Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1360-1372. doi:10.7150/thno.16532 This issue Cite
Research Paper
Nucleolin-targeted Extracellular Vesicles as a Versatile Platform for Biologics Delivery to Breast Cancer
Yayu Wang1*, Xiaojia Chen1*, Baoqing Tian1*, Jiafan Liu1, Li Yang1, Lilan Zeng2, Tianfen Chen2, An Hong1 ![]() , Xiaogang Wang1
, Xiaogang Wang1 ![]()
1. Institute of Biomedicine & Department of Cell Biology, Jinan University; National Engineering Research Center of Genetic Medicine; Guangdong Provincial Key Laboratory of Bioengineering Medicine, Guangzhou, China;
2. Department of Chemistry, Jinan University, Guangzhou, China.
* These authors contributed equally to this work.
Received 2016-6-19; Accepted 2017-1-30; Published 2017-3-22
Abstract

Small interfering RNAs (siRNA)/microRNAs (miRNA) have promising therapeutic potential, yet their clinical application has been hampered by the lack of appropriate delivery systems. Herein, we employed extracellular vesicles (EVs) as a targeted delivery system for small RNAs. EVs are cell-derived small vesicles that participate in cell-to-cell communication for protein and RNA delivery. We used the aptamer AS1411-modified EVs for targeted delivery of siRNA/microRNA to breast cancer tissues. Tumor targeting was facilitated via AS1411 binding to nucleolin, which is highly expressed on the surface membrane of breast cancer cells. This delivery vesicle targeted let-7 miRNA delivery to MDA-MB-231 cells in vitro as confirmed with fluorescent microscopic imaging and flow cytometry. Also, intravenously delivered AS1411-EVs loaded with miRNA let-7 labeled with the fluorescent marker, Cy5, selectively targeted tumor tissues in tumor-bearing mice and inhibited tumor growth. Importantly, the modified EVs were well tolerated and showed no evidence of nonspecific side effects or immune response. Thus, the RNAi nanoplatform is versatile and can deliver siRNA or miRNA to breast cancer cells both in vitro and in vivo. Our results suggest that the AS1411-EVs have a great potential as drug delivery vehicles to treat cancers.
Keywords: Extracellular vesicles (EVs), Aptamer, Tumor-targeted delivery system, siRNA, microRNA.
Introduction
Nucleic acid drugs include oligonucleotides, aptamers, and nucleic acid vaccines [1, 2]. Numerous studies suggest that oligonucleotide drugs have promise for treating cancer and other diseases [3-7]. For example, miRNA lethal-7 (let-7) plays a critical role in cell proliferation, differentiation, and apoptosis in mammals. The let-7 gene was first identified in the nematode as a key developmental regulator and became one of the earliest known microRNAs [8]. Its expression is significantly different between normal and tumor tissues. In normal tissues, it inhibits malignant growth of cancer cells by reducing MYC and RAS expression. Evidence suggests that restoration of let-7 expression may be a useful therapeutic option for treating cancers [9, 10]. Also, siRNA target-specific gene silencing has been attempted for cancer therapy because cancers often overexpress a specific growth factor as well as rely on relevant receptor proteins for their growth and progression [11]. Use of siRNAs to specifically silence genes for treating cancers or other diseases may have value [12-17], but successful implementation of nucleic acid drugs as new therapeutic tools largely depends on the appropriate delivery system.
The efficiency and safety of drug delivery by current methods remain a challenge. Viruses can mediate long-term gene silencing by integrating small hairpin RNA expression cassettes into the genome and can be engineered to be tissue-specific. The drawbacks, however, are genetic deregulation and oncogene activation by insertional mutagenesis [18]. It was previously reported that lipid nanoparticle-delivered siRNA escaped from endosomes into the cytosol at low efficiency [19]. Therefore, new delivery agents are needed that combine a high degree of target specificity with efficient delivery and minimal immunogenicity.
EVs consist of a lipid bilayer membrane containing various proteins, RNA, and DNA. There are subpopulations of EVs including apoptotic bodies (1,000-5,000 nm), intermediate-sized microvesicles (200-1,000 nm), and exosomes (30-150 nm). Their classification usually dependens upon their size and specific biogenesis [20]. The best studied of these are exosomes which are released by most if not all cell types [21, 22]. In 2007, Valadi's group first reported that EVs contain endogenous RNAs [23]. These nano-size bubble-like particles function as vehicles between cell-to-cell trafficking to release proteins, mRNAs, and miRNAs for intercellular communication. EVs as drug delivery vehicles may have advantages. First, EVs can be derived from the patient's cells implying low immunogenicity. EVs are widely distributed in extracellular fluids and likely have longer circulating times which may translate into improved efficacy. EVs may fuse with the plasma membrane of the target cell releasing contents into the cytosol [24]. These advantages suggest that EVs are natural carriers for nucleic acid drug delivery and can be exploited as ideal drug delivery systems for therapeutic applications in cancer. To date, most studies have exploited EVs as delivery systems for transfer of siRNAs [25-27] and microRNA [28] as therapeutic cargo. Few studies exist to describe other types of therapeutic cargo by exosomal vesicles [29, 30]. Problems with tissue or cell targeting have been addressed by modifying the EVs to express a targeting peptide on the EVs surface membrane [28, 29, 31] but the scale-up production is complicated and time consuming. Furthermore, yields of engineered EVs are not suitable for clinical applications [29].
AS1411, a DNA aptamer that binds with high affinity to nucleolin (overexpressed on breast cancer cell plasma membranes) is the first one that was assessed in oncology clinical trials [32]. As a tumor-targeting ligand, AS1411 shows promise because it inhibits tumor activity and has low systemic toxicity according to a phase II trial [33]. EVs are rich in lipids—predominantly cholesterol, sphingolipids, phospholipids and bisphosphates [34]. Cholesterol for example, has a strong affinity for the EVs membrane. Motivated by this rationale, we employed cholesterol for conjugating the AS1411 aptamer with EVs to confer tumor targeting ability. We then assessed the ability of AS1411 aptamer-modified EVs to deliver let-7 miRNA or VEGF siRNA to nucleolin-expressing cancer tissues. We observed the high antitumor activity of the AS1411-EVs loaded with the lethal-7 gene microRNA precursor, let-7, or VEGF siRNA in vitro and in vivo. The results suggest that our work may provide a versatile delivery platform for nucleic acid drugs in cancer treatment.
Materials and methods
Materials
100kD ultrafiltration membrane was purchased from Merck Millipore. Total Exosome Isolation Reagent (No.4478359) was purchased from Invitrogen. Electroporation cuvettes were obtained from BIO-RAD. Transwell inserts (BD Falcon; 353097) were acquired from Becton Dickinson. Dynabeads® was purchased from Life Technologies. 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8) reduction assay kit was purchased from Dojindo Molecular Technologies. Anti-KRAS (sc-30), anti-nucleolin (MS-3) and anti-c-Myc (sc-40) antibodies were from Santa-Cruz, anti-VEGF antibody (BS6496) was from Bioworld, anti-β-actin (4967s) antibody was from Cell Signaling Technology. Anti-EEA1 (3288) and anti-RAB5 (3547) antibodies were acquired from Cell Signaling Technology. Cholesterol- polypeptide (Cholesterol- IIASTIGGIFG SSTTQSGGGG) was purchased from Top-peptide Biotechnology. NH2-AS1411 (NH2-GGTGGTGGTGGTTGTGGTGGTGGTGG) and NH2-C-AS1411 (NH2-ATCGATCGATCGATCGATCGATCGAT) was obtained from Sangon Biotechnology. siRNA-VEGF (sense: 5′-GCACAUAGGAGAGAUGAGCUU-3′ and antisense: 5′-AAGCUCAUCUCUCCUAUGUGCUG-3′)[35] and let-7 were purchased from Guangzhou RIBOBIO company.
Cells
The cell lines, MDA-MB-231 and C2C12, were purchased from the American Type Culture Collection (ATCC). MDA-MB-231 cells were cultured in RPMI 1640 basic medium (Life Technologies, USA) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, USA) and antibiotics. C2C12 cells were cultured in in Dulbecco's Modified Eagle's Media (DMEM) basic medium (Life Technologies, USA) supplemented with 10% FBS. All cells were maintained in 5% CO2 at 37°C.
Isolation of dendritic cells and purification of EVs
Primary dendritic cells were generated from the bone marrow of the murine 4T1 breast tumor model and cultured in DMEM Glutamax (Life Technologies, USA); 10% FBS and antibiotics; and supplemented with 10 ng/ml murine GM-CSF and IL-4 (Sigma-Aldrich, USA). To remove most of the EVs present in the FBS, it was spun at 100,000 g for 60 minutes before adding to DMEM. The cell culture medium was changed every two days. The cell culture medium was harvested, centrifuged at 12,000 g for 45 minutes to remove cell debris, ultrafiltered through a 100 kDa membrane to concentrate the EVs-containing medium, and washed once with PBS. Subsequently, 1/2 volume of Total Exosome Isolation Reagent and mixed well by vortexing until a homogenous solution was formed. The samples were incubated at 4°C overnight and then centrifuged at 4°C at 12,000 g for 1 hour. The supernatant was discarded and the purified EVs were resuspended in PBS buffer and stored at -80°C.
Synthesis of T-AS1411
10 mg of the cholesterol-modified peptide, 27 mg EDC (1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride), and 16 mg NHS (N-Hydroxysuccinimide; 1-hydroxypyrrolidine-2,5-dione) were dissolved in 1 mL Milli-Q water and stirred for 15 min before adding 50 μL of 100 M NH2-AS1411. The mixed solution was reconstituted in a final volume of 10 mL with Milli-Q water. After 24 h, the mixed solution was dialyzed against Milli-Q water for 12 h to remove the excess AS1411 and polypeptide. The product was named T-AS1411. So T-AS1411 was synthesized after AS1411 was modified by covalently conjugating polypeptides with cholesterol.
Transmission electron microscopy
15 μg of AS1411-EVs were fixed with 4% paraformaldehyde for 30 minutes at room temperature, treated in an ultrasonic bath for 2 minutes, and then vortexed for 1 minute. Subsequently, the sample was dropped onto UV-treated EM (electron microscopy) grids and kept at room temperature for 30 minutes to dry. The samples were stained with 2% uranyl acetate for 15 minutes, the excess uranyl acetate was removed and the EVs were examined by transmission electron microscope (TEM) using Philips Tecani 10 (Philips, Eindhoven, Netherlands) at 100 kV.
Loading of siRNA/miRNA into AS1411-EVs by electroporation
150 μg AS1411-EVs derived from dendritic cells and 150 μg siRNA/miRNA were gently mixed in 200 mL of opti-MEMTM reduced serum medium. The mixture was transferred into an ice cold 0.4 cm electroporation cuvette and electroporated by the Eppendorf Multiporator at 0.7 kV using a 350 microsecond pulse 20 times as previously described [25]. The electroporated mixture was placed at 37°C for 30 minutes. Finally, the mixture was concentrated and washed on a 100kD ultrafiltration membrane to remove naked siRNA/miRNA.
Efficiency of miRNA loading into AS1411-EVs by Real-time PCR (Q-PCR) assay
To test the loading efficiency by electroporation, nonconserved Caenorhabditis elegans miRNA, cel-miR-67 (5′-UCACAACCUCCUAGAAAGAGUAGA-3′), was loaded in AS1411-EVs by electroporation. The mixture was divided into two halves. RNase was added to one half for 30 minutes at 37°C to digest the miRNA in the outer AS1411-EVs membrane; the other did not undergo RNAase digestion and served as a control. The samples were homogenized in Trizol Reagent (Life Technologies, USA) and equal amounts of cel-miR-39 (5′-UCACCGGGUGUAAAUCAGCUUG-3′) were added to lysed samples for technical normalization. miRNA samples were reverse transcribed and the AS1411-EVs internal cel-miRNA-67 was subjected to Q-PCR.
To address the issue that electroporation could potentially cause siRNA/miRNA aggregation [36] and the aggregated miRNA may be resistant to RNAse, we set up a negative control. For this, just only cel-miR-67 self was used for electroporation and analyzed as the following described. The loading efficiency was calculated as (%)= 100 X(Iafter digest - Icontrol)/ (Ibefore digest ) where Iafter digest was the quantity of cel-miRNA-67 in the group that the mixture of AS1411-EVs and cle-miRNA-67 was subjected to electroporation and subsequent RNAase digestion; the Ibefore digest was the quantity of the cel-miRNA-67 in the group that the mixture of AS1411-EVs and cle-miRNA-67 was subjected to electroporation but did not undergo RNAase digestion; the Icontrol was the quantity of aggregated cel-miRNA-67 in the negative control group where just only cle-miRNA-67 self was electroporated and underwent RNAase digestion.
Identification of AS1411-EVs-let-7-Cy3 by Dynabeads® capture
The let-7-Cy3 miRNA was loaded into the AS1411-EVs by electroporation. The AS1411-EVs-let-7-Cy3 were incubated with CD63 antibodies conjugated to Dynabeads® at room temperature for 1 hour. The fluorescence signal was then analyzed by fluorescence microscopy.
Snythesis and characterization of AS1411-EVs
To combine EVs with AS1411, 10 μL EVs (10 μg/μL) and 40 μL T-AS1411 (10 μM) were gently mixed in 100μL PBS. The mixture was incubated at 4°C overnight. The characterization of size and zeta potential of EVs and AS1411-EVs was performed with the Malvern Zetasizer Nano ZS (Malvern Instruments Ltd, UK).
Quantification of released miRNA by Q-PCR
The miRNA cel-miR-67 was loaded into AS1411-EVs by electroporation and divided into divided into equal amounts, which were cultured in 200 μL PBS or serum at 37°C, digested by RNAase at 0, 2, 4, 8, 12, 24 h, and then homogenized in Trizol. Finally, we analyzed the AS1411-EVs internal cel-miRNA-67 by Q-PCR. Since electroporation could potentially cause siRNA/miRNA aggregation, a negative control was set up with only electroporation of cel-miR-67 self and RNAse digestion in corresponding time. The aggregation cel-miRNA-67 was then analyzed by Q-PCR.
Western blot analysis
Cells were lysed in lysis buffer (50 mM Tris, pH 7.5, 250 mM NaCl, 0.1% SDS, 2 mM dithiothreitol, 0.5% NP-40, 1 mM PMSF and protease inhibitor cocktail) on ice for 30 minutes. Protein fractions were collected by centrifugation at 13,000 g at 4°C for 10 minutes and the supernatant was used for examining the protein concentration by BCA kit (Sigma-Aldrich). Samples were diluted in SDS-PAGE sample buffer and denatured for 5 minutes at 95°C. Equal amounts of the protein were subjected to SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% BSA and incubated with specific antibodies overnight at 4°C. Subsequently, horseradish peroxidase labeled secondary antibody was added and visualized using an enhanced ECL kit. Relative protein expression was analyzed by Image-Pro Plus software version 6.0 (Media Cybernetics, Inc., Rockville, MD, USA)[37].
Cell viability assay
Cell viability was assessed by using the 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8) reduction assay kit according to the manufacturer's instructions. MDA-MB-231 cells were plated in 96-well plates at a cell density of 1.5 × 103 cells per well in 100 μL of complete medium (RPMI1640 basic medium+10% FBS). The following day, the medium was replaced with reagents and incubated for 48 hours. Subsequently, WST-8 was added to the culture for 2 hours at 37°C and absorbance of each well was measured at 450 nm wavelength by enzyme standard instrument (Thermo Scientific, USA).
Cell migration assay
The transwell inserts with a pore diameter of 8 μm were placed in a 24-well culture plate containing cell culture medium (750 μL). The upper chambers were filled with 500 μL cell culture medium containing MDA-MB-231 cells, which were previously treated with AS1411-EVs-let-7, for 24 hours at a concentration of 5 ×104 cells/mL. The cells were cultured for 48 hours at 37°C following which the cells that remained on the top of the filter were removed. The cells that migrated to the lower surface were fixed for 30 minutes by 4% paraformaldehyde and then stained by crystal violet. The transwell inserts were washed with PBS and the cells were calculated by fluorescence microscopy.
Q-PCR assay
The samples were homogenized in Trizol Reagent (Life Technologies, USA) and the RNA was isolated and purified using the protocols provided by the manufacturer. RNA concentration was determined by the NanoDrop-2000 spectrophotometer (Thermo Scientific, USA). Reverse transcription for mRNA into cDNA was performed using Prime Script®RT reagent kit (Takara, Japan). We then performed Real-Time PCR using SYBR Prime Script RT-PCR kit (Takara, Japan). PCR primers for 18s rRNA were, Forward: GTAACCCGTTGAACCCCATT and Reverse: CCATCCAATCGGTAGTAGCG [38] and those for VEGF were Forward: GCCAGCACATAGGAGAGATGAGC and Reverse: CAAGGCTCACAGTGATTTTCTGG [39]. miRNA samples were reverse transcribed using the stem-loop reverse transcriptase primer purchased from Guangzhou RIBOBIO (RIBOBIO, PRC) [40]. Real-Time PCR was performed using SYBR Prime Script RT-PCR kit (Takara, Japan). All assays were performed in triplicate according to the protocols provided by the manufacturer. Relative expression was determined using comparative Ct (2-∆∆Ct) method [41].
Immunofluorescence
For in vitro immunofluorescence analysis, cells were fixed in 4% paraformaldehyde at room temperature for 15 minutes and then washed 3 times for 5 minutes each with PBS. Subsequently, cells were incubated for 10 minutes in permeabilization solution (PBS; 0.25% Triton X-100) and then washed again with PBS 3 times for 5 minutes each. The cells were blocked in blocking solution (PBS; 1% BSA; 0.1% Tween 20) for 30 minutes, incubated overnight at 4°C with primary antibodies, anti-EEA1 (Cell Signaling Technology; 3288); and anti-RAB5 (Cell Signaling Technology; 3547) in blocking solution, and washed intensively 5 times for 5 minutes each with PBST. . FITC-labeled secondary antibody was then applied for 1 hour at room temperature following which the cells were stained with DAPI (staining of nuclei) for 10 minutes. The images were acquired on a confocal microscope (Zeiss LSM 700, Germany).
In vivo antitumor effect study
Six week old female BALB/c nude mice were purchased from the Animal Experiment Center of Sun Yat-Sen University. 3.0 × 106 MDA-MB-231 cells in 200 μL 1640 medium were injected subcutaneously into the right flanks of the mice. The tumors were allowed to grow up to 0.8 cm3 (volume = length × width2/2). The mice were divided into 7 groups (n = 6 mice/group): PBS, free let-7, free EVs, free T-AS1411, AS1411-EVs, nontargeted EVs-let-7, or nucleolin-targeted AS1411-EVs-let-7 groups. Each animal was injected intravenously with solutions (let-7 per dose, 150 μg, iv) every other day and measured for tumor volume at the same time. At the end of the experiment (day 25), the tumor tissue was excised and imaged.
In vivo targeting of AS1411-EVs
The AS1411-EVs were loaded with let-7-Cy5 by electroporation and purified by a 100kD ultrafiltration membrane. AS1411-EVs-let-7-Cy5 (50 μg EVs) were injected intravenously into MDA-MB-231 tumor-bearing nude mice. After 4.5 hours, Cy5 fluorescence in the entire body of the mouse was acquired by In-Vivo Xtreme (Bruker, Germany). After the mouse was sacrificed, Cy5 fluorescence was quantified in tumor, brain, liver, heart, spleen and lung tissues. Radiant efficiency was measured using MI SE software (Bruker, Germany) and normalized by tissue volume.
Statistics
All numerical data were expressed as the Mean ± S.D. Statistical differences between two groups were determined by the Student's t test. P < 0.05 was considered statistically significant.
Results
Synthesis and characterization of AS1411-EVs system
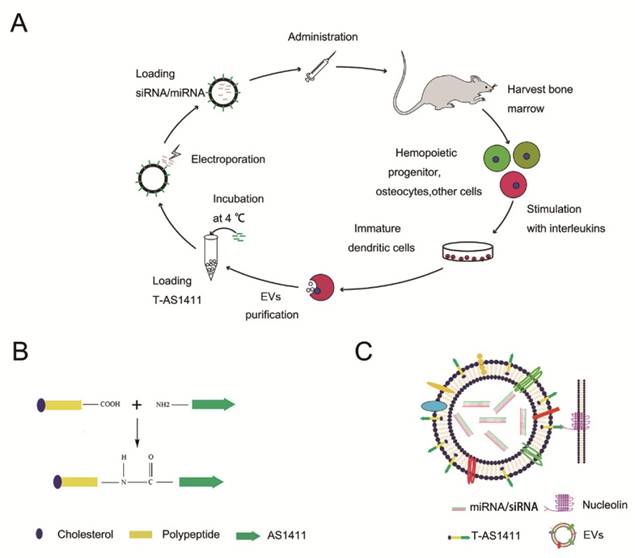
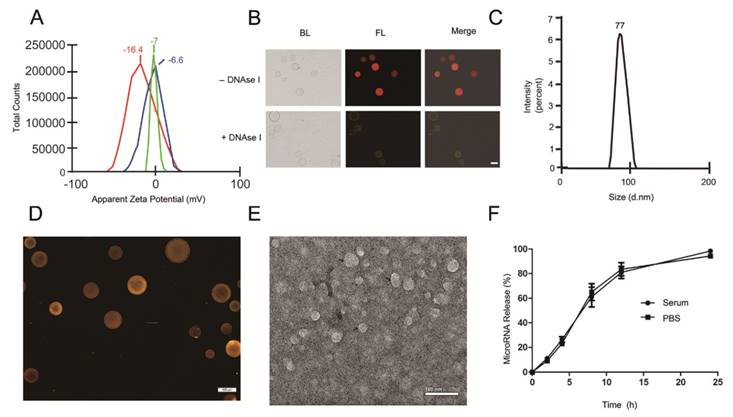
With the goal of developing a strategy to use biological nanoparticles of cellular origin for targeted drug-delivery, we designed and synthesized AS1411-EVs for nucleolin-specific delivery. In our study approach, illustrated in Figure 1A and detailed below, we isolated monocytes from bone marrow of C57BL/6 mice harboring breast tumors. These monocytes differentiated into immature dendritic cells (DCs) in the presence of GM-CSF and IL-4 (10 ng/mL). We then purified EVs from culture supernatants of immature dendritic cells. These EVs were characterized by Western blots and TEM (Fig. S1A, B). Subsequently, we combined the T-AS1411, the targeting ligand for cancer, with the EVs membrane using cholesterol affinity. The drug (miRNA/siRNA) was loaded into the AS1411-EVs by electroporation. Then, intravenously injected AS1411-EVs delivered miRNA/siRNA to mice harboring breast tumors for cancer therapy. (Fig. 1A). To confer EVs targeting capabilities, we designed the tumor-targeting ligand, T-AS1411, which could bind with the EVs membrane by cholesterol hydrophobic interaction. T-AS1411 was synthesized by covalently conjugating polypeptides with cholesterol onto amine-functionalized AS1411 through the -COOH group (Fig. 1B). The polypeptide had two domains: hydrophobic (IIASTIGGIFG) and hydrophilic (SSTTQSGGGG) and could make AS1411 stay at the outer surface membrane of EVs [42]. The AS1411 aptamer confers tumor targeting ability on the delivery system by binding to nucleolin (Fig. 1C).We analyzed the efficiency of miRNA loading into AS1411-EVs by Q-PCR. The results showed that approximately 3% miRNA was loaded into the AS1411-EVs (Fig. S2). To confirm the successful conjugation of the negatively charged aptamer with the EVs, we measured the zeta potential of the EVs and the AS1411-conjugated EVs. The results showed that the zeta potential was -6.6 mV and -16.4 mV for EVs and the AS1411-EVs, respectively (Fig. 2A); digestion of the AS1411-EVs with DNase I increased the zeta potential to -7 mV (Fig. 2A). We then modified the T-AS1411 with Cy3 and incubated with EVs for conjugation. Next, we captured the AS1411-EVs by beads and incubated with DNase I following which the red fluorescence signal almost disappeared (Fig. 2B). This indicated that T-AS1411 was mostly bound to the outer surface of the EVs membrane. Nanoparticle tracking analysis (NTA) revealed that AS1411-EVs had a mean diameter of 77 nm (Fig. 2C). As shown in Fig. 2D, let-7-Cy3 could be loaded into AS1411-EVs by electroporation as confirmed with fluorescent microscopy. The morphology of the AS1411-EVs was spherical and intact as demonstrated by TEM (Fig. 2E). Also, the efflux of miRNA from the AS1411-EVs was determined after incubation in PBS or mouse serum. To minimize the experimental background, we chose Cel-miRNA-67 (Caenorhabditis elegans-derived miRNA not expressed in mammals) [43] for analyzing the efflux of miRNA from the AS1411-EVs as opposed to human or mouse let 7. After 2 h of incubation, Cel-miRNA-67 gene release was approximately 11% in serum and 9% in PBS, respectively. After a 24 h incubation, cel-miRNA-67 release was saturated at approximately 98% in serum and 94% in PBS, respectively (Fig. 2F).
Design of T-AS1411 and modification on the external surface of EVs for targeted delivery: A. Schematic representation of production, harvest, modification, and loading of drug and re-administration of targeted EVs for gene delivery. B. Schematic illustration for synthesis of T-AS1411. C. Schematic illustration of T-AS1411 conjugated EVs by cholesterol and encapsulating the nucleic acid drugs. The schematic represents the mechanism of affinity of AS1411-EVs with nucleolin-positive cancer.
Characterization of AS1411-EVs for breast cancer targeting: A. Zeta potential distribution of EVs (blue), AS1411-EVs (red) and AS1411-EVs digested by DNase I (green) measured by nanoparticle tracking analysis. B. Fluorescence microscopy analysis of the T-AS1411 stable binding with EVs membrane outer surface. T-AS1411 was modified by Cy3 and bound to EVs via cholesterol affinity. The Cy3-AS1411-EVs were digested by DNase I and then captured by Dynabeads® with CD63 antibody (down). The resulting fluorescence signal was very weak. However, control group Cy3-AS1411-EVs not digested by DNase I (up) had a very strong fluorescence signal. (BL: bright light, FL: fluorescence light, Scale bars: 100 μm). C. Size distribution of AS1411-EVs measured by nanoparticle tracking analysis. The peak diameter was at 77 nm for AS1411-EVs. D. AS1411-EVs-let-7-Cy3 was captured by beads and the fluorescence signal detected by fluorescence microscopy. The red fluorescence signal showed successful electroporation of let-7-Cy3 into EVs (Scale bars: 100 μm). E. Transmission electron microscopy of AS1411-EVs-let-7. The morphology of AS1411-EVs-let-7 was intact and the size was approximately 30-100 nm (Arrows indicate AS1411-EVs-let-7, Scale bars: 100 nm). F. Release kinetics of encapsulated cel-miRNA-67 from the AS1411-EVs as determined by incubation for up to 24 hours in PBS or mouse serum at 37°C (n=3) (Each bar represents the mean ±SD of three replicates).
The size of the targeted delivery system, AS1411-EVs, used in our study was approximately 80 nm suitable for passive targeting via enhanced permeability and retention (EPR) [44]. To determine whether AS1411-EVs could effectively deliver nucleic acid drugs to breast cancer cells in vitro, we incubated MDA-MB-231 cells with AS1411-EVs-let-7-Cy3 for 30 min and 1h. The presence of let-7-Cy3 in the cytosol was assessed using confocal microscopy. The results showed let-7 in the cytosol of MDA-MB-231 cells. Also, there was little miRNA co-localization with early endosome proteins EEA1 and RAB5 (Fig. S3A, B).
Nucleolin-dependent tumor targeting of modified EVs in vitro
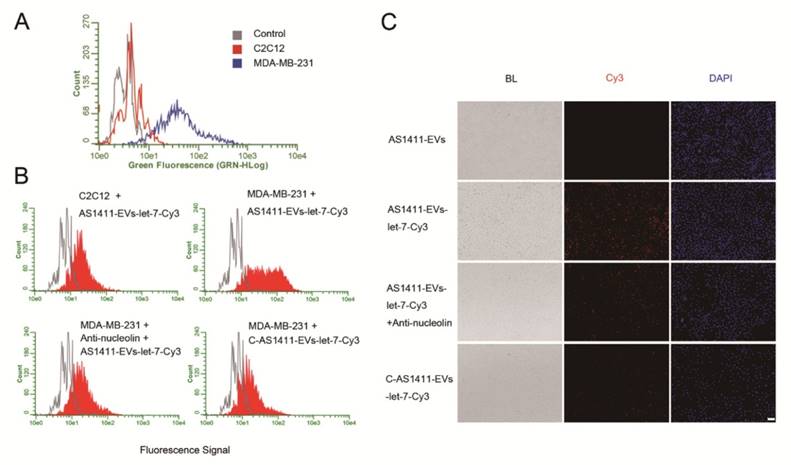
We next examined whether AS1411-EVs bound to recipient cells in a nucleolin-dependent manner. We first evaluated nucleolin expression in C2C12 cells (nucleolin negative cells) [45] and MDA-MB-231 human breast cancer cells (nucleolin positive cells) [46]. Flow cytometry results showed that MDA-MB-231 had higher nucleolin expression levels than C2C12 cells (Fig. 3A). To determine whether AS1411-EVs were taken up by cells expressing nucleolin, AS1411-EVs were loaded with let-7-Cy3 (red fluorescence) and added to cultures of the nucleolin-positive and -negative cell lines. The flow cytometry results showed that AS1411-EVs-let-7-Cy3 bound to MDA-MB-231 cells more efficiently than to C2C12 cells (Fig. 3B). However, when the nucleolin-positive breast cancer cells were blocked by nucleolin antibody, the enhanced delivery of AS1411-EVs-let-7-Cy3 apparently declined (Fig. 3B, 3C. Fig. S4). Thus, the targeted delivery system taken up by the cells appeared to reflect their nucleolin expression levels. Next, we replaced the nucleotide sequence of AS1411 with a random sequence, C-AS1411 (5'-ATCGATCGATCGATCGATCGATCGAT-3') as the control group. As expected, the C-AS1411-EVs-let-7-Cy3 had a much lower efficiency of binding to cancer cells (Fig. 3B, 3C) demonstrating the specificity of AS1411-EVs to target nucleolin-expressing cancer cells.
Nucleolin-dependent tumor targeting of modified EVs in vitro. A. Flow cytometry analysis of nucleolin expression on the surface of MDA-MB231 and C2C12 cells. B. Uptake of AS1411-EVs-let-7-Cy3 detected by flow cytometry in C2C12 and MDA-MB-231 cells. AS1411-EVs or C-AS1411-EVs (control group in MDA-MB-231) loaded with let-7-Cy3 were incubated with the cells at 37 °C for 1 h. Anti-nucleolin antibody was also used in MDA-MB-231 cells for blocking the target-specific binding. The fluorescent signal was relatively stronger in the MDA-MB-231 cells incubated with AS1411-EVs-let-7-Cy3. C. Cell-associated AS1411-EVs-let-7-Cy3 was detected in MDA-MB-231 cells using fluorescence microscopy. Cells were treated with PBS then co-cultured for 1 h with AS1411-EVs, AS1411-EVs-let-7-Cy3 or C-AS1411-EVs-let-7-Cy3. Cells were also treated with anti-nucleolin antibody then co-cultured for 1 h with AS1411-EVs-let-7-Cy3. (Scale bar = 100 μm).
In vitro targeted delivery of miRNA using AS1411-EVs
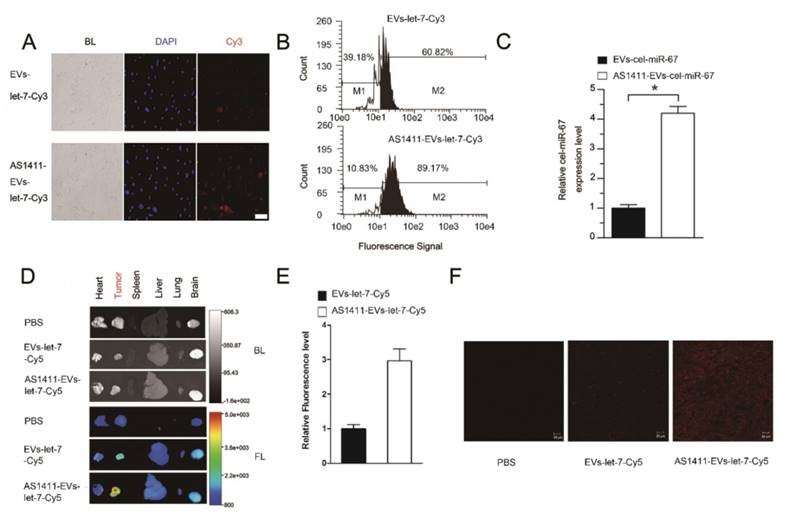
To confirm the more efficient delivery of AS1411-EVs to nucleolin-positive cancer cells, MDA-MB-231 human breast cancer cells were treated with EVs-let-7-Cy3 or AS1411-EVs-let-7-Cy3 for 45 minutes at 37°C. Fluorescent microscopic analysis revealed a brighter red fluorescence on the cell surface in the AS1411-EVs-let-7-Cy3 treated group compared with the EVs-let-7-Cy3 treated cells (Fig. 4A). More efficient binding of AS1411-EVs-let-7-Cy3 to MDA-MB-231 cells than EVs-let-7-Cy3 was also evident by flow cytometry analysis (Fig. 4B). Also, Q-PCR data suggested that cel-miR-67 expression level in MDA-MB-231 cells was much higher after AS1411-EVs -miR-67 treatment. Taken together, these analyses showed a ~4 times greater delivery efficiency of the AS144-EVs was than EVs alone (Fig. 4C).
Breast cancer-specific targeting of AS1411-EVs. A. Representative images by fluorescence microscopy of breast cancer after incubation with equal amount EVs-let-7-Cy3 (top) and AS1411-EVs-let-7-Cy3 (bottom) for 45 minutes. (Scale bar = 100 μm). B. Flow cytometric analysis of EVs-let-7 (top) and AS1411-EVs-let-7 (bottom) taken up by MDA-MB-231 cells after incubation for 45 minutes. The percentages represent fraction of tumor cells encapsulating Cy3-labeled let-7 (Cy3 positive tumor cells) AS1411-EVs-let-7-Cy3 showed significantly stronger binding ability to breast cancer cells compared with EVs-let-7-Cy3. C. Q-PCR analysis of cel-miRNA-67 level in MDA-MB-231 cells incubated with equal amounts of EVs-cel-miR-67 or AS1411-EVs-cel-miR-67 for 45 minutes. D. Ex vivo fluorescence imaging of major organs from tumor-bearing mice 4.5 h after intravenous injection with 50μg of AS1411-EVs-let-7-Cy5 (bottom) or EVs-let-7-Cy5 (middle) or PBS (top). In AS1411-EVs-let-7-Cy5 group, tumor tissue had strong fluorescence signals, whereas other organs had weak signals. In EVs-let-7-Cy5 group, tumor tissue had a weak fluorescence signal. (BL, bright light. FL, fluorescence light). E. Quantification of average fluorescence signal intensity of the tumor in figure D by MI SE software (fluorescence signal of AS1411-EVs-let-7-Cy5 minus PBS control vs. fluorescence signal of EVs-let-7-Cy5 minus PBS control), Data are presented as the mean ± s.e.m. (n=3). F. Confocal microscopic analysis of tumor sections in figure D shows the distribution of miRNA in cancer cells treated with PBS, EVs-miRNA-Cy5 and AS1411-EVs-miRNA-Cy5. (Scale bar = 20 μm).
In vivo targeting of AS1411-EVs to mouse tumors
To assess tumor targeting of the AS1411-EVs in vivo, we established an MDA-MB-231 tumor-bearing nude mouse model by subcutaneous administration of breast cancer cells. AS1411-EVs and EVs containing let-7-Cy5 were injected (iv) into MDA-MB-231-tumor-bearing mice. Mice in the control group received PBS injections (iv). Localization of let-7 was monitored by in vivo imaging at 4.5 h after administration of EVs. Mice were sacrificed and tumors and other organs were harvested. Ex vivo fluorescent imaging data revealed strong fluorescent signals in tumor tissues compared to other non-cancerous tissues in mice treated with AS1411-EVs-let-7-Cy5. Weaker fluorescence was noted in tumor tissues of mice treated with EVs-let-7-Cy5 (Fig. 4D, E). We also assessed AS1411-EVs-let-7-Cy5 distribution by confocal microscopy and detected strong fluorescent signals in most cells in the tumor sections. In contrast, weak fluorescence was present in tumor sections from mice injected with EVs-let-7 (Fig. 4F).
Functional delivery of siRNA-VEGF via the AS1411-EVs in vitro
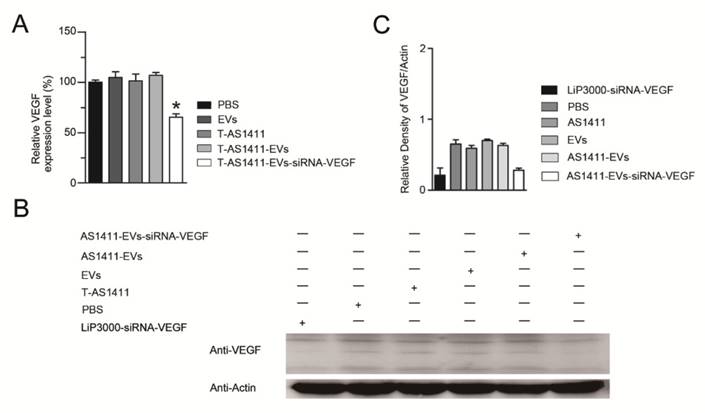
Since AS1411-EVs could efficiently deliver exogenous small RNAs into cells in vitro, we studied delivery of siRNA into human breast cancer cells to induce gene silencing. We loaded VEGF siRNA into the AS1411-EVs by electroporation and incubated this with MDA-MB-231 cells. After 48 h, measurement of VEGF expression with Q-PCR and Western blotting showed that AS1411-EVs containing VEGF siRNA reduced both VEGF mRNA and protein expression (Fig. 5A, B, C).
Functional delivery of siRNA-VEGF by AS1411-EVs in vitro. A, B. The MDA-MB-231 cells were treated with PBS, EVs, T-AS111, AS1411-EVs and AS1411-EVs-siRNA-VEGF (150 μg) for 48 hours and the expression levels of VEGF were examined by (A) Q-PCR and (B) Western blotting. C. Quantification of VEGF protein expression level (shown in B) in various treatment groups by Image-Pro Plus software. Statistical analyses were performed using the Student's t test. *, P< 0.05.
In vitro antitumor effects of AS1411-EVs-let-7
Since AS1411-EVs could deliver siRNA to cells and could inhibit the target protein expression in vitro, we assessed whether AS1411-EVs delivery of exogenous microRNA into cells could also have an antitumor effect. First, we analyzed the ability of AS1411-EVs-let-7 to inhibit breast cancer cell proliferation. MDA-MB-231 cells were treated with PBS, EVs, AS1411-EVs, T-AS1411, AS1411-EVs-miR-67 or AS1411-EVs-let-7 for 48 h following which cell viability was measured with a WST-8 assay kit. AS1411-EVs-let-7 inhibited cell proliferation compared to PBS (Fig. 6A). Cell growth was not inhibited in the T-AS1411, EVs, AS1411-EVs or AS1411-EVs-miR-67 treated groups. This indicated that there was no toxicity at this concentration of T-AS1411 and EVs.
Functional delivery of let-7 by AS1411-EVs in vitro and in vivo. MDA-MB-231 cells were incubated with PBS, EVs, T-AS111, AS1411-EVs, AS1411-EVs-miR-67 and AS1411-EVs-let-7 (30 μg) for 48 hours. (A) AS1411-EVs-let-7 inhibited tumor growth; no cytotoxicity was observed in various control groups. Each bar represents the mean ±SD of three replicates.*, P< 0.05. (B). Cell migration was assessed using transwell assay. AS1411-EVs-let-7 (150 μg) inhibited cell migration remarkably, but almost had no effect in various control groups. (C). Western blot analysis of the expression level of let-7-targeted proteins, KRAS and cMyc which were down-regulated in AS1411-EVs- group, but had no effect in various control groups. (D). Mice bearing tumors (~0.8 cm3) were injected intravenously with different reagents (PBS, T-AS1411, EVs, AS1411-EVs, EVs-let-7, AS1411-EVs-let-7 (150 μg) every other day for a total of 12 injections. The AS1411-EVs-let-7 and EVs-let-7 groups show inhibition of tumor growth compared to other control groups. with a more prominent effect in the AS1411-EVs-let-7 group. Statistical analyses were performed using the Student's t test. *, P< 0.05 (n =6).
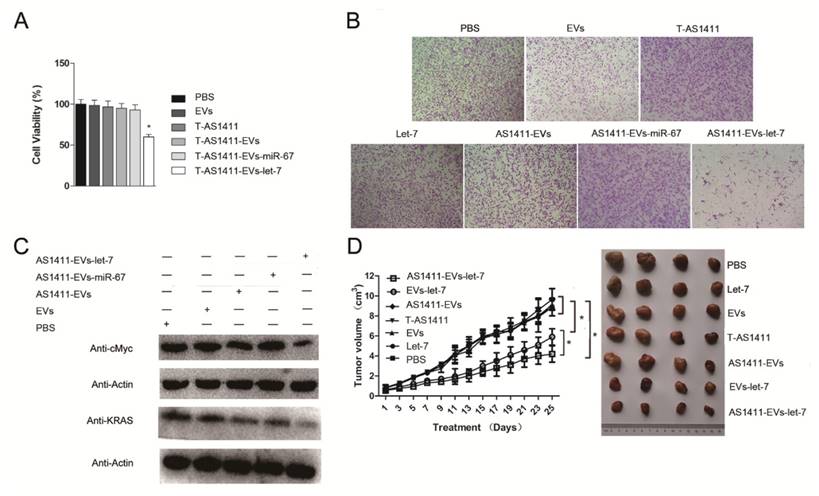
Next, we measured migration of cells treated with the control medium, free EVs, free T-AS1411, free let-7, AS1411-EVs, AS1411-EVs-miR-67 or AS1411-EVs-let-7 after 48 h using a transwell migration assay. Fewer cells migrated through the transwell in the AS1411-EVs-let-7 treated group compared to PBS treated cells (Fig. 6B). Treatment with free T-AS1411, free EVs, free let-7, AS1411-EVs or AS1411-EVs-miR-67 did not change migration compared to controls. To establish the mechanism underlying inhibition of migration, we evaluated let-7 target gene silencing in breast cancer cells treated with PBS, EVs, AS1411-EVs, AS1411-EVs-miR-67 or AS1411-EVs-let-7 for 48 h by Western blotting. The results indicated that c-MYC and K-RAS protein expression was significantly suppressed (Fig. 6C).
In vivo antitumor effects of AS1411-EVs-let-7
MDA-MB-231 cells were inoculated in nude mice. Two weeks after inoculation, tumor-bearing mice were divided into 7 groups (n = 6 mice/group): Each group received via tail vein injections PBS, free let-7, free EVs, free T-AS1411, AS1411-EVs, non-targeted EVs-let-7, or nucleolin-targeted AS1411-EVs-let-7. Mice were treated (let-7 per dose, 150 μg, iv) every other day for a total of 12 injections. Treatment started when tumor volume reached ~0.8 cm3. Tumor volumes and animal weights were monitored throughout treatment and tumor growth inhibition was determined on the final day. For mice injected with AS1411-EVs-let-7, tumor growth was delayed significantly compared to all other groups. Tumor growth inhibition in the AS1411-EVs-let-7-treated group compared to PBS and EVs-let-7 group was 57% and 29%, respectively. Next, tumors were harvested from animals in each group and assessed for therapeutic efficacy (Fig. 6D). Consistent with the delay in tumor growth; excised tumor volumes in nucleolin-targeted AS1411-EVs-let-7-treated animals were smaller than those in other treatment groups.
Safety of the AS1411-EVs-let-7
We measured the in vivo toxicity of the AS1411-EVs delivery system, especially with regard to cardiac, hepatic and renal damage. Mice were injected with PBS or AS1411-EVs-let-7 every other day for 4 days. Subsequently, animals were sacrificed and heart, liver, spleen and kidney were removed and stained with hematoxylin and eosin. There was no obvious tissue damage in AS1411-EVs-let-7-treated mice compared with controls (Fig. 7A).
Evaluation of the toxicity profile of AS1411-EVs-let-7. A. H&E staining of various organs. Scale bar = 100 μm). B. Serum markers of organ damage. Each bar represents means with SD of three replicates. NS, not significant, CK-MB: creatine kinase-MB isoenzyme; ALT: alanine aminotransferase; AST: aspartate aminotransferase; BUN: blood urea nitrogen. C. Serum-associated inflammatory cytokines (TNF-α and INF-α). No significant difference noted between the 2 treatment groups.
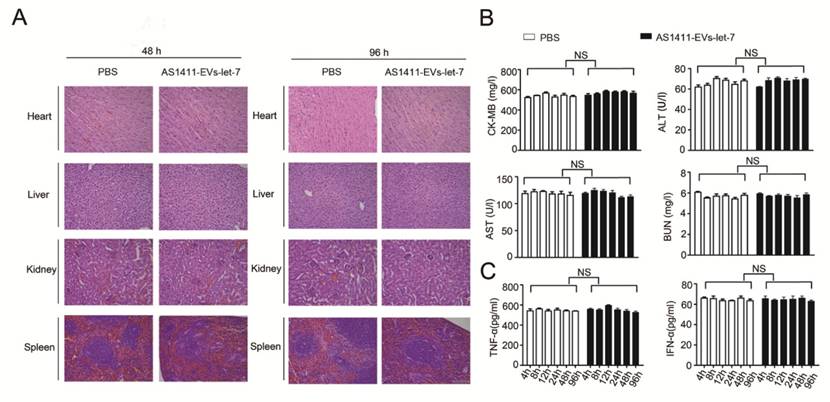
Furthermore, serum collected from mice treated with PBS or AS1411-EVs-let-7 was assessed for creatine kinase MB isoenzyme (CK-MB) and aspartate aminotransferase (AST), common cardiac damage markers. We also measured alanine aminotransferase (ALT) for hepatotoxicity as well as blood urea nitrogen (BUN) for renal toxicity. All serum chemistries of mice treated with AS1411-EVs-let-7 were similar to those of controls (Fig. 7B).
To evaluate the AS1411-EVs-let-7-induced possible immune response, we measured serum-associated inflammatory cytokines such as INF-α and TNF-α in mice treated with AS1411-EVs-let-7 or PBS. No statistically significant differences among groups in any of the tested parameters were observed (Fig. 7C). Thus, our data suggested that AS1411-modified EVs are safe and effective nucleic acid drug delivery carriers for in vivo therapy.
Discussion
Although delivery of biologics (siRNA, miRNA) have promising applications with respect to cancer treatment [47, 48], clinical use of small RNA molecules is presently limited due to low efficiency, toxicity, and lack of a cancer-specific delivery system. The delivery of biologics is restricted by numerous barriers, such as toxicity, inadequate cellular uptake, immunogenicity, poorly defined in vivo characteristics, renal clearance, degradation by nucleases, elimination by phagocytic immune cells, and poor endosomal release. Virus particles have inherent capabilities to penetrate into the cells for the easy transfer of nucleic acids, whereas non-viral particulates get transported to the cell by either physical (electroporation, gene gun) or chemical (utilizing lipoplexes and polyplexes) means for delivery. Typically, viral vectors can achieve high transduction efficiencies in vitro, however their potential to trigger mutagenesis and induce host immune responses are some other known bottlenecks. Examples of viral vectors typically are complexes of nucleic acids with cationic lipids (lipoplexes) and cationic polymers (polyplexes). Non-viral vectors based on nanometer-sized particles have shown great promise because of their low immunogenicity; however, poor transfection rate is a challenge that must be addressed in vivo [49, 50]. Recently, EVs have been used to deliver drugs as natural nano-sized vesicles but few studies exist to describe targeted delivery of the therapeutic cargo. The most popular approach was based on the expression of specific ligands or peptides in the outer section of EVs transmembrane protein, such as lactadherin [51, 52], lysosome-associated membrane protein-2b (LAMP2b) [25, 29], glycosylphosphatidylinositol (GPI) [53] and platelet-derived growth factor receptor (PDGFR) [54]. Koppers-Lalic and his collaborators reported that fusion of membrane proteins with specific viral proteins can also direct EVs towards specific target cells [55]. But these methods were not suitable for large-scale modified EVs production for clinical application [56].
Recently, other methods to modify the EVs without having to manipulate EVs producing cells have been explored. For example, Kooijmans et al reported that polyethyleneglycol (PEG) polymer chains could link cell specific peptides to the EVs surface [57]. Jang et al reported that EVs derived from UG37 and Raw264.7 cells exhibited high level of tumor-targeting ability. The authors hypothesized that EVs membrane expresses protein LFA-1 which can bind to endothelial cell adhesion molecules (CAMs) [30]. Also, Hongzhao and colleagues used multiple superparamagnetic nanoparticles to anchor onto each reticulocytes (RTC)-derived EVs by Tf-Tf receptor interaction [58]. By taking advantage of these properties, they generated EVs with a tumor-targeting ability.
Here, we developed a modified EVs vesicle by introducing aptamer AS1411 in the EVs surface membrane (Figure 1A). Our results provided evidence that the vesicles could enhance cancer-targeted delivery in vitro (Figure 4A, B, C). Furthermore, analysis of the small RNA delivery system revealed inhibition of triple-negative breast cancer cells growth both in vitro and in vivo with no noticeable toxicity or immunogenicity (Figure 3, Figure 4, Figure 6). Thus, aptamer-modified EVs can likely be used for developing drug delivery/tissue targeting strategies.
Although we successfully targeted tissues with the aptamer-modified EVs as a drug delivery system, there are a few outstanding issues that need to be addressed in future studies. First, clinical application of EVs has been limited by inefficient drug loading approaches [59, 60]. We used electroporation to load nucleic acid drugs into EVs according to published methods [25, 27]. However, in our experience, drug loading by electroporation was less than previously reported. Other methods such as simple incubation, chemical transfection, or activation of EVs donor cells had low efficiency or were too time-consuming [26, 27, 61-64]. Thus, additional loading methods are required to maximize this technology. Second, a fraction of the EVs used in this study could originate from the FBS in the culture medium despite centrifugation at 100,000 g. Although it does not change the overall conclusions in the present study, the possible contamination with EVs from FBS in the in vitro studies should be minimized or eliminated. And third, the AS1411-EVs could also deliver drugs to other normal organs such as the heart and brain (Figures 4D). The EVs membrane is rich in adhesion-relation proteins such as integrins and ICAMI-1 [65]. These proteins, especially integrins, mediate cell adhesion and uptake by cells [66]. If these adhesion-relation proteins were blocked with small molecule inhibitors such as the RGD ligand [67, 68], EVs delivery off-target effects may be reduced.
Conclusion
In summary, we used the cancer-targeting ligand, AS1411, to modify EVs to generate a tumor-targeting small nucleic acid delivery system. The AS1411-EVs had a high affinity for breast cancer cells and effectively delivered small nucleic acids into breast cancer cells. Our approach offers low systemic toxicity and may have a great potential for future clinical applications.
Acknowledgements
This work was supported by National Natural Science Foundation Projects (81370971 and 81473131), Guangdong Natural Science Funds (2014A030313358), the Major Project in Guangdong Province of Science (2014KZDXM011), and Guangdong Natural Science Funds for Distinguished Young Scholar (S2013050013880).
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Opalinska JB, Gewirtz AM. Nucleic-acid therapeutics: basic principles and recent applications. Nat Rev Drug Discov. 2002;1:503-14
2. Wong JP. Nucleic acid-based drugs against emerging zoonotic viruses. Future Med Chem. 2015;7:1709-1719
3. Gleave ME, Monia BP. Antisense therapy for cancer. Nat Rev Cancer. 2005;5:468-79
4. Izquierdo M. Short interfering RNAs as a tool for cancer gene therapy. Cancer Gene Ther. 2005;12:217-27
5. Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. 2012;6:590-610
6. Yang T, Zhao P, Rong Z, Li B, Xue H, You J. et al. Anti-tumor Efficiency of Lipid-coated Cisplatin Nanoparticles Co-loaded with MicroRNA-375. Theranostics. 2016;6:142-54
7. Bertoli G, Cava C, Castiglioni I. MicroRNAs: New Biomarkers for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Breast Cancer. Theranostics. 2015;5:1122-43
8. Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE. et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901-6
9. Barh D, Malhotra R, Ravi B, Sindhurani P. MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010;17:70-80
10. Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17:F19-36
11. Haussecker D, Kay MA. RNA interference. Drugging RNAi. Science. 2015;347:1069-70
12. Lee SJ, Yook S, Yhee JY, Yoon HY, Kim MG, Ku SH. et al. Co-delivery of VEGF and Bcl-2 dual-targeted siRNA polymer using a single nanoparticle for synergistic anti-cancer effects in vivo. J Control Release. 2015;220:p.631-641
13. Feng Q, Yu MZ, Wang JC, Hou WJ, Gao LY, Ma XF. et al. Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core-shell nanoparticles. Biomaterials. 2014;35:5028-38
14. Yang C, Nilsson L, Cheema MU, Wang Y, Frokiaer J, Gao S. et al. Chitosan/siRNA nanoparticles targeting cyclooxygenase type 2 attenuate unilateral ureteral obstruction-induced kidney injury in mice. Theranostics. 2015;5:110-23
15. Gao S, Hein S, Dagnaes-Hansen F, Weyer K, Yang C, Nielsen R. et al. Megalin-mediated specific uptake of chitosan/siRNA nanoparticles in mouse kidney proximal tubule epithelial cells enables AQP1 gene silencing. Theranostics. 2014;4:1039-51
16. Lee J, Saw PE, Gujrati V, Lee Y, Kim H, Kang S. et al. Mono-arginine Cholesterol-based Small Lipid Nanoparticles as a Systemic siRNA Delivery Platform for Effective Cancer Therapy. Theranostics. 2016;6:192-203
17. Cheng Q, Du L, Meng L, Han S, Wei T, Wang X. et al. The Promising Nanocarrier for Doxorubicin and siRNA Co-delivery by PDMAEMA-based Amphiphilic Nanomicelles. ACS Appl Mater Interfaces. 2016;8:4347-56
18. Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573-87
19. Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U. et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638-46
20. Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581-93
21. Thery C, Zitvogel L, Amigorena S. Exosomes: Composition, biogenesis and function. Nat Rev Immunol. 2002;2:569-79
22. Villasante A, Marturano-Kruik A, Ambati SR, Liu Z, Godier-Furnemont A, Parsa H. et al. Recapitulating the Size and Cargo of Tumor Exosomes in a Tissue-Engineered Model. Theranostics. 2016;6:1119-30
23. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654-9
24. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan MLG, Karlsson JM. et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119:756-66
25. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341-5
26. Wahlgren J, Karlson TD, Brisslert M, Sani FV, Telemo E, Sunnerhagen P. et al. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012:40
27. Shtam TA, Kovalev RA, Varfolomeeva EY, Makarov EM, Kil YV, Filatov MV. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun Signal. 2013:11
28. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N. et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol Ther. 2013;21:185-91
29. Tian YH, Li SP, Song J, Ji TJ, Zhu MT, Anderson GJ. et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383-90
30. Jang SC, Kim OY, Yoon CM, Choi DS, Roh TY, Park J. et al. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano. 2013;7:7698-710
31. Alvarez-Erviti L, Seow YQ, Yin HF, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341-U179
32. Mongelard F, Bouvet P. AS-1411, a guanosine-rich oligonucleotide aptamer targeting nucleolin for the potential treatment of cancer, including acute myeloid leukemia. Curr Opin Mol Ther. 2010;12:107-14
33. Rosenberg JE, Bambury RM, Van Allen EM, Drabkin HA, Lara PN, Harzstark AL. et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Invest New Drug. 2014;32:178-87
34. Lin J, Li J, Huang B, Liu J, Chen X, Chen XM. et al. Exosomes: novel biomarkers for clinical diagnosis. TheScientificWorldJournal. 2015;2015:657086
35. Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ. et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3:406-17
36. Kooijmans SA, Stremersch S, Braeckmans K, de Smedt SC, Hendrix A, Wood MJ. et al. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release. 2013;172:229-38
37. Phillips JL, Tolan DR, Hausman RE. Antisense inhibition of R-cognin expression modulates differentiation of retinal neurons in vitro. Mol Vis. 1997;3:12
38. Zhang HT, Sun ZG, Wei WJ, Liu ZH, Fleming J, Zhang S. et al. Identification of Serum microRNA Biomarkers for Tuberculosis Using RNA-seq. PloS One. 2014:9
39. Darland DC, Cain JT, Berosik MA, Saint-Geniez M, Odens PW, Schaubhut GJ. et al. Vascular endothelial growth factor (VEGF) isoform regulation of early forebrain development. Dev Biol. 2011;358:9-22
40. Chen CF, Ridzon DA, Broomer AJ, Zhou ZH, Lee DH, Nguyen JT. et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005:33
41. Yuan JS, Reed A, Chen F, Stewart CN. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006:7
42. Milovanovic D, Honigmann A, Koike S, Gottfert F, Pahler G, Junius M. et al. Hydrophobic mismatch sorts SNARE proteins into distinct membrane domains. Nat Commun. 2015:6
43. Buller B, Liu X, Wang X, Zhang RL, Zhang L, Hozeska-Solgot A. et al. MicroRNA-21 protects neurons from ischemic death. FEBS J. 2010;277:4299-307
44. Gao W, Wang Z, Lv L, Yin D, Chen D, Han Z. et al. Photodynamic Therapy Induced Enhancement of Tumor Vasculature Permeability Using an Upconversion Nanoconstruct for Improved Intratumoral Nanoparticle Delivery in Deep Tissues. Theranostics. 2016;6:1131-44
45. Li L, Hou J, Liu X, Guo Y, Wu Y, Zhang L. et al. Nucleolin-targeting liposomes guided by aptamer AS1411 for the delivery of siRNA for the treatment of malignant melanomas. Biomaterials. 2014;35:3840-50
46. Hovanessian AG, Soundaramourty C, El Khoury D, Nondier I, Svab J, Krust B. Surface Expressed Nucleolin Is Constantly Induced in Tumor Cells to Mediate Calcium-Dependent Ligand Internalization. PloS One. 2010:5
47. Pille JY, Denoyelle C, Varet J, Bertrand JR, Soria J, Opolon P. et al. Anti-RhoA and anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol Ther. 2005;11:267-74
48. Majumder S, Miller TE, Ghoshal K, Ramaswamy B, Shapiro CL, Roy S. et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27(kip1). J Biol Chem. 2008;283(44):29897-29903
49. Midoux P, Pichon C, Yaouanc JJ, Jaffres PA. Chemical vectors for gene delivery: a current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br J Pharmacol. 2009;157:166-78
50. Koynova R, Tenchov B. Recent patents in cationic lipid carriers for delivery of nucleic acids. Recent Pat DNA Gene Seq. 2011;5:8-27
51. Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A. et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68:1228-35
52. Hartman ZC, Wei J, Glass OK, Guo H, Lei G, Yang X-Y. et al. Increasing vaccine potency through exosome antigen targeting. Vaccine. 2011;29:9361-7
53. Kooijmans SA, Aleza CG, Roffler SR, van Solinge WW, Vader P, Schiffelers RM. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J Extracell Vesicles. 2016;5:31053
54. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N. et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21:185-91
55. Koppers-Lalic D, Hogenboom MM, Middeldorp JM, Pegtel DM. Virus-modified exosomes for targeted RNA delivery; a new approach in nanomedicine. Adv Drug Deliv Rev. 2013;65:348-56
56. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ. et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383-90
57. Kooijmans SA, Fliervoet LA, van der Meel R, Fens MH, Heijnen HF, van Bergen En Henegouwen PM. et al. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release. 2016;224:77-85
58. Qi H, Liu C, Long L, Ren Y, Zhang S, Chang X. et al. Blood Exosomes Endowed with Magnetic and Targeting Properties for Cancer Therapy. ACS Nano. 2016;10:3323-33
59. Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles - Endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta. 2014;1846:75-87
60. Kooijmans SA, Stremersch S, Braeckmans K, de Smedt SC, Hendrix A, Wood MJ. et al. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release. 2013;172:229-38
61. Bryniarski K, Ptak W, Jayakumar A, Pullmann K, Caplan MJ, Chairoungdua A. et al. Antigen-specific, antibody-coated, exosome-like nanovesicles deliver suppressor T-cell microRNA-150 to effector T cells to inhibit contact sensitivity. J Allergy Clin Immunol. 2013;132:170-U287
62. Zhuang XY, Xiang XY, Grizzle W, Sun DM, Zhang SQ, Axtell RC. et al. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-Inflammatory Drugs From the Nasal Region to the Brain. Mol Ther. 2011;19(10):1769-1779
63. Chen L, Charrier A, Zhou Y, Chen RJ, Yu B, Agarwal K. et al. Epigenetic Regulation of Connective Tissue Growth Factor by MicroRNA-214 Delivery in Exosomes From Mouse or Human Hepatic Stellate Cells. Hepatology. 2014;59:1118-29
64. Zhang YJ, Liu DQ, Chen X, Li J, Li LM, Bian Z. et al. Secreted Monocytic miR-150 Enhances Targeted Endothelial Cell Migration. Mol Cell. 2010;39:133-44
65. Tan A, Rajadas J, Seifalian AM. Exosomes as nano-theranostic delivery platforms for gene therapy. Adv Drug Deliv Rev. 2013;65:357-67
66. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M. et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329-35
67. Takagi J. Structural basis for ligand recognition by RGD (Arg-Gly-Asp)-dependent integrins. Biochem Soc Trans. 2004;32:403-6
68. Chen H, Niu G, Wu H, Chen X. Clinical Application of Radiolabeled RGD Peptides for PET Imaging of Integrin alphavbeta3. Theranostics. 2016;6:78-92
Author contact
![]() Corresponding authors: Xiaogang Wang, Department of Cell Biology & Institute of Biomedicine, Jinan University, 601 Huangpu Avenue. West, Guangzhou, 510632, China Phone: 86-020-85226059 Fax: 86-020-85226059 E-mail: txg_wangedu.cn or An Hong, Department of Cell Biology & Institute of Biomedicine, Jinan University, 601 Huangpu Avenue. West, Guangzhou, 510632, China Phone: 86-020-85221983 Fax: 86-020-85221983 E-mail: thaedu.cn
Corresponding authors: Xiaogang Wang, Department of Cell Biology & Institute of Biomedicine, Jinan University, 601 Huangpu Avenue. West, Guangzhou, 510632, China Phone: 86-020-85226059 Fax: 86-020-85226059 E-mail: txg_wangedu.cn or An Hong, Department of Cell Biology & Institute of Biomedicine, Jinan University, 601 Huangpu Avenue. West, Guangzhou, 510632, China Phone: 86-020-85221983 Fax: 86-020-85221983 E-mail: thaedu.cn