Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(12):2141-2160. doi:10.7150/thno.16184 This issue Cite
Research Paper
A New Concept of Enhancing Immuno-Chemotherapeutic Effects Against B16F10 Tumor via Systemic Administration by Taking Advantages of the Limitation of EPR Effect
Yuting Yang1, Xiaowei Tai1, Kairong Shi1, Shaobo Ruan1, Yue Qiu1, Zhirong Zhang1, Bing Xiang2 ![]() , Qin He1
, Qin He1 ![]()
1. Key Laboratory of Drug Targeting and Drug Delivery Systems, West China School of Pharmacy, Sichuan University, Block 3, 17 Southern Renmin Road, Chengdu 610041, P. R. China
2. Department of hematology, West China Hospital, Sichuan university, No. 17, Block 3, Southern Renmin Road, Chengdu 610041, P. R. China.
Received 2016-5-16; Accepted 2016-8-4; Published 2016-9-12
Abstract

The enhanced permeability and retention (EPR) effect has been comfortably accepted, and extensively assumed as a keystone in the research on tumor-targeted drug delivery system. Due to the unsatisfied tumor-targeting efficiency of EPR effect being one conspicuous drawback, nanocarriers that merely relying on EPR effect are difficult to access the tumor tissue and consequently trigger efficient tumor therapy in clinic. In the present contribution, we break up the shackles of EPR effect on nanocarriers thanks to their universal distribution characteristic. We successfully design a paclitaxel (PTX) and alpha-galactosylceramide (αGC) co-loaded TH peptide (AGYLLGHINLHHLAHL(Aib)HHIL-Cys) -modified liposome (PTX/αGC-TH-Lip) and introduce a new concept of immuno-chemotherapy combination via accumulation of these liposomes at both spleen and tumor sites naturally and simultaneously. The PTX-initiated cytotoxicity attacks tumor cells at tumor sites, meanwhile, the αGC-triggered antitumor immune response emerges at spleen tissue. Different to the case that liposomes are loaded with sole drug, in this concept two therapeutic processes effectively reinforce each other, thereby elevating the tumor therapy efficiency significantly. The data demonstrates that the PTX/αGC-TH-Lip not only possess therapeutic effect against highly malignant B16F10 melanoma tumor, but also adjust the in vivo immune status and induce a more remarkable systemic antitumor immunity that could further suppress the growth of tumor at distant site. This work exhibits the capability of the PTX/αGC-TH-Lip in improving immune-chemotherapy against tumor after systemic administration.
Keywords: Liposome, Distribution characteristic, Paclitaxel, Alpha-galactosylceramide, Cancer immune-chemotherapy.
1. Introduction
Even considerable efforts have been devoted to tumor therapy, it remains one of the most intractable diseases accounting for about 8 million deaths globally per annum [1]. The appalling number reflects the complexity of tumor development, as such, an efficient and effective solution is being highly demanded. Tumor-oriented nanotechnology has arisen as an attractive strategy to precisely deliver cargoes to disease sites and simultaneously decrease off-target effect, which is attributed to the hyper-permeability of the blood vessels in tumor and the dysfunction of intratumoral lymphatics [2, 3]. This glorified phenomenon termed the “enhanced permeability and retention” (EPR) effect is regarded as a keystone in the research on tumor-targeted drug delivery [4]. However, the tumor targeting efficiency of nanocarriers based on EPR effect is not adequately satisfying to meet our expectation for tumor therapy in clinic [5]. To surmount this limitation, numerous novel and improved approaches for cancer diagnosis and therapy have been attempted, among which the construction of nanoparticles for active targeting has drawn particular attention [6]. A mushrooming number of active targeting systems have been constructed to enhance the accumulation rate of drugs in tumor tissues by engineering individual nanoparticles with differed features, including geometry, surface chemistry, ligand type and ligand density [7, 8]. Nevertheless, one unchangeable factor that limits the effectiveness of active tumor targeting is the mononuclear phagocytic system (MPS), because of which the majority of the intravenously injected nanocarriers tend to accumulate at liver and spleen [9]. After systemic administration, a large proportion of the nanocarriers were totally wasted due to their low possibility to reach the tumor sites in vivo, let alone the therapeutic effect. Therefore, our strategy, in a novel way, is to incorporate MPS and convert this limitation into a conduit to channel the immunotherapy into our tumor treatment scheme combined with chemotherapy.
Previously we reported the TH peptide modified liposomes could efficiently deliver PTX (PTX-TH-Lip) to tumor sites [10]. The cell-penetrating capability of TH (AGYLLGHINLHHLAHL(Aib)HHIL-Cys) could be only revealed within acidified microenvironment due to the protonation effect of imidazole ring [11]. The PTX-TH-Lip leaded to stronger inhibition against tumor growth comparing to conventional long-circulating liposomes (PEG-Lip). Meanwhile, the considerable accumulation of PTX-TH-Lip at the spleen and liver also draw our attention. We carefully selected a unique immunologic adjuvant αGC to be incorporated with PTX into the TH-modified liposome. αGC, as a glycolipid antigen, has been proved to specially stimulate invariant Natural Killer-like T (iNKT) cells after being presented on CD1d molecules in antigen presenting cells (APCs). Subsequently, it activates both innate and adaptive immunity via the secretion of large amounts of interferon (IFN)-γ from activated iNKT cells, which is expected to exert antitumor effects independent of the specific class of tumor [12]. Notably, the iNKT cells as the target of αGC were found in high numbers in spleen, liver and bone marrow [13], suggesting that the destination of αGC delivery in vivo should be the spleen and liver, particularly the spleen where abundant APCs existed [14]. Nevertheless, αGC could not be applied in soluble form due to the induction of long-term anergy of iNKT cells after a single i.v. administration. The reason is that B cells, unlike dendritic cells (DCs) and macrophages, lack proper secondary and tertiary signals, and therefore offer non-selective presentation [15, 16]. Efforts to solve this problem have been greatly laid on nano-formulated αGC, due to their preferable uptake by dendritic cells [15, 17]. Prakash et al. reported that nanoparticle-formulated αGC could repeatedly stimulate iNKT cells without inducing hyporesponsiveness after systemic administration [15]. Takashi et al. also verified that R8-modified αGC-loaded liposomes could improve the accumulation of αGC in spleen and exert impressive anti-metastasis potential in the B16F10 melanoma model [14]. Therefore, we assumed that our TH-modified liposomes also could shield αGC from the uptake by B cells and subsequently avert the anergy of iNKT cells.
Thus in this study, we designed the (PTX/αGC-TH-Lip) based on the following hypothesis: there are two major directions for intravenously injected PTX/αGC-TH-Lip during the circulation. On one hand, a small part of them gradually accumulate at the tumor sites via EPR effect where the TH peptide could assist these liposomes to penetrate tumor cells under acidified tumor microenvironment, killing the tumor cells directly. On the other hand, a majority of the liposomes finally settle at the spleen and liver where the αGC would be presented by CD1d-expressing APCs and get recognized by iNKT cells, triggering the following antitumor immune response. In addition, PTX-mediated cell death would help to release tumor-associated antigens into the surrounding milieu [18], which could further strengthen the specific antitumor immunity stimulated by αGC, embodied as an improved tumor-specific CD8+ T cell cytotoxicity. Mounting evidence indicated that PTX could also affect immune system as an immune adjuvant more than an inhibitor of microtubule polymerization [19, 20]. It was also reported that PTX could enhance tumor vaccine efficacy in low doses [21], and nanoparticle-formulated PTX could retain its immune stimulatory activity and induce the DC maturation and cytokine production [22]. Therefrom, we believe that PTX could be advantageous to αGC presentation and the downstream immune response after co-delivery with αGC to the spleen tissue by PTX/αGC-TH-Lip. By this means, the liposomes that should have been wasted in spleen could be reutilized, introducing the antitumor immunity into our scheme for tumor therapy incorporated with the conventional tumor-targeting chemotherapy. In other words, the cytotoxicity against tumor cells initiated by PTX at tumor sites and the antitumor immune response provoked by αGC at spleen tissue could function simultaneously and reinforce each other, leading to a better antitumor effect. To verify the proposed novel concept, we managed a number of experiments on the PTX/αGC-TH-Lip, comprising the investigation of whether the co-entrapped PTX and αGC would impair each other; whether the PTX/αGC-TH-Lip would prevent the hyporesponsiveness of iNKT cells induced by free αGC; the αGC presentation and alterations of immune status and also the evaluation of the antitumor effects and anti-metastasis potentials of PTX/αGC-TH-Lip in the B16F10 melanoma model.
2. Materials and Methods
2.1 Materials
TH peptide with a terminal cysteine (AGYLLGHINLHHLAHL(Aib)HHIL-Cys) was synthesized according to the standard solid phase peptide synthesis by China peptides Co. Ltd. (Shanghai, China). SPC was purchased from Shanghai Taiwei Chemical Company. Cholesterol was purchased from Chengdu Kelong Chemical Company. DSPE-PEG2000 and DSPE-PEG2000-Mal were purchased from Shanghai Advanced Vehicle Technology L.T.D. Co. Paclitaxel was purchased from AP Pharmaceutical Co. Ltd. (Chongqing, China). αGC (KRN7000) was purchased from Cayman chemical(Ann Arbor, Michigan 48108 USA), Anti-mouse alpha GalCer:CD1d Complex, rat anti-mouse I-A/I-E, FITC-label anti-mouse CD86, FITC-labeled anti-mouse CD80, FITC-labeled anti-mouse CD40, FITC-labeled anti-mouse CD14 PE-labeled anti-mouse CD11c, FITC-labeled anti-mouse CD8α and FITC-labeled anti-mouse NK1.1 were purchased from eBioscience (San Diego, CA, USA).FITC-labeled goat anti-mouse secondary antibody and Rhodamine labeled goat anti-rat secondary antibody were purchased from ZSGB-BIO (Beijing, China). IFN-γ Elisa kit, IL-2 Elisa kit and IL-4 Elisa kit were purchased from R&D system (Minneapolis, USA). 1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine, 4-chlorob-enzenesulfonate salt (DiD) were purchased from Biotium (Hayward, CA, USA).
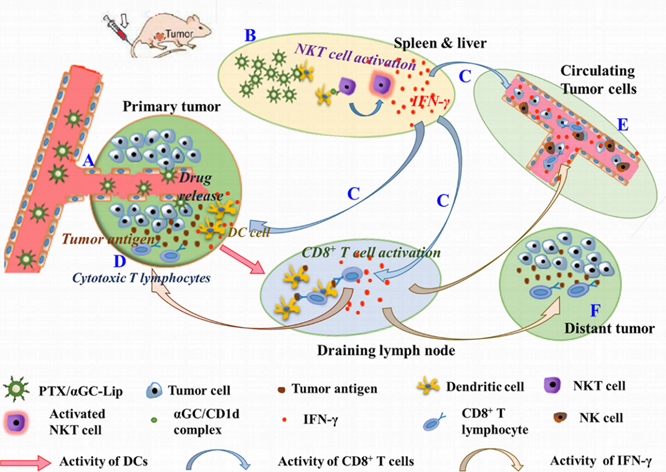
Diagram of PTX/αGC-TH-Lip mediated combination of chemotherapy and immunotherapy against tumor. The intravenously injected PTX/αGC-TH-Lip will function at two major sites during circulating in vivo: A. A relatively small part of the liposomes will accumulate at tumor sites (primary tumor), with PTX killing the tumor cells directly. B. A large proportion of the liposomes will finally settle at the spleen and liver, where the αGC would be presented on the CD1d molecules in APCs and further activate the iNKT cells, leading to the ongoing production of large amounts of IFN-γ. C. The IFN-γ could subsequently activate other cell types, such as NK cells and macrophages in the innate immune system as well as CD8+ T cells in the acquired immune system. D. The PTX-induced tumor cell death in tumor sites could boost the tumor-associated antigens release and the tumor antigen presentation process is stimulated by IFN-γ secreted from iNKT cells, bringing about an amplified tumor-specific CD8+ T cells cytolytic effect. E. The activated NK cells, macrophages and CD8+ T cells would contribute a lot to eliminating the circulating tumor cells and ease metastasis status. F. The tumor-specific CD8+ T cells enter the systemic circulation and will be recruited to tumors at distant sites to trigger the “effector phase” of the adaptive immune response.
2.2 Cells and animals
B16F10 murine melanoma cells and DC2.4 cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% FBS, 100 U/mL streptomycin, and 100 U/mL penicillin at 37 °C in a humidified 5% CO2 atmosphere.
Female C57BL/6 (6 to 8 weeks old) mice were purchased from experiment animal center of Sichuan University (P.R. China). All animal experiments were performed in accordance with the principles of care and use of laboratory animals and were approved by the experiment animal administrative committee of Sichuan University.
2.3 Preparation and characterization of liposomes
Before the preparation of liposomes, DSPE-PEG2000-TH was synthesized in accordance with the process described in the previous study of our laboratory [10]. Briefly, we dissolved DSPE-PEG2000-Mal (3 μmol) and Cys-TH (4.5 μmol) in the mixture of chloroform and methanol (v/v =2:1) containing triethylamine (6 μmol). Then we allowed the above mixed solution to react in the presence of argon for about 24 h in darkness under room temperature. The thin film hydration method was adopted to prepare the liposome. Lipid compositions of the prepared liposomes were as follows: Cholesterol / SPC / DSPE-PEG2000 / DSPE-PEG2000-TH (molar ratio = 33:59:2:6). All the components were dissolved in the mixture of chloroform and methanol (v/v = 2:1). Then the organic solvent was removed by rotary evaporation, and the formed film was dried and stored in vacuum overnight. 1.0 mL of 10 mmol/L Hepes (pH 7.4) was added to the lipid film, and the mixture was hydrated under 37 °C for 30 min with mild oscillation. Then it was further intermittently sonicated by a probe sonicator at 80 W for 100 s to produce the TH peptide modified liposome (TH-Lip). In the preparation of the conventional liposome (without the TH modification), the DSPE-PEG2000-TH would be replaced by commensurable DSPE-PEG2000.
PTX-loaded, DiD-loaded, or αGC-loaded liposomes were prepared with appropriate amount of DiD, PTX or αGC added to the lipid organic solution before the rotary evaporation, respectively. The entrapment efficiency of PTX was determined by HPLC (Agilent1200, USA). For αGC appears to act as a lipid (like SPC and chol), the entrapment ratio of αGC into a liposome is considered to be 100% [14]. Malvern Zetasizer Nano ZS90 (Malvern Instruments Ltd., UK) was used to determine the mean size and zeta potential of different groups of liposomes.
Besides, free PTX and αGC was prepared as follows: PTX was dissolved in a mixture of ethanol-cremophor ELP 35 with a volume ratio of 1:1, and was diluted to required concentrations with 10 mmol/L Hepes. Soluble αGC was prepared by dissolving it into 10 mmol/L Hepes containing 5.6% sucrose, 0.75% l-histidine and 0.5% Tween 20 and heated at 80 °C for several minutes [14]. In all experiments, free drugs indicated the mixture of free PTX and αGC, in which the ratio of PTX to αGC was consistent with that of the PTX/αGC-TH-Lip.
2.4 In vitro stability of liposomes in serum and PTX release study
The TH peptide modified PTX-loaded liposome was previously constructed in our lab [10]. The purpose we repeated these basic in vitro studies was to ensure that αGC had little influence on the liposome stability and PTX release efficiency. The turbidity variations of liposomes were monitored to evaluate the serum stability of liposomes during incubation with 50% fetal bovine serum (FBS). Briefly, different liposomes were mixed with equal volume of FBS and the mixtures were incubated under 37 °C with gentle oscillating for 48 h. 200 μL of the sample was transferred to a quartz cuvette by pipette and the transmittance at 750 nm was measured by a microplate reader (Thermo Scientific Varioskan Flash, USA) at predetermined time points (0 h, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h and 48 h).
In vitro PTX release study was conducted with the dialysis method. 50 ml PBS (pH 7.4) containing 0.1% Tween 80 (v/v) was utilized as the release medium. 0.6mL PTX-loaded liposome or free PTX was added into cellulose acetate dialysis tubes (MWCO 8-14 kDa), which were tightly sealed and then immersed into the release medium, followed by mild shaking at 50 rpm under 37 °C for 24 h. At predetermined time points, 0.1 mL release medium was sampled and the release medium ought to be replenished by equal volume of fresh medium. Then the samples were diluted with acetonitrile and the concentrations of PTX were analyzed by HPLC.
2.5 In vitro cytotoxicity study
The cytotoxicity of every PTX-preparation was measured by MTT assay. B16F10 cells were seeded into a 96-well plate in quintuplicate at a density of 2×103 cell/well and cultured for 24 h. PTX/αGC-TH-Lip, PTX-TH-Lip + IFN-γ and PTX-TH-Lip were diluted to predetermined concentrations and added to each well for 24 h incubation. The final concentration of αGC entrapped in the liposomes was 1.25 μg/mL after dilution, and free IFN-γ was adopted as an additive with final concentration of 25 ng/mL. PTX concentration varied from 0.1 μg/mL to 20 μg/mL. After incubation, 20 μL of MTT solution (5 mg/mL in PBS) was added into each well and further incubated for 4 h under 37 °C. Then the medium was removed, and the formazan was dissolved by 150 μL dimethyl sulfoxide. The absorbance at 570 nm was then measured. The cells only treated with medium were evaluated as controls. Cell viability (%) was calculated by the following formula: cell viability (%) = Atreated/Acontrol × 100%, where Atreated and Acontrol represented the absorbance of treated cells and control cells, respectively.
2.6 In-vitro uptake of PTX/αGC-TH-Lip by DC2.4 cells
2.6.1 Flow cytometry study
1×106 DC2.4 cells were seeded per well onto a 6-well plate in 1 mL of DMEM supplemented with 10% FBS and 1% antibiotic solution, and allowed for attachment to the plate under 37 °C and 5% CO2 for 24 h. The CFPE-labeled liposomes were then added at a final CFPE concentration of 2 μg/mL, and liposome-free culture medium was applied as the control group. After incubation for 6 h, the cells were washed with cold PBS for three times, and resuspended in 0.3 mL PBS after being trypsinized. The fluorescent intensity of cells was measured by a flow cytometer (Cytomics FC 500, Beckman Coulter, USA), with the excitation wavelength at 495 nm and the emission wavelength at 515 nm. 1×104 cells were recorded for each sample.
2.6.2 Confocal laser scanning microscopy (CLSM)
DC2.4 cells were plated at a density of 1×105 cells/well on cover slip in a 6-well plate. After incubation for 24 h, CFPE-labeled liposomes were applied to each well and allowed for further incubation for 6 h. 30 min before the end of the incubation, Lysotracker Red was added to each well. Then the cells were washed with cold PBS for three times and fixed with 4% paraformaldehyde at room temperature for 15 min, following by nuclei staining with DAPI for 5 min. Cells were observed by a laser scanning confocal microscope (TCS SP5 AOBS confocal microscopy system, Leica, Germany).
2.7 Ex-vivo organ distribution study of the liposomes
DiD-loaded liposomes were prepared in accordance with the process described in Section 2.3 and were injected to C57BL/6 mice intravenously at a dose of 500 μg DiD/kg. The mice were sacrificed at predetermined time points (1 h, 4 h, and 24 h after injection), and the hearts, livers, spleens, lungs, kidneys and tumors were harvested. All the organs were imaged with IVI Spectrum system (Caliper, Hopkington, MA, USA).
2.8 Measurement of IFN-γ level in serum after αGC stimulation in vivo
C57BL/6 mice bearing B16F10 melanoma tumor were employed in this study, while the average volume of grown tumors was controlled between 50-100 mm3 before any further action. 20 mice were selected and assigned into four groups randomly, and αGC-TH-Lip with different αGC concentration (from 0 to 25 μg/mL) was intravenously injected into mice of each group (injection volume: 200 μL). 20 h later, blood samples were collected through heart puncture with syringes and centrifuged at 3000 rpm for 5 min after the blood clotting. The supernatants were collected as serum samples.
Another 10 mice were randomly divided into two groups (n=5), free αGC and αGC-TH-Lip were intravenously administrated respectively as the first treatment at a dose of 5 μg αGC. 7 days later, the mice of each group received the second αGC challenges to analyze the recall response of iNKT cells. Serum samples were respectively prepared after the first and second treatments.
In the case of PTX and αGC were incorporated into the liposome, we also evaluated whether PTX would get involved in the iNKT cell activation induced by αGC. A series of liposomes were prepared with the PTX concentration ranging from 0 to 0.6 mg/mL, while the αGC concentration was restricted at 25 μg/mL. 7 groups of tumor bearing mice were employed and each group (n=5) was given 200 μL of different PTX/αGC-TH-Lip. Serum samples were harvested as described above.
Besides, whether the TH-modification on the liposome could affect the activation process of iNKT cell was also evaluated. 2 groups (n=5) of tumor-bearing mice were employed and respectively given 200 μL of αGC-TH-Lip and αGC-Lip with αGC concentration at 25 μg/mL. Serum samples were harvested as described above.
An ELISA kit (R&D systems, Basel, Switzerland) was utilized to measure the IFN-γ level in serum. These experiments were conducted in strict accordance with the manufacturer's instruction.
2.9 αGC presentation on CD1d in antigen presenting cells in vivo
Free αGC or αGC liposome preparations were intravenously injected at a dose of 5 μg. Free αGC was prepared as described in Section 2.3. 18 h after administration, the spleens were collected, and splenocytes were prepared by using the Spleen Dissociation kit (Miltenyi Biotec Germany). 1×107 splenocytes were incubated with anti-mouse αGC/CD1d Complex and anti-mouse I-A/I-E at 4 °C for 2 h, the mixture was centrifuged and the supernatant was discarded to remove the redundant first antibody, followed by further staining with the FITC-labeled goat anti-mouse secondary antibody and Rhodamine labeled goat anti-rat secondary antibody. The fluorescence of stained cells was measured by a flow cytometer (Cytomics FC 500, Beckman Coulter, USA).
2.10 Evaluation of in-vivo immune status after intravenous administration of PTX/αGC-TH-Lip
B16F10 melanoma tumor-bearing mice (C57BL/6) were also employed in this study. 7 days after tumor implantation, animals with an average tumor volume of 50-100 mm3 were selected and divided into 5 groups randomly (n=5). Hepes group, free drugs group (Free PTX+ Free αGC), PTX-TH-Lip group, αGC-TH-Lip group and PTX/αGC-TH-Lip group. Mice of each group were treated with i.v. injections twice, the mice were sacrificed 7 days after the last dose and their spleens were collected. Splenocytes suspensions were prepared by using the Spleen Dissociation kit (Miltenyi Biotec Germany). The cells were labeled with fluorescent marker antibodies for specific cell types: PE anti-mouse CD11c for Dendritic Cells, FITC anti-mouse CD8α for cytotoxic T lymphocytes, FITC anti-mouse NK1.1 for natural killer cells, FITC anti-mouse CD86 was applied in the analysis of maturation marker of Dendritic Cells.
2.11 The polarization of Th1/Th2 after administration of PTX/αGC-TH-Lip
The polarized Th1 and Th2 responses were investigated by determining the secretion of different cytokines. We regarded the level of IL-2 and IL-4 as the representatives of the Th differentiation into Th1 and Th2 subsets, respectively. The C57BL/6 mice were challenged with B16F10 tumor and grouped as done previously (n=5). The mice of each group were dosed on day 0 and day 3, and on day 6 and day 9 blood samples were collected through heart puncture with syringes and centrifuged at 3000 rpm for 5 min after the blood clotting. ELISA kits (R&D systems, Basel, Switzerland) were utilized to measure the IL-2 and IL-4 level in serum in strict accordance with the manufacturer's instruction.
2.12 Antitumor efficacy
2.12.1 Evaluation of the therapeutic effect of PTX/αGC-TH-Lip on B16F10 tumor
B16F10 melanoma cells were inoculated into the right flank of 6-8 weeks old C57BL/6 mice. 7 days after tumor implantation, animals with an average tumor volume of 50-100 mm3 were selected and divided into 5 groups randomly (n=7): Hepes group, free drugs group (Free PTX+ Free αGC), PTX-TH-Lip group, αGC-TH-Lip group and PTX/αGC-TH-Lip group. The mice of each group were dosed intravenously on Day 7, 10, 13 and 16, and the tumor volumes were measured with a vernier caliper every two days. According to the earlier results we obtained, the administration dosage of PTX and αGC was finalized at 3 mg/kg and 5 μg per mouse, respectively. On the 23rd day after tumor implantation, the tumors were resected from the euthanized mice and processed for paraffin sections and HE (hematoxylin and eosin) staining.
2.12.2 Cytotoxicity assay of cytotoxic T lymphocytes
In order to obtain the activated splenocytes as effector cell, 1 × 106 B16F10 cells were subcutaneously inoculated into the left flank of C57BL/6 mice on day 0. The average volume of tumors was approximately 100 mm3 on day 10, the mice were divided into 5 groups randomly to receive the following treatments: Hepes, Free drugs (Free PTX+ Free αGC), PTX-TH-Lip, αGC-TH-Lip and PTX/αGC-TH-Lip. Unless otherwise indicated, PTX was dosing at 3 mg/kg, and αGC was injected at a dose of 5 μg per mouse. The second treatment with the same dosing regimens was given to each group on day 15. 3 days later, the mice were sacrificed and the spleens were harvested. The Spleen Dissociation kit (Miltenyi Biotec Germany) were utilized to prepare the splenocytes solution, which were further incubated with mitomycin C-treated B16F10 tumor cells for 72 h at 37 °C under 5% CO2 with mouse IL-2 added into the culture media. After stimulation, viable splenocytes were used as effector cells for the measurement of specific cytolytic activity. And B16F10 cells were used as target cells. The cytotoxicity of cytotoxic T lymphocytes was tested by the lactate dehydrogenase (LDH) assay according to the previous method. 2×104 B16F10 cells/well were distributed in triplicate into a 96-well plate in 100 μL complete DMEM medium supplemented with 2% FBS. The stimulated splenocytes diluted in the same medium were added to target cells in triplicate at different E/T (effector/target) cell ratios of 20:1, 5:1, and 1:1 in a final volume of 200 μL. Spontaneous release was obtained from wells where target cells were cultured with medium alone. The maximal release of LDH was calculated from wells where target cells were treated with lysis buffer. The cell mixtures were incubated at 37 °C under 5% CO2. After 6 h, the supernatant was collected and tested for LDH in strict accordance with the manufacturer's instruction (Beyotime Shanghai China).
2.12.3 Evaluation of the inhibiting effect of PTX/αGC-TH-Lip on distant tumor
Tumor-specific inhibition efficacies of different preparations were also evaluated in animal model. The establishment of tumor in C57BL/6 mice with B16F10 cells was identical to the aforementioned method. A week after the tumor implantation, mice whose tumor volume approximated at 50-100 mm3 were selected and divided into 5 groups (n=7): Hepes group, free drugs group, PTX-TH-Lip group, αGC-TH-Lip group and PTX/αGC-TH-Lip group. The mice in each group received different treatments four times with 48 h dosing intervals. The primary tumors (right flank) were resected 7 days after the last injection, and meanwhile 2×106 B16F10 cells were subcutaneously inoculated into the left flank. Once being visible, the sizes of left tumors were recorded every two days.
2.13 Anti-tumor effects in lung metastatic tumor models
1×106 B16F10 cells suspended in 100 μL of PBS were injected into the tail veins of C57BL/6 mice on day 1. Then the mice were separated into 5 groups (n=5), and different groups received different PTX and αGC treatment only once on day 4. On day 15, one animal from each group was randomly selected and sacrificed, so did on day 18 and 24. The lung tissues were collected and the macroscopic tumor nodules on the whole surface were counted.
2.14 Statistical analysis
All the data were presented as mean ± standard deviation. Statistical comparisons were performed by analysis of variance (ANOVA) for multiple groups, and p value <0.05 and 0.01 were considered indications of statistical difference and statistically significant difference respectively.
3. Results
3.1 The construction of PTX/αGC-TH-loaded liposome
The preparation of TH-modified PTX-loaded liposome was performed by a mature method in our group [10]. However, it would be interesting to see if this reported method still works in the preparation of PTX and αGC co-loaded TH-modified liposome.
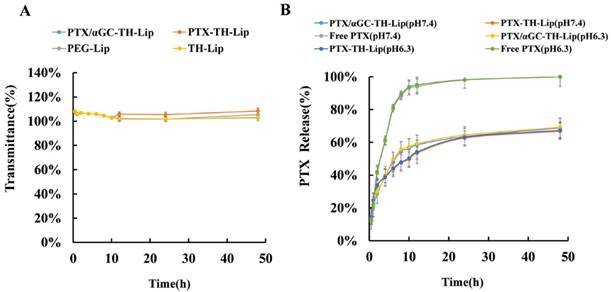
Particle sizes and zeta potentials of PTX/αGC-TH-Lip and PTX-TH-Lip in different pH (pH 7.4 and pH 6.0) are listed in Table 1. Both parameters exhibit no significant variation, regardless of αGC being added or not. And the entrapment efficiencies of PTX in both kinds of liposomes are above 90%. Moreover, the serum stability of liposomes and PTX release efficacy were also investigated following the established method to confirm that the construction of a sound liposome was unaffected by the new lipid composition (αGC). 50% FBS was employed to mimic the in vivo environment, and the transmittance at 750 nm showed little change for both PTX/αGC-TH-Lip and PTX-TH-Lip over 48 h (Figure 1A). The results suggested that there was no significant aggregation in the presence of FBS. In order to obviate the effect associated with the ligand or the liposome itself, two empty liposomes (TH-Lip and PEG-Lip) were applied as control. The in vitro PTX release properties in different pH were then assessed with a dialysis method. The results indicated that PTX-TH-Lip and PTX/αGC-TH-Lip exhibited a similar sustained drug release behavior over 48 h, while free PTX carried out a more rapid release in the media within only 10 h (Figure 1B). In short, no obvious impact initiated by the addition of αGC was observed in the preparation of liposomes.
The characterization of PTX/αGC-TH-Lip. (A) The variations in turbidity (represented by transmittance) of PTX/αGC-TH-Lip, PTX-TH-Lip, TH-Lip and PEG-Lip in 50% FBS (n=3, mean ± SD). (B) The PTX release profiles of free PTX, PTX/αGC-TH-Lip and PTX-TH-Lip in PBS over 48 h under different pH conditions (n=3, mean ± SD).
The particle sizes and zeta potentials of PTX-TH-Lip and PTX/αGC-TH-Lip under different pH conditions. The “PTX” and “PTX/αGC” prefixes indicate PTX-loaded and PTX/αGC co-loaded liposomes (n=3, mean ± SD).
| Liposome | Size (nm) | PDI | zeta-potential (mV) |
|---|---|---|---|
| PTX-TH-Lip (pH6.0) | 117.9 ± 4.6 | 0.228 ± 0.04 | 3.34 ± 0.5 |
| PTX-TH-Lip (pH7.4) | 116.2 ± 3.2 | 0.226 ± 0.07 | -6.68 ± 0.7 |
| PTX/αGC-TH-Lip (pH6.0) | 119.2 ± 5.7 | 0.188 ± 0.03 | 3.18 ± 0.3 |
| PTX/αGC-TH-Lip (pH7.4) | 113.9 ± 3.6 | 0.216 ± 0.05 | -8.27 ± 0.6 |
3.2 Organ distribution and tumor delivery of liposomes
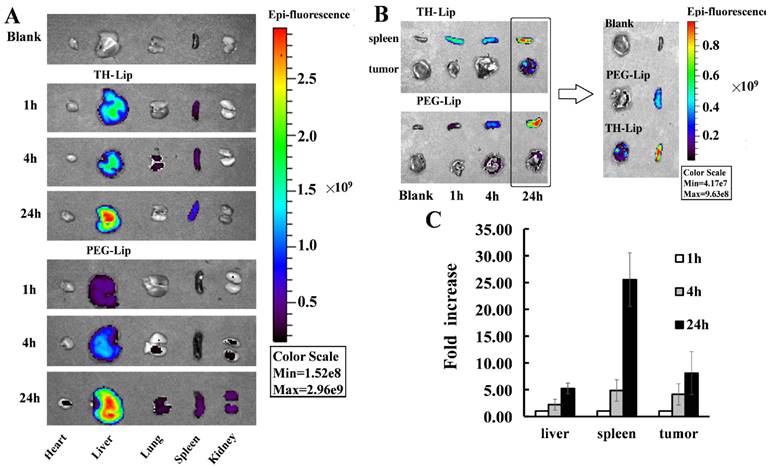
The B16F10 tumor bearing mice were established, DiD as one near-infrared probe was used to label the liposomes. First, the original distribution behavior of the liposomes in different organs and tumors were investigated. Second, we tested if the TH peptide modification could alter the distribution behavior of the liposome. Two groups of mice were injected with TH-Lip and PEG-Lip respectively. One mouse of each group was sacrificed at predetermined time points, and dissected organs and tumors were collected for imaging. As shown in Figure 2A, the fluorescence signals of both TH-Lip and PEG-Lip became significantly stronger in livers and spleens from 1 h to 24 h while much weaker (by more than one order of magnitude) signals were observed for both TH-Lip and PEG-Lip in tumors, although there was a slight increase within the same time window (Figure 2B). That is to say, moderate improvement in tumor targeting efficiency of ligand-modified liposomes could never disguise the inevitable large accumulation in liver and spleen [9]. Then, we particularly compared the distribution patterns between these two groups 24 h post-injection. It is manifest that the fluorescence signals of TH-Lip group were stronger than that of PEG-Lip group in both spleens and tumors (Figure 2B), suggesting that TH-Lip could achieve relatively higher accumulation within tumor tissues after surface charge conversion (from negative to positive) under the tumor microenvironment. Moreover, the TH peptide modification may enable the liposomes to be easily recognized by the mononuclear phagocyte system, leading to an advanced transfer of liposomes to the biggest immune organ—spleen, which was a plausible explanation for the stronger fluorescence signals of TH-Lip group. Additionally, TH-Lip and PEG-Lip exhibited parallel distribution status in other organs, due to their comparable PEGylation degree, similar particle size and close surface charge (data not shown) [10]. As the PTX/αGC-TH-Lip was TH modified, the fold increase of fluorescence signals of TH-Lip in livers, spleens and tumors at different time points was estimated by semi-quantitative analysis - the fluorescence intensity ratio of a specific time point to 1 h. Of note, the fluorescence signal in the spleen (25-fold at 24 h) displayed a much more rapid increase compared to that in liver (5-fold at 24 h) or the tumor (9-fold at 24 h) (Figure 2C). This demonstrated the existence of sustainable and relatively high level accumulation of the liposomes in spleen for at least 24 h. All these results are consistent with our preliminary perspective: liposomes that injected into the circulation were unable to massively accumulate in the tumor sites only by EPR effect, while the uptake of liposomes by spleen or liver was also impressive. Inspired by this distribution phenomenon, we constructed the PTX/αGC co-loaded liposome, in which αGC played a major role in spleen tissue and triggered the downstream immune response against tumor, and meanwhile, PTX took the lead and exerted its antitumor effect directly at tumor sites. Combining both aspects into the regimen for treating tumor, better anti-tumor effect could be expected compared with liposomes carrying either drug alone (αGC or PTX).
The accumulation of liposomes in organs and tumors. (A) Ex vivo analysis of organs from B16F10 tumor bearing mice by NIR fluorescence imaging at 1 h, 4 h and 24 h post-injection of DiD-loaded TH-Lip (upper) or PEG-Lip (lower). (B) NIR fluorescence imaging of spleens and tumors from B16F10 tumor bearing mice at 1 h, 4 h and 24 h post-injection of DiD-loaded TH-Lip (upper) or PEG-Lip (lower), and the comparison of the fluorescence intensity in spleen and tumor at 24 h between DiD-loaded TH-Lip and PEG-Lip. (C) The fold increase of fluorescence intensity in liver, spleen and tumor (the fluorescence intensity of each tissue at 4 h or 24 h / that at 1 h). Values are the mean ± SD (n=3).
3.3 IFN-γ released from iNKT cells stimulated by αGC in vivo
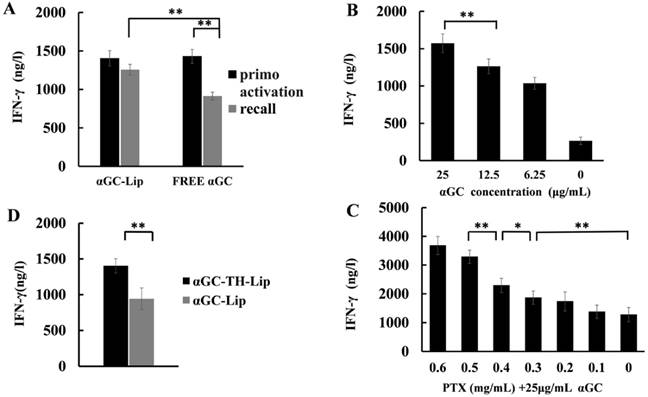
As mentioned in the introduction, the iNKT cells exhibited a diminished response to αGC stimulation after repeated i.v. injection, ascribed to the non-selective presentation of αGC by B cells, together with the down-regulation of the TCR expression on the surface of iNKT cells [15, 16]. Fortunately, some previous studies had pointed out that nanoparticle formulated αGC could avoid the hyporesponsiveness of iNKT cells after restimulation. Thus in this work, analogous sheltering effects of our liposomes would be the prerequisite for our following studies. As shown in Figure 3A, the IFN-γ production that implied the iNKT cells activation status was positively correlated with the concentration of αGC entrapped in liposome in the range from 0 to 25 μg/mL. Taking this result and some relevant research outcomes into account [14, 15, 23], 25 μg/mL (5 μg per mouse) was determined as the final concentration of αGC in all the in vivo experiments. In terms of the iNKT cells reactivation assay, the IFN-γ level of αGC-TH-Lip group remained high after the second injection. In sharp contrast, the IFN-γ production of free αGC group was significantly reduced after the readminstration (Figure 3B), which denoted that this liposome-formulated αGC could efficiently overcome the anergy of iNKT cells induced by soluble αGC. Given PTX and αGC were entrapped together in the liposome, we need to clarify the role of PTX in the αGC-induced activation of iNKT cells. 7 groups of B16F10 tumor bearing mice (n=5) were injected with a series of PTX/αGC-TH-Lip with PTX concentration ranging from 0 to 0.6 mg/mL. Interestingly, the IFN-γ secretion status 20 h post-injection of each group presented a growing trend as the PTX concentration rised (Figure 3C). Especially when PTX concentration in the liposome was over 0.3 mg/mL (0.3-0.6 mg/mL), the IFN-γ production was significantly higher compared to that of the PTX concentration in lower range (0-0.2 mg/mL). This behavior indicated that PTX exhibited moderate synergistic interaction with αGC, instead of weakening the capacity of αGC to activate iNKT cells. PTX, as one of the classical anti-tumor agents, was verified to be an immunologic adjuvant and arouses contribution to immune responses [20, 21, 24]. This is the most plausible explanation for the enhanced IFN-γ production from iNKT cells in the presence of αGC and PTX, compared to liposomes that were loaded with αGC alone. Except for the PTX, we also evaluated whether the TH peptide modification also influenced the enhanced IFN-γ secretion (Figure 3D). Interestingly, it showed that the TH modification was conducive to the activation on iNKT cells, resulting in increased IFN-γ production compared to conventional liposome (without TH modification). In words, the PTX/αGC-TH-Lip could not only repeatedly stimulate iNKT cells without leading to anergy, but also significantly promote the IFN-γ production in comparison with αGC-TH-Lip alone.
The secretion of IFN-γ after intravenous administration of αGC formulations or free αGC. (A) The production of IFN-γ after intravenous administration of αGC-loaded liposomes with the concentration of αGC ranging from 0 to 25 μg/mL. (B) The production of IFN-γ after primary and secondary administration of liposome-formulated αGC or free αGC (αGC concentration was 25 μg/mL, and the injection volume was 200 μL), the following operation was the same as above. (C) IFN-γ production after application of different PTX/αGC-TH-Lips in which the concentration of PTX was adjusted to 0, 0.1, 0.2, 0.3, 0.4, 0.5 and 0.6 mg/mL, respectively (αGC concentration was 25 μg/mL, and the injection volume was 200 μL). (D) IFN-γ production after application of αGC-TH-Lips or αGC-Lips. (n=5, *P < 0.05, **P < 0.01).
3.4 αGC presentation on CD1d in APCs in vivo
The presentation of αGC on CD1d molecules in APCs in spleen after different treatments was also evaluated. The names of different formulations represent the modification status of different liposomes; for example, the αGC-TH-Lip means the TH-modified liposomes loaded with αGC only, whilst the PTX/αGC-Lip indicates the liposomes loaded with both PTX and αGC without the TH modification. The splenocytes harvested from each group were stained with anti-αGC/CD1d and anti-major histocompatibility complex (MHC) class II antibodies, followed by further staining with related fluorescent secondary antibodies. Figure 4 illustrates the typical dot diagrams of splenocytes from each group. The top right area demonstrates the distribution of cells co-stained with anti-αGC/CD1d and anti-MHC-II antibodies. Compared with free αGC (Figure 4B), αGC loaded liposomes (Figure 4C-E) were better at promoting αGC presentation on CD1d in APCs, indicating the liposome formulation was beneficial to the αGC presentation process. In comparison to αGC-TH-Lip group (Figure 4C), PTX/αGC-TH-Lip group (Figure 4E) displayed a significantly increased number of co-stained cells, which indicated the fact that PTX can effectively stimulate the αGC presentation on CD1d by APCs as an immunologic adjuvant [19, 22, 24].
αGC presentation on CD1d molecules in antigen presenting cells (APCs). APCs presenting αGC on CD1d were identified as MHC-II+αGC/CD1d+ cells. (A-E) The typical dot plots of the splenocyte derived from non-treated mouse, free αGC treated mouse αGC-TH-Lip treated mouse, PTX/αGC-Lip treated mouse and PTX/αGC-TH-Lip treated mouse. The numbers in the top right area indicate the percentage of MHC-II+ αGC /CD1d+ cells. (F) The quantitative analysis of flow cytometry data. Values are the mean ± SD (n=3, *P <0.05).
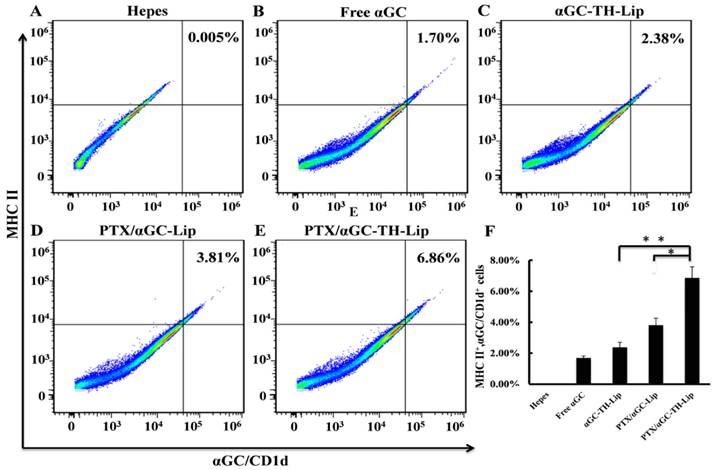
Furthermore, the PTX/αGC-TH-Lip (Figure 4E) also induced a higher percentage of co-stained cells than PTX/αGC-Lip group (Figure 4D), offering a new notion that TH modification could not only promote the tumor cell penetration but also conduce to αGC presentation. Quantitative analysis of the above flow cytometry data is illustrated in Figure 4F. It is worth noting that the amount of co-stained cells of PTX/αGC-TH-Lip group was higher by a factor of 3.2 and 1.74 than that of αGC-TH-Lip group and PTX/αGC-Lip, respectively. Hence, these results indicated that when the liposomes involuntarily accumulated in spleen after systemic administration, the PTX/αGC-TH-Lip was the most competent one to deliver αGC to APCs and enhance αGC presentation.
3.5 In vitro study of cellular uptake of TH-modified liposome by DC2.4 cell
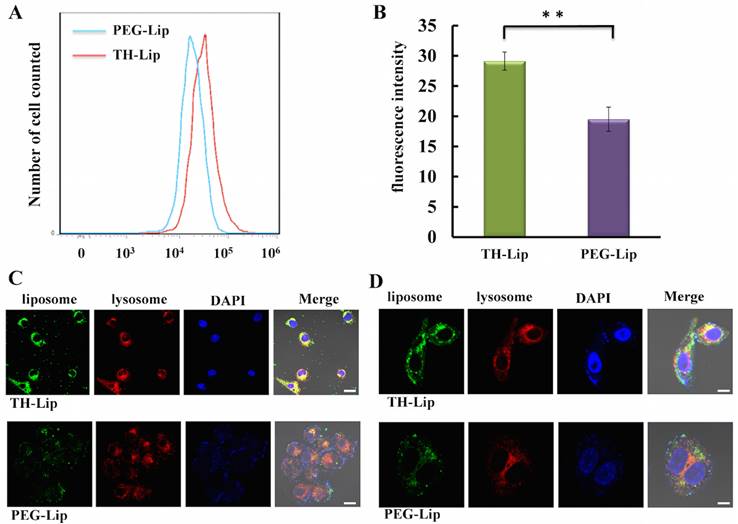
From the obtained data, it is speculated that there is a close correlation between the increased accumulation of liposomes in spleen (Figure 2B), the promoted IFN-γ secretion (Figure 3D), the enhanced αGC presentation status in APCs (Figure 4D-E) and the TH modification. Therefore, we studied the cellular uptake of TH-Lip and PEG-Lip on DC2.4 (a murine dendritic cell line) to verify our speculation. As shown in Figure 5A, TH-Lip exhibited obviously higher cellular uptake than PEG-Lip, which indicated the existence of TH peptide on the surface could render the liposomes more susceptible to dendritic cells. The quantitative analysis of flow cytometry data was displayed in Figure 5B. The CLSM pictures further demonstrated the cellular trafficking of TH-Lip and PEG-Lip. After 6 h incubation, the liposome fluorescence (green) of TH-Lip could be clearly observed in the DC2.4 cells, while the fluorescence of PEG-Lip was randomly scattered around the cell membrane (Figure 5C). Furthermore, the TH-Lip group also exhibited increased overlapping of fluorescence-labeled liposomes (green) and stained lysosomes (red) compared to PEG-Lip, which denoted the enhanced cellular intake of TH-Lip (Figure 5C). Underpinned by the cellular uptake study, we could preliminarily conclude that the TH modification was conducive to the uptake of liposomes by APCs, which could further promote the αGC presentation in the case of αGC-loaded liposomes.
Cellular uptake of CFPE-labeled TH-Lip and PEG-Lip on DC2.4 cell. (A) The fluorescence peak of TH-Lip and PEG-Lip on flow cytometry histogram. (B) The quantitative result of the fluorescence intensity in DC2.4 cells incubated with TH-Lip or PEG-Lip. Values are the mean ± SD (n=5, **P <0.01). (C) — (D) The confocal images of cellular uptake of TH-Lip and PEG-Lip with different magnification. Scale bars represent 20 μm and 5 μm, respectively.
3.6 BMDCs maturation after the activation of iNKT cells
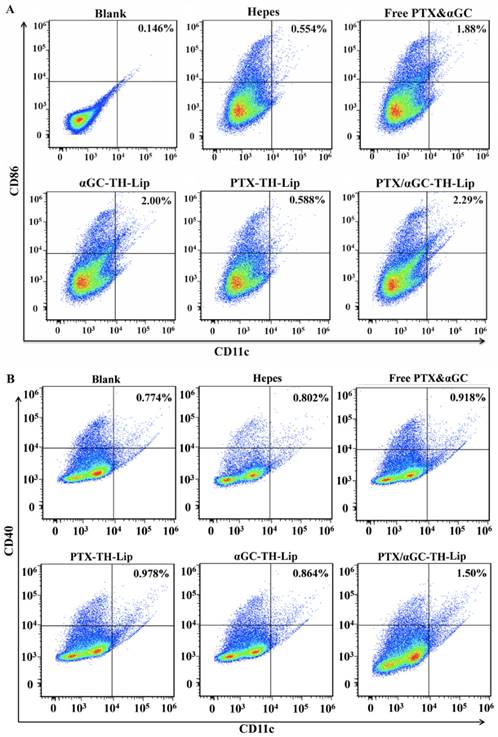
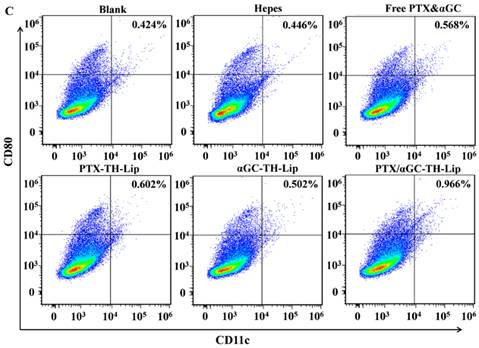
In this study, the aim of activating iNKT cells by delivering αGC to spleen was to stimulate the cellular immunity against tumor antigen. Thus, the maturation of bone marrow derived dendritic cells (BMDCs) became particularly important, which was essential for the tumor antigen presentation [25]. To evaluate whether the abundant IFN-γ secreted from activated iNKT cells could contribute to the maturation process of BMDCs, the expression status of maturation markers CD86, CD80, CD40 were analyzed on the CD11c+ dendritic cells from spleen [26]. Figure 6 illustrates the typical dot diagrams of splenocytes from each group. The top right area demonstrates the distribution of CD11c+ cells stained with anti-CD86 (Figure 6A), anti-CD40 (Figure 6B) and anti-CD80 (Figure 6C) antibodies. Treatments with αGC including Free PTX&αGC, αGC-TH-Lip and PTX/αGC-TH-Lip significantly induce higher percentage of co-stained BMDCs compared with treatments without αGC, such as Hepes and PTX-TH-Lip. This suggested that the production of IFN-γ from iNKT cells could boost the maturity of BMDCs and further benefit the antigen presentation process. Additionally, the PTX/αGC-TH-Lip group demonstrated a relatively higher level of co-stained cells in the comparison with αGC-TH-Lip group, which was probably attributed to the adjuvant effect of PTX. In one word, the PTX/αGC-TH-Lip showed the best immune stimulating ability in contrast to all the control groups.
The expression status of CD86, CD40 and CD80 on CD11c+ dendritic cells from spleens. Diagrams are the typical dot plots of the splenocyte derived from non-treated mouse, Hepes treated, free PTX&αGC treated mouse, αGC-TH-Lip treated mouse, PTX-TH-Lip treated mouse and PTX/αGC-TH-Lip treated mouse. The numbers in the top right area indicate the percentage of CD86+CD11c+cells (A), CD40+CD11c+cells (B) and CD80+CD11c+cells (C).
3.7 In vivo status of immune cells
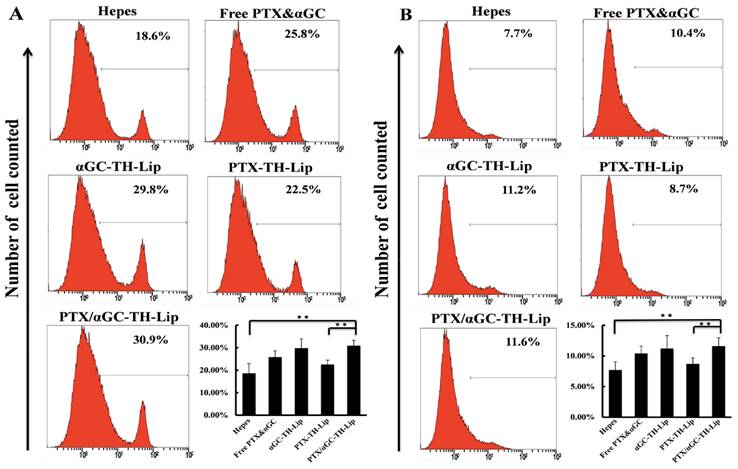
To further test the in vivo immuno-stimulation after treatment with different preparations, we focused on two classes of cells that are closely related to tumoricidal action. One class is the CD8α+ T cells that dominate cytotoxicity and always plays a vital role in the immune-killing of tumor cells [27]. The other class, like NK cells and macrophages, could participate in eliminating the circulating tumor cells and inhibit the metastasis of primary tumors [28, 29]. Therefore, we measured the relative populations of CD8α+ T cells, NK cells and macrophages in spleens after different treatments to evaluate the in vivo immune status associated with the suppression of tumor development. Figure 7A and 7B illustrate the percentages of CD8α+ T cells and NK cells in the splenocytes, respectively. After the treatments with αGC (including Free PTC&αGC, αGC-TH-Lip, PTX/αGC-TH-Lip), the relative quantities of CD8α+ T cells and NK cells were significantly increased compared with those treatments without αGC (including Hepes and PTX-TH-Lip). It reveals that the IFN-γ secreted from αGC-activated iNKT cells was also capable of inducing the proliferation of CD8α+ T cells, NK cells and macrophages. This adjustment of immunity condition could pave the way for the further antitumor effects combined with chemotherapy. The percentage of macrophages in splenocytes was displayed in (Figure S2).
Relative populations of immune cells (CD8α+ T cells, NK cells) in spleens at Day 7 after twice systemic administrations of samples. (A) The relative amount of CD8α+ T cells in spleens. (B) The relative amount of NK cells in spleens.
The CD4+ T helper (Th) cells were also investigated. They could be classified into two functionally distinct Th cell subsets, known as Th1 and Th2 cells, which are responsible for both humoral and cell-mediated immunity [30, 31]. Notably, the therapeutic strategy based on tumor-specific CD8+ T cells cytotoxicity benefits from the promotion of Th1 polarization, due to the fact that Th1 cells are involved in the CD8+ T cells activation and play an essential role in CTL responses [32, 33]. The pivotal cytokine induced by the PTX/αGC-TH-Lip in vivo was IFN-γ, which is also regarded as one of the typical Th1 type cytokines. Thus, we further evaluated the Th1/Th2 polarization by measuring the cytokines that were respectively produced. IL-2 and IL-4 were selected to reflect Th1- and Th2-dominated responses, respectively [31]. Figure S1A demonstrates the IL-2 level (upper) and IL-4 level (lower) measured on the 3rd day after different treatments. The IL-2 concentration of PTX/αGC-TH-Lip group was significantly higher than control groups, indicating IFN-γ benefited the Th cell differentiation into Th1 cell type. While the IL-4 level of each group showed little difference, implying that the Th2 polarization was not stimulated after injection of αGC preparations. Figure S1B displays the IL-2 (upper) and IL-4 (lower) levels measured on the 6th day after the intravenous administrations. The general trend of IL-2 levels among all the groups remained the same, but the PTX/αGC-TH-Lip aroused a more durable IL-2 secretion compared with αGC-TH-Lip, suggesting the PTX existed as adjuvant at spleen could indeed make a positive difference in the activation process of iNKT cells by αGC. In contrast, there was no observable alteration in the production of IL-4 among all the groups compared with the previous result. We can concisely conclude that preparations with αGC are able to favor the Th1 development from the Th precursor via the IFN-γ secretion, which not only exhibited the different cytokine release pattern, but also stimulated the antitumor cellular immunity mediated by CTL. Besides, with the assistance of PTX and the liposome entrapment, the PTX/αGC-TH-Lip showed the best inductive effect in Th1 differentiation compared to αGC-TH-Lip and Free PTX& αGC.
3.9 Antitumor efficacies
3.9.1 The therapeutic effect of PTX/αGC-TH-Lip on B16F10 melanoma tumor
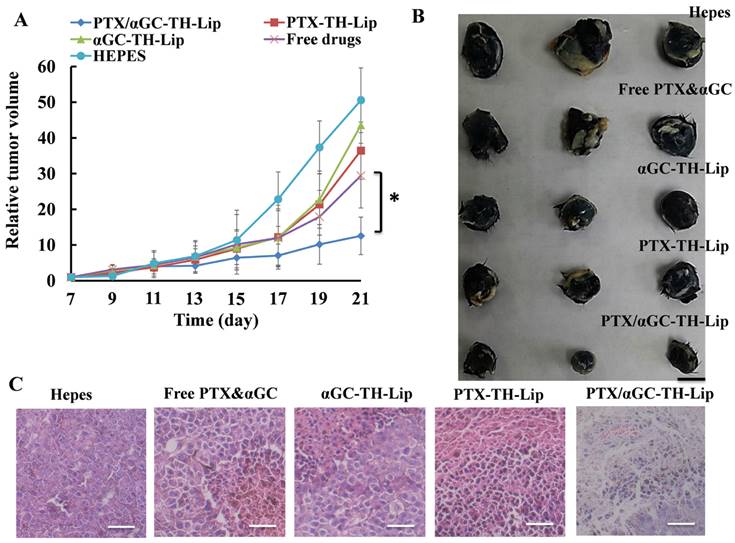
To appraise the in vivo antitumor effect of PTX/αGC-TH-Lip, B16F10 cells were inoculated in the right flank of C57BL/6 mice and applied treatments 7 days later. The tumor volumes were recorded in the following two weeks. Apparently PTX/αGC-TH-Lip exhibited the most excellent tumor inhibition effect by significantly delaying the tumor growth compared with free drugs or liposomes loaded with either drug alone (Figure 8A). Figure 8B is the photograph of the tumors at the end of the treatments. It is clearly observed that the tumors of PTX/αGC-TH-Lip group were the smallest among all the groups, intuitively demonstrating the prominent antitumor ability of PTX/αGC-TH-Lip. In addition, the H&E staining pictures of paraffin-embedded tumor sections from each group were consistent with the results obtained above as well. Incompact cells, shrunk nuclei and damaged morphology were observed in the PTX/αGC-TH-Lip group (Figure 8C), which reflected more advanced cell apoptosis was induced by PTX/αGC-TH-Lip after systemic administration.
The anti-tumor assay of different liposome preparations or free drugs on B16F10 tumor bearing C57BL/6 mice. (A) Tumor growth curves of mice receiving different in vivo treatments (n=7, mean ± SD). (B) Photographs of tumors at the end of treatment. (C) HE staining of tumor tissues after treatment. a: Hepes; b: αGC-TH-Lip; c: Free drugs; d: PTX -Lip; e: PTX/αGC-TH-Lip. Scale bars represent 50 μm.
Moreover, the changes in body weight of mice in each group during the dosing process were also recorded as an indication for safety, with no significant body weight changes observed in all the groups (Figure S3). Kaplan-Meier survival curves demonstrated a prolonged survival time of the PTX/αGC-TH-Lip (35 days) that was significantly longer than other groups (about 25 days), further suggesting its potential in anti-tumor activities (Figure S4).
Since αGC and PTX can be co-delivered to the tumor sites in vivo, it is of great necessity to shed insight into any possible detrimental influence of αGC on PTX antitumor effect. Once αGC worked as an adjuvant after systemic administration, PTX was in fact collaborating with various cytokines, particularly the IFN-γ, to resist tumor development. Before the in vivo experiments we analyzed the influence of αGC and IFN-γ themselves on the antitumor effect of PTX by MTT assay (Figure S5). The PTX/αGC-TH-Lip, in which αGC was incorporated with PTX, revealed a similar anti-proliferation effect with PTX-TH-Lip. IFN-γ was added after PTX-TH-Lip was obtained, and the mixture displayed no distinct change in B16F10 cytostasis compared to PTX-TH-Lip itself. Therefore, we are convinced that under in vitro condition αGC and IFN-γ could neither contribute to the anti-proliferation efficacy of PTX, nor impair it.
3.9.2 The prophylactic effect of PTX/αGC-TH-Lip on distant B16F10 tumor
We asserted that the synergetic action between PTX and αGC exhibited in the in vivo antitumor experiment was essentially attributed to the joint effects of tumor immunotherapy and chemotherapy. In other words, the chemotherapeutic effect mediated by PTX at tumor sites and the immune response against tumor cells initiated by αGC at spleen made a concerted effort in achieving remarkable delay of tumor growth. The CD8+ T cells mediated cytotoxicity constantly plays an important part in the immune-killing of tumor cells [22, 34, 35], which significantly inhibits the development of primary tumor and also affords a natural protection from relapse (the distant tumor in this case) [25, 27]. Interestingly, the iNKT cell system activated by PTX/αGC-TH-Lip could act as a functional bridge between innate and adaptive immunity [12] to give rise to an indirect stimulation of PTX/αGC-TH-Lip on the tumor-specific CD8+ T cells cytotoxicity.
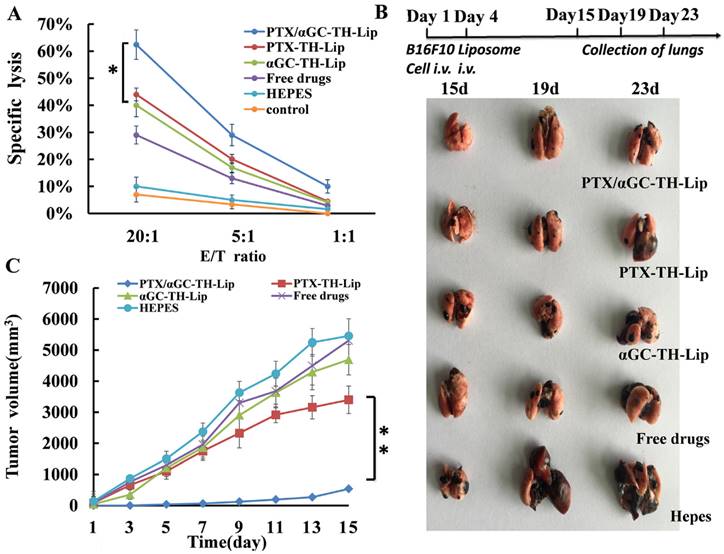
To verify this, we firstly evaluated the in vitro tumor cytolytic ability of splenocytes collected from the tumor-bearing mice after different treatments to test the effector function of cytotoxic T lymphocytes directly. At all three E/T (effector-to-target) cell ratios, PTX/αGC-TH-Lip treatments displayed the most remarkable cytotoxicity against B16F10 tumor cells (Figure 9A). However, both of the PTX-TH-Lip and αGC-TH-Lip were unable to optimize the tumor cytolysis activities of CD8+ T cells, which corroborated our knowledge that PTX can boost tumor antigen release by causing apoptosis in tumor cells and further lead to a more advanced tumor specific CD8+ T cell cytotoxicity with the stimulation of αGC. Secondly, we conducted another in vivo experiment to verify the enhanced T cell response: B16F10 cells were inoculated in the right flank of mice (same grouping as previous experiment) to establish the primary tumors, and each group received corresponding treatment for four times after the tumors grew to 100 mm3. 3 days after the last injection, the primary tumor was resected with another tumor implanted in the left flank of mice as the distant tumor. It was selected as Day 1 when any tumor at distant site became visible, and from then on the tumor volumes of all the groups were measured every other day in the following fortnight. As shown in Figure 9C, the tumors of PTX/αGC-TH-Lip group kept immeasurable until Day 9, while in other groups the tumor clearly appeared around Day 3. This indicated that the tumor-specific immune response was utmost stimulated by PTX/αGC-TH-Lip among all groups. Meanwhile, it was also noted that the αGC-TH-Lip failed to delay the tumor growth at distant sites, suggesting that unilaterally activating iNKT cells in spleen was unable to avoid the tumor proliferation after a second implantation. In other words, even from the immunotherapy perspective, the existence of PTX was still desirable, because PTX not only worked as an immunologic adjuvant but also induced the death of tumor cell and the release of tumor antigen. An ideal cancer therapy should sequentially eradicate the treated primary tumors and induce a systemic antitumor immunity to control metastatic tumors [36]. PTX/αGC-TH-Lip has been proved to be a promising anti-tumor drug delivery system that could satisfy both of the rigid requirements. Although PTX and αGC function at the different tissues, they still reciprocally interact with each other to dramatically enhance the overall antitumor efficiency.
(A) The Cytotoxicity Assay of CD8+ T lymphocytes against B16F10 cells. B16F10 tumor-bearing mice were given systemic injection of Hepes or different formulations containing 5 μg of αGC at the 10th day and 15th day after tumor inoculation. The splenocytes were collected a week later followed by mitomycin C-treated B16F10 tumor cells and mIL-2 (5 U/mL) stimulation in vitro. Cytotoxic activity of the splenocytes was determined by lactate dehydrogenase (LDH) assay. “E/T ratio” represents effector/target cell ratio. (n=3, mean ± SD). (B) Therapeutic effects on experimental lung metastasis model with B16F10 melanoma. The diagram above indicates the experimental scheme. The lung tissues were collected at day 15, day 19 and day 23, respectively. The metastasis statuses are exhibited in the picture below. (C) Growth of the distant (contralateral to the primary tumor) untreated tumor over time. (n=7, mean ± SD).
3.10 Anti-metastasis effect of PTX/αGC-TH-Lip
Takashi et al. had constructed an R8-modified αGC loaded liposome, which could significantly improve the lung metastasis status of intravenously grafted B16F10 cells by eliminating the circulating tumor cells [12, 14, 37]. According to the obtained results, we are convinced that PTX/αGC-TH-Lip is competent to fulfill antitumor effect. Therefore, it would be interesting to see if PTX/αGC-TH-Lip also features capability of restraining the lung metastasis of B16F10 cells.
The mice were inoculated with B16F10 cells intravenously at day 1 and received different treatments 4 days' post-inoculation. Then we randomly selected one mouse from each treated group on Day 15, Day 19 and Day 23 respectively and harvested the lung tissue. The status of lung metastasis is shown in Figure 9B. According to our macroscopic observation, there were only some minute nodules on the surface of lungs collected from PTX/αGC-TH-Lip group at multiple time points, although the appearance of lungs remained normal. But in other groups, the status of lung metastasis was obviously severe. Compared with the Hepes group, the mice treated with liposomes loaded with either drug (PTX or αGC) alone or soluble mixture of PTX and αGC exhibited relatively controlled lung metastasis. However, the tumors on lungs in these three groups were far more than small nodules at the last dissection (23rd day), and also it displayed a worsening trend that the lungs became black and swollen. Herein, we conclude that PTX/αGC-TH-Lip also has notable advantage over other control groups in the inhibition of the lung metastasis of B16F10 cells.
4. Discussion
In this work, we designed a special tumor therapy strategy that exploited the original distribution characteristic of the liposomes by combining the anti-tumor immunotherapy with tumor-targeting chemotherapy. Numerous tumor-oriented strategies have recently been attempted to enhance the tumor targeting efficiency via a spectrum of nanocarrier engineering methodologies including geometry, surface chemistry, ligand type, and ligand density [8, 9]. However, a general concern about these nanotherapeutics is that EPR effect, as the basis for the use of nanoparticles in cancer [2, 4, 38], is actually not sufficiently competent to fulfill our expectation in tumor-targeting of nanocarriers [5]. There is even another theory pointing out that the EPR effect, in certain degrees, would pose a barrier to the effective delivery of large therapeutic agents [39, 40]. The hyper-permeable blood vessels of tumor would induce excessive fluid loss from the vascular to the interstitial space inevitably, which reduces tumor perfusion and further hinder the systemic delivery of nanoparticles [41, 42]. To overcome the inertial logic of tumor targeting nanotheraputics based on EPR effect, we herein introduce a new scheme by simultaneously utilizing the liposomes that were naturally delivered to the spleen and liver as well as accumulating in the tumor tissues. Instead of just concentrating on the promotion of the tumor retention of liposomes, we tactfully rescued the liposomes that should have been wasted in conventional methods, and recycled them for more rational usage.
To date, plenty of immunotherapy strategies against tumor have undergone extensive development and clinical testing, from nonspecific immunostimulation to the production of exquisitely specific genetically modified T cells [34]. The tumor-associated antigens, which cannot be regarded as foreign or non-self substance, consist of various altered proteins and overexpressed proteins that emerge during the development of spontaneous tumors [34]. Mutated β-catenin found in melanoma, cyclin-dependent kinase 4 in renal cancer, caspase 8 in head and neck cancer as well as the carcinoembryonic antigen or HER2 were all typical examples [43-45]. These so-called tumor antigens could be presented to CD8+ or CD4+ T lymphocytes and elicit adaptive immune responses [46, 47]. Bearing this in mind, several vaccine strategies have been attempted, while most of the phase 3 trials failed to show a demonstrable benefit, as the tumor-induced immunosuppression assumed to be a critical barrier. In contrast, the non-antigen-specific immunotherapy approaches aiming at the activating innate arm of immune system could exert antitumor effect independent of the specific class of tumor, albeit the tumor inhibition effect is limited [48, 49]. As such, we decided to apply the non-specific immunostimulation to our liposome-based drug delivery system.
Immunologic adjuvants as the mainstay of non-specific immunostimulation could significantly amplify tumor vaccine potency by improving the humoral and/or cell-mediated immune response. Among various immunologic adjuvants, alpha-galactosylceramide (αGC) as a lipid antigen is a distinctive adjuvant, possessing the ability to selectively activate the iNKT cell via the CD1d-mediated presentation process in APCs. αGC had been proven to be able to effectively suppress a variety of tumors in hepatic and lung metastasis models after systemic administration [23, 50, 51]. The only obstacle is the dissolved αGC could induce long-term iNKT anergy due to the presentation by circulating CD1d expressing B cells after repeated injection. This feature seriously limits the application of αGC in antitumor treatment [17, 52]. Researchers proposed a possible solution with the use of nanoparticle-formulation, which was expected to provide certain shield effect on αGC, avoiding hyporesponsiveness of iNKT cells [14, 15, 53]. In our study the liposome-formulated αGC it is also possible to stimulate iNKT cells repeatedly and effectively in vivo. Standalone chemotherapy or immunotherapy always has inevitable limitations, including systemic side-effects, insufficient immuno-stimulation, tumor directed tolerance of immune cells, lack of systemic and tumor specific immune response and tumor recurrence [18]. Combination of both approaches could bypass the disadvantages and reinforce the advantages of each other. Michael A et al. reported that ionizing radiation therapy had systemic immunologic effects [54]. Liangran Guo et al. proposed a promising strategy to combine photothermal therapy with immunotherapy, rendering the tumor residues and metastases more susceptible to immune-mediated killing [36]. Anushree Seth et al. introduced the excellent synergetic effect of poly (γ-glutamic acid) (γ-PGA) based combination of anti-cancer drug (paclitaxel) and immune-stimulatory agent (imiquimod) against solid tumor [18].
In this case we utilize PTX as the chemotherapeutic agent to be combined with αGC, because PTX is not only a classical chemotherapeutic agent against different categories of tumors, but it also affects the immune system as an immune adjuvant role [24, 55]. Hence, when PTX/αGC co-loaded liposomes are injected intravenously, the αGC could trigger the IFN-γ production and further elevate the tumor-killing immune response, synergistic with PTX cytotoxicity; on the other hand, PTX also could contribute to αGC presentation process as well as the downstream immune response as an immune adjuvant. Coupled with the distribution characteristic of liposome, the intravenously administered PTX/αGC-TH-Lip would be distributed over the body initially, and a largish proportion of them would gradually accumulate at tumor sites, spleen and liver. The PTX/αGC-TH-Lip settling at different tissues would function differently, with PTX and αGC leading the way respectively at tumor sites and spleen tissue. The introduced antitumor effects were subsequently synergistic in both macroscopic and microscopic level.
Above all, we tested the shelter effect of the liposome in delivering αGC via the iNKT cells reactivation assay, the IFN-γ level of αGC-TH-Lip group remained high after the second injection, in contrast to the IFN-γ production of free αGC group that was significantly reduced after the readminstration (Figure 3B), which provided the basis for the further experiments. Compared with the control groups, the significant advantages of PTX/αGC-TH-Lip had been highlighted in various experiments, which should be attributed to the mutual aid between PTX and αGC. The PTX/αGC-TH-Lip treated group exhibited increased IFN-γ production (Figure 3C), higher αGC presentation on APCs (Figure 4E), and higher degree of BMDC maturity (Figure 6) in the comparison with αGC-TH-Lip, confirming the undeniable adjuvant role of PTX. Besides, we were also attracted by another interesting phenomenon: the TH-modification on the liposome appeared to be conducive to the immune-stimulating effect of αGC, which was embodied as higher accumulation of liposomes in spleen (Figure 2B), enhanced IFN-γ secretion (Figure 3D), and promoted αGC presentation (Figure 4E). We speculated that the increase of ligand density (in our case the TH peptide modification) on the surface might render the liposome more susceptible to the APCs, which was further verified in cellular uptake study quantitatively and qualitatively on DC2.4 cells (Figure 5). Since the cytotoxic effect of PTX had been extensively proved, we then mainly focused on the antitumor immunity provoked by αGC in the following mechanism studies. The PTX/αGC-TH-Lip treated mice exhibited the most significant regulations in immune status compared with other control groups, including the BMDC maturation status (Figure 6), the relative populations of CD8α+ T cells, NK cells (Figure 7) macrophages (Figure S2) and the Th1/Th2 polarization (Figure S1). Analyzing these results, the remarkable ability of PTX/αGC-TH-Lip in inducing cellular immune responses not only ascribed to the existence of αGC but also got benefited by PTX adjuvant effect as well as the liposome formulation. Last but not least, we determined the antitumor ability of PTX/αGC-TH-Lip through a series of in vivo experiments: the inhibition of primary tumor confirmed our initial idea about the cooperation of PTX/αGC-TH-Lip accumulated at different tissues. The PTX/αGC-TH-Lip could significantly delay the tumor growth compared with liposomes loaded with PTX or αGC alone (Figure 8). In addition, the PTX/αGC-TH-Lip also exerted an abscopal effect by suppressing tumor growth at a distant site, which is probably related to the agitation of systemic antitumor immunity (Figure 9C). The result of anti-metastasis experiment attested the ability of PTX/αGC-TH-Lip to induce immune response that could favor the elimination of circulating tumor cells (Figure 9B), echoing the upregulation of NK cells (Figure 7B).
Given that each component of PTX/αGC-TH-Lip could play different roles at different tissues (tumor or spleen), we concluded that the PTX/αGC-TH-Lip could be a cost-effective strategy in tumor immuno-chemotherapy, and underscore the practicability and necessity of combination of chemotherapy and immunotherapy in antitumor treatment.
5. Conclusion
We have demonstrated the utility of a liposome-based combined approach to deliver the unique immunologic adjuvant (αGC) to the spleen tissue and the prominent chemotherapy drug (PTX) to the tumor tissues simultaneously. Taking all the results together, we could clarify that the PTX/αGC-TH-Lip was a promising strategy in tumor immuno-chemotherapy, and showed enormous potential for both therapeutic and prophylactic anti-tumor therapy. We believe the concept of this combined approach could also be applicable for other proper anti-cancer drugs and immune-stimulating agents to rescue the liposomes that would have been wasted.
Supplementary Material
Supplementary figures.
Acknowledgements
The work was funded by the National Natural Science Foundation of China (81373337) and the National Basic Research Program of China (973 Program, 2013CB932504).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30
2. Stylianopoulos T, Jain RK. Design considerations for nanotherapeutics in oncology. Nanomedicine. 2015;11(8):1893-907
3. Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161-171
4. Park K. Questions on the role of the EPR effect in tumor targeting. J Control Release. 2013;172:391
5. Hollis CP, Weiss HL, Leggas M, Evers BM, Gemeinhart RA, Li T. Biodistribution and bioimaging studies of hybrid paclitaxel nanocrystals: lessons learned of the EPR effect and image-guided drug delivery. J Control Release. 2013;172:12-21
6. Lin KY, Kwon EJ, Lo JH, Bhatia SN. Drug-induced amplification of nanoparticle targeting to tumors. Nano Today. 2014;9:550-9
7. Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug discovery. 2010;9:615-27
8. Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615-26
9. Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496-504
10. Zhang Q, Tang J, Fu L, Ran R, Liu Y, Yuan M. et al. A pH-responsive alpha-helical cell penetrating peptide-mediated liposomal delivery system. Biomaterials. 2013;34:7980-93
11. Zhang W, Song J, Zhang B, Liu L, Wang K, Wang R. Design of acid-activated cell penetrating peptide for delivery of active molecules into cancer cells. Bioconjug Chem. 2011;22:1410-5
12. Taniguchi M, Seino K, Nakayama T. The NKT cell system: bridging innate and acquired immunity. Nat Immunol. 2003;4:1164-5
13. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297-336
14. Nakamura T, Yamazaki D, Yamauchi J, Harashima H. The nanoparticulation by octaarginine-modified liposome improves alpha-galactosylceramide-mediated antitumor therapy via systemic administration. J Control Release. 2013;171:216-24
15. Thapa P, Zhang G, Xia C, Gelbard A, Overwijk WW, Liu C. et al. Nanoparticle formulated alpha-galactosylceramide activates NKT cells without inducing anergy. Vaccine. 2009;27:3484-8
16. Bontkes HJ, Moreno M, Hangalapura B, Lindenberg JJ, de Groot J, Lougheed S. et al. Attenuation of invariant natural killer T-cell anergy induction through intradermal delivery of alpha-galactosylceramide. Clin Immunol. 2010;136:364-74
17. Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat Immunol. 2002;3:867-74
18. Seth A, Heo MB, Lim YT. Poly (gamma-glutamic acid) based combination of water-insoluble paclitaxel and TLR7 agonist for chemo-immunotherapy. Biomaterials. 2014;35:7992-8001
19. Chan OT, Yang LX. The immunological effects of taxanes. Cancer Immunol Immunother. 2000;49:181-5
20. Kawasaki K, Akashi S, Shimazu R, Yoshida T, Miyake K, Nishijima M. Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J Biol Chem. 2000;275:2251-4
21. Garnett CT, Schlom J, Hodge JW. Combination of docetaxel and recombinant vaccine enhances T-cell responses and antitumor activity: effects of docetaxel on immune enhancement. Clin Cancer Res. 2008;14:3536-44
22. Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials. 2014;35:814-24
23. Uldrich AP, Crowe NY, Kyparissoudis K, Pellicci DG, Zhan Y, Lew AM. et al. NKT cell stimulation with glycolipid antigen in vivo: costimulation-dependent expansion, Bim-dependent contraction, and hyporesponsiveness to further antigenic challenge. J Immunol. 2005;175:3092-101
24. Yuan L, Wu L, Chen J, Wu Q, Hu S. Paclitaxel acts as an adjuvant to promote both Th1 and Th2 immune responses induced by ovalbumin in mice. Vaccine. 2010;28:4402-10
25. Quaratino S, Duddy LP, Londei M. Fully competent dendritic cells as inducers of T cell anergy in autoimmunity. Proc Natl Acad Sci U S A. 2000;97:10911-6
26. White K, Kearns P, Toth I, Hook S. Increased adjuvant activity of minimal CD8 T cell peptides incorporated into lipid-core-peptides. Immunol Cell Biol. 2004;82:517-22
27. Tscharke DC, Croft NP, Doherty PC, La Gruta NL. Sizing up the key determinants of the CD8+ T cell response. Nat Rev Immunol. 2015;15(11):705-16
28. Hanna N. Role of natural killer cells in control of cancer metastasis. Cancer Metastasis Rev. 1982;1:45-64
29. Liu X, Chen Q, Yan J, Wang Y, Zhu C, Chen C. et al. MiRNA-296-3p-ICAM-1 axis promotes metastasis of prostate cancer by possible enhancing survival of natural killer cell-resistant circulating tumour cells. Cell Death Dis. 2013;4:e928
30. Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med. 2002;8:567-73
31. D'elios M, Del Prete G. Th1/Th2 balance in human disease. Transplant Proc: Elsevier. 1998;30(5):2373-7
32. Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD. et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88-93
33. Sung Hee Y, Ok YS, Jung Yong P, Hee Yeun W, Eun Kyung K, Hyun Jung S. et al. Selective addition of CXCR3(+) CCR4(-) CD4(+) Th1 cells enhances generation of cytotoxic T cells by dendritic cells in vitro. Exp Mol Med. 2009;41:161-70
34. Saied A, Pillarisetty VG, Katz SC. Immunotherapy for solid tumors-a review for surgeons. J Surg Res. 2014;187:525-35
35. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480-9
36. Guo L, Yan DD, Yang D, Li Y, Wang X, Zalewski O. et al. Combinatorial photothermal and immuno cancer therapy using chitosan-coated hollow copper sulfide nanoparticles. ACS Nano. 2014;8:5670-81
37. Gul N, Babes L, Siegmund K, Korthouwer R, Bogels M, Braster R. et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. 2014;124:812-23
38. Taniguchi M, Seino K-i, Nakayama T. The NKT cell system: bridging innate and acquired immunity. Nat Immunol. 2003;4:1164-5
39. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653-64
40. Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev. 2012;64:353-65
41. Netti PA, Roberge S, Boucher Y, Baxter LT, Jain RK. Effect of transvascular fluid exchange on pressure-flow relationship in tumors: a proposed mechanism for tumor blood flow heterogeneity. Microvasc Res. 1996;52:27-46
42. Stylianopoulos T, Jain RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci U S A. 2013;110:18632-7
43. Rosenberg SA, Kawakami Y, Robbins PF, Wang R. Identification of the genes encoding cancer antigens: implications for cancer immunotherapy. Adv Cancer Res. 1996;70:145-77
44. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT. et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323-37
45. Nukaya I, Yasumoto M, Iwasaki T, Ideno M, Sette A, Celis E. et al. Identification of HLA-A24 epitope peptides of carcinoembryonic antigen which induce tumor-reactive cytotoxic T lymphocyte. Int J Cancer. 1999;80:92-7
46. Czerniecki BJ, Koski GK, Koldovsky U, Xu S, Cohen PA, Mick R. et al. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res. 2007;67:1842-52
47. Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulieres D. et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol. 2005;23:6674-81
48. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154-8
49. Foa R, Guarini A, Gansbacher B. IL2 treatment for cancer: from biology to gene therapy. Br J Cancer. 1992;66:992-8
50. Subrahmanyam P, Webb TJ. Boosting the Immune Response: The Use of iNKT cell ligands as vaccine adjuvants. Frontiers in biology. 2012;7:436-44
51. Zhou D, Mattner J, Cantu C 3rd, Schrantz N, Yin N, Gao Y. et al. Lysosomal glycosphingolipid recognition by NKT cells. Science. 2004;306:1786-9
52. Macho-Fernandez E, Cruz LJ, Ghinnagow R, Fontaine J, Bialecki E, Frisch B. et al. Targeted delivery of alpha-galactosylceramide to CD8alpha+ dendritic cells optimizes type I NKT cell-based antitumor responses. J Immunol. 2014;193:961-9
53. McKee SJ, Young VL, Clow F, Hayman CM, Baird MA, Hermans IF. et al. Virus-like particles and alpha-galactosylceramide form a self-adjuvanting composite particle that elicits anti-tumor responses. J Control Release. 2012;159:338-45
54. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S. et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366:925-31
55. Manthey CL, Perera PY, Salkowski CA, Vogel SN. Taxol provides a second signal for murine macrophage tumoricidal activity. J Immunol. 1994;152:825-31
Author contact
![]() Corresponding authors: Tel. /fax: +86 28 85502532. E-mail addresses: qinheedu.cn (Q. He), xiang7199com (B. Xiang).
Corresponding authors: Tel. /fax: +86 28 85502532. E-mail addresses: qinheedu.cn (Q. He), xiang7199com (B. Xiang).