Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(2):253-269. doi:10.7150/thno.16681 This issue Cite
Research Paper
Enhancing Nanoparticle Accumulation and Retention in Desmoplastic Tumors via Vascular Disruption for Internal Radiation Therapy
Andrew B. Satterlee1, 2, Juan D. Rojas2, Paul A. Dayton2, Leaf Huang1,2 ![]()
1. Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7571, USA.
2. UNC and NCSU Joint Department of Biomedical Engineering, Chapel Hill, NC 27599.
Received 2016-7-1; Accepted 2016-9-1; Published 2017-1-1
Abstract

Aggressive, desmoplastic tumors are notoriously difficult to treat because of their extensive stroma, high interstitial pressure, and resistant tumor microenvironment. We have developed a combination therapy that can significantly slow the growth of large, stroma-rich tumors by causing massive apoptosis in the tumor center while simultaneously increasing nanoparticle uptake through a treatment-induced increase in the accumulation and retention of nanoparticles in the tumor. The vascular disrupting agent Combretastatin A-4 Phosphate (CA4P) is able to increase the accumulation of radiation-containing nanoparticles for internal radiation therapy, and the retention of these delivered radioisotopes is maintained over several days. We use ultrasound to measure the effect of CA4P in live tumor-bearing mice, and we encapsulate the radio-theranostic isotope 177Lutetium as a therapeutic agent as well as a means to measure nanoparticle accumulation and retention in the tumor. This combination therapy induces prolonged apoptosis in the tumor, decreasing both the fibroblast and total cell density and allowing further tumor growth inhibition using a cisplatin-containing nanoparticle.
Keywords: Cancer, Theranostic, Nanoparticle, CA4P, EPR, Desmoplastic, Cisplatin.
Introduction
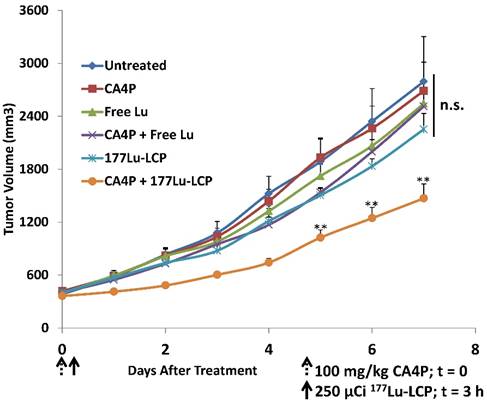
We have previously published on the antitumor efficacy of our 177Lutetium (177Lu)-loaded Lipid-Calcium-Phosphate (177Lu-LCP) nanoparticles, which provide internal radiation therapy via the beta decay of 177Lu as well as live animal imaging via 177Lu's gamma decay [1]. One dose of these nanoparticles generated significant and prolonged tumor growth inhibition in two subcutaneous xenograft tumor models with initial tumor volumes of 100-150 mm3. The success of this approach prompted us to gauge whether the antitumor effect could be maintained in larger tumors, which present a number of additional, clinically relevant challenges to delivery and treatment success, including an increased growth rate, a more mature and resistant stromal structure/microenvironment, and increased interstitial pressure that can decrease nanoparticle accumulation. Successful strategies against these large, aggressive, and often desmoplastic (stroma-rich) tumors may therefore translate more effectively into positive patient outcomes in the clinic. The UMUC3 human bladder cancer model, supplemented with murine NIH/3T3 fibroblasts, is histologically similar to desmoplastic bladder cancer seen in the clinic and responds to treatment similarly to patient bladder cancer as well [2]. Although smaller UMUC3/3T3 tumors were shown to be sensitive to 177Lu-LCP, we show in Figure 4 of this paper that in our large UMUC3/3T3 tumor model (initial tumor volumes ~300-400 mm^3; Volume = (Length x Width x Depth)/2), 177Lu-LCP monotherapy no longer induces a significant antitumor effect. We therefore designed a combination therapy in which the small molecule drug and radiosensitizer [3, 4] Combretastatin A-4 Phosphate (CA4P) is used alongside our radioactive nanoparticles to induce a multifaceted synergistic effect.
In vivo tumor growth inhibition in UMUC3/3T3 stroma-rich bladder cancer subcutaneous xenograft tumor model. On Day 0, 100 mg/kg CA4P was administered i.p. 3 h before i.v. treatment with 250 μCi 177Lu, either as 177LuCl3 or formulated as 177Lu-LCP. No subsequent doses were given. Initial tumor sizes = 300-400 mm3; n = 5-7; **p<0.01 compared to 177Lu-LCP.
The microtubule inhibitor CA4P works by binding to the alpha-beta tubulin heterodimer in the cell cytoplasm and preventing microtubule polymerization. Because cellular spindle fibers are composed of microtubules, this action has a cytotoxic effect on actively dividing cells in vitro, preventing separation of chromosomes during mitosis and leading to cell death via mitotic catastrophe [5].
Treatment with the vascular disrupting agent CA4P also generates a tumor-selective mechanism of cytotoxicity in vivo [3, 6, 7]. During tumor growth, angiogenesis of new blood vessel endothelium occurs, and these endothelial cells must maintain a flat structure in order to provide a stable and open blood vessel lumen. While the cytoskeletons of mature vasculature, such as those found in healthy tissue, are composed of actin, the cytoskeletons of newly formed tumor vasculature are composed of microtubules [3, 8]. It has been hypothesized that when microtubule polymerization is inhibited by CA4P, the flat structure of only the tumor endothelial cells is compromised, causing them to become more round and occlude the lumen of the blood vessel [9]. This decreases blood flow to the center of the tumor, depriving the cells of oxygen and nutrients. In extreme cases, the destruction of blood vessel integrity has been shown to cause hemorrhage and subsequent necrosis of the surrounding region. Because mature, healthy vasculature has a cytoskeleton made of actin and healthy cells do not frequently divide, CA4P has very low toxicity to healthy tissue and is tolerable in mice at doses well above 100 mg/kg [6].
It is important to note that vascular disrupting agents such as CA4P are distinct from anti-angiogenic agents. Anti-angiogenic agents are used to prevent the formation of new blood vessels and are most effective when treating small tumors or metastases in order to prevent significant blood vessel formation and growth [6]. In contrast, vascular disrupting agents work to destroy existing blood vessels in the tumor, and are particularly suited for treating large, resistant tumors by causing secondary necrosis in the middle of the tumor [10].
A recent publication that has used CA4P in combination with nanoparticles has reported that the CA4P-induced vessel occlusion limited nanoparticle accumulation to the outer rim of the tumor [11], but we have alternatively found that the intratumoral hemorrhage induced by CA4P allows an increased accumulation of nanoparticles throughout the tumor. This additional dose was retained in the tumor over several days, and the sustained higher dose combined with the destructive effects of CA4P significantly inhibited tumor growth even in our large, stroma-rich tumor model.
Materials and Methods
Materials
177LuCl3 and 90Yttrium (90Y)Cl3 were purchased from PerkinElmer (Waltham, MA). 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA), and 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) were purchased from Avanti Polar Lipids (Alabaster, AL). N-(Carbonyl-methoxypolyethyleneglycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine, sodium salt (DSPE-PEG2000) was purchased from NOF America Corporation (White Plains, NY). DSPE-PEG2000-Anisamide (DSPE-PEG-AA) was synthesized in our lab as described previously [12]. Other chemicals and antibodies were purchased from Sigma-Aldrich (St. Louis, MO) or Abcam (Cambridge, MA).
Cell Lines
Four different cell lines were used in the experiments described below: UMUC3 human bladder cancer cells, NIH/3T3 (3T3) murine fibroblasts, 4T1 murine breast cancer cells, and B16F10 murine melanoma cells. In some experiments, UMUC3 cells that had been stably transfected with green fluorescence protein (GFP) and 3T3 cells that had been stably transfected with red fluorescence protein (RFP) were used.
Experimental Animals
Female athymic nude mice were used for UMUC3/3T3 tumor experiments presented in this manuscript. Balb/c mice were used as syngeneic hosts for 4T1 tumor inoculation, C57Bl/6 mice were used as syngeneic mice for B16F10 tumor inoculation, and CD-1 mice were used for toxicity studies. The nude mice were purchased from the National Cancer Institute (Bethesda, MD) and bred at the Division of Laboratory Animal Medicine at the University of North Carolina-Chapel Hill. The Balb/c, C57Bl/6, and CD-1 mice were purchased from Charles River labs. All work performed on these animals was approved by the Institutional Animal Care and Use Committee at the University of North Carolina-Chapel Hill, protocol #14-045.
177Lu-LCP, 90Y-LCP, and LPC Fabrication and Characterization
The Lipid-Platinum-Chloride (LPC) nanoparticles were fabricated exactly as described in the literature [13]. LPC encapsulation of CDDP was determined by nuclear magnetic resonance and X-ray photoelectron spectroscopy, and drug loading of cisplatin (cis-diamminedichloridoplatinum(II), or CDDP) was determined by Inductively Coupled Plasma Mass Spectroscopy (ICP-MS). 177Lu/90Y-LCP fabrication was slightly modified from what is described in a previous publication [1]. Two reverse microemulsions were prepared in round-bottom flasks. Both microemulsion oil phases contained a mixture of 71% Cyclohexane and 29% Igepal CO-520. For a batch size of 1.67 mL oil phase per flask, 50 μL of 200 mM (NH4)2PO4 was added to one flask and 50 μL of 2.5 M CaCl2 with up to five additional μL of 177LuCl3 or 90YCl3 in 0.05 M HCl was added to the other under stirring. After mixing separately for five min, the phosphate-containing emulsion was added directly to the calcium-containing emulsion. After five more min, 26.6 μL of 25 mg/mL DOPA dissolved in chloroform was added to the mixture. After 20 more min of mixing, 3.34 mL of 100% ethanol was added to break the emulsion, and the mixture was stirred for 10 more minutes before centrifugation at 12,600g for 15 min. The supernatant was then removed (~15% of input 177Lu was removed here) and ethanol was again added to wash the pelleted cores. After addition of ethanol, the vial(s) were vortexed to resuspend the nanoparticle cores and centrifuged at 12,600g for 10 min. After removing the supernatant, the organic-soluble cores were dissolved in chloroform and centrifuged again to remove any insoluble salts or particles that had not been well-coated with DOPA (~5% of input removed here). Purifying the LCP cores in these two ways ensured that 177Lu or 90Y were only present inside the LCP. Outer leaflet lipids (OLLs) dissolved in chloroform were then added to the chloroform-soluble cores (40:40:18:2 DOTAP:cholesterol:DSPE-PEG2000:DSPE-PEG2000-AA mol:mol:mol) (0.11:1 total OLL:initial oil phase v:v; all lipids at 20mM concentration). In experiments using fluorescent 1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindocarbocyanine Perchlorate (DiI)-labeled LCP, DiI dissolved in chloroform was added to the cores with the OLLs (1:100 DiI:total OLL mol:mol). Chloroform was evaporated until the particles and lipids coated the vial in a lipid film. Particles were then rehydrated to the injection volume with 5% glucose in water at 60 oC. The solution was vortexed and sonicated liberally before injection into mice. The anisamide ligand was used in all formulations to target the sigma receptor overexpressed in UMUC3 cells [14].
In experiments using DiI-labeled Lu-LCP, sucrose gradient ultracentrifugation (50,000g for 6 h) was used to purify DiI-Lu-LCP from excess DiI that had incorporated into excess free liposomes.
Encapsulation efficiency of 177Lu into 177Lu-LCP was measured by a Model AA2010 “Nucleus” Gamma Scintillation Counter or a Capintec Radioisotope Calibrator CRC-127R and calculated as (signal from 177Lu in LCP cores)/(signal from 177Lu in input aqueous phase). Although 90Y is a pure beta emitter, secondary emissions were able to be used to calculate the encapsulation efficiency of large doses of 90Y-LCP using the CRC-127R. Encapsulation efficiencies for 177Lu and 90Y into LCP were both found to be ~80-85% of the input. The encapsulation efficiency of CDDP into LPC was only ~20% of the input, but the % drug loading of CDDP into LPC nanoparticles was ~80%, consistent with the literature[13]. 177Lu-LCP, 90Y-LCP, and LPC hydrodynamic diameters, polydispersity indices (PDI), and zeta potentials were measured using a Malvern Nano ZS dynamic light scattering instrument. Particle size was corroborated using transmission electron microscopy (TEM).
The stability of LCP after sonication and vortexing was quantified using ultracentrifugation across a water-sucrose boundary, as nanoparticles in water should move down across the sucrose gradient during centrifugation due to their high density, while free 177Lu atoms should remain at the top layer, only slowly diffusing across the sucrose barrier. Three different groups were tested, each in duplicate. In the 177Lu-LCP group receiving vortexing and sonication, water was added to the lipid film to hydrate the particles, followed by ten seconds of vortexing and ten seconds of sonication. In the 177Lu-LCP group receiving no vortexing or sonication, water was added to hydrate the particles and the water was gently swirled in the vial. In the free 177Lu group, 177LuCl3 was diluted in water. Each sample of 250 μl was carefully added on top of a layer of 10% w/v sucrose in water in an ultracentrifuge tube. The samples were spun at 50,000 RPM for 5 h, and then the top 400 μl of liquid was collected from each tube. The percentage of total 177Lu signal in the top 400 μl was compared across all groups.
To quantify the release of 177Lu from 177Lu-LCP in pH 7.4 tris-buffered saline at 37oC, 177Lu-LCP was dialyzed using 3K molecular weight cutoff Slide-A-Lyzer mini dialysis devices (Thermo Fisher, Waltham, MA) and the un-dialyzed supernatant was periodically measured for 177Lu activity using gamma scintillation.
Cell Viability Assay
UMUC3 and 3T3 cells were seeded at a density of 2,500 cells/well in a 96-well culture plate. UMUC3 cells were incubated in 200 µL/well of Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum and 1% penicillin/streptomycin, and 3T3 cells were incubated in 200 µL/well DMEM containing 10% bovine calf serum and 1% penicillin/streptomycin. 24 h later, escalating doses of CA4P were added to the cell culture media, n = 6 for each cell type. After 48 h, 20 μL of working 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) solution was added to each well. ~2 h later, the media was carefully aspirated and replaced with 200 μL Dimethyl Sulfoxide (DMSO) in order to solubilize the MTT product. The DMSO was mixed thoroughly to ensure complete dissolution of the product, and each well was measured for absorbance at 570 nm. Cell viability was calculated as percent of untreated wells.
CA4P Vessel Occlusion
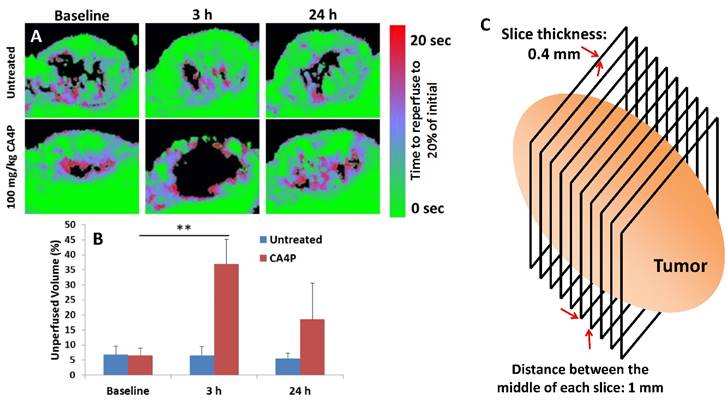
Nude mice bearing subcutaneous UMUC3/3T3 tumors were sedated with isofluorane and an intravenous catheter was inserted into the tail vein. Mice were transferred to a heated stage and ultrasound gel was used to couple a 15L8 ultrasound transducer to the tissue. An Acuson Sequoia 512 ultrasound system (Siemens Corporation, Washington DC, USA) was used to capture contrast images and parametric maps of blood perfusion. Preclinical ultrasound microbubble contrast agents composed of a phospholipid shell and a perfluorocarbon core (diameter ~ 2 μm) were manufactured in house and injected into circulation for the contrast imaging experiments. Microbubbles were continuously infused until a steady state concentration in the blood was reached. A quantitative measure of perfusion was obtained using a technique called “Destruction Reperfusion”, where bubbles are cleared from the imaging plane using high-amplitude ultrasound pulses, and the tissue is monitored as microbubbles replenish the tissue [15, 16]. Parametric perfusion maps were generated using proprietary Sequoia software; the time necessary for each pixel to regain 20% of its original intensity before destruction was measured and displayed in a colormap ranging between fast (green) and slow (red) perfusion as can be seen in the figure. The reperfusion was monitored for 20 seconds after destruction, and a 3D perfusion map was captured by sweeping the transducer across the tumor and performing destruction reperfusion at several locations, using a 1mm step size. The thickness of each section was 0.4 mm, equal to the width of the ultrasound beam. The area that was not reperfused in that time frame, shown in black, was quantified and averaged for all sections in four separate tumors. After this initial baseline value was recorded, the mouse was treated intraperitoneally (i.p.) with 100 mg/kg CA4P. At t = 3 h and 24 h after treatment, the same four mice were again imaged in the same manner as before.
Effects of CA4P on UMUC3/3T3 Tumors
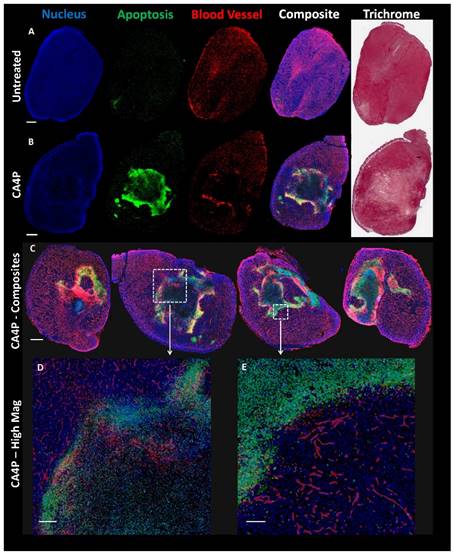
Mice bearing UMUC3/3T3 tumors ~300-400 mm3 in volume were either treated i.p. with 100 mg/kg CA4P in 200 μL of phosphate-buffered saline (PBS) or left untreated. 24 h after treatment, the mice were sacrificed and their tumors were dissected. Tumors were fixed in 10% formalin for 48 h, embedded in paraffin, and sectioned. Three adjacent sections were stained for apoptosis (using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), Promega), blood vessels (CD-31 immunofluorescence, Abcam), and Masson's Trichrome. The Aperio Scanscope at UNC's Translational Pathology lab was used to acquire images of the entire tumor section. Sections were overlaid using ImageJ.
Tumor Growth Inhibition Studies
Four different tumor growth inhibition experiments were performed in this study. In all cases, initial tumor volumes for each group averaged ~300-400 mm3, calculated as Volume = (Length x Width x Depth)/2. Digital calipers were used to measure the greatest diameter of the tumor (length), the greatest diameter in the perpendicular dimension (width), and the greatest thickness of the tumor in the direction perpendicular to both length and width (depth). To measure depth, mice were first restrained by the tail while standing on their cage. The tumor was rocked to the side using the hand holding the tail, and the other hand was used to measure the tumor's depth using the digital calipers. This method was facile and reproducible. Five million UMUC3 cells and 2.5 million 3T3 cells were inoculated in a total volume of 100 μL and in a 1:3 matrigel:PBS solution (Corning Matrigel Matrix, High Concentration). Tumor volumes were measured daily. In groups receiving CA4P, CA4P was always dissolved in PBS and administered i.p. in a total of 200 ul. Groups receiving 177Lu-LCP always received a dose of 250 microCuries (μCi) administered i.v. via the tail vein. Groups receiving 90Y-LCP always received a dose of 125 μCi administered i.v. via the tail vein. Groups receiving a combination therapy always received CA4P 3 h before LCP.
Tumor Accumulation and Retention of 177Lu
UMUC3/3T3, 4T1, and B16F10 tumors were inoculated in nude, Balb/c, and C57Bl/6 mice, respectively. When tumors reached ~300-500 mm3, half were treated with CA4P followed by a known amount of 177Lu-LCP at t = 3 h, and half were treated with 177Lu-LCP only. Twenty-four h later, the mice were sacrificed and their tumors were dissected. Tumor accumulation of 177Lu was quantified via gamma scintillation. After quantification, the tumors were incubated in formamide for 48 h at 60 oC to show the blood in the tumor centers that had been treated with CA4P. Separate UMUC3/3T3 tumors were treated with purified DiI-Lu-LCP and dissected 24 h after treatment. These tumors were fixed for 48 h in 10% formalin at 4 oC, incubated in 30% sucrose in PBS overnight, and then frozen in optimal cutting temperature (OCT) reagent. After cutting into the middle of each tumor, a photograph was taken to show the extent of the blood-filled region, and two adjacent sections were cut. One section was imaged to show the distribution of DiI in the tumor, and the second section was stained with Masson's trichrome to show the morphology of the section.
Measurement of 177Lu-LCP retained in the tumor over several days was also quantified by gamma scintillation of dissected tumors once those tumors reached a volume of 1000 mm3.
Prolonged Tumor Necrosis
Mice bearing UMUC3/3T3 tumors ~300-400 mm3 in volume were treated with 100 mg/kg CA4P only or with 177Lu-LCP or 90Y-LCP 3 h after CA4P treatment. Four days after treatment, mice were sacrificed and tumors were dissected. Tumors were cut into 3-5 pieces to facilitate fixation and were fixed in 10% formalin for 48 h at 4 oC before being embedded in paraffin. Tumor sections were cut and two sections 8 µm apart were stained in two different ways: the first was stained for apoptosis with the TUNEL assay and the second was 3,3'-Diaminobenzidine (DAB)-stained using immunohistochemistry (IHC) against the Ki-67 proliferation marker. The images taken from these two nearby sections were overlaid using ImageJ in order to compare distribution of apoptosis and proliferation.
Treatment-Induced Changes in Microenvironment
Mice were inoculated with UMUC3 cells stably expressing green fluorescence protein and 3T3 cells stably expressing red fluorescence protein in the same manner as described above. Treatment began when tumors reached a volume of ~300-400 mm3. On day seven after treatment, the tumors were dissected, cut, and fixed in 10% formalin for 48 h, followed by incubation in 1:1 PBS:OCT overnight and freezing in 100% OCT. Representative tumors were sectioned and two adjacent sections were used for quantification. One section was stained with 4'6-Diamidino-2-Phenylindole (DAPI) only using Prolong Gold Antifade mounting medium (Thermo Fisher, Waltham, MA) and was used to compare the distribution of green UMUC3 cells and orange 3T3 cells. The other section was DAB-stained using IHC against cleaved Caspase 3 to measure apoptosis. In order to gain appropriate resolution for quantification, multiple random fields (~4 mm2) from multiple tumors were used to quantify fibroblast density, total cell density, and apoptotic cells.
Additional Treatment with Cisplatin-Containing LPC
Mice were inoculated with UMUC3/3T3 tumors as described above. At the time of study initiation, there were two groups comprised of tumors ~300-400 mm3 in volume. One group remained untreated, while the other group received 100 mg/kg CA4P + 125 μCi 90Y-LCP. When the untreated group reached an average volume of ~ 800 mm3, half of the tumors received 1.5 mg/kg cisplatin formulated in the LPC nanoparticle, and the other half of the tumors received no additional treatment. When the group receiving CA4P + 90Y-LCP reached an average volume of ~ 800 mm3, half of those tumors received 1.5 mg/kg cisplatin formulated in the LPC nanoparticle, and the other half of the tumors received no additional treatment. Tumor volumes were measured daily and mice were sacrificed t = 10 days after the initial treatment.
To test nanoparticle accumulation in previously treated and untreated tumors, 300-400 mm3 tumors were treated with CA4P + 90Y-LCP or left untreated, and six days later all tumors were injected with 177Lu-LCP. 24 h later, the mice were sacrificed and the tumor accumulation of 177Lu was measured via gamma scintillation counting.
Toxicity Studies
CD-1 mice were treated with several combination therapies or appropriate controls and were sacrificed 10 days after treatment. Blood and organs were dissected. Serum levels of AST, ALT, BUN, and creatinine were measured, and organs were stained with H&E to measure any changes in tissue morphology. Whole blood was measured for changes in white blood cell count (WBC), hematocrit (HCT), mean cell volume (MCV), red blood cell count (RBC), hemoglobin count (HGB), and platelet count (PLT).
Results
CA4P Monotherapy
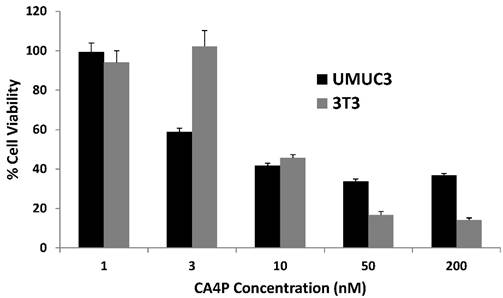
We first desired to gauge the relative toxicity of CA4P on UMUC3 and 3T3 cells in vitro, and an MTT cell proliferation assay was conducted after the cells were incubated with escalating doses of CA4P for 48 h. Both cell lines responded at low doses, both with a half maximal inhibitory concentration (IC50) around 10nM (p < 0.0001 compared to untreated cells for UMUC3 and 3T3 cells), indicating the potency of microtubule polymerization inhibition on both cell types.
The effects of 100 mg/kg CA4P on the blood vessels of large (300-400 mm3), stroma-rich UMUC3/3T3 subcutaneous xenograft tumors in vivo were also determined. We used ultrasound microbubble contrast agents to measure the perfusion of blood through tumors before and after i.p. administration of CA4P. Microbubbles with diameter ~2 μm were continuously intravenously administered via syringe pump during imaging, and an ultrasound transducer recorded the distribution of the bubbles in several 0.4 mm-thick sections across the entire length of the tumor. The way the transducer moved across the tumor is depicted as a cartoon in Figure 2C. High-amplitude ultrasound was used to pop the bubbles in each section (termed microbubble destruction), and time-dependent reperfusion of microbubbles back into the section was measured over a span of 20 s (Figure 2A). Areas that were quickly reperfused with microbubbles are shown in green in Fig 2A, while areas that took a longer time for microbubbles to reperfuse are shown in red. Any area that microbubbles did not reperfuse to 20% of the steady-state density in the first 20 s after destruction is shown as black. The black areas from each imaged section across a tumor were averaged, and the quantification is shown in Fig 2B. Significant blood vessel occlusion occurred 3 h after treatment, and after 24 h, some but not all of the vessel perfusion had normalized. Preliminary data (not pictured) showed that vessel occlusion was greater at t = 3 h after treatment than it was at t = 1 h or t = 7 h after treatment. Figure S1 shows videos of real-time microbubble reperfusion into the middle section of representative treated and untreated tumors at t = 3 h, as well as images of the microbubble reperfusion pattern in all sections across a single tumor at t = 20s after microbubble destruction. As reported in literature, the outer rim of each treated tumor at t = 3 h did not show significantly decreased perfusion. It is hypothesized that this occurs because these cells can be fed by more mature vasculature in the surrounding healthy tissue.
UMUC3 and 3T3 proliferation under CA4P dosing in vitro. Drug incubated with cells for 48 h. Cell viability expressed as percent of untreated control group. n = 6.
CA4P vessel occlusion. Nude mice bearing subcutaneous UMUC3/3T3 tumors were imaged using a microbubble ultrasound contrast agent at a baseline level and at t = 3 h and 24 h after i.p. administration of 100 mg/kg CA4P. Untreated group not given CA4P. Microbubbles estimate speed of blood perfusion in the tumor. A) Middle section of a representative tumor from each group and time point. B) Quantification of tumor volume that was not perfused to 20% of initial microbubble concentration 20 sec after destruction, n = 4. **p < 0.01; Baseline CA4P compared to CA4P at t = 3 h. C) Image acquisition scheme.
The decrease in blood perfusion caused by CA4P generated areas of significant apoptosis 24 h after treatment. Figure 3A (top row of Figure 3) shows a low magnification view of untreated tumor sections stained in different manners. From left to right, sections are shown with DAPI nuclear stain (blue), TUNEL apoptosis stain (green), CD-31 blood vessel stain (red), a composite of DAPI, TUNEL, and CD-31, and Masson's trichrome stain. Figure 3B directly below shows a tumor section 24 h after treatment with 100 mg/kg CA4P. The middle of this tumor contains a large amount of apoptosis as well as a loss of blood vessel structure. Figure 3C shows additional sections of tumors treated with CA4P, each with varying degrees of apoptosis in response to the CA4P treatment. High-magnification composite images in Fig 3D and 3E also show a lower cell density and reduced vessel structure in apoptotic regions.
Effects of CA4P on UMUC3/3T3 Tumors. A) Adjacent untreated tumor sections stained with DAPI (cell nucleus, blue), TUNEL (apoptosis, green), CD-31 immunostain (blood vessels, red), or Masson's Trichrome. Composite image formed from overlay of DAPI, TUNEL, and CD-31 stains. Scale bar (A, B, C) = 1 mm. B) Adjacent sections of tumor 24 h after mouse was treated with 100 mg/kg CA4P i.p. Stainings match those in (A). C) Composite image of four representative treated tumor samples 24 h after 100 mg/kg CA4P. D) Zoomed composite image; scale bar = 200 µm. E) Zoomed composite image; scale bar = 100 µm.
CA4P + 177Lu-LCP Combination Therapy
Despite the in vitro and in vivo cytotoxic effects of CA4P, treatment with the drug alone is completely ineffective against our large, aggressive UMUC3/3T3 tumor model. Monotherapy with 177Lu-LCP also does not generate any significant tumor growth inhibition, although when 177Lu-LCP is given intravenously (i.v.) three hours after CA4P is given i.p.—the time at which vessel occlusion is highest—tumor growth is significantly inhibited compared to 177Lu-LCP alone (Figure 4). In a previous publication, we have shown that LCP containing the non-radioactive 175Lu also provides no therapeutic effect.
CA4P increases the “enhanced permeability and retention” (EPR) of tumors
After observing such a marked decrease in tumor growth rate, we attempted to determine how CA4P and 177Lu-LCP were able to work synergistically. One reason why 177Lu-LCP alone is ineffective may be because in large tumors, the high interstitial pressure limits accumulation of nanoparticles into tumors [17]. We found that while the accumulation of 177Lu-LCP was quite low in larger UMUC3/3T3 tumors receiving 177Lu-LCP alone, if 177Lu-LCP was given 3 h after 100 mg/kg CA4P, its accumulation significantly increased (Fig 5A). In two additional large tumor models—a 4T1 orthotopic breast cancer model syngeneically grown in Balb/c mice and a B16F10 subcutaneous melanoma model syngeneically grown in C57Bl/6 mice—CA4P induced higher tumor accumulation, although for tumor models with higher 177Lu-LCP accumulation during monotherapy, the increase in tumor accumulation after CA4P was less pronounced. This could be related to the natural differences in vessel permeability among the models. While the hard, dense UMUC3/3T3 and 4T1 tumors can be described as “stroma-vessel” [18] types in which blood vessels grow within a stroma/fibroblast structure, the soft B16F10 tumors grow much less densely as “tumor-vessel” type tumors with leakier vasculature, which may contribute to their innately high tumor accumulation and insensitivity to changes in nanoparticle accumulation after treatment with CA4P.
Tumor Accumulation of 177Lu. A) Subcutaneous tumor accumulation of 177Lu 24 h after i.v. administration of 177Lu-LCP with or without 100 mg/kg CA4P given i.p. 3 h earlier. UMUC3/3T3 tumors inoculated in nude mice; n = 8. 4T1 tumors inoculated in Balb/c mice; n = 4. B16F10 tumors inoculated in C57BL/6 mice; n = 4-5. B) Image of UMUC3/3T3 tumors untreated (left) or 24 h after treatment with 100mg/kg CA4P i.p. (right); Scale bars ~ 5mm. C) Three tumor types incubated in formamide for 48 h at 60 oC show blood in their centers when treated with CA4P; scale bar ~5mm. D) UMUC3/3T3 tumor 24 h after treatment with 100 mg/kg CA4P; left panel: photograph of tumor tissue frozen in OCT during sectioning, bloody area outlined in black; middle panel: distribution of DiI-177Lu-LCP in tumor; right panel: H&E stained section adjacent to section taken for middle panel. Scale bar = 1 mm. E) Quantification of DiI-177Lu-LCP in bloody and non-bloody regions of tumor sections; n = 4.
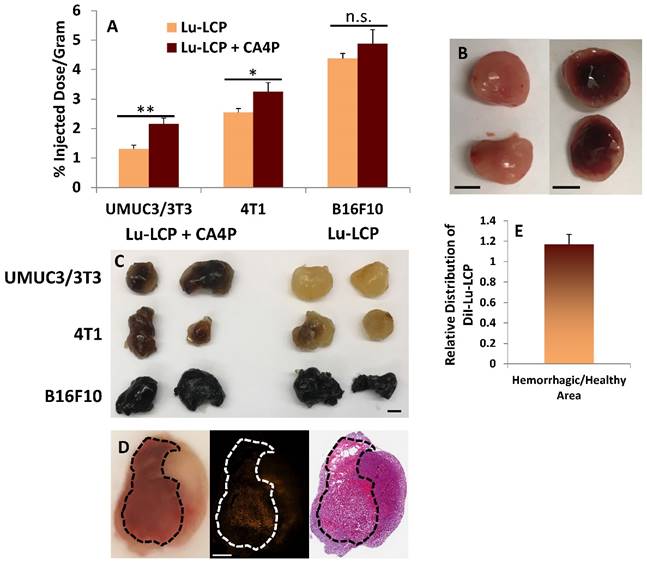
Upon cutting a CA4P-treated UMUC3/3T3 tumor in half at t = 24 h (Fig 5B, right image), a large portion of the tumor interior had become filled with blood, further suggesting that CA4P had indeed compromised the integrity of the blood vessels and caused a massive hemorrhage in the tumor. The resulting poorly-perfused pool of blood would certainly present as was observed in our ultrasound experiments, as the rapid and orderly vascular flow to and from the tumor had been destroyed.
After the 177Lu-LCP accumulation was quantified in the tumors used in Fig 5A, the tumors were soaked in formamide for 48 h at 60 oC in order to show the extent of additional blood stuck in the tumors. Representative tumors in Fig 5C show that blood pools in both UMUC3/3T3 and 4T1 tumors after treatment with CA4P, but does not in untreated tumors of a similar size. The black melanocytes in the B16F10 tumors do not allow us to observe the effect of CA4P, but it is known that untreated B16F10 tumors are naturally soft and bloody, a fact that was verified in this study as well. Again, this could be why B16F10 tumors naturally allow greater 177Lu-LCP accumulation and are less affected by CA4P.
The distribution of 177Lu-LCP in the bloody and non-bloody regions of CA4P-treated tumors was also compared. We desired to know whether CA4P would affect the vessels' permeability to nanoparticles, and whether a significant amount of additional 177Lu-LCP would distribute into the blood pool. The lipid bilayer of 177Lu-LCP was therefore labeled with the fluorescent small molecule DiI—known to faithfully remain in the labeled lipid bilayers [19, 20]—and the nanoparticles were purified using sucrose gradient centrifugation. t = 3 h after treatment with CA4P, DiI-177Lu-CA4P was given to mice bearing UMUC3/3T3 tumors, and 24 h later the mice were sacrificed and their tumors were dissected, processed, and frozen in OCT. After cutting into the middle of each tumor, a photograph was taken to show the extent of the blood-filled region (Fig 5D, left image), and two adjacent sections were cut. One section was imaged to show the distribution of DiI in the tumor (middle image), and the second section was stained with Masson's trichrome to show the morphology of the section (right image). The average distribution of nanoparticles in all imaged sections (Fig 5E, n = 4) showed that there was slightly more DiI signal per area in bloody regions, but in a paired t-test comparing DiI distribution in each tumor, this difference was not significant (p = 0.19). This means that the additional 177Lu-LCP that accumulated after CA4P treatment distributed relatively evenly throughout the tumor and not only in the regions where the vasculature became permeable to blood cells. In those regions where blood vessels were not permanently compromised, a temporary loss of the vessel endothelial cells' flat structure—induced by CA4P's inhibition of microtubule polymerization—may have created gaps between the cells and allowed the vessels to become more permeable to nanoparticles, leading to higher nanoparticle accumulation in non-bloody regions as well.
Even though some nanoparticles are delivered to the apoptotic center of the tumor, internal radiation therapy is able to deliver treatment to faraway cells, increasing the utility of those particles delivered to the middle of the tumor. Additionally, because 177Lu slowly decays independent of its environment (t1/2 = 6.6 days), if the radiation remains in the tumor, it may prevent regrowth of tumor tissue.
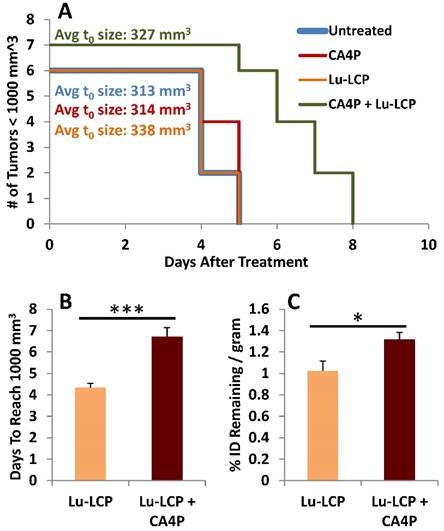
At this point, we had determined that CA4P induces enhanced accumulation of 177Lu-LCP, but had not determined whether this additional accumulation could be retained over a significant length of time. It could be that as the pooled blood is filtered out of the tumor over time, it would take many of the particles and radioactive cargo with it; however, because 177Lu decays slowly over time, to be most effective, it should remain in the tumor as long as possible, continuously giving a low dose of radiation to the tumor. To test this, mice bearing UMUC3/3T3 tumors ~300-400 mm3 were treated with 100 mg/kg CA4P + 250 μCi 177Lu-LCP or controls. Tumor volumes were measured daily, and the number of days it took for each tumor to reach 1,000 mm3 was determined. When a tumor reached 1,000 mm3, the mouse was sacrificed and its tumor was dissected, weighed, and assessed for radioactivity. We again found that CA4P + 177Lu-LCP significantly increased time to reach 1,000 mm3 compared to controls (Fig 6A and B), but more importantly, at the time each tumor reached 1,000 mm3, those tumors that had received CA4P + 177Lu-LCP still contained significantly more 177Lu per gram of tumor than those that had received 177Lu-LCP alone, even though it took on average ~30% longer for those tumors receiving CA4P + 177Lu-LCP to reach 1,000 mm3. This means that those tumors were in fact able to retain the increased accumulation of 177Lu over an extended period of time. The increased vessel permeability leading to the increased accumulation of 177Lu-LCP combined with the sustained retention of 177Lu in tumors means that CA4P does indeed increase the EPR of 177Lu-LCP.
Tumor Growth to 1,000 mm3. A) Kaplan-Meier plot of mouse tumor growth to 1,000 mm3, n = 6-7. B) Median time to reach 1,000 mm3 in mice receiving 250 μCi 177Lu-LCP with or without 100 mg/kg CA4P i.p. C) Tumor accumulation of 177Lu when tumors reach 1,000 mm3.
Choosing the most effective radionuclide
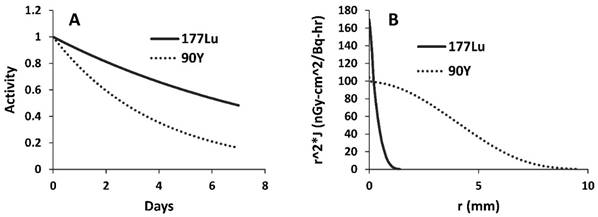
177Lu is a relatively new radioisotope that is advancing toward the clinic in a variety of applications. 177Lu is a good model isotope because its 6.6 day half-life provides the researcher a relatively long time to perform experiments before it decays, and the gamma component of its decay allows simple quantification of signal as well as live animal imaging using SPECT [1]. Despite these advantages, 177Lu has a fairly low beta energy (max = 0.5 Megaelectron-volts (MeV)) with an average path length in tissue of only 200-300 μm [21]. In a large, stroma-rich tumor model, it may be better to use an isotope with a more cytotoxic, higher energy beta emission that can reach farther away from the source, especially considering the heterogeneous and high stromal delivery of nanoparticles and the fact that treatment with CA4P may further induce regions of higher and lower nanoparticle density. 177Lu was therefore compared to 90Yttrium (90Y), a clinically used radioisotope [22, 23] which has a max beta energy of 2.28 MeV and a half-life of 2.67 days. Figure 7A compares the relative decay of 177Lu and 90Y over a period of seven days, while Figure 7B compares the previously calculated dose kernels for point sources of the two isotopes in units of absorbed dose (nanogray per decay per h over a given surface area) [24, 25]. While 90Y decays more quickly than 177Lu, its higher beta energy allows more energy to reach farther away from the source and impart more energy per interaction.
Comparison of 177Lu and 90Y. A) Different half-lives result in quicker drop-off of activity over time for 90Y. B) Graphical display of dose kernels representing absorbed dose at increasing distances from a point source in an infinite water medium; decay products from 90Y are better able to impart energy to targets at longer distances from the source.
Y is a trivalent transition metal that sits just above Lu in group 3 of the periodic table, and is therefore very chemically similar to Lu. Not surprisingly, 90Y can also be easily encapsulated into LCP at the same high encapsulation efficiency as 177Lu (80-85%), and changing the LCP cargo from 177Lu to 90Y does not change the morphology of the nanoparticles (Supplementary Fig S2A and S2B). The injected activity per mouse of 90Y-LCP was set at 125 μCi, half the dose of 177Lu-LCP. This is common in publications comparing 90Y and 177Lu, often because the maximal tolerable dose for the delivered 177Lu is approximately twice as high as that of 90Y [26, 27]. In addition to displaying TEM images for LCP, Figure S2 also reports the size, polydispersity index, zeta potential, and stability of LCP nanoparticles.
Combination treatment prolongs tumor necrosis. (A), top row: four days after CA4P monotherapy or combination treatment with 250 μCi 177Lu-LCP or 125 μCi 90Y-LCP, tumors from several mice were dissected, sectioned, and stained for apoptotic cells (TUNEL, green). Middle row: sections 8 μm away from TUNEL sections were stained with proliferation marker Ki-67 using IHC (brown). Bottom Row: Ki-67 (yellow) overlaid with TUNEL (green). Scale bar = 2 mm. B) Quantification of TUNEL-positive area, n = 8 (CA4P), 30 (CA4P + 177Lu-LCP), 15 (CA4P + 90Y-LCP).
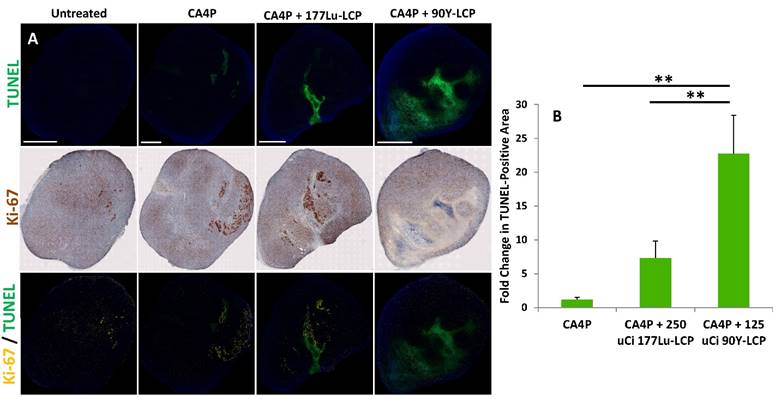
In order to compare the efficacy of 177Lu and 90Y while also determining how their delivery 3 h after CA4P might affect the regrowth of apoptotic areas initially generated by CA4P, we measured the amount of apoptosis in large tumors four days after treatment with CA4P alone, or after combination therapy with CA4P + 177Lu-LCP or CA4P + 90Y-LCP. After four days, the apoptosis caused by CA4P alone had been nearly completely healed and replaced with a high concentration of cells staining positively for the proliferation marker Ki-67, while significant apoptosis in the tumor centers still remained after combination treatment with CA4P + 90Y-LCP. After treatment with CA4P + 177Lu-LCP, there was a large variation in the apoptotic and proliferative areas of various tumors. There was an increase in apoptotic area when compared to CA4P treatment alone, but this increase was not statistically significant. These results suggested that 90Y-LCP would be more effective at inhibiting tumor growth than 177Lu-LCP, and may do so at least in part by maintaining the apoptotic region generated by CA4P.
Tumor Growth Inhibition with 90Y-LCP and LPC
In the following tumor growth inhibition study, UMUC3 cells stably expressing green fluorescence protein and 3T3 cells stably expressing red fluorescence protein (UMUC3-GFP/3T3-RFP) were inoculated into mice. Treatment with CA4P + 90Y-LCP was compared against CA4P + 177Lu-LCP, using 90Y-LCP monotherapy as a control alongside the untreated group. CA4P + 90Y-LCP tumors grew significantly more slowly than any other group, including CA4P + 177Lu-LCP (Fig 9A). At the study endpoint, the tumors were dissected and processed for frozen sectioning. Two adjacent sections were stained: the first section was stained with DAPI, and fluorescence imaging was used to determine the distribution of UMUC3-GFP and 3T3-RFP cells in the tumor (Fig 9B and 9C); the second section was stained using IHC against cleaved Caspase 3 to quantify apoptosis (Fig 9D). The average relative fibroblast density, total cell density, and apoptotic cell density across groups was calculated and showed that even seven days after treatment with 90Y-LCP + CA4P, the tumors still contained significant amounts of apoptosis and the total cell density in the tumor had been decreased by 25%. In addition, the fibroblast density had been greatly reduced, eliminating the aggressive and resistant “tumor nest” structure observed in many desmoplastic tumors (Fig 9E).
Mice bearing UMUC3-GFP/3T3-RFP tumors of volume 300 - 400 mm3 were treated as shown in A), n = 5-6 mice. On Day 7, tumors were harvested, processed, sectioned in OCT, and stained. B) Representative tumors shown from each group; green = UMUC3-GFP, orange = 3T3-RFP. Scale bar = 2 mm. C) 3T3-RFP signal isolated to show distribution. D) Caspase 3 distribution in adjacent section to (B). E) Quantification of fibroblast density, total cell density, and apoptosis from each group. Image resolution for quantification was enhanced by quantifying several random areas at higher magnification, ~4 mm2/section. n = 9-13. ***p < 0.001; *p < 0.05.
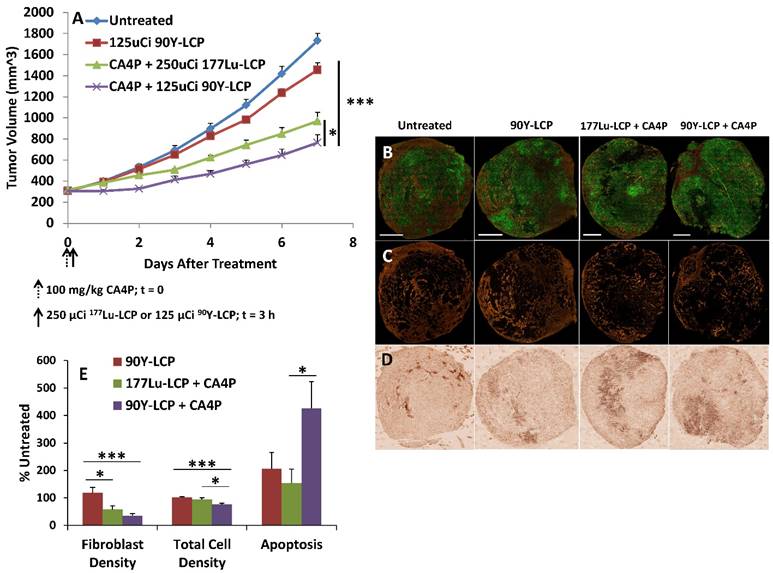
Despite these changes to the tumor microenvironment, the tumors continued to slowly grow, although it is clear that their microenvironment had become much more susceptible to additional treatment. The lower cell density had undoubtedly decreased the tumor's interstitial pressure, and the loss of fibroblast structure had removed the physical barrier to the remaining tumor cells. With the tumors in this weakened state, it was hypothesized that treatment using a potent nanoformulated drug may further decrease the growth rate of these now very large tumors. Nanoparticles composed nearly entirely of cisplatin (termed Lipid-Platinum-Chloride particles, or LPC [13, 20]; characterized in supplementary Fig S2 and in much greater detail in the referenced publications) have been shown to be highly effective against a variety of tumors, but have been ineffective in treating large, desmoplastic tumors such as this stroma-rich bladder cancer model. Although cisplatin is generally a potent and effective drug, the fibroblasts in large UMUC3/3T3 tumors physically prevent the nanoparticles from reaching the tumor nest in addition to secreting the resistance-forming chemokine Wnt16b in response to treatment [2]. In our final tumor inhibition experiment, we examined whether two low doses of LPC given once tumors had grown very large (~800 mm3) could significantly slow the growth of untreated tumors or tumors that had previously been given 100 mg/kg CA4P + 125 μCi 90Y-LCP (Fig 10).
Mice bearing UMUC3/3T3 tumors of volume 300 - 400 mm3 were either treated with 100 mg/kg CA4P + 125 μCi 90Y-LCP or left untreated (n = 10). When tumors reached ~800 mm3, half of the mice from each group (n = 5) were treated with 1.5 mg/kg CDDP formulated in LPC nanoparticles on two consecutive days. A) Tumor volumes recorded daily. B) All four groups normalized to the volume of each individual tumor at the time of initial CDDP dose. *p < 0.05; ***p < 0.001.
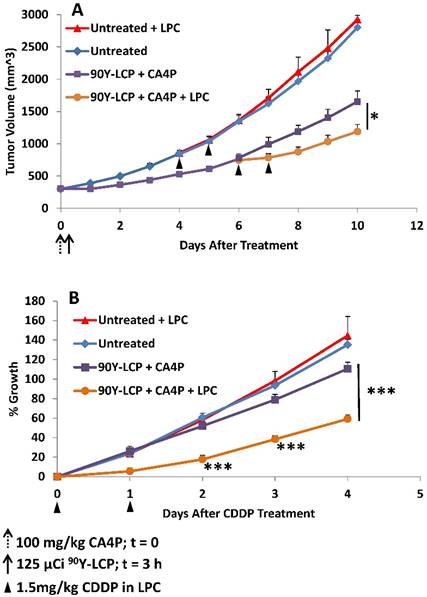
To fairly compare the efficacy of LPC on previously treated and untreated tumors, LPC was initially given when the average volume of tumors within each group reached ~800 mm3. At those points, half of the untreated tumors and half of the previously treated tumors received a total of two doses of 1.5 mg/kg LPC, administered i.v. through tail vein injection on two consecutive days (Fig 10A). Groups were divided evenly to maintain the average tumor volume of each group.
All tumors were then normalized to the date at which they received LPC and their volume on that date (Fig 10B). The rate of tumor growth from the date of initial LPC treatment (or paired untreated controls) was then quantified. In tumors that initially received CA4P + 90Y-LCP, a significant difference in growth rate was observed between those tumors that received additional LPC treatment and tumors that received no additional treatment. In contrast, in tumors that initially received no treatment, additional treatment with LPC induced no distinguishable change in growth rate compared to completely untreated tumors. This suggests that the treatment-induced changes in the tumor microenvironment shown in Fig 9 allowed LPC to provide further tumor growth inhibition even in very large tumors. By increasing the dose of LPC or the duration of treatment, further tumor growth inhibition can be expected.
To test our hypothesis that these weakened tumors were still more susceptible to increased nanoparticle accumulation—whether because of lower total cell density or otherwise—we treated 300-400 mm3 tumors with CA4P + 90Y-LCP, six days later injected 177Lu-LCP, and 24 h after that, sacrificed the mice and measured the tumor accumulation of 177Lu via gamma scintillation counting. We compared the accumulation of 177Lu in these tumors to the accumulation of 177Lu in previously untreated very large (~800 mm3) tumors that had been dissected 24 h after the mice received 177Lu-LCP. After normalizing for tumor mass, we found that those tumors that had been previously treated with CA4P + 90Y-LCP had accumulated, on average, 40% more 177Lu than those tumors with no prior treatment, although this difference was not statistically significant (p ~ 0.1).
Toxicity Considerations
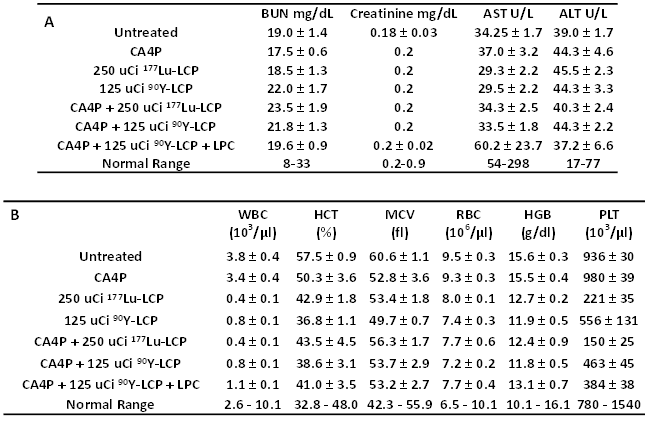
Lastly, we determined if there was any toxicity induced by these radiation and combination treatments. Because internal radiotherapy imparts a slow, continuous dose to tissues, the cumulative toxicity generated ten days after treatment was measured. There was no observed renal or hepatic toxicity observed (Table 1A), but as expected, the radiation treatments induced a tolerable decrease in white blood cell count and platelet count. Interestingly, the treatments incorporating 90Y induced less toxicity than the treatments using 177Lu, even though the 90Y was more effective at inhibiting tumor growth. No toxicity was observed in H&E-stained organ sections (Supplementary Fig S3).
Toxicity studies, measured 10 days after treatment in immunocompetent CD-1 mice to test cumulative radiation toxicity: A) Serum protein values for all treatment groups, measuring blood urea nitrogen (BUN), creatinine, alanine aminotransferase (ALT), and aspartate aminotransferase (AST). B) Whole blood analysis for all treatment groups, measuring white blood cell count (WBC), hematocrit (HCT), mean cell volume (MCV), red blood cell count (RBC), hemoglobin count (HGB), and platelet count (PLT).
Discussion
While a recent study has shown that vessel occlusion caused by CA4P prevents nanoparticles from entering the tumor center [11], we have shown that CA4P induces an increased EPR effect, allowing a greater accumulation and retention of nanoparticles throughout the tumor. We have observed that the increase in nanoparticle accumulation comes alongside vascular hemorrhage in the tumor, and it makes sense that if blood cells can enter the tumor center, the much smaller nanoparticles also slowly flowing through the broken vasculature would not be kept at the tumor periphery. We have also observed that the pooled blood in tumors dissected 24 h after CA4P treatment had not yet coagulated, suggesting the presence of at least a slow flow of blood capable of bringing nanoparticles into the tumor. What, then, could cause the discrepancy in the data? The seemingly contradictory study also reports vascular hemorrhage, but they administer their nanoparticles at the same time that they administer the CA4P. It could be that the immediate vascular occlusion effect of CA4P, which occurs before any hemorrhage begins, stops all flow into the center of the tumor, trapping the nanoparticles on the outer rim. If their particles quickly clear from circulation, by the time vascular hemorrhage begins, there are no particles left to enter the inner regions of the tumor. In contrast, our nanoparticles are given 3 h after CA4P treatment, at which point vascular hemorrhage may have already begun, allowing an increased amount of particles into the tumor.
If this is the case, it is worth discussing whether one approach is more effective than another. This answer must be given in light of the tumor model being studied. In the recent article, CA4P alone is able to cause vascular hemorrhage across nearly the entire tumor in just four hours, and only a small portion of the tumor requires nanoparticle treatment. Only having a small portion of the tumor available for nanoparticle deposition may greatly reduce the overall tumor accumulation of the particle, but in this case, the authors show that this is all that is needed. In our case, although ultrasound images suggest that a large percentage of the tumor is not well perfused 3 h after CA4P treatment, some of this vessel occlusion is transient. The ability of vasculature in UMUC3/3T3 tumors to partially repair after treatment with CA4P may be due in part to their stroma-vessel structure, as the organized fibroblasts which line the vessel endothelium may provide enough structural support to allow vessel repair. In addition, the TUNEL-positive area 24 h after treatment is variable in size, and is also able to be replaced with proliferating cells over time. In this case, we have shown that the heightened nanoparticle accumulation and retention in the tumor is vital for effective tumor inhibition. Strategies to deal with these resistant and aggressive tumors may be different from those aiming to treat tumors with less stroma.
It is important to note that the large tumor model used in this study was chosen not only because of its size, but because its size provided it with characteristics that are difficult to treat. The difficulty of slowing down the growth of an aggressive, desmoplastic tumor with a mature microenvironment requires a synergistic approach that provides the right treatments at the right times. CA4P simultaneously causes cell death in a significant portion of the tumor and induces an increased EPR in the tumor. The cell death in the middle of the tumor is maintained by 90Y-LCP, which also presents cytotoxicity to the surviving cells in the tumor. Changes in the tumor microenvironment in response to this combination therapy then allow treatment with LPC nanoparticles, which are completely ineffective in tumors that have not been previously treated. Although these treatments do not cause tumor remission, they generate significant tumor growth inhibition in this tumor model that will grow tenfold in ten days if left untreated.
Of course, clinical implementation of this combination therapy must be modified to minimize systemic toxicity. While intravenous administration of 90Y-LCP presents a good model of internal radiation therapy's effectiveness in combination with CA4P, the well-known uptake of nanoparticles into clearing organs presents a compelling argument against this injection route. This subject has been recently reviewed [28], and perhaps the best future use for 177Lu/90Y-LCP is in selective internal radiation therapy (SIRT) [29, 30]. SIRT locally delivers 90Y-microspheres around primary or metastatic tumors in the liver via a catheter placed into the hepatic artery. The large size of the microspheres prevents them from entering the tumor capillaries, so a larger dose must be given in order to provide treatment to all tumor cells, resulting in off-target toxicity to the liver and lung. Local delivery of 177Lu/90Y-LCP via hepatic catheter may result in better tumor penetration than 90Y-microspheres and less off-target dose than systemic administration of LCP, increasing the dose to the tumor while decreasing off-target effects. Combination therapy with CA4P against desmoplastic tumors in the liver would not only cause massive apoptosis in the tumor centers, but also induce an increased accumulation of nanoparticles in the tumor and prime the microenvironment for further treatment.
Conclusion
In conclusion, we have generated significant tumor growth inhibition in a large, desmoplastic tumor model by using CA4P to increase the tumor EPR and allow increased accumulation and retention of radioactive nanoparticles for internal radiation therapy. This combination therapy decreases the fibroblast and overall cellular density in the tumor, allowing effective treatment with a second nanoparticle system containing cisplatin—an approach which may also be effective as a local combination therapy against desmoplastic cancers in the liver.
Supplementary Material
Supplementary figures.
Abbreviations
CA4P: Combretastatin A-4 Phosphate
DOPA: 1,2-dioleoyl-sn-glycero-3-phosphate
DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane
DSPE-PEG2000: N-(Carbonyl-methoxypolyethyleneglycol2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine, sodium salt
ICP-MS: Inductively Coupled Plasma Mass Spectroscopy
IHC: Immunohistochemistry
LCP: Lipid-Calcium-Phosphate
LPC: Lipid-Platinum-Chloride
Lu: Lutetium
MTT: (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide)
TEM: Transmission Electron Microscopy
TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling
Y: Yttrium
Acknowledgements
ABS was supported by the National Science Foundation's Graduate Research Fellowship Program and the Dissertation Completion Fellowship from the Graduate School at the University of North Carolina at Chapel Hill. The work was supported by NIH grants CA149363, CA151652 and CA149387.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Satterlee AB, Yuan H, Huang L. A radio-theranostic nanoparticle with high specific drug loading for cancer therapy and imaging. Journal of controlled release: official journal of the Controlled Release Society. 2015;217:170-82
2. Miao L, Wang Y, Lin CM, Xiong Y, Chen N, Zhang L. et al. Nanoparticle modulation of the tumor microenvironment enhances therapeutic efficacy of cisplatin. Journal of controlled release: official journal of the Controlled Release Society. 2015;217:27-41
3. Siemann DW, Chaplin DJ, Walicke PA. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert opinion on investigational drugs. 2009;18:189-97
4. Ng QS, Goh V, Carnell D, Meer K, Padhani AR, Saunders MI. et al. Tumor antivascular effects of radiotherapy combined with combretastatin a4 phosphate in human non-small-cell lung cancer. International journal of radiation oncology, biology, physics. 2007;67:1375-80
5. Tozer GM, Kanthou C, Parkins CS, Hill SA. The biology of the combretastatins as tumour vascular targeting agents. International journal of experimental pathology. 2002;83:21-38
6. Clemenson C, Chargari C, Deutsch E. Combination of vascular disrupting agents and ionizing radiation. Critical reviews in oncology/hematology. 2013;86:143-60
7. Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer research. 1997;57:1829-34
8. Daenen LG, Roodhart JM, Shaked Y, Voest EE. Vascular disrupting agents (VDAs) in anticancer therapy. Current clinical pharmacology. 2010;5:178-85
9. Galbraith SM, Chaplin DJ, Lee F, Stratford MR, Locke RJ, Vojnovic B. et al. Effects of combretastatin A4 phosphate on endothelial cell morphology in vitro and relationship to tumour vascular targeting activity in vivo. Anticancer research. 2001;21:93-102
10. Siemann DW, Bibby MC, Dark GG, Dicker AP, Eskens FA, Horsman MR. et al. Differentiation and definition of vascular-targeted therapies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:416-20
11. Song W, Tang Z, Zhang D, Yu H, Chen X. Coadministration of Vascular Disrupting Agents and Nanomedicines to Eradicate Tumors from Peripheral and Central Regions. Small. 2015;11:3755-61
12. Banerjee R, Tyagi P, Li S, Huang L. Anisamide-targeted stealth liposomes: a potent carrier for targeting doxorubicin to human prostate cancer cells. International journal of cancer Journal international du cancer. 2004;112:693-700
13. Guo S, Miao L, Wang Y, Huang L. Unmodified drug used as a material to construct nanoparticles: delivery of cisplatin for enhanced anti-cancer therapy. Journal of controlled release: official journal of the Controlled Release Society. 2014;174:137-42
14. Miao L, Guo S, Zhang J, Kim WY, Huang L. Nanoparticles with Precise Ratiometric Co-Loading and Co-Delivery of Gemcitabine Monophosphate and Cisplatin for Treatment of Bladder Cancer. Advanced functional materials. 2014;24:6601-11
15. Kogan P, Johnson KA, Feingold S, Garrett N, Guracar I, Arendshorst WJ. et al. Validation of dynamic contrast-enhanced ultrasound in rodent kidneys as an absolute quantitative method for measuring blood perfusion. Ultrasound in medicine & biology. 2011;37:900-8
16. Feingold S, Gessner R, Guracar IM, Dayton PA. Quantitative volumetric perfusion mapping of the microvasculature using contrast ultrasound. Investigative radiology. 2010;45:669-74
17. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nature reviews Clinical oncology. 2010;7:653-64
18. Smith NR, Baker D, Farren M, Pommier A, Swann R, Wang X. et al. Tumor stromal architecture can define the intrinsic tumor response to VEGF-targeted therapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:6943-56
19. Honig MG, Hume RI. Dil and diO: versatile fluorescent dyes for neuronal labelling and pathway tracing. Trends in neurosciences. 1989;12:333-5 40-1
20. Guo S, Wang Y, Miao L, Xu Z, Lin CM, Zhang Y. et al. Lipid-coated Cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy. ACS nano. 2013;7:9896-904
21. Sofou S. Radionuclide carriers for targeting of cancer. International journal of nanomedicine. 2008;3:181-99
22. Reagan PM, Baran A, Kelly JL, Barr PM, Casulo C, Chengazi VU. et al. Consolidative Radioimmunotherapy After Chemoimmunotherapy in Patients With Histologic Transformation of Indolent Non-Hodgkin Lymphoma. Clinical lymphoma, myeloma & leukemia. 2016
23. Lau WY, Teoh YL, Win KM, Lee RC, de Villa VH, Kim YH. et al. Current role of selective internal radiation with yttrium-90 in liver tumors. Future Oncol. 2016;12:1193-204
24. Wendt R. Beta-ray dose kernels for Ho-166 and Lu-177. The Journal of Nuclear Medicine. 2010;51:1428
25. Cross WGF. et al. Tables of beta-ray dose distributions in water. Atomic Energy of Canada Limited. 1992
26. Stein R, Govindan SV, Chen S, Reed L, Richel H, Griffiths GL. et al. Radioimmunotherapy of a human lung cancer xenograft with monoclonal antibody RS7: evaluation of (177)Lu and comparison of its efficacy with that of (90)Y and residualizing (131)I. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2001;42:967-74
27. Shi J, Fan D, Dong C, Liu H, Jia B, Zhao H. et al. Anti-tumor effect of integrin targeted (177)Lu-3PRGD2 and combined therapy with Endostar. Theranostics. 2014;4:256-66
28. Satterlee AB, Huang L. Current and Future Theranostic Applications of the Lipid-Calcium-Phosphate Nanoparticle Platform. Theranostics. 2016;6:918-29
29. Kolligs FT, Bilbao JI, Jakobs T, Inarrairaegui M, Nagel JM, Rodriguez M. et al. Pilot randomized trial of selective internal radiation therapy vs. chemoembolization in unresectable hepatocellular carcinoma. Liver international: official journal of the International Association for the Study of the Liver. 2015;35:1715-21
30. Lau WY, Ho S, Leung TW, Chan M, Ho R, Johnson PJ. et al. Selective internal radiation therapy for nonresectable hepatocellular carcinoma with intraarterial infusion of 90yttrium microspheres. International journal of radiation oncology, biology, physics. 1998;40:583-92
Author contact
![]() Corresponding author: Tel.: 919 843 0736 E-mail address: leafhedu
Corresponding author: Tel.: 919 843 0736 E-mail address: leafhedu