Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2017; 7(2):308-318. doi:10.7150/thno.17155 This issue Cite
Research Paper
Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor
Greta Hultqvist ![]() , Stina Syvänen, Xiaotian T Fang, Lars Lannfelt, Dag Sehlin
, Stina Syvänen, Xiaotian T Fang, Lars Lannfelt, Dag Sehlin
Department of Public Health and Caring Sciences/Geriatrics, Uppsala University, Rudbeck Laboratory, 75185 Uppsala, Sweden.
Received 2016-8-10; Accepted 2016-10-19; Published 2017-1-1
Citation:
Hultqvist G, Syvänen S, Fang XT, Lannfelt L, Sehlin D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics 2017; 7(2):308-318. doi:10.7150/thno.17155. https://www.thno.org/v07p0308.htm
Other stylesAbstract

The blood-brain barrier (BBB) is an obstacle for antibody passage into the brain, impeding the development of immunotherapy and antibody-based diagnostics for brain disorders. In the present study, we have developed a brain shuttle for active transport of antibodies across the BBB by receptor-mediated transcytosis. We have thus recombinantly fused two single-chain variable fragments (scFv) of the transferrin receptor (TfR) antibody 8D3 to the light chains of mAb158, an antibody selectively binding to Aβ protofibrils, which are involved in the pathogenesis of Alzheimer's disease (AD). Despite the two TfR binders, a monovalent interaction with TfR was achieved due to the short linkers that sterically hinder bivalent binding to the TfR dimer. The design enabled efficient receptor-mediated brain uptake of the fusion protein. Two hours after administration, brain concentrations were 2-3% of the injected dose per gram brain, comparable to small molecular drugs and 80-fold higher than unmodified mAb158. After three days, fusion protein concentrations in AD transgenic mouse brains were 9-fold higher than in wild type mice, demonstrating high in vivo specificity. Thus, our innovative recombinant design markedly increases mAb158 brain uptake, which makes it a strong candidate for improved Aβ immunotherapy and as a PET radioligand for early diagnosis and evaluation of treatment effect in AD. Moreover, this approach could be applied to any target within the brain.
Keywords: BBB shuttle /TfR /antibodies / Alzheimer's disease/ immunotherapy/PET.
Introduction
The blood-brain barrier (BBB) is comprised of tightly connected endothelial cells and serves as a protector of the brain by keeping both small substances and larger molecules like proteins outside the brain. Although essential for brain homeostasis, the presence of this barrier is an obstacle to treatment and diagnosis of diseases in the brain. Immunotherapy is rapidly growing and several therapeutic antibodies have reached the clinic in recent years. This development has not been seen for brain disorders, as only around 0.1% of peripherally administered antibodies will enter the brain due to the BBB [1]. However, active transport from blood across the BBB into the brain has been described for some proteins. One such example is transferrin that transports iron to the brain via the transferrin receptor (TfR) expressed at the BBB. Also, other proteins that bind to the TfR can be endocytosed on the luminal side and transported across the BBB and this mechanism can be employed to actively transport molecules into the brain [2,3]. It is essential that the protein/antibody can dissociate from the TfR on the brain side, and when released, that it can bind to its intra-brain target. (Fig 1A). Dissociation is more likely when the overall affinity to TfR is moderate or low [4]. Bivalent binding to TfR, naturally occurring as a homodimer, has also been found to be disadvantageous [5]. Bivalent binding decreases the rate of dissociation from the TfR since the binding of the first antigen will bring a second binding-site closer to its antigen. This in turn increases the probability of binding and hence the binding time, i.e. the avidity effect.
In Alzheimer's disease (AD), the most common neurodegenerative disorder, several trials have been conducted with antibodies targeting the self-aggregating protein amyloid-β (Aβ). Although amyloid plaques, consisting of fibrillar deposits of Aβ, are a hallmark of AD, soluble Aβ, in particular oligomers and protofibrils, measured in post mortem AD brain tissue and CSF, have been shown to correlate better with disease progression [6-8], and to be harmful to synapses and neurons [6,9-15]. Soluble Aβ assemblies are therefore a suitable target for immunotherapy and diagnostics with Aβ specific antibodies. BAN2401, a humanized version of the well-studied murine anti-Aβ protofibril antibody mAb158 [16-18], is currently evaluated in phase 2b clinical trials as an anti-Aβ therapy for AD. This antibody has a much lower affinity for Aβ-monomers than for protofibrils and binds with moderate affinity to insoluble Aβ fibrils [19]. In addition, it has no affinity for the Aβ protein precursor (AβPP). RmAb158 is a recombinant version of mAb158 with identical Aβ binding properties.
We have recently shown that a bispecific protein based on mAb158, chemically conjugated to the TfR antibody 8D3, displayed 20-fold higher brain-to-blood concentrations than mAb158 at 3 days' post injection [19]. The study further showed that a radiolabeled version of the bispecific protein could be used for in vivo quantification of Aβ protofibrils in the brain in two mouse models of AD with positron emission tomography (PET). However, chemical conjugation, as used in the previous study, results in a heterogeneous mixture of fusion proteins randomly linked together at different positions, which is not suitable for a clinical application. In addition, the use of the complete 8D3 antibody results in bivalent TfR binding, which has been shown to be suboptimal for transcytosis [5].
To adapt the fusion protein for future use in the clinic and to modify its TfR binding properties for optimal BBB passage, we have, in the present study, recombinantly produced a bispecific fusion protein based on RmAb158 with a single chain fragment variable (scFv) of 8D3 (scFv8D3) attached to the C-terminal end of the RmAb158 light chains. This novel design greatly improved transfer across the BBB, with higher or equal brain uptake compared to previously reported BBB shuttles and with a less marked decline in uptake at therapeutic doses [4,5,20,21].
Results
Generation and characterization of a recombinant bispecific brain shuttle
A single chain variable fragment (scFv), comprising the heavy and light chain variable regions of 8D3 [2] connected to each other by a linker, was attached to the C-terminus of each of the RmAb158 light chains via a short peptide linker (Fig 1B). This linker was designed to avoid formation of alpha helices, and the number of hydrophilic amino acids was selected to avoid formation of a hydrophobic core. Amino acids with small side chains were incorporated in the linker to ensure flexibility. The purpose of the short length of the linker was to tie the scFv closely to RmAb158, so that a bivalent binding to TfR would be sterically difficult (Fig 1C). Protein expression in Expi293 cells resulted in yields around 15-30 mg antibody per liter of transfected cell culture. The purified protein was analyzed with SDS-PAGE on which a single band was observed (Fig 2A), i.e. RmAb158-scFv8D3 was pure and the same batch was used in all in vitro and in vivo studies described below.
To evaluate the functionality of the generated recombinant fusion protein, in vitro binding analyses were performed. For efficient BBB transcytosis and release in the brain, the shuttle should preferably interact with its receptor through a monovalent binding. TfR binding was assessed with a competition TfR ELISA, where plates were coated with a high concentration of recombinant TfR protein to mimic the possibility to achieve a bivalent binding, with higher avidity as a read-out for that. A biotinylated scFv of 8D3 was subjected to competition by serially diluted scFv8D3, RmAb158-scFv8D3, 8D3 and our previously generated, chemically fused 8D3-F(ab')2-h158. As predicted, RmAb158-scFv8D3 displayed a 10-fold lower avidity compared with the entire 8D3 antibody and 8D3-F(ab')2-h158, with estimated Kd values of 8.0, 0.80 and 0.69 nM respectively. ScFv8D3, which is monovalent by definition, displayed a Kd value of 15 nM, i.e. approximately 2-fold higher than RmAb158-scFv8D3. Since the recombinant fusion protein has two possible binding sites per molecule, there is no actual difference in binding between scFv8D3 and RmAb158-scFv8D3, strongly suggesting a monovalent interaction with TfR.
The fusion protein's selectivity to different Aβ species was studied with an inhibition ELISA, where the ability of Aβ monomers and protofibrils to inhibit the signal in an indirect Aβ ELISA approximates the affinity to the tested antigen (Fig 2C). RmAb158-scFv8D3 showed a near 500-fold stronger binding to protofibrils than to monomers, with median inhibitory concentrations (IC50) of 0.85 and 400 nM, respectively, similar to RmAb158, with IC50 of 1.2 nM for protofibrils and 1.2 µM for monomers. The widely used Aβ antibody 6E10 served as control, displaying no difference in binding to the different Aβ preparations.
In vivo studies of brain distribution with radiolabeled RmAb158-scFv8D3
RmAb158-scFv8D3 was radiolabeled with I125 and I124 in yields around 70% for in vivo studies in mice. The specific activity was 0.4±0.1 MBq/µg (79±19 MBq/nmol) for [125I]RmAb158-scFv8D3 and 1.2±0.7 MBq/µg (261±145 MBq/nmol) for [124I]RmAb158-scFv8D3.
To assess the ability of the scFv8D3 moiety to enable TfR mediated transcytosis in vivo, wt mice were administered with tracer doses (~0.05 mg/kg) of [125I]RmAb158 or [125I]RmAb158-scFv8D3 and the brains were isolated 2 h after injections. The brain concentrations, expressed as per cent of injected dose per gram brain tissue (% ID/g, Equation 1) were 0.028±0.010 and 2.20±0.92 for [125I]RmAb158 and [125I]RmAb158-scFv8D3, respectively. Thus, the scFv8D3 modification lead to an 80-fold increase in brain concentrations at this time point. To confirm that TfR was responsible for the uptake, 10 mg/kg of non-labeled native 8D3 was co-injected with [125I]RmAb158-scFv8D3 (i.e. around 100 times surplus of 8D3). This led to a brain concentration of 0.089±0.029, i.e. only 3-fold higher than [125I]RmAb158, demonstrating a substantial blocking of fusion protein binding to endothelial TfR (Fig 3A). The brain-to-blood concentration ratio (Equation 2) was 0.0010±0.00013 and 0.15±0.10 for the two ligands, respectively, and 0.0036±0.0009 when 8D3 was co-administered with [125I]RmAb158-scFv8D3 (Fig 3B). Hence the brain-to-blood ratio, which takes into account available ligand in blood, was increased 140-fold by the scFv8D3 modification of RmAb158 and showed a similar blocking effect on TfR binding upon addition of 8D3.
Figure 1
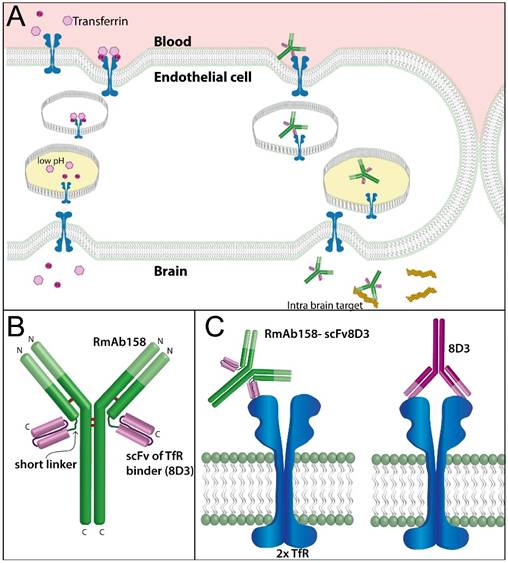
Design and uptake mechanism of the BBB shuttle. (A). Transferrin is transported across the BBB by binding to iron and the TfR. The complex is endocytosed and with an altered pH in the endosomes the affinity changes so that transferrin and iron are released in the endosomes and will eventually reach the brain parenchyma. An antibody that binds monovalently to the TfR, will according to the same mechanism also be released in the endosomes. Bivalent TfR binders have higher affinity to the TfR and hence less antibody will be released, and as a consequence, more antibody will instead be degraded. When the antibody is released in the brain parenchyma it can find and bind to its intra-brain target. (B). Schematic picture of the RmAb158-scFv8D3 fusion protein design. RmAb158 binds to Aβ protofibrils, involved in AD pathogenesis. The scFv of 8D3, which binds to TfR, is attached to the C-terminus of the RmAb158 light chain with short linkers. (C). The short linker length between RmAb158 and scFv8D3 combined with the placement of scFv8D3 on the C-terminus of the light chains ensures that there cannot be bivalent binding to the TfR dimer (left). The presence of two rather than one scFv8D3 will increase the concentration of TfR binders and hence increase the likelihood of uptake. In contrast, 8D3 can bind bivalently to the TfR dimer (right).
Figure 2
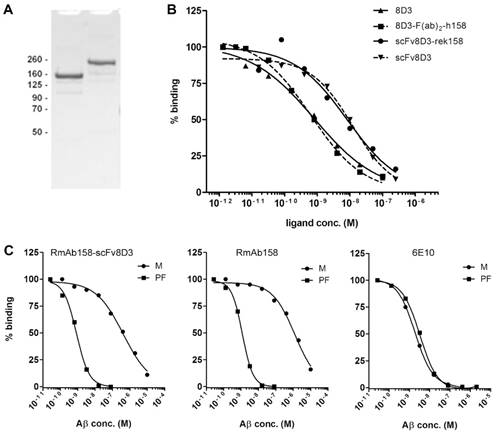
Characterisation of the binding properties of the brain shuttle. (A). SDS-PAGE of RmAb158 and RmAb158-scFv8D3, displaying a single band of each antibody with an approximate size of 160 and 210 kDa, respectively. (B). TfR competition ELISA, demonstrating that 8D3, in line with the chemically conjugated fusion protein 8D3-F(ab')2-h158 [19], binds bivalently to TfR, and displays 10-fold stronger binding compared with RmAb158-scFv8D3. A scFv of 8D3 displays an even weaker TfR binding due to its single binding site. (C). Inhibition ELISA, displaying that RmAb158-scFv8D3 retains a high, selective binding to Aβ protofibrils (PF) over monomers (M) comparable to RmAb158, whereas the control antibody 6E10 binds equally well to both Aβ species. Representative graphs from three independent experiments.
To further investigate the pharmacokinetics and brain distribution, a tracer dose (~0.05 mg/kg) of radiolabeled RmAb158-scFv8D3 was administered to young (8-9 months) and old (18-24 months) wt and tg-ArcSwe mice. The majority (n=14) of the mice were euthanized 3 days' post injection, while two old tg-ArcSwe mice were kept under investigation until 6 days after injection and one old tg-ArcSwe was euthanized at day 10 after injection.
Three days after administration of RmAb158-scFv8D3, a 9-fold higher brain concentration of recombinant fusion protein was found in old tg-ArcSwe mice (%ID/g = 0.69±0.17) compared with old wt mice (%ID/g = 0.078±0.012) (Fig 4A). This equals a 5-fold increase in comparison with the chemically generated fusion protein 8D3-F(ab')2-h158 (Fig 4B). The brain concentrations of RmAb158-scFv8D3 decreased only slightly during the next 7 days after administration, since the antibody was bound to its target, abundantly present in the brain of old tg-ArcSwe mice (Fig 4C). The half-life in blood based on samples obtained between 3 h and 3 days was 18.6±1.5 h. From 1 day to 3 days, i.e. during the elimination phase, the half-life was 24.4±3.3 h (Fig 4C). There was no difference in blood concentrations or half-life between transgenic and wt mice.
Figure 3
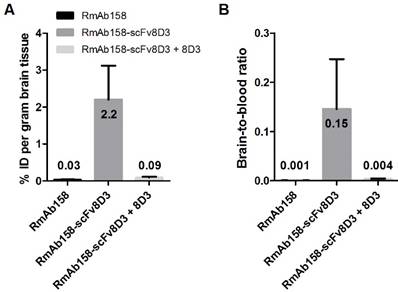
Increased brain uptake of the brain shuttle connected to the RmAb158 antibody in wt mice. (A). Ex vivo experiment displaying 80-fold difference in brain concentration of [125I]RmAb158 and [125I]RmAb158-scFv8D3 in wt mice 2 h post injection. When co-injected with 10 mg/kg 8D3, the difference is reduced to 3-fold. (B). Brain-to-blood concentration ratio in the same experiment. The ratio is 140-fold higher for [125I]RmAb158-scFv8D3 than for [125I]RmAb158, whereas only a 4-fold difference remains when 8D3 is co-administered. (n=3 for each group).
Figure 4
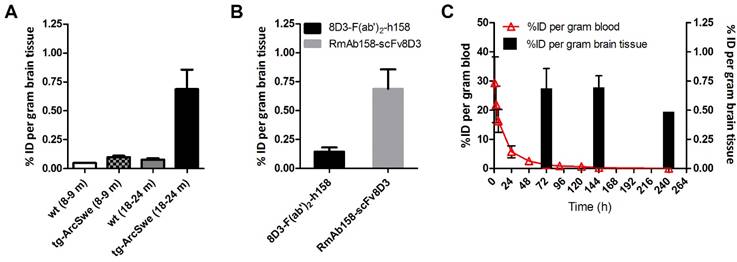
High brain retention of RmAb158-scFv8D3 in transgenic mice. (A). Ex vivo quantified brain concentration of [124/125I]RmAb158-scFv8D3 in old (18-24 months) and young (8-9 months) wt and tg-ArcSwe mice 3 days post injection (n=3, 4, 4 and 6 for young wt, young tg-ArcSwe, old wt respective old tg-ArcSwe). (B). Comparison of ex vivo brain concentration of the chemically fused 8D3-F(ab')2-h158 (n=5) and the recombinant RmAb158-scFv8D3 (n=6) in 18 months old tg-ArcSwe mice 3 days post injection. (C). Ex vivo quantified brain concentration of [124/125I]RmAb158-scFv8D3 in old (18-24 months) tg-ArcSwe mice at 3, 6 and 10 days post administration in comparison with blood concentration over the same time period. Elimination from brain was much slower than elimination from blood (n=8).
Peripheral biodistribution of RmAb158-scFv8D3
The distribution of [125/124I]RmAb158-scFv8D3 in peripheral organs, 3-10 days post injection, is shown in Fig 5. The tissue concentrations in most organs decreased somewhat faster than the concentrations in blood, explaining the relatively low concentrations in excretory organs such as liver and kidney at these time points. Spleen displayed the highest uptake and although the concentrations in the spleen decreased, the decrease was slower than the elimination of [125/124I]RmAb158-scFv8D3 from the other organs, but still faster than elimination from blood. In contrast, the tg-ArcSwe brain, where the ligand binds to the abundant Aβ pathology, displayed high values at all time points with a slow decrease and an estimated half-life of approximately two weeks.
Figure 5
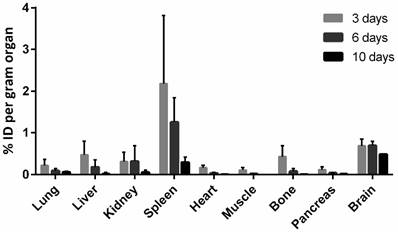
Biodistribution of RmAb158-scFv8D3. Biodistribution of [124/125I]RmAb158-scFv8D3 in different peripheral organs quantified as per cent of injected dose per gram organ at 3 (n=14), 6 (n=4) and 10 (n=10) days after administration. Brain concentrations in old tg-ArcSwe mice from fig 4C are included for comparison.
Evaluation of RmAb158-scFv8D3 brain uptake and retention with PET
We have previously shown, in both in vivo and ex vivo experiments, that mAb158 and different variants of the antibody specifically target Aβ pathology in tg-ArcSwe mice [16,17,19,22]. To specifically visualize Aβ pathology, animals were PET-scanned at the time of [124I]RmAb158-scFv8D3 injection or at 3, 6 or 10 days post injection. PET data acquired during the first 0-60 min after injection were quantified in relation to a blood sample obtained directly after the scan, under the assumption that 3% of the brain volume is blood [23]. This experiment showed that 72% of the PET signal at this time point was derived from [124I]RmAb158-scFv8D3 associated with the brain tissue, suggesting a high concentration in brain was obtained almost immediately following injection (Fig 6A). This is indicative of active transport into the brain, unlike unmodified mAb158 which has a peak in brain concentration 3 days' post injection [17]. Three days after injection, the recombinant fusion protein concentration had decreased to about 25% and to 10% of what had been observed in the scan at injection time in the brains of tg-ArcSwe and wt mice, respectively. At later time points the brain concentrations measured by PET was only somewhat decreased in old tg-ArcSwe animals while there was almost no [124I]RmAb158-scFv8D3 left in the brain of wt and young tg-ArcSwe mice (Fig 6B). Because of the brain's relatively high content of blood, the PET signal in the brain is influenced by the blood concentration of recombinant fusion protein, which decreases significantly during the first three days after injection, thus explaining the large drop in PET signal between the first and second scans. This suggests that [124I]RmAb158-scFv8D3 that is not bound to Aβ in the brain is in equilibrium with blood and is actively transported out of the brain. Compared with the previously generated, chemically conjugated fusion protein, brain concentration was markedly increased 3 days' post injection (Fig 6C) and, due to the improved brain uptake, Aβ pathology could be visualized also in young mice (Fig 6D).
PET data of amyloid levels in the brain, assessed with e.g. [11C]PIB, is often quantified as the brain-to-cerebellum concentration ratio, as the cerebellum is largely spared from Aβ pathology and can act as a reference region. The concentration ratios, using cerebellum as the reference, of whole brain, thalamus, striatum, hippocampus and cortex, quantified by PET, are shown in Fig 7. There was a clear distinction already at three days' post injection with no overlap in ratios between old tg-ArcSwe and the other groups. At six days' post injection, both young and old tg-ArcSwe mice could be distinguished from wt mice without Aβ protofibril accumulation in the brain. This suggests that the [124I]RmAb158-scFv8D3 binding in the brain is indeed specific to the Aβ pathology, as previously proven for the chemically conjugated fusion protein [19].
Evaluation of RmAb158 and RmAb158-scFv8D3 brain uptake at therapeutic doses
All of the brain uptake experiments described above were carried out using tracer doses of RmAb158 or RmAb158-scFv8D3, i.e. around 0.05 mg/kg. A second set of experiments was performed using the same amount of radioactivity as in the earlier experiments but with the addition of unlabeled RmAb158 or RmAb158-scFv8D3 at doses of 10 mg/kg. The brain concentrations in relation to amount of RmAb158 administered after therapeutic dosing, expressed as %ID/g brain and brain-to-blood concentration ratio (Fig 8), was the same as those obtained after tracer dosing (Fig 3). Hence, the transport across the BBB was linear with dose. However, for RmAb158-scFv8D3 endothelial TfR binding seemed to be saturated, yielding lower brain concentrations in relation to dose and systemic concentrations when therapeutic doses were administered (Fig 8). At 2h post injection the increase in uptake of RmAb158-scFv8D3 was 10 times compared to RmAb158. However, when 10 mg/kg of non-labeled native 8D3 was co-injected with [125I]RmAb158-scFv8D3, as described earlier (Fig 3A), only a 3-fold increase could be measured, demonstrating that the 8D3 binds stronger to the endothelial TfR (Fig 3A) than RmAb158-scFv8D3.
Figure 6
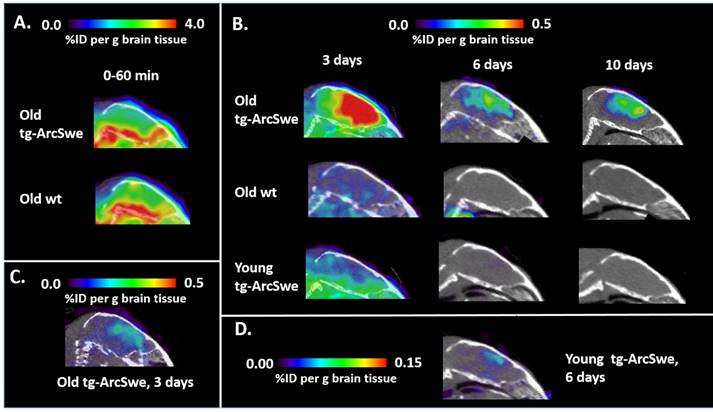
Improved sensitivity in PET imaging of Aβ pathology with RmAb158-scFv8D3 as a ligand. Sagittal PET images obtained after administration of [124I]RmAb158-scFv8D3. (A). PET image in old tg-ArcSwe and wild-type (wt) mice obtained at 0-60 min post administration of [124I]RmAb158-scFv8D3. Radioactivity concentration is similar in all brain regions. The same scale as in the other figures could not be used as the radioactivity was much higher during this early time point. (B). PET images obtained in old tg-ArcSwe, old wt and in young tg-ArcSwe mice at 3, 6 and 10 days post injection of [124I]RmAb158-scFv8D3. (C). PET image images obtained from an old tg-ArcSwe injected with the previously generated chemically conjugated fusion protein [19] using the same colour scale as in B to allow for comparison of brain concentrations. (D). PET image of a young tg-ArcSwe mouse at 6 days post injection (same as in B) displayed using a colour scale with a lower threshold. Cortical brain uptake of [124I]RmAb158-scFv8D3 can be then visualized with PET also at this early stage of disease progression.
Figure 7
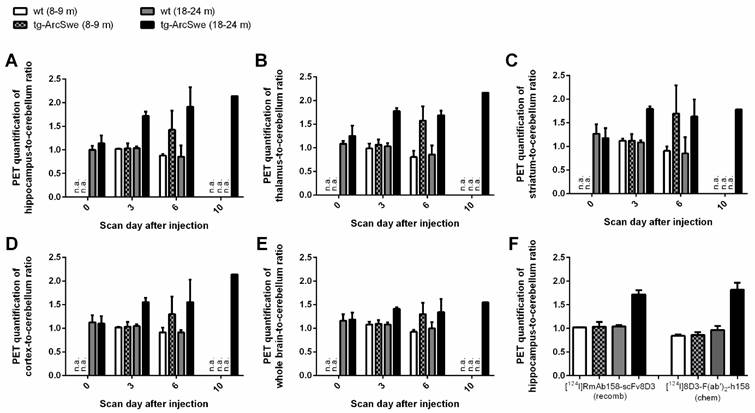
PET quantification of RmAb158-scFv8D3 concentration in wt and transgenic mouse brains. PET based brain region-to-cerebellum concentration ratios in hippocampus (A), thalamus (B), striatum (C), cortex (D) and whole brain (E). A comparison to chemically conjugated fusion protein is shown in (F). On day 0 some animal groups were not investigated and on day 10, cerebellum levels were below the limit of quantification in wt and young tg-ArcSwe mice, and hence, ratios could not be calculated. (n=3-5 per group except for 10 days when n=1. Total number of animals is 24 and each animal had one or two PET scans).
Figure 8
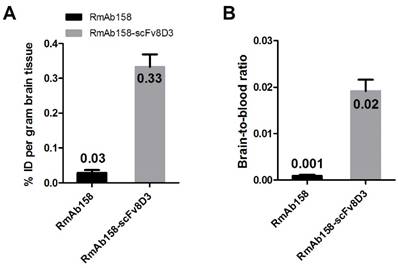
Uptake of the brain shuttle connected to the RmAb158 antibody at therapeutic doses. (A). Ex vivo experiment using therapeutic doses of RmAb158 and RmAb158-scFv8D3 displaying 10-fold difference in brain of the two ligands in wt mice 2 h post injection. (B). Brain-to-blood concentration ratio in the same experiment displaying a 20-fold difference between the two proteins. (n=5 for each group).
The aim of the present study was to develop a recombinant shuttle for efficient BBB transport of antibodies. Such shuttle can be used to facilitate immunotherapy, for example as a treatment for neurodegenerative diseases or brain tumors. The shuttle can also be applied for antibody-based diagnostics of brain diseases. To illustrate the capabilities of the brain shuttle we used an antibody, RmAb158, with a selective affinity for Aβ protofibrils, suggested to play an important role in AD. The brain shuttle consisted of moieties binding to the TfR. We and others have previously used the TfR binding antibody 8D3 to increase BBB uptake of antibodies [5,19,20]. In the present study, we recombinantly attached the scFv8D3 to the C-terminus of each of the RmAb158 light chains with short a linker to sterically hinder bivalent binding to the TfR dimer. Hence, despite having two TfR binding sites, RmAb158-scFv8D3 binds monovalently to TfR, as opposed to the high avidity, bivalent binding of the entire 8D3 antibody. Niewoehner et al [5] constructed a somewhat similar fusion protein consisting of an anti-Aβ antibody with scFab-8D3 attached to the two C-termini of the heavy chains using longer linkers. With no steric hinder, the two scFab-8D3 could simultaneously bind to TfR, resulting in bivalent and/or high avidity binding. However, linking Fab-8D3 to only one of the two heavy chains resulted in a considerably improved distribution into the brain parenchyma [5].
The two scFv8D3 connected to the RmAb158 will double the amount of available binding sites to TfR compared with bifunctionalization of only one heavy/light chain. In addition, the generation of an antibody consisting of two identical light chains has a lower risk for mismatches and will result in a more homogeneous preparation compared with the production of an antibody with two different light chains (one with and one without scFv8D3). Thus, by maintaining monovalent binding despite having two TfR binding scFv, the efficiency of transport across the BBB may increase. To our knowledge, a monovalent binding with two TfR binding sites has not been achieved with any previous design of the TfR binder moiety. This was accomplished by the use of the novel short linkers and placement of the TfR binders on the C-terminal of the light chain which sterically hinders bivalent binding.
We have previously described the brain pharmacokinetics of the chemically coupled fusion protein 8D3-F(ab')2-h158 used as a PET ligand. The new recombinant bispecific fusion protein RmAb158-scFv8D3 generated in the present study displayed higher brain and blood concentrations throughout the study period compared to the chemically conjugated version. The longer half-life in blood of RmAb158-scFv8D3 (24 h), compared with the chemically conjugated fusion protein (15 h), can probably be attributed to the intact murine Fc domain of the recombinant version. As a consequence of this difference, PET images obtained with the chemically coupled fusion protein (Fig 6C) displayed a background signal similar to images obtained at 6 or 10 days with [124I]RmAb158-scFv8D3 (Fig 6B). In contrast, the specific signal generated from binding to Aβ pathology in the brain was markedly increased with the newly developed ligand due to its higher brain uptake. Clearly, the increased brain concentrations are an advantage both from a therapeutic and diagnostic perspective. The region specific-to-cerebellum ratios of [124I]RmAb158-scFv8D3 distinctly distinguished mice with Aβ protofibril pathology from mice without Aβ protofibril accumulation (Fig 7) and the ratios were similar to those found with the chemically conjugated protein (Fig 7F). However, in contrast to the chemically conjugated fusion protein, [124I]RmAb158-scFv8D3 could detect pathology also in 8 month old tg-ArcSwe mice, where Aβ plaque formation just started, demonstrating its efficient brain uptake and high in vivo specificity to Aβ pathology.
It has previously been reported that the uptake of antibody with a TfR shuttle relative to a naked antibody is higher at trace dosing, compared to therapeutic dosing, suggesting that the TfR system gets saturated at therapeutic doses. Yu et al. reported that at trace dosing, the uptake of anti-TfRA antibody was increased 40 times compared to the naked antibody 1 h post injection, but only 1.4 times at therapeutic doses [4]. The same group has also developed a BBB shuttle targeting CD98 that displayed the same dose dependency. At trace dosing the uptake was 80 times higher than antibody without shuttle but at therapeutic dosing it was only 3.2 times higher [21]. Boado et al. described a shuttle that showed 43 times increased transport across the BBB at trace doses compared to a reference antibody, but did not report the difference at therapeutic doses [24]. In the present study, we also observed decreased relative uptake of RmAb158-scFv8D3 at therapeutic compared to trace dosing, but not to the same extent. While our TfR shuttle increased brain uptake of RmAb158 by 80-fold 2 h post injection at trace dosing, the uptake was still 10 times higher at therapeutic dosing. When RmAb158-scFv8D3 was co-injected with a therapeutic dose of 8D3, binding bivalently to TfR, the fusion protein brain uptake was reduced to 3-fold higher than RmAb158 (Fig 3), further supporting that monovalent binding increases brain uptake.
In conclusion, we have developed a BBB shuttle which is actively transported across the BBB, more efficiently than any previously reported BBB shuttle at both trace and therapeutic doses. The enhanced brain uptake was achieved with a novel design to obtain monovalent interactions with TfR despite having two binding sites. The retained affinity and selectivity to Aβ protofibrils of the tested bispecific antibody, demonstrated both in vitro and in vivo, illustrates that the antibody format has the potential to increase the efficacy of immunotherapy or diagnostic applications of any antibody targeting proteins within the CNS.
Methods
Cloning RmAb158-scFv8D3
Both the heavy and light chain of the expressed antibody were cloned into the vector pcDNA3.4 with signal peptides on the N-terminal. The 8D3 sequence [2] was made into a scFv with the heavy chain variable fragment as the N-terminal part and the light chain variable fragment on the C-terminal. The heavy and light chains were separated by an 18 amino acid long glycine/serine-rich amino acid linker (GSTSGGGSGGGSGGGGSS), used for instance in [25]. The scFv 8D3 was then connected to the C-terminus of the RmAb158 light chain with an in house designed linker (APGSYTGSAPG). Prolines were added at the beginning and at the end of the linker to ensure that alpha helices are not extended, since they cannot donate the amide hydrogen bond needed in alpha helices. Polar amino acids like serine and threonine were added to ensure that the linker is hydrophilic, and the smaller amino acid glycine was added to ensure flexibility.
Expression and purification of RmAb158-scFv8D3
The recombinant fusion protein, RmAb158-scFv8D3, was expressed using the Expi293 cells (ThermoFisher) transiently transfected with the heavy and light chain pcDNA3.4 vectors using polyethylenimine (PEI) as transfection reagent and valproic acid (VPA) as a cell cycle inhibitor [26]. RmAb158-scFv8D3 was purified on a protein G column, and eluted with an increasing gradient of 0.7% HAc. ScFv8D3, including a His-tag, was expressed with the same system and purified with a nickel column.
In vitro analysis of RmAb158-scFv8D3
To confirm the size and integrity of RmAb158-scFv8D3, it was analyzed with SDS-PAGE. RmAb158 and RmAb158-scFv8D3 were mixed with Bolt® LDS sample buffer, without reducing agent, and directly loaded onto a 10% Bolt Bis-Tris Plus gel (ThermoFisher), run for 22 min at 200 V, washed in dH2O and stained with Page blue (Fermentas). A Chameleon pre-stained protein marker (Li-Cor) was used as a molecular weight standard.
A competition ELISA was used to assess the ability of RmAb158-scFv8D3 to bind to TfR in comparison with the chemically fused 8D3-F(ab')2-h158, 8D3 and a scFv fragment of 8D3. 96-well plates were coated over night at +4°C with 50 ng/well of recombinant transferrin receptor protein (Sinobiological, Beijing, China) and blocked with BSA. Serially diluted antibody was incubated on the plates for 2 h on a shaker in competition with 2.5 nM of biotinylated scFv8D3, then detected with horseradish peroxidase (HRP) conjugated streptavidin (Mabtech AB, Nacka Strand, Sweden) and K blue aqueous TMB substrate (Neogen Corp., Lexington, KY, USA) and read with a spectrophotometer at 450 nm. All antibody dilutions were made in ELISA incubation buffer (PBS with 0.1% BSA, 0.05% Tween, and 0.15% Kathon).
To assess specific binding to Aβ monomers and protofibrils in solution, RmAb158-scFv8D3 was analyzed with an inhibition ELISA in comparison with RmAb158 and the Aβ antibody 6E10 (Covance), as previously described [16,19]. 96-well plates were coated for 2 h at +4°C with 45 ng/well of Aβ protofibrils, followed by 1 h blocking with BSA. Serially diluted Aβ monomers or protofibrils were preincubated 1 h with a fixed concentration of antibody (RmAb158-scFv8D3 and RmAb158 - 155 pM; 6E10 - 1.25 nM) in a non-binding 96-well plate. The Aβ-antibody solution was then transferred to the Aβ coated plates and incubated for 15 min, followed by detection with HRP-conjugated anti-mouse-IgG-F(ab')2 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and K blue aqueous TMB substrate (Neogen Corp.) and read with a spectrophotometer at 450 nm. All Aβ and antibody dilutions were made in ELISA incubation buffer. Aβ preparations were made as previously described [17].
Animals
The tg-ArcSwe mouse model, harbouring the Arctic (AβPP E693G) and Swedish (AβPP KM670/671NL) mutations and maintained on a C57BL/6 background, shows elevated levels of soluble Aβ protofibrils already at a very young age and abundant and rapidly developing plaque pathology starting at around 6 months of age. Both males and females were used and littermates were used as control animals (wt). The animals were housed with free access to food and water in rooms with controlled temperature and humidity in an animal facility at Uppsala University. All procedures described in this paper were approved by the Uppsala County Animal Ethics board (#C17/14), following the rules and regulations of the Swedish Animal Welfare Agency and were in compliance with the European Communities Council Directive of 22 September 2010 (2010/63/EU). All efforts were made to minimize animal suffering and to reduce the number of animals used.
Radiochemistry
The bispecific RmAb158-scFv8D3 was labelled with iodine-125 ( 125I) for ex vivo experiments and iodine-124 (124I) for PET experiments using direct radioiodination [27], The method is based on electrophilic attack of the phenolic ring of tyrosine residues by in situ oxidized iodine. Briefly, for 125I-labelling, 120 pmoles of antibody or recombinant fusion protein (assumed Mw 210 kDa), 125I stock solution (Perkin-Elmer Inc., Waltham, MA, USA) and 5 µg Chloramine-T (Sigma Aldrich, Stockholm, Sweden) were mixed in PBS to a final volume of 110 µl. The reaction was allowed to proceed for 90 s and subsequently quenched by addition of double molar excess of sodium metabisulfite (Sigma Aldrich) and dilution to 500 µl in PBS. For 124I-labelling, 60 µl 124I stock solution (Perkin-Elmer Inc.) was pre-incubated 15 min with 12 µl 50 µM NaI before addition of 240 pmoles of recombinant fusion proteins and 40 µg Chloramine-T, mixed in PBS to a final volume of 420 µl. The reaction was allowed to proceed for 120 s and subsequently quenched by addition of 80 µl of sodium metabisulfite in PBS (1 mg/ml). The radiolabeled proteins were purified from free iodine and low-molecular weight components with a disposable NAP-5 size exclusion column, Mw cut-off 5 kDa (GE Healthcare AB, Uppsala, Sweden), according to the manufacturer's instructions and eluted in 1 ml of PBS. The yield was calculated based on the added radioactivity and the radioactivity in the purified radioligand solution. Labelling was always performed less than 2 h prior to each study.
Ex vivo studies
Mice were intravenously (i.v.) injected with 0.44±0.03 MBq [125I]RmAb158 (n=3) or 0.89±0.26 MBq [125I]RmAb158-scFv8D3 (n=11), which equals a dose of 0.05 mg/kg. Blood samples (8 µl) were obtained from the tail vein at 0.5, 1, 2, 3, 4, 6, 8, 24 and 48 h after injection and when mice were euthanized 3 days' post injection. A second group of animals (n=3 for each ligand) were euthanized 2 h after injection and here we also added a group of animals (n=3) injected with 0.05 mg/kg of [125I]RmAb158-scFv8D3 together with 10 mg/kg of 8D3. A separate group of wt mice were injected with 10 mg/kg RmAb158 (n=5) and 13.3 mg/kg RmAb158-scFv8D3 (n=5), containing 1.5% radiolabeled protein for detection, and euthanized 2 h after injection. Animals were perfused with 50 ml physiological saline during 2 min immediately following a terminal blood sample obtained from the heart. After perfusion, brain was isolated and the cerebellum was separated from the rest of the brain before the brain tissue samples were frozen on dry ice. Liver, lung, heart, spleen, kidney, pancreas, muscle and femur were also isolated. Radioactivity in blood, brain, cerebellum and isolated organs was measured with a γ-counter (1480 Wizard™, Wallac Oy, Turku, Finland). The brain, cerebellum and blood concentrations, quantified as % of injected dose per gram tissue (% ID/g), were calculated as following:
% ID/g = Measured radioactivity per gram tissue (or blood) / Injected radioactivity (eq.1)
In addition, the tissue-to-blood concentration ratio was calculated as following:
Tissue-to-blood concentration ratio= Measured radioactivity per gram tissue / Measured radioactivity per gram blood (eq.2)
PET studies
The day before injection of [124I]RmAb158-scFv8D3 animals were given water supplemented with 0.2% NaI to reduce thyroidal uptake of 124I. Mice (n=24) were intravenously (i.v.) injected with 6.7±2.6 MBq [124I]RmAb158-scFv8D3. Blood samples (8 µl) were obtained from the tail vein at 1, 3, 6, 24, 48, 72, 144 and 240 h after injection (no samples were obtained at 144 and 240 hours for animals that were euthanized 3 days post administration). Mice had either one or two PET scans starting at the time of injection, at 3 days, 6 days or 10 days after administration of [124I]RmAb158-scFv8D3. The animals were placed in the gantry of the animal PET/CT scanner (Triumph Trimodality System, TriFoil Imaging, Inc., Northridge, CA, USA) with the head in the centre of the Field of View (FOV, 8.0 cm) and then scanned in list mode during 60 min followed by a computed tomography (CT) examination for 3 min. Animals were euthanized after the final PET scan according to the same procedure as described above for the ex vivo studies including a terminal blood sample from the heart. Radioactivity in blood, brain, cerebellum and isolated organs was measured with a well counter (GE Healthcare, Uppsala, Sweden) and %ID/g and tissue-to-blood concentrations were calculated according to Equations 1 and 2.
The PET data were reconstructed using the ordered subsets expectation-maximization (OSEM) 3D algorithm (20 iterations). The CT raw files were reconstructed using Filter Back Projection (FBP). All subsequent processing of the PET and CT images were performed in imaging software Amide 1.0.4 [28]. The CT scan was manually aligned with a T2 weighted, magnetic resonance imaging (MRI) based mouse brain atlas [29] containing outlined regions of interests for hippocampus, striatum, thalamus, cortex and cerebellum. The PET image was then aligned with the CT, and thus, the MRI-atlas was also aligned with the PET data. The PET data shown in Fig 6 are summed images, i.e. representing the average activity during the whole PET scan. The PET data was quantified as a concentration ratio of the radioactivity in 5 regions of interest (whole brain, cortex, hippocampus, thalamus and striatum) to that in cerebellum.
Ex vivo data shown in Fig 4 and 5 are based on both 124I labelled and 125I labeled recombinant fusion protein.
Abbreviations
BBB: blood-brain barrier; TfR: transferrin receptor; AD: Alzheimer's disease; scFv: single chain fragment variable; Aβ: amyloid-β; AβPP: Aβ protein precursor; PET: positron emission tomography; wt: wild-type; tg-ArcSwe: AβPP transgenic mice with the Arctic and Swedish AβPP mutations; PEI: polyethylenimine; VPA: valproic acid.
Acknowledgements
We are grateful to BioArctic Neuroscience AB for experimental assistance with protein expression and for sharing the RmAb158 sequence. We would also like to thank S. Estrada, V. Asplund and R.K. Selvaraju for technical assistance with PET scanning and L. Nilsson who developed the mouse models. This work was supported by grants from the Swedish Research Council (#2012-1593, #2012-2172), Alzheimerfonden, Swedish Brain Foundation, Åhlén-stiftelsen, Stohnes stiftelse, Stiftelsen för Gamla tjänarinnor, Magnus Bergwalls stiftelse and Uppsala Berzelii Technology Centre for Neurodiagnostics.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bard F, Cannon C, Barbour R, Burke R-L, Games D, Grajeda H. et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916-919
2. Boado RJ, Zhang Y, Wang Y, Pardridge WM. Engineering and expression of a chimeric transferrin receptor monoclonal antibody for blood-brain barrier delivery in the mouse. Biotechnol Bioeng. 2009;102:1251-1258
3. Pardridge WM. Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin Drug Deliv. 2015;12:207-222
4. Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y. et al. Boosting Brain Uptake of a Therapeutic Antibody by Reducing Its Affinity for a Transcytosis Target. Sci Transl Med. 2011;3:84ra44-84ra44
5. Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P. et al. Increased Brain Penetration and Potency of a Therapeutic Antibody Using a Monovalent Molecular Shuttle. Neuron. 2014;81:49-60
6. Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM. et al. Amyloid-beta Oligomerization in Alzheimer Dementia vs. High Pathology Controls. Ann Neurol. 2013;73:104-119
7. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Konrad Vbeyreuther. et al. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860-866
8. Näslund J, Haroutunian V, Mohs R. et al. COrrelation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA. 2000;283:1571-1577
9. Fowler SW, Chiang ACA, Savjani RR, Larson ME, Sherman MA, Schuler DR. et al. Genetic Modulation of Soluble Aβ Rescues Cognitive and Synaptic Impairment in a Mouse Model of Alzheimer's Disease. J Neurosci. 2014;34:7871-7885
10. Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M. et al. High-molecular-weight β-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J. 2010;24:2716-2726
11. Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A. et al. Amyloid β Protein Dimer-Containing Human CSF Disrupts Synaptic Plasticity: Prevention by Systemic Passive Immunization. J Neurosci. 2008;28:4231-4237
12. Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C. et al. The “Arctic” APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat Neurosci. 2001;4:887-893
13. Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F. et al. Large Aggregates Are the Major Soluble Aβ Species in AD Brain Fractionated with Density Gradient Ultracentrifugation. PLoS ONE. 2012;7:e32014
14. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I. et al. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837-842
15. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS. et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535-539
16. Englund H, Sehlin D, Johansson A-S, Nilsson LNG, Gellerfors P, Paulie S. et al. Sensitive ELISA detection of amyloid-β protofibrils in biological samples. J Neurochem. 2007;103:334-345
17. Magnusson K, Sehlin D, Syvänen S, Svedberg MM, Philipson O, Söderberg L. et al. Specific Uptake of an Amyloid-β Protofibril-Binding Antibody-Tracer in AβPP Transgenic Mouse Brain. J Alzheimers Dis. 2013;37:29-40
18. Sehlin D, Hedlund M, Lord A, Englund H, Gellerfors P, Paulie S. et al. Heavy-Chain Complementarity-Determining Regions Determine Conformation Selectivity of Anti-Aβ Antibodies. Neurodegener Dis. 2011;8:117-123
19. Sehlin D, Fang XT, Cato L, Antoni G, Lannfelt L, Syvänen S. Antibody-based PET imaging of amyloid beta in mouse models of Alzheimer/'s disease. Nat Commun. 2016;7:10759
20. Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1959-1972
21. Zuchero YJY, Chen X, Bien-Ly N, Bumbaca D, Tong RK, Gao X. et al. Discovery of Novel Blood-Brain Barrier Targets to Enhance Brain Uptake of Therapeutic Antibodies. Neuron. 2016;89:70-82
22. Lord A, Gumucio A, Englund H, Sehlin D, Sundquist VS, Söderberg L. et al. An amyloid-β protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2009;36:425-434
23. Todd MM, Weeks JB, Warner DS. Microwave fixation for the determination of cerebral blood volume in rats. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 1993;13:328-336
24. Boado RJ, Zhou Q-H, Lu JZ, Hui EK-W, Pardridge WM. Pharmacokinetics and Brain Uptake of a Genetically Engineered Bifunctional Fusion Antibody Targeting the Mouse Transferrin Receptor. Mol Pharm. 2010;7:237-244
25. Wu AM, Tan GJ, Sherman MA, Clarke P, Olafsen T, Forman SJ. et al. Multimerization of a chimeric anti-CD20 single-chain Fv-Fc fusion protein is mediated through variable domain exchange. Protein Eng. 2001;14:1025-1033
26. Backliwal G, Hildinger M, Kuettel I, Delegrange F, Hacker DL, Wurm FM. Valproic acid: A viable alternative to sodium butyrate for enhancing protein expression in mammalian cell cultures. Biotechnol Bioeng. 2008;101:182-189
27. Greenwood FC, Hunter WM, Glover JS. The preparation of 131I-labelled human growth hormone of high specific radioactivity. Biochem J. 1963;89:114-123
28. Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging. 2003;2:131-137
29. Ma Y, Hof PR, Grant SC, Blackband SJ, Bennett R, Slatest L. et al. A three-dimensional digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Neuroscience. 2005;135:1203-1215
Author contact
![]() Corresponding author: G.H. (email: greta.hultqvistuu.se, phone: +46 70 2253522).
Corresponding author: G.H. (email: greta.hultqvistuu.se, phone: +46 70 2253522).
Citation styles
APA
Hultqvist, G., Syvänen, S., Fang, X.T., Lannfelt, L., Sehlin, D. (2017). Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics, 7(2), 308-318. https://doi.org/10.7150/thno.17155.
ACS
Hultqvist, G.; Syvänen, S.; Fang, X.T.; Lannfelt, L.; Sehlin, D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics 2017, 7 (2), 308-318. DOI: 10.7150/thno.17155.
NLM
Hultqvist G, Syvänen S, Fang XT, Lannfelt L, Sehlin D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics 2017; 7(2):308-318. doi:10.7150/thno.17155. https://www.thno.org/v07p0308.htm
CSE
Hultqvist G, Syvänen S, Fang XT, Lannfelt L, Sehlin D. 2017. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics. 7(2):308-318.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.