Plus) on ABI-7900 Real-Time PCR Detection System (Applied Biosystems). U6 was used as an internal control for miRNA template normalization.
Pharmacokinetics of miRNA
miR-433 antagomir or the scramble control (Ribobio, Guangzhou, China) was prepared in PBS and administered via tail vein at a dose of 7.5 mg/kg for each mice. Subsequently, mice were sacrificed and plasma and heart tissues were collected immediately at different time points of 5, 10, 15, 30, 60, 120, 240, 480, 1320 and 1440 minutes after injection (n=5 per group for each time point) [27]. miR-433 expression levels in plasma and heart samples were determined using qRT-PCRs as described above.
Immunofluorescence and EdU Staining
Cardiac fibroblasts were fixed in 4% paraformaldehyde (PFA) for 20 min at room temperature. Cells were then permeabilized with 0.2% Triton X-100 for 20 min and blocked with 10% goat serum in PBS-Tween for 1 h at room temperature. Subsequently, cardiac fibroblasts were incubated with α-SMA-Cy3 antibody (1:500, Sigma, St. Louis, MO, USA) diluted in 10% goat serum overnight at 4°C. To detect proliferation, EdU assays were performed using Click-iT Plus EdU Alexa Fluor 488 Imaging Kit (Invitrogen) according to manufacturer's instructions. Cell nuclei were counterstained with DAPI and the number of EdU-positive nuclei was calculated. Fifteen fields/sample (200 x magnification) were viewed under a confocal microscope (Carl Zeiss, Thuringia, Germany).
Sections of heart samples were cut at a thickness of 5-6 μm. Subsequently, the sections were fixed in 4% PFA for 20 min at room temperature, permeabilized with 0.2% Triton X-100 for 20 min, and then blocked with 10% goat serum in PBS-Tween for 1 h at room temperature. Next, the sections were incubated with diluted primary antibodies at 4°C overnight; the following antibodies were used: α-SMA-Cy3 antibody (1:500, Sigma), Vimentin antibody (1:100, Abcam), Ki67 antibody (1:100, Abcam), and pHH3 antibody (1:100, Abcam). After three washes with PBS for 5 min each, the sections were incubated with secondary antibodies or other dyes at room temperature for 2 h. Fifteen fields/sample (400x magnification) were viewed under a confocal microscope (Carl Zeiss).
Western Blotting Analysis
Cardiac fibroblasts and heart samples were lysed using RIPA buffer (Beyotime Institute of Biotechnology, Nantong, China), which contained a protease inhibitor cocktail (Sigma). The concentration of protein samples was evaluated by Bicinchoninic Acid Protein Assay Kit (Thermo Fisher, Waltham, MA, USA). Equal amounts of protein were separated in SDS-PAGE and blotted onto PVDF membranes. The primary antibodies used were from the following sources: α-SMA (1:1000, Sigma), TGF-β (1:1000, Cell Signaling Technology, Boston, MA, USA), p38 (1:1000, CST), p-p38 (1:1000, CST), ERK (1:1000, CST), p-ERK (1:1000, CST), p-Smad3 (1:1000, CST), Smad3 (1:1000, CST), JNK1 (1:1000, CST), AZIN1 (1:500, Proteintech, Wuhan, China), CTGF (1:500, Proteintech, Wuhan, China), Col1a1 (1:500, Proteintech), Col3a1 (1:500, Proteintech), MMP2 (1:500, Proteintech), MMP9 (1:500, Proteintech) and GAPDH (1:10000, Kangchen, Shanghai, China). All proteins were visualized by ECL Chemiluminescence Kit (Thermo Fisher) and the quantification of each band was performed using Imagelab Software (Bio-Rad) with GAPDH as a loading control.
Luciferase Reporter Assay
A fragment of the 3'UTRs of AZIN1 or JNK1 containing the target site of miR-433 was obtained by PCR amplification and then cloned into the pGL3-Basic Vector (Promega, Madison, WI, USA) to generate the AZIN1 or JNK1 wt-luc vector. The AZIN1 or JNK1 mutant-luc vector was generated by using the MutaBest kit (Takara, Tokyo, Japan). Forty-eight hours after transfection, luciferase activities were measured using a dual luciferase reporter assay system (Promega) following a standard procedure.
Echocardiography
Three weeks after the injection of miR-433 antagomir, mice were anesthetized with 1.5-2% isoflurane and then evaluated by Vevo 2100 echocardiography (VisualSonics Inc, Toronto, Ontario, Canada) with a 30 MHz central frequency scan head to detect cardiac function. The following parameters were measured from M-mode images taken from the parasternal short-axis view at papillary muscle level: left ventricular fractional shortening (FS) and left ventricular ejection fraction (EF). The left ventricle internal diameter (LVID), interventricular septum (IVS), and left ventricle posterior wall (LVPW) in diastole or systole were also measured. At least three measurements were obtained and averaged for each mouse.
TTC staining
At 3 days' post LAD ligation, mice were anesthetized with intraperitoneal injection of 0.5 mg/g tribromoethanol. Subsequently, 1 ml Evans blue (BioSharp, Anhui, China) was slowly injected into inferior vena and the heart was removed immediately. After storage for 15 minutes at -20°C, the heart was cut into 5 transverse slices at 1 mm thickness across the long axis. The slices were then stained with 1% triphenyltetrazolium chloride (TTC, Amresco, OHIO, USA) in PBS for 10 min at 37°C following which the slices were fixed with 4% PFA and analyzed. The final infarct size was calculated by Image J Software (National Institutes of Health).
Masson's Trichrome Staining
Heart samples were fixed in 4% PFA and then embedded in paraffin. Five μm-thick sections were subjected to Masson's trichrome staining following a standard procedure. Images of the left ventricular area of each section were taken by Nikon model (200x magnification) with Spot Insight camera. Image J Software (National Institutes of Health) was used to quantify fibrotic region in each section. The percentage of fibrosis was measured as fibrosis areas/total left ventricular areas x 100%.
Collagen content assay
A quantitative dye-binding method was used to determine the collagen content. Analysis of heart tissues was performed using Sircol assay (Biocolor, Carrickfergus, UK) according to manufacturer's instructions. In this assay, each heart sample was weighed and homogenized with pepsin. The BioTek Software (Hercules, CA, USA) was used to quantify collagen content in each sample.
Statistical Analysis
Data were presented as mean ± SE. A Student's t-test, Chi-squares test or one-way ANOVA followed by Bonferroni's post-hoc test was used to compare the one-way layout data when appropriate. P values less than 0.05 were considered to be statistically different. All analyses were performed using GraphPad Prism 5.
Results
miR-433 is Increased in Cardiac Fibrosis
miRNA arrays were used to determine aberrant expressions of miRNAs, which might contribute to cardiac fibrosis in the post-MI ventricle at a time point notable for prominent fibrosis. A total of 26 miRNAs were found to be dysregulated (Fold change >2.0; P<0.05; Figure 1A and Supplemental Table 2). Interestingly, the top 3 dysregulated miRNAs including miR-34b-3p, 34c-5p, and 34c-3p belong to the miR-34 family, whose inhibition has been shown to attenuate pathological cardiac remodeling [13]. Since miR-433 (number fourth) has previously been reported to participate in kidney and liver fibrosis [21, 22] but has not so far been explored in the myocardium and during cardiac fibrosis, we explored its function further.
miR-433 is increased in cardiac fibrosis. A, dysregulated miRNAs in hearts from 21 days post-myocardial infarction (MI) versus sham control mice (n=4); B, upregulated miR-433 in ventricle samples from 21 days post-MI mice (n=4), a rodent model of doxorubicin (Dox)-induced cardiomyopathy (n=6), and human dilated cardiomyopathy (n=4); C-D, increased miR-433 in two in vitro cardiac fibrosis model induced either by TGF-β or Angiotensin II (n=6); E, expression of miR-433 in neonatal cardiac fibroblasts (NRCF) compared to cardiomyocytes (NRCM) (n=6); F, markers for pathological hypertrophy (ANP, BNP and Myh7) and extracellular matrix proteins (CTGF, TSP-1, Col1a1 and Col3a1) in cardiomyocytes with miR-433 overexpression (n=6). Scale bar: 50 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
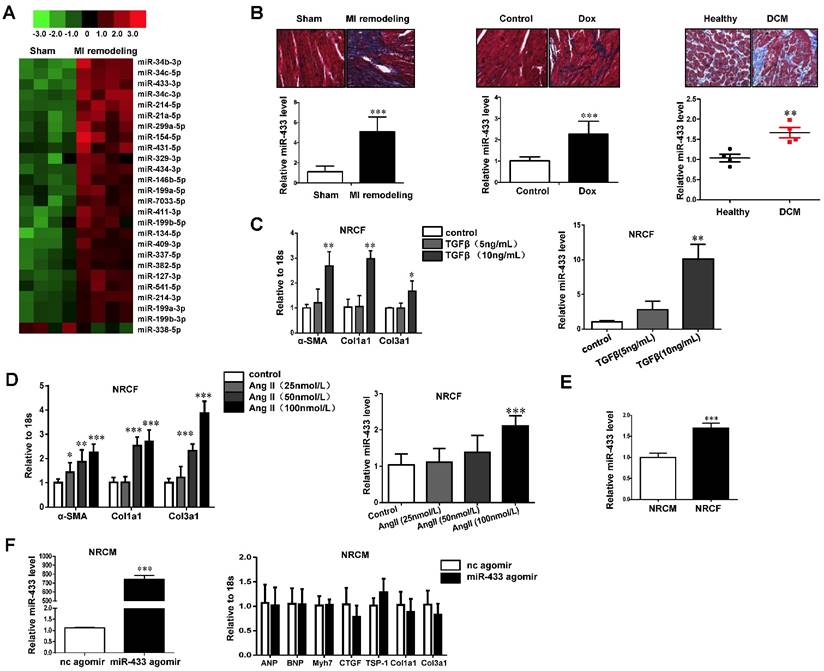
Based on the qRT-PCR analysis, we confirmed that miR-433 was upregulated in heart samples with fibrosis from mice 3 weeks post-MI (Figure 1B). To exclude the possibility that increased miR-433 is specific to cardiac fibrosis post-MI, we also determined its expression in doxorubicin-induced cardiomyopathy rodent model and in human dilated cardiomyopathy (DCM) (Figure 1B). The clinical information and echocardiography parameters for DCM patients are presented in Supplemental Table 3. The DCM sample size is small due to the difficulty of acquiring human heart tissues. Interestingly, miR-433 was consistently upregulated in all three models, i.e., in heart tissues with fibrosis, in doxorubicin-induced cardiomyopathy, and in patients with DCM (Figure 1B). Thus, there appeared to be a strong correlation between the presence of cardiac fibrosis and an increase in miR-433 expression in several different cardiac diseases. Furthermore, miR-433 was also increased in cultured neonatal rat cardiac fibrosis models stimulated by TGF-β or Ang II (Figure 1C-D). Taken together, these data supported a potential role for miR-433 in cardiac fibrosis.
In vivo Inhibition of miR-433 Preserves Cardiac Function and Prevents Fibrosis
Next, we determined the relative expression level of miR-433 in isolated neonatal rat cardiac fibroblasts versus cardiomyocytes, and demonstrated higher expression level in fibroblasts compared to cardiomyocytes (Figure 1E). Forced expression of miR-433 in cardiomyocytes did not lead to an elevation of markers for pathological hypertrophy (ANP, BNP, and Myh7) or extracellular matrix proteins (CTGF, TSP-1, Col1a1 and Col3a1) (Figure 1F) supporting a more prominent role for miR-433 in fibroblasts rather than cardiomyocytes.
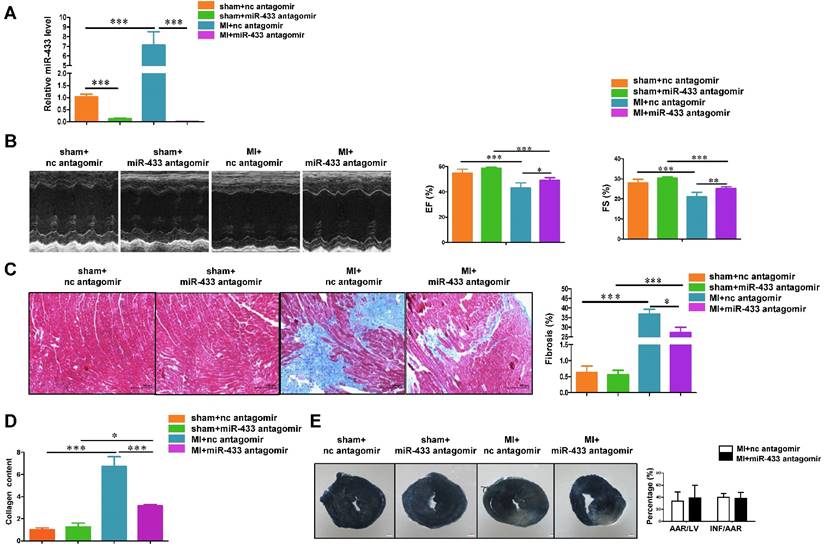
To evaluate the effect of miR-433 inhibition on cardiac fibrosis, we administrated miR-433 antagomir in mice via tail vein to downregulate miR-433 in vivo. First, the pharmacokinetic analysis for miR-433 antagomir was performed by measuring miR-433 expression level in both plasma and heart samples at different time points after mice were administrated with a single bolus of miR-433 antagomir at the dose of 7.5 mg/kg as previously reported [27]. The pharmacokinetic analysis showed that miR-433 was significantly downregulated in plasma and heart samples at 10 min post injection maintaining the low expression level thereafter (Supplemental Figure 1). Next, to explore whether antagonizing miR-433 attenuates cardiac fibrosis and preserves ventricular function post-MI, we treated mice with miR-433 antagomir or scrambled negative control via tail vein injection for 3 consecutive days and subjected them to MI or sham surgery. Then mice were sacrificed 3 weeks after MI and the loss of miR-433 in the heart was confirmed by qRT-PCRs (Figure 2A). Echocardiography showed that miR-433 antagomir preserved cardiac function including FS and EF (Figure 2B), and also reversed MI-induced increase in systolic left ventricle internal diameter (LVID;s) and diastolic left ventricle internal diameter (LVID;d) as shown in Supplemental Table 4. Importantly, inhibition of miR-433 also attenuated cardiac fibrosis as evidenced by reduced collagen deposition and content in MI heart tissues (Figure 2C-D). In particular, we evaluated the effect of miR-433 inhibition on cardiac infarction 3 days after MI; the purpose was to determine whether miR-433 inhibition predominantly protects against cardiac fibrosis in the remodeling phase after MI or prevents cardiac infarction in the acute phase after MI. Based on TTC staining, there was no difference in the infarct size between mice treated with miR-433 antagomir or negative control, strongly suggesting that miR-433 inhibition predominantly protects against cardiac fibrosis in the remodeling phase after MI (Figure 2E).
Antagonizing miR-433 attenuates cardiac fibrosis and preserves ventricular function post-myocardial infarction. A, decreased miR-433 in hearts from mice treated with miR-433 antagomir (n=6); B, preserved left ventricular fractional shortening (FS) and ejection fraction (EF); C, reduced cardiac fibrosis; D, decreased collagen content in myocardial infarction (MI) with miR-433 inhibition, as evidenced by echocardiography (n=6), Masson's trichrome staining (n=4), and Sircol assay (n=4); E, no difference in the infarct size between mice treated with miR-433 antagomir or negative control 3 days post-MI (n=7). Scale bar: 100 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
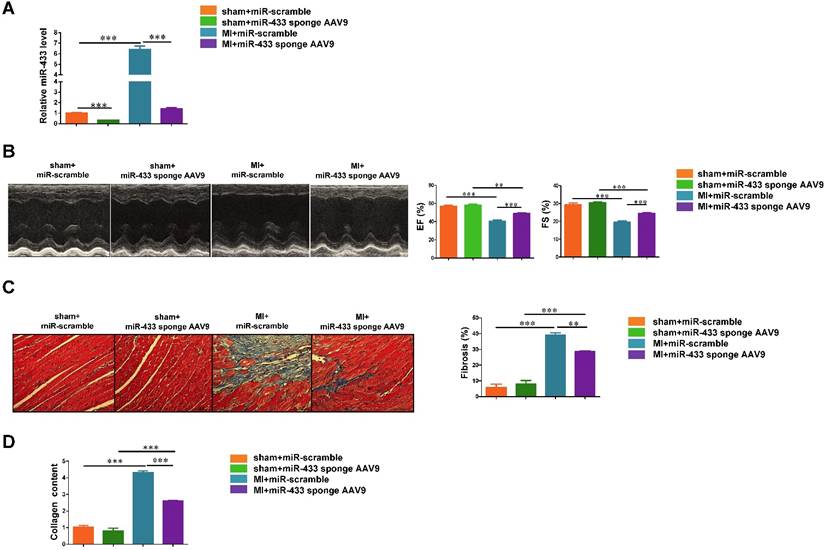
To further confirm the effect of miR-433 inhibition in preventing cardiac fibrosis, we used a cardiotropic AAV9 delivery system to achieve cardiac inhibition of miR-433 in vivo. Mice received a single-bolus tail vein injection of either miR-433 sponge AAV9 or miR-scramble. After 1 week, mice were subjected to LAD ligation and sacrificed at 3 weeks post-MI. Using qRT-PCR, we confirmed that miR-433 sponge AAV9 efficiently reduced miR-433 expression level in heart tissues (Figure 3A). Furthermore, our data showed that AAV9-mediated inhibition of miR-433 could significantly preserve left ventricular EF and FS (Figure 3B), and reduce increased systolic LVID and diastolic LVID in mice 3 weeks post-MI (Supplemental Table 5). Cardiac inhibition of miR-433 also reduced collagen deposition and collagen content in hearts post-MI (Figure 3C-D). These data provide strong evidence that inhibition of miR-433 has cardioprotective effect against fibrosis.
Cardiac inhibition of miR-433 via AAV9 attenuates cardiac fibrosis and preserves ventricular function post-myocardial infarction. A, decreased miR-433 in hearts from mice treated with miR-433 sponge AAV9 (n=6); B, preserved left ventricular fractional shortening (FS) and ejection fraction (EF); C, reduced cardiac fibrosis; D, decreased collagen content in myocardial infarction (MI) interfered with miR-433 sponge AAV9, as evidenced by echocardiography (n=6), Masson's trichrome staining (n=4), and Sircol assay (n=4). Scale bar: 100 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
Inhibition of miR-433 Attenuates Cardiac Fibroblast Proliferation and Myofibroblast Differentiation In Vivo and In Vitro
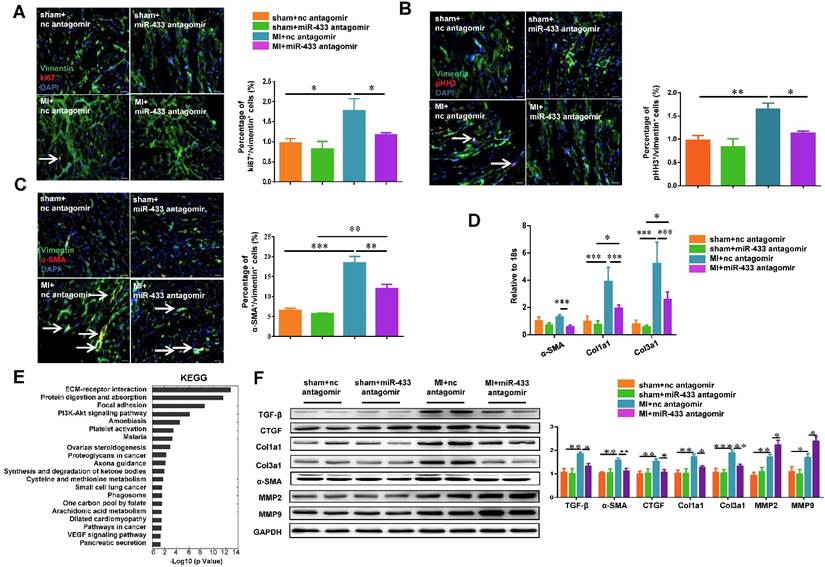
The transformation of fibroblasts into myofibroblasts is a critical event in the genesis of cardiac fibrosis [28, 29]. We determined the effects of miR-433 inhibition on cardiac fibroblasts proliferation and their differentiation into myofibroblasts in both post-MI mice and cultured cardiac fibroblasts. Based on the heart samples from in vivo experiments, immunofluorescence analysis revealed that antagonizing miR-433 decreased cardiac fibroblast proliferation as evidenced by reduced Ki-67/Vimentin or phospho-HistoneH3 (pHH3)/Vimentin double positive cells (Figure 4A-B). Furthermore, miR-433 inhibition also attenuated the differentiation of cardiac fibroblasts into myofibroblasts as shown by decreased number of cells double-positive for α-SMA and Vimentin (Figure 4C). Consistent with this, the expression levels of α-SMA, Col1a1, and Col3a1 in the ventricle following MI were also attenuated by miR-433 inhibition (Figure 4D). Agilent gene arrays were used to compare the difference of gene expressions between ventricle samples from miR-433 antagomir or scrambled negative control post-MI (Supplemental Tables 6-7). The KEGG pathway analysis based on dysregulated genes showed that extracellular matrix (ECM) receptor interaction was the most affected pathway (Figure 4E). Also, the protein levels of pro-fibrotic genes (TGF-β, α-SMA, CTGF, Col1a1, and Col3a1) were decreased, while genes responsible for collagen degradation (MMP2 and MMP9) were further increased by miR-433 inhibition in post-MI hearts (Figure 4F). Similar results were obtained for fibrosis-associated genes in miR-433 sponge AAV9-treated MI mice (Supplemental Figure 2).
Antagonizing miR-433 attenuates cardiac fibroblasts proliferation and their differentiation into myofibroblasts in vivo. A-B, decreased cardiac fibroblasts proliferation; C, reduced differentiation into myofibroblasts in myocardial infarction (MI) with miR-433 inhibition, as determined by immunofluorescent staining for Vimentin and Ki-67 or pHH3 or α-SMA (n=4); D, decreased α-SMA, Col1a1, and Col3a1 in MI mice with miR-433 inhibition (n=4); E, Agilent gene arrays and KEGG pathway analysis identified extracellular matrix (ECM) receptor interaction as the most affected pathway in MI hearts with miR-433 inhibition (n=4); F, decreased TGF-β, CTGF, Col1a1, Col3a1 and α-SMA and increased MMP2 and MMP9 after treatment with miR-433 antagomir in MI mice (n=4). Scale bar: 20 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
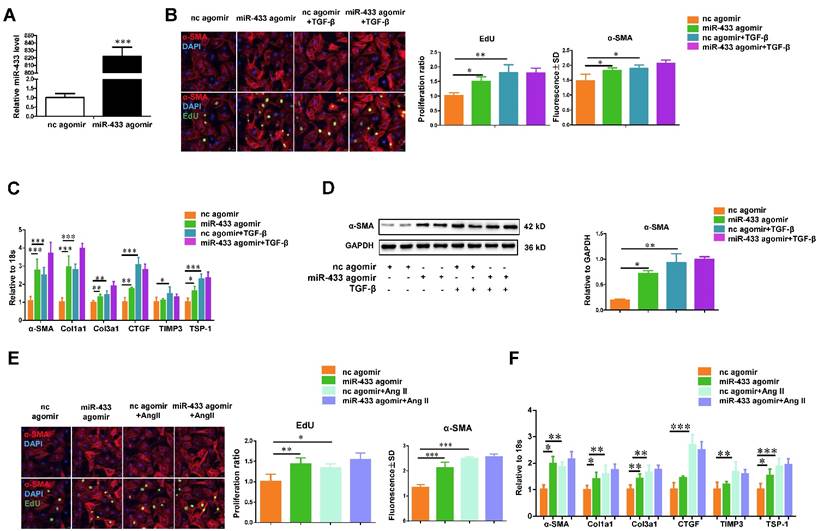
To gain mechanistic insight into the role of miR-433 in regulating fibrosis, we investigated the effect of miR-433 overexpression in cardiac fibroblasts in vitro. miR-433 overexpression promoted proliferation and differentiation of cardiac fibroblasts, as evidenced by an increase in EdU and α-SMA staining and increased expression levels of α-SMA, Col1a1, Col3a1, CTGF, and TSP-1 (Figure 5). However, up-regulation of miR-433 failed to further enhance cardiac fibroblasts proliferation and differentiation in the presence of either TGF-β or Ang II stimulation (Figure 5). Contrary to the effects of miR-433 overexpression, inhibition of miR-433 decreased cardiac fibroblasts proliferation and differentiation (Figure 6). Collectively, these data indicate that inhibition of miR-433 attenuates proliferation of cardiac fibroblasts and their differentiation into myofibroblasts both in vitro and in vivo.
miR-433 promotes cardiac fibroblasts proliferation and their differentiation into myofibroblasts in vitro.A, increased miR-433 in cardiac fibroblasts treated with miR-433 agomir (n=6); B, enhanced cardiac fibroblasts proliferation and their differentiation into myofibroblasts (n=4); C-D, upregulated fibrosis-related genes in cardiac fibroblasts with miR-433 overexpression in the absence of TGF-β stimulation, as evidenced by EdU/α-SMA staining (n=4), qRT-PCR (n=6), and Western blot analysis (n=4); E-F, upregulated fibrosis-related genes in cardiac fibroblasts with miR-433 overexpression in the absence of Angiotensin II stimulation. Scale bar: 50 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
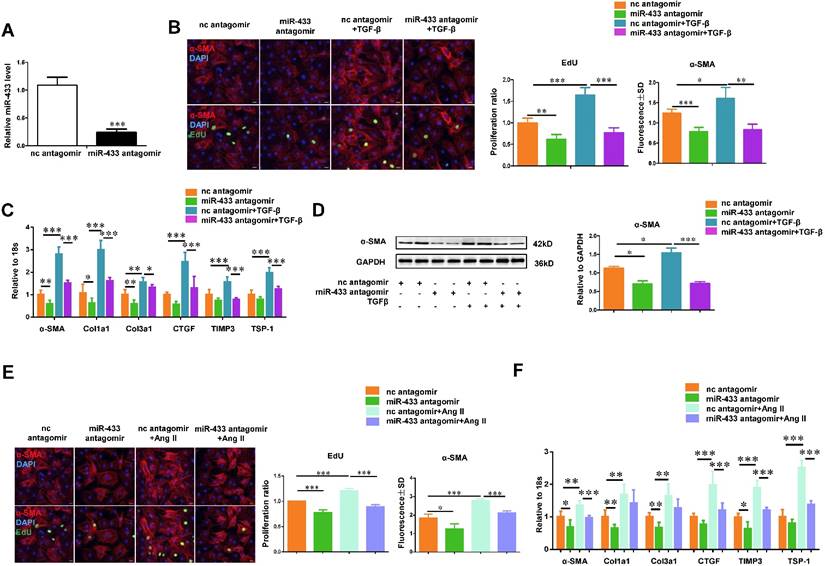
Inhibition of miR-433 attenuates TGF-β/Ang II-induced cardiac fibroblasts proliferation and their differentiation into myofibroblasts in vitro. A, decreased miR-433 in cardiac fibroblasts treated with miR-433 antagomir (n=6); B, reduced cardiac fibroblasts proliferation and their differentiation into myofibroblasts (n=4); C-D, downregulated fibrosis-related genes in cardiac fibroblasts with miR-433 inhibition regardless of TGF-β stimulation, as evidenced by EdU/α-SMA staining (n=4), qRT-PCR (n=6), and Western blot analysis (n=4); E-F, downregulated fibrosis-related genes in cardiac fibroblasts with miR-433 inhibition regardless of Angiotensin II stimulation. Scale bar: 50 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
AZIN1 and JNK1 are Identified as Two Target Genes of miR-433
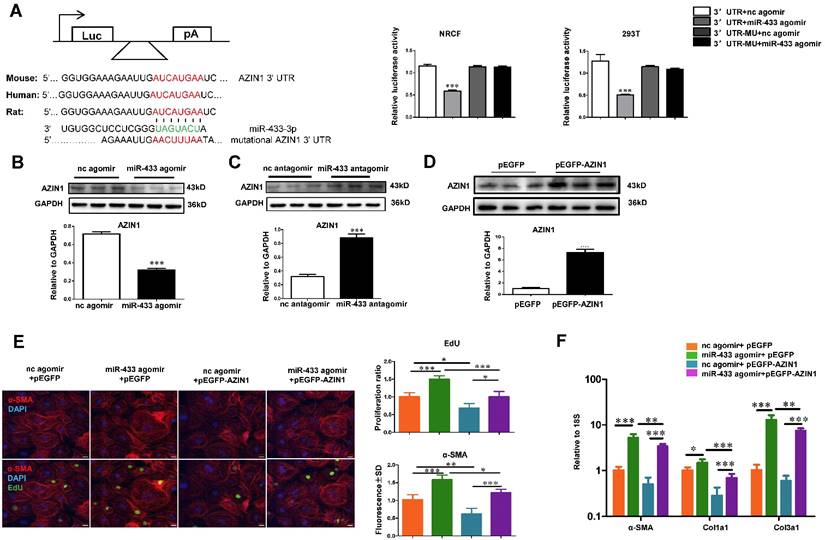
AZIN1 is reported to be a target gene of miR-433 in renal fibrosis [21]. However, its role in cardiac fibroblasts is not known. We first performed luciferase reporter assays to confirm that miR-433 could directly target the 3'UTR of AZIN1 in both 293T cells and cardiac fibroblasts (Figure 7A). Next, we investigated whether AZIN1 could potentially mediate the effects of miR-433 in cardiac fibrosis. In cardiac fibroblasts, the expression level of AZIN1 was decreased by miR-433 agomir but increased by miR-433 antagomir as determined by Western blotting (Figure 7B-C), indicating that miR-433 could regulate endogenous AZIN1 expression levels. We next used AZIN1 overexpression plasmid to determine AZIN1's role in mediating the effect of miR-433 on cardiac fibroblasts proliferation and differentiation into myofibroblasts. Our data illustrated that overexpression of AZIN1 could attenuate the pro-fibrotic effect of miR-433 agomir on cardiac fibroblasts (Figure. 7D-F). Also, AZIN1 knockdown via siRNA failed to have an additive effect on fibroblast proliferation and myofibroblast differentiation in cells co-treated with miR-433 agomir (Supplemental Figure 3). These data strongly suggest that AZIN1 is a target gene of miR-433 mediating its effect in cardiac fibrosis.
AZIN1 is a target gene of miR-433 in cardiac fibroblasts. A, Targetscan and Luciferase reporter assays identified AZIN1 as a direct target gene of miR-433 (n=6); B-C, AZIN1 was negatively regulated by miR-433 in cardiac fibroblasts (n=3); D-F, Overexpression of AZIN1 via pEGFP plasmid attenuated the pro-fibrotic effect of miR-433 agomir on cardiac fibroblasts proliferation and their differentiation into myofibroblasts. n=3 for Western blot, n=4 for EdU and α-SMA staining, n=6 for qRT-PCR. Scale bar: 50 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
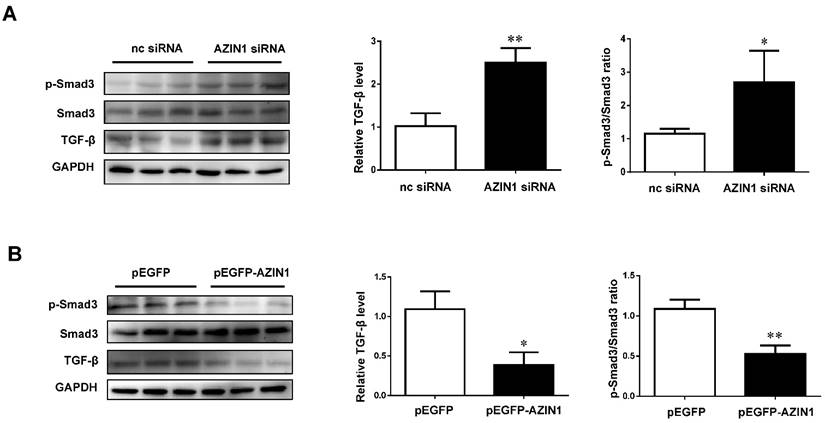
As AZIN1 was previously reported to be linked to TGF-β signaling in both kidney and liver fibrosis [21, 22], we further examined the modulatory effect of AZIN1 on TGF-β and its downstream effector Smad3. Our data revealed that knockdown of AZIN1 could upregulate TGF-β expression and activate Smad3 phosphorylation, while overexpressing AZIN1 had an opposite effect (Figure 8), indicating a potential relationship between AZIN1 and TGF-β/Smad3 signaling in the regulation of cardiac fibrosis.
AZIN1 inactivates TGF-β/Smad3 signaling in cardiac fibroblasts. A, Knockdown of AZIN1 could upregulate TGF-β expression and activate Smad3 phosphorylation (n=3); B, Overexpression of AZIN1 had opposite effects (n=3). *, P<0.05, **, P<0.01, ***, P<0.001 versus respective control.
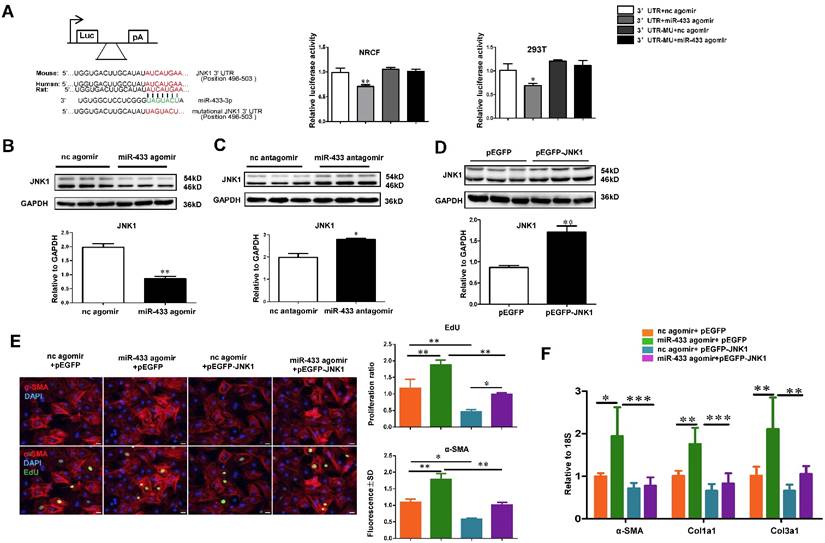
Besides AZIN1, bioinformatic analysis using Targetscan indicated that JNK1 might be an additional potential target gene of miR-433 (Figure 9A). Luciferase reporter assays further confirmed that miR-433 led to a reduction in luciferase activity for the wild-type 3'UTR construct for JNK1, but had no effect when the miR-433 binding site in the JNK1 3'UTR was mutated, implying that JNK1 is a direct target of miR-433 (Figure 9A). To check if miR-433 could regulate endogenous JNK1 expression in cardiac fibroblasts, miR-433 agomir, antagomir, or their negative controls were transfected into cardiac fibroblasts. As determined by Western blotting, miR-433 agomir decreased, while miR-433 antagomir increased JNK1 expression (Figure 9B-C), confirming that miR-433 could regulate endogenous JNK1 expression levels in cardiac fibroblasts. In addition, we used JNK1 overexpression plasmid to determine its role in the miR-433-mediated cardiac fibroblasts proliferation and differentiation into myofibroblasts. Our results clearly indicated that overexpression of JNK1 could attenuate the pro-fibrotic effect of miR-433 agomir on cardiac fibroblasts (Figure 9D-F). JNK1 knockdown via siRNA, on the other hand, did not further increase fibroblast proliferation, though myofibroblast differentiation was slightly enhanced in cells co-treated with miR-433 agomir (Supplemental Figure 4). These data identify JNK1 as a novel target gene of miR-433 contributing to cardiac fibroblast proliferation and myofibroblast differentiation.
JNK1 is a novel target gene of miR-433 in cardiac fibroblasts. A, Targetscan and Luciferase reporter assays identified JNK1 as a direct target gene of miR-433 (n=6); B-C, JNK1 was negatively regulated by miR-433 in cardiac fibroblasts (n=3); D-F, Overexpression of JNK1 via pEGFP plasmid attenuated the pro-fibrotic effect of miR-433 agomir on cardiac fibroblasts proliferation and their differentiation into myofibroblasts. n=3 for Western blot, n=4 for EdU and α-SMA staining, n=6 for qRT-PCR. Scale bar: 50 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
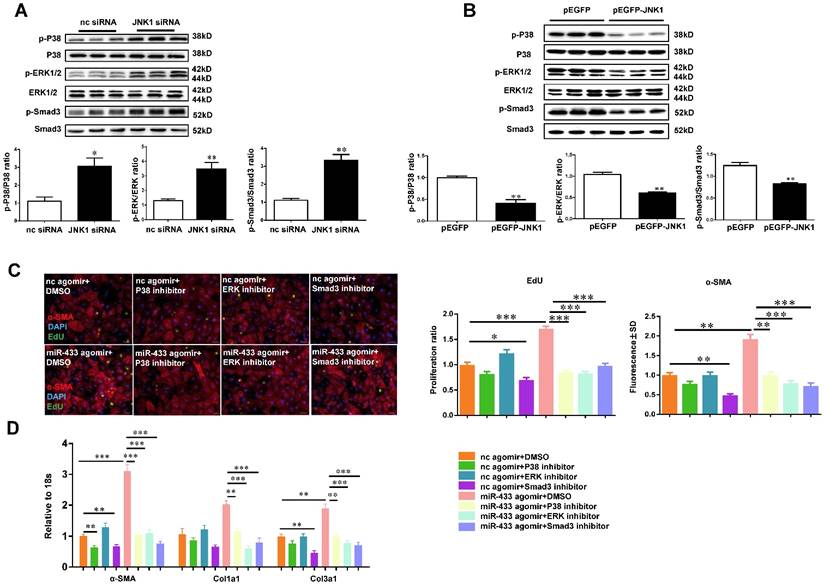
As a member of mitogen-activated protein kinase (MAPK) family, JNK1 may have functional cross-talk with two other members of MAPK family, namely ERK and p38 kinase [30, 31]. To confirm this, JNK1 was knocked down by siRNA and the expression levels of ERK and p38 kinase were determined by Western blotting. We observed that JNK1 knockdown significantly activated ERK and p38 kinase as evidenced by increased ratios of p-ERK/ERK and p-p38/p38, paralleling with the activation of Smad3 (Figure 10A). However, the introduction of the JNK1 overexpression plasmid resulted in reduced phosphorylation levels of ERK1/2, p38, and Smad3 (Figure 10B). Interestingly, inhibition of p38, ERK or Smad3 could block the positive effects of miR-433 agomir on cardiac fibroblasts proliferation and differentiation, as determined by α-SMA and EdU staining, and reduce the expression levels of α-SMA, Col1a1, and Col3a1 (Figure 10 C-D).
JNK1 knockdown significantly activates ERK and p38 kinase. A, JNK1 knockdown via siRNA increased ERK, p38, and Smad3 phosphorylation (n=3); B, Overexpression of JNK1 via pEGFP plasmid had opposite effects (n=3); C-D, reduced cardiac fibroblasts proliferation and their differentiation into myofibroblasts in cells treated with miR-433 agomir and inhibitor of p38, ERK, or Smad3. n=4 for EdU and α-SMA staining, n=6 for qRT-PCR. Scale bar: 50 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
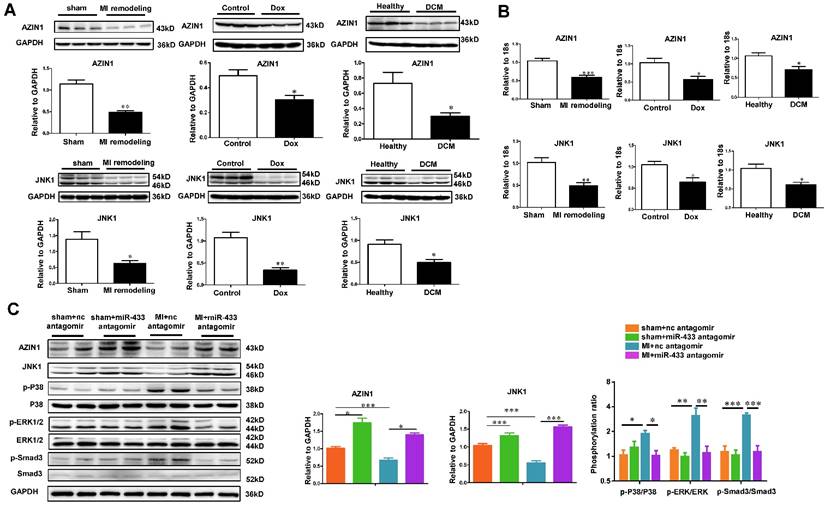
To examine whether AZIN1 and JNK1 could be regulated during cardiac fibrosis and/or miR-433 inhibition, we first examined their expression levels in vivo in the heart samples from post-MI rodent model, doxorubicin-induced cardiomyopathy rodent model, and human dilated cardiomyopathy. Consistently, both AZIN1 and JNK1 were down-regulated in these three fibrotic conditions at both protein and mRNA levels (Figure 11A-B). We next tested whether AZIN1 and JNK1 were increased in ventricle samples of miR-433 antagomir-injected mice. As determined by gene arrays, we did not detect changes of AZIN1 and JNK1 at the mRNA level. However, considering the fact that miRNAs regulate their target genes mostly at posttranscriptional levels, we also determined AZIN1 and JNK1 protein levels by Western blotting. Our results clearly showed that treatment with miR-433 antagomir increased the expression of AZIN1 and JNK1 in the presence or absence of MI (Figure 11C). These data are consistent with the hypothesis that one or both genes are target genes of miR-433 in vivo. Interestingly, during MI, miR-433 antagomir inactivated ERK and p38 kinase as evidenced by the decreased ratio of p-ERK/ERK and p-p38/p38, together with Smad3 (Figure 11C).
AZIN1 and JNK1 are downregulated by miR-433 antagomir in vivo. A-B, downregulated AZIN1 and JNK1 in ventricular samples from 21 day post-myocardial infarction (MI) mice, rodent model of doxorubicin (Dox)-induced cardiomyopathy, and human dilated cardiomyopathy (DCM). For Western blot, n=3. For qRT-PCR, n=3 for mice and n=4 for patients; C, Upregulated AZIN1 and JNK1, accompanied by an inactivation of ERK, p38, and Smad3 phosphorylation in MI hearts with miR-433 inhibition (n=4). *, P<0.05, **, P<0.01, ***, P<0.001 versus respective controls.
Discussion
Myocardial fibrosis is a common hallmark in a variety of cardiomyopathies [4]. Consequently, anti-fibrotic therapies are increasingly considered as an extremely promising approach for the treatment of heart failure [3]. Unfortunately, effective strategies to attenuate cardiac fibrosis are not available [2]. Aberrant expression of various miRNAs has been shown to play a crucial role in cardiac fibrosis and heart failure [1, 32]. These small non-coding miRNAs with conserved sequences have become promising therapeutic candidates from a drug development standpoint [7]. Recently, manipulating miRNAs for developing anti-fibrotic therapies has emerged as a novel treatment strategy for fibrotic changes [1, 33].
According to the miRBase 21 release, 1881 miRNAs have been identified in humans. Numerous studies have demonstrated the involvement of many of these miRNAs in vital cellular processes. However, the role of miRNAs in the heart and especially for cardiac fibrosis is unclear. It has been suggested that dysregulated miRNAs such as miR-21 and miR-29b, contribute to cardiac fibrosis [16, 18]. Using miRNA arrays, we identified elevated levels of miR-433 in post-MI cardiac fibrosis. The same modulation was also observed in other cardiac pathologies, including doxorubicin-induced cardiomyopathy in a rodent model, and in a limited number of human DCM samples, indicating that upregulation of miR-433 might be a common feature of adverse cardiac remodeling. Besides cardiac pathologies, miR-433 has been reported to be downregulated in human gastric carcinoma. Ectopic expression of miR-433 in the gastric cancer cell line HGC-27 could inhibit cellular proliferation, migration, invasion, and cell cycle progression [34]. miR-433 also inhibits liver cancer cell migration and oral squamous cell carcinoma (OSCC) cell growth and metastasis [35, 36], indicating that miR-433 acts as a tumor suppressor. In other studies, miR-433 has been shown to promote renal fibrosis and also TGF-β-dependent fibrogenesis in liver and kidney [21, 22]. In another report, miR-433 has been described to promote resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells [37]. These data point to the complex tissue- and cell-based specific roles of miR-433 in various cancers.
The role of miR-433 in the heart and during cardiac fibrosis had not been investigated previously. The proliferation and transformation of cardiac fibroblasts into myofibroblasts are key events for cardiac fibrosis [29]. Fibroblast proliferation and myofibroblast differentiation can be differentially regulated by growth factors such as TGF-β, EGF, PDGF, CTGF, and IGF [38]. Herein, we demonstrated that miR-433 over-expression enhanced both cardiac fibroblast proliferation and their differentiation into myofibroblasts, whereas inhibition of miR-433 attenuated these processes, indicating the critical stimulatory effect of miR-433 on cardiac fibroblast activation. We also observed that miR-433 was enriched in cardiac fibroblasts compared to cardiomyocytes. Furthermore, overexpression of miR-433 in cardiomyocytes does not appear to play a role in cardiomyocyte biology, as seen by the lack of effect on markers for pathological hypertrophy and extracellular matrix proteins.
A previously reported target gene of miR-433 in renal fibrosis, AZIN1, has been linked to TGF-β signaling in both kidney and liver fibrosis [21, 22]. It is an ornithine decarboxylase (ODC) homolog that binds to antizyme with a higher affinity [21, 22]. Suppression of AZIN1 expression results in antizyme repression followed by a decline of polyamine levels and consequent activation of the TGF-β signaling pathway to promote fibrosis [21, 22]. To date, very little information is available on the role of AZIN1 in cardiac pathologies. In this study, AZIN1 appeared to be responsible for the effects of miR-433 in cardiac fibroblasts. It was downregulated in the heart tissues from post-MI mice, doxorubicin-induced cardiomyopathy rodent model, and human dilated cardiomyopathy, indicating its potential role in the diseased myocardium with fibrosis. Furthermore, knockdown of AZIN1 could promote proliferation and differentiation of cardiac fibroblasts into myofibroblasts accompanied with an activation of TGF-β/Smad3 signaling pathway. However, the direct relationship between AZIN1 and TGF-β1 and their functional roles in the regulation of cardiac fibrosis needs to be further clarified through the function-rescue assay. Taken together, these results suggest that AZIN1 is a target gene of miR-433 in cardiac fibrosis and also provide evidence for the functional role of AZIN1 in the heart that needs to be explored in the future.
Besides AZIN1, based on bioinformatic analysis and experimental validation, JNK1 was identified as a novel target gene of miR-433 in cardiac fibroblasts. Jun NH2-terminal kinases, including three isoforms (JNK1, JNK2, and JNK3), belong to the MAPK family and play major roles in development, cell proliferation, differentiation, and apoptosis [31, 39]. JNK1 and JNK2 are abundant in myocardium while JNK3 is most abundant in the brain [31, 39]. In this study, luciferase assays demonstrated that JNK1 was a direct target of miR-433 and Western blot analysis confirmed that miR-433 could endogenously regulate JNK1 expression in cardiac fibroblasts. Functional studies in cardiac fibroblasts further indicated that reduction of JNK1 was responsible for the pro-fibrotic effects of miR-433 in cardiac fibroblasts. Furthermore, JNK1 may also have a functional cross-talk with ERK and p38 kinase, two other members of MAPK family [30]. ERK and p38 kinase pathways were activated while JNK1 was inhibited in the heart samples from post-MI mice, doxorubicin-induced cardiomyopathy rodent model, and human dilated cardiomyopathy. Also, reduction of JNK1 in cardiac fibroblasts activated ERK and p38 kinase and inhibition of ERK and p38 kinase attenuated the biological effects of miR-433 agomir on the proliferation and differentiation of cardiac fibroblasts. Collectively, these results suggest that miR-433 downregulates JNK1 and subsequently activates ERK and p38 kinase promoting cardiac fibrosis.
The protective effects of miR-433 inhibition against cardiac fibrosis were confirmed by antagonizing miR-433 or inhibiting miR-433 via cardiotropic AAV9, which attenuated cardiac fibrosis and preserved ventricular function post-MI. Although several lines of evidence presented here strongly supports the functional role of miR-433 in regulating cardiac fibrosis, more rigorous approaches are required to support this contention. These may include intra-myocardial rather than systemic delivery with a cardiac fibroblast-specific promoter and/or using a miR-433 transgenic mouse model created by using the cardiac fibroblast-specific promoter.
It is of note that cardiac fibrosis was decreased with the miR-433 antagomir but not abolished indicating the involvement of other pathways. For example, some clustered miRNAs of miR-433 including miR-431, miR-434 and miR-127 were also elevated in our initial miRNA array based on fibrotic heart samples post-MI. These miRNAs might work coordinately to promote cardiac fibrosis. It would also be interesting to further determine in vivo therapeutic roles for each of miR-433 targets, alone or in combination, by gain-of-function and loss-of-function studies. Furthermore, the therapeutic effects of miR-433 reduction on cardiac fibrosis in an established model need to be determined in the future. Last but not least, as cardiac fibrosis in the acute phase post-MI may protect the ischemic heart from structural rupture [40], the effect as well as the safety of miR-433 inhibition in the treatment of cardiac fibrosis must be carefully evaluated during the early phase post-MI. Notably, the data from the present study demonstrated that antagonizing miR-433 in vivo did not impact the infarct size 3 days after MI surgery suggesting that inhibition of miR-433 does not affect infarct size during the early phase post-MI.
In summary, our study has shown that miR-433 is induced by cardiac fibrosis, subsequently reducing the expression of AZIN1 and JNK1. Decreased AZIN1 activates TGF-β1 pathway while down-regulated JNK1 leads to activation of ERK and p38 kinase stimulating Smad3 and ultimately leading to cardiac fibrosis
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (81570362 and 81200169 to JJ Xiao, 81370332 and 81170201 to XL Li, 81270314 and 81470515 to JH Xu, 81472158 to L Che, 81400647 to YH Bei, 81370362 to JC Zhong), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD20102013 to XL Li), the National Basic Research Program of China (2014CB542300), the National Major Research Plan Training Program (91339108), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant (20152509), Shanghai Medical Guide Project from Shanghai Science and Technology Committee (134119a3000 to JH Xu), Natural Science Foundation of Shanghai (14ZR1437900 to L. Che), the Netherlands Cardiovascular Research Initiative (CVON): the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science (to JPG Sluijter) and the National Institutes of Health (NCATS Grant UH3 TR000901 to S Das). Dr XL Li is an Associate Fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine.
Author Contributions
J.X. designed the study, instructed all experiments and drafted the manuscript. X.L. participated in the design of the study and coordination of the whole work. L.T., Y.B., P.C., Z.L., S.F., H.Z., J.X. and L.C. performed the experiments and analyzed the data. X.C., X.B., J.Z., J.PG.S., S.D. helped to perform the experiments, provided technical assistance and revised the manuscript.
Supplementary Material
Supplementary tables and figures.
Competing Interests
The authors declare no competing financial interests.
References
1. Thum T. Noncoding rnas and myocardial fibrosis. Nat Rev Cardioy. 2014;11:655-63
2. Leask A. Getting to the heart of the matter: New insights into cardiac fibrosis. Cir Res. 2015;116:1269-76
3. Segura AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev. 2014;19:173-85
4. Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: Ultimate and proximate causes. J Clin Invest. 2014;124:4673-7
5. Thum T, Lorenzen JM. Cardiac fibrosis revisited by microrna therapeutics. Circulation. 2012;126:800-2
6. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR. et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993-1004
7. Olson EN. Micrornas as therapeutic targets and biomarkers of cardiovascular disease. Sci Transl Med. 2014;6:239ps3
8. Quiat D, Olson EN. Micrornas in cardiovascular disease: From pathogenesis to prevention and treatment. J Clin Invest. 2013;123:11-8
9. Mendell JT, Olson EN. Micrornas in stress signaling and human disease. Cell. 2012;148:1172-87
10. Piccoli MT, Gupta SK, Thum T. Noncoding rnas as regulators of cardiomyocyte proliferation and death. J Mol Cell Cardiol. 2015;89:59-67
11. Tao H, Yang JJ, Shi KH. Non-coding rnas as direct and indirect modulators of epigenetic mechanism regulation of cardiac fibrosis. Expert Opin Ther Targets. 2015;19:707-16
12. Kumarswamy R, Thum T. Non-coding rnas in cardiac remodeling and heart failure. Cir Res. 2013;113:676-89
13. Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H. et al. Therapeutic inhibition of the mir-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci U S A. 2012;109:17615-20
14. Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S. et al. Microrna-34a regulates cardiac ageing and function. Nature. 2013;495:107-10
15. Jazbutyte V, Fiedler J, Kneitz S, Galuppo P, Just A, Holzmann A. et al. Microrna-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr). 2013;35:747-62
16. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M. et al. Microrna-21 contributes to myocardial disease by stimulating map kinase signalling in fibroblasts. Nature. 2008;456:980-4
17. Wei C, Kim IK, Kumar S, Jayasinghe S, Hong N, Castoldi G. et al. Nf-kappab mediated mir-26a regulation in cardiac fibrosis. J Cell Physiol. 2013;228:1433-42
18. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. Mir-29b as a therapeutic agent for angiotensin ii-induced cardiac fibrosis by targeting tgf-beta/smad3 signaling. Mol Ther. 2014;22:974-85
19. Wang J, Huang W, Xu R, Nie Y, Cao X, Meng J. et al. Microrna-24 regulates cardiac fibrosis after myocardial infarction. J Cell Mol Med. 2012;16:2150-60
20. Tijsen AJ, van der Made I, van den Hoogenhof MM, Wijnen WJ, van Deel ED, de Groot NE. et al. The microrna-15 family inhibits the tgfbeta-pathway in the heart. Cardiovasc Res. 2014;104:61-71
21. Li R, Chung AC, Dong Y, Yang W, Zhong X, Lan HY. The microrna mir-433 promotes renal fibrosis by amplifying the tgf-beta/smad3-azin1 pathway. Kidney Int. 2013;84:1129-44
22. Espinosa-Diez C, Fierro-Fernández M, Sánchez-Gómez F, Rodríguez-Pascual F, Alique M, Ruiz-Ortega M. et al. Targeting of gamma-glutamyl-cysteine ligase by mir-433 reduces glutathione biosynthesis and promotes tgf-beta-dependent fibrogenesis. Antioxid Redox Signal. 2015;23:1092-105
23. Wahlquist C, Jeong D, Rojas-Muñoz A, Kho C, Lee A, Mitsuyama S. et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508:531-5
24. Aoyama Y, Kobayashi K, Morishita Y, Maeda K, Murohara T. Wnt11 gene therapy with adeno-associated virus 9 improves the survival of mice with myocarditis induced by coxsackievirus B3 through the suppression of the inflammatory reaction. J Mol Cell Cardiol. 2015;84:45-51
25. Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073-80
26. Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S. et al. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. 2013;2:e000078
27. Wang H, Chiu M, Xie Z, Chiu M, Liu Z, Chen P. et al. Synthetic microRNA cassette dosing: pharmacokinetics, tissue distribution and bioactivity. Mol Pharm. 2012;9:1638-44
28. Goldsmith EC, Bradshaw AD, Spinale FG. Cellular mechanisms of tissue fibrosis. 2. Contributory pathways leading to myocardial fibrosis: Moving beyond collagen expression. Am J Physiol Cell Physiol. 2013;304:C393-402
29. Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardioy. 2013;10:15-26
30. Peng T, Zhang T, Lu X, Feng Q. Jnk1/c-fos inhibits cardiomyocyte tnf-alpha expression via a negative crosstalk with erk and p38 mapk in endotoxaemia. Cardiovasc Res. 2009;81:733-41
31. Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507-46
32. Melman YF, Shah R, Das S. Micrornas in heart failure: Is the picture becoming less mirky? Cir Heart Fail. 2014;7:203-14
33. van Rooij E, Olson EN. Microrna therapeutics for cardiovascular disease: Opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860-72
34. Guo LH, Li H, Wang F, Yu J, He JS. The tumor suppressor roles of mir-433 and mir-127 in gastric cancer. Int J Mol Sci. 2013;14:14171-84
35. Yang Z, Tsuchiya H, Zhang Y, Hartnett ME, Wang L. Microrna-433 inhibits liver cancer cell migration by repressing the protein expression and function of camp response element-binding protein. J Biol Chem. 2013;288:28893-9
36. Tao L, Shen S, Fu S, Fang H, Wang X, Das S. et al. Traditional chinese medication qiliqiangxin attenuates cardiac remodeling after acute myocardial infarction in mice. Sci Rep. 2015;5:8374
37. Weiner-Gorzel K, Dempsey E, Milewska M, McGoldrick A, Toh V, Walsh A. et al. Overexpression of the microrna mir-433 promotes resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells. Cancer Med. 2015;4:745-58
38. Grotendorst GR, Rahmanie H, Duncan MR. Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB J. 2004;18:469-79
39. Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116:1413-23
40. Fan Z, Guan J. Antifibrotic therapies to control cardiac fibrosis. Biomater Res. 2016;20:13
Author contact
![]() Corresponding authors: Dr. Xinli Li Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, China; Tel: 0086-25-84352775; Fax: 0086-25-84352775; E-mail: xinli3267_njcom Or Dr. Junjie Xiao Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, 333 Nan Chen Road, Shanghai 200444, China; Tel: 0086-21-66138131; Fax: 0086-21-66138131; E-mail: junjiexiaoedu.cn.
Corresponding authors: Dr. Xinli Li Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, China; Tel: 0086-25-84352775; Fax: 0086-25-84352775; E-mail: xinli3267_njcom Or Dr. Junjie Xiao Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, 333 Nan Chen Road, Shanghai 200444, China; Tel: 0086-21-66138131; Fax: 0086-21-66138131; E-mail: junjiexiaoedu.cn.