Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
5. Conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(2):344-356. doi:10.7150/thno.16562 This issue Cite
Research Paper
Inhibition by Multifunctional Magnetic Nanoparticles Loaded with Alpha-Synuclein RNAi Plasmid in a Parkinson's Disease Model
Shuiqin Niu1,2*, Ling-Kun Zhang1*, Li Zhang1, Siyi Zhuang1, Xiuyu Zhan1, Wu-Ya Chen1, Shiwei Du1, Liang Yin1, Rong You1, Chu-Hua Li1 ![]() , Yan-Qing Guan1, 2
, Yan-Qing Guan1, 2 ![]()
1. School of Life Science, South China Normal University, Guangzhou 510631, China.
2. MOE Key Laboratory of Laser Life Science & Institute of Laser Life Science, College of Biophotonics, South China Normal University, Guangzhou, 510631, China.
* These authors contributed equally to this work and should be considered co-first author.
Received 2016-6-21; Accepted 2016-10-20; Published 2017-1-1
Abstract

Lewy bodies are considered as the main pathological characteristics of Parkinson's disease (PD). The major component of Lewy bodies is α-synuclein (α-syn). The use of gene therapy that targeting and effectively interfere with the expression of α-syn in neurons has received tremendous attention. In this study, we used magnetic Fe3O4 nanoparticles coated with oleic acid molecules as a nano-carrier. N-isopropylacrylamide derivative (NIPAm-AA) was photo-immobilized onto the oleic acid molecules, and shRNA (short hairpin RNA) was absorbed. The same method was used to absorb nerve growth factor (NGF) to NIPAm-AA to specifically promote neuronal uptake via NGF receptor-mediated endocytosis. Additionally, shRNA plasmid could be released into neurons because of the temperature and pH sensitivity of NIPAm-AA interference with α-syn synthesis. We investigated apoptosis in neurons with abrogated α-syn expression in vitro and in vivo. The results demonstrated that multifunctional superparamagnetic nanoparticles carrying shRNA for α-syn could provide effective repair in a PD model.
Keywords: Fe3O4 nanoparticles, shRNA, RNAi, α-synuclein, Parkinson's disease.
1. Introduction
Parkinson's disease (PD) is the second most common age-related neurodegenerative disease [1]. Neuropathologically, PD is characterized by the selective demise of dopaminergic neurons mainly in the substantia nigra pars compacta, accompanied by the formation of intracytoplasmic inclusions known as Lewy bodies which stain positively for α-syn [2,3]. Although PD is currently believed to be caused by complex interactions between genetic abnormalities, environmental toxins, mitochondrial dysfunction and other cellular processes, the major parkinsonian symptoms result from the massive demise of dopaminergic neurons [2,4].
Genetic studies have provided valuable insight into the pathological mechanisms underlying PD [5]. Mutations in 6 genes (SNCA, Parkin, DJ-1, PINK1, LRKK2, and ATP13A2) have been shown to cause familial PD [6]. Furthermore, not only multiplications but also duplications and triplications of the normal α-syn gene can increase the propensity of α-syn to accumulate and accumulate within cells [7,8]. The discovery of α-syn both as a genetic cause of disease and as the major component of LBs in sporadic and familial cases of PD has strengthened the link between sporadic and hereditary PD forms and the possibility of an underlying mechanism that is common to the development of the disease [9].
Over the previous decade, several innovative gene therapies for PD have been shown to be relatively safe and well-tolerated, suggesting that gene therapy may be a viable treatment option for PD in the future [10]. RNA interference (RNAi) is one method that can be used to reduce SNCA gene expression. A viral vector is one kind of gene delivery method that can be used to deliver RNAi, and this method has been successfully implemented in preclinical studies to reduce the level of SNCA both in vitro and in vivo [11,12].
However, some gene therapy trials utilizing adenoviral, herpes simplex virus and adenovirus vectors for the treatment of many neurological diseases resulted in fatal inflammatory responses [13,14]. In addition, a major limitation of gene therapy for neurological disorders is delivering the gene across the blood-brain barrier (BBB). Almost all virus vectors are incapable of crossing the BBB [15]. Nanometric objects chosen for their biocompatibility and biodegradation have provided evidence for their potential use as nonviral carriers for drug delivery to the brain with the possibility of targeting specific brain tissue according to their composition and structure [16,17].
Among the nanometer targeting drugs, magnetic nanoparticles (MNPs) are superior to most other nano carriers because of their small particle size, large specific surface, and superparamagnetic behaviors, among other properties, providing the possibility of overcoming the drawbacks of traditional administrations [18-20]. The long-chain oleic acid (OA) polymer and its salts are utilized for the stabilization of iron oxide nanoparticles from aggregation and for effective drug delivery [21,22].
However, N-isopropylacrylamide (NIPAm) is one kind of nano polymer materials, that exhibits a low critical solution temperature (LCST), thermoresponsiveness and pH sensitivity. These characteristics of NIPAm can permit targeting and controlled release. Such materials are usually prepared by combining NIPAm with a pH-sensitive polymer such as acrylic acid (AA) via a copolymerization roadmap [23]. In recent years, the NIPAm involved thermoresponsive magnetic nanoparticles have also been assessed [24].
Moreover, studies have revealed that Nerve growth factor (NGF) and its cognate receptor tyrosine kinases are important regulators of neuronal survival and differentiation. Recent studies have revealed that the internalization and trafficking of neurotrophin receptors (Trks) play critical roles in neurotrophin-mediated signaling [25]. Increasing evidence from studies of PD has highlighted the importance of NGF for the promotion of cancer cell uptake via NGF receptor-mediated endocytosis [26]. One model that has been used to study PD consists of the administration of 1-methyl-4-phenyl-pyridinium (MPP+), which induces cytotoxicity in PC12, HT1080, HEK-293 and SH-SY5Y cell lines and results in the development of a PD phenotype in mice. Some research also suggests that adult male C57BL/6 mice weighing approximately 25-30 g are the preferred strain for the administration of 1-methyl-4-phenyl- 1,2,3,6- tetrahydropyridine (MPTP) [29].
Because α-syn is tightly linked to the pathogenesis of PD, interference with α-syn expression in neurons has recently received widespread attention. In the present study, we report a novel gene delivery technique that utilizes the temperature and pH sensitivity properties of NIPAm-AA to carry and release shRNA. Furthermore, photo-immobilized NGF is utilized to specifically promote PC12 cellular uptake through NGF receptor-mediated endocytosis. We combined gene therapy methods with cell targeting and a drug-controlled release system to prevent the overexpression of α-syn. This novel nano gene delivery system provides a novel strategy for PD treatment, especially in terms of targeting and control.
2. Materials and methods
2.1. Synthesis of AzPhNGF
The photoactive NGF (abbreviated AzPhNGF) was synthesized in our laboratory following the procedure presented in Figure 1 of Ref [30]. All treatments were conducted in dark room. In a separate sequence, NGF at 15 g·mmol-1 was added to 3 ml of DMF/PBS (4:1) (Sigma), which included 61.5 mg. 0.2 mmol-1 N-(4-azidobenzoyloxy) succinimide (Sigma). After stiring for 48h at 4℃, the NGF derivatives were purified using a dialysis membrane (Millipore Molecut II, 10000) for 72 h. The prepared AzPhNGF were stored at 4℃.
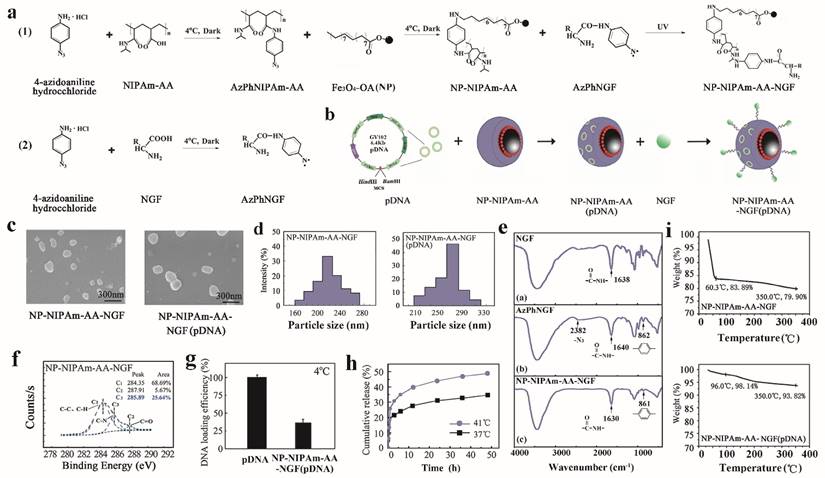
Preparation and characteristics. (a), Molecular structures and chemical processes for preparing NP-NIPAm-AA-NGF and NP-NIPAm-AA-NGF (pDNA). (b), The general stereo morphology of the nanoparticles. (c), SEM images of NP-NIPAm-AA-NGF and NP-NIPAm- AA-NGF (pDNA). The black bar stands for 300 nm. (d), size distribution. (e), FTIR spectra of NGF, AzPhNGF and NP-NIPAm-AA-NGF. (f), The electron spectroscopy for the chemical analysis of NP-NIPAm-AA-NGF. (g), DNA loading efficiency at 4℃. (h), The cumulative release of NP-NIPAm-AA-NGF (pDNA) at 37℃ and 41℃, and (i) thermal stability analysis of NP-NIPAm-AA-NGF and NP-NIPAm- AA-NGF (pDNA).
2.2. Synthesis of NP-NIPAm-AA
The oleic acid-coated magnetic nanoparticles (Fe3O4-OA, NP) and NP-NIPAm-AA were synthesized in our laboratory following a previously reported procedure [31].
Nanoparticles were synthesized by alkaline co-precipitation of FeSO4·7H2O and FeCl3 ·6H2O salts. After precipitation, oleic acid was added into the reaction chamber with HCl solution to adjust the solution pH value to pH=3 at 70℃. Finally, the nanoparticles were washed using absolute ethyl alcohol and acetone under a magnetic field. The oleic acid coated Fe3O4 nanoparticles (Fe3O4-OA) were synthesized.
For synthesis of the NP-NIPAm-AA samples, we first prepared N-isopropylacrylamide derivative (NIPAm-AA). Then, the NIPAm-AA was bonded to the Fe3O4-OA by irradiated with a UV lamp (125 W) for 10 s at the distance of 10 cm, resulting in the formation of NP-NIPAm-AA.
2.3. Synthesis of NP-NIPAm-AA (pDNA)
Five milligrams of NP-NIPAm-AA was dissolved in PBS. Next, 1.5 ml of 600 mg·mL-1 plasmid DNA in PBS was incubated with the NP-NIPAm-AA for 72 h at 4℃, allowing the sufficient adsorption of the pDNA. The water solution was then transferred to dialysis bag at 4℃ for 72 h to remove unabsorbed pDNA. The NP-NIPAm-AA (pDNA) was then obtained.
2.4. Synthesis of NP-NIPAm-AA and NP-NIPAm-AA (pDNA) with AzPhNGF
The initially prepared AzPhNGF (10 ng) and NP-NIPAm-AA (pDNA) (or NP-NIPAm-AA (1,000 ng)) were added to the PBS solution for stabilization for 48 h, and then the following treatments were applied. NP-NIPAm-AA (pDNA) or NP-NIPAm-AA together with AzPhNGF were transferred to a culture vessel and rotated at 100 rpm, followed by irradiation with a UV lamp (125 W) for 10-60 s positioned at a distance of 10 cm. Depending on the features of the highly active azido group, the AzPhNGF was immobilized on the surface of NP-NIPAm-AA (pDNA) (or NP-NIPAm-AA) (prior to this procedure, all of the treatments were prepared in the dark). The initially prepared MNPs were thoroughly purified in phosphate buffered saline without Ca2+ and Mg2+ (PBS (-), pH 7.4) by membrane dialysis (Millipore Molecut II, 10000) for 72 h and then stored at 4℃.
2.5. Microscopic surface and functional group analysis
After the photo-immobilization procedure, it is critical to ascertain whether the photoactive AzPhNGF are immobilized onto the surface of the OA-MNPs. Figure 1e,f shows the Fourier transform infrared spectroscope (FTIR) (TENSOR27, Bruker, Germany) images of the 3 powdered samples (NGF, AzPhNGF and NP-NIPAm-AA-AzPhNGF) and the electron spectroscopy images for their chemical analysis (ESCA, Escalab MKII, VG Scientific Co., East Grinstead, UK). Scanning electron microscopy (SEM) (JEM-100 CXII) and the particle size distribution were evaluated by dynamic light scattering (DLS) using the Zetasizer Nano-ZS90, Malvern Instruments Ltd, England, as shown in Figure 1c,d.
2.6. Screening of DNA
The pDNA was sufficiently absorbed by the NP-NIPAm-AA-NGF in PBS buffer solution after 3 days of mixing at 4℃. The nanoparticles were isolated from the mixture by centrifugation (in the supernatant). The concentration of pDNA in the supernatant was determined using GeneQuant. Finally, the loading dose was calculated by screening the pDNA concentration in the supernatant and the original concentration (Figure 1g).
2.7. DNA release efficiency
The pDNA nanoparticles were loaded by stirring in PBS buffer solution at 37℃ and 41℃ for 3 days. At several time points, 1 ml of solution was collected from the mixture to obtain the supernatant via high speed centrifugation. Subsequently, the GeneQuant method was used to quantify the pDNA content in the supernatant. Finally, we calculated the pDNA release efficiency and tested the higher pDNA concentration in the supernatant, as shown in Figure 1h.
2.8. Evaluation of thermal stability
Thermal gravimetric analyses (TGA) was performed by thermal gravimetric analyzer (TG209F1, NETZSCH, Germany) under a steady flow of static air at a heating rate of 10℃·min-1 in the range of 25℃ to 300℃. The reference standard was an empty crucible. All presented data are mean values (Figure 1i).
2.9. Establishment of a model of Parkinson's disease in vitro
The PC12 cells obtained from Sun Yat-Sen University were seeded at 1×105 cells·mL-1 in 24-well polystyrene (PSt) culture plates. After stimulation for 24 h, 72 h and 144 h with different concentrations of MPP+, we obtained the optimal concentration of MPP+ (1-methyl-4-phenylpyridinium ion) for induction of PC12 cell apoptosis. After applying the drugs, all cells were cultured under the same conditions (37℃, 5% CO2). Conversion of the number of surviving to dead cells can indirectly reflect the rate of MPP+-induced PC12 cell apoptosis, as shown in Figure 2a ((a)-(c)).
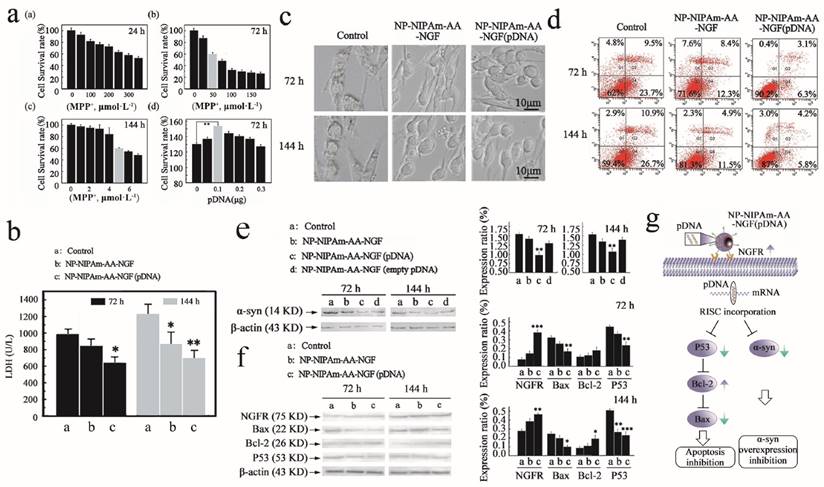
Measured efficacy data in vitro. (a), Cellular apoptosis model induced by MPP+ at 24 h, 72 h and 144 h, and pDNA concentration screening. (b), The LDH expression determined using the LDH detection kit for the control, NP-NIPAm-AA-NGF and NP-NIPAm-AA-NGF (pDNA) group at 72 h and 144 h. (c), Cell morphology evaluated by light microscopy (black bars, 10 μm). (d), Cell mortality data assessed by flow cytometry in the three groups: control, NP-NIPAm-AA-NGF, and NP-NIPAm-AA-NGF (pDNA) at 72 h and 144 h. (e), Protein expression of α-syn by western blot analysis in the control, NP-NIPAm-AA-NGF, NP-NIPAm-AA-NGF (pDNA) and NP-NIPAm-AA-NGF (empty pDNA) groups at 72 h and 144 h. (f), Protein expression of NGFR, Bax, Bcl-2, and P53 determined by western blot analysis in the control, NP-NIPAm-AA-NGF and NP-NIPAm-AA-NGF (pDNA) groups at 72 h and 144 h. The blots were re-probed to detect β-actin as a control to confirm equal protein loading. Protein expression determined using Image pro-plus 6.0. (g). The possible molecular mechanisms for multifunctional nano-biomaterials release α-syn interference plasmid to inhibit the synthesis of α-syn. The relative levels are plotted at a significance of p < 0.05 indicated by *, 0.001 < p < 0.01 indicated by **, and p < 0.001 indicated by ***, in comparison to the control group.
2.10. Screen for optimal dose of loaded pDNA
To screen the optimal dose of nanoparticle-loaded pDNA, an experimental duration of 72 h was used, at which time the nanoparticles demonstrated the best efficacy. To each well containing a concentration of 50 μmol · L-1 · well-1 MMT+, 0 μg, 0.05 μg, 0.1 μg, 0.15 μg, 0.2 μg, 0.25 μg or 0.3 μg pDNA was added, as shown in Figure 2a (d).
2.11. Detection of lactate dehydrogenase (LDH)
After stimulation for 72 h and 144 h, floating and adherent cells were combined, and cell viability is determined using the trypan blue dye exclusion method. To determine the DNA content, 1×106 cells were fixed and permeabilized in 70% ethanol, washed with phosphate-buffered saline (PBS, pH 7.4), and treated with RNase (40 U·mL-1). The LDH kit was used to determine the content of LDH, as shown in Figure 2b.
2.12. Cell morphology
Three groups of PC12 cells (NP-NIPAm-AA, NP-NIPAm-AA-NGF, NP-NIPAm-AA- NGF (pDNA)) were seeded at 1×106 cells·mL-1 in 24-well PSt culture plates for 72 h and 144 h. After stimulation, the morphology and inner structure of the PC12 cells were characterized by light microscopy (NIKON, Ti-U, Japan) as shown in Figure 2c.
2.13. Cell cycle arrest
After 72 h and 144 h of stimulation, floating and adherent cells were combined, and cell viability was determined using the trypan blue dye exclusion method. To assess the DNA content, 1×106 cells were fixed and permeabilized in 70% ethanol, washed with PBS (pH 7.4), treated with RNase (40 U·ml-1) and stained with propidium iodide (PI) (50 mg·ml-1). Flow cytometry (FACS Aria, BD Biosciences, USA) analysis was performed as shown in Figure 2d.
2.14. Western blot analysis for PC12 cells
The PC12 cells were stimulated for equivalent periods of time and lysed in extraction buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 5 mM EDTA [pH 8.0]). The protein samples were separated by SDS-PAGE (10%) and electro-transferred onto a nitrocellulose (NC) membrane (Boster Biotechnology Co., Ltd., China). Protein expression was evaluated using antibodies against P53, Bax, Bcl-2, NGFR and α-syn (Boster Biotechnology Co., Ltd., China). The blots were incubated with the appropriate secondary antibodies conjugated to alkaline phosphatase (AP) peroxidase (Boster Biological Technology Co., Ltd., China). The protein levels were normalized by re-probing the blots with antibody against β-actin (Boster Biological Technology Co., Ltd., China) as shown in Figure 2e,f.
2.15. PD animal model constructing and treatment
Male C57BL/6 mice (7-8 weeks) obtained from Sun Yat-sen University. 48 mice were used, of which 24 mice were treated with saline and 24 mice were treated with the MPTP (4 mg·kg-1, Sigma) intraperitoneally (i.p.) at 24 h intervals, for 15 consecutive days. Mice were killed 3 days after saline and MPTP administration. After the PD model was successfully established, comparative efficacy studies were performed by dividing the animals into 3 groups including: 1) saline i.p. + saline i.p., 2) MPTP i.p. + saline i.p., and 3) MPTP i.p. + NP (0.1 μg·20 g-1) i.p. The mice were killed 2 days after NPs treatment.
2.16. Gait analysis
Front and back paws were painted with red and blue gouache, respectively, and the animals were placed on a dark runway (20 cm wide, 100 cm long, with walls 10 cm high walls) to run. The mice were subjected to 3 training trials per day for 5 consecutive days for acclimatization to the environment. A single test trial was performed, and stride length was measured as the distance between successive paw prints as shown in Figure 3b.
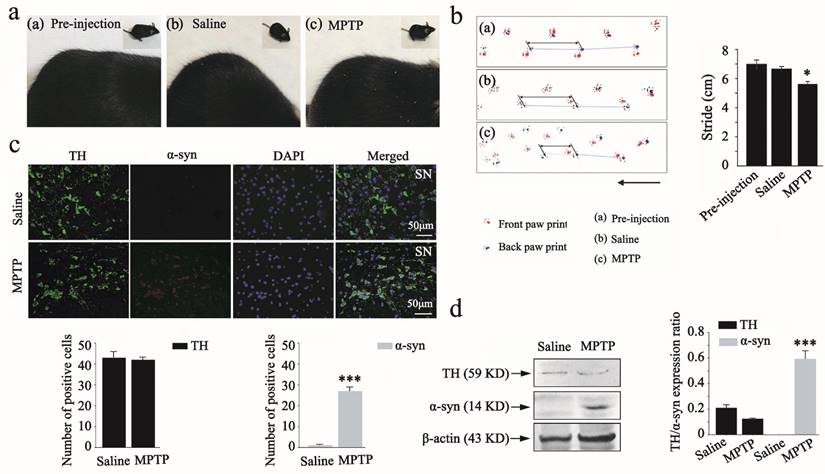
Parkinson disease animal model constructing. (a), Photos for C57BL/6 mice fur in the pre-injection, saline, and MPTP administration groups. (b), Back stride and front stride measures of walking gait in the pre-injection, saline and MPTP administration groups. The Student's t test was used for walking gait in comparison to pre-injection group. (c), Immunofluorescence for TH (green) and α-syn (red) in substantia nigra of saline and MPTP administrated groups, and DAPI (blue) staining for nucleus. The number of positive cells was determined using Image pro-plus 6.0. (d), Protein expression of α-syn and TH by western blot analysis in the saline and MPTP groups. The test was plotted with the significance p < 0.05 indicated by *, 0.001 < p < 0.01 indicated by **, and p < 0.001 indicated by ***, in comparison to the saline group (n=8).
2.17. Open field test
Mice were maintained in a light quiet place for acclimatization for 10 min. All procedures were conducted in a square open field chamber (35 cm × 35 cm). Behavior was monitored via a grid of invisible infrared light beams on top of the chamber for 15 min as shown in Figure 4a-c. Data were collected and analyzed using analyzer software. The chamber was cleaned with 70% ethanol after each mouse completed a session.
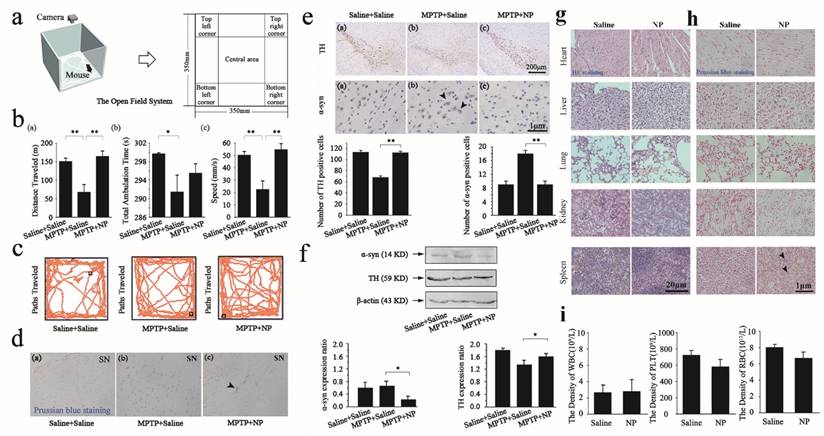
Measured efficacy and toxicities data in vivo. (a) Schematic representation of the open field system. (b-c), Distance traveled, total ambulation time, and speeds of mice (b) and paths traversed (c) in the open-field during consecutive 15min by saline+saline group mice, MPTP+saline group mice and MPTP+NP group mice. (d), Prussian blue staining of substantia nigra in three groups: saline+saline, MPTP+saline, and MPTP+NP. The white arrow means that the MNPs are distributed in substantia nigra. (e), Immunohistochemical for TH and α-syn in substantia nigra of three groups: saline+saline, MPTP+saline and MPTP+NP groups respectively. The black arrow means for α-syn positive cells. Number of positive cells determined using Image pro-plus 6.0. (f), Protein expression of α-syn and TH by western blot analysis in the saline+saline, MPTP+saline and MPTP+NP groups. The blots were re-probed to detect β-actin as a control to confirm equal protein loading. The relative levels are plotted at a significance of p<0.05 indicated by *, 0.001<p<0.01 indicated by **, and p<0.001 indicated by ***, in comparison to the MPTP+saline group. The bars stand for the standard deviations (n=8). (g), HE staining of heart, liver, spleen, lung and kidney in two groups: saline and NP. (h), Prussian blue staining of heart, liver, spleen, lung and kidney in two groups: saline and NP. (i), Serological and toxicology test of C57BL/6 mice, including the density of white blood cell (up), the density of platelet (middle), and the density of red blood cell (down) in two groups: saline and NP. The bars stand for the standard deviations (n=8).
2.18. Immunofluorescence
Brains were fixed in 4% paraformaldehyde and embedded in paraffin. The brains were then cut into 3 μm coronal sections with a paraffin microtome. Sections containing substantia nigra regions were subjected to immunostaining. Endogenous peroxidase activity was quenched by incubation in 1% hydrogen peroxide in methanol for 30 min and then cleared in PBS for 5 min. The sections were blocked for 30 min with bovine serum albumin (Sigma-Aldrich, BSA) diluted in PBS. These sections were incubated with primary antibody against tyrosine hydroxylase (TH) (Abcam, 1:400) and α-syn protein (Boster, 1:400) overnight at 4℃.
After washing in PBS, the sections were incubated in fluorescein isothiocyanate (FITC) and (1-(5-carboxypentyl)-1'-propylindocarbocyanine halide N-hydroxysuccinimidyl ester (Cy3) goat anti-rabbit IgG antibody (Boster, 1:1,000) for 1 h at room temperature (25℃). The sections were subsequently washed with PBS and viewed under a fluorescence microscope (NIKON, Ti-U, Japan), as shown in Figure 3c.
2.19. Western blot for animal studies
After the animal studies, brain tissues of the mice were harvested, washed with cold PBS [10 mmol·L-1 (pH 7.4)], and lysed with ice-cold lysis buffer for 1 h on ice. The lysates were centrifuged in 13,000 rpm for 10 min at 4℃ and the supernatants were used for western blot analysis. The protein expression in vivo was also assessed using the antibodies to TH and α-syn. The results are summarized in Figure 3d and Figure 4f.
2.20. Prussian blue staining
Paraffin embedded brain tissue was placed in water. Hydrochloric acid and potassium ferrocyanide solution at a volume ratio was freshly prepared as the working solution. The sections were immersed in working solution for 10-30 min. After the reaction, the sections were immersed in distilled water for 3 times for 3 min each, and then rinsed thoroughly. Nuclear fast red solution was added to the specimens, which were then covered for 5-10 min. After the reaction, the sections were immersed in distilled water for 3 times for 3 min each. The sections were then immersed in 95% alcohol and xylene (Aladdin) for 2 min for dehydration. Mounting medium was placed on the sections and covered with a coverslip. The distribution of NPs assessed by Prussian blue (Yeasen) staining was observed under a microscope as shown in Figure 4d,h.
2.21. Immunohistochemistry
Brains were fixed in 4 % paraformaldehyde and embedded in paraffin. The brains were cut into 3 μm coronal sections with a paraffin microtome. Sections containing substantia nigra regions were subjected to immunostaining. Endogenous peroxidase activity was quenched by incubation in 1% hydrogen peroxide in methanol for 30 min and then cleared in PBS for 5 min. The sections were blocked for 30 min with BSA diluted in PBS. These sections were incubated with primary antibody against TH (Abcam, 1:400) and α-syn protein (Boster, 1:400) for overnight at 4℃. After washing with PBS, the sections were incubated in biotinylated goat anti-rabbit IgG antibody (1:1,000) for 1 h at room temperature (25℃), and subsequently washed with PBS and viewed under a light microscope (NIKON, Ti-U Japan), as shown in Figure 4e.
2.22. Hematoxylin eosin (HE) staining
Five normal tissues (heart, spleen, lung, kidney, and liver) were dissected. The tissues were fixed for 12 h with 10% neutral buffered formalin fixative, and 80% ethanol containing a small amount of eosin (ScyTek) was added for 1 min. A set of graded alcohols (95%, 95%, 100%, 100%, 100%) was used to dehydrate the tissues for 15 min. These tissues were then dipped in liquid paraffin 3 times for 30 min each, frozen, and removed from the plastic centrifuge tubes. The maximum surface of the tissues was embedded in paraffin wax, and then 3 μm serial sections were obtained. Dewaxing was completed using xylene, and the samples were washed using graded ethanol and distilled water. HE staining was used to assess the morphology and NPs toxicity as showed in Figure 4g.
2.23. Serological detection
Sterilized scissors were used to obtain blood samples from the heads of NPs-treated mice. For each animal, 1.5 mL of blood was obtained and preserved in eppendorf (EP) tubes with anticoagulant. After collecting the blood, all samples were sent to the south hospital (Guangzhou, China) for serum evaluation, including the white blood cells (WBC), blood platelets (PLT), and red blood cells (RBC). The serological detection results are shown in Figure 4i.
2.24. Quantitative reverse-transcriptase PCR
Briefly, total ribonucleic acid (RNA) was extracted from 100 mg of midbrain tissue from the PD mouse. Reverse transcription was performed using the RevertAid First Strand cDNA Synthesis Kit (Thermo) with anchored-oligo(dT)18 primer. Gene expression was determined by ready-to-use amplification primer mixes for RT-PCR (Invitrogen Biotechnology Co., LTD) and FastStart Universal SYBR Green Master(Rox) (Roche Diagnostics). The expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was determined in the same way in all samples. For the relative quantification of α-syn, or TH gene expression, the expression of each target gene was normalized to the expression of GAPDH in the same sample.
The following oligonucleotide primers were used: M-GAPDH-S: 5'-GTTCCTACCCCCAATGTGTCC-3'; M-GAPDH-A: 5'-TAGCCCAAGATGCCCTTCAGT-3'; M-α-SY N-S: 5'-TGTCAAGAAGGACCAGATGGG-3'; M-α-SYN-A: 5'-TTTCATAAGCCTCACTGCCAG-3'; M-TH-S: 5'-CAGAAGAGCCGTCTCAGAGC-3'; M-TH-A 5'-CCTCGAATACCACAGCCTCC-3'.
2.25. Statistical analysis
Statistical results were obtained using the statistical software SPSS 17.0. One-way analysis of variance (ANOVA) was used to analyze statistical differences between groups under different conditions, and the Student's t-test was performed. P<0.05 was considered statistically significant (in vitro, n=3; in vivo, n=8).
3. Results
3.1. Synthesis and characterization of NGF, AzPhNGF, NP-NIPAm-AA-NGF and NP- NIPAm-AA-NGF (pDNA)
A schematic diagram of the substrate preparation for the initially preparation of magnetic nanometer Fe3O4 particle drugs is shown in Figure 1a,b. Three kinds of MNPs were prepared in our experiments: NP-NIPAm-AA, NP-NIPAm-AA-NGF, and NP-NIPAm-AA-NGF (pDNA).
The samples were prepared using the liquid photo-immobilization method [30]. First, we examined the chemical bonding between the functional groups of the drug molecules and NPs upon photo-immobilization using Fourier transform infrared spectroscopy (FTIR). The FTIR data for NGF, AzPhNGF and NP-NIPAm-AA-NGF are summarized in Figure 1e ((a)-(c)). The appearance of 2 peaks from the azido groups (~2382 cm-1) and a 1, 4-substituted benzene ring (~862 cm-1) in AzPhNGF indicated the successful synthesis of photoactive NGF. The absence of an azido group peak in NP-NIPAm-AA-NGF suggested that the liquid immobilization in the present scheme had been completed and that AzPhNGF had been successfully immobilized on the surface of NP-NIPAm-AA, because the azido groups of the photoactive NGF were very active under UV irradiation. These photoactive molecules will interact with nitrogen radicals and capture hydrogen from carbon to form C-N covalent bonds, leading to the disappearance of methyl and methylene.
Additional evidence for the chemical composition and bonding of NP-NIPAm-AA-NGF calculated from the electron spectroscopy for chemical analysis (ESCA) survey scan spectra are shown in Figure 1f. The volume fraction of C-N bonds for NP-NIPAm-AA has been shown to be enhanced from zero up to 10.09% after the surface modification step [31]. For NP-NIPAm-AA-NGF, the fitting analysis of chemical bonding revealed the addition of C-N bonds are added. Furthermore, the volume fraction of the C-N bonds reached 25.64% after the surface modification procedure, demonstrating that liquid immobilization was successfully completed and that attachment of AzPhNIPAm and AzPhNGF were attached to the NPs surface.
3.2. Hydrogel adsorption and release efficiencies
The pDNA and nanoparticles were sufficiently mixed in PBS for 3 days at 4 ℃. After centrifugation; the concentration of pDNA in the supernatant was detected using GeneQuant. The results showed that the loading dose of the hydrogels for pDNA was approximately 36%, as shown in Figure 1g.
Furthermore, a suitable amount of fully loaded pDNA nanoparticles (NP-NIPAm-AA (pDNA)) was stirred at 37℃ and 41℃ in PBS buffer solution. At different time intervals (0 min, 15 min, 30 min, 45 min, 1 h, 3 h, 6 h, 12 h, 24 h, 36 h and 48 h), 1 mL of solution was obtained from the mixture to collect the supernatant by high speed centrifugation. GeneQuant was then used to quantify the pDNA content in the supernatant. The release kinetics of loading the pDNA nanoparticles at 37℃ and 41℃ are shown in Figure 1h.
Based on the diagram, we can conclude that the magnetic hydrogel (NP-NIPAm-AA) released more pDNA at 41℃ than at 37℃ over 48 h. At the same time, the overall release rate of pDNA at 41℃ was 48.63%, which was higher than the total release rate observed at 37℃ (only 34.35%). However, the results indirectly demonstrated that the hydrogels had undergone successful grafting to the nanoparticles and that the magnetic hydrogels displayed good adsorption at an environmental temperature (e.g., 37℃) lower than the lower critical solution temperature (LCST). In contrast, when the environmental temperature was higher than the LCST (e.g., 41℃), a larger amount of pDNA was released from the hydrogels. These results provide strong evidence for the use of magnetic carrier hydrogel-loaded pDNA that is targeted for transport and controlled release.
3.3. Characterization of NP-NIPAm-AA-NGF and NP-NIPAm-AA-NGF (pDNA)
Subsequently, the morphology, size distributions and thermal stability of NP-NIPAm-AA-NGF and NP-NIPAm-AA-NGF (pDNA) were assessed. The SEM images and corresponding size distributions evaluated from the DLS data are presented in Figure 1c,d and Table 1. All of the NPs were almost round in morphology and displayed a homogeneous distribution. The NP-NIPAm-AA-NGF (pDNA) (~290 nm) displayed a larger size than NP-NIPAm-AA-NGF (~220 nm), indicating that these particles were successfully co-immobilized on NGF and embedded with pDNA. The thermal stability of the NPs was evaluated by measuring the percentage of weight loss during TGA, as shown in Figure 1i. TGA showed that NP-NIPAm-AA-NGF had a weight loss of 20.1wt% at temperatures below 350℃. However, the weight loss of NP-NIPAm-AA-NGF (pDNA) declined to 6.18wt% below 350℃ after the pDNA loading, demonstrating that the thermal stability of NP-NIPAm-AA-NGF (pDNA) was enhanced.
The mean particle size of different drug delivery systems.
| Particals | Fe3O4-OA(NP) | NP-NIPAm-AA | NP-NIPAm-AA-NGF | NP-NIPAm-AA-NGF(pDNA) |
|---|---|---|---|---|
| Z-Average (nm) | 111.846 | 216.008 | 224.600 | 290.400 |
| Particals | Fe3O4-OA(NP) | NP-NIPAm-AA | NP-NIPAm-AA-NGF | NP-NIPAm-AA-NGF(pDNA) |
| Z-Average (nm) | 111.846 | 216.008 | 224.600 | 290.400 |
3.4. Molecular mechanism of cell death in vitro
Next, we performed an experiment to evaluate the biological effects of the nano-biomaterials. In the study establishing the PD model in vitro, we screened out the optimum nano-drug concentration. Figure 2a ((a)-(c)) shows the different survival rates of MPP+-induced PC12 cells after 24 h, 72 h and 144 h. The diagram reveals that the cells had different degrees of apoptosis after exposure to MPP+. We selected an apoptosis rate of 40% and a 24 h drug concentration of 250 μmol·L-1 MPP+ per well for the in vitro cell model. Similarly, we chose 50 μmol·L-1 and 5 μmol·L-1 as the 72 h and 144 h drug concentrations, respectively.
To screen the optimal dose of nanoparticle-loaded pDNA, an experimental duration of 72 h was used, at which time the nanoparticles demonstrated the best efficacy. Based on the PC12 cell survival rate shown in Figure 2a (d), nanoparticles at a concentration of 0.01 μg·μL-1 pDNA clearly had the strongest ability to prevent PC12 cell apoptosis.
Subsequently, we investigated the expression of LDH in the cell cultures, and the results are summarized in Figure 2b. Indeed, the NP-NIPAm-AA-NGF (pDNA) groups displayed lower LDH expression levels compared with the NP-NIPAm-AA-NGF and control groups at both 72 h and 144 h. This result demonstrated that the cells treated with NP-NIPAm-AA-NGF (pDNA) had less damage in response to MPP+.
In addition, the morphology of the PC12 cells is shown in Figure 2c,d. Light microscopy revealed that the cells were undergoing programmed cell death, demonstrating highly condensed nuclear chromatin with chromatin blocks following exposure to NP-NIPAm-AA for 72 h and 144 h. However, the cell morphology improved in response to treatment with NP-NIPAm-AA-NGF. Moreover, the improved integrity, greater number of cells with improved morphology, and decrease in apoptotic bodies indicated that treatment with NP-NIPAm-AA-NGF (pDNA) better programmed cell repair than treatment with NP-NIPAm-AA and NP-NIPAm-AA-NGF.
Concomitantly, at 72 h and 144 h, the treated control group (NP-NIPAm-AA) and NP-NIPAm-AA-NGF displayed cell mortality rates of 33.2% and 37.6% and of 20.7% and 16.4%, respectively, while those for NP-NIPAm-AA-NGF (pDNA) were only 9.4% and 10%, respectively, as shown in Figure 2d. These results demonstrated the superior performance of NP-NIPAm-AA-NGF (pDNA) compared with the other treatments for the inhibition of PC12 cell apoptosis induced by MPP+. Moreover, nanoparticles had better effects in preventing cell apoptosis at 72 h than at 144 h. The accumulation of cellular metabolites could promote cell apoptosis at 144 h.
The blank plasmid control experiment illustrated that pDNA could successfully interfere with the synthesis of α-syn successfully after being delivered into PC12 cells at 72 h and 144 h. Figure 2e indicates that NP-NIPAm-AA-NGF (pDNA) could significantly down-regulate the expression of α-syn compared with the other 3 groups. However, NP-NIPAm-AA-NGF (empty pDNA) and NP-NIPAm-AA-NGF treatments induced a greater down-regulation of α-syn than the control groups due to the ability of NGF to reduce α-syn expression to some extent.
We also analyzed the activity of NGFR (NGF receptor), Bax, Bcl-2 and P53 in response to treatment with the 3 nanoparticles at 72 h and 144 h, respectively (as shown in Figure 2f). The expression levels of NGFR and Bcl-2 (75 kDa and 26 kDa, respectively) showed a more significant up-regulation of NP-NIPAm-AA-NGF (pDNA) at 72 h and 144 h in comparison to the other 2 groups. The NGFR data suggested a greater activation of the NGF control capability on the outer membrane of PC12 cells in response to NP-NIPAm-AA-NGF (pDNA). However, NP-NIPAm-AA-NGF (pDNA) treatment induced a greater down-regulation of Bax and P53 (22 kDa and 53 kDa, respectively) at 72 h and 144 h. These results indicated that NP-NIPAm-AA-NGF (pDNA) may interfere with the expression of α-syn and subsequently hinder programmed cell death by up-regulating the expression of Bcl-2 and down-regulating the expression of Bax and P53.
3.5. Activities and toxicities in vivo
To investigate whether the MPTP-induced chronic PD model construction was successful, we observed morphological changes in the fur of mice and performed the walking gait test. Several significant symptoms were observed in the MPTP administration group, such as raised and rough fur (Figure 3a). When the walking gait test was conducted, the results showed that MPTP administration induced a slight reduction in stride length. Statistically significant differences were also observed between the pre-injection and MPTP group (Figure 3b).
We further examined the population of TH and α-syn positive dopaminergic neurons in mouse substantia nigra by immunofluorescence analysis. The double staining showed a reverse correlation between TH and α-syn expression in the substantia nigra after MPTP administration (Figure 3c). There was also a statistically significant difference between saline and MPTP group. These results indicated that the MPTP-induced chronic PD model was successful. To further support the reliability of the MPTP-induced PD model, we also analyzed the activity of TH and α-syn in response to saline and MPTP administration by western blot analysis (as shown in Figure 3d). The expression levels of TH (59 kDa) showed a slight down-regulation in the MPTP group in comparison to the saline group. However, MPTP administration induced a significant up-regulation of α-syn (14 kDa) in comparison to the saline group. An additional statistical analysis was also performed to assess changes in the expression of TH and α-syn after MPTP administration, as determined by western blot analysis.
After the PD model was successfully established, we performed comparative efficacy studies by dividing the animals into 3 groups including 1) saline i.p. + saline i.p., 2) MPTP i.p. + saline i.p., and 3) MPTP i.p. + NP i.p..
Firstly, the overall locomotor activity and spatial distribution of mice were observed in an open field for 15min. The open field system consists of a box that is separated into different areas and equipped with a camera. Figure 4a shows the open field system, which contains different areas in which mouse behaviors are recorded. As shown in Figure 4b (a), the MPTP+saline mice exhibited a significant reduction of locomotion distance compared with the saline+saline group, which indicated that MPTP induced PD in mice. A significant difference was also observed between the MPTP+saline group and the MPTP+NP treated group in the distance travelled after NPs surgery. Mice in the MPTP+NP group were more active than those in the MPTP+saline group in the open field. The same differences were also reflected in the total ambulation time and speed (as shown in Figure 4b (b) and (c)). Moreover, Figure 4c shows the paths traveled by the saline+saline group, MPTP+saline group and MPTP+NP group in the open field. The results indicated that NPs had the potential to improve PD motor dysfunction.
The BBB is consistently the main obstacle in the treatment of the central nervous system (CNS). We performed Prussian blue staining to confirm the presence of NPs in the substantia nigra as proof that our NPs could cross the BBB. The blue color of ther NPs and the red color of the cell nucleus is evident in Figure 4d. According to the images, Prussian blue staining of NPs was observed in the NPs treated groups, confirming that the NPs had been effectively delivered across the BBB to the brain (Figure 4d (c)).
Furthermore, we performed immunohistochemical staining and western blot analysis of TH and α-syn in the 4 groups to evaluate the efficacy of NPs. Immunohistochemical staining of TH and α-syn in the substantia nigra is shown in Figure 4e. A decrease in the population of TH-positive dopaminergic neurons and an increase in the number of α-syn positive dopaminergic neurons were observed in the MPTP+saline group, revealing that MPTP induced overexpression of α-syn and down-regulation of TH as a result of dopaminergic neuronal damage. However, the number of TH-positive cells indicated a significant increase in the MPTP+NP group. Concomitantly, there was a significant decrease in α-syn-positive neurons that were treated with NPs. These results indicated that NPs had the potential to prevent dopamine neurons degeneration, as reflected by the up-regulation of TH and the down-regulation of α-syn. In agreement with the immunohistochemical staining results, an up-regulation of TH and a down-regulation of α-syn in brain tissue were also determined by western blot analysis (Figure 4f).
We further assessed the toxicity in the saline and NP groups in vivo by HE staining and Prussian blue staining of the heart, liver, spleen, lung and kidney tissues, and by counting the WBC, PLT, and RBC, respectively. As shown in Figure 4g, HE staining revealed that NPs could not cause organ damage, which indicated that they were safe for PD treatment. Next, we presented the obtained results from the Prussian blue staining to show the distribution of NPs in different organs. The blue color for NPs and the red color for cell nuclei are evident in the Figure 4h, which revealed a minimal NP distribution in organs other than the spleen. Moreover, previous research had shown that the liver and spleen are considered 2 dominant organs for the biodistribution and metabolism of nanoparticles [32]. To quantify the toxicity of NPs, the next important step was the assessment of standard hematologic parameters. The assessment of the WBC, PLT, and RBC counts in the NPs treated group on day 12 provided weak evidence for leukopenia or associated toxicities (Figure 4i).
To assess whether the efficacy of NPs alters the time of NPs treatment, we used quantitative real-time PCR (qPCR) to measure the mRNA expression of α-syn and TH genes in various groups of mice treated for 5 days, 10 days and 15 days. The results are summarized in Table 2. Indeed, NPs caused a remarkable decrease in the mRNA levels of α-syn and enhancement of TH mRNA levels compared with the MPTP + saline group. These results indicated that the efficacy of the treatment improved with increasing durations.
The expression levels of α-syn and TH via treated by NPs or not in the midbrain of Parkinson mouse model.
| Time | 5 days | 10 days | 15 days | |||
|---|---|---|---|---|---|---|
| Groups | MPTP i.p.+saline i.p | MPTP i.p. +NP i.p. | MPTP i.p.+saline i.p | MPTP i.p.+NP i.p. | MPTP i.p.+saline i.p | MPTP i.p.+NP i.p. |
| α-syn | 0.96 | 0.84* | 1.14 | 0.96 | 1.25 | 0.26** |
| TH | 1.43 | 16.39*** | 0.90 | 4.4*** | 0.20 | 1.20** |
The relative levels are plotted at a significance of 0.001<p<0.01 indicated by **, and p<0.001 indicated by ***, in comparison to the MPTP+saline group, n=8.
4. Discussion
In recent years, nanocarriers have been developed for the targeted transport of genes in gene therapy [33]. RNAi has been widely applied for tumors, virus infection, nervous system disease and other diseases as an effective gene therapy technique [34,35]. In addition, the limitations of traditional PD treatments necessitate the discovery of new and effective therapeutic interventions [36,37]. Therefore, the successful design of a targeted gene therapy system will provide a highly efficient and low-toxicity treatment method for PD.
Three main methods have been applied for the treatment of PD, including surgical treatment, drug therapy and gene therapy [38,39]. Traditional PD treatments still depend on dopamine replacement therapy (DRT), and levodopa is the only drug to effectively extend life expectancy [40]. However, highly disabling fluctuations and sleep attacks induced by the agonist are serious side effects associated with the long-term use of DRT [41,42]. SNCA is a therapeutic target for PD in gene therapy [43]. Therefore, several new methods to suppress SNCA gene expression have been explored. For example, natural substances (cinnamon extract precipitation), peptides, prolyl oligopeptidase inhibitors, siRNA and shRNA have been examined as candidates for the treatment of PD [44-46].
A central challenge of gene therapy associated with successful RNA interference involves techniques to efficiently deliver genetic carriers to target cells without inducing significant toxicity and side effects [47]. Viral vectors are widely used in gene therapy. However, studies have revealed that viral vectors for the treatment of many neurological diseases have dangerous adverse effects: they can apparently trigger potent immunogenicity, mutagenesis, and other biohazards, weakening their application [48]. A recent topic has focused on new non-viral vectors. Nanoparticles display prominent superiority for gene therapy in nervous system diseases; they are able to specifically target cells, avoid gene integration and are less immunogenic [49]. However, magnetic nanoparticles used as gene delivery system have rarely been reported, particularly in PD.
In the present work, we took advantage of the characteristics and performance of multifunctional magnetic nanoparticles that were functionalized with NGF (small particle size, large specific surface, coupling of high capacity, magnetic response, superparamagnetic, and NGF receptor-mediated endocytosis). Their nontoxic nature and potential for nanoparticle synthesis combined with RNA interference technology makes these particles an ideal targeted gene therapy system for PD.
Patients with PD present degenerative apoptosis in the substantia nigra pars compacta, and the pH of the cytoplasmic matrix of apoptotic cells is lower than that in normal tissue cells, demonstrating intracellular acidification. The pH sensitivity of the hydrogel can automatically direct hydrogel accumulation at the apoptotic cell surface to achieve therapeutic targeting [50-52]. In addition, because the acidified intracellular environment (pH of approximately 5.0-6.0) is lower than that of the extracellular milieu, the hydrogel can release shRNA via changes in hydrogel volume.
In this study, we developed a novel inner/outer nanoparticle that carries shRNA plasmid to interfere with α-syn synthesis. We demonstrated that these superparamagnetic nanoparticles can provide effective repair in a PD model in vitro and in vivo and further inhibit apoptosis. As a result, α-syn expression is reduced, thus preventing the toxic effects of α-syn on the cell and suppressing apoptosis. Our results are consistent with the present experiments and the desired purpose of the study. Further investigations should focus on elucidating the underlying mechanisms and signal transduction pathways associated with these novel and powerful magnetic nanoparticles.
5. Conclusions
In summary, we developed a novel inner/outer doubt-targeted nanoparticle (NP-NIPAm-AA-NGF(pDNA)) with the size of 290 nm via co-precipitation, self-assembled, photo-immobilization, and physical adsorption, which carried shRNA plasmid to interfere with α-synuclein. We demonstrated that these superparamagnetic nanoparticles can provide effective repair in a PD model in vitro and in vivo and inhibit further apoptosis. Future investigations should focus on elucidating the underlying mechanisms and signal transduction pathways associated with these novel and powerful magnetic nanoparticles.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31170919, 31370967), the Science and Technology Planning Project of Guangdong Province (No.2015A020212033), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2014), China, the Innovation Project of Graduate School of South China Normal University (2014ssxm24), China, and the Innovative Entrepreneurial Training for college students of South China Normal University (234).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ozansoy M, Başak AN. The central theme of Parkinson's disease: alpha-synuclein. Mol Neurobiol. 2013;47:460-5
2. Desplats P, Patel P, Kosberg K, Mante M, Patrick C, Rockenstein E. et al. Combined exposure to Maneb and Paraquat alters transcriptional regulation of neurogenesisrelated genes in mice models of Parkinson's disease. Mol Neurodegener. 2012;7:49-58
3. Vekrellis K, Rideout HJ, Stefanis L. Neurobiology of alpha-synuclein. Mol Neurobiol. 2004;30:1-21
4. Agid Y. Parkinson's disease: pathophysiology. Lancet. 1991;337:1321-4
5. Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson's disease. Annu Rev Genom Hum G. 2011;12:301-25
6. Bekris LM, Mata IF, Zabetian CP, Geriatr J. The genetics of parkinson disease. Psychiat Neurol. 2010;23:228-42
7. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J. et al. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841
8. Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33-8
9. Oliveras-Salvá M, Van der Perren A, Casadei N, Stroobants S, Nuber S, Hooge RD. et al. rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol Neurodegener. 2013;8:44-57
10. Allen PJ, Feigin A. Gene-based therapies in Parkinson's disease. Neurotherapeutics. 2014;11:60-7
11. Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. P Natl Acad Sci USA. 2009;106:13052-7
12. Khodr CE, Sapru MK, Pedapati J. An α-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson's disease, but displays toxicity in dopamine neurons. Brain Res. 2011;1395:94-107
13. Witt J, Marks WJ. An update on gene therapy in Parkinson's disease. Curr Neurol Neuro Sci. 2011;11:362-70
14. Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP. et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148-58
15. Maguire CA, Ramirez SH, Merkel SF, Sena-Esteves M, Breakefield XO. Gene therapy for the nervous system: challenges and new strategies. Neurotherapeutics. 2014;11:817-39
16. Garcia-Garcia E, Andrieux K, Gil S, Couvreur P. Colloidal carriers and blood-brain barrier (BBB) translocation: a way to deliver drugs to the brain. Int J Pharm. 2005;298:274-92
17. Stojanov K, Zuhorn I, Dierckx R. Imaging of cells and nanoparticles: implications for drug delivery to the brain. Pharm Res-dordr. 2012;12:3213-34
18. Hua MY, Liu HL, Yang HW, Chen PY, Tsai RY, Huang CY. et al. The effectiveness of a magnetic nanoparticle-based delivery system for BCNU in the treatment of gliomas. Biomaterials. 2011;32:516-27
19. Sun C, Du K, Fang C, Bhattarai N, Veiseh O, Kievit F. et al. PEG-mediated synthesis of highly dispersive multifunctional superparamagnetic nanoparticles: their physicochemical properties and function in vivo. ACS Nano. 2010;4:2402-10
20. Veiseh O, Gunn JW, Zhang M. Design and fabrication of magnetic nanoparticles for targeted drug delivery and imaging. Adv Drug Deliver Rev. 2010;62:284-304
21. Namiki Y, Namiki T, Yoshida H, Ishii Y, Tsubota A, Koido S. et al. A novel magnetic crystal-lipid nanostructure for magnetically guided in vivo gene delivery. Nat Nanotechnol. 2009;4:598-606
22. Nunes J, Herlihy KP, Mair L, Superfine R, DeSimon JM. Multifunctional shape and size specific magneto-polymer composite particles. Nano Lett. 2010;10:1113-9
23. Zhang F, Wang CC. Preparation of P (NIPAm-co-AA) microcontainers surface-anchored with magnetic nanoparticles. Langmuir. 2009;25:8255-62
24. Gong Y, Dai J, Li H, Wang X, Xiong H, Zhang Q. et al. Magnetic, fluorescent, and thermo-responsive poly(MMA-NIPAM-Tb(AA)3Phen)/Fe3O4 multifunctional nanospheres prepared by emulsifier-free emulsion polymerization. J Biomater Appl. 2015;30:201-11
25. Wan J, Cheung AY, Fu WY, Wu C, Zhang M, Mobley WC. et al. Endophilin B1 as a novel regulator of nerve growth factor/TrkA trafficking and neurite outgrowth. J Neurosci. 2008;28:9002-12
26. Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Therape. 2013;138:155-75
27. De Oliveria DM, Barreto G, de Andrade DV, Saraceno E, Aon-Bertolino L, Capani F. et al. Cytoprotective effect of Valeriana officinalis extract on an in vitro experimental model of Parkinson disease. Neurochem Res. 2009;34:215-20
28. Wu KC, Lu YH, Peng YH, Tsai TF, Kao YH, Yang HT. et al. Decreased expression of organic cation transporters, Oct1 and Oct2, in brain microvessels and its implication to MPTP-induced dopaminergic toxicity in aged mice. J Cerebr Blood F Met. 2015;35:37-47
29. Vernice JL, Serge P. Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc. 2007;2:141-51
30. Guan YQ, Zheng Z, Huang Z, Li Z, Niu S, Liu JM. Powerful inner/outer controlled multi-target magnetic nanoparticle drug carrier prepared by liquid photo-immobilization. Sci Rep. 2014;4:4990-8
31. Zhan XY, Guan YQ. Design of magnetic nanoparticles for hepatocellular carcinoma treatment using the control mechanisms of cell internal nucleus and external membrane. J Mater Chem B. 2015;3:4191-204
32. Zhang XD, Wu HY, Wu D, Wang YY, Chang JH, Zhai ZB. et al. Toxicologic effects of gold nanoparticles in vivo by different administration routes. Int J Nanomed. 2010;5:771-81
33. He Q, Zhang J, Shi J, Zhu Z, Zhang L, Bu W. et al. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials. 2010;31:1085-92
34. Rosenholm JM, Sahlgren C, Linden M. Towards multifunctional targeted drug delivery systems using mesoporous silica nanoparticles-opportunities & challenges. Nanoscale. 2010;2:1870-83
35. Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T. et al. Codelivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrugresistant cancer cells. Small. 2009;5:2673-7
36. Li F, Harmer P, Fitzgerald K, Eckstrom E, Stock R, Galver J. et al. Tai chi and postural stability in patients with Parkinson's disease. New Engl J Med. 2012;366:511-9
37. Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT. et al. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's diseases. P Natl Acad Sci USA. 2006;103:4246-51
38. Barker RA, Barrett J, Mason SL, Bjorklund A. Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson's disease. Lancet Neurol. 2013;12:84-91
39. Schapira AH, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet. 2014;384:545-55
40. Cao XB, Guan Q, Xu Y, Wang L, Sun SG. Mechanism of over-activation in direct pathway mediated by dopamine D (1) receptor in rats with levodopa-induced dyskinesia. Neurosci Bull. 2006;22:159-64
41. Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A. et al. Levodopa and the progression of Parkinson's disease. New Engl J Med. 2004;351:2498-508
42. Hobson DE, Lang AE, Martin WR, Razmy A, Rivest J, Fleming J. Excessive daytime sleepiness and sudden-onset sleep in Parkinson disease: a survey by the Canadian Movement Disorders Group. JAMA. 2002;287:455-63
43. Chesselet MF, In vivo alpha-synuclein overexpression in rodents. a useful model of Parkinson's disease. Exp Neurol. 2008;209:22-7
44. Shaltiel-Karyo R, Davidi D, Frenkel-Pinter M, Ovadia M, Segal D, Gazit E. Differential inhibition of α-synuclein oligomeric and fibrillar assembly in parkinson's disease model by cinnamon extract. BBA-Biomembranes. 2012;1820:1628-35
45. Myöhänen TT, Hannula MJ, Elzen RV, Gerard M, Van Der Veken P, García-Horsman JA. et al. A prolyl oligopeptidase inhibitor, KYP-2047, reduces alpha-synuclein protein levels and aggregates in cellular and animal models of Parkinson's disease. Br J Pharmacol. 2012;166:1097-113
46. Khodra CE, Sapru MK, Pedapati J, Han Y, West NC, Kells AP. et al. An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson's disease, but displays toxicity in dopamine neurons. Brain Res. 2011;1395:94-107
47. Liu YY, Yang XY, Li Z, Liu L, Cheng D, Wang Y. et al. Characterization of polyethylene glycol-polyethyleneimine as a vector for alpha-synuclein siRNA delivery to PC12 cells for Parkinson's disease. CNS Neurosci Ther. 2014;20:76-85
48. Huang PI, Lo WL, Cherng JY, Chien Y, Chiou GY, Chiou SH. Non-viral delivery of RNA interference targeting cancer cells in cancer gene therapy. Curr Gene Ther. 2012;12:275-84
49. Liang Y, Liu Z, Shuai X, Wang W, Liu J, Bi W. et al. Delivery of cationic polymer-siRNA nanoparticles for gene therapies in neural regeneration. Biochem Biophy Res Co. 2012;421:690-5
50. Akiyama Y, Mori T, Katayama Y. et al. The effects of PEG grafting level and injection dose on gold nanorod biodistribution in the tumor-bearing mice. J Control Release. 2009;139:81-4
51. Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK. Delivery of molecular and nanoscale medicine to tumors: Transport barriers and strategies. Annu Rev Chem Biomol. 2011;2:281-98
52. Hoare T, Timko BP, Santamaria J, Goya GF, Irusta S, Lau S. et al. Magnetically triggered nanocomposite membranes. a versatile platform for triggered drug release. Nano Lett. 2011;11:1395-400
Author contact
![]() Corresponding author: Chuhua Li, School of Life Science, South China Normal University, Guangzhou 510631, China. E-mail address: lichedu.cn (C. H. Li). Yan-Qing Guan, College of Biophotonics, South China Normal University, Guangzhou 510631, China. Tel.: (+86-20)85211241; E-mail address: guanyqedu.cn (Y. Q. Guan).
Corresponding author: Chuhua Li, School of Life Science, South China Normal University, Guangzhou 510631, China. E-mail address: lichedu.cn (C. H. Li). Yan-Qing Guan, College of Biophotonics, South China Normal University, Guangzhou 510631, China. Tel.: (+86-20)85211241; E-mail address: guanyqedu.cn (Y. Q. Guan).