Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(12):2114-2128. doi:10.7150/thno.16614 This issue Cite
Research Paper
Gelatin-based Hydrogel Degradation and Tissue Interaction in vivo: Insights from Multimodal Preclinical Imaging in Immunocompetent Nude Mice
Christoph Tondera1,2, Sandra Hauser1, Anne Krüger-Genge3, Friedrich Jung3,4, Axel T. Neffe3,4, Andreas Lendlein3,4, Robert Klopfleisch5, Jörg Steinbach2,6, Christin Neuber1, Jens Pietzsch1,2 ![]()
1. Helmholtz-Zentrum Dresden-Rossendorf, Institute of Radiopharmaceutical Cancer Research, Department of Radiopharmaceutical and Chemical Biology, Dresden, Germany;
2. Technische Universität Dresden, Department of Chemistry and Food Chemistry, Dresden, Germany;
3. Institute of Biomaterial Science and Berlin-Brandenburg Centre for Regenerative Therapies, Helmholtz-Zentrum Geesthacht, Teltow, Germany;
4. Helmholtz Virtual Institute “Multifunctional Biomaterials for Medicine”, Teltow and Berlin;
5. Freie Universität Berlin, Institute of Veterinary Pathology, Berlin, Germany;
6. Helmholtz-Zentrum Dresden-Rossendorf, Institute of Radiopharmaceutical Cancer Research, Dresden, Germany.
Received 2016-6-27; Accepted 2016-8-5; Published 2016-9-12
Abstract

Hydrogels based on gelatin have evolved as promising multifunctional biomaterials. Gelatin is crosslinked with lysine diisocyanate ethyl ester (LDI) and the molar ratio of gelatin and LDI in the starting material mixture determines elastic properties of the resulting hydrogel. In order to investigate the clinical potential of these biopolymers, hydrogels with different ratios of gelatin and diisocyanate (3-fold (G10_LNCO3) and 8-fold (G10_LNCO8) molar excess of isocyanate groups) were subcutaneously implanted in mice (uni- or bilateral implantation). Degradation and biomaterial-tissue-interaction were investigated in vivo (MRI, optical imaging, PET) and ex vivo (autoradiography, histology, serum analysis). Multimodal imaging revealed that the number of covalent net points correlates well with degradation time, which allows for targeted modification of hydrogels based on properties of the tissue to be replaced. Importantly, the degradation time was also dependent on the number of implants per animal. Despite local mechanisms of tissue remodeling no adverse tissue responses could be observed neither locally nor systemically. Finally, this preclinical investigation in immunocompetent mice clearly demonstrated a complete restoration of the original healthy tissue.
Keywords: Autoradiography ex vivo, Biomaterials, Computed tomography, Magnetic resonance imaging, Optical imaging, Positron emission tomography.
Introduction
In recent years, hydrogel-based biomaterials have evolved as highly versatile and promising tools for clinical [1] and research [2] applications. Different hydrogels are produced depending on the specific need, such as soft tissue replacement, organ patches, or drug delivery systems. Hydrogels with different physical properties can also be designed and implanted in non-self-healing critical size defects, temporarily replacing the extracellular matrix (ECM), and assisting the healing process for example in bony structures [3]. The material of choice should be degraded in rates suitable for the envisioned application, allowing optimal ingrowth of cells and supporting the wound healing process. Ideally, physical properties such as Young's and compression moduli of the material match those of the tissue to be replaced and help to guide cells to promote tissue regeneration. Tailoring the physicochemical properties of polymers at the start of their application as well as during their degradation may be reached by the formation of polymer networks. For this purpose, biopolymers derived from ECM are attractive materials, as they are degradable, offer sites for cell adhesion, and are generally highly biocompatible [4]. Current clinical applications for soft tissue replacement focus mainly on the use of collagen or differentially cross-linked collagen [5]. However, immunogenic responses to collagen-based implants have been reported [6]. In addition, cross-linking agents needed for network formation may show toxic side effects [7] during material degradation processes [8], which could induce adverse tissue reactions, such as strong inflammatory and immunogenic responses, thereby adversely affecting the healing process and successful tissue restoration [9,10].
Previous research has shown that gelatin, which is the partial thermally and chemically degraded product of collagen, possesses lower immunogenicity than collagen [11], and can be stabilized through the introduction of covalent net points [12]. Gelatin as highly biocompatible, biodegradable, low immunogenic, and low-cost material is therefore highly attractive for future clinical applications e.g. in soft tissue replacement strategies [13].
The use of gelatin-based hydrogels stabilized through reaction with lysine diisocyanate ethyl ester (LDI) [14] has already yielded promising in vitro results in the interaction with mesenchymal stem cells [15] and arterial endothelial cells [16]. First results from a pilot animal experiment using an immunocompetent nude mouse (SKH1) model further revealed a high degree of biocompatibility together with a specific degradation response of the cross-linked gelatin over 35 days. This has been demonstrated by histological (Masson Goldner staining) and immunohistochemical (cyclooxygenase-2 staining) examinations of sections from explants of hydrogels with surrounding tissue ex vivo [16]. For a better understanding of intra-individual physiological processes as well as potential long lasting and late effects, it is necessary to employ in vivo imaging techniques depicting the degradation of the implant, biomaterial-tissue-interactions, and systemic reactions in a quantitative manner, and to correlate these findings with ex vivo techniques showing local cellular behavior. Thereby, multimodal and multiscale techniques contribute to a better understanding of the biological response to the hydrogels and will provide selection criteria for potential therapeutic applications.
This comprehensive small animal multimodal and multiscale imaging study in SKH1 mice conducted in vivo and ex vivo aimed at detailed elucidation of hydrogel degradation, tissue response, integration, and systemic response at different time points (1, 7, 14, 21, 35 days) up to 84 days post subcutaneous implantation of two hydrogels with different numbers of covalent and physical net points.
Methods
Hydrogel preparation. Sterile, dried hydrogel implants of gelatin-based networks G10_LNCO3 and G10_LNCO8 were synthesized as described before [14,15], and quality control was achieved by controlling the rheological and swelling behavior of the films as well as their FT-IR spectra. Briefly, hydrogels were synthesized by reacting a 10% (w/v) aqueous gelatin solution (from porcine skin, 200 bloom, type A, low endotoxin content, GELITA) in the presence of 1% (w/v) poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (Pluronic® F-108, SigmaAldrich) with different amounts of ethyl lysine diisocyanate ethyl ester (LDI; 3-fold and 8-fold molar excess of NCO groups compared to NH2 groups of the gelatin). Two gelatin-based hydrogels with different degree of crosslinking, G10_LNCO3 and G10_LNCO8, were obtained.
Hydrogel implantation. Animal experiments were performed in accordance with the guidelines of the German Regulations for Animal Welfare. The protocol was approved by the local Ethical Committee for Animal Experiments (reference number 24-9168.11-4/2013-1). The implantation procedure was done as described previously [16]. Briefly, female immunocompetent SKH1-Elite mice were purchased from Charles River. SKH1 mice (age 8-15 weeks, weight 22-30 g) were implanted subcutaneously (s.c.) at the lower back with one hydrogel piece of G10_LNCO3 (left side) or G10_LNCO8 (right side) or with both G10_LNCO3 and G10_LNCO8 at the left and right side, respectively. Mice were anesthetized using 8% (v/v) desfluran (Baxter). A small incision (< 5 mm length) was made on the dorsal area. Subcutaneous skin pockets were formed posterior to incision using surgical scissors. The hydrogel pieces (1-2 × 3 × 10 mm size, 1 h swollen in PBS, sterile, endotoxin free) were implanted and the wound was closed using spray dressing. Sham-operated animals followed the same procedure except for the actual hydrogel implantation. All animals received the analgesic Rimadyl (5 mg/kg) as single injection.
Volume determination measurements. Hydrogel implant volume was determined in vivo using dedicated 7 T small animal magnetic resonance imaging (MRI, Bruker) with a T2-weighted measuring (TRARE) sequence. Relaxation time was 38 ms. Resolution was 156 µm in x-y-direction and as determined by the slice thickness 800 µm in z-direction. For quantification of the hydrogel volume the software ROVER (ABX GmbH) was used. To distinguish water (water-1H) from fatty acid (fat/fatty acid/lipid-1H) chemical environment protons spectral MRI analysis was performed as published elsewhere [17]. Of importance, no MRI contrast agents were used. To verify the MRI volume quantification a computed tomography-based (NanoScan CT, Mediso) ex vivo method was used. For this purpose, hydrogels together with surrounding tissue were surgically removed at different time points representing different material sizes for both hydrogels. The tissue samples were fixed for 24 h in 4% (w/v) paraformaldehyde (PFA) at room temperature, incubated for 3 days in 20% (w/v) sucrose solution at 4°C, and afterwards stored at -65°C. For ex vivo CT measurements the tissue samples of a subgroup of double-implanted animals (n=4) were dehydrated by incubation in a graded series of 30%, 50%, and 70% (v/v) ethanol for 24 h each concentration. Staining was performed in 0.3% (w/v) of phosphotungstic acid (PTA) in 70% (v/v) ethanol for 49 days. For visualization using CT, samples were measured with a resolution of 42 µm in xyz. The volume was quantified using ROVER (ABX GmbH).
In vivo luminescence and fluorescence imaging. For luminescence and fluorescence measurements the small animal optical imaging device in vivo Xtreme (Bruker) was used. The detection of reactive oxygen species was performed using the luminescent probe L-012 (Wako Chemicals). Animals were injected intraperitoneally (i.p.) with 1.5 mg/100 µl L-012 in 0.9% (w/v) sodium chloride (60 mg/kg) and luminescence was measured 30 min after injection using 4 × 4 binning and 5 min exposure time. COX-2 was visualized using the fluorescent probe XenoLight RediJect (Perkin Elmer, 570/600 nm). 100 µl of the solution were injected i.p. and fluorescence was measured 3 h post injection. To induce an inflammatory response, which served as positive control for both L-012 and XenoLight RediJect, 12‑O‑Tetradecanoylphorbol-13-acetate (TPA) was used [18,19]. Animals were injected with 100 µl of a 100 µM TPA solution behind the ear and in the lower dorsal area on the right side on day 1 and day 2. On day 3 animals were again injected with 50 µl of the same TPA solution. In parallel, the same amount of 0.9% (w/v) sodium chloride solution was injected on the left side of the animals as a negative control. MMP activity was determined using MMP-Sense 680 (Perkin Elmer, 650/700 nm). 24 h after intravenous (i.v.) injection of 100 µl MMP-Sense 680, fluorescence was assessed using optical imaging.
Exposure time for fluorescence images was 1 s. As reference, a channel with no specific fluorescence GFP (480/535 nm) was chosen and an image with an exposure time of 4 s was acquired.
For the acquisition and quantification of images Bruker Molecular Imaging software version 7.2 was used. For luminescence images, net luminescence intensity was determined. To minimize quantification of unspecific auto-fluorescence, fluorescence images of MMP activity measurements were first divided by the reference channel. Subsequently, net fluorescence intensity was quantified.
PET imaging. Dynamic small animal positron emission tomography (PET) after i.v. injection of [18F]fluorodeoxyglucose ([18F]FDG) was performed in mice implanted with either G10_LNCO3 or G10_LNCO8 and mice implanted with both G10_LNCO3 and G10_LNCO8 on day 14, 21, and 35 after implantation. PET acquisition was performed using NanoScan PET/CT scanner (Mediso) and the protocol given in detail elsewhere [20]. In brief, PET acquisition was started 20 s before infusion of [18F]FDG (10 MBq in 200 µl 0.9% NaCl) through a needle catheter into a lateral tail vein. Emission data were collected continuously for 60 min. The 3D list mode data were sorted into sinograms with 32 frames (15 × 10 s, 5 × 30 s, 5 × 60 s, 4 × 300 s, 3 × 600 s). The data were decay, scatter, and attenuation (X-ray) corrected. The frames were reconstructed by maximum a posteriori (MAP) method with 4 iterations and 6 subsets. Image volume data were converted to Siemens ECAT7 format, co-registered to MRI data, and analyzed by ROVER software (ABX GmbH). Based on MRI data, masks were determined around G10_LNCO3 or G10_LNCO8 and three-dimensional regions of interest (ROI) were defined by thresholding PET data within these masks. In cases where thresholding of PET data was impossible due to insufficient activity or close proximity to a high [18F]FDG accumulating organ such as the kidney, thresholding was performed on MRI data and the ROI was translated to PET data. Standardized uptake values (SUVs) were calculated over the ROI as a ratio of activity concentration at time t and injected dose at time of injection (t0) divided by body weight. Following PET scanning (60 min p.i.) at day 35, mice were sacrificed and subjected to ex vivo whole-body autoradiography.
Serum sample preparation and analysis. At selected time points, whole blood samples were collected by heart puncture of 4 anesthetized mice for each group and allowed to clot for 30 min at room temperature. Samples were centrifuged at 2,000 × g for 15 min at 4°C. The supernatant was transferred into a fresh tube and snap frozen in liquid nitrogen immediately. Serum was stored at -65°C.
Cytokine concentrations of serum samples were quantified using Bioplex Pro™ Mouse Cytokine 26-plex Assay from Bio-Rad and MMP concentrations were quantified using MILLIPLEX® Map (Mouse MMP Magnetic Bead Panel 1 and 2, Luminex®).
Histological analysis. For histological analysis 3 animals per group were sacrificed at the selected time points. The hydrogels were surgically removed including the surrounding tissue. The tissues were fixed in 4% (w/v) PFA for 24 h at room temperature and incubated for 3 days in 20% (w/v) sucrose in phosphate buffered saline (PBS) at 4°C solution. Tissue samples were snap frozen and stored at -65°C. For cryosectioning samples were cut through the middle of the remaining hydrogel piece. Samples were embedded in 7.5% (w/v) gelatin solution with 20% (w/v) sucrose in PBS, frozen, and cut at 10 µm thickness using a cryostat at -30°C. Histological samples were stained by hematoxylin & eosin (H&E) using standard protocols. Specific tissue response was visualized using immunohistochemical staining for different cell markers, which are summarized in Table 1.
Antibodies used for immunohistochemical staining.
| Antibody | Catalog no. | Dilution | Species | Antigen retrieval | Cell or tissue type |
|---|---|---|---|---|---|
| Primary antibody | |||||
| CD31 (abcam) | ab28364 | 1:75 | Rabbit | Citrate buffer | Blood vessels [21] |
| CD34 (abcam) | ab81289 | 1:200 | Rabbit | Citrate buffer | Progenitor cells [22,23] |
| CD68 (AbD Serotec) | MCA-1957 | 1:100 | Rat | - | Macrophages [24] |
| CD206 (abcam) | ab64693 | 1:100 | Rabbit | Citrate buffer | M2-macrophages [25] |
| Involucrin (abcam) | ab28057 | 1:1500 | Rabbit | Citrate buffer | Keratinocytes [26] |
| Ki67 (abcam) | ab15580 | 1:200 | Rabbit | Citrate buffer | Proliferatory cells [27] |
| Lrig-1 (abcam) | ab36707 | 1:200 | Rabbit | Citrate buffer | Fibroblast stem cells [28] |
| S100A4 (Thermo Scientific) | RB-9411 | 1:100 | Rabbit | Citrate buffer | Fibroblasts [29] |
| VEGFR-2 (abcam) | ab39256 | 1:100 | Rabbit | Citrate buffer | Newly formed blood vessels [21] |
| Isotype-control | |||||
| Rabbit polyclonal IgG (abcam) | ab27478 | Concentration equal to primary antibody | Rabbit | Equal to primary antibody | |
| Normal rat IgG (Santa Cruz) | sc-2026 | Concentration equal to primary antibody | Rat | Equal to primary antibody | |
| Secondary antibody | |||||
| Goat anti-rabbit (Dianova) | 111-065-003 | 1:200 | Goat | ||
| Rabbit anti-rat (Dianova) | 312-066-045 | 1:100 | Rabbit | ||
| Goat anti-rabbit Alexa-Fluor 647 (Life Technologies™) | A21245 | 1:200 | Goat | ||
Except for VEGFR-2 staining, where a fluorescent secondary antibody (Alexa-Fluor 647) was used, biotinylated secondary antibodies were used. After incubation with ExtrAvidin peroxidase (Sigma‑Aldrich), staining was visualized using AEC substrate kit (BD Biosciences). Sections were counterstained using Mayer's hematoxylin. Fluorescence staining of VEGFR-2 sections was counterstained with Hoechst 33258 (Life Technologies™). Images were acquired using AxioImager.A1 and the appropriate software package AxioVision (Carl Zeiss). Fluorescent images were acquired using a laser scanning confocal microscope (IX83, Olympus) and the appropriate software FluoView (Olympus).
Quantification of immunohistochemical staining was performed for sections of three different animals per time point using ImageJ/FIJI [30]. To this end, the color threshold plugin was used. RGB values were set for cell nuclei and for immunohistochemically positively stained areas. Subsequently, the plugin “analyze particles” was applied. The positively stained area was divided by area of cell nuclei for AEC staining or by number of cell nuclei for fluorescent staining.
To measure capsule thickness of the implanted hydrogels van Gieson's staining was performed using standard protocols. Cell nuclei were stained using Weigert's iron hematoxylin solution. Collagen was stained using Van-Gieson-mixture consisting of saturated picric acid and 1% (w/v) thiazine red. For the measurement of capsule thickness AxioVision (Carl Zeiss) software was used. Capsule thickness was measured for sections of three different animals per time point. For each section 5 points on each capsule-side (the skin- and muscle-side) around the implant were measured.
For pathohistological investigation organs (inguinal lymph nodes, kidneys, liver, and spleen) were surgically removed and fixed in 4% (w/v) PFA for 24 h at room temperature. The organs were washed for 1 h in PBS to remove the PFA and stored in PBS with 0.1% (w/v) sodium azide until paraffin blocks were prepared. All organs were cut into 1 µm thick sections and stained by H&E for subsequent pathological examination.
Statistical analysis. Statistical significance of measured degradation volumes, in vivo MMP activity, serum cytokine levels and serum MMP levels was calculated using two-way ANOVA followed by a Bonferroni post hoc test using Prism 6 (GraphPad Software). Statistical significance was assumed for p < 0.1 in case of histological stainings and p < 0.01, p < 0.05 for all other statistical analyses. Descriptive data were expressed as means and standard deviations. For correlation between datasets Pearson correlation coefficient was calculated.
Results
Quantification of hydrogel degradation in vivo: methodological considerations
To determine the volume of implanted gelatin-based hydrogels, magnetic resonance imaging (MRI) with a specifically designed T2 weighted measuring sequence was performed. Both G10_LNCO3 and G10_LNCO8, two gelatin-based hydrogels with different crosslinking levels and therefore different surface structures, physical and mechanical properties [14,15] (Fig. 1a, Table 2), could be clearly visualized by MRI (Fig. 1b).
Hydrogel implantation and visualization. (a) Synthesis scheme of gelatin-based hydrogels G10_LNCO. Gelatin was reacted with 3- or 8-fold excess of L-lysine diisocyanate ethyl ester (LDI) related to amine-groups in gelatin for G10_LNCO3 and G10_LNCO8 (upper left panel). Confocal microscopy images reveal different surface structures of the different hydrogels (upper right panel). Implantation workflow is shown in the lower panel. First mice skin is disinfected, second incision and skin pocket is formed, third pre-swollen hydrogel is implanted, fourth incision is closed by the use of spray-plaster. (b) Representative axial MRI images on day 1 and day 35 after implantation. After drawing a volume of interest around the material (red sphere) and applying a threshold (middle panel) the volume of the hydrogels could be calculated. Hematoxylin & Eosin (H&E) staining confirmed difference in degradation of G10_LNCO3 and G10_LNCO8 35 days after implantation (right panel).
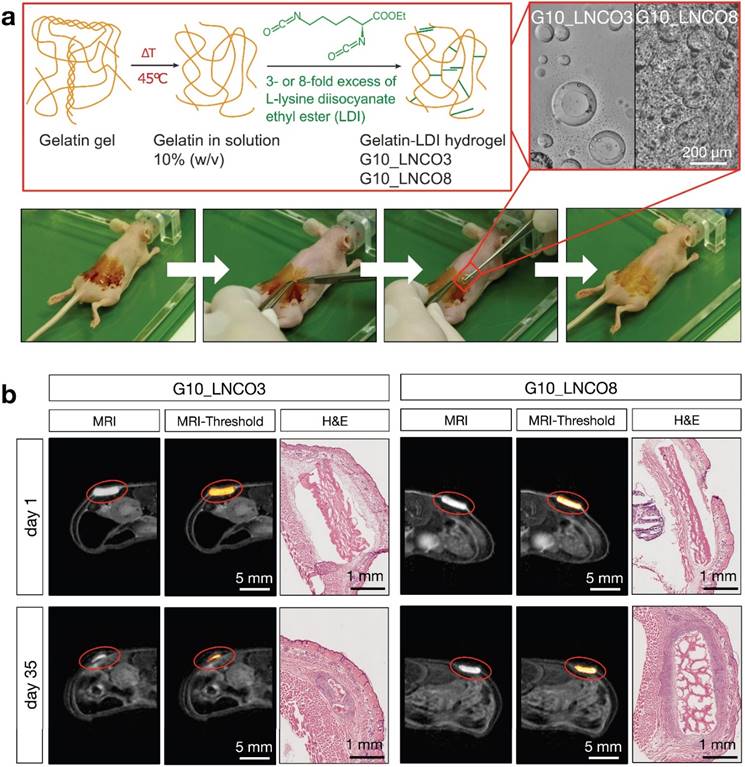
Physical and mechanical properties of G10_LNCO3 and G10_LNCO8
| G10_LNCO3 | G10_LNCO8 | |
|---|---|---|
| Young's modulus | 13 ± 3 kPa | 55 ± 11 kPa |
| Elongation at break | 43 ± 25% | 30 ± 11% |
| Tensile strength | 7 ± 3 kPa | 19 ± 11 kPa |
Ex vivo cryosections revealed similar sizes for the hydrogels compared to the sizes at the MRI images at the different time points (Fig. 1b). To determine whether the volume measured by MRI represents the actual volume of the hydrogel, spectral MRI analysis was performed to distinguish free water molecules from non-covalently bound water and fatty acid chemical environment protons [17] (Fig. S1a). Spectra of both materials showed only one prominent peak, assigned to the free water signal. Therefore, protons measured within the hydrogel are in chemical environment of free water, which is in accordance with the situation in a swollen hydrogel [31,32]. Computed tomography (CT) as an additional imaging method was applied to verify the MRI volume quantification (Fig. S1b). Due to similar densities of the gelatin-based hydrogels compared to the surrounding tissue the material could not be visualized and, therefore, not be quantified directly using small animal CT measurements in vivo. Consequently, volume of implanted hydrogels was initially quantified at different time points after implantation using MRI. Immediately after performing MRI measurements, a subgroup of double-implanted animals (n=4) was sacrificed. The implanted material as well as the surrounding tissue was surgically removed and explants were further processed for phosphotungstic acid (PTA) staining. PTA binds to fibrin, collagen, and other proteins that are ubiquitously present in connective tissues throughout all organs, allowing analysis of soft tissue using CT ex vivo [33,34]. Therefore, we hypothesized that PTA would also stain the hydrogels, or more precisely the remaining covalently cross-linked gelatin-LDI scaffolds. Supporting this hypothesis, the original implants and, according to progress of degradation, their remains could be clearly visualized and quantified using CT after PTA staining (Fig. S1c). Volumes quantified using the ex vivo PTA-CT method were lower than volumes initially measured by MRI by a factor of approximately 10. This factor is explained by dehydration of hydrogels during the staining procedure. However, both quantification methods showed strong correlation (r=0.9691, P<0.0001) at all time points (Fig. S1d). Taken together, the combination of PTA-CT and MRI led to the conclusion that our proposed MRI volume analysis method is indeed reliable for studying size and degradation behavior of implanted gelatin-based hydrogels in vivo.
Hydrogel degradation and tissue interaction in vivo
Successful establishment of the MRI quantification method allowed for assessment of the volumes of implanted hydrogels over the whole degradation period. To this end, and to investigate whether a different number of hydrogel implants could cause different degradation rates, animals were implanted with either G10_LNCO3 or G10_LNCO8 (unilateral/single implantation, Fig. 2a, Fig. S2a) or with both G10_LNCO3 and G10_LNCO8 (bilateral/double implantation, Fig. 2b, Fig. S2b). Corresponding to the hydrolytic degradation profile, G10_LNCO8, the hydrogel with the higher degree of covalent and physical net points, showed slower degradation in vivo in both experimental situations compared to G10_LNCO3. After 63 days less than 5% of the initial volume of the hydrogel remained in the body. In contrast, G10_LNCO3 was almost completely (V < 5% of initial volume) degraded after 35 days. Importantly, a difference in degradation rates between animals bearing one implant (single-implanted) or two implants (double-implanted) was observed. The double-implanted animals showed higher initial degradation of G10_LNCO3 and a higher degradation of G10_LNCO8 at the later phase between day 35 and day 42 after implantation (Fig. 2a, 2b, Fig. S2c).
Quantification of hydrogel degradation and local MMP-activity. Degradation behavior of G10_LNCO3 and G10_LNCO8 could be observed noninvasively using dedicated small animal MRI with a specialized T2 measuring sequence. Volume was quantified after applying a threshold. Volumes are given as % of initial volume. Degradation behavior of G10_LNCO3 and G10_LNCO8 in (a) single-implanted (n=6-7) and (b) double-implanted animals (n=10). In vivo quantification of MMP activity for (c) single-implanted and (d) double-implanted animals. Mean + s.d. * G10_LNCO3 vs. G10_LNCO8, # G10_LNCO3 vs. Sham, ° G10_LNCO8 vs. Sham, * p < 0.05, ** p < 0.01.
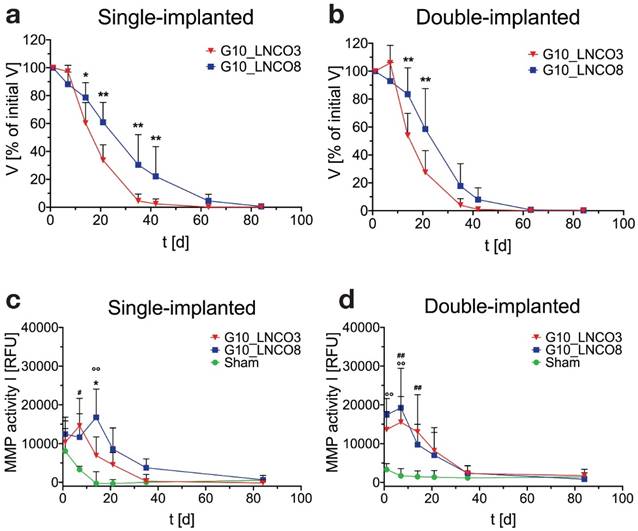
Important mechanisms for degradation of the gelatin-based hydrogels are hydrolysis and enzymatic degradation [16]. To investigate to which extent enzymatic degradation plays a role in vivo, and to detect potential differences between the two hydrogel-species, MMP activity measurements were performed in vivo. In vitro experiments investigating the direct contact between endothelial cells (HAEC), macrophages (activated THP-1) and the hydrogels showed no elevation of MMP levels in the cell culture supernatant [16]. However, MMP activity itself was not measured in vitro. In vivo, single-implanted animals showed material dependent regulation of MMP activity over time. In animals single-implanted with G10_LNCO3, high MMP activity at 1 and 7 days after implantation was observed, followed by a continuous decrease of activity levels over time (Fig 2c). In contrast, MMP activity after unilateral implantation of G10_LNCO8 peaked at 14 days after implantation, which correlates well with volume differences quantified by MRI (Fig. 2a, 2b). Double-implanted animals showed an MMP activity time course similar to mice single-implanted with G10_LNCO3 (Fig. 2d).
Additionally, our data show a reciprocal influence of implanted hydrogels on each other. This was shown for degradation and local MMP activity at the implantation site of double-implanted compared to single-implanted animals. Due to the observed differences between single- and double-implanted mice with regard to hydrogel degradation and MMP activity, subsequent experiments for determining biomaterial-tissue-interaction and systemic response of the respective hydrogels ex vivo were carried out using only single-implanted animals.
To further investigate potential inflammatory reactions, in vivo optical imaging of double-implanted animals using a fluorescent COX-2 inhibitor [35] and a reactive oxygen species (ROS) detecting substance (L-012) [36,37] were performed. Only minor inflammation limited to the incision site on day 1 and partly on day 7 after implantation were observed when compared to control animals (Fig. S3a, S3b, S3c, S3d). Neither elevated COX-2 synthesis nor ROS production could be detected at the site of the gelatin-based hydrogel.
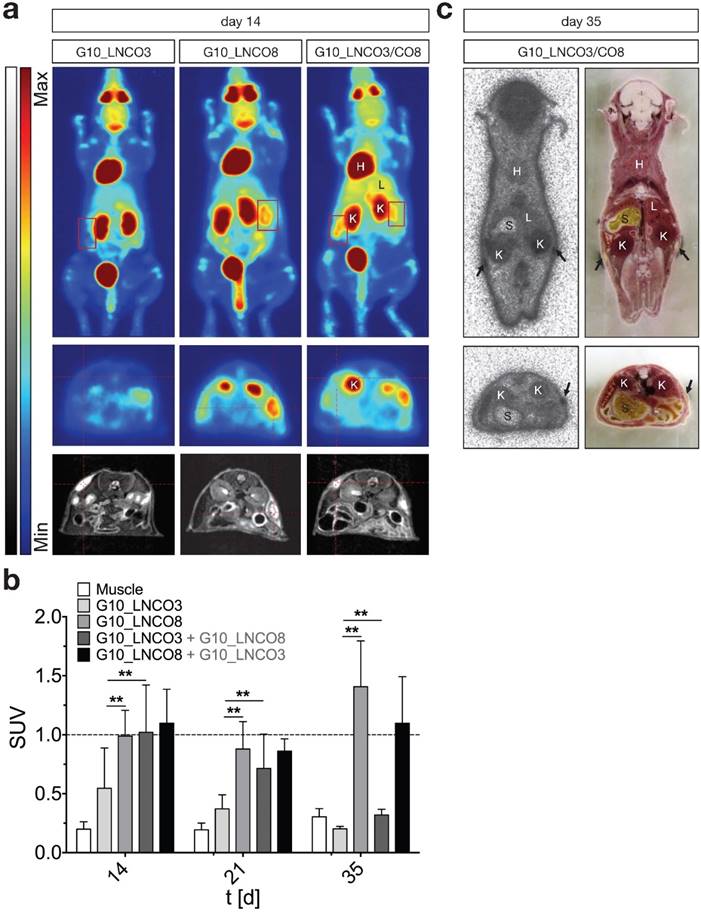
To assess whether implantation of G10_LNCO3 and/or G10_LNCO8 induced proliferation or inflammation, two processes detectable by increased trapping of [18F]fluorodeoxyglucose ([18F]FDG), dynamic small animal PET was performed 14, 21, and 35 days after implantation. Figure 3a shows maximum intensity projections (MIP, 0-60 min, upper panel) and transversal PET projections (lower panel, 30-60 min) co-registered to MRI projections of representative mice 14 days after implantation of either G10_LNCO3 (left panel) or G10_LNCO8 (mid panel) as well as of both G10_LNCO3 and G10_LNCO8 (right panel). Single-implanted G10_LNCO3 was characterized by only marginal [18F]FDG trapping, whereas significant radiotracer accumulation could be observed at implantation sites of single-implanted G10_LNCO8 and of double-implanted animals. Sequential measurements of single-implanted mice on day 21 and 35 after implantation revealed that in comparison to day 14, the mean SUV 30-60 min post injection (p.i.) of G10_LNCO3 was similar to mean SUV of muscle tissue, which indicates a decreased level of [18F]FDG trapping (Fig. 3b, Fig. S4a). This effect was partially attributed to degradation of G10_LNCO3. By contrast, SUV of G10_LNCO8 first decreased from day 14 to 21, but then reached a maximum 35 days after implantation. Interestingly, in contrast to single-implanted animals, double-implanted mice achieved substantial [18F]FDG trapping on day 14 for both hydrogels, which is similar to levels at the implantation site of single-implanted G10_LNCO8. However, despite the similar mean SUVs on day 14 and 21 for both implants in double-implanted mice, no [18F]FDG trapping could be observed on day 35 at the implantation site of G10_LNCO3, in contrast to G10_LNCO8 (Fig. 3b). Accordance of [18F]FDG PET signal and localization of G10_LNCO3 and G10_LNCO8 was further confirmed by whole-body autoradiography (Fig. 3c).
[18F]FDG PET imaging and quantification. (a) Maximum intensity projection, coronal (0-60 min p.i., upper panel) and transversal projection (mid panel, 30-60 min p.i.) of dynamic PET experiments with [18F]FDG coregistered to MRI projection (lower panel) 14 days after implantation of either G10_LNCO3 (left panel) or G10_LNCO8 (mid panel) as well as of both G10_LNCO3 and G10_LNCO8 (right panel). (b) Mean SUV of [18F]FDG (30‑60 min p.i.) on day 14, 21, and 35 after hydrogel implantation. (c) Autoradiography of double-implanted mice 35 days after implantation (the upper panel). Transversal section is shown in the lower panel. Left side shows autoradiography. Whole animal cryo-sections are shown on the right side. Mean + s.d. * p < 0.05, ** p < 0.01.
Local effects of hydrogel degradation and tissue interaction ex vivo
Specific tissue-hydrogel-interactions on microscopic scale were evaluated by immunohistochemical studies (Fig. S5a). Quantification of the pan-macrophage marker CD68 revealed a two‑peaked accumulation of macrophages on day 7 and 21 for G10_LNCO3 and on day 14 and 35 for G10_LNCO8 (Fig. 4a). Significant differences in macrophage accumulation were found between G10_LNCO3 and sham-operated mice at the same time points and between G10_LNCO8 and sham-operated mice on day 14 and 21. CD206, which characterizes M2-macrophages (M2Φ), showed similar peaks for both materials as observed with CD68. The highest amount of M2Φ for G10_LNCO3 and G10_LNCO8 was observed 21 and 35 days after implantation, respectively (Fig. 4b).
Histological quantification. Histological quantification of positive-stained area compared to counterstaining of cell nuclei of single-implanted animals. Time course for (a) CD68 as pan-macrophage marker, (b) CD206 as M2-macrophage marker, (c) Ki67 for proliferation, (d) S100A4 as fibroblast marker, (e) CD31 as blood vessel marker, and (f) VEGFR-2 for new born blood vessels (n=3 different animals). Quantification was applied using color thresholds at mosaic images of whole, centric 10 µm slices of the hydrogel implant and the surrounding tissue. Mean + s.d. * G10_LNCO3 vs. G10_LNCO8, # G10_LNCO3 vs. Sham, ° G10_LNCO8 vs. Sham p < 0.1.
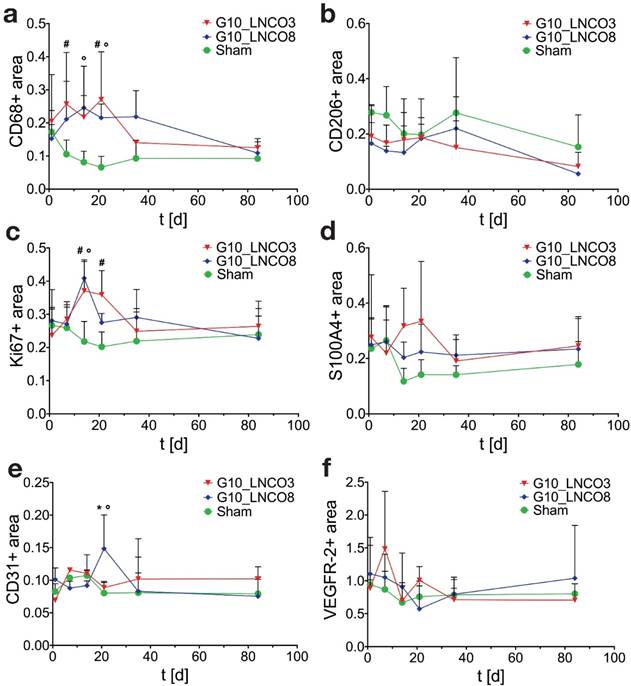
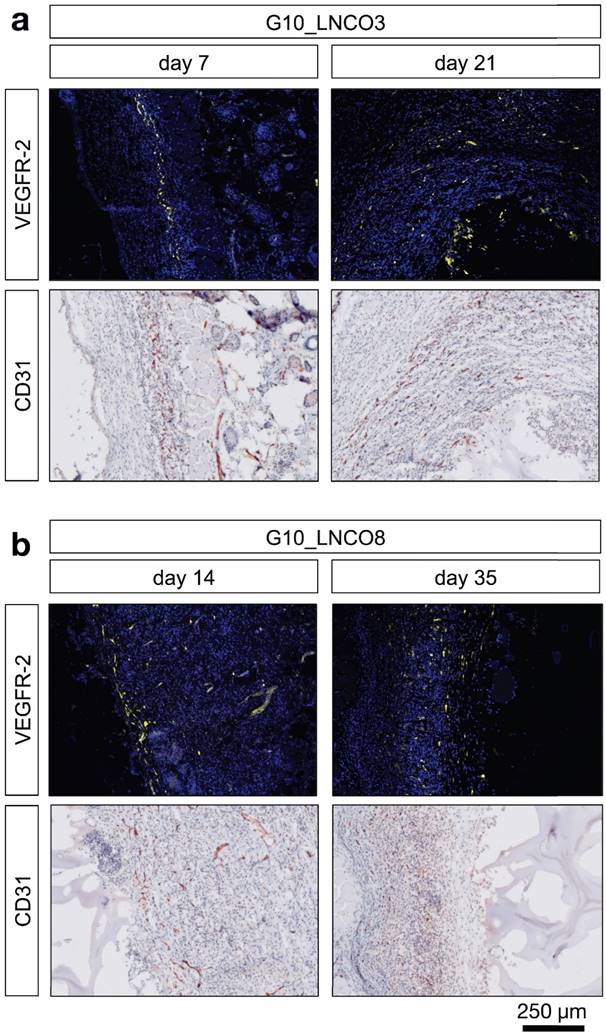
Furthermore, additional cell types associated with wound repair and skin regeneration, such as keratinocytes and progenitor cells, as indicated by staining of involucrin and CD34, accumulated around the implanted hydrogels (Fig. S5b, S5c). However, a high amount of involucrin- and CD34-positive cells was also found around the skin pocket in sham-operated animals. Following initial macrophage accumulation, fibroblasts play an important role during tissue restoration processes [38]. Quantification of the fibroblast stem cell marker Lrig-1 is shown in Fig. S5d. Only a slightly elevated amount of fibroblast stem cells could be observed for G10_LNCO3 on day 7 compared to sham-operated mice. For G10_LNCO8 a higher amount of Lrig-1 positive cells compared to sham controls could be observed from day 14 onwards. Even though different amounts of stem cells were found for G10_LNCO3 and G10_LNCO8, the proliferative response around the hydrogel implants appeared to be of similar intensity, as indicated by Ki67-positive cells (Fig. 4c). Noticeably, G10_LNCO3 increased proliferation from days 14 to 21, compared to a single peak of proliferation at day 14 for G10_LNCO8. Consequently, a greater amount of S100A4-positive fibroblasts at the implant site of G10_LNCO3 was found (Fig 4d). The increased amount of fibroblasts led to formation of a collagenous capsule around the hydrogels (Fig. S5e). The capsule around G10_LNCO3 was significantly thinner than the capsule around G10_LNCO8. Both materials showed the highest capsule thickness 14 days after implantation followed by a continuous decrease. On day 84 no significant difference in capsule thickness between implanted animals and sham controls could be detected. A frequently encountered complication of current implantation strategies is the fact that elevated levels of fibroblasts and collagenous ECM lead to 'irreversible' fibrotic encapsulation of the material, thereby hindering vessel ingrowth and supply of newly formed tissue with O2 and nutrients [39]. Hence, formation of new blood vessels within the implantation site was investigated as well. CD31, a well-established endothelial marker, was used to identify blood vessels within the tissue. Quantification revealed increased CD31 expression at the implantation site of G10_LNCO8 compared to G10_LNCO3 and sham control on day 21 (Fig. 4e). However, using the angiogenesis marker VEGFR-2, no significant increase could be detected (Fig. 4f). This is due to the high amount of cells found at the implant site compared to sham controls, which displayed less cells but still showed positively stained areas for CD31 and VEGFR-2 as skin is a well‑vascularized organ. Although quantification showed no significant differences, images (Fig. 5a, 5b, Fig. S5a) clearly depict a high density of CD31-positive blood vessels and VEGFR-2-positive (newly formed) blood vessels after implantation of G10_LNCO3 and G10_LNCO8, respectively. Already 7 days (G10_LNCO3) and 14 days (G10_LNCO8) after implantation a ring of VEGFR-2-positive newly formed blood vessels was observed around both hydrogels (Fig. 5a, 5b). Until day 21 the amount of VEGFR-2- and CD31-positive cells constantly increased. Blood vessels grew through the capsule to reach the tissue-hydrogel-interface, but only rarely sprouted into the material itself. 84 days after implantation blood vessels receded again and no difference in appearance could be observed between previously implanted and sham-operated mice.
Angiogenesis at the biomaterial-tissue-interface. Representative histological images of angiogenic blood vessels (VEGFR-2) and all blood vessels (CD31) are shown at the first and last time point of detection around the hydrogels (a) 7 days (left) and 21 days (right) after implantation for G10_LNCO3 and (b) 14 days (left) and 35 days (right) after implantation for G10_LNCO8.
Systemic effects of hydrogel degradation and tissue interaction ex vivo
Potential systemic effects of hydrogel degradation and tissue interaction were investigated by quantifying serum MMP levels and serum cytokine levels as well as performing pathohistological examinations of inguinal lymph nodes, spleen, liver, and kidneys.
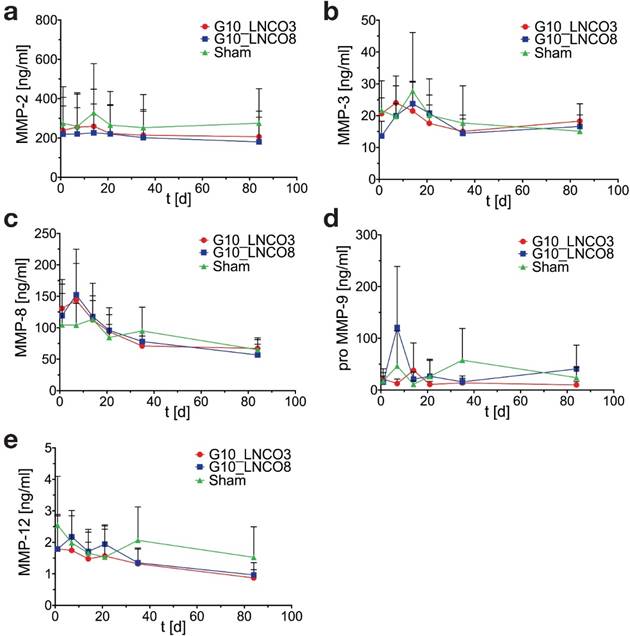
Serum MMP concentrations showed no significant elevation compared to sham control mice in case of MMP-2 (gelatinase A), MMP-3 (stromelysin), MMP-8 (neutrophil collagenase), proMMP-9 (gelatinase B), and MMP-12 (macrophage metalloelastase) (Fig. 6a, 6b, 6c, 6d, 6e). A slightly but not significant elevation of MMP-8 in implanted mice could be observed 7 days after implantation (Fig. 6c). Analysis of serum cytokine levels revealed no effect of hydrogel implantation on anti‑inflammatory cytokines IL-Ra, IL-10, IL-13 as well as on pro-inflammatory cytokines IL-1β, IL-6, IFN-γ, and TNF-α during degradation of both hydrogels (Fig. S6).
Serum MMP-levels. Time course of (a) MMP-2, (b) MMP-3, (c) MMP-8, (d) proMMP‑9, and (e) MMP-12 serum concentrations of single-implanted mice (n=4 different animals). Mean + s.d.
Moreover, pathohistological examination of lymphoid organs such as inguinal lymph nodes and spleen as well as of liver and kidney, which play a role in detoxification and excretion of foreign materials and material degradation products, were performed. No pathological abnormalities during and after the complete degradation of both materials (day 84) compared to the sham control mice could be observed (Fig. S7).
Discussion
This work describes a comprehensive small animal imaging study in immunocompetent SKH1 nude mice in vivo and ex vivo implanted with two gelatin hydrogels, stabilized through reaction with different amounts of LDI (single- and double-implanted or only single-implanted, respectively). As major result, this first preclinical investigation revealed that both hydrogels resulted in complete restoration of the original healthy condition (restitutio ad integrum). Despite all necessary caution and considering limitations, e.g., of the model, mode of implantation, and duration of the study, the results strongly suggest the potential for clinical use of LDI-stabilized gelatin hydrogels for soft tissue regeneration. As second major aspect, this investigation established a preclinical, multimodal, and multiscale imaging platform suitable for studying different hydrogels of interest.
Degradation of gelatin-based hydrogels in vivo was visualized and quantified using 7 T MRI with dedicated T2 sequences. Correct volume quantification using MRI was confirmed using an ex vivo CT-based method of PTA-stained tissue samples [34]. The strong correlation between both datasets at all time points further confirmed the high robustness of our MRI-based quantification method. Therefore, MRI is the method of choice for noninvasive investigation of hydrogel degradation in vivo. Degradation behavior of G10_LNCO3 and G10_LNCO8 clearly revealed that kinetics depend on the number of net points. A higher amount of LDI, which correlates with an increased number of covalent and physical net points, led to an increase in degradation time from 35 days for G10_LNCO3 up to 63 days for G10_LNCO8 until complete (V < 5% of initial volume) degradation was achieved. In case of the gelatin-based hydrogels tested in this study, G10_LNCO3 showed a higher initial degradation, whereas G10_LNCO8 was degraded faster at later time points. Hydrogels with higher net point density likely restrict diffusion of proteases. The later increase of degradation rates in these hydrogels may be explained by an initial primarily hydrolytic degradation, enabling a higher rate of diffusion of proteases at later time points, which then accelerate the rate of degradation. In vivo MMP activity measurements revealed a different time course for double-implanted compared to single-implanted mice. Implantation of both G10_LNCO3 and G10_LNCO8 on a single animal led to an earlier activation of MMPs at the implant site. MMP activity in general was slightly elevated in double-implanted mice. Single-implanted animals showed later activation of MMPs at the implant site, especially for G10_LNCO8. This suggests that, in response to surgery, material, or material degradation products, the enzymatic degradation of materials will occur more rapidly in double-implanted animals than in single-implanted animals. This finding is substantiated by the fact that [18F]FDG-PET revealed a great difference between single- and double-implanted animals. The increased trapping of [18F]FDG can serve as an indicator for different metabolic states of cells and tissues, such as proliferation, inflammation, macrophage accumulation, or neovascularization [40,41]. In single-implanted animals a great difference in [18F]FDG trapping of G10_LNCO3 (low) and G10_LNCO8 (high) in the time course of 14 to 35 days after implantation was observed. For double-implanted mice this difference was completely absent for day 14 and 21 after implantation. Both implantation sites showed a high accumulation of [18F]FDG. This could be due to a higher activation of M1Φ around the hydrogels in the double-implanted animals, which consequently caused higher MMP release and faster initial degradation of the lower cross-linked G10_LNCO3 and a fast degradation of G10_LNCO8 at later time points. Single-implanted animals on the other hand showed first macrophage accumulation indicated by the pan-macrophage staining (likely M1Φ) around G10_LNCO3 on day 7 decreasing until day 14 and around G10_LNCO8 on day 14 decreasing after day 35.
The results suggest that multiple implants per animal not only cause systemic effects but also alter the local material-tissue interaction, potentially due to the greater area of material-tissue interface. This raises the question on how reliable data obtained from multiple implant animal models are [37,42]. Therefore, it is of great importance to use single-implanted mice to investigate degradation itself and also degradation-specific responses by MRI, MMP in vivo measurements, and [18F]FDG-PET measurements. Due to the great differences observed between single- and double-implanted mice, subsequent studies investigating hydrogel tissue interaction at microscopic level as well as any systemic responses to the implants (MMP levels, cytokine levels) were carried out using single-implanted mice only.
Immunohistochemical data revealed no difference in the amount of pan- or M1-macrophages, however, their temporal regulation was significantly different. G10_LNCO3 showed faster accumulation of macrophages from day 7 until degradation was completed on day 35. For G10_LNCO8, the accumulation of macrophages started from day 14 and lasted until complete degradation on day 84. The two peaks in macrophage quantification and accordance of the second peak to accumulation of M2Φ lead to the conclusion that macrophages act in a two-step process during degradation of the two used hydrogels. First, M1-like macrophages arrive at the biomaterial implant and start degradation via secretion of MMPs and other proteases [43]. Next, M1Φ switch to M2Φ [44] at the partially degraded implant to start tissue regeneration. As proposed by van Putten et al. 2013 [45] the classical M1/M2 differentiation model for macrophages lacks many different intermediate types of macrophages. Therefore, CD68 pan-macrophage staining could also indicate different intermediate states of macrophages, which require further characterization by different immunochemical co-staining like iNOS for M1Φ [44].
During the degradation process, MMPs also play an important role in promoting sprouting of new blood vessels [46,47] and also gelatin itself has been reported to have pro-angiogenic properties [48]. CD31 and VEGFR-2 immunohistochemistry revealed a pro-angiogenic effect of both materials in similar intensity only differing in a time-dependent manner. Until now, great success in tissue engineering has been achieved in the field of 'medium' vascularized tissues such as skin or cartilage [49,50] compared to 'highly' vascularized organs such as heart or liver. As a consequence, bioactive gelatin-based hydrogels, which induce angiogenesis, are promising candidates for tissue engineering of these 'highly' vascularized organs [51].
Formation of granulation tissue, which normally follows the action of macrophages by proliferation of fibroblasts [38], was also observed around the gelatin-based hydrogels. The amount of proliferation and subsequent amount of fibroblasts was slightly higher for the lower cross-linked hydrogel. However, both hydrogels showed a minor increase of fibroblasts compared to sham-operated animals, which lead to the conclusion that only a minor amount of collagen and granulation tissue was formed. The observed elevated capsule thickness does not take cellular and collagen density into account. Histological data revealed that the capsule showed less dense collagen but a high level of newly formed blood vessels, indicating a temporary fibrous encapsulation of the hydrogel by the organism [52-54], which disappears after degradation of the hydrogel without formation of scar tissue [48].
The hydrogels showed high biocompatibility as no systemic effects, such as elevated cytokine or serum MMP levels could be detected. Furthermore, pathological examination of liver, spleen, kidneys, and inguinal lymph nodes revealed no morphological changes/necrosis during and after complete degradation of implanted materials (day 35 and 84).
In conclusion, this small animal multimodal imaging study presents two novel LDI-cross-linked gelatin-based biomaterials to be well tailorable for specific degradation rates without causing any adverse local tissue reactions or systemic response neither in vivo nor ex vivo on a cellular level.
Both studied materials finally enabled restitutio ad integrum in SKH1 immunocompetent nude mice as preclinical model. Hydrogel-tissue interactions were highly similar for both hydrogels and only showed differences in the time required for degradation, which was strictly dependent on the excess of LDI in comparison to free amine groups during preparation of the hydrogels. Additionally, due to the different mechanical properties of the hydrogels published in former studies [14,16] and the high biocompatibility observed in this study, gelatin-based hydrogels offer a promising strategy for material-induced tissue regeneration in several medical indications. Hydrogels with lower Young's moduli may be suitable for skin, mammary, or liver tissue, while hydrogels with higher Young's moduli may enable applications in the repair of muscle, cartilage, or pre-calcified bone. The hydrogels can be produced relatively easy, fast, and cheap, while showing superior biocompatibility [4,14,16].
Conclusion
Despite all necessary caution and considering study limitations as discussed above, this study strongly suggests a potential clinical use of hydrogels based on LDI-modified gelatin with different quantity of covalent and physical net points in a versatile way, e.g., as organ patches or soft tissues replacement materials. Moreover, sophisticated multi-modal imaging both will impel finding of 'tailored' solutions for novel hydrogels and, from a translational point of view, will essentially contribute to and forecast success of treatment. The very early implementation of multi-modal imaging approaches in biocompatibility testing of novel materials is needed to overcome the existing divergence between research findings on good in vitro performance but failure in translation to clinical application.
Supplementary Material
Supplementary figures.
Acknowledgements
The excellent technical assistance of Aline Morgenegg, Catharina Heinig, Johanna Pufe, Mareike Barth, and Sebastian Meister (Dresden-Rossendorf) as well as of Jessica Reinert, Nicole Hüttig, Tim P. Gebauer, Peter Viszokai, and Radovan Vukicevic (Teltow) is greatly acknowledged. We also thank Birgit Belter, Ph.D., and Ralf Bergmann, Ph.D., for their expert advice and many fruitful discussions. The authors further wish to thank the staff of the Production of Radiopharmaceuticals Department at the Institute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf for production of [18F]FDG. The authors thank the Helmholtz Association for funding of this work through Helmholtz-Portfolio Topic "Technologie und Medizin - Multimodale Bildgebung zur Aufklärung des In-vivo-Verhaltens von polymeren Biomaterialien". Sandra Hauser (née Ullm) was recipient (2011-2014) of a fellowship by the Europäische Sozialfonds (ESF). Anne Krüger-Genge was recipient of a fellowship of the Helmholtz Postdoc Program 2012 (PD-064). The authors also wish to thank Viktoria Bosak who assisted in the proof-reading of the final version of the manuscript.
Author Contributions
C.T., S.H., A.K.-G., and C.N. performed the experiments and analyzed the data. A.T.N. and A.L provided the hydrogels and contributed the analysis of material behavior. R.K. performed the pathological examinations. C.T., S.H., AK.-G, F.J., A.L., A.T.N., J.S., C.N., and J.P. designed the study, interpreted the data and wrote the paper. All authors read the paper and contributed to its final form.
Competing Interest
The authors declare no competing financial interests.
References
1. Rosiak JM, Yoshii F. Hydrogels and their medical applications. Nucl Instruments Methods Phys Res Sect B Beam Interact with Mater Atoms. 1999;151:56-64
2. Kopeček J. Hydrogel biomaterials: A smart future? Biomaterials. 2007;28:5185-5192
3. Neffe AT, Pierce BF, Tronci G. et al. One Step Creation of Multifunctional 3D Architectured Hydrogels Inducing Bone Regeneration. Adv Mater. 2015;27:1738-1744
4. Sartori S, Chiono V, Tonda-Turo C. et al. Biomimetic polyurethanes in nano and regenerative medicine. J Mater Chem B. 2014;2:5128-5144
5. Patino MG, Neiders ME, Andreana S. et al. Collagen as an implantable material in medicine and dentistry. J Oral Implantol. 2002;28:220-225
6. Lynn a. K, Yannas I V, Bonfield W. Antigenicity and immunogenicity of collagen. J Biomed Mater Res - Part B Appl Biomater. 2004;71:343-354
7. Olde Damink LHH, Dijkstra PJ, Luyn MJ a. et al. Glutaraldehyde as a crosslinking agent for collagen-based biomaterials. J Mater Sci Mater Med. 1995;6:460-472
8. van Luyn MJ, van Wachem PB, Olde Damink LH. et al. Secondary cytotoxicity of cross-linked dermal sheep collagens during repeated exposure to human fibroblasts. Biomaterials. 1992;13:1017-1024
9. Franz S, Rammelt S, Scharnweber D. et al. Immune responses to implants - A review of the implications for the design of immunomodulatory biomaterials. Biomaterials. 2011;32:6692-6709
10. Lin TH, Tamaki Y, Pajarinen J. et al. Chronic inflammation in biomaterial-induced periprosthetic osteolysis: NF-κB as a therapeutic target. Acta Biomater. 2014;10:1-10
11. Elzoghby AO, Samy WM, Elgindy N a. Protein-based nanocarriers as promising drug and gene delivery systems. J Control Release. 2012;161:38-49
12. Young S, Wong M, Tabata Y. et al. Gelatin as a delivery vehicle for the controlled release of bioactive molecules. J Control Release. 2005;109:256-274
13. Su K, Wang C. Recent advances in the use of gelatin in biomedical research. Biotechnol Lett. 2015
14. Tronci G, Neffe AT, Pierce BF. et al. An entropy-elastic gelatin-based hydrogel system. J Mater Chem. 2010;20:8875-8884
15. Pierce BF, Pittermann E, Ma N. et al. Viability of human mesenchymal stem cells seeded on crosslinked entropy-elastic gelatin-based hydrogels. Macromol Biosci. 2012;12:312-21
16. Ullm S, Krüger A, Tondera C. et al. Biocompatibility and inflammatory response in vitro and in vivo to gelatin-based biomaterials with tailorable elastic properties. Biomaterials. 2014;35:9755-9766
17. Strobel K, van den Hoff J, Pietzsch J. Localized proton magnetic resonance spectroscopy of lipids in adipose tissue at high spatial resolution in mice in vivo. J Lipid Res. 2008;49:473-480
18. Kundu JK, Shin YK, Kim SH. et al. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-κB in mouse skin by blocking IκB kinase activity. Carcinogenesis. 2006;27:1465-1474
19. Kundu JK, Shin YK, Surh YJ. Resveratrol modulates phorbol ester-induced pro-inflammatory signal transduction pathways in mouse skin in vivo: NF-κB and AP-1 as prime targets. Biochem Pharmacol. 2006;72:1506-1515
20. Kniess T, Laube M, Bergmann R. et al. Radiosynthesis of a 18F-labeled 2,3-diarylsubstituted indole via McMurry coupling for functional characterization of cyclooxygenase-2 (COX-2) in vitro and in vivo. Bioorganic Med Chem. 2012;20:3410-3421
21. Hayashi M, Majumdar A, Li X. et al. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun. 2013;4:1-15
22. Sidney LE, Branch MJ, Dunphy SE. et al. Concise review : Evidence for CD34 as a common marker for diverse progenitors. Stem Cells. 2014;32:1380-1389
23. Ghadially R. 25 years of epidermal stem cell research. J Invest Dermatol. 2012;132:797-810
24. Badylak SF, Valentin JE, Ravindra AK. et al. Macrophage phenotype as a determinant of biologic scaffold remodeling. Tissue Eng Part A. 2008;14:1835-1842
25. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015. 2015
26. Sonkoly E, Wei T, Pavez Loriè E. et al. Protein kinase C-dependent upregulation of miR-203 induces the differentiation of human keratinocytes. J Invest Dermatol. 2010;130:124-134
27. Scholzen T, Gerdes J. The Ki-67 protein: From the known and the unknown. J Cell Physiol. 2000;182:311-322
28. Driskell RR, Watt FM. Understanding fibroblast heterogeneity in the skin. Trends Cell Biol. 2014;25:92-99
29. Zhang J, Chen L, Liu X. et al. Fibroblast-specific protein 1/S100A4-positive cells prevent carcinoma through collagen production and encapsulation of carcinogens. Cancer Res. 2013;73:2770-2781
30. Schindelin J, Arganda-Carreras I, Frise E. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676-682
31. Peppas NA, Bures P, Leobandung W. et al. Hydrogels in pharmaceutical formulations. Eur J Pharm Biopharm. 2000;50:27-46
32. Hoffman AS. Hydrogels for biomedical applications. Adv Drug Deliv Rev. 2012;64:13-28
33. Nemetschek TH. Topochemistry of the Binding of Phosphotungstic Acid to Collagen. J Mol Biol. 1979:67-83
34. Metscher BD. MicroCT for comparative morphology: simple staining methods allow high-contrast 3D imaging of diverse non-mineralized animal tissues. BMC Physiol. 2009:9
35. Uddin MJ, Crews BC, Blobaum AL. et al. Selective visualization of cyclooxygenase-2 in inflammation and cancer by targeted fluorescent imaging agents. Cancer Res. 2010;70:3618-3627
36. Nishinaka Y, Aramaki Y, Yoshida H. et al. A new sensitive chemiluminescence probe L-012, for measuring the production of superoxide anion by cells. Biochem Biophys Res Commun. 1993;193:554-559
37. Liu WF, Ma M, Bratlie KM. et al. Real-time in vivo detection of biomaterial-induced reactive oxygen species. Biomaterials. 2011;32:1796-801
38. Anderson JM. Biological responses to materials. Annu Rev Mater Res. 2001;31:81-110
39. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-257
40. Van Waarde A, Cobben DCP, Suurmeijer AJH. et al. Selectivity of 18 F-FLT and 18 F-FDG for differentiating tumor from inflammation in a rodent model. J Nucl Med. 2004;45:695-700
41. Wu C, Li F, Niu G. et al. PET imaging of inflammation biomarkers. Theranostics. 2013;3:448-466
42. Bratlie KM, Dang TT, Lyle S. et al. Rapid biocompatibility analysis of materials via in vivo fluorescence imaging of mouse models. PLoS One. 2010;5:e10032
43. Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20:86-100
44. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014:6
45. Van Putten SM, Ploeger DT a, Popa ER. et al. Macrophage phenotypes in the collagen-induced foreign body reaction in rats. Acta Biomater. 2013;9:6502-6510
46. Haas TL, Milkiewicz M, Davis SJ. et al. Matrix metalloproteinase activity is required for activity-induced angiogenesis in rat skeletal muscle. Am J Physiol Heart Circ Physiol. 2000;279:H1540-H1547
47. Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9:267-285
48. Dreesmann L, Ahlers M, Schlosshauer B. The pro-angiogenic characteristics of a cross-linked gelatin matrix. Biomaterials. 2007;28:5536-5543
49. Priya SG, Jungvid H, Kumar A. Skin tissue engineering for tissue repair and regeneration. Tissue Eng Part B Rev. 2008;14:105-118
50. Temenoff JS, Mikos AG. Review: Tissue engineering for regeneration of articular cartilage. Biomaterials. 2000;21:431-440
51. Moon JJ, Saik JE, Poche RA. et al. Biomimetic hydrogels with pro-angiogenic properties. Biomaterials. 2010;31:3840-3847
52. Morais JM, Papadimitrakopoulos F, Burgess DJ. Biomaterials/tissue interactions: possible solutions to overcome foreign body response. AAPS J. 2010;12:188-196
53. Gretzer C, Emanuelsson L, Liljensten E. et al. The inflammatory cell influx and cytokines changes during transition from acute inflammation to fibrous repair around implanted materials. J Biomater Sci Polym Ed. 2006;17:669-687
54. Luttikhuizen DT, Hamsen MC, Van Luyn MJA. Cellular and Molecular Dynamics in the Foreign Body Reaction. Tissue Eng. 2006;12:1955-1970
Author contact
![]() Corresponding author: Phone: +49 351 260 2622; Fax: +49 351 260 12622; j.pietzschde (J. Pietzsch).
Corresponding author: Phone: +49 351 260 2622; Fax: +49 351 260 12622; j.pietzschde (J. Pietzsch).