Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(2):452-465. doi:10.7150/thno.16677 This issue Cite
Research Paper
Dual-targeted and pH-sensitive Doxorubicin Prodrug-Microbubble Complex with Ultrasound for Tumor Treatment
Wanxian Luo1,2*, Ge Wen3*, Li Yang4, Jiao Tang2, Jianguo Wang2, Jihui Wang2, Shiyu Zhang1, Li Zhang1, Fei Ma1, Liling Xiao1, Ying Wang2 ![]() , Yingjia Li1
, Yingjia Li1 ![]()
1. Department of Medicine Ultrasonics, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China;
2. Guangdong Provincial Key laboratory of cancer immunotherapy research, Cancer Research Institute, School of Basic Medical Science, Southern Medical University, Guangzhou 510515, China;
3. Imaging center, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China;
4. Department of Pharmacy, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China.
*These authors contributed equally to this work.
Received 2016-7-1; Accepted 2016-10-25; Published 2017-1-5
Abstract

In this study, we investigated the potential of a dual-targeted pH-sensitive doxorubicin prodrug-microbubble complex (DPMC) in ultrasound (US)-assisted antitumor therapy. The doxorubicin prodrug (DP) consists of a succinylated-heparin carrier conjugated with doxorubicin (DOX) via hydrazone linkage and decorated with dual targeting ligands, folate and cRGD peptide. Combination of microbubble (MB) and DP, generated via avidin-biotin binding, promoted intracellular accumulation and improved therapeutic efficiency assisted by US cavitation and sonoporation. Aggregates of prepared DP were observed with an inhomogeneous size distribution (average diameters: 149.6±29.8 nm and 1036.2±38.8 nm, PDI: 1.0) while DPMC exhibited a uniform distribution (average diameter: 5.804±2.1 μm), facilitating its usage for drug delivery. Notably, upon US exposure, DPMC was disrupted and aggregated DP dispersed into homogeneous small-sized nanoparticles (average diameter: 128.6±42.3 nm, PDI: 0.21). DPMC could target to angiogenic endothelial cells in tumor region via αvβ3-mediated recognition and subsequently facilitate its specific binding to tumor cells mediated via recognition of folate receptor (FR) after US exposure. In vitro experiments showed higher tumor specificity and killing ability of DPMC with US than free DOX and DP for breast cancer MCF-7 cells. Furthermore, significant accumulation and specificity for tumor tissues of DPMC with US were detected using in vivo fluorescence and ultrasound molecular imaging, indicating its potential to integrate tumor imaging and therapy. In particular, through inducing apoptosis, inhibiting cell proliferation and antagonizing angiogenesis, DPMC with US produced higher tumor inhibition rates than DOX or DPMC without US in MCF-7 xenograft tumor-bearing mice while inducing no obvious body weight loss. Our strategy provides an effective platform for the delivery of large-sized or aggregated particles to tumor sites, thereby extending their therapeutic applications in vivo.
Keywords: Dual-targeted, pH-sensitive, Doxorubicin prodrug, Microbubble complex, Ultrasound.
Introduction
Doxorubicin hydrochloride (DOX), an anticancer agent belonging to the anthracycline class, is a leading clinical cytotoxic drug for breast cancer. However, the therapeutic efficacy of free DOX is compromised by various side-effects, including severe cardiotoxicity, nephrotoxicity and myelosuppression [1]. Development of novel strategies to improve therapeutic efficacy and reduce side-effects is therefore crucial for the successful treatment of breast cancer.
The pH value of normal tissue is around 7.4, while that of tumor tissue is as low as 6.0, due to hypoxia and high lactate metabolism of the tumor microenvironment [2, 3]. Here, we prepared a pH-sensitive prodrug composed of DOX and dual-targeted ligands, cRGD and folate, using heparin as the backbone. cRGD was decorated with polyethyleneglycol (PEG), with the aim of extending circulating time and escaping the reticuloendothelial system (RES) in vivo [4]. Through recognition of αvβ3, the prodrug could specifically target to angiogenic endothelial cells in tumor region, and subsequently, selectively bind to tumor cells via recognition of FR after US exposure [5-7]. Simultaneously, DOX was conjugated to heparin via a pH-sensitive hydrazone linkage to facilitate its release in the tumor microenvironment [8]. Although DP with high drug-loading capability exerts a greater anti-cancer effect than free DOX in vitro, DP is inclined to aggregate and enlarge in size, which is unfavorable for drug dispersion and presents a significant obstacle for penetration into solid tumors.
Ultrasound-targeted microbubble destruction (UTMD) is an adjuvant modality for drug delivery to localize intratumoral drug release and enhance intracellular drug accumulation. The inertial acoustic cavitation of microbubbles (MBs), including bubble implosion, microstreaming, shock waves and microjets, causes sonoporation (pore forming), which greatly improves intracellular uptake of drugs at the target site [9]. Aggregation is a common issue during preparation of the nanoparticle drug delivery system due to the interaction force between particles, which may limit its penetration into solid tumors. In view of previous study that nanoparticles can be fragmented into smaller pieces under laser irradiation to promote drug release [10], we combined aggregated DP and MB and explored whether these large-sized drug-loaded particles could be disrupted to facilitate intracellular uptake into tumor cells, assisted by US as an external force.
In the current study, aggregated dual-targeted pH-sensitive DP was conjugated with MB via an avidin-biotin bridge to generate a DOX prodrug-MB complex (DPMC). We examined the morphological changes of DPMC before and after US destruction, then focused on validating its tumor targeting specificity and imaging ability using in vivo fluorescence and ultrasound molecular imaging analyses. In particular, the anti-tumor efficacy of the complex with and without US was evaluated, both in vitro and in vivo. For prodrugs with significant cytotoxicity but relatively larger sizes, the newly generated complex assisted by US represents a promising approach to decrease size and integrate tumor imaging and therapy, providing an alternative therapeutic anti-tumor strategy.
Materials and methods
Materials
Doxorubicin hydrochloride (DOX) was purchased from Sangon Biotech Co., Ltd. (Shanghai, China). Heparin sodium salt (Mn=1.25 kDa, 189 U/mg) was obtained from Sinopharm Chemical Reagent Co. (Shanghai, China) and succinylated in our lab. Adipic dihydrazide (ADH), 1-ethyl-3,3-dimethylaminopropylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Medpep Co. (Shanghai, China). Polyethylene glycol (PEG, MW 5069 Da) was synthesized by AppliChem Co. (Darmstadt, Germany). Cyclic RGD peptide (cRGD) was synthesized by Bohin biotechnology Co. Ltd. (Xiamen, China). Cyanine5.5 amine (Cy5.5) was purchased from Lumiprobe LLC (Florida, USA). Dialysis Membrane (MWCO 3,500) was obtained from Pharmacia (Piscataway, NJ, USA). Folate-NH2 and Biotin-NH2 were prepared in our laboratory. Biotinylated MBs were donated by the Department of Pharmacy (Nanfang Hospital, China). Human breast carcinoma cell line MCF-7 and lung cancer cell line A549 were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China). All other chemicals and reagents were purchased from Sigma Co. (St. Louis, MO, USA).
Synthesis of the DOX prodrug and DOX prodrug-MB complex
Synthesis of the DOX prodrug
PEG-cRGD was prepared using a previously described method. Briefly, PEG (506.9 mg, 0.1 mmol) and cRGD (75 mg, 0.2 mmol) were dissolved in dry dimethylfumarate (DMF) with the addition of EDC (40 mg, 0.2 mmol) and reacted with catalytic triethylamine overnight at 50°C. The mixture was evaporated to remove DMF. Trifluoroacetic acid (TFA) was added and reacted at room temperature for 1 h, the solution evaporated, and neutralized with triethylamine. The mixture was re-evaporated and the product obtained.
To prepare Heparin-Folate-PEG-cRGD-DOX prodrugs, succinylated-heparin (50 mg) was stirred in dry dimethyl sulfoxide (DMSO) with gentle heating. Folate-NH2 (6 mg), Biotin-NH2 (4.5 mg), PEG-cRGD (2 mg of cRGD), EDC (14 mg) and NHS (11 mg) were added and reacted at room temperature for 24 h. The mixture was dialyzed using a dialysis membrane for 48 h. Adipic acid dihydrazide (ADH; 2 mg, 0.01 mmol) and EDC (4 mg, 0.02 mmol) were added and reacted in water solution at room temperature for 6 h. Following the addition of corresponding amounts of DOX (6 mg, 0.01 mmol), the mixture was reacted under acid conditions overnight. The mixture was subsequently neutralized and subjected to membrane dialysis for a further 48 h, and the product obtained as lyophilized red powder.
Conjugation of biotinylated DOX prodrug to biotinylated MB
The complex was prepared with the aid of avidin-biotin staining using a previously described method [9]. Briefly, A given amount of avidin (final concentration of 0.3 mg/mL) per 108 MBs was added to biotinylated MBs and incubated on ice for 30 min. MBs were washed with double-distilled water three times to remove unreacted avidin, and the corresponding amounts of prodrugs added and incubated for a further 30 min. Complexes were washed 3-4 more times to remove free prodrugs. Cy5.5-labeled complexes were synthesized using a similar procedure.
Characterization of the DOX prodrug and DOX prodrug-MB complex
Characterization of the DOX prodrug
Particle size and zeta potential of DP were assessed using Dynamic laser light scattering (DLS, Zetasizer Nano-ZS, Malvern, UK). Samples were dissolved in deionized water to generate a solution with a final concentration of 0.1 mg/mL. Procedures were conducted at 25oC after equilibration for 10 s according to the manufacturer's instructions. Morphology and particle size of DP were further assessed via transmission electronic microscopy (TEM, Hitachi HC-1, Tokyo, Japan) with an accelerating voltage of 80 kV. Prior to examination, a drop of DP solution was deposited on carbon-coated formvar copper grids for 2 min. The grid was blotted with filter paper to remove excess aqueous solution and dried at room temperature. Samples were subsequently stained with phosphotungstic acid and air-dried. The 1H NMR spectra of DP were determined by a Bruker-400 MHz NMR in DMSO-d6 and D2O, while its FT-IR spectrum was analysed using infrared detection. To evaluate the drug loading content (DLC), DP was dissolved in PBS and analyzed via UV-vis spectrophotometry (UV-2401PC) using a standard curve constructed from DOX/PBS solutions with different DOX concentrations. UV-vis analysis was conducted at a wavelength of 480 nm and the whole procedure performed in the dark. DLC was calculated according to the following formula: DLC (%) = ((weight of loaded drug)/(total weight of DP))x100%. The weight of folate on DP was also calculated by UV spectrometer based on a folate standard curve of concentration-absorption at 280 nm. The loading content of cRGD was determined by a BCA protein kit at 570 nm. All values were calculated as the average of at least three independent samples.
Characterization of the DOX prodrug-MB complex
DPMC with US exposure was additionally characterized via dynamic light scattering (DLS) and transmission electron microscopy (TEM). Briefly, 1 mL DPMC in a 2.0 mL Eppendorf tube was placed in a water tank. Exposure was achieved with 20 mm US probe of a therapeutic US system (CZ906A, Chongqing Medical University, Chongqing, China), which was placed 2 cm away from the Eppendorf tube below the water surface. Irradiation parameters were set as follows: 1 MHz, 2 % duty cycle, duration of 1 min and US intensity of 1 W/cm2 [11, 12]. To characterize the morphologic differences between DPMC with and without US exposure, the suspension was mounted on a slide with a coverslip and visualized using confocal microscopy. The size distribution of DPMC was further estimated using a Coulter Multisizer (Beckman Coulter, Brea, California, USA). To determine the drug loading content of DPMC, the amount of DP that bound to MBs was estimated by removing unbound DP via centrifugation at 400 g for 3 min [9]. Unloaded DOX in the collected solution was quantified at 480 nm with a UV-Vis spectrophotometer, and the drug loading content calculated using the formula: DLC (%) = (total added drug - unloaded drug)/total amount of MB.
In vitro release of doxorubicin
The release profiles of DOX from DP were examined using dialysis [1]. Briefly, 5 mg of freeze-dried DP without US, DPMC with or without US exposure applied according to the above parameters was dissolved in 5 mL phosphate buffered saline solution (PBS) at different pHs (5.0 and 7.4) and sealed in a dialysis bag. Samples was dialyzed against 50 mL corresponding buffers and incubated in a horizontal shaker at 37oC under constant shaking (150 rpm). At predetermined time intervals, 2 mL of release medium was removed for drug concentration measurement and replenished with an equal volume of fresh medium. The amount of released DOX was analyzed at 480 nm under a UV/Vis spectrophotometer. Release of free DOX was conducted under the same conditions as control. All experiments were conducted in the dark and performed in triplicate.
Cell culture
The human breast carcinoma cell line MCF-7 and lung cancer cell line A549, were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China). Cells were cultured in RPMI 1640 (HyClone, Logan, UT, USA) supplemented with 10% (v/v) fetal bovine serum (FBS, HyClone), 100 IU/mL penicillin and 100 mg/mL streptomycin sulfate. All cells were cultured at 37oC in 5% CO2.
Cellular uptake
The cellular uptake behavior and the intracellular distribution of DOX and DOX-loaded prodrug were analyzed using both flow cytometry and confocal laser scanning microscopy (CLSM). For flow cytometry, MCF-7 cells were seeded into six-well culture plates at a density of 5×105 cells/well and incubated overnight. To investigate the cellular uptake of carrier with single targeted ligand and dual ligands, the medium was replaced with fresh medium containing H-F-DOX, H-RGD-DOX and H-F-RGD-DOX at a DOX concentration of 2 μg/mL. For quantitative evaluation of the cellular uptake of DOX, MCF-7 cells were incubated according to the above methods. The medium was replaced with fresh medium containing free DOX, DP and DPMC at a DOX concentration of 2 μg/mL. Simultaneously, cells treated with DPMC were irradiated with US as for the cytotoxicity assay. Untreated cells were used as the blank control. After a 4 h incubation period, cells were washed with cold PBS and harvested. The fluorescence intensity of DOX was measured using a flow cytometer (FACSCalibur, BD Biosciences, Franklin Lakes, NJ, USA) with a FL2-H filter. For CLSM studies, MCF-7 cells (1×105) were seeded onto glass coverslips placed in 6-well plates and cultured overnight. Cells were treated with free DOX, pH-insensitive prodrug, DP and DPMC with US for 4 h (DOX concentration 2 μg/mL). To evaluate the specific binding of DPMC to tumor cells with US exposure, FR-positive MCF-7 cells and FR-negative A549 cells (1×105) were seeded onto glass coverslips as the above method, then cells were incubated with small-sized nanoparticles from DPMC disrupted by US for 5 mins (DOX concentration 2 μg/mL). Then, cells were washed and fixed with 4% (w/v) para-formaldehyde followed by Hochest33342 staining. Coverslips were placed onto glass microscope slides, the fluorescence images were observed by confocal microscope (Olympus FV1000, Japan).
In vitro cytotoxicity assays
The cytotoxicity of prepared DP and DPMC with US was evaluated using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. MCF-7 cells were routinely cultured before seeding into 96-well plates at a density of 5×103 cells/well and incubated overnight. The medium was replaced with fresh medium containing free DOX, DP and DPMC to generate final DOX concentrations of 0.1, 0.25, 0.5, 1 and 3 μg/mL, respectively. Cells treated with the same number of MBs with US were used as control. Cells treated with DPMC and MBs were irradiated using US with the probe covered with coupling agent and placed on the bottom of the plate according to the above parameters. The medium was substituted with complete medium, followed by incubation for 48 h. The culture medium was removed and 20 μL MTT solution (dissolved in RPMI1640 to a final concentration of 0.5 mg/mL) added for another 4 h. Next, the medium was replaced with 150 μL dimethyl sulfoxide (DMSO) per well to dissolve formazan crystals generated by living cells, and absorbance monitored using a microplate reader (Bio-Tek ELx800) at a wavelength of 490 nm. Untreated cells were used as the control. Relative cell viability (%) was calculated by comparing absorbance with that of control cells. IC50 values were determined from the dose-effect curve and expressed as concentration (ng/mL) of DOX equivalency. Experiments were performed in triplicate.
Mouse tumor model
Female BALB/c nude mice (4-6 weeks old, 16-20 g) were purchased from the Department of Experimental Animals of Sun Yat-sen University (Guangdong, China), and received care in compliance with the guidelines of the Guide for the Care and Use of Laboratory Animals. All procedures were approved by the ethics committee of the Southern Medical University, Chinese Academy of Sciences. To generate the breast xenograft tumor model, MCF-7 cells (1×106) suspended in 50 µL PBS were injected subcutaneously into the flank region of each mouse.
Ultrasound molecular imagingImaging protocol
The imaging session was carried out with a Philips iU22 ultrasound system using a 10 MHz ultrasound probe, according to a previous protocol [13]. Briefly, prior to the imaging session, each animal was restrained on a flat platform and anesthetized with 10% hydral. The ultrasound probe was placed gently on the top of the tumor covered with 5 mm thick commercial diagnostic ultrasound gel and the orientation aligned along the longest axis of the tumor. After a single bolus injection of 0.2 mL blank MBs or DPMC (1 × 109/mL) through the tail vein, the process was continuously monitored via ultrasonography in B-mode with low MI value (0.07). After 4 min, targeted MBs generally adhered firmly to the tumor site. Blank MBs or DPMC were destroyed using the Flash mode of the ultrasound system with a relatively higher MI value (0.64). The process was monitored, starting from injection to at least 30 s after Flash destruction (Figure 5A). To minimize bias from repetitive injections in the same mice, injections were separated by at least 30 min to allow clearance of MBs from previous injections.
Schematic illustration of the image and analysis protocol (A). After monitoring for 4 min, targeted MBs were considered to adhere to the tumor site firmly. Blank MBs or DPMC were destroyed by the Flash mode of the ultrasound system. The process was monitored, starting from injection to at least 30 s after Flash destruction. The video intensity (VI) from adherent MBs (Targeted VI) was assessed by calculating the average pre- and post-destruction VI, and subtracting average post- from pre-destruction intensity. Ultrasound molecular images representing intensity from adherent blank MBs or DPMC displayed as color maps overlaid on B-mode images (B). Yellow line represents the region of interest (ROI). Scale bar represents 1 cm. Image signals are quantitatively presented (*** p<0.001) (C).
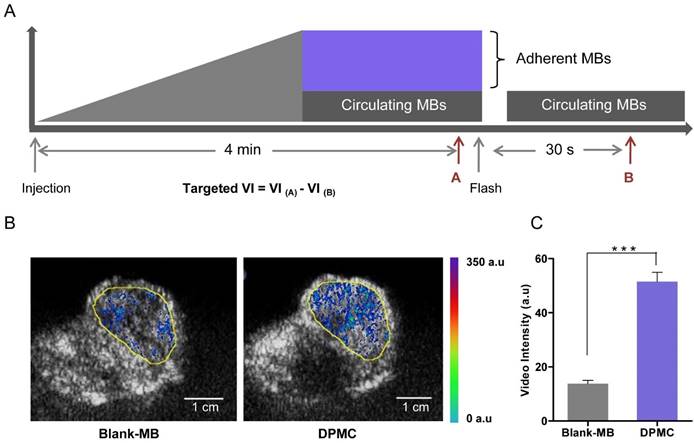
Imaging data analysis
After each imaging session, datasets from all mice were analyzed offline in a random order with commercially available software for myocardial contrast echocardiography (MCE, Florida, USA). Analysis was performed by one independent reader blinded to the type of MB (DPMC versus blank-MB). Regions of interest (ROI) were drawn over the tumor site and vedio intensity (VI) from adherent MB assessed by calculating average pre- and post-destruction vedio intensities and subtracting the average post- from pre-destruction intensity (Figure 5A) [13]. Images representing intensity from adherent MBs (molecular imaging signals) were displayed as color maps overlaid on B-mode images, automatically generated by MCE. The scale for the color maps was constant for all images.
In vivo fluorescence imaging analysis
Free Cy-5.5, Cy-5.5-loaded DPMC and Cy-5.5-loaded DPMC with US (similar absorption intensity as Cy5.5, 200 µL) were intravenously injected into tumor xenograft mice. The DPMC with US exposure group was irradiated with US after the injection as described earlier. At 0.5, 12, 24, 48 and 72 h post-injection, biodistribution in MCF-7 tumor-bearing mice was visualized using an in vivo fluorescence imaging system (IVIS Lumina II, Caliper, Boston, USA), with excitation at 640 nm and emission at 695-770 nm. Mice under anesthesia via isoflurane inhalation were automatically moved into the imaging chamber for scanning.
In vivo antitumor efficacy
Tumor volumes were measured using a caliper and calculated using the formula: V = (L × W2) / 2, whereby length (L) is the longest diameter and width (W) the shortest diameter perpendicular to length. After a tumor volume of ~30 mm2 was reached, tumor-bearing mice were randomized into four groups (5 mice/group): PBS (Control), free DOX (DOX), DPMC without US (DPMC) and DPMC with US (DPMC+US). Each mouse was intravenously injected with the corresponding formulations at 2.5 mg DOX equivalent per kg within a final volume of 200 µL through the tail vein five times every 4 days. For the DPMC+US group, tumors were irradiated with US after every injection under the following settings: 1 MHz, 2 % duty cycle, duration of 1 min and intensity of 2 W/cm2 [11, 12]. At the end of the experimental period, all mice were sacrificed, and tumors harvested and weighed. Throughout the experiment, body weights of mice and tumor volumes were measured every other day.
Histological and immunohistochemical analyses
At the end of the experiment, tumors from different groups were collected and washed with PBS, fixed in 4% formaldehyde and embedded in paraffin. Embedded tissues were cut into 4 mm slices for immunohistochemical analyses of Capase 3 (1:50 dilution in 5% bovine serum albumin (BSA); Cell Signaling Technology, Danvers, Massachusetts, USA), Ki67 (1:200 dilution in 5% BSA; ABclonal, Woburn, MA, USA), and CD34 (1:200 dilution in 5% BSA; Abnova, Taipei, Taiwan). Tissue slices were visualized under an optical microscope (Nikon Eclipse 80i, Tokyo, Japan) and subsequently analyzed with Image J2x software (National Institutes of Health, Bethesda, Maryland, USA). The ratio of cells staining positive for Capase 3 and Ki67 in each image was determined as a ratio of apoptotic or proliferative cell number to total tumor cell number. For CD34 staining, microvessel density (MVD) was quantified by counting the number of capillaries per microscopic field within five random fields in the hot spot per slide at 400x magnification.
Statistical analysis
Statistical analysis was conducted with SPSS software (version 19.0). Data are presented as means ± SD. Significant differences between groups were determined using Student's t-test, one-way ANOVA and Repeated Measures ANOVA. Data were considered statistically significant at p<0.05. All experiments were repeated at least three times.
Results and discussion
Design and characterization of the DOX prodrug and DOX prodrug-MB complex
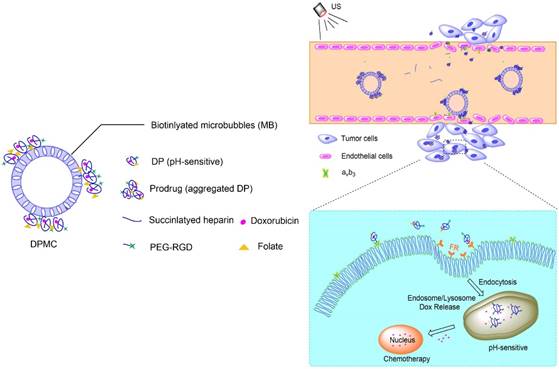
The pH value of normal tissue is around 7.4 while that of tumor could be as low as 6.0 [1]. pH-sensitive DOX-loaded nanoparticles exhibited significant antitumor potential due to their selective release in tumor and relative stability in blood circulation, as reported previously [4, 14, 15]. Hydrazone linkage, a pH-responsive bond, has been widely used in the design of anticancer drug delivery systems to facilitate drug release and improve anti-tumor efficiency [16]. Recent development of the heparin-based drug delivery system has had a significant impact on the tumor therapy field owing to the biocompatibility and biological activities of heparin [17]. Previous studies have demonstrated selective binding of the cRGD peptide to αvβ3 integrin, a principal marker of angiogenesis that is overexpressed in angiogenic endothelial cells of various malignant tumors and some of the tumor cells, including breast cancer cells [5, 18]. Another targeting ligand used for the preparation of our complex was folate, which specifically targets the folate receptor (FR) that is significantly upregulated in malignant tumor cells [6, 7]. In our study, the first step involved the preparation of pH-sensitive DP. In brief, DOX was engineered to the heparin backbone via hydrazone linkage to promote drug release, and the terminal carboxyl groups of heparin decorated with dual targeting ligands, PEG-cRGD and folate, facilitating its specific binding to angiogenic endothelial cells and tumor cells in target sites. The flexible spacer, PEG, was connected with cRGD conjugated to heparin, with a view to extending the retention time in blood and avoiding premature clearance of DP via escape from the RES [4]. Furthermore, DP was biotinylated by biotinamide to facilitate combination with the ultrasound-mediated delivery system. Subsequently, MB was incorporated in the lipid shell of the complex via avidin-biotin binding to generate the DOX prodrug-MB complex (DPMC).
Complex with micron size is expected to be limited in vascular without US radiation thereby reducing its side effect to non-targeted tissue. However, endothelial gaps of tumor range from 100 to 700 nm, and thus penetration of tumor tissues is evidently difficult for large-sized particles. In other words, particles with sizes of about 10-300 nm should display better permeation and accumulation in tumor tissues, instead of rapid elimination [19, 20]. In our experiments, DPMC was designed to selectively target to angiogenic endothelial cells in the tumor region via RGD-αvβ3 integrin mediated recognition. In addition, disruption using localized US cavitation resulted in DP dispersion into uniform small-sized nanoparticles, thereby facilitating its penetration into tumor interstitium through temporary endothelial gaps. Subsequently, small-sized DP could specifically bind tumor cells mediated via recognition of FR. Release of DOX into nuclei was pH-triggered, inducing higher cytotoxicity (Figure 1). The process resulted in better dispersion of prodrugs with larger sizes, thus enhancing the feasibility of their application in tumor therapy. On the other hand, DPMC can be used as acoustic probes for tumor imaging. The accumulation of complexes and accurate position of drugs in tumor sites in vivo can be observed and tracked by US imaging system, indicating their important role in integrating tumor imaging and therapy [9].
Schematic illustration of US combined with DPMC to deliver DOX into nuclei and induce cytotoxicity. Using US cavitation and sonoporation, DPMCs were disrupted, facilitating drugs penetration into the tumor sites through temporary gaps in the endothelium. DPs were targeted to tumor cells, followed by release of pH-triggered DOX into nuclei.
As shown in Figure 2A, TEM images revealed approximately spherical, small-sized DP, along with aggregated morphology, which may be attributed to high drug loading weight and particle aggregation via noncovalent forces [4]. Similarly, corresponding DLS revealed inhomogeneous size distribution (average diameter: 149.6±29.8 nm and 1036.2±38.8 nm) and the polydispersity index (PDI) was as large as 1.0 (Table 1). However, the average diameter of the prepared DPMC was 5.804±2.1 µm, as estimated using a Coulter counter, clearly indicating better dispersion than DP alone. Compared with aggregated DP, application of external US induced fragmentation of DPMC into uniformly smaller particles (average diameter: 128.6±42.3 nm, PDI: 0.21), as measured with DLS, corresponding to TEM findings. The morphology of DPMC was additionally confirmed via confocal microscopy (Figure 2B). The surface of MB was clearly surrounded by DP with red fluorescence before US dispersion, suggesting that preparation of DPMC was able to prevent DP from further aggregating. After US radiation, DPMCs were disrupted into small fragments displaying red fluorescence (Figure 2B), observed as nanoparticles in post-US TEM images (Figure 2A). Previous research showed that nanoparticles can be disrupted into smaller fragments under heat, visualized using TEM [10], indicating that external forces, including ultrasound and thermal effects, are capable of affecting particle behavior in aqueous solution. In addition, the DOX loading content of DP detected via UV-vis analysis was 18.9%, which was higher than that of particles investigated previously [21, 22]. Estimation of the amount of DP bound to MBs following removal of unbound DP via centrifugation showed that 75% DP attached to MBs. When 1×107 MBs were mixed with 50 µg DP, the loading content of DPMC was ~70.9 µg DOX/108 MBs. 1HNMR spectra, FT-IR spectrum and quantitative analysis of DP were shown in supplementary information.
Characterization of DP and DPMC. (A) TEM images and DLS graphs of DP pre-destruction with US (left) and DPMC post-destruction with US (right). (B) Confocal laser scanning microscopy images and corresponding bright field images of DPMC pre-destruction (left) and post-destruction (right) by US. DPMCs were visualized using the fluorescence of bound DOX. Data are presented as means ± standard deviation (n=3).
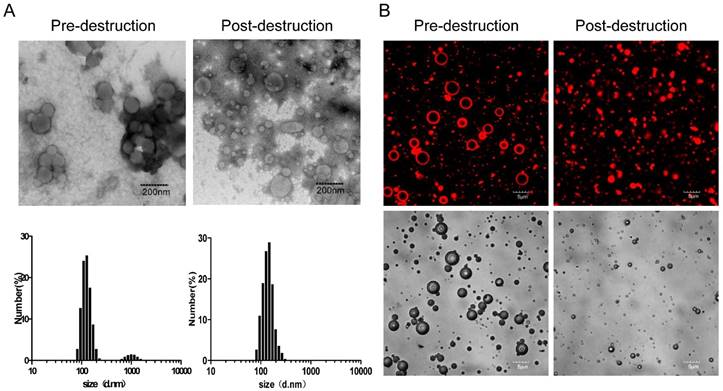
Characterization of DP and DPMC determined by DLS, UV-vis analysis and Coulter counter.
| Size(nm) | PDI | Zeta-potential (mV) | DOX loading content | ||
|---|---|---|---|---|---|
| DP | Pre-destruction | 149.6±29.8 | 1.0 | -19.8±4.5 | 18.9%(w/w) |
| 1036.2±38.8 | |||||
| Post-destruction | 128.6±42.3 | 0.21 | -20.6±3.4 | ||
| DPMC | 5804±2100 | 70.9μg DOX/108 MBs |
In vitro release of doxorubicin
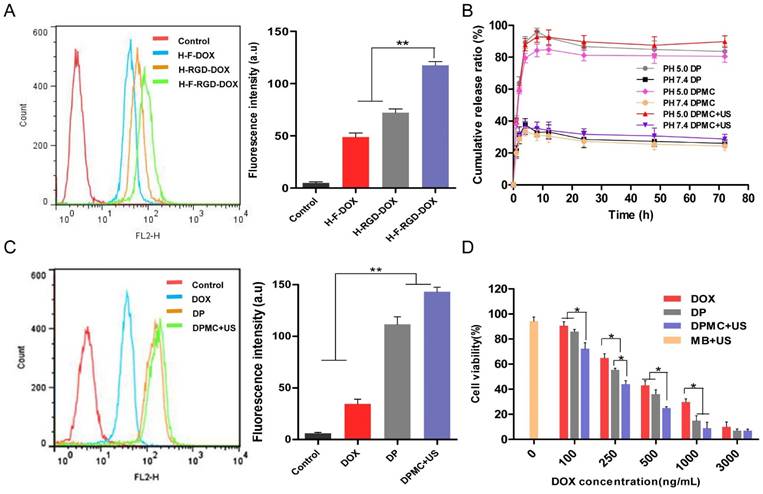
To evaluate the pH sensitivity of DP and DPMC with or without US, an in vitro DOX release experiment was performed at 37°C in PBS at pH 7.4 and 5.0 (Figure 3B). Compared with 30% obtained at pH 7.4, cumulative release of DOX from DP and DPMC with or without US at pH 5.0 was estimated as 84%, 90% and 81% after 8 h respectively, indicating comparative stability in neutral conditions. The release rate was dramatically improved due to hydrolysis of hydrazone, accelerating DOX liberation at decreasing pH values, consistent with large numbers of recent studies in pH-responsive drug delivery system [1]. In addition, compared to DP, the release rate of DPMC was slightly higher following US exposure. The data clearly demonstrate that the introduction of MB into DP does not hinder its pH responsiveness, but rather, promotes DOX release due to US cavitation, providing a foundation for its application in vivo within an acidic tumor environment.
Flow cytometry and corresponding histogram profiles of MCF-7 cells incubated with H-F-DOX, H-RGD-DOX and H-F-RGD-DOX (A) and DOX, DP and DPMC with US (C). In vitro release of DOX from DP and DPMC with or without US after incubation at 37oC in phosphate buffer (pH 5.0 and 7.4) (B). Cytotoxicity of MCF-7 cells incubated with MB with US, DOX, DP and DPMC with US (D). Values represent means ± SD (n=3).
Cellular uptake
Flow cytometry was conducted to investigate the cellular uptake of carrier with single ligand and dual ligands, with untreated cells as the control (Figure 3A). The fluorescence of DOX allowed its direct use to assay cellular uptake with no requirement for additional markers. The data indicates that the fluorescence intensity of H-F-RGD-DOX was higher than that of H-F-DOX and H-RGD-DOX, confirming the effectiveness of dual-ligand mediated targeting since their corresponding receptors, αvβ3 integrin and FR, are overexpressed in malignant tumor cells [5-7]. In the evaluation of cellular uptake of DOX, DP and DPMC with US, we found that the fluorescence intensity of DP was higher than that of free DOX, indicating that the dual ligand-targeting effect and pH-sensitive properties of DP contribute to enhanced cellular internalization (Figure 3C). Moreover, DPMC with US exhibited the greatest internalization ability among the three groups. Based on the collective findings, we suggest that US disruption of the complex into smaller particles facilitates its cellular uptake and drug liberation. Additionally, cell membrane permeability enhanced by US sonoporation was not negligible.
Intracellular distribution of different groups was detected by confocal microscopy (Figure 4A). The fluorescence of DOX was visualized in cytoplasm in cells treated with pH-insensitive prodrug, while that of cells treated with free DOX, DP and DPMC with US were visualized mainly in nuclei. This finding might be due to the different mechanisms of the cellular uptake of free DOX, pH-sensitive and pH-insensitive DOX-loaded prodrugs. The pH-insensitive DOX-loaded prodrugs were taken up by tumor cells via receptor-medicated endocytosis, while free DOX was transported into cells via a passive diffusion mechanism and used as a DNA intercalator in nuclei [23, 24]. For pH-sensitive DOX-loaded prodrugs, DOX was easier to be released in microenvironment of tumor cells and enhanced its penetration into nuclei through triggering endosomal escape [8, 17]. Furthermore, in pH-sensitive group, fluorescence of DOX in cells treated with additional US was higher than that of non-US treating group. The observation indicated that US promoted intracellular uptake of DOX resulting in much more nuclear accumulation, which might be due to not only the improvement of cell membrane permeability but also the good dispersion of DOX-prodrug caused by US cavitation and sonoporation [25].
Confocal microscope images of MCF-7 cells treated with free DOX (DOX), pH-insensitive prodrug (pH-insensitive), DP (pH-sensitive) and DPMC with US (pH-sensitive+US) for 4 h. Scale bar represents 20 μm (A) . Confocal microscope images of MCF-7 cells and A549 cells incubated with small-sized nanoparticles from DPMC disrupted by US for 5 mins. Scale bar represents 10 μm (B).
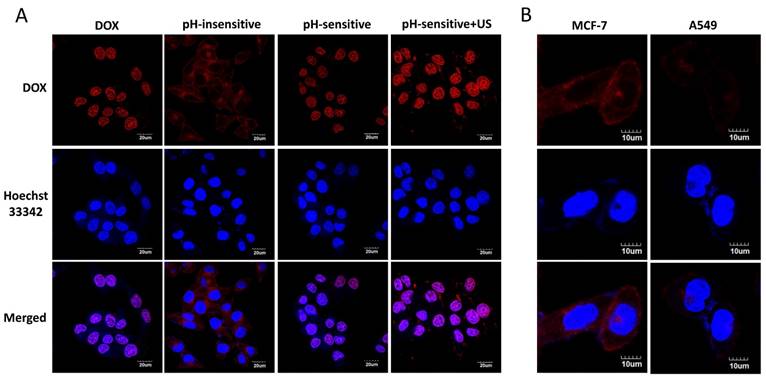
In the evaluation of specific binding of DPMC to tumor cells with US exposure, the fluorescence attached to FR-positive MCF-7 cells was distinctly higher than that of FR-negative A549 cells, providing evidence that FR-mediated specific targeting to tumor cells contributed significantly to the endocytosis of DPMC (Figure 4B).
In vitro cytotoxicity
The in vitro cytotoxicity of DP and DPMC with US, compared with free DOX, was estimated in MCF-7 cells using the MTT assay, while cells treated MBs with US were used as control (Figure 3D). After 48 h incubation of the particles with cells, cytotoxicity was increased in the order of free DOX < DP < DPMC with US (IC50: 310.35 ng/mL, 217.43 ng/mL, 120.23 ng/mL, respectively). The ascending cytotoxicity of DP may be attributed to the dual ligand-specific targeting and pH-triggered release of DOX in the tumor environment. Owing to the sonoporation effect of US, cell membrane permeability could be enhanced, contributing to improved cytotoxicity and drug delivery efficiency to target cells [12]. On the other hand, cell viability of the group treated with MBs with US was about 94%, confirming that the toxicity was not caused by the setting US condition. In conclusion, DPMC with US exhibited the greatest cytotoxicity among the three formulations, which could be attributed to the fact that US cavitation disrupt the aggregated prodrugs into small uniform particles, improve DOX release and further enhance its cellular internalization.
Ultrasound molecular imaging
cRGD selectively binds to αvβ3 integrin overexpressed in angiogenic endothelial cells, promoting specific targeting of cRGD-loaded MBs to tumor sites [5]. Recent research on a mouse model of breast cancer in vivo using ultrasound molecular imaging revealed significantly greater adhesion ability of cRGD-MB to tumor sites, compared to non-targeted MB. Moreover, imaging signals were obviously decreased after using an anti-αv monoclonal antibody, validating the utility of ultrasound molecular imaging in demonstrating the targeting ability of cRGD-loaded MB [26]. Accordingly, we speculated that DPMC composed of cRGD and folate upon flash effect should exhibit superior targeting ability, compared to blank MBs. The targeting ability of DPMC using ultrasound molecular imaging was investigated, compared with that of blank MBs. In this experiment, videos were recorded for the whole process and datasets of all mice analyzed offline in a random order using commercially available software (MCE). Images representing intensities from adherent blank MBs or DPMC, displayed as color maps overlaid on B-mode images, were automatically generated using MCE (Figure 5B). The color intensity localized in the tumor region (yellow circle) from mice treated with DPMC was obviously higher than that from mice treated with blank MBs. Simultaneous video intensities for the images are presented quantitatively in Figure 5C. Significant differences were observed between the two groups. The results indicate that the proportion of DPMC adhering to the tumor site is significantly higher than that of blank MBs (p<0.001), providing further evidence that DPMC exhibits better tumor targeting imaging ability, consistent with earlier related reports [26]. The existing of MBs in complex makes it possible to monitor the position of the prodrug in vivo using US imaging system in real time. Herein, the targeted complexes can be used as acoustic probes for imaging and thereby integrate tumor imaging and therapy [9].
In vivo fluorescent imaging
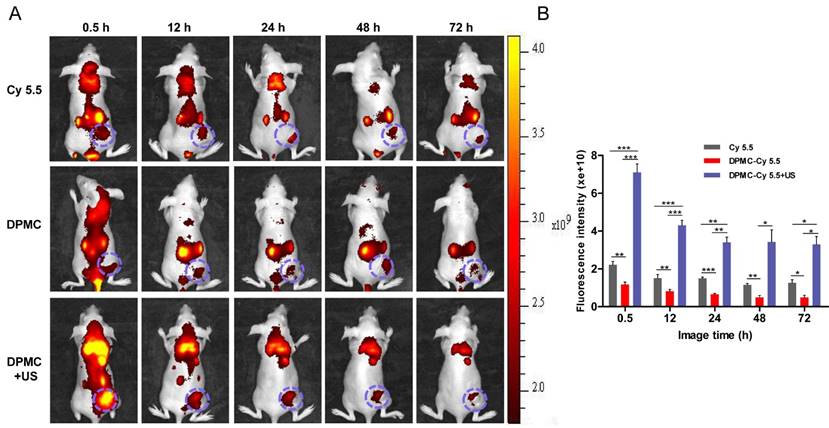
Fluorescent intensity detection of MCF-7 tumor-bearing mice was performed to evaluate the tumor targeting efficiency of the complex in vivo. As shown in Figure 6, mice treated with Cy5.5-labeled DPMC combined with US presented higher accumulation of the complex at the tumor site, compared with other groups (p<0.001). And the strongest fluorescence intensity was detected within the initial 0.5 h after intravenous injection. We presume that upon reaching the tumor region, DPMC is fragmented by localized US, leading to smaller prodrug size and further enhancing its penetration into tumor tissue. Compared with the non-US exposed group, Cy5.5 loaded in DPMC assisted by US could be more efficiently delivered to the tumor site, resulting in higher and earlier initial accumulation. On the other hand, PEG in DP could extend circulation time in vivo through decreasing opsonin adsorption and RES recognition [27, 28]. Receptor-mediated endocytosis may additionally be responsible for improving cellular uptake and retention of DP, in accordance with previous reports [24]. In the absence of US stimulation, a slight fluorescence signal from Cy5.5-labeled DPMC was detected in the tumor region, which may be attributable to its targeting effect mediated by recognition of αvβ3 integrin. A fluorescence signal from free Cy5.5 was additionally visualized at the tumor site, possibly due to passive diffusion of small molecules.
In vivo fluorescence images of tumor-bearing mice administered Cy 5.5-labeled DPMC with US, Cy 5.5-labeled DPMC and Cy 5.5 at different time-points (A). Quantification of the in vivo tumor fluorescence accumulation of different formulations expressed as fluorescence per mm2 of tumor(B). Data are presented as mean values ± SD (n=5).
In vivo antitumor efficacy
Prepared DP inclined to aggregate with increasing drug loading weight, hindering its penetration into tumor tissue and compromising therapeutic efficiency in vivo. In the study of drug delivery system, drugs loaded in MBs are easily released and extravasated into tumor tissues, leading to enhanced antitumor efficiency upon US exposure. Additionally, drug-loaded MBs are not likely to rupture and penetrate into tissues not exposed to US, resulting in reduction of side-effects to normal tissue [29]. However, the drug loading capacity of MBs with lipid monolayer shells is limited due to the gas cores and thin lipid shells [30], and thus increased drug payload in each bubble is desirable. Accordingly, we combined DP with high drug-loading capacity with MB to generate homogeneous DPMC beneficial for delivery and therapy in vivo with the assistance of US.
To estimate antitumor activity in vivo, DPMC, DPMC combined with US and free DOX were intravenously injected into mice bearing MCF-7 breast xenograft tumors, using saline-injected mice as the control. Considering its aggregation property, DP without filtered was not suitable for intravenous therapy as a positive control group. The formulations with or without US (2.5 mg equivalent DOX/kg mice) were applied at four-day intervals for 20 days. It was observed that the control group showed a progressive increase in tumor volume while tumors in all drug-treated groups displayed growth retardation. The DOX-treated group displayed greater antitumor efficiency than mice treated with DPMC without US exposure. One possible explanation for this finding is that small molecules easily enter cells via free diffusion while DPMC with micron sizes are restricted in blood vessels, hardly penetrating the interstitial spaces in the absence of US stimulation. Notably, treatment with DPMC assisted by US led to a higher tumor inhibition rate than DOX alone, suggesting that US physically exerts a positive influence on drug release and cell membrane permeability, consistent with previous reports [9]. Significant differences were observed between groups treated with DPMC in the presence or absence of US (p<0.01) (Figure 7A). The body weights of tumor-bearing mice were simultaneously recorded. The DOX group showed an obvious weight decrease, compared to the control, suggesting a certain extent of systemic toxicity (p<0.05). No obvious body weight shifts were observed for the group exposed to DPMC without US. Interestingly, the group exposed to DPMC plus US presented better drug tolerability, possibly due to localized DOX release and accumulation in tumor regions, leading to reduced side- effects (Figure 7B).
In vivo tumor growth inhibition of DPMC with or without US. Comparison of the tumor inhibition effect of DPMC with US versus DPMC without US, DOX and saline in a breast tumor model (n=5). DPMC with US achieved significant tumor inhibition (***p<0.001, compared to saline; ** p< 0.01, compared to DPMC without US) (A). During the treatment period, mice administered DPMC with US showed no significant body loss compared to those given saline, but differences were observed in relation to the DOX treatment group (* p< 0.05) (B). At the end of the experiment, tumor tissues were collected from each sacrificed animal after 20 days of treatment, photographed (D) and weighed (*** p<0.001, compared to saline and DPMC without US; * p< 0.05, compared to DOX) (C).
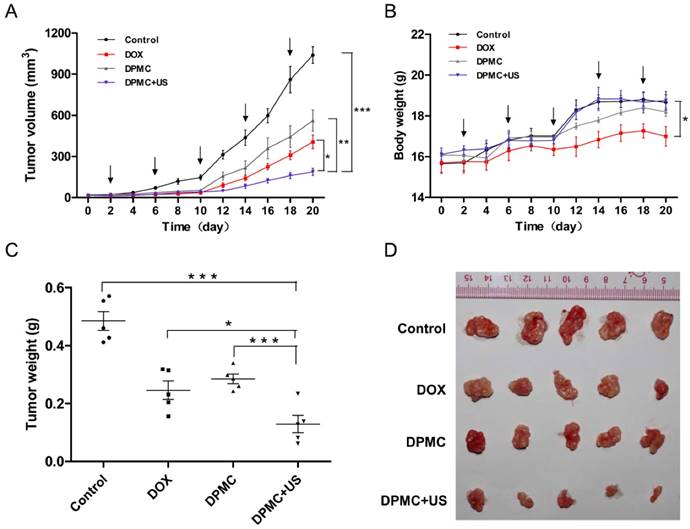
All tumors of the treated mice were excised, photographed and weighed at the end of the experimental period (Figure 7C and D). Tumor weights from mice treated with DPMC along with US exposure were significantly lower than those from control (p<0.001), free DOX- (p<0.05) and DPMC-treated mice (p<0.001), consistent with the data on tumor volumes (Figure 7A). Our results collectively indicate that DPMC with US presents excellent antitumor efficacy and relative low systemic toxicity, which may be attributable to not only US cavitation and sonoporation but also the dual-targeted effect and pH sensitivity of DP [31, 32], further confirming the superiority of the combination treatment strategy.
Immunohistochemical analysis
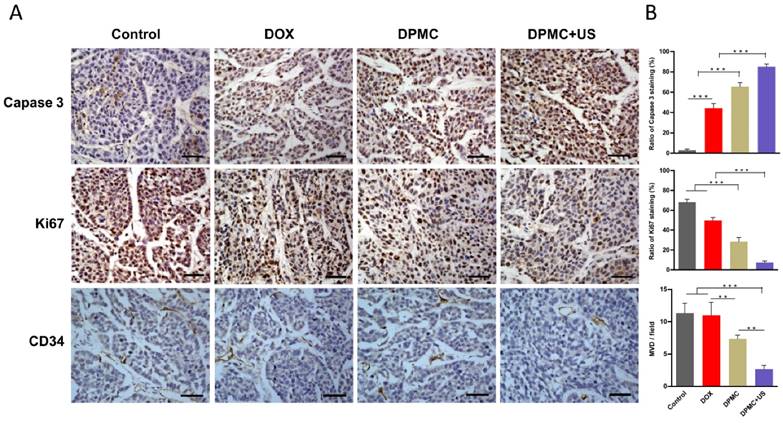
Immunohistochemical analysis of tumors was conducted to evaluate apoptosis, proliferation and angiogenesis. Staining for caspase-3 and Ki-67, commonly used metabolic markers of apoptosis and proliferation, was performed to determine antitumor efficacy. Compared to the control group, tumor tissues of mice exposed to DPMC combined with US showed relatively higher levels of capase-3-positive and lower levels of Ki-67-positive cells (Figure 8A). Significant differences were observed relative to the other groups (p<0.001), clearly indicating that DPMC with US is considerably more effective in inducing apoptosis and inhibiting tumor cell proliferation (Figure 8B). Angiogenesis is known to play a major role in tumor growth, invasion and metastasis in solid tumors [33, 34]. Antiangiogenic therapy may therefore suppress the blood supply and trigger apoptosis of tumor vascular endothelial cells, leading to antitumor effects. As a specific ligand of DP, cRGD played an important role in the anti-angiogenic effect of our complex by blocking αvβ3 integrin. CD34 (a marker of vessel endothelium) staining was further performed to determine angiogenesis inhibition. As shown in Figure 8B, DPMC with US exerted the greatest antiangiogenic effect, and the corresponding microvessel density (MVD) per field was significantly lower than that of other treated groups (p<0.01), which may be attributed to enhancement of the targeting effect of cRGD by localized US and consequent improvement of angiogenesis inhibition ability. Moreover, relatively few microvessels were observed in the group treated with DPMC without US, compared with the DOX and control groups, since cRGD incorporated into DPMC could suppress angiogenesis to some extent, even without US stimulation. Relatively higher MVD was observed in the free DOX and control groups, with no significant differences. One possibility for this finding is that the equivalent dosages of DOX administered to all the groups were insufficient to exert anti-angiogenic effects unless decorated with targeted ligands and applied with the assistance of US, in accordance with safety evaluation data. Our immunohistochemical findings suggest that various mechanisms contribute to the antitumor effects of DPMC assisted by US, including enhancement of cell apoptosis, inhibition of cell proliferation and antagonism of angiogenesis.
Histological analysis of tumors from mice treated with different formulations. Images (A) and corresponding quantification (B) of Caspase-3, Ki67 and CD34 staining of tumors treated with saline, DOX, DPMC without US and DPMC with US. Scale bar represents 100 μm.
Conclusion
Here, we successfully engineered a dual-targeted pH-sensitive DOX prodrug displaying high drug-loading capacity, excellent tumor targeting specificity and release ability, but relatively large sizes due to aggregation, which may limit its application in tumor therapy. A combination of DP and MB generated via an avidin-biotin bridge presented uniform distribution. Importantly, with the assistance of localized US, the DPMC could be fragmented into smaller nanoparticles, facilitating intracellular accumulation and antitumor activity in vivo. Despite its important usage as acoustic probes for tumor imaging, the complex exerted significant antitumor efficacy through inducing apoptosis, inhibiting cell proliferation and antagonizing angiogenesis in conjunction with US exposure. Our data collectively highlight the significant potential of this strategy to improve the delivery of large-sized or aggregated particles to tumor sites and integrate tumor imaging and therapy, thereby extending their applications in vivo.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The work was granted by Natural Science Foundation of China (No. 81671709, 81371559, 30670580); Science and Technology Planning Project of Guangdong Province (No. 2013B010404024); Natural Science Foundation of Guangdong Provinces (No. 2014A030313347). The authors were grateful for the support from Guangzhou Key Laboatory of Tumor Immunology Research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ruan S, Yuan M, Zhang L, Hu G, Chen J, Cun X. et al. Tumor microenvironment sensitive doxorubicin delivery and release to glioma using angiopep-2 decorated gold nanoparticles. Biomaterials. 2015;37:425-35 doi: 10.1016/j.biomaterials.2014.10.007
2. Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev. 2012;64:353-65 doi: 10.1016/j.addr.2012.09.011
3. Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Gold nanoparticles with a monolayer of doxorubicin-conjugated amphiphilic block copolymer for tumor-targeted drug delivery. Biomaterials. 2009;30:6065-75 doi:10.1016/j.biomaterials.2009.07.048
4. She W, Luo K, Zhang C, Wang G, Geng Y, Li L. et al. The potential of self-assembled, pH-responsive nanoparticles of mPEGylated peptide dendron-doxorubicin conjugates for cancer therapy. Biomaterials. 2013;34:1613-23 doi: 10.1016/j.biomaterials.2012.11.007
5. Chakravarty R, Chakraborty S, Dash A. Molecular Imaging of Breast Cancer: Role of RGD Peptides. Mini Rev Med Chem. 2015;15:1073-94
6. Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13:256-62 doi: 10.1016/j.cbpa.2009.03.022
7. Xia W, Low PS. Folate-targeted therapies for cancer. J Med Chem. 2010;53:6811-24 doi: 10.1021/jm100509v
8. Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S. et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS nano. 2013;7:5858-69 doi: 10.1021/nn4010796
9. Yan F, Li L, Deng Z, Jin Q, Chen J, Yang W. et al. Paclitaxel-liposome-microbubble complexes as ultrasound-triggered therapeutic drug delivery carriers. J Control Release. 2013;166:246-55 doi: 10.1016/j.jconrel.2012.12.025
10. Zheng M, Yue C, Ma Y, Gong P, Zhao P, Zheng C. et al. Single-step assembly of DOX/ICG loaded lipid-polymer nanoparticles for highly effective chemo-photothermal combination therapy. ACS nano. 2013;7:2056-67 doi: 10.1021/nn400334y
11. Yin T, Wang P, Li J, Wang Y, Zheng B, Zheng R. et al. Tumor-penetrating codelivery of siRNA and paclitaxel with ultrasound-responsive nanobubbles hetero-assembled from polymeric micelles and liposomes. Biomaterials. 2014;35:5932-43 doi: 10.1016/j.biomaterials.2014.03.072
12. Yin T, Wang P, Li J, Zheng R, Zheng B, Cheng D. et al. Ultrasound-sensitive siRNA-loaded nanobubbles formed by hetero-assembly of polymeric micelles and liposomes and their therapeutic effect in gliomas. Biomaterials. 2013;34:4532-43 doi: 10.1016/j.biomaterials.2013.02.067
13. Bachawal SV, Jensen KC, Lutz AM, Gambhir SS, Tranquart F, Tian L. et al. Earlier detection of breast cancer with ultrasound molecular imaging in a transgenic mouse model. Cancer Res. 2013;73:1689-98 doi: 10.1158/0008-5472.CAN-12-3391
14. Li M, Tang Z, Lv S, Song W, Hong H, Jing X. et al. Cisplatin crosslinked pH-sensitive nanoparticles for efficient delivery of doxorubicin. Biomaterials. 2014;35:3851-64 doi: 10.1016/j.biomaterials.2014.01.018
15. Dai J, Lin S, Cheng D, Zou S, Shuai X. Interlayer-crosslinked micelle with partially hydrated core showing reduction and pH dual sensitivity for pinpointed intracellular drug release. Angew Chem Int Ed Engl. 2011;50:9404-8 doi: 10.1002/anie.201103806
16. Zhou Z, Li L, Yang Y, Xu X, Huang Y. Tumor targeting by pH-sensitive, biodegradable, cross-linked N-(2-hydroxypropyl) methacrylamide copolymer micelles. Biomaterials. 2014;35:6622-35 doi: 10.1016/j.biomaterials.2014.04.059
17. Li Y, Wen G, Wang D, Zhang X, Lu Y, Wang J. et al. A complementary strategy for enhancement of nanoparticle intracellular uptake. Pharm Res. 2014;31:2054-64 doi: 10.1007/s11095-014-1307-5
18. Schottelius M, Laufer B, Kessler H, Wester HJ. Ligands for mapping alphavbeta3-integrin expression in vivo. Acc Chem Res. 2009;42:969-80 doi: 10.1021/ar800243b
19. Li Y, He H, Jia X, Lu WL, Lou J, Wei Y. A dual-targeting nanocarrier based on poly(amidoamine) dendrimers conjugated with transferrin and tamoxifen for treating brain gliomas. Biomaterials. 2012;33:3899-908 doi: 10.1016/j.biomaterials.2012.02.004
20. Crampton HL, Simanek EE. Dendrimers as drug delivery vehicles: non-covalent interactions of bioactive compounds with dendrimers. Polym Int. 2007;56:489-96
21. Bandekar A, Karve S, Chang MY, Mu Q, Rotolo J, Sofou S. Antitumor efficacy following the intracellular and interstitial release of liposomal doxorubicin. Biomaterials. 2012;33:4345-52 doi: 10.1016/j.biomaterials.2012.02.039
22. Pang Z, Gao H, Yu Y, Guo L, Chen J, Pan S. et al. Enhanced intracellular delivery and chemotherapy for glioma rats by transferrin-conjugated biodegradable polymersomes loaded with doxorubicin. Bioconjug Chem. 2011;22:1171-80 doi: 10.1021/bc200062q
23. Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydrazone bond for tumor-targeted drug delivery. Biomaterials. 2009;30:5757-66 doi: 10.1016/j.biomaterials.2009.07.020
24. Lee Y, Park SY, Mok H, Park TG. Synthesis, characterization, antitumor activity of pluronic mimicking copolymer micelles conjugated with doxorubicin via acid-cleavable linkage. Bioconjug Chem. 2008;19:525-31 doi: 10.1021/bc700382z
25. Yoon YI, Kwon YS, Cho HS, Heo SH, Park KS, Park SG. et al. Ultrasound-mediated gene and drug delivery using a microbubble-liposome particle system. Theranostics. 2014;4:1133-44 doi: 10.7150/thno.9945
26. Anderson CR, Hu X, Zhang H, Tlaxca J, Decleves AE, Houghtaling R. et al. Ultrasound molecular imaging of tumor angiogenesis with an integrin targeted microbubble contrast agent. Invest Radiol. 2011;46:215-24 doi: 10.1097/RLI.0b013e3182034fed
27. Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv Drug Deliv Rev. 2009;61:428-37 doi: 10.1016/j.addr.2009.03.009
28. Owens DE 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93-102
29. Lentacker I, Geers B, Demeester J, De Smedt SC, Sanders NN. Design and evaluation of doxorubicin-containing microbubbles for ultrasound-triggered doxorubicin delivery: cytotoxicity and mechanisms involved. Mol Ther. 2010;18:101-8 doi: 10.1038/mt.2009.160
30. Klibanov AL, Shevchenko TI, Raju BI, Seip R, Chin CT. Ultrasound-triggered release of materials entrapped in microbubble-liposome constructs: a tool for targeted drug delivery. J Control Release. 2010;148:13-7 doi: 10.1016/j.jconrel.2010.07.115
31. Shao K, Ding N, Huang S, Ren S, Zhang Y, Kuang Y. et al. Smart nanodevice combined tumor-specific vector with cellular microenvironment-triggered property for highly effective antiglioma therapy. ACS nano. 2014;8:1191-203 doi: 10.1021/nn406285x
32. Kopechek JA, Carson AR, McTiernan CF, Chen X, Hasjim B, Lavery L. et al. Ultrasound Targeted Microbubble Destruction-Mediated Delivery of a Transcription Factor Decoy Inhibits STAT3 Signaling and Tumor Growth. Theranostics. 2015;5:1378-87 doi: 10.7150/thno.12822
33. Chen H, Niu G, Wu H, Chen X. Clinical Application of Radiolabeled RGD Peptides for PET Imaging of Integrin alphavbeta3. Theranostics. 2016;6:78-92 doi: 10.7150/thno.13242
34. Melemenidis S, Jefferson A, Ruparelia N, Akhtar AM, Xie J, Allen D. et al. Molecular magnetic resonance imaging of angiogenesis in vivo using polyvalent cyclic RGD-iron oxide microparticle conjugates. Theranostics. 2015;5:515-29 doi: 10.7150/thno.10319
Author contact
![]() Corresponding author: Yingjia Li, Department of Medicine Ultrasonics, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China. Phone: +86 020 61642100; E-mail: lyjiaedu.cn. Ying Wang, Guangdong Provincial Key laboratory of cancer immunotherapy research, Cancer Research Institute, School of Basic Medical Science, Southern Medical University, Guangzhou 510515, China. Phone: +86 020 62789410; E-mail: ningmengquancom.
Corresponding author: Yingjia Li, Department of Medicine Ultrasonics, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China. Phone: +86 020 61642100; E-mail: lyjiaedu.cn. Ying Wang, Guangdong Provincial Key laboratory of cancer immunotherapy research, Cancer Research Institute, School of Basic Medical Science, Southern Medical University, Guangzhou 510515, China. Phone: +86 020 62789410; E-mail: ningmengquancom.