Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(12):2278-2291. doi:10.7150/thno.15898 This issue Cite
Research Paper
Highly Effective Auger-Electron Therapy in an Orthotopic Glioblastoma Xenograft Model using Convection-Enhanced Delivery
Helge Thisgaard1,2,# ![]() , Bo Halle2,3,4,#, Charlotte Aaberg-Jessen1,3, Birgitte Brinkmann Olsen1, Anne Sofie Nautrup Therkelsen1, Johan Hygum Dam1, Niels Langkjær1, Sune Munthe2,3,4, Kjell Någren1, Poul Flemming Høilund-Carlsen1,2*, Bjarne Winther Kristensen2,3*
, Bo Halle2,3,4,#, Charlotte Aaberg-Jessen1,3, Birgitte Brinkmann Olsen1, Anne Sofie Nautrup Therkelsen1, Johan Hygum Dam1, Niels Langkjær1, Sune Munthe2,3,4, Kjell Någren1, Poul Flemming Høilund-Carlsen1,2*, Bjarne Winther Kristensen2,3*
1. PET & Cyclotron Unit, Department of Nuclear Medicine, Odense University Hospital, Odense, Denmark;
2. Department of Clinical Research, University of Southern Denmark, Odense, Denmark;
3. Department of Pathology, Odense University Hospital, Odense, Denmark;
4. Department of Neurosurgery, Odense University Hospital, Odense, Denmark.
# These authors contributed equally to the work.
* Shared last authorship.
Received 2016-4-19; Accepted 2016-8-30; Published 2016-9-29
Abstract

Glioblastoma, the most common and malignant primary brain tumor, always recurs after standard treatment. Therefore, promising new therapeutic approaches are needed. Short-range Auger-electron-emitters carry the ability of causing highly damaging radiation effects in cells. The aim of this study was to test the effect of [125I]5-Iodo-2'-deoxyuridine (125I-UdR, a radioactive Auger-electron-emitting thymidine analogue) Auger-therapy on immature glioblastoma spheroid cultures and orthotopic xenografted glioblastoma-bearing rats, the latter by means of convection-enhanced delivery (CED). Moreover, we aimed to determine if the therapeutic effect could be enhanced when combining 125I-UdR therapy with the currently used first-line chemotherapeutic agent temozolomide. 125I-UdR significantly decreased glioblastoma cell viability and migration in vitro and the cell viability was further decreased by co-treatment with methotrexate and/or temozolomide. Intratumoral CED of methotrexate and 125I-UdR with and without concomitant systemic temozolomide chemotherapy significantly reduced the tumor burden in orthotopically xenografted glioblastoma-bearing nude rats. Thus, 100% (8/8) of the animals survived the entire observation period of 180 days when subjected to the combined Auger-chemotherapy while 57% (4/7) survived after the Auger-therapy alone. No animals (0/8) treated with temozolomide alone survived longer than 50 days. Blood samples and post-mortem histology showed no signs of dose-limiting adverse effects. In conclusion, the multidrug approach consisting of CED of methotrexate and 125I-UdR with concomitant systemic temozolomide was safe and very effective leading to 100% survival in an orthotopic xenograft glioblastoma model. Therefore, this therapeutic strategy may be a promising option for future glioblastoma therapy.
Keywords: Glioblastoma, Auger-electron therapy, convection-enhanced delivery, [125I]5-Iodo-2'-deoxyuridine, temozolomide.
Introduction
Glioblastomas (GBMs) - the most common and malignant primary brain tumor - always recur after standard treatment consisting of surgery, radiation and chemotherapy. The overall median survival is only approximately 15 months following surgical resection, adjuvant external radiotherapy and concomitant and adjuvant alkylating chemotherapy using temozolomide (TMZ) [1, 2]. Due to their invasive nature, GBMs are non-curable by surgery and side effects as well as poor blood-brain-barrier (BBB) penetration of most drugs are dose-limiting for conventional radio- and chemotherapy [3]. Sub-lethal doses may, therefore, favor escape of cancer cells from standard therapy and, thus, new therapeutic strategies to eradicate GBMs are needed.
A novel promising therapeutic tool that may circumvent conventional resistance comprises short-range Auger-electron-emitters (AEEs). The decay of AEEs creates multiple, highly localized ionizations (medium to high linear energy transfer (LET): 4-26 keV/µm) possible of causing highly damaging radiation effects in cells that may result in superior properties for AEE-based therapeutic strategies [4, 5]. The primary radiobiological effect responsible for cell killing is believed to be creation of DNA double-strand breaks, especially if the AEE is bound to the DNA [6], though other possible targets have been suggested lately [7, 8]. This is a consequence of the emission of an electron cascade in each radioactive decay, with all electrons having low energies (eV to keV) and thus short ranges in biological tissue. For the majority of the electrons in the cascade their range is typically limited to less than the diameter of a single cell (2-500 nm). Therefore, AEEs are able to provide an extremely high local radiation dose, which may minimize toxic adverse effects to normal tissues due to cross-fire irradiation. However, to achieve this, selective delivery of the AEEs to the genomic DNA of the cancer cells is required. Hence, we hypothesized that using the highly toxic AEE compound [125I]5-Iodo-2'-deoxyuridine, we could effectively eradicate GBM cells in vitro and in vivo.
Since 125I-UdR is nonspecifically taken up by all DNA-synthesizing cells in the body [9] combined with a very short systemic biological half-life [10], it has to be delivered locally in the central nervous system to induce a significant antitumor effect in brain tumors and avoid systemic adverse effects. In several clinical trials (reviewed by Vogelbaum & Iannotti [11]), new compounds for the treatment of GBMs have been administered by so-called convection-enhanced delivery (CED) [12]. This approach involves continuous infusion of a drug under positive pressure. In practice, one or several catheters connected to an infusion pump are placed in the brain parenchyma, most often in areas of residual tumor. This bypasses the BBB and a high concentration of the drug is obtained in the brain with no or very little systemic toxicity [11, 12]. Therefore, the CED method was chosen for 125I-UdR administration. The radiotherapeutic effects of 125I-UdR can be further increased by exposing the tumor cells to methotrexate (MTX) prior to the 125I-UdR, which increases the percentage of cells undergoing DNA replication and, simultaneously, increases the DNA-incorporation of 125I by thymidylate synthetase inhibition [13].
The overall aim of this study was to test the effect and safety profile of 125I-UdR therapy in vitro and in vivo on immature GBM spheroid cultures (GSCs) [14-16] and orthotopic xenografted GBM-bearing rats, respectively. Moreover, we aimed to determine if a further therapeutic effect was achieved when combining 125I-UdR therapy with the currently used first-line chemotherapeutic agent TMZ [1].
Material and Methods
Chemicals and Radiosyntheses
125I-UdR was either purchased from Perkin Elmer (Skovlunde, Denmark) or prepared essentially as described by Wang et al. [17] using trimethylstannyl-2'-deoxyuridine as precursor. The final product was sterilized by passing the eluate through a 0.22 µm sterile filter into a capped vial. MTX (Sigma-Aldrich, Copenhagen, Denmark) and TMZ (Sigma-Aldrich) was prepared according to the manufacturer's instructions. MTX (1 mM) and TMZ (129 mM) were stored at -20 °C and diluted in fresh medium for in vitro studies right before each use.
The amino-acid tracer [11C]methylaminoisobutyric acid ([11C]MeAIB) was prepared as described previously [18].
Cell Culture
Cells were cultured as free-floating spheroids in serum-free medium at 36°C in a humidified incubator with 5% CO2 [19]. Two immature GSCs, passage 9-12, designated T78 and T87, were utilized. These were both derived from males in 2009 and 2010, respectively. They were established and characterized, as previously described [19, 20] in our laboratory according to approval by the Regional Scientific Ethical Committee (approval number S-VF-20040102). These GSCs have the ability to form new spheroids at clonal density, a karyotype typical of GBMs, and the ability to form highly invasive tumors upon orthotopic xenografting. Moreover, they differentiate into cells expressing neuronal, astrocytic and oligodendrocyte markers upon culturing in serum-containing medium. Both GSCs were derived from mutated isocitrate dehydrogenase 1 (mIDH1) negative tumors representing primary GBMs [21] and both have a hypermethylated O6-methylguanine-DNA methyltransferase (MGMT) promoter region indicating sensitivity to TMZ [22].
Viability Assay
T78 and T87 cells cultured as spheroids were trypsinized and seeded in 96-well plates in serum-free medium (1,000 cells/well). The next day, the cells were incubated in either increasing activities of 125I-UdR (0-3 kBq/ml) or the “cold”, non-radioactive, but chemically identical, 127I-UdR (12 pg/ml, corresponding to the mass concentration of 3 kBq/ml 125I-UdR, Sigma-Aldrich). To investigate the additional effect of MTX and TMZ co-exposure, cells were incubated with 125I-UdR as above with 0.01 μM MTX added or with 0.01 μM MTX plus equipotent concentrations of TMZ added, i.e., 10 μM and 100 μM TMZ for T78 and T87 cells, respectively. At day 7, 20 μl of CellTiter Blue (Promega, Nacka, Sweden) was added and the cells were returned to the incubator for 6 hours prior to recording of the absorbance in a Elx800 microplate reader (BioTek, Brondby, Denmark).
Migration Assay
Geltrex (Gibco, Naerum, Denmark) and serum-free medium was mixed (1+49) and 1.4 ml was added to each well in 12-well plates. Coated plates were incubated over night at 36°C and the following morning the supernatant was aspirated. One spheroid (100-200 µm) was embedded in the coating per well. After incubating the plate for 75 minutes at 36°C, 800 µl serum-free medium was added. When spheroids started migrating (designated day 0) additionally 200 µl of serum-free medium (serving as untreated controls), 127I-UdR or 125I-UdR was added resulting in a final concentration of 0.3 µg/ml or 3 kBq/ml, respectively. Images were obtained daily from day 0 through day 5 using a Leica DM IRB inverted phase-contrast microscope, a Leica DFC300FX camera, and the Leica Application Suite version 2.6.0 RI computer software. The spheroid radius was determined (http://imagej.nih.gov/ij/) and the migration speed/day was calculated.
Nuclear Uptake and Clonogenic Assay
The amount of radioactivity in the cell nuclei was determined by incubating T87 cells with 0, 0.5, 1, 2 and 3 kBq/ml 125I-UdR, respectively, for 24 h. Cells were counted, washed in PBS and fractionated using the Nuclei EZ Prep Nuclei isolation kit (Sigma-Aldrich) as described previously [23].
For the clonogenic assay, T87 cells were seeded into poly-L-Lysine coated 6-well plates and incubated with the same 125I-UdR activity concentrations as above for 24 h followed by 7 d incubation in serum-free medium. Cells were stained with 0.05% Crystal Violet and colonies larger than 50 cells were counted using the colony counter from the ImageQuantTL software (GE Healthcare, Brondby, Denmark). An estimate of the absorbed radiation dose to the cell nuclei was calculated using the MIRD cellular approach [24] from the measured mean activity uptake per cell nucleus. Since 125I-UdR is taken up by the cells and bound to the DNA indefinitely and the half-life of 125I is long compared to the duration of the assay, the activity per cell nucleus decreases over time primarily due to cell division. Hence, the effective half-life was assumed to be equal to the T87 cell doubling time, which was measured to be 22 h. By integrating the area under the time-activity curve per nucleus, the total number of cumulative decays was found. Only the self-dose from activity in the nucleus was considered, since the dose contribution from activity in the cytoplasm was negligible as is the cross-dose contribution for 125I-UdR [25]. The mean absorbed radiation dose to the nucleus could then be calculated by multiplying the cumulative decays in the nucleus by the tabulated S-value, S(N←N) = 2.12*10-3 Gy/(Bq s) [24], for a measured mean cell nucleus radius of approximately 6 µm.
In Vivo Experimental Setup
To test the effect and safety of 125I-UdR-based Auger-electron therapy with and without concomitant TMZ, an in vivo experiment was set up as illustrated in Fig. 1.
Schematic view of the in vivo study in orthotopic xenografted GBM-bearing rats.
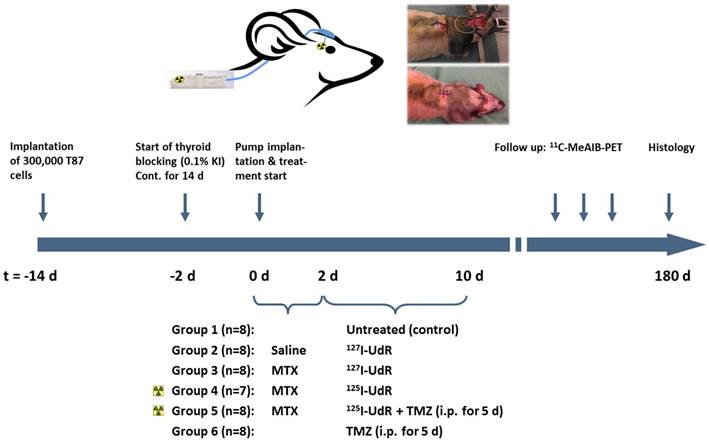
Animal Model and In Vivo Auger-Therapy
All animal procedures were conducted in accordance with the obtained approval from the national Animal Experiments Inspectorate (permission J. Nr. 2008/561-1572). Male athymic nude rats (n=48) aged six weeks (Hsd:RH-Foxn1rnu, Harlan Laboratories, Venray, Nederlands) were anaesthetized subcutaneously (mixture of Hypnorm and Midazolam) and placed in a small animal stereotaxic instrument (Model 900, David Kopf Instruments, Tujunga, USA). Through a burr hole placed one mm anteriorly and two mm laterally to bregma a 2 µl cell suspension of 300,000 T87 single cells in Hank's Balanced Salt Solution (Gibco) supplemented with 0.9% glucose (SAD 500 mg/ml) was slowly injected at a depth of 3.5 mm. Two weeks later, SMP-200 iPRECIO® Micro Infusion Pumps (Data Sciences International, MC s'Hertogenbosch, Nederlands) were implanted subcutaneously in 32 rats, since preliminary experiments had shown establishment of tumors at this time point. Rats were anaesthetized and fixated as described above and the midline incision was re-incised and the burr hole was identified. A 4 mm pump connector cannula (PlasticsOne, Roanoke, Virginia, USA) was then connected to the pump catheter and implanted with the tip of the cannula in the center of the bulk tumor. Pumps were programmed to instant mode, constant flow mode and 5 µl/hour infusion rate and loaded with isotone saline (n=8, group 2) or 0.1 mM MTX (n=24, group 3, 4 and 5). This concentration of infused MTX has previously been shown to effectively increase the DNA-incorporation of 125I in intrathecal tumor cells in rats [13]. As one rat with an MTX loaded pump never woke up after surgery it was censored from the study. Two days later the 31 rats were anaesthetized using isoflurane, and residual saline or MTX was extracted from the pump reservoirs. The reservoirs were re-filled with 960 µl of 0.3 µg/ml 127I-UdR (n=16, group 2 and 3, corresponding to approximately the same number of I-UdR molecules as for the 125I-UdR-groups) or with the 125I-UdR activity (n=15, group 4 and 5), enabling 8 days of continuous infusion and a cumulated infused 125I-UdR activity of 83 ± 7 MBq. Initiated on the same day, eight of these rats (group 5) had a daily intraperitoneal (IP) injection of 200 mg/m2 TMZ (Sigma-Aldrich) dissolved in 10% DMSO (Sigma-Aldrich) v/v 0.9% NaCl for five consecutive days. Two additional groups received either no treatment (n=8, group 1, serving as control group) or systemic TMZ treatment (n=8, group 6) by daily IP injections of 200 mg/m2 TMZ for five consecutive days as above.
The rats were fed ad libitum and to block the thyroid gland, potassium iodine (KI, Sigma-Aldrich) was added to the drinking water (1 mg/ml) from the day of pump implantation and the following 14 days. At the end of the observation period (180 days after completion of treatment) or once animals reached the pre-determined humane endpoints (neurological deficits or >20% weight-loss) they were euthanized.
Distribution Evaluation by SPECT/CT
The intracranial distribution of infused 125I-UdR was evaluated on the last day of infusion using a Siemens Inveon Small Animal PET/SPECT/CT scanner. The animals (n=4) were anesthetized using isoflurane and subsequently SPECT scanned for approximately 42 min. The images were reconstructed using the OSEM3D algorithm. In one of the animals a low-dose CT scan was performed in combination with the SPECT scan.
Tumor Growth Evaluation by PET
For tumor growth evaluation, the animals were PET scanned biweekly in the initial phase and later every third week using the Inveon PET scanner. Briefly, the animals were anesthetized using isoflurane and injected via the tail vein with 50 MBq [11C]MeAIB, a metabolically stable alanine analogue targeting the amino-acid transport system A [26, 27]. Forty minutes after tracer injection pairs of animals were placed head-to-head in the scanner and a 14 min. static scan was performed. The acquired data were reconstructed using the OSEM2D algorithm without scatter and attenuation correction in the Inveon Acquisition Workplace software module (Siemens, Knoxville, USA).
Blood Sampling
Blood samples were taken 3 weeks pre-treatment, and 3, 5, 13 and 26 weeks post-treatment to evaluate serological signs of potential toxicity. Due to limitations in blood collection volume, rats from each treatment group were divided in two groups; one for thyroid and one for hepatic, renal and bone marrow toxicity evaluation.
During anesthesia, blood samples were collected via the tail vein. Thyroid function was evaluated using total T3 and T4 ELISA kits (Nordic Biosite, Copenhagen, Denmark) on plasma samples. Plasma levels of alanine aminotransferase (ALT), creatinine and blood urea nitrogen (BUN) were analyzed on a Cobas Mira Plus (Roche Diagnostics, Hvidovre, Denmark) analyzer. Whole blood levels of potassium and sodium were analyzed on a GEM Premier 3000 analyzer (Instrumentation Laboratory, Massachusetts, USA). Hemoglobin (Hb), white blood cell (WBC) and platelet counts were determined in whole blood using a scil Vet ABC Hematology Analyzer and the rat species-specific smart card (scil animal care company, Viernheim, Germany). All analyses were carried out in accordance with the manufacturers' instructions.
Histology
Brain, thyroid gland, liver and kidneys were removed post-mortem and fixed in 4% formaldehyde. After fixation, brains were divided in 1 mm thick coronal sections using a rat brain matrix (PlaticsOne) followed by paraffin embedment. Thyroid gland, liver and kidneys were paraffin embedded as well. Three µm sections were cut and all sections were stained with hematoxylin and eosin (HE) for morphological evaluation. To evaluate the human-derived brain tumors, brain sections were immunohistochemically stained with an antibody targeting human vimentin. This was carried out using the AutostainerPlus (Dako, Glostrup, Denmark) as follows. Sections were deparaffinized and heat-induced epitope retrieval was performed in a buffer solution consisting of 10 mM Tris-base and 0.5 mM ethylene glycol tetraacetic acid, pH 9. After blocking of endogenous peroxidase activity by incubation in 1.5% hydrogen peroxide, sections were incubated with primary antibody against human vimentin (clone SP20, Nordic Biosite, 1+200) for 60 minutes. The antigen-antibody complex was detected using PowerVision (Novocastra, Newcastle upon Tyne, UK) and visualized with 3,3′-diaminobenzidine as chromogen. Sections were then counterstained with Mayers Haematoxylin, and coverslips were mounted using AquaTex. Omission of primary antibodies served as negative controls and controls for non-specific staining related to the detection systems. All slides were scanned on a Hamamatsu Digital Slide Scanner (Hamamatsu, Japan). Additionally, kidney sections were periodic acid-Schiff (PAS) and liver PAS-diastase stained to complement the morphological evaluation.
Statistical Analysis
In vitro and blood sample data were analyzed with unpaired, two-tailed Student's t-tests or two-way ANOVA with Holm-Sidak's correction for multiple comparisons in GraphPad Prism. A p-value of < 0.05 was considered statistically significant. The Kaplan-Meier-survival data were analyzed using the log-rank (Mantel-Cox) test correcting for multiple comparisons. A Bonferroni-corrected p-value of < 0.05/9 was considered statistically significant.
Results
Cell Viability and Spheroid Migration In Vitro
In vitro, the effects of 125I-UdR were tested on two immature GSCs. As seen in Fig. 2A, an activity-dependent reduction in cell survival was found in both cell lines. The cell survival was significantly decreased (p<0.0001) in T78 for all 125I-UdR activity concentrations used, when compared to untreated controls, while 1000 Bq/ml or more were required in T87 GSCs to obtain a significant therapeutic effect. This difference in the response to the 125I-UdR therapy between the T78 and T87 GSCs, with T87 being the most resistant, was highly significant (p<0.0001). In both cell lines, the therapeutic effects of 125I-UdR were also significantly higher compared to the chemically identical, but nonradioactive, 127I-UdR for most activity concentrations tested. Co-treatment with MTX significantly increased the therapeutic effect of 125I-UdR in the T87 GSCs at activity concentrations between 250 and 1000 Bq/ml (p<0.0001), while significance was only reached at one activity concentration (100 Bq/ml) in T78 GSCs. By combining MTX and 125I-UdR with an equipotent amount of TMZ, a significant further increase in the therapeutic effect was observed for both cell lines compared to MTX + 125I-UdR or 125I-UdR alone, except at the highest activity concentrations tested (Fig. 2A).
Antiproliferative effects in T78 and T87 GBM cells (A) exposed to increasing activity concentrations of 125I-UdR, MTX+125I-UdR or MTX+125I-UdR+TMZ or corresponding groups with stable 127I-UdR, and anti-migratory effects (B) of T78 and T87 spheroids exposed to either 127I-UdR or 125I-UdR. Error bars represent SEM for three independent CellTiter Blue assays (n=4) or five independent wells per group in the migration assay. NS: p > 0.05; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001.
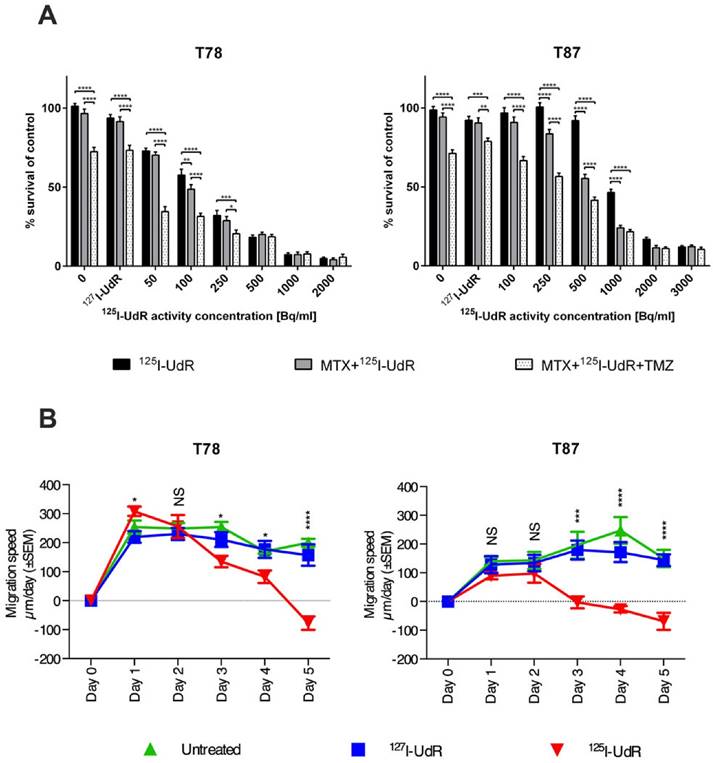
To test the anti-migratory effect of 125I-UdR a 2D migration assay based on migration of spheroids on a coating of reduced growth factor basement membrane extracts was applied. As opposed to many other migration assays, it does not rely on serum-based chemo-attraction and may, therefore, imitate the in vivo migration better. As illustrated (Fig. 2B + supplementary Fig S1), spheroid migration was significantly reduced by 125I-UdR in T78 and T87 GSCs from day three, compared to 127I-UdR treated spheroids serving as control.
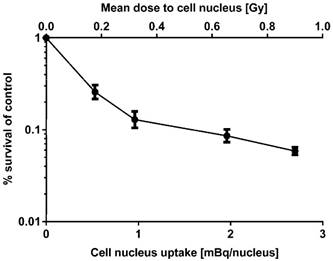
Clonogenic survival was measured for 125I-UdR in the T87 GBM cell line only, as this cell line was chosen for the following in vivo therapy in rats. As can be seen in Fig. 3, the surviving fraction decreased as function of an increasing nuclear uptake of 125I-UdR and resulting increasing absorbed radiation dose to the nuclei of the GBM cells.
Clonogenic survival of T87 GBM cells as a function of the mean cell nucleus uptake of 125I-UdR and resulting mean absorbed dose to the cell nuclei. Error bars represent SD for three independent assays (n=3).
125I-UdR Intracerebral Distribution
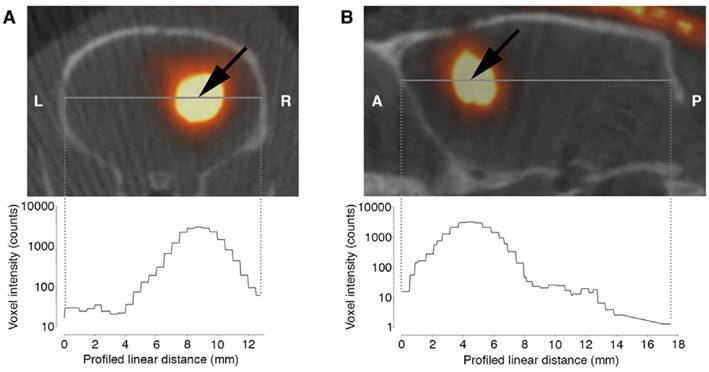
By intratumoral CED of 125I-UdR for 8 days in orthotopic GBM-bearing rats, a high concentration of 125I was found in the tumor region determined by combined SPECT/CT (Fig. 4, n=1) or SPECT alone (n=3, data not shown). Moreover, a small fraction of the 125I could be seen in the contralateral hemisphere and in the posterior part of the brain, allowing for irradiation of possible distantly located migrating GBM cells.
Axial (A) and sagittal (B) SPECT/CT images of a rat after 125I-UdR infusion showing the 125I activity distribution in the brain. The black arrow indicates the isocenter of delivery. L:left, R:right, A:anterior, P:posterior.
In Vivo Effects
The 47 orthotopic xenografted T87 GBM-bearing rats were treated with: 1) no treatment (control), 2) 127I-UdR by CED, 3) neoadjuvant MTX + 127I-UdR by CED, 4) neoadjuvant MTX + 125I-UdR by CED, 5) neoadjuvant MTX + 125I-UdR by CED + concomitant systemic TMZ, or 6) systemic TMZ as monotherapy, as outlined in Fig. 1. Tumors rapidly progressed in all animals in group 1, 2 and 3, as determined by [11C]MeAIB PET (Fig. 5A). The median survival in these groups were 24, 21 and 30.5 days, respectively, after treatment start (Fig. 6). In group 1 and 3, no animals survived longer than 35 days after initiation of treatment due to massive tumor burden, while in group 2 the longest survival time was only 23 days. Tumor progression in group 4 was strongly delayed and only three out of seven animals in this group developed a PET-detectable tumor during the 180 days of observation. The time from therapy initiation to the first PET-based tumor detection in this group was 91 days and four animals (4/7, 57%) survived the entire observation period. None of the animals in group 5 developed a PET-detectable tumor during the 180-day observation period and all animals (8/8, 100%) survived this period. The median survival in group 6 subjected to TMZ monotherapy was 47 days with no animals surviving longer than 50 days after initiation of treatment.
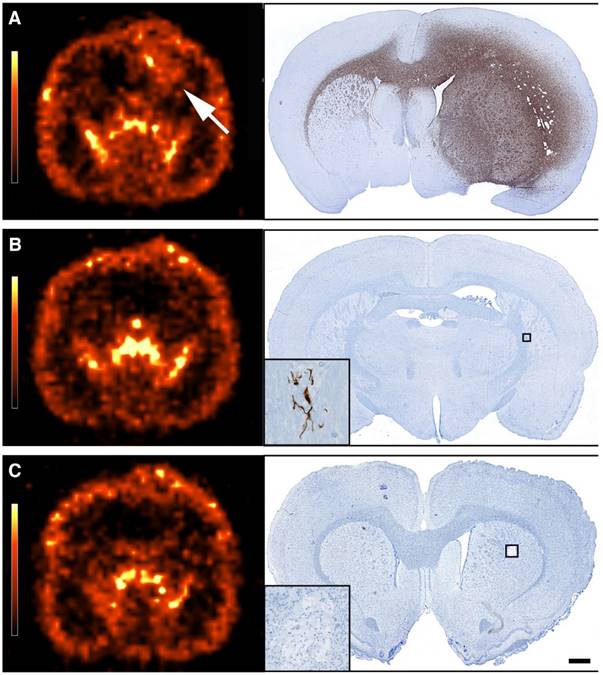
Transaxial [11C]MeAIB PET images and corresponding post-mortem immunohistochemical anti-human vimentin stained coronal brain sections obtained 1-2 days later. (A) PET image of a rat from group 4 showing a very large tumor (white arrow), confirmed by histology. (B-C) PET images of rats from group 4 and 5 scanned 178 days after treatment start showing no PET detectable tumors. Histology showed empty tumor beds with either only a few GBM cells located in the ipsilateral hemisphere (B) or no identifiable GBM cells (C). Scale bar 1 mm.
Kaplan-Meier plot showing that the survival benefit of neoadjuvant MTX + 125I-UdR as stand-alone Auger-therapy (group 4) or with concomitant, systemic TMZ chemotherapy (group 5) was highly significant compared with the non-radioactive, but chemically identical treatment MTX + 127I-UdR (group 3, p=0.0001 and p<0.0001, respectively) or untreated controls (group 1, both p<0.0001). The Auger-therapy was also significantly better than systemic TMZ-chemotherapy alone (group 6, p=0.0001).
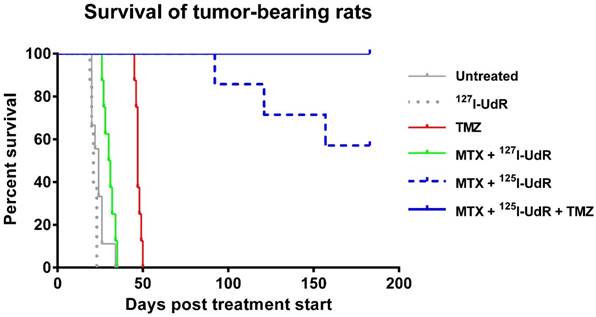
The survival benefit of neoadjuvant MTX + 125I-UdR as stand-alone Auger-therapy (group 4) was highly significant compared with the non-radioactive, but chemically identical treatment MTX + 127I-UdR (group 3, p=0.0001) or untreated controls (group 1, p<0.0001). Importantly, the Auger-therapy was also significantly better than the systemic TMZ-chemotherapy alone (group 6, p=0.0001). The survival benefit of neoadjuvant MTX + 125I-UdR with concomitant, systemic TMZ (group 5) was also highly significant (p<0.0001) compared with these groups. However, the added therapeutic effect of the concomitant TMZ treatment (group 5) compared to the Auger-therapy (group 4) alone was not found significant (p=0.04).
Post-mortem histology of the brains revealed that all animals in group 1, 2, 3 and 6 had very large tumors (Fig. 5A) explaining their short survival. The three animals in group 4, not surviving the entire observation period, had large tumors as well. The remaining animals in this group had an empty tumor-bed (the tissue surrounding the original bulk tumor) with a few GBM cells located in the ipsilateral hemisphere (Fig. 5B). No distant migratory cells could be identified. In group 5, five animals had the same histological appearance as just described, whereas no vimentin positive GBM cells could be detected in brains of the remaining three animals (Fig. 5C).
Adverse Effects
The local administration of MTX and 125I-UdR by CED as well as IP administration of TMZ could potentially give rise to local and/or systemic adverse effects. To evaluate this, blood samples (Fig. 7) were examined and post-mortem histological analyses performed (Fig. 8).
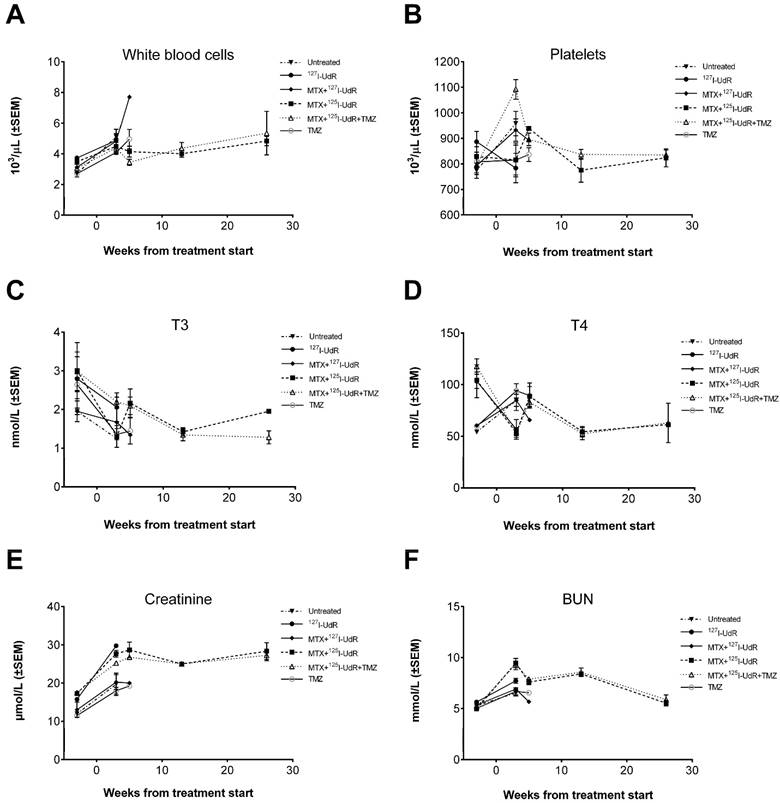
Evaluation of adverse effects by blood samples before and after treatment. (A) White blood cell and (B) platelet levels. (C) Plasma total T3 and (D) total T4 thyroid hormone blood levels. (E) Plasma creatinine and (F) BUN blood levels.
Evaluation of adverse effects by histology. (A) Thyroid gland HE staining from group 2 showing normofollicular architecture. (B) Similar HE staining from group 4 showing microfollicular architecture and colloids with scalloped margins (black arrow). (C) Liver PAS-diastase staining from group 2 showing normal liver architecture and no signs of hepatocyte necrosis, haemorrhage or inflammation. (D) Liver PAS-diastase staining from group 5, showing a few necrotic hepatocytes (black arrow). The liver architecture was normal and without haemorrhages. (E-F) Kidney PAS staining from group 2 and 4 showing no histopathologic findings. Scale bar 100 µm.
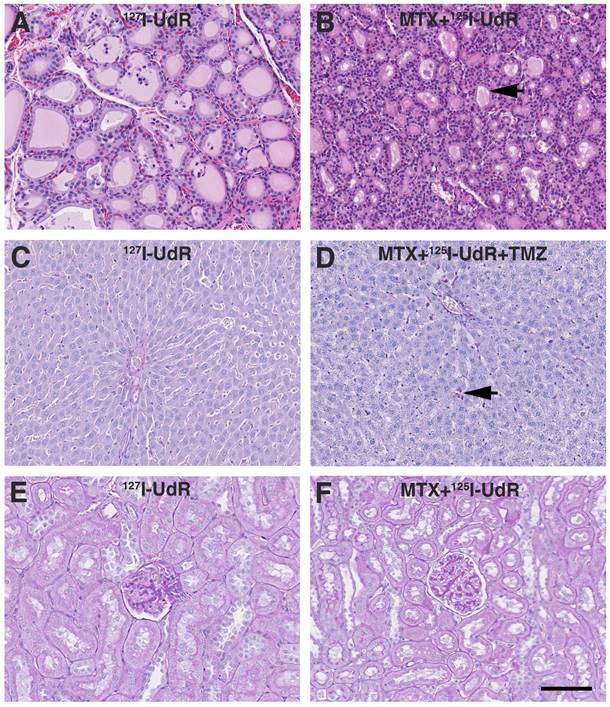
In general, no animal presented with neurological deficits or signs of discomfort indicating brain-related adverse effects due to the infused drugs or the CED procedure itself. All animals reaching the humane endpoint of >20% weight-loss had large tumors explaining their terminal weight-loss. No relevant histopathological findings were observed in the non-tumor infiltrated brain parenchyma. Total T3 and T4 levels in the Auger-therapy groups diminished to some extend post-treatment, but the drop stabilized from 13 to 26 weeks' post-treatment (Fig. 7). Histologically, animals in group 4 and 5 presented with microfollicular thyroid gland architecture and colloids with scalloped margins, indicative of stimulated and active glands (Fig. 8). Normofollicular architecture was found in the remaining groups. Hepatic function was evaluated by plasma ALT and BUN. No increase in ALT was observed in any of the groups (supplementary Fig. S2). BUN, which encompasses both hepatic (production) and renal (excretion) functions, increased after treatment start, but returned to baseline at 26 weeks. In all groups the liver architecture was normal. There were no signs of hepatocyte necrosis, haemorrhage or inflammation in all groups except group 5. In this group, a few cases of slight portal tract edema were observed and focally a few necrotic hepatocytes were present (Fig. 8). Renal function was assessed by plasma creatinine and whole blood potassium and sodium and to some degree plasma BUN. In all groups, creatinine initially increased (Fig. 7), whereas potassium decreased after treatment start while sodium remained stable throughout the study (Supplementary Fig. S2). In all groups, the kidneys appeared histologically normal without signs of tubular or glomerular damage or interstitial fibrosis (Fig. 8). Bone marrow function was assessed by whole blood Hb, WBC and platelets. No sign of bone marrow suppression was observed.
Discussion
As hypothesized, the use of the DNA-incorporating AEE compound 125I-UdR alone or in combination with TMZ effectively eradicated GBM cells by high-LET Auger-electron cascades. As seen in Fig. 2, viability and migration were significantly reduced in vitro in immature GSCs using very low activity concentrations of 125I-UdR and by concomitant TMZ treatment further cytotoxicity was achieved. This illustrates the therapeutic potential of DNA-incorporated AEEs, which has been documented in many cell lines [28, 29]. 125I decays via electron capture accompanied by a high degree of internal conversion, which results in nearly two Auger-electron cascades with on average approximately 20 electrons per decay. This gives rise to a very high radiation dose close to the decay site and high LET values (4-26 keV/µm) approaching those observed with α-particles (~80 keV/µm). The complex double-strand breaks created by these Auger-electron cascades from DNA-incorporated AEEs are very difficult for the cells to repair leading to cell death or loss of proliferative capacity [4].
The profound therapeutic response observed in vivo in the two 125I-UdR treated groups (Fig. 6) was confirmed by immunohistochemical vimentin staining as no residual bulk tumors were left (Fig. 5B-C). However, in 7/7 (100 %) rats in group 4 and 5/8 (63 %) rats in group 5, a few tumor cells were found near the original tumor-bed (Fig. 5B) 180 days after treatment start. These cells could represent cancer stem cells that survived the treatment [30, 31]. Alternatively, the remaining cells may represent non-clonogenic cells that have lost their ability to divide and, thus, from a radiobiological point of view could be regarded as “dead” cells, i.e. not being able to repopulate the tumor [32].
In the in vivo study, the infused 125I-UdR activity concentration was approximately 4-6 orders of magnitude higher than in the in vitro studies. The high concentration was chosen to ensure that distantly located migratory GBM cells were exposed to 125I-UdR. Moreover, 125I-UdR was delivered by CED, relying on a pressure-driven bulk flow created by a small pressure gradient [12], to increase the regional distribution as compared to simple passive diffusion [11]. This was confirmed by SPECT/CT, showing that 125I-UdR was distributed far away from the catheter tip (Fig. 4). As expected, SPECT/CT showed a decline in 125I activity concentration in the contralateral hemisphere and the caudal part of the brain compared to the isocenter of delivery. For that reason, more than one catheter has been applied in the majority of clinical CED trials [33, 34]. Technically, this is difficult in small animals as opposed to a full-sized human brain, where it will likely be required to obtain a reliable and widespread MTX + 125I-UdR intracerebral distribution. Barua et al. recently demonstrated in a glioblastoma patient, that it is possible to obtain a carboplatin distribution covering both the tumor volume and peri-tumoral penumbra using a transcutaneous bone-anchored port for CED [35]. Hence, CED of 125I-UdR including neoadjuvant MTX leading to a full tumor coverage of the compounds should be clinically achievable. By using the imaging analogue, 124I-UdR, which is chemically and thus, biologically identical to the therapeutic compound, it will be possible to determine the distribution and kinetics of the compound in the brain of the patient using PET imaging and to optimize the CED parameters prior to the therapy. Therefore, a patient-specific treatment may be possible using the theranostic pair 124/125I-UdR.
Infusion of 125I-UdR into rat brain tumors has previously been performed by Kassis et al., who used the syngeneic 9L rat gliosarcoma model and infused 6.9-8.8 MBq 125I-UdR intracerebrally for 1.5 or 6 days with a 10-fold lower infusion rate than in the current study [36]. They observed the best therapeutic response in animals with cells inoculated the day before and, thus, being without an established tumor. The median survival increased from 18 to 26 days and 20% of the 125I-UdR treated animals survived more than 85 days. In animals with established tumors, the median survival increased from 24 to 28.5 days. The explanation for the pronounced increase in therapeutic response in the current study, using animals with established tumors, is probably multifaceted. A much higher amount of cumulated activity was infused (approximately 80 MBq vs. 8.8 MBq), a 10-fold higher infusion rate was applied and the infusion period was prolonged to 8 days. This presumably results in a higher and a wider intracerebral activity distribution allowing eradication of both bulk tumor and distantly located GBM cells leading to a better effect. The prolonged infusion increases the probability that all tumor cells will undergo DNA synthesis during the infusion period, and thus, incorporate 125I-UdR. Finally, we applied neoadjuvant MTX, which is known to increase the percentage of cells undergoing DNA replication and, simultaneously, to increase the DNA-incorporation of 125I-UdR by thymidylate synthetase inhibition, leading to a better therapeutic effect in vivo as investigated by Kassis et al. [13, 37]. The authors found that pre-infusion of MTX prior to 125I-UdR therapy of spinal cord tumors in rats, enhanced the 125I-UdR tumor uptake a factor of 2 compared to 125I-UdR alone and led to a longer median survival. In the present study, 57% in group 4 became long-term survivors, and with the addition of TMZ the survival rate increased to 100% in group 5 (Fig. 6). This emphasizes the importance of not only local, but also regional, intracerebral distribution of high activity concentrations of 125I-UdR, which is obtainable by CED as opposed to local implant techniques [12, 38]. Additionally, the pronounced effect observed in group 5 of targeting the tumor cell DNA using two drugs (125I-UdR and TMZ) with different DNA targeting mechanisms became evident. A therapeutic effect of MTX itself was not anticipated in the present study. The increase in median survival with MTX + 127I-UdR was only approximately 6 days compared to untreated controls (non-significant, p=0.02). In comparison, systemic TMZ treatment alone increased the median survival 23 days (p<0.0001) compared to untreated controls. Hence, the considerable survival rate observed in the current study using 125I-UdR Auger-therapy emphasizes the therapeutic potential of this novel type of GBM-treatment compared to the currently used first-line chemotherapeutic agent TMZ. In GBM patients, the addition of concomitant TMZ chemotherapy to external radiotherapy increases the median survival from 12.1 to 14.6 months [39], so the proposed Auger-therapy could potentially have a significant impact on future GBM treatment - either alone or in combination with TMZ and external radiotherapy.
No animals showed objective signs of neurotoxicity. The same was reported in, to our knowledge, the only existing study on administration of 125I-UdR into the human central nervous system [40]. 125I-UdR entering the systemic circulation has a short biological half-life of only five minutes due to rapid catabolization and dehalogenation resulting in 125I-uracil and free 125I- [10]. None of these compounds are incorporated into the genomic DNA during cell division in normal tissue (as the 125I-UdR), which is required for high-LET-like radiotoxic effects and, hence, no severe adverse effects were anticipated. In line with this, blood parameters evaluating the bone marrow, harboring some of the most rapidly dividing cells in the body, remained unaffected. However, since athymic nude rats lacking mature T cells were used in the current study, further studies in immunocompetent rats should ideally be performed to fully characterize the bone marrow toxicity profile for the treatment.
The kidneys are another important potential dose-limiting organ, because of their excretion of free 125I. The treatment resulted in an increase in creatinine (Fig. 7), which remained, but apparently with little functional impact on the kidneys judged by the course of BUN and the excretion of potassium and sodium, and the lack of histological changes. However, to fully characterize the renal toxicity profile for the treatment, long-term studies (up to 12 months) in healthy rats will be required since late effects may show up at time points later than the six months used in the current study. To prevent uptake of free 125I- in the thyroid gland, the drinking water was supplemented with KI. Histologically, animals from group 4 and 5 had microfollicular thyroid architecture and colloids with scalloped margins (Fig. 8), which are signs of stimulated and active glands. This probably indicates that the thyroid blockings were not complete, leading to some uptake of free 125I-. However, the drop in total T3 and T4 blood levels in these groups during the study was considered irrelevant since the blood levels at the late time points were in the same range as those found for the untreated group 1. Furthermore, a drop in total T3 and T4 was also observed in some of the non-radioactive groups, and thus, not related to radiation exposure. Finally, signs of hepatotoxicity were assessed. In group 5, a few animals presented with slight portal tract edema, and focally a few necrotic hepatocytes were present (Fig. 8). In the remaining groups, histology was inconspicuous. In contrast to MTX, which is known to be highly hepatotoxic [41], TMZ has only been reported casuistically to induce liver injury (toxic hepatitis) [42]. However, the histological changes observed in group 5 were only very subtle and in comparison, no changes were found in group 6 subjected to TMZ monotherapy. In addition, blood samples in all groups showed no evidence of sustained toxic effects and, therefore, we regard the treatments as being non-hepatotoxic. Summarized, CED of MTX + 125I-UdR in combination with concomitant systemic TMZ proved safe without signs of dose-limiting adverse effects.
Conclusion
In conclusion, the multidrug approach including CED of MTX and the AEE-compound 125I-UdR in combination with systemic TMZ was safe and very effective in the orthotopic xenograft GBM model, leading to 100% survival. This suggests that this AEE-based therapeutic strategy may be a promising new option in the treatment of patients suffering from - presently incurable - GBMs.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by grants from Odense University Hospital, Region of Southern Denmark, Foundation of Family Hede Nielsen, Merchant M. Brogaard & Wife's Memorial Foundation, Director Kurt Bønnelycke & Wife's Mrs Grethe Bønnelycke's Foundation, A. J. Andersen & Wife's Foundation. Foundation of Family Erichsen, Carl J. Beckers Foundation, P. A. Messerschmidt & Wife's Foundation and the Grant of Master of Laws Torkil Steenbeck.
We thank Sönke Detlefsen, Niels Marcussen and Stine Larsen, the Department of Pathology, Odense University Hospital, for the histological examination and Christina Baun, Department of Nuclear Medicine and Helle Wohlleben and Tanja Højgaard, Department of Pathology, Odense University Hospital, for their technical assistance.
Competing Interests
The authors H. Thisgaard, B. Halle, C. Aaberg-Jessen, P. F. Høilund-Carlsen and B. W. Kristensen have filed a patent application on the subject matter, described in this publication. The other authors disclosed no potential conflicts of interest.
References
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96
2. Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA. et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma A Randomized Clinical Trial. Jama-J Am Med Assoc. 2015;314:2535-43
3. Mrugala MM. Advances and challenges in the treatment of glioblastoma: a clinician's perspective. Discov Med. 2013;15:221-30
4. Kassis AI. Therapeutic radionuclides: biophysical and radiobiologic principles. Semin Nucl Med. 2008;38:358-66
5. Olsen BB, Thisgaard H, Vogel S, Thomassen M, Kruse TA, Needham D. et al. Novel radioisotope-based nanomedical approaches. Eur J Nanomed. 2013;5:181-93
6. Kassis AI. Molecular and cellular radiobiological effects of Auger emitting radionuclides. Radiat Prot Dosimetry. 2011;143:241-7
7. Pouget JP, Santoro L, Raymond L, Chouin N, Bardies M, Bascoul-Mollevi C. et al. Cell membrane is a more sensitive target than cytoplasm to dense ionization produced by auger electrons. Radiat Res. 2008;170:192-200
8. Maucksch U, Runge R, Wunderlich G, Freudenberg R, Naumann A, Kotzerke J. Comparison of the radiotoxicity of the 99mTc-labeled compounds 99mTc-pertechnetate, 99mTc-HMPAO and 99mTc-MIBI. Int J Radiat Biol. 2016:1-9
9. Hampton EG, Eidinoff ML. Administration of 5-Iododeoxyuridine-I131 in the Mouse and Rat. Cancer Res. 1961;21:345-52
10. Klecker RW Jr, Jenkins JF, Kinsella TJ, Fine RL, Strong JM, Collins JM. Clinical pharmacology of 5-iodo-2'-deoxyuridine and 5-iodouracil and endogenous pyrimidine modulation. Clin Pharmacol Ther. 1985;38:45-51
11. Vogelbaum MA, Iannotti CA. Convection-enhanced delivery of therapeutic agents into the brain. Handb Clin Neurol. 2012;104:355-62
12. Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91:2076-80
13. Kassis AI, Dahman BA, Adelstein SJ. In vivo therapy of neoplastic meningitis with methotrexate and 5- I-125 iodo-2 '-deoxyuridine. Acta Oncol. 2000;39:731-7
14. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391-403
15. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J. et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821-8
16. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T. et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401
17. Wang K, Adelstein SJ, Kassis AI. DMSO increases radioiodination yield of radiopharmaceuticals. Appl Radiat Isot. 2008;66:50-9
18. Dam JH, Nagren K. Good manufacturing practice production of the system A amino acid transport tracer C-11 MeAIB on a commercial synthesis module. J Labelled Compd Rad. 2014;57:61-4
19. Jensen SS, Aaberg-Jessen C, Andersen C, Schroder HD, Kristensen BW. Glioma Spheroids Obtained via Ultrasonic Aspiration Are Viable and Express Stem Cell Markers: A New Tissue Resource for Glioma Research. Neurosurgery. 2013;73:868-86
20. Kolenda J, Jensen SS, Aaberg-Jessen C, Christensen K, Andersen C, Brunner N. et al. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J Neurooncol. 2011;103:43-58
21. Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009;100:2235-41
22. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V. et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350-4
23. Thisgaard H, Olsen BB, Dam JH, Bollen P, Mollenhauer J, Høilund-Carlsen PF. Evaluation of Cobalt-Labeled Octreotide Analogs for Molecular Imaging and Auger Electron-Based Radionuclide Therapy. J Nucl Med. 2014;55:1311-6
24. Goddu SM, Howell RW, Lionel GB, Bolch WE, Rao DV. MIRD Cellular S Values. Reston, VA: Society of Nuclear Medicine. 1997
25. Howell RW, Rao DV, Hou DY, Narra VR, Sastry KSR. The Question of Relative Biological Effectiveness and Quality Factor for Auger Emitters Incorporated into Proliferating Mammalian Cells. Radiat Res. 1991;128:282-92
26. Dam JH, Halle B, Thisgaard H, Hvidsten S, Kristensen BW, Nagren K. Fully automated radiosynthesis and formulation of C-11 MeAIB applied for in vivo imaging of glioblastoma. J Labelled Compd Rad. 2013;56:S112
27. Halle B, Thisgaard H, Hvidsten S, Dam JH, Aaberg-Jessen C, Thykjaer AS. et al. Estimation of Tumor Volumes by C-11-MeAIB and F-18-FDG PET in an Orthotopic Glioblastoma Rat Model. J Nucl Med. 2015;56:1562-8
28. Adelstein SJ, Kassis AI, Bodei L, Mariani G. Radiotoxicity of iodine-125 and other Auger-electron-emitting radionuclides: Background to therapy. Cancer Biother Radio. 2003;18:301-16
29. Elmroth K, Stenerlöw B. DNA-Incorporated125I Induces more than one Double-Strand Break per Decay in Mammalian Cells. Radiat Res. 2005;163:369-73
30. Altaner C. Glioblastoma and stem cells. Neoplasma. 2008;55:369-74
31. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
32. Hall EJ. Radiobiology for the radiologist. 6. ed. ed: Lippincott Williams and Wilkins, Philadelphia, USA. 2006
33. Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G. et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010;12:871-81
34. Sampson JH, Akabani G, Archer GE, Berger MS, Coleman RE, Friedman AH. et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro Oncol. 2008;10:320-9
35. Barua NU, Hopkins K, Woolley M, O'Sullivan S, Harrison R, Edwards RJ. et al. A novel implantable catheter system with transcutaneous port for intermittent convection-enhanced delivery of carboplatin for recurrent glioblastoma. Drug Deliv. 2015:1-7
36. Kassis AI, Wen PY, Van den Abbeele AD, Baranowska-Kortylewicz J, Makrigiorgos GM, Metz KR. et al. 5- I-125 iodo-2 '-deoxyuridine in the radiotherapy of brain tumors in rats. J Nucl Med. 1998;39:1148-54
37. Kassis AI, Kirichian AM, Wang K, Semnani ES, Adelstein SJ. Therapeutic potential of 5-[125I]iodo-2'-deoxyuridine and methotrexate in the treatment of advanced neoplastic meningitis. Int J Radiat Biol. 2004;80:941-6
38. Juratli TA, Schackert G, Krex D. Current status of local therapy in malignant gliomas-a clinical review of three selected approaches. Pharmacol Ther. 2013;139:341-58
39. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459-66
40. Rebischung C, Hoffmann D, Stefani L, Desruet MD, Wang K, Adelstein SJ. et al. First human treatment of resistant neoplastic meningitis by intrathecal administration of MTX plus (125)IUdR. Int J Radiat Biol. 2008;84:1123-9
41. Dalaklioglu S, Genc GE, Aksoy NH, Akcit F, Gumuslu S. Resveratrol ameliorates methotrexate-induced hepatotoxicity in rats via inhibition of lipid peroxidation. Hum Exp Toxicol. 2013;32:662-71
42. Sarganas G, Orzechowski HD, Klimpel A, Thomae M, Kauffmann W, Herbst H. et al. Severe sustained cholestatic hepatitis following temozolomide in a patient with glioblastoma multiforme: case study and review of data from the FDA adverse event reporting system. Neuro Oncol. 2012;14:541-6
Author contact
![]() Corresponding author: Dr. Helge Thisgaard, PhD. Department of Nuclear Medicine, Odense University Hospital, Sdr. Boulevard 29, DK-5000 Odense, Denmark. Email: Helge.Thisgaarddk Phone: +45 2138 0417 Fax: +45 6590 6192.
Corresponding author: Dr. Helge Thisgaard, PhD. Department of Nuclear Medicine, Odense University Hospital, Sdr. Boulevard 29, DK-5000 Odense, Denmark. Email: Helge.Thisgaarddk Phone: +45 2138 0417 Fax: +45 6590 6192.