Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Supplementary Material
Acknowledgements
Conflicts of interest
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1333-1345. doi:10.7150/thno.17092 This issue Cite
Research Paper
Interleukin 3- receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth
Daniele Bellavia1*, Stefania Raimondo2*, Giovanna Calabrese3, Stefano Forte3, Marta Cristaldi2, Agostina Patinella2, Lorenzo Memeo3, 4, Mauro Manno5, Samuele Raccosta5, Patrizia Diana6, Girolamo Cirrincione6, Gianluca Giavaresi1,7, Francesca Monteleone2, Simona Fontana2, Giacomo De Leo2, Riccardo Alessandro2 ![]()
1. Rizzoli Orthopedic Institute, Innovative Technological Platform for Tissue Engineering, Theranostics and Oncology, Palermo 90133, Italy;
2. Dipartimento di Biopatologia e Biotecnologie Mediche, University of Palermo, Palermo 90133, Italy;
3. IOM Ricerca, Viagrande, Catania, Italy;
4. Dipartimento di Oncologia Sperimentale, Istituto Oncologico del Mediterraneo, Viagrande, Catania, Italy;
5. National Research Council of Italy, Institute of Biophysics, Palermo, Italy;
6. Dipartimento di Scienze e Tecnologie Biologiche Chimiche e Farmaceutiche, University of Palermo, Palermo 90123, Italy;
7. Rizzoli Orthopedic Institute, Laboratory of Preclinical and Surgical Studies, Bologna 40136, Italy.
* These authors contributed equally to this work
Received 2016-8-3; Accepted 2017-1-3; Published 2017-3-16
Abstract

Despite Imatinib (IM), a selective inhibitor of Bcr-Abl, having led to improved prognosis in Chronic Myeloid Leukemia (CML) patients, acquired resistance and long-term adverse effects is still being encountered. There is, therefore, urgent need to develop alternative strategies to overcome drug resistance.
According to the molecules expressed on their surface, exosomes can target specific cells. Exosomes can also be loaded with a variety of molecules, thereby acting as a vehicle for the delivery of therapeutic agents.
In this study, we engineered HEK293T cells to express the exosomal protein Lamp2b, fused to a fragment of Interleukin 3 (IL3). The IL3 receptor (IL3-R) is overexpressed in CML blasts compared to normal hematopoietic cells and thus is able to act as a receptor target in a cancer drug delivery system. Here we show that IL3L exosomes, loaded with Imatinib or with BCR-ABL siRNA, are able to target CML cells and inhibit in vitro and in vivo cancer cell growth.
Keywords: Chronic Myeloid Leukemia, Engineered exosomes, Drug delivery, Drug resistance, Interleukin 3.
Introduction
Chronic Myelogenous Leukemia (CML) is a myeloproliferative neoplasia characterized by the reciprocal translocation t(9:22) (q34:q11) [1], which results in the expression of the chimeric Bcr-Abl oncoprotein with constitutive tyrosine kinase activity. Bcr-Abl kinase drives the pathogenesis of the disease through the phosphorylation and activation of a broad range of downstream substrates which play critical roles in cellular signal transduction. This ultimately leads to cell growth, inhibition of apoptosis and altered cell adhesion [2].
A radical change in CML therapy took place in the early 90s with the discovery of Imatinib mesylate (IM) [3]: first line therapy for patients due to its specific action against Bcr-Abl protein tyrosine kinase activity. This “targeted” approach has improved the 10-year survival rate of patients from approximately 20% to 80-90%.
For those patients who fail standard treatment, an increasing number of "second" (dasatinib and nilotinib) and "third" (bosutinib and ponatinib) generation tyrosine kinase inhibitors (TKIs) have been developed in recent years, now commonly used in the treatment of CML patients [4].
Despite the fact that TKIs are effective agents in CML treatment, the development of pharmacological resistance poses a limit to their therapeutic potential. This is further compounded by the need to increase administered doses, which then have long-term side effects, such as cardiac toxicity, altered bone and mineral metabolism and hypothyroidism [5, 6]. Acquired resistance can be caused by different mechanisms: overexpression of the Bcr-Abl protein or successive mutations in the BCR-ABL gene that determine low binding of the Imatinib [7]. Thus, there is an urgent need to develop alternative strategies to overcome drug resistance [8].
As a strategy for targeting neoplastic cells, the application of nucleic acid-based inhibitors of gene expression, such as RNA interference (RNAi), has been proposed in the treatment of CML [9, 10].
However, the clinical application of RNA-based therapy has been hampered by a lack of appropriate delivery systems [11].
An ideal drug delivery system should be able to direct the drug to a specific type of cells, thereby improving the biological effectiveness and reducing the toxicity of the therapy. As a drug delivery systems, liposomes, synthetic vesicles with a lipid bilayer membrane, are the most well-investigated systems due to their application in cancer therapy [12].
Exosomes are small vesicles, 40-100 nm in diameter, which are initially formed within the endosomal compartment and, subsequently secreted when a multi-vesicular body (MVB) fuses with the plasma membrane. These vesicles are released by a number of cell types including cancer cells and are considered messengers in intercellular communication [13-16]; recently, a wealth of information has been gained in recent years which considers these vesicles as new vehicle types, containing cytokines, growth factors, adhesion molecules and nucleic acids [17].
Interest in using exosomes, as an alternative to liposomes, in therapeutic approaches is an even more recent development and is based on surmounting evidence of their clinical applicability. Exosomes are an extremely promising therapeutic tool for numerous diseases given their ability to shuttle small molecules between cells. In particular, due to their endogenous origin, exosomes avoid immune recognition and clearance compared to exogenous nanovesicles. Furthermore, exosomes have a complex protein membrane composition that contributes to efficient cellular uptake [18, 19].
Recent studies have demonstrated that exosomes have a tissue specific tropism, according to the molecules expressed on their surface, which can be used to target them to specific tissues and/or organs [20]. Furthermore, exosomes or exosome mimetic nanoparticles can be engineered and loaded with various molecules (drugs, small molecules or siRNA) and targeted to specific organs [21-24].
The first example of functional delivery of siRNA using exosomes was developed by Alvarez-Erviti et al [24]; the authors demonstrated that engineered exosomes with RVG peptide (portion of G protein of rabdovirus) specifically recognize neuronal cells, thus overcoming the blood-brain barrier.
In this study we generated targeted exosomes able to deliver Imatinib or BCR-ABL siRNA to CML cells in order to overcome pharmacological resistance (Figure 1A). It is known that interleukin-3 receptor (IL3-R) is overexpressed on CML and AML (acute myeloid leukemia) blasts while expressed at low levels or absent on normal hematopoietic stem cells [25-27], suggesting that IL3-R could serve as a receptor target in a cancer drug delivery system using exosomes to deliver drugs or siRNA to inhibit BCR-ABL. Here we show that modified exosomes, containing IL3-Lamp2B, and loaded with Imatinib, are able to specifically target tumor cells in vivo, leading to a the reduction in tumor size. Furthermore, we showed that modified exosomes are able to deliver functional BCR-ABL siRNA towards Imatinib-resistant CML cells.
Schematic representation of IL3-R-targeted exosomes: (A) HEK293T cells were transfected with a plasmid containing the recombinant human protein IL3-Lamp2b. Transfected cells were treated with Imatinib in order to generate Imatinib-containing IL3L exosomes or, transfected with BCR-ABL siRNA in order to generate siRNA-containing IL3L exosomes. The efficacy of engineered exosomes were tested in vitro and in vivo on Imatinib- sensitive or -resistant CML cells. (B) Schematic representation of Pinco plasmid construct containing IL3-Lamp2b.
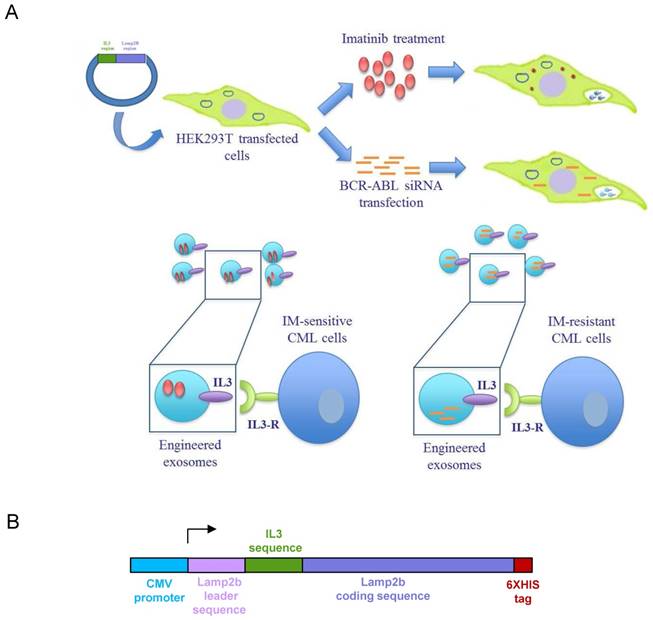
Results
Characterization of IL3-Lamp2b Exosomes
In order to produce IL3-Lamp2b (IL3L) expressing exosomes, we generated a Pinco plasmid containing the recombinant human Lamp2b gene fused with a human interleukin-3 gene fragment. A 6XHis tag in the C-terminus of the protein (Figure 1B) was then added so as to confirm the expression of the recombinant protein. HEK293T were selected as exosome producing cells due to their high transfection efficacy and the amount of exosomes released [28]. HEK293T cells, transfected with recombinant Pinco plasmid, and exosomes released from these cells expressed IL3-Lamp2b as shown by western blot analysis (Figure 2A).
Generation and characterization of IL3-Lamp2b exosomes. (A) Western blot analysis was performed on HEK293T cells, transfected or not with IL3L, and on released exosomes. Protein levels of IL3-Lamp2b (using 6XHis tag), Alix, CD81 and Tsg101 were evaluated. (B) Exosome size distribution was determined by DLS analysis. (C) Exosome particles were visualized by Atomic Force Microscopy (large square 2 μm, small square 500 nm). The apparent heights are colour coded as in the side bar. (D) Western blot analysis was performed on LAMA84, K562 and K562R cell lines. Protein levels of IL3-Rα (Cd123) were evaluated. Blots were stripped and subsequently re-probed with an antibody against Gapdh to ensure equal loading. (E) Analysis at confocal microscopy of LAMA84 (upper panel) or K562R (lower panel) cells treated, for 3h and 6h, with 10μg/ml of HEK293T-derived exosomes (Exo) and IL3L-HEK293T-derived exosomes (IL3L Exo). Nuclei were stained with Hoechst (blue); exosomes were labeled with PKH26 (red).
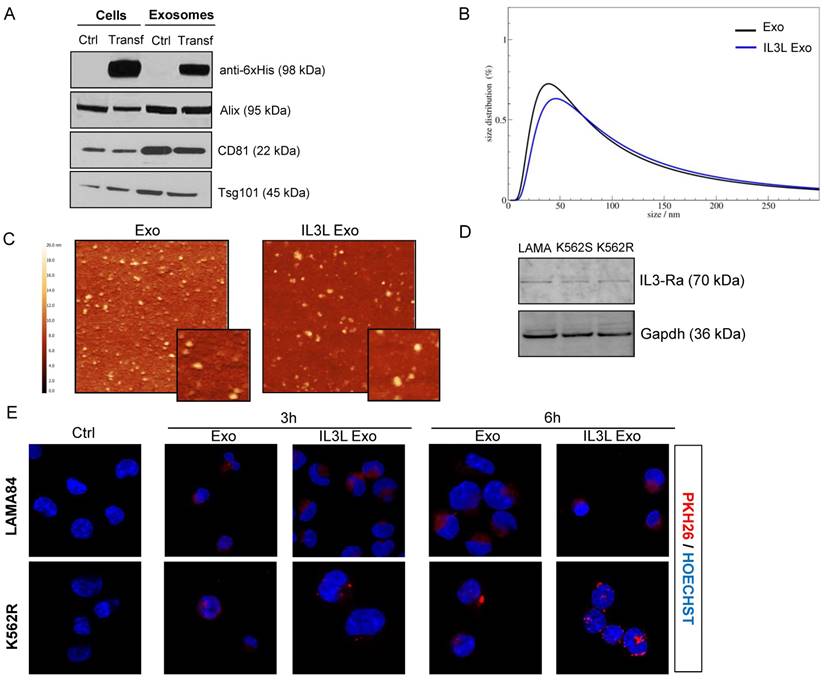
For confirmation that the transfection did not alter exosome composition, we evaluated the expression of different exosomal markers: Alix, CD81 and Tsg101, which are expressed in modified exosomes, as shown in Figure 2A. Quantitative proteomic analysis of Exo and IL3L Exo was also performed and we found that only the 27% of the 340 identified proteins (Table S1) were significantly differentially represented (FC≥2; p≤0.05) in the two exosome populations; 52% of these were structural constituent of ribosomes (Figure S1).
Exosome size distribution (ranging between 30-60 nm), and morphology were analyzed using dynamic light scattering (DLS) and atomic force microscopy (AFM) (Figure 2B and 2C).
On the whole, the data suggest that exosome modification does not alter their size or molecular properties. Western blot analysis then confirmed that CML cell lines express IL3-R (Figure 2D), as reported in literature [27].
In order to determine whether HEK293T-derived exosomes, either expressing IL3-Lamp2b or not, are internalized by human cancer cells, exosomes were labeled with the lipophilic dye PKH26. LAMA84 and K562R cells, treated at 37°C with 10 μg/ml of exosomes for 3 or 6hrs, internalized nanovesicles as shown in Figure 2E and in Figure S2A. Significantly higher fluorescence intensity was detected for IL3L Exo treated cells compared to cells treated with Exo control. Significantly higher fluorescence intensity was also found in cells treated for 6hrs in comparison with 3hrs, independent of the type of exosome. The uptake was blocked after incubation at 4°C (Figure S2B), thus confirming that exosome uptake was mediated by a biologically active process.
In vitro effect of Imatinib-loaded IL3-Lamp2b exosomes
We have examined the possibility of loading engineered exosomes with Imatinib through the direct treatment of HEK293T cells with 0.1, 0.5 and 1 μM of the drug. Exosomes were isolated from the conditioned medium, their Imatinib content quantified (as indicated in Materials and Methods) and used for in vitro and in vivo studies. The final drug quantities in the exosomes of samples obtained from cells treated with 0.1, 0.5 and 1 μM of Imatinib were as follows: 32.55±2.05, 265.3±6.08, 461.65±34.43 nmol/100 μL respectively.
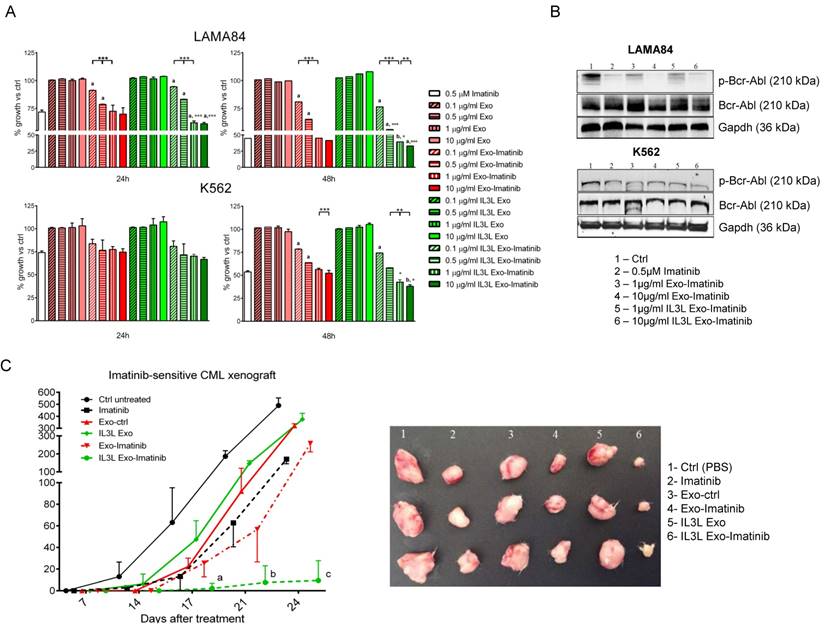
In order to test whether Imatinib-loaded exosomes (isolated from the conditioned medium of cells treated with Imatinib 0.5 μM) were effective in killing CML cells, we treated LAMA84 and K562 cell lines for 24hrs or 48hrs with 0.1, 0.5, 1 or 10 μg/ml of exosomes engineered with and without IL3-Lamp2b, and loaded with and without Imatinib. As shown in Figure 3A, time and dose-dependent reduced viability of LAMA84 (upper panel) or K562 (lower panel) cells treated with Imatinib loaded exosomes (targeted and not) was observed. The effects, which were witnessed following treatment with 1 or 10 μg/ml, were comparable to those obtained with Imatinib, used as the positive control. A slightly greater reduction in cell viability was observed in cells treated with IL3-Lamp2b exosomes loaded with Imatinib. No difference in cell death compared to controls was observed when unloaded exosomes were used.
In vitro and in vivo effects of Imatinib loaded IL3L-exosomes. (A) LAMA84 (upper panel) and K562 (lower panel) growth was measured by MTT assay after 24, 48 h of treatment with Imatinib (positive control), with 0.1, 0.5, 1 or 10 μg/ml of exosomes derived from HEK293T cells, containing or not Imatinib, and with 0.1, 0.5, 1 or 10 μg/ml of exosomes derived from IL3L-HEK293T cells, containing or not Imatinib. The values were plotted as % of growth vs Ctrl (untreated cells - dot line). Each point represents the mean ± SD of three independent experiments (**, p < 0.005; ***, p < 0.0005): versus Imatinib (a, p < 0.0005; b, p < 0.005); versus Exo-Imatinib (°, p < 0.05; °°°, p < 0.0005). (B) Western blot analysis was performed on LAMA84 and K562 cell lines treated for 24h with Imatinib, with 1 or 10 μg/ml Imatinib loaded exosomes derived from HEK293T cells or from IL3L-HEK293T cells. Protein levels of p-Bcr-Abl or Bcr-Abl were evaluated. Blots were stripped and subsequently re-probed with an antibody against Gapdh to ensure equal loading. (C) Comparison of the median tumor volume was an index of the antitumor efficacy of Imatinib loaded exosomes. Significant differences in terms of tumor volume were observed: IL3L Exo-Imatinib versus IL3L Exo at 17 days (a, p < 0.05), IL3L Exo-Imatinib versus IL3L Exo and Imatinib at 21 days (b, p < 0.005), and IL3L Exo-Imatinib versus IL3L Exo, Exo-Imatinib and Imatinib at 24 days (c, p < 0.0005).
To verify in vitro the binding specificity of exosomes expressing IL3L to IL3-R positive cells, we performed a competition assay using IL3L Exo-Imatinib, with increasing amount of empty IL3L Exo. As shown in Supplementary Figure S2C, we observed a decreased effect of IL3L Exo-Imatinib on cell viability, depending on the increased amount of empty IL3L Exo. The effect of IL3L Exo- Imatinib was maintained when control exosomes were used as competitors.
In order to correlate the antiproliferative effects of Imatinib-loaded exosomes on CML cells with Bcr-Abl activity, cells treated with engineered exosomes were harvested and subjected to immunoblotting with antibodies against phosphorylated Bcr-Abl. As shown in Figure 3B, the treatment of CML cells with Imatinib-loaded exosomes was able to decrease the phosphorylation of Bcr-Abl in a dose dependent manner, without altering total protein level.
In vivo effect of Imatinib loaded IL3-Lamp2b exosomes
The ability of Imatinib-loaded targeted exosomes to reduce tumor growth was also tested in an in vivo tumor xenograft model. LAMA84 cells were inoculated subcutaneously in NOD/SCID mice; one week post cell injection, mice were treated intraperitoneally (IP) twice a week with vehicle (PBS), Imatinib (50mg/kg) and 100 μg of exosomes released by HEK293T cells (Exo or IL3L-Exo) loaded with or without Imatinib. At the end of the treatment regimen, mice were sacrificed and the tumors removed. Figure 3C and Figure S3A show that tumor growth was reduced in mice treated with Imatinib (positive control), as well as in mice treated with Imatinib-loaded exosomes. Strikingly, a marked reduction in tumor size was observed in mice treated with Imatinib-loaded IL3L exosomes, leading to the formation of smaller tumors compared to mice treated with Imatinib-loaded exosomes. There were no statistically significant differences between mice treated with PBS (ctrl), with control exosomes or with IL3L exosomes.
In vitro effect of IL3-Lamp2b exosomes loaded with BCR-ABL siRNA
As the clinical application of RNA-based therapy for the treatment of CML [9] has been hampered by the lack of an appropriate delivery systems [11, 29], we next examined the possibility of loading engineered exosomes with BCR-ABL specific siRNA to test their functional activity towards Imatinib sensitive and resistant CML cells. We transfected HEK293T cell lines, engineered with or without IL3L, with siRNAs; exosomes were isolated from the conditioned medium 24hrs after transfection and used in both in vitro and in vivo studies.
In order to test whether siRNA-loaded exosomes showed functional activity in inhibiting Imatinib sensitive and resistant CML cell growth, we treated LAMA84, IM-sensitive or resistant K562 cell lines for 24, 48 and 72hrs with 1 μg/ml of engineered exosomes (IL3L exo), loaded with either BCR-ABL siRNA or scrambled siRNA. As shown in Figure 4A, we observed a time dependent reduced viability of the three cell lines treated with BCR-ABL siRNA-loaded exosomes. As expected, Imatinib treatment did not alter K562R cell growth. No differences compared to controls were observed in scrambled siRNA-loaded engineered exosomes.
In vitro and in vivo effects of BCR-ABL siRNA loaded IL3L-exosomes. (A) LAMA84, K562 and Imatinib resistant K562 cell growth was measured by MTT assay after 24, 48 or 72 h of treatment with Imatinib (on the right side of each histograms) or with 1 μg/ml of exosomes derived from IL3L-HEK293T containing scrambled siRNA or BCR-ABL siRNA. The values were plotted as % of growth vs Ctrl (untreated cells - dot line). Each point represents the mean ± SD of three independent experiments. Statistically significant differences were found between 1 μg/ml IL3L Exo with BCR-ABL and 1 μg/ml IL3L Exo with scramble values (Sidak test: ***, p < 0.0005). In addition, significant decrease in MTT value was observed in each cell type overtime (Sidak test: a, 48h versus 24h - p < 0.0005; b, 72h versus 48h - p < 0.0005). (B) Real-time PCR analysis was performed on CML cell lines treated for 24 or 48 h with Imatinib (on the right side of each histograms) or with 1 μg/ml of exosomes derived from IL3L-HEK293T containing scrambled siRNA or BCR-ABL siRNA. The values were plotted as fold change compared to control (untreated cells - dot line). Each point represents the mean ± SD of three independent experiments. GLM highlighted a significant influence on PCR data by the treatment effect (***, p < 0.0005), but not by experimental time effect. (C) Western blot analysis was performed on CML cell lines treated for 72h with Imatinib or with 1 μg/ml of exosomes derived from IL3L-HEK293T containing scrambled siRNA or BCR-ABL siRNA. Protein levels of Bcr-Abl were evaluated. Blots were stripped and subsequently re-probed with an antibody against Gapdh to ensure equal loading. (D) Comparison of the mean tumor volume was an index of the anti-tumor efficacy of IL3L-HEK293T derived exosomes containing BCR-ABL siRNA. Significant differences in terms of tumor volume were observed at 28 days: IL3L BCR-ABL siRNA versus Ctrl untreated (a, p < 0.0005), IL3L Exo-scrambled siRNA and Imatinib (b, p < 0.005), and versus Exo-scrambled siRNA (c, p < 0.005).
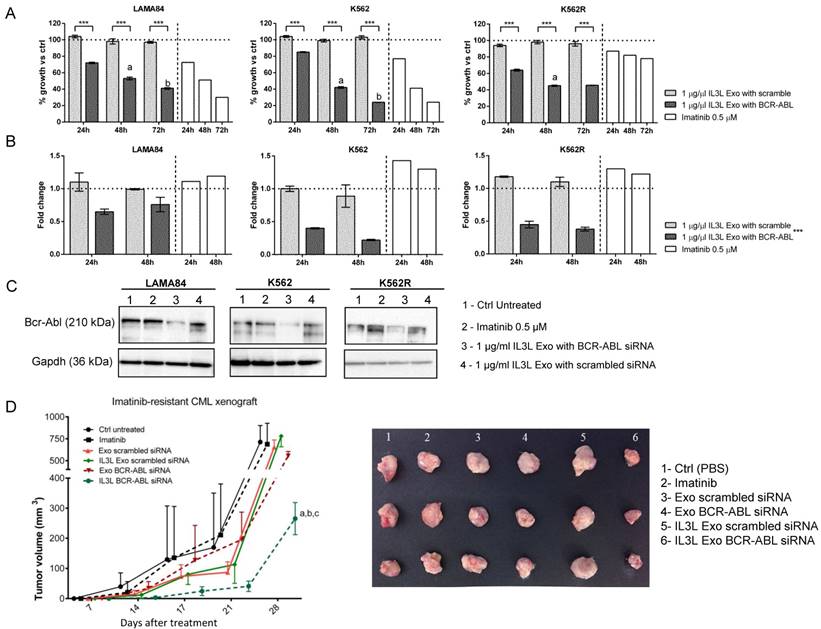
In order to correlate the anti-proliferative effects of BCR-ABL siRNA-loaded exosomes on CML cells with siRNA-mediated Bcr-Abl reduction, cells treated with engineered exosomes were harvested and subjected to Real-Time PCR and western blot analysis with antibodies directed against Bcr-Abl. As shown in Figure 4B, the treatment of CML cells with BCR-ABL siRNA-loaded exosomes was able to decrease Bcr-Abl both at the mRNA (24hrs and 48hrs, Figure 4B) and protein level (72hrs, Figure 4C). No differences in Bcr-Abl levels with respect to controls were observed in cells treated with scrambled siRNA-loaded exosomes.
In vivo effect of IL3-Lamp2b exosomes loaded with BCR-ABL siRNA
In order to test whether IL3L-exosomes were able to deliver functional BCR-ABL siRNA in vivo, K562R cells were inoculated subcutaneously into NOD/SCID mice; one week after cell injection, mice were treated intraperitoneally (IP) twice a week with vehicle (PBS), Imatinib (50mg/kg), 10 μg of HEK293T derived exosomes expressing IL3L and loaded with BCR-ABL siRNAs or scrambled siRNA. At the end of treatment regimen, mice were sacrificed. As reported in Figure 4D and in Figure S3B, the treatment of mice bearing Imatinib-resistant CML cells with IL3L-exosomes containing BCR-ABL siRNA (IL3L Exo BCR-ABL siRNA) determined slower tumor growth compared to control mice (PBS) and to mice treated with control exosomes containing BCR-ABL siRNAs (Exo BCR-ABL siRNA). No differences were observed in mice treated with exosomes containing scrambled siRNAs when compared to the control. Our data suggest that functional BCR-ABL siRNA delivery to the tumor mass by targeted exosomes is possible. Real Time PCR analysis on tumor biopsies showed that the inhibition of tumor growth by targeted exosomes is correlated to a reduction in BCR-ABL mRNA (Figure S4A).
This study highlights a possible new approach to overcome pharmacological resistance in CML by using siRNAs appropriately delivered with exosomes.
In vivo distribution of engineered exosomes
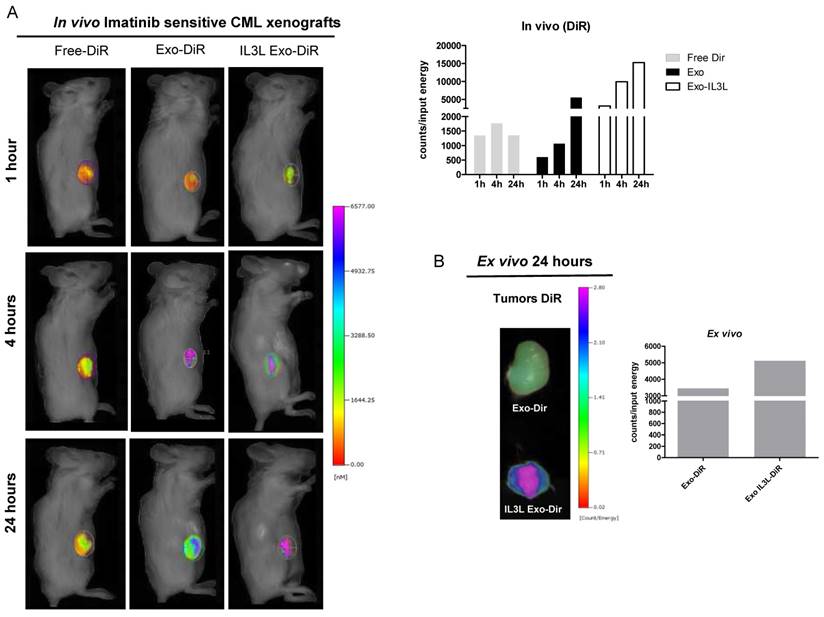
For the purpose of further investigating the in vivo distribution of engineered exosomes, vesicles were labeled with the lipophilic fluorescent tracer DiR. To assess whether engineered exosomes were, in fact, able to reach the tumor site, NOD/SCID mice bearing CML xenografts were treated intraperitoneally with fluorescent tracer DiR (Free-DiR), with PBS, with control exosomes (Exo) or with engineered exosomes (IL3L Exo), labeled with DiR.
As shown in Figure 5A (Imatinib-sensitive CML xenograft) IL3L labeled-exosomes quickly reached tumor tissue and accumulated starting at 1 hr and continuing up to the 24 hr time point, while an increase in the signal corresponding to the accumulation of control exosomes compared to Free-DiR was observed only at the 24hr time point. Similarly, data reported in Figure 6A (Imatinib-resistant CML xenograft) showed that both exosomes accumulated in tumor tissues starting at 4hrs; however, engineered exosomes expressing IL3L reached the tumor site in greater abundance.
In vivo distribution of engineered exosomes in Imatinib sensitive mouse xenograft. (A) NOD/SCID mice bearing Imatinib sensitive CML xenograft tumors in the right flank were injected intraperitoneally with Free-DiR, 100 μg of HEK293T exosomes-DiR, 100 μg of IL3L-HEK293T exosomes-DiR in a volume of 150 μl PBS. Mice were imaged at 1, 4 and 24 h post injection. A scale of the radiance efficiency is presented to the right of mice image. Histogram represents in vivo quantification of in vivo fluorescence (B) Tumors were excised and imaged after 24 h. A scale of the radiance efficiency is presented to the right. Histogram represents ex vivo quantification of tumor fluorescence.
In vivo distribution of engineered exosomes in Imatinib resistant mouse xenograft. (A) NOD/SCID mice bearing Imatinib resistant CML xenograft tumors in the right flank were injected intraperitoneally with Free-DiR, 10 μg of HEK293T exosomes-DiR, 10 μg of IL3L-HEK293T exosomes-DiR in a volume of 150 μl PBS. Mice were imaged at 1, 4 and 24 h post injection. A scale of the radiance efficiency is presented to the right of mice image. Histogram represents in vivo quantification of in vivo fluorescence (B) Tumors were excised and imaged after 24 h. A scale of the radiance efficiency is presented to the right. Histogram represents ex vivo quantification of tumor fluorescence.
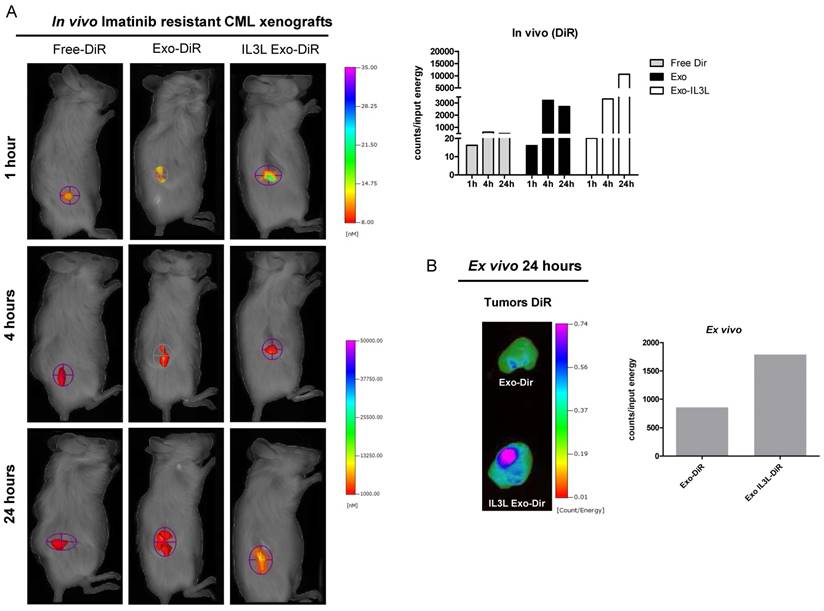
Organ analysis excised 24hrs post injection showed significant absorption by liver, spleen and partially by kidneys (Figure S1C) of both Free-DiR and exosomes-Dir. Analysis of tumors (from mice injected with Free-DiR, with exosomes-DiR or with IL3L exosomes-DiR) indicated that modified exosomes were internalized by tumors and remained in the tumor mass; a weaker signal, however, was observed in the tumors from mice treated with unmodified exosomes (Figure 5B and 6B). Generally speaking, these results clearly suggest that targeted exosomes reach tumor sites in vivo, implying that they are suitable vehicles for the specific delivery of drugs or siRNAs to the tumor site.
Discussion
Although the development and clinical use of Imatinib and third tyrosine kinase inhibitors has revolutionized treatment and improved prognosis in affected patients, the development of pharmacological resistance still remains a problem which urgently needs to be addressed. Alternative methods may consist of a short interfering RNA (siRNA)-based therapy to downregulate the aberrant protein responsible for the neoplastic transformation [30, 31]. Although this approach is highly promising, RNAi for leukemia has reached preclinical trials only in very few cases [32].
The key challenge in the clinical application of siRNAs has been their stability and efficient delivery; such efficiency would require delivery to malignant cells without degradation. In this paper, we propose a new approach to convey Imatinib or BCR-ABL siRNA to CML cells by using exosomes as a delivery system. Exosomes are natural carriers of various molecule species (nucleic acid, proteins, lipids, metabolites) between cells and literature on their use in drug delivery is increasing; thus suggesting that the approach is highly feasible. A number of research groups have demonstrated that it is possible to convey therapeutic RNA molecules through exosomes to target cancer cells or other diseases, such as neurodegenerative disorders [21, 24, 33, 34]. One of the first successful trials of the system was described by Alvarez-Erviti and colleagues: they engineered dendritic cells to express the exosomal protein Lamp2b fused to the neuron-specific RVG peptide in order to deliver siRNAs targeting BACE1, a protein involved in Alzheimer's disease pathogenesis [24]. One year later, Ohno demonstrated that exosomes could also be used to deliver let-7a miRNA to EGFR-expressing breast cancer cells and xenografts, establishing that this approach is functional both in vitro and in vivo [21]. Furthermore, recent evidences underlines the possibility of delivering drugs, such as doxorubicin [35], through exosomes, thereby leading to a reduction in the toxicity of the cancer therapy and to the improvements in drug solubility. Kim and colleagues used exosomes to deliver paclitaxel or doxorubicin in order to overcome multiple drug resistance in lung cancer [36]. In addition, Sun showed that exosomes could enhance the anti-inflammatory activity of curcumin [37].
The benefit of using exosomes as a drug delivery system lies in the fact that they can be specifically targeted to a particular cell type by engineering exosome-producer cells. Nievergall and colleagues have reported that the IL3 Receptor (CD123) is overexpressed in CD34+CD38- CML cells compared to normal CD34+CD38- cells and that its expression level increases with disease progression [25]. Furthermore, Frolova and collaborators have established an efficient IL3-R-targeted system for inducing apoptosis in CML cells by creating a diphtheria toxin- IL3 fusion protein [38].
Based on these data, we considered IL3-R an attractive therapeutic target and so generated an IL3-R-targeted drug delivery system. First we demonstrated the ability to generate exosomes that express IL3 by creating a fusion protein with Lamp2b, a well-characterized exosomal membrane protein [39]. In vitro confocal microscopy analysis showed that CML cells internalize exosomes; in vivo imaging assays also show that IL3L-exosomes are more rapidly internalized by both Imatinib-sensitive and Imatinib-resistant xenografts. This would seem to confirm the efficiency of our IL3-R-targeted delivery approach. Data then led us to use engineered vesicles in the delivery of Imatinib to CML cells. This approach would overcome systemic toxicity associated with prolonged administration of the drug. A targeted drug delivery system would enable drugs to be directed to targeted tissues, thereby requiring lower doses. As clear from Imatinib quantification in the exosomes, doses of Imatinib in the drug-loaded exosomes used in in vitro experiments were 37 times lower than those required for Imatinib treatment alone. As shown, so as to generate IL3L exosomes containing Imatinib, we treated exosome-producing cell lines with 0.5 μM Imatinib and, as described by in vivo tumor growth analysis, we showed that IL3L-resulting exosomes are able to inhibit xenograft growth more efficiently than Imatinib alone or than control exosomes containing Imatinib. In vivo imaging, showing the rapid internalization of IL3L exosomes, supports these data.
Test results encouraged us to use the same approach in the delivery of siRNAs to Imatinib-resistant cells. This system could be highly important in clinical application and could be employed to overcome pharmacological resistance in tumors, such as in CML, where preclinical studies on the use of siRNA-based therapy gave promising data but the lack of an appropriate delivery system led to reduced clinical application. Here we show for the first time that it is possible to generate IL3L exosomes containing siRNAs to BCR-ABL; engineered exosomes are internalized by Imatinib resistant CML cells and xenografts, and lead to a reduction in tumor growth in an efficient manner.
Data we show are very promising and provide a rational base for the use of exosomes expressing a fragment of IL3 in a receptor-targeted therapy approach for use in CML patients and, in particular, in order to overcome pharmacological resistance. A future step, providing information on the clinical applicability of this approach, will be to engineer exosomes from immature dendritic cells (DCs) or mesenchymal stem cells (MSCs). The use of exosomes from DCs will prevent any in vivo immunological stimulating activity, while the use of MSCs will ensure an adequate amount of nanovesicles for clinical studies.
Finally, considering the in vivo heterogeneity of a tumor, which leads to differing cellular response to therapies, the use exosomes to deliver a combination of drugs and siRNAs is also conceivable. Further studies are needed to test the feasibility of this combined approach.
While focusing on CML, results from this study could have an impact on other types of tumor, such as acute myeloid leukemia, where IL3R is abundantly expressed.
Methods
Ethics Statement
The study was conducted in compliance with Italian and European laws concerning animal experiments. The research protocol "Sviluppo e caratterizzazione di modelli animali di xenotrapianto di tumori solidi, cellule tumorali e staminali tumorali da neoplasie umane" was authorized by Italian Ministry of Health (2014/01/13) according to Legislative Decree 116/92 and was performed according to Legislative Decree 26/2014.
Cell cultures and reagents
HEK293T cells were obtained from ATCC (Manassas, VA, USA) and grown in DMEM High Glucose supplemented with 10% fetal bovine serum. For exosome loading, cells were treated with 0.5 μM of Imatinib. Chronic Myelogenous Leukemia cell lines, LAMA84, K562 and Imatinib-resistant K562 cells (K562R), kindly provided by Dr. Paolo Vigneri (University of Catania), were cultured in RPMI 1640 medium (Euroclone, UK) supplemented with 10% fetal bovine serum (Euroclone, UK). Imatinib Mesylate (Selleckchem, Munich, Germany) was prepared as a 500 μM stock solution in sterile phosphate-buffered saline (PBS).
IL3- Lamp2b Plasmid construction and transfection protocol
We developed a Pinco-based construct that over-expresses a fusion protein (IL3-Lamp2b) that contains human Lamp2B protein and a portion of human Interleukin 3 protein in the extracellular domain as described[40]. The 6XHis tag has been added to the carboxy-terminal region of IL3-Lamp2b recombinant protein.
HEK293T cells were seeded in a 6-well plate; after 24 hours, cell transfection was performed with 2.5 μg of IL3-Lamp2b plasmid DNA according Lipofectamin 3000 protocol (Life Technologies, California, US). The cells were expanded and then selected with 1 μg/ml of puromicin for 4 weeks.
SiRNA Cell transfections
HEK293T and IL3L-HEK293T cells were transfected with 100 μM of scrambled siRNA or BCR-ABL siRNAs (Dharmacon RNA Technologies, Lafayette, CO) using Lipofectamine RNAiMAX Transfection Reagent (Life Technologies) following manufacturer's indications.
Exosome preparation
Exosomes released by HEK293T cells and by IL3L-HEK293T cells, treated or not with 0.5 μM Imatinib for 24h or transfected with BCR-ABL or scrambled siRNAs, cultured in presence of FBS previously ultracentrifuged (vesicle free media), were isolated from conditioned culture medium by differential centrifugation, as previously described[41]. Exosome protein content was determined with the Bradford assay (Pierce, Rockford, IL, USA).
Proteomic analysis: sample preparation, SWATH-MS and data analysis
Exosomes were dissolved in 50% TFE/PBS and subjected to tryptic digestion. Samples were prepared and subjected to DDA and SWATH analysis. A deep description of tryptic digestion protocol and DDA/SWATH procedures are reported in Supplementary Material and Methods. DDA raw files were combined and searched against the human database to generate the reference spectral library which was used for SWATH data processing and quantification. The protein list with FDR lower than 5% generated by analyzing SWATH data with PeakView 2.2, was exported to MarkerView for statistical data analysis using a pairwise t-test. Four technical replicates were performed for each exosome population and Fold Change (FC) IL3L Exo vs Exo thresholds at 2 with an adjusted p-value inferior to 0.05 were used to consider a protein up or down-regulated. The Gene Ontology (GO) of proteins differentially regulated in IL3L Exo was performed using the stand-alone enrichment analysis tool FunRich. (Functional Enrichment analysis tool; http://www.funrich.org).
Atomic Force Microscopy (AFM) analysis
Substrate functionalization: glass sliced were functionalized according to the following treatment: (i) cleaned by immersion in boiling acetone for a few minutes, washed with Milli-Q water, dried in a stream of high-purity nitrogen and exposed to UV rays (30W Hg lamp) in order to expose the hydroxyl groups of silica; (ii) treated with a 0.25 M (3-aminopropyl)-triethoxysilane (APTES) in chloroform solution for 3 minutes at room temperature, and then rinsed with chloroform and dried with nitrogen; (iii) treated with 0.4 M glutaraldehyde aqueous solution for 3 minutes at room temperature and then rinsed with Milli-Q water and dried with nitrogen. AFM vesicle imaging: a vesicle solution diluted in PBS to a final concentration of 1 ng/μl was deposited into APTES/gluteraldehyde functionalized glass slides and incubated for 24h. Tapping mode AFM measurements were carried out in liquid by using a Nanowizard III scanning probe microscope (JPK Instruments AG, Germany) equipped with a 12-um scanner, and AC40 (Bruker) silicon cantilevers (spring constant 0.1 N/m, typical tip radius 7 nm, resonance frequency 33kHz, oscillation amplitude 7nm, scan rate 1.5 Hz).
Quantification of Imatinib Mesylate in exosomes
For generating standard curve, Imatinib Mesylate was prepared as a 250 μM stock solution in sterile PBS. The calibration curve was carried out using different aliquots of standard solution diluted with PBS to give Imatinib final concentrations of 12.5, 2.50, 1.25, 0.62, 0.31, and 0.155 μM. All absorption spectral measurements of each solution were measured at absorption maximum of 321.7 nm using a Beckman DU-640 spectrophotometer as described in Bende et al[42]. Samples of 2 μg/μL of exosomes (final volume 100 μL) obtained from cells treated with Imatinib (0.1, 0.5 and 1 μM) were diluted to 600 μL and sonicated for 5 minutes. Exosome obtained from untreated cells were used as blank. The absorbance was measured for each sample.
Uptake of exosomes by LAMA84 and K562R cells
Exosomes were isolated as described above and labeled with PKH26 (Sigma-Aldrich, St. Louis, MO, USA) for 10 min at room temperature. Labeled vesicles were washed twice in PBS and suspended in complete medium. LAMA84 and K562R cells were treated with 10 μg/ml of labeled vesicles for 3 or 6 hours at 4 or 37°C. Nuclei were stained with Hoechst 3342 (Molecular probes, Life Technologies) and analyzed by confocal microscopy. The analysis of fluorescence intensity was performed using IMAGE-J software (http://imagej.nih.gov/ij/).
CML mouse xenograft
Female NOD/ SCID mice, four-to-five weeks old, were purchased from Harlan (Harlan Laboratories, Indianapolis, Indiana) and acclimated for a week prior to experimentation. Mice received filtered water and sterilized diet ad libitum. Each mouse was inoculated subcutaneously in the right flank with viable single human LAMA84 cells or K562R cells (2 x 107) suspended in 0.2 ml of PBS. The day of injection was considered as Day 0. On Day 7, when tumors were palpable, mice carrying LAMA84 xenografts were randomly assigned to six groups of five and were treated with: (a) PBS (Ctrl), (b) Imatinib (50mg/kg), (c) HEK293T-derived exosomes (Exo-ctrl, 100 μg/mouse), (d) IM-loaded HEK293T-derived exosomes (Exo-Imatinib, 100 μg/mouse), (e) IL3L/HEK293T -derived exosomes (IL3L Exo, 100 μg/mouse) or with (f) IM-loaded IL3L/HEK293T -derived exosomes (IL3L Exo-Imatinib, 100 μg/mouse).
Mice carrying K562R xenografts were randomly assigned to six groups of five and were treated with: (a) PBS (Ctrl), (b) Imatinib (50mg/kg), (c) scrambled siRNA transfected HEK293T-derived exosomes (Exo scrambled siRNA, 10 μg/mouse), (d) BCR-ABL siRNA transfected HEK293T-derived exosomes (Exo BCR-ABL siRNA, 10 μg/mouse), (d) scrambled siRNA transfected IL3L/HEK293T-derived exosomes (IL3L Exo scrambled siRNA, 10 μg/mouse), (e) BCR-ABL siRNA transfected IL3L/HEK293T-derived exosomes (IL3L Exo BCR-ABL siRNA, 10 μg/mouse).
All treatments were administered intraperitoneally and repeated twice a week for three weeks. Xenograft tumors were measured and mice were weighed twice a week. Tumor volume was determined by caliper with the following formula: (length x width2) /2. Animals were euthanized at day 24 and the tumors resected.
In vivo exosomes distribution
Exosome labeling procedure: DiR was used to fluorescently label the lipid bilayer of HEK293T-derived exosomes or IL3L/HEK293T-derived exosomes as previously described [15].
In vivo imaging: for the detection of fluorescence, mice bearing CML (LAMA84 or K562R) xenografts were anesthetized and then injected IP with Free DiR or DiR labeled-exosomes. Mice images were acquired after 1h, 4h or 24h from injection (FMT 2500X, PerkinElmer LifeSciences). Following fluorescent background subtraction, images were analyzed and scaled after completion of all acquisitions, using appropriate computer software (TrueQuant; PerkinElmer LifeSciences). Following the last acquisition, the animals were sacrificed and the organs (spleen, liver, kidneys and tumor) were collected and acquired with same imaging system. The scale bar was expressed as Count/ Input Energy.
Statistical analysis
Statistical analysis was performed by using IBM® SPSS® Statistics 23 software. Data are reported as mean ± standard deviation (SD) at a significant level p < 0.05. After having verified normal distribution (Shapiro-Wilk test) and homoschedasticity of data (Levene test), a general linear model (GLM) was used by considering treatment and experimental time as fixed effects, followed by Sidak post hoc test to evaluate significant differences among cell viability results and among RT-PCR results. For cell viability results, the following comparisons were taken into account because considered biological relevant:
- 1 μg/ml IL3L Exo-Imatinib, 10 μg/ml IL3L Exo-Imatinib, 1 μg/ml Exo-Imatinib and 10 μg/ml Exo-Imatinib treaments versus 0.5 μM Imatinib;
- 1 μg/ml IL3L Exo-Imatinib and 10 μg/ml IL3L Exo-Imatinib versus 1 μg/ml Exo-Imatinib and 10 μg/ml Exo-Imatinib treatments.
Regarding the analysis of volume tumor data, a GLM (General Linear Model) procedure for repeated measures (experimental time) was applied. The sample size n=30 for each experiment was calculated with a priori power analysis by considering an effect size f=0.25, α=0.05 and 1-β=0.80.
Supplementary Material
Additional File 1Supplementary methods and figures.
Supplementary table S1.
Acknowledgements
We would like to thank Dr. Giampiero Santocono for in vivo experiment support. This work was supported by a grant from the Associazione Italiana per la Ricerca sul Cancro (AIRC) to Riccardo Alessandro; University of Palermo to Riccardo Alessandro and Giacomo De Leo. This study was also developed with the contribution of National Operational Programme for Research and Competitiveness 2007-2013- PON01_01059 “Sviluppo di una piattaforma tecnologica per il trattamento non invasivo di patologie oncologiche e infettive basate sull'uso di ultrasuoni focalizzati (FUS)”. Stefania Raimondo was supported by a “Fondazione Veronesi” and a “FIRC” (Fondazione Italiana Ricerca sul Cancro) fellowship.
Authorship contributions
D.B., S.R., R.A. designed the experiments and wrote the manuscript. D.B., S.R. performed and analyzed the experiments. S.R., G.C., S.F. performed in vivo experiments, M.M., S.R., performed DLS analysis, P.D., G.C. performed Imatinib quantification in exosomes, S.F., F.M. performed proteomic analysis, G.G. performed statistical analysis, L.M., G.D.L. provided critical feedback to the manuscript.
Conflicts of interest
The authors have declared that no competing interest exists. The content is object of Italian Patent Application No. 102016000038900 filed on April 15th 2016.
References
1. Faderl S, Kantarjian HM, Talpaz M. Chronic myelogenous leukemia: update on biology and treatment. Oncology (Williston Park). 1999;13:169-80 discussion 81, 84
2. Smith DL, Burthem J, Whetton AD. Molecular pathogenesis of chronic myeloid leukaemia. Expert Rev Mol Med. 2003;5:1-27
3. Druker BJ, Lydon NB. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest. 2000;105:3-7
4. Jabbour E, Kantarjian H, Cortes J. Use of second- and third-generation tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia: an evolving treatment paradigm. Clin Lymphoma Myeloma Leuk. 2015;15:323-34
5. Mughal TI, Schrieber A. Principal long-term adverse effects of imatinib in patients with chronic myeloid leukemia in chronic phase. Biologics. 2010;4:315-23
6. Farmer S, Horvath-Puho E, Vestergaard H, Hermann AP, Frederiksen H. Chronic myeloproliferative neoplasms and risk of osteoporotic fractures; a nationwide population-based cohort study. Br J Haematol. 2013;163:603-10
7. Mahon FX, Deininger MW, Schultheis B, Chabrol J, Reiffers J, Goldman JM. et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070-9
8. Pinilla-Ibarz J, Sweet K, Emole J, Fradley M. Long-term BCR-ABL1 Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia. Anticancer Res. 2015;35:6355-64
9. Koldehoff M, Zakrzewski JL, Beelen DW, Elmaagacli AH. Additive antileukemia effects by GFI1B- and BCR-ABL-specific siRNA in advanced phase chronic myeloid leukemic cells. Cancer Gene Ther. 2013;20:421-7
10. Zhao RC, McIvor RS, Griffin JD, Verfaillie CM. Gene therapy for chronic myelogenous leukemia (CML): a retroviral vector that renders hematopoietic progenitors methotrexate-resistant and CML progenitors functionally normal and nontumorigenic in vivo. Blood. 1997;90:4687-98
11. Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19:60-71
12. Yingchoncharoen P, Kalinowski DS, Richardson DR. Lipid-Based Drug Delivery Systems in Cancer Therapy: What Is Available and What Is Yet to Come. Pharmacol Rev. 2016;68:701-87
13. Raimondo S, Corrado C, Raimondi L, De Leo G, Alessandro R. Role of Extracellular Vesicles in Hematological Malignancies. Biomed Res Int. 2015;2015:821613
14. Corrado C, Raimondo S, Chiesi A, Ciccia F, De Leo G, Alessandro R. Exosomes as intercellular signaling organelles involved in health and disease: basic science and clinical applications. Int J Mol Sci. 2013;14:5338-66
15. Raimondo S, Naselli F, Fontana S, Monteleone F, Lo Dico A, Saieva L. et al. Citrus limon-derived nanovesicles inhibit cancer cell proliferation and suppress CML xenograft growth by inducing TRAIL-mediated cell death. Oncotarget. 2015;6:19514-27
16. Conigliaro A, Costa V, Lo Dico A, Saieva L, Buccheri S, Dieli F. et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol Cancer. 2015;14:155
17. Simons M, Raposo G. Exosomes-vesicular carriers for intercellular communication. Curr Opin Cell Biol. 2009;21:575-81
18. El Andaloussi S, Lakhal S, Mager I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev. 2013;65:391-7
19. Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles - endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta. 2014;1846:75-87
20. Hu G, Drescher KM, Chen XM. Exosomal miRNAs: Biological Properties and Therapeutic Potential. Front Genet. 2012;3:56
21. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N. et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21:185-91
22. Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z. et al. Exosomes as drug delivery vehicles for Parkinson's disease therapy. J Control Release. 2015;207:18-30
23. Saari H, Lazaro-Ibanez E, Viitala T, Vuorimaa-Laukkanen E, Siljander P, Yliperttula M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J Control Release. 2015
24. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341-5
25. Nievergall E, Ramshaw HS, Yong AS, Biondo M, Busfield SJ, Vairo G. et al. Monoclonal antibody targeting of IL-3 receptor alpha with CSL362 effectively depletes CML progenitor and stem cells. Blood. 2014;123:1218-28
26. Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P. et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100:2980-8
27. Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014;2:4
28. Li J, Chen X, Yi J, Liu Y, Li D, Wang J. et al. Identification and Characterization of 293T Cell-Derived Exosomes by Profiling the Protein, mRNA and MicroRNA Components. PLoS One. 2016;11:e0163043
29. Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711-9
30. Mendonca LS, Moreira JN, de Lima MC, Simoes S. Co-encapsulation of anti-BCR-ABL siRNA and imatinib mesylate in transferrin receptor-targeted sterically stabilized liposomes for chronic myeloid leukemia treatment. Biotechnol Bioeng. 2010;107:884-93
31. Landry B, Valencia-Serna J, Gul-Uludag H, Jiang X, Janowska-Wieczorek A, Brandwein J. et al. Progress in RNAi-mediated Molecular Therapy of Acute and Chronic Myeloid Leukemia. Mol Ther Nucleic Acids. 2015;4:e240
32. Koldehoff M, Steckel NK, Beelen DW, Elmaagacli AH. Therapeutic application of small interfering RNA directed against bcr-abl transcripts to a patient with imatinib-resistant chronic myeloid leukaemia. Clin Exp Med. 2007;7:47-55
33. Greco KA, Franzen CA, Foreman KE, Flanigan RC, Kuo PC, Gupta GN. PLK-1 Silencing in Bladder Cancer by siRNA Delivered With Exosomes. Urology. 2016;91:241 e1-7
34. Cooper JM, Wiklander PB, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M. et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord. 2014;29:1476-85
35. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ. et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383-90
36. Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL. et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine. 2016;12:655-64
37. Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C. et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. 2010;18:1606-14
38. Frolova O, Benito J, Brooks C, Wang RY, Korchin B, Rowinsky EK. et al. SL-401 and SL-501, targeted therapeutics directed at the interleukin-3 receptor, inhibit the growth of leukaemic cells and stem cells in advanced phase chronic myeloid leukaemia. Br J Haematol. 2014;166:862-74
39. Simhadri VR, Reiners KS, Hansen HP, Topolar D, Simhadri VL, Nohroudi K. et al. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS One. 2008;3:e3377
40. El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C. et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012;7:2112-26
41. Raimondo S, Saieva L, Corrado C, Fontana S, Flugy A, Rizzo A. et al. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun Signal. 2015;13:8
42. Bende G, Kollipara S, Sekar V, Saha R. UV-spectrophotometric determination of imatinib mesylate and its application in solubility studies. Pharmazie. 2008;63:641-5
Author contact
![]() Corresponding author: Riccardo Alessandro, Dipartimento di Biopatologia e Biotecnologie Mediche, University of Palermo, Palermo 90133, Italy. E-mail: riccardo.alessandroit, phone: +390916554603
Corresponding author: Riccardo Alessandro, Dipartimento di Biopatologia e Biotecnologie Mediche, University of Palermo, Palermo 90133, Italy. E-mail: riccardo.alessandroit, phone: +390916554603