Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(6):1612-1632. doi:10.7150/thno.17573 This issue Cite
Research Paper
Lung Cancer-Targeting Peptides with Multi-subtype Indication for Combinational Drug Delivery and Molecular Imaging
Yi-Hsuan Chi1, 2, Jong-Kai Hsiao3, Ming-Huang Lin4*, Chen Chang4*, Chun-Hsin Lan1*, Han-Chung Wu1, 2 ![]()
1. Institute of Cellular and Organismic Biology, Academia Sinica, Taipei, Taiwan;
2. Graduate Institute of Pathology, College of Medicine, National Taiwan University, Taipei, Taiwan;
3. Department of Medical Imaging, Taipei Tzu-Chi Hospital, New Taipei City, Taiwan;
4. Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan.
* These authors contributed equally to this work.
Received 2016-9-14; Accepted 2017-1-30; Published 2017-4-10
Abstract

Lung cancer is the leading cause of cancer-related death worldwide. Most targeted drugs approved for lung cancer treatment are tyrosine kinase inhibitors (TKIs) directed against EGFR or ALK, and are used mainly for adenocarcinoma. At present, there is no effective or tailored targeting agent for large cell carcinoma (LCC) or small cell lung cancer (SCLC). Therefore, we aimed to identify targeting peptides with diagnostic and therapeutic utility that possess broad subtype specificity for SCLC and non-small cell lung cancer (NSCLC). We performed phage display biopanning of H460 LCC cells to select broad-spectrum lung cancer-binding peptides, since LCC has recently been categorized as an undifferentiated tumor type within other histological subcategories of lung cancer. Three targeting phages (HPC1, HPC2, and HPC4) and their respective displayed peptides (HSP1, HSP2, and HSP4) were able to bind to both SCLC and NSCLC cell lines, as well as clinical specimens, but not to normal pneumonic tissues. In vivo optical imaging of phage homing and magnetic resonance imaging (MRI) of peptide-SPIONs revealed that HSP1 was the most favorable probe for multimodal molecular imaging. Using HSP1-SPION, the T2-weighted MR signal of H460 xenografts was decreased up to 42%. In contrast to the tight binding of HSP1 to cancer cell surfaces, HSP4 was preferentially endocytosed and intracellular drug delivery was thereby effected, significantly improving the therapeutic index of liposomal drug in vivo. Liposomal doxorubicin (LD) conjugated to HSP1, HSP2, or HSP4 had significantly greater therapeutic efficacy than non-targeting liposomal drugs in NSCLC (H460 and H1993) animal models. Combined therapy with an HSP4-conjugated stable formulation of liposomal vinorelbine (sLV) further improved median overall survival (131 vs. 84 days; P = 0.0248), even in aggressive A549 orthotopic models. Overall, these peptides have the potential to guide a wide variety of tailored theranostic agents for targeting therapeutics, non-invasive imaging, or clinical detection of SCLC and NSCLC.
Keywords: Small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), targeting peptides, liposomal drugs, molecular imaging, magnetic resonance imaging (MRI), theranostics.
Introduction
Lung cancer is the leading cause of cancer-related mortality in both men and women, accounting for about 27% of all cancer deaths [1]. For treatment purposes, lung cancer can be histopathologically classified as small-cell lung carcinoma (SCLC; 15%) and non-small-cell lung carcinoma (NSCLC; 84%), with the latter further divided into large cell carcinoma (LCC), adenocarcinoma, and squamous cell carcinoma (SCC). Adenocarcinomas often have glandular histology, distinguished from SCCs by squamous differentiation. Carcinomas that do not express adenocarcinoma or SCC biomarkers, and are not glandular or squamous, are classified as LCC [2]. Recent large-scale systematic analyses have characterized genetic alterations in 1255 clinically annotated lung tumors and incorporated genomic information into lung cancer taxonomy, resulting in the reclassification of LCC as a heterogeneous undifferentiated group with genetic similarity to adenocarcinoma, SCC, or neuroendocrine (SCLC) [3, 4].
Currently available targeted therapies, such as erlotinib or crizotinib, are tyrosine kinase inhibitors (TKIs) for treatment of carcinoma (mainly lung adenocarcinoma) bearing EGFR mutations or EML4-ALK fusions [5, 6]. In contrast, potential inhibitors which target cells with aberrations more common to SCC, such as amplified FGFR, mutated SOX2 or mutated DDR2, are currently under clinical or preclinical investigation [7]. However, most patients develop acquired resistance within 9-12 months of treatment with the aforementioned TKIs [8, 9]. Since LCC exhibits no consistent genetic alterations [3], there is no tailored targeting agent to date. The current first-line treatment for LCC is pemetrexed, a non-targeted cytotoxic agent approved for treating non-SCC [10]. Unfortunately, targeted therapies for SCLC are no more efficacious than cisplatin and etoposide treatment [11, 12]. Overall, the 5-year survival rates for SCLC and NSCLC remain low, at 7% and 21%, respectively [13]. Currently, there are no effective or druggable cell-surface markers for LCC or SCLC. To develop diagnostics and targeted drug delivery systems (TDDS) for both SCLC and NSCLC, we aimed to identify broad spectrum lung cancer-targeting peptides through biopanning of an undifferentiated cell type, LCC. We hypothesized that targeting peptides which bind to undifferentiated LCC cells have potential to bind to other subtypes of lung cancer as well.
Although peptide ligands are often flexible and have lower affinity to receptors compared to antibodies, the use of peptides in theranostic nanomedicine is advantageous due to their smaller size, reduced immunogenicity, high multivalency, deep tumor penetration, ease of synthesis, and lower production costs [14-16]. Early detection and diagnosis of primary or metastatic lung cancer can significantly improve patient prognosis [13, 17]. Although chest X-ray and low-dose spiral computed tomography (CT) have been used clinically to detect lung nodules as small as 1-2 mm, the exposure to radiation after repeated scanning poses a serious health risk [18, 19]. Additionally, over 90% of pulmonary nodules (< 2 cm in diameter) detected by chest radiography are noncancerous and may be caused by infections, calcification, inflammation, fibrosis, normal vasculature shadow, autoimmune disease, benign tumors, or other reasons [20, 21]. Thus, the identified nodules must be further examined by biopsy or positron emission tomography (PET) to differentiate between benign or malignant growths [20, 21]. Moreover, low spatial resolution (> 5 mm) of PET and the use of radiolabeled glucose limit its application [22, 23]. To develop more specific and safer diagnostics for lung cancer, we used dextran-coated iron oxide nanoparticles as a T2 magnetic resonance imaging (MRI) contrast agent for peptide-mediated tumor tracing. MRI is a non-invasive tomographic technique that does not use ionizing radiation and has high spatial resolution (25-100 μm) [24] making it a relatively safe and informative clinical diagnostic tool [25, 26]. Gadolinium-chelating molecules, the main T1 contrast agent used in clinical MRI [27, 28], exhibit poor physiological stability, have uncontrollable pharmacokinetics, and elevate risk for nephrogenic systemic fibrosis (NSF) [29, 30] However, several iron oxide nanoparticles with higher biocompatibility and strong T2 effects have been approved for clinical use [27, 31, 32]. In addition, super-paramagnetic iron oxide nanoparticles (SPIONs) are tunable in size [33, 34] and shape [35, 36], and their surface properties can be modified [27, 37]; further, they can be coupled with targeting moieties, making them an ideal MR probe for molecular imaging [15].
Encapsulation of anticancer drugs in nanocarriers, such as liposomes, markedly improves their pharmacokinetic profiles by increasing the drug payload, prolonging serum half-life, and reducing exposure to normal tissues [38]. Nanosized drug delivery systems (DDS) utilize passive targeting, via the enhanced permeability and retention (EPR) effect, to accumulate in tumor tissue [39-41]. However, nanodrugs are no more effective than free drugs in treating blood cancers [42], metastatic tumors, or some solid tumors [39, 40]. TDDS, which couple disease-specific ligands to the surface of nanoparticles, have been employed as an advanced approach for active tumor delivery [43, 44]. Targeting moieties, such as peptides or antibodies, selectively bind to tumor-specific antigens and then trigger receptor-mediated endocytosis for controlled release of payloads to the target cells [45-49]. In addition, the released drugs will diffuse from target sites and kill neighboring tumor-associated cells through the bystander effect.
PEGylated liposomal doxorubicin (Doxil/Caelyx) is the most commonly used liposomal drug, and has been approved for the treatment of Kaposi's sarcoma, refractory ovarian cancer, breast cancer, and multiple myeloma [50-52], making it an ideal proof-of-concept tool for testing novel targeting ligands. On the other hand, encapsulation of the NSCLC treatment, vinorelbine [53, 54], has been technically challenging, due to issues related to stabilizing the hydrophobic vinorelbine molecules within the nanocarrier [55]. A recently developed liposomal vinorelbine formulation (NanoVNB®, Taiwan Liposomal Company) currently in clinical trials for colorectal cancer [51, 56], was prepared by using sucrose octasulfate as a stabilizer in a lipid composition of distearoylphosphatidylcholine/cholesterol/poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) (3:2:0.015 molar ratio), resulting in a half-life of 9.4 hr [57]. Polyethylene glycol (PEG) modification of liposomes confers several advantages, including resistance to opsonization, evasion of mononuclear phagocytic system clearance, decreased renal elimination rate, and prolonged circulation time [58]. However, early difficulties in preparing highly PEGylated (DSPE-PEG) liposomal vinorelbine using available gradient-based loading strategies resulted in accelerated drug release and poor encapsulation efficiencies [57, 59]. Our approach utilized an ammonium 5-sulfosalicylate gradient to load vinorelbine into vesicles at an appropriate drug-to-lipid ratio, even for higher PEG-DSPE compositions (10-fold mole parts more than NanoVNB®), giving rise to a modified stable formulation of liposomal vinorelbine (sLV) with a half-life of more than 14 hr. Our group demonstrated the efficacy of sLV against colon cancer in 2015 [60], and in the current study the preclinical efficacy of sLV formulations with conjugated lung cancer targeting peptides was evaluated for the treatment of NSCLC.
Although several studies on lung cancer-binding peptides have been published, most of the identified peptides were either found using differentiated adenocarcinoma cell types [61, 62] or are not robust due to partial NSCLC selectivity, absence of specific consensus motifs, or a lack of in vivo verification [63]. In the present study, three novel peptides, HSP1, HSP2, and HSP4, with theranostic potential in both SCLC and NSCLC, have been verified and used for preclinical drug delivery, molecular imaging, and clinical immunohistochemical analyses.
Methods
Cell lines and cultures
NL20 human bronchial epithelial cells and NCI-H460, NCI-H661, NCI-H1993, NCI-H441, NCI-H520, NCI-H1688, and A549 human lung cancer cell lines were purchased from the American Type Culture Collection (ATCC) and authenticated by ATCC based on their DNA profiles, cytogenetic analyses, and isoenzymology. These cells were cultured according to ATCC's protocols and passaged for fewer than 6 months after resuscitation. CL1-5 cells were established by Chu et al. [64] and routinely verified (based on their growth, morphology, and lack of mycoplasma contamination) in our laboratory. Human normal nasal mucosal epithelial (NNM) cells were a primary culture derived from a nasal polyp [65], and were grown in DMEM.
Phage display biopanning procedures
H460 cells (human lung LCC) were incubated with UV-treated inactive control helper phage (insertless phage). The Ph.D.-12TM (New England BioLabs) phage display peptide library, which initially contained 5 × 1010 plaque-forming units (pfu), was then added. After washing, the bound phages were eluted with a lysis buffer [150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), pH 7.4] on ice. This eluted phage pool was amplified and titrated in an Escherichia coli ER2738 culture (New England BioLabs). The recovered phages were used as input for the next round of panning, as described previously [65]. In the fourth and fifth round of biopanning, phage clones were randomly selected to be cultured for subsequent ELISA screening.
Peptide synthesis and labeling
The synthetic targeting peptides HSP1 (GAMHLPWHMGTL), HSP2 (NPWEEQGYRYSM), and HSP4 (NNPWREMMYIEI), as well as control peptide (Ctrl P) (KATGHSLKTVMQ) or scramble peptides ScP1 (LGHPMATMWLGH), ScP2 (MYQEPSRWGENY), and ScP4 (IMEWNEYIMRPN) corresponding to HSP1, HSP2, and HSP4, were all prepared by Fmoc SPPS using a CEM Liberty automated microwave peptide synthesizer, and purified using reverse-phase high-performance liquid chromatography to 95% purity. For flow cytometry and cellular immunofluorescence assays (IFA), fluorescein isothiocyanate (FITC) was conjugated to the C-terminus of each peptide by attaching it to the ε-amino group (NH2) of an additional lysine at the C-terminus. Peptide synthesis, conjugation, and purification were performed by the Peptide Synthesis Core Facility of the Institute of Cellular and Organismic Biology, Academia Sinica (Taipei, Taiwan).
Flow cytometry analysis
Lung cancer cell lines and control cells were collected using enzyme-free cell dissociation buffer (Gibco, CA, USA), followed by a 30-min blocking step at 4°C. The cells were subsequently incubated with 20 μg/mL FITC-conjugated HSP1, HSP2, HSP4, or Ctrl P at 4°C for 1 hr. After washing thrice by centrifugation at 300 g for 3 min, the cells were resuspended and analyzed using a flow cytometer (Becton Dickinson). The blocking step, peptide incubation, cell washing, and suspension were all performed using ice-cold fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline [PBS] and 1-5% bovine serum albumin (BSA) or 1-5% fetal bovine serum [FBS]).
Immunohistochemical staining of human surgical specimens
Paraffin tissue sections from eleven cases of lung adenocarcinoma and ten cases of lung SCC were obtained from the tissue bank of National Taiwan University Hospital (NTUH), with the approval of the Institutional Review Board of NTUH (IRB9461702021). Written informed consent was obtained, and all human tissue samples were coded for anonymity. To increase the case number and histopathological subtypes of lung cancer specimens, we also obtained commercial tissue microarray sections (US Biomax, Inc.) consisting of a total of 120 cases of lung adenocarcinoma, SCC, LCC, SCLC, etc., with the approval of the Academia Sinica Institutional Review Board (AS-IRB03-102103). To examine localization of phage bound to lung cancer tissues, the tissues were incubated with HPC1, HPC2, HPC4, or control phage (2~5 × 108 pfu/μl). After washing with PBS, sections were treated with anti-M13 mouse mAb (GE Healthcare) for 1 hr at room temperature. Following washing steps, a biotin-free super sensitive polymer-HRP detection system (Biogenex) was used to detect immunoreactivity. The slides were lightly counterstained with hematoxylin, mounted with Aquatex (Merck), and examined by light microscopy.
Ethics Statement for animal experiments
All animal care and handling during the experimentation was carried out in strict accordance with the guidelines of the Academia Sinica Institutional Animal Care and Use Committee. The animal experimental protocols were approved by the Committee on the Ethics of Animal Experiments of Academia Sinica (Permit Number: MMi-ZOOWH2009102 for therapy and pharmacokinetic studies and Permit Number: 15-09-853 for in vivo imaging studies). For mouse experiments, the mice were monitored daily, and human endpoints were used such as weight loss (> 20% of body weight) or the mouse activity was assessed (hunching, stationary, ruffled fur, and poor grooming). At the end of the experiments, the mice were sacrificed by CO2 asphyxiation, and all efforts were made to minimize the animal suffering.
In vivo tumor homing assay and optical imaging
Severe combined immunodeficiency (SCID) mice bearing size-matched H460 lung cancer xenografts (approximately 300 mm3) were intravenously injected with 2 × 109 pfu of targeting or control phage. After 8 min of circulation, the mice were sacrificed and perfused with 50 mL PBS to wash out the unbound phage. Subsequently, the xenograft tumors and mouse organs were dissected and homogenized. The phage bound to each tissue sample was recovered by the adding ER2738 bacteria, and then the mixture was titrated onto IPTG/X-Gal agar plates. For the in vivo whole body imaging, the HPC1, HPC2, HPC4, and control phage were labeled with a fluorescent dye, HiLyte FluorTM 750 acid NHS ester (HL750, AnaSpec), using the NHS functional group. The established H460 xenograft model mice were intravenously injected with 5 × 109 pfu of HL750-labeled targeting or control phage. The fluorescence imaging of the mice and tissues was performed using the Xenogen IVIS200 imaging system (excitation: 710/760 nm; emission: 810/875 nm) at indicated time points. The fluorescence intensity of the tissues was calculated by subtracting the background using the Living Image software (Xenogen).
Synthesis and characterization of peptide-conjugated SPIONs
The parental Dex-Fe3O4 (SPIONs) and peptide-conjugated SPIONs were synthesized by MagQu company (Taiwan). By oxidizing the dextran coating of SPIONs with NaIO4 to create aldehyde groups, the peptides were conjugated to the outer shell of SPIONs covalently [66, 67]. The dynamic light scattering (DLS) measurement indicated that the average hydrodynamic size of peptide-conjugated SPIONs was 140 nm.
In vivo MR imaging using targeting peptide-Dex-Fe3O4 nanoparticles for tumor tracing
SCID mice bearing size-matched H460 xenografts were intravenously injected with HSP1-Dex-Fe3O4, HSP2-Dex-Fe3O4, HSP4-Dex-Fe3O4, or Ctrl P-Dex-Fe3O4 nanoparticles (8 mg/kg mouse body weight). The MR images were acquired pre-contrast and at various time points (3, 6, and 24 hr) post-contrast. All mice were scanned using T2-weighted imaging with a 5 × 5 cm field of view (FOV) and a 256 × 256 matrix size at every 1 mm slice thickness. The T2-weighted fast spin-echo sequence was utilized with a repetition time (TR) = 4000 ms; effective echo times (TEeff) = 80 ms; and number of excitation (NEX) = 6. All MRI experiments were performed with a 4.7 T Biospec 47/40 spectrometer (Bruker, Germany) with a volume coil used as radio frequency (RF) transmitter and signal receiver. The maximum cross-sectional area of the tumor region was selected as the region of interest (ROI) for signal-to-noise ratio (SNR) quantification using MRVision software version 1.6.6 (MRVision, USA), and plotted against time. Tumor volumes were measured by plotting the area of whole tumor region of each slice and that of summarized total region of all tumor slices. Tumor volume measurements, T2 pseudo-color mapping, and 3D image reconstruction were achieved using Avizo 6.1 software (FEI Company).
Prussian blue staining and histological analysis
H460 lung cancer cells were seeded at a density of 2 × 104 cells/well on cover slides (1 × 1 cm) and allowed to grow for 24 hrs. Next, culture cells were fixed with 2% paraformaldehyde, and then incubated with HSP1-Dex-Fe3O4, HSP2-Dex-Fe3O4, HSP4-Dex-Fe3O4, or Ctrl P-Dex-Fe3O4 nanoparticles (10 μg /ml) for 1 hr at room temperature. Slides were washed and stained with routine Prussian blue reagents (1 : 1 mixture of 4% potassium ferrocyanide and 4% hydrochloric acid) for 30 min at room temperature, followed by counter staining for 5 min with Nuclear Fast Red solution (Sigma-Aldrich Corporation).
Tumor-bearing mice were sacrificed after MR imaging studies, and H460 tumor tissue were then fixed with 4% paraformaldehyde and embedded in paraffin. Sections (5 μm) were cut from representative tumors. Xenograft tumor tissues were stained with Prussian blue and Nuclear Fast Red to examine iron localization. All sections were dehydrated, photographed, and quantified.
Preparation of LD and liposomal SRB (LSRB)
Distearoylphosphatidylcholine (DSPC)/cholesterol/mPEG2000-DSPE (3:2:0.3 molar ratio) and DSPC/cholesterol/PEG-PE/sulforhodamine B-DSPE (3:2:0.18:0.06 molar ratio) lipid mixtures were used to encapsulate doxorubicin and sulforhodamine B (SRB), respectively. Dried lipid films were hydrated at 60°C in 250 mM ammonium sulfate for doxorubicin and ddH2O for SRB, and were extruded through polycarbonate membrane filters with a pore size of 0.08 μm using high-pressure extrusion equipment (Lipex Biomembranes, Vancouver, British Columbia) at 55°C. Doxorubicin was loaded into the liposomes at a drug-to-phospholipid ratio of 100 mg/mol. Unencapsulated chemicals were removed by Sephadex G50 (GE Healthcare) gel filtration chromatography, with elution performed with HEPES buffer. The phospholipid concentration of the liposome was quantified using the phosphorus assay of Bartlett [68]. Doxorubicin and SRB concentrations were determined by measuring Excitation/Emission wavelengths of fluorescence at 485/590 and 520/570 nm, respectively, using a spectrofluorometer (Spectra Max M5, Molecular Devices).
Preparation of sLV
The PEGylated liposomes composed of DSPC, cholesterol, and mPEG2000-DSPE that were used to encapsulate vinorelbine (3:2:0.15 molar ratio) were prepared using a lipid film hydration method. Liposomes ranging in size from 80 to 100 nm were prepared by hydrating the lipid films at 60°C in 300 mM ammonium salts of 5-sulfosalicylic acid solution, and then they were extruded through polycarbonate membrane filters with a 0.08-μm pore size using high-pressure extrusion equipment (Lipex Biomembranes, Vancouver, British Columbia) at 55°C. Vinorelbine was loaded into the liposomes using the ammonium 5-sulfosalicylate gradient at a drug-to-phospholipid ratio of 350 mg/mol [60]. Sephadex G50 (GE Healthcare) gel filtration chromatography was used to remove the unencapsulated free drug. The phospholipid concentrations and final encapsulation rate of the liposome were estimated using the phosphorus assay of Barlett [68]. Vinorelbine concentrations in the eluent fractions were determined using HPLC.
Synthesis of targeted peptide-PEG-DSPE conjugates and incorporation into liposomal nanoparticles
Synthetic peptides (HSP1, HSP2, or HSP4) were coupled to NHS-PEG-DSPE [N-hydroxysuccinimido-carboxyl-polyethylene glycol (MW, 3400)-derived distearoylphosphatidyl ethanolamine] (NOF Corporation, Japan) at a 1.1:1 molar ratio. Targeting peptide-PEG-DSPE conjugates were purified by Sephadex G-15 (GE Healthcare) gel filtration chromatography, and were subsequently dried through lyophilization. The conjugates were quantitatively analyzed by HPLC and qualitatively analyzed by MALDI-TOF-MS (BRUKER microflex). Incorporation of PEGylated peptides into the outer monolayer of liposomal nanoparticles (LD, sLV or LSRB) was performed using a previously described “post-insertion” method [65] instead of the “co-dissolving” method.
Characterization of liposomal nanoparticles
The mean particle diameters of liposomes (LD, sLV, or LSRB) were measured by DLS at 25°C using a Malvern Zetasizer Nano ZS (Malvern Instruments, Worcestershire, United Kingdom) with a laser at a wavelength of 633 nm and a 90° scattering angle. The zeta-potential of the liposomes was determined using laser Doppler electrophoresis on a Malvern Zetasizer Nano ZS. Liposome structure was visualized with a cryo-TEM (Tecnai F20, Philips) operating at 200 kV. Specimens were plated on porous carbon film-covered 300-mesh copper grids (HC300-Cu, PELCO). The grids were blotted at 100% humidity and 4 °C for 3 s, and then rapidly frozen in liquid ethane cooled by liquid nitrogen using a Vitrobot (FEI). The grids were introduced into the high-vacuum of the electron microscope column. The low dose condition for each exposure was ∼20 e Å-2. The liposomes in the holes of the carbon film were observed under cryo-TEM using a 70 μm objective aperture. Images were acquired at 10 k or 50 k magnification and were recorded using a 4 k × 4 k CCD camera (Gatan UltraScan 4000).
Uptake of targeting peptide-conjugated LSRB or LD by human lung cancer cells
H460 and H1993 cells were grown on 24-well plates to 90% confluency, and HSP1, 2, or 4-LSRB, or LSRB in complete culture medium were added, at the indicated concentrations. Cells were subsequently incubated at 37°C, and washed with PBS at selected time points (10, 30, 60, 120, 240, 480, 960, and 1440 minutes). Non-internalized LSRB on the cell surface was removed by adding 0.1 M Glycine, pH 2.8, for 10 min. Cells were then lysed with 200 µl 1% Triton X-100. Uptake of low concentrations of LD by H1993 cells was examined using the same protocol. For extraction of SRB or doxorubicin, 300 µl IPA (0.75 N HCl in isopropanol) was added to the lysate and shaken for 30 min. Following centrifugation of the lysate at 12,000 rpm for 5 min, the amount of uptake was determined by measuring Excitation/Emission wavelengths of fluorescence (at 520/570 nm for SRB and 485/590 nm for doxorubicin) using a spectrofluorometer (SpectraMax M5, Molecular Devices). Concentrations of SRB and doxorubicin were calculated by interpolation using a standard curve.
Animal models for the study of ligand-targeted therapy
Female SCID mice between 4 and 6 weeks of age were subcutaneously injected in the dorsolateral flank with human NSCLC cells. Mice with size-matched tumors (approximately 75 mm3 for small tumor; 300 or 500 mm3 for large tumor) were then randomly assigned to different treatment groups, and were injected intravenously with LD, targeting peptide (HSP1, HSP2, or HSP4)-conjugated LD, free doxorubicin (FD), or equivalent volumes of saline. The dosages of drugs and administration time courses differ depending on the experiment, and are described in the respective figure legends. Mouse body weights and tumor sizes were measured twice a week. Tumor volumes were calculated according to the formula: length × (width)2 × 0.52.
Detailed procedures for the establishment of orthotopic lung cancer models and measurement of luciferase expression in cancer are described as follows. SCID mice (6-weeks-old) were anesthetized with isofloruane mixed with oxygen, and placed in the right decubitus position. The skin overlying the left chest wall in the mid-axillary line was prepared with alcohol, and the underlying chest wall and intercostal spaces were visualized. Luciferase-overexpressing H460 or A549 cells (5 × 105 cells) in 50 μl serum-free media plus Matrigel Matrix (BD Biosciences, MA, USA) (2:1) were injected into the left lateral thorax, at the lateral dorsal axillary line. After tumor injection, the mice were turned to the left decubitus position and observed for 45 to 60 min until fully recovered. Luciferase-expressing cancer cells were imaged and quantified using the IVIS200 system (Xenogen Corporation, Alameda, CA) at 10 minutes after i.p. injection of luciferin (Caliper Life Sciences), prior to each drug administration.
Pharmacokinetic and biodistribution studies
SCID mice bearing H460 lung cancer xenografts (~300 mm3) were injected with a single dose of FD, LD, or targeting (HSP1, HSP2, or HSP4) LD at 2 mg/kg in the tail vein. At 1 and 24 hr post-injection, blood samples were collected through submaxillary punctures, and mice were then anaesthetized and sacrificed (three mice/group). Following the perfusion of 50 mL of PBS through the heart, xenograft tumors and organs (brain, lung, heart, liver, and kidney) were dissected, weighed, and homogenized to calculate the amounts of doxorubicin in tissues. Total doxorubicin was quantified by measuring fluorescence at λEx/Em = 485/590 nm using a spectrofluorometer (SpectraMax M5, Molecular Devices).
Statistical analysis
Two-sided unpaired Student's t-test was used to calculate P values. P < 0.05 was considered significant for all analyses.
Results
Identification of three novel peptides that bind to several histopathological subtypes of human lung cancer
In this study, we used a phage displayed random peptide library to isolate phages that bind to H460 LCC cells. After five rounds of affinity selection (biopanning), the titer of bound phage increased by up to 9-fold (Figure S1A). Forty-seven phage clones with high affinity to H460 cells were selected from the fifth round of biopanning, and the binding activity of these clones to other NSCLC cell lines (including human lung adenocarcinoma H1993, CL1-5, A549, murine Lewis lung carcinoma 3LL, and human normal nasal mucosal epithelial NNM cells) was examined (Figure S1B). By sequencing the thirteen phage clones with the highest lung cancer binding affinity but with the weakest reactivity to normal cells, we identified two distinctive groups with differing consensus sequences (Table 1). The high occurrence of the GAMHLPWHMGTL sequence (displayed by HPC1, HPC5, and HPC13) is indicative of its specificity for H460 cells.
Alignment of phage displayed peptide sequences selected by H460 cells.
| Phage clone | Phage displayed peptide sequence* | Frequency | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HPC1, 5, 13 | G | A | M | H | L | P | W | H | M | G | T | L | 3/13 | |||
| HPC12 | G | A | M | H | L | S | W | H | M | G | T | H | 1/13 | |||
| HPC10 | D | P | M | H | N | N | W | H | S | S | P | I | 1/13 | |||
| HPC9 | G | L | D | H | L | W | W | S | S | Q | T | P | 1/13 | |||
| HPC2 | N | P | W | E | E | Q | G | Y | R | Y | S | M | 1/13 | |||
| HPC3 | N | P | W | N | E | M | W | F | Q | T | S | R | 1/13 | |||
| HPC4 | N | N | P | W | R | E | M | M | Y | I | E | I | 1/13 | |||
| HPC6 | W | A | D | M | M | T | S | V | T | P | W | L | 1/13 | |||
| HPC7 | S | E | F | P | R | S | W | D | M | E | T | N | 1/13 | |||
| HPC8 | Q | H | Y | E | T | L | A | F | R | P | K | H | 1/13 | |||
| HPC11 | A | T | Y | N | S | V | N | R | H | S | A | V | 1/13 | |||
Of the 47 phage clones randomly selected in the fifth round of biopanning, 13 phage clones with higher H460 binding affinity were identified; the displayed peptides of these clones were sequenced and aligned. *Phage displayed consensus amino acids are shown in boldface.
We proceeded to compare the binding intensity of these two groups of phages to different lung cancers, by examining the binding patterns of HPC2, HPC3, HPC4, and HPC6 to H460, H1993, CL1-5, A549, and 3LL by cellular ELISA. As shown in Figure S2A, HPC2, HPC3, and HPC4, all of which contain the NPW-E motif, exhibited divergent patterns of binding to different cells, while the binding patterns of HPC3, HPC4, and HPC6, all of which contain a W-EMM motif, exhibited greater similarity. This suggests that W-EMM-mimetic motifs play a more prominent role than the NPW-E motif in binding of HPC3 and HPC4 to lung cancers. Phage clones that contain the MHL-W consensus sequence also showed similar binding patterns (Figure S2B). Based on these findings, we chose to focus on HPC1, HPC2, and HPC4 for further study, as representatives of the MHL-W, NPW-E, and W-EMM motifs, respectively.
To determine whether the peptide sequences displayed on HPC1, HPC2, and HPC4 are able to bind lung cancer cells, we synthesized HSP1, HSP2, and HSP4 peptides, which have the amino acid sequences GAMHLPWHMGTL, NPWEEQGYRYSM, and NNPWREMMYIEI, respectively. SP in HSP refers to the Synthetic Peptide displayed by the relevant HPC phage. To test whether HSP1, HSP2, and HSP4 bind to target molecules expressed on the surface of lung cancer cells, we analyzed the surface binding activities of each FITC-conjugated peptide by flow cytometry (Figure 1A) and immunofluorescent staining (Figure S3). The FACS data revealed that all three FITC-labeled peptides exhibited prominent binding to cell lines from several pathological subtypes of human lung cancer, including LCC (H460 and H661), adenocarcinoma (H1993, H441, CL1-5, and A549), SCC (H520), and small cell carcinoma (H1688), but not to human normal bronchial epithelial cells (NL20) (Figure 1A). Furthermore, HSP1, HSP2, and HSP4 show different patterns of fluorescence intensity in various lung cancer cells, suggesting that these peptides may target different molecules on the cell surface or exhibit different binding affinities for the same target. Cellular IFA were used to reveal that FITC-labeled HSP1, HSP2, and HSP4, but not FITC-labeled control, can bind to H460 LCC cells and H1993 adenocarcinoma cells (Figure S3A). FITC-labeled scrambled peptides ScP1, ScP2, and ScP4 corresponding to HSP1, HSP2, and HSP4 respectively, were also used to verify the binding specificity and showed no binding to H460 cells (Figure S4). The FITC-positive cells represent cells expressing peptide target molecules. Thus, we calculated the percentages of positively-stained H460 and H1993 cells for each peptide (Figure S3B). The proportion of target-expressing cells relative to entire populations, and receptor densities on the cell surface were quantified. It is worth noting that for H1993 cells, HSP4 exhibited higher receptor density on the cell surface (as evidenced by its stronger fluorescent intensity in the images), despite having a lower overall positive rate as compared to the other peptides. In addition, HSP4 showed the highest reactivity to H460 cells, both in terms of percentage of positive cells and receptor density.
Three novel peptides and their respective phage clones showed different binding activities to various histopathological subtypes of human lung cancer. (A) FACS data indicating that HSP1, HSP2, and HSP4 are able to bind to several human lung cancer cell lines, but not normal bronchial epithelial cells. These three peptides showed different binding patterns in SCLC and NSCLC cell lines. NSCLC includes adenocarcinoma, SCC, and LCC. FITC-conjugated Ctrl P served as a negative control. (B) Immunohistochemical staining of human NSCLC and SCLC clinical specimens using HPC1, HPC2, and HPC4 phage clones (2~5 × 108 pfu/μl). Normal bronchiole was not detected by these targeting phages. The same titer of helper phage was used as negative control. The red arrow indicates plasma membrane localization of HPC1 and HPC4 in adenocarcinoma. Scale bar, 25 μm.
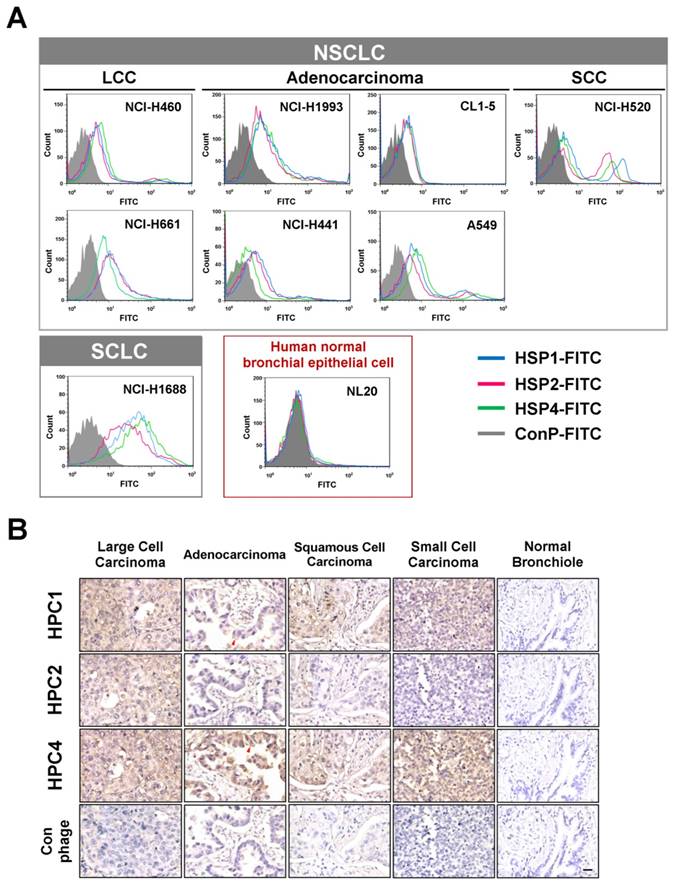
Binding activities of HPC1, HPC2, and HPC4 to clinical surgical specimens of human lung cancer
Validation of targeting using clinical samples is one of the most significant hurdles in drug development. Here, we examined whether HPC1, HPC2, or HPC4 react with several different types of human lung cancer specimens, including adenocarcinoma, papillary adenocarcinoma, bronchioloalveolar carcinoma (BAC), SCC, LCC, and SCLC. Table S1A lists the percentages of binding for HPC1, HPC2, and HPC4 for several types of lung cancer. In general, HPC4 displayed the best reactivity (>80%) for almost all types of lung cancers, followed by HPC1 (>50%). Moreover, HPC1, HPC2, and HPC4 also recognized metastatic adenocarcinoma or SCC from lung (Table S1B), but exhibited no reactivity with normal lung tissue or cancer-adjacent normal lung tissue (Table S1C). High (Figure 1B) and low magnification IHC images (Figure S5) showed clear tumor staining, but poor binding to tumor-adjacent stroma by HPC1 and HPC4. Of equal importance, none of the targeting phages bound bronchiole, alveoli, or pseudostratified columnar epithelium in normal lung. Thus, these data demonstrate that HPC1, HPC2, and HPC4 can recognize NSCLC and SCLC surgical specimens, without cross-reacting to normal pneumonic tissues.
Table S2 shows detailed information regarding the pathological diagnosis, tumor stage and grade of specimins that were used to evaluate HPC1, HPC2, and HPC4 binding. While HPC4 exhibits clear binding in almost all cases of lung cancer, without stage or grade differences, HPC1 shows a higher reactive intensity in higher grade (poorly differentiated) papillary adenocarcinoma (Table S2B) and SCC (Table S2D).
In vivo tumor homing and optical imaging of HPC1, HPC2, and HPC4
To investigate the targeting ability of the selected phage clones in vivo, we intravenously injected each clone into mice bearing H460-derived tumor xenografts. After perfusion, we measured phage titers in tumor and normal organs [49, 69] (Figure 2A and Figure S6). Tumor homing ability was estimated from the phage titer ratio of tumor to normal organs, and compared to that of control phage. In the first group of phages sharing a consensus sequence (HPC2, 3, 4, 6), HPC2, HPC3, and HPC4 all exhibited prominent tumor homing, whereas less tumor localization was observed for HPC6 in vivo (Figure 2A). Furthermore, among the three phage clones (HPC3, 4, 6) with similar binding patterns toward several lung cancer cell lines (Figure S2A), HPC4 showed better binding intensity to A549 cells than HPC3 and HPC6. Therefore, HPC2 and HPC4 were chosen to represent the NPW and W-EMM motifs for further study. Examination of phages with the MHL-W consensus sequence (Figure S2B) revealed that HPC1, but not HPC9, possesses considerable tumor homing ability (Figure 2A).
Verification of the tumor homing ability of H460-targeting phage in vivo. (A) HPC2, HPC3, and HPC4 exhibited the greatest tumor homing ability among the four phage clones of group 1, while HPC1 possessed the best ability in group 2 (n=3). Phage clones were grouped based on the presence of consensus sequences. (B) Optical imaging of whole body and dissected organs from HPC1-HL750-injected mice, as compared to those from the control phage group at 24 hr post-injection. (C) Tissue distribution of HiLyte Fluor 750-labeled phage was examined at 24 hr post-injection, and signal intensities of tumor and organs were measured using IVIS200 software. *, P<0.05; * * *, P<0.001 (n=3).
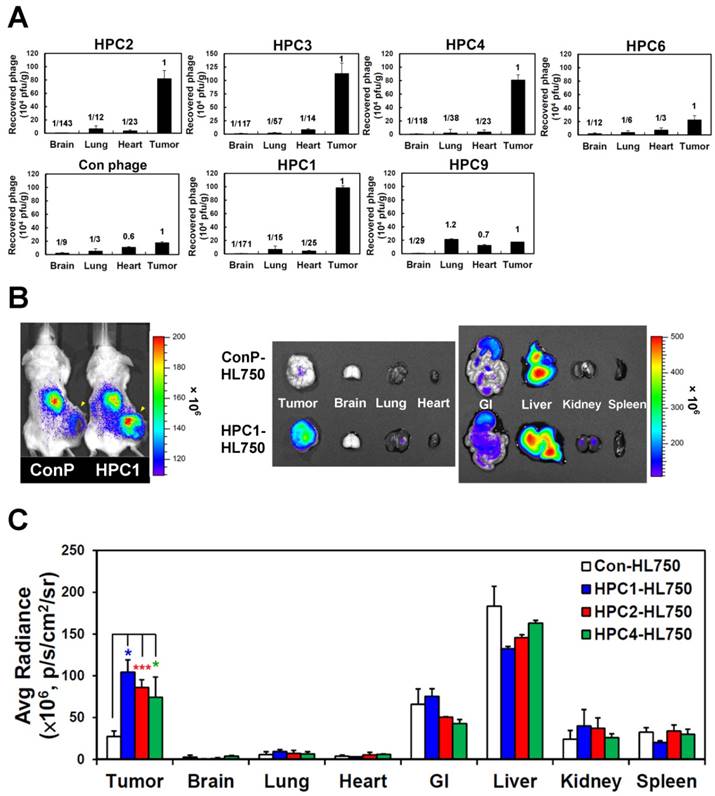
Next, the HL750 fluorescence dye-labeled phages were systemically administered to mice for whole-body imaging. SCID mice bearing size-matched H460 xenografts were i.v. injected with HPC1-HL750, HPC2-HL750, HPC4-HL750, or control phage-HL750, and serially monitored by IVIS200. Accumulation of the targeting phage in tumor tissue was apparent after 6 hr, and was optimal at 24 hr post-injection compared to the control phage group. Whole body optical images of mice and dissected tissues were captured at 24 hr post-injection, as shown in Figure 2B. The tumor fluorescent intensities from HPC1 were maximal and were about 4-fold higher than those in the control phage group (Figure 2C), while the tumor fluorescent intensities were about 3-fold higher for HPC2 and HPC4 compared to those of the control phage group. These results indicate that HPC1, HPC2, and HPC4 possess substantial tumor-homing ability.
In vivo MR imaging of HSP1-, HSP2-, and HSP4-conjugated SPIONs for tumor tracing
SPIONs are highly biocompatible and less toxic T2 MRI contrast agents, and thus are ideal for clinical translation [15]. Targeting peptides HSP1, HSP2, HSP4, and Ctrl P were separately conjugated to parental Dex-Fe3O4 nanoparticles in order to examine tumor targeting capacity. We first tested the tumor binding activity of peptide-conjugated SPIONs by Prussian blue staining in vitro. HSP1-, HSP2-, and HSP4-Dex-Fe3O4 nanoparticles bound to fixed H460 cells (resulting in strong blue signals), whereas Ctrl P-Dex-Fe3O4 nanoparticles did not (Figure S7).
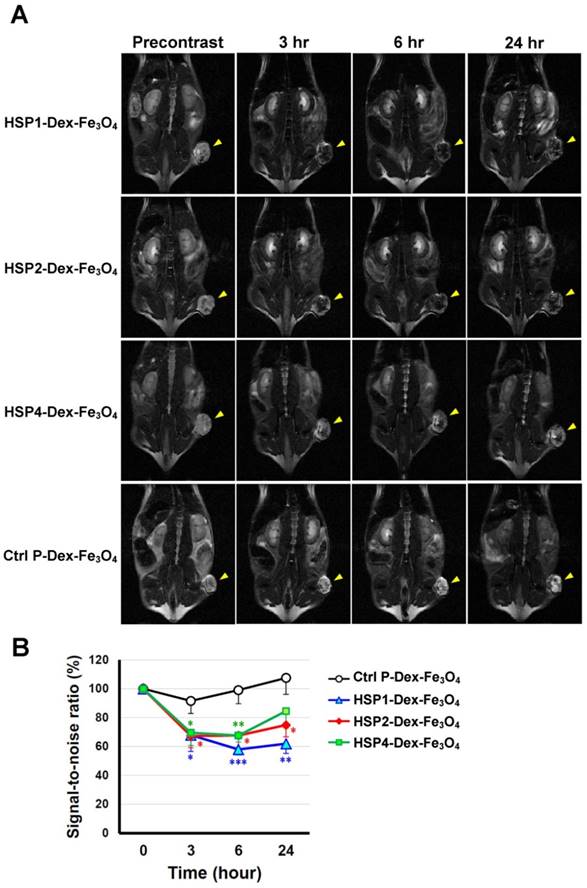
In vivo MR imaging of targeting peptide-Dex-Fe3O4 nanoparticles (final hydrodiameter of ~140 nm) was performed to examine tumor specificity in H460 xenografts; similar-sized Ctrl P-Dex-Fe3O4 nanoparticles were used as a non-targeting control (Figure 3). In general, contrast agents with superparamagnetism shorten T2 relaxation times. In this time-course experiment, T2-weighted fast spin-echo sequence was used to examine each time point. Examination of the T2-weighted imaging kinetics revealed that all of the targeted-SPIONs exhibited significant negative contrast enhancements at tumor sites, as compared to Ctrl P-SPIONs (Figure 3). The non-targeting Ctrl P-SPIONs group also showed a minor signal drop at 3 hr, probably due to passive targeting via the EPR effect, but recovered to baseline as soon as 6 hr post-injection (Figure 3) through blood-clearance. HSP1-Dex-Fe3O4 exhibited the most obvious negative contrast effect in tumor tissue, with the lowest signal (42% reduction) observed at 6 hr post-injection (Figure 3).
T2-weighted MR imaging of HSP1-, HSP2-, and HSP4-Dex-Fe3O4 nanoparticles in H460 xenografts. (A) SCID mice bearing similarly-sized H460 xenografts (approximately 120 mm3 in volume) were subjected to 4.7 T MRI analyses after i.v. injection with HSP1-, HSP2-, HSP4-, or Ctrl P-Dex-Fe3O4 nanoparticles. T2-weighted imaging was performed before injection, and at 3 hr, 6 hr, and 24 hr post-injection. Yellow arrows indicate subcutaneous tumors in the right thigh. (B) Quantitative analysis based on the T2 relative signal-to-noise ratio of the ROI. HSP1-, HSP2-, and HSP4-Dex-Fe3O4 nanoparticles exhibited significant targeting effect in the tumor regions as compared to Ctrl P-Dex-Fe3O4 nanoparticles. Data points, mean; error bars, SD; *, P<0.05; * * *, P<0.001 (n=3).
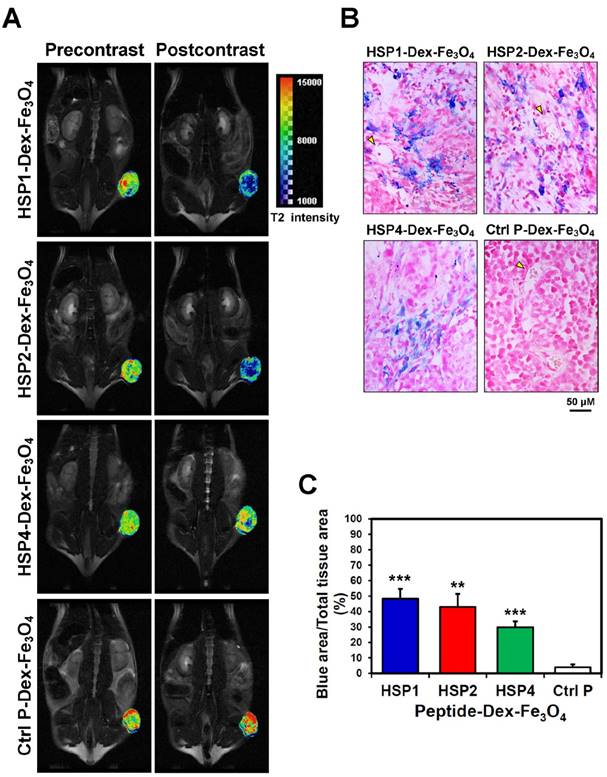
By pseudo-coloring the T2 maps of 6 hr post-injection images (Figure 4A), we visualized the subtle distribution patterns of SPIONs in tumor tissue. The hypointense region colored in blue accounts for about 4/5, 3/4, and 1/4 of the whole tumor area in HSP1-, HSP2-, and HSP4-Dex-Fe3O4-treated groups, respectively. Histological analyses of H460 tumor tissue after MRI experiments revealed that targeted-SPIONs had extravasated from the blood vessels and were bound to the surrounding cancer cells (as can be observed by the presence of Prussian blue reaction products) (Figure 4B). In particular, HSP1- and HSP2-SPIONs exhibited deeper tumor penetration, while an indistinct signal was observed in the control SPION-treated group. The distribution area of Prussian blue reaction products (Figure 4B-C) was highly correlated with the MRI hypointense signals.
Visualization of the detailed tumor distribution of targeted SPIONs by T2 color mapping and histological analyses. (A) The T2-weighted MR images were subjected to pseudo-color mapping to reveal detailed signal changes in tumor tissues. Remarkable signal reduction can be seen in blue, corresponding to roughly 4/5, 3/4, and 1/4 of tumor area at 6 hr post-contrast with HSP1-, HSP2-, and HSP4-Dex-Fe3O4 nanoparticles, respectively. (B) Histological analyses of H460 tumor tissue specimens acquired 24 hrs after injection of HSP1-, HSP2-, HSP4-,or Ctrl P-Dex-Fe3O4 nanoparticles. Sections were stained with Prussian blue to detect Fe deposition, and were counterstained with Nuclear Fast Red. Targeting peptides enhanced extravasation of SPIONs out of blood vessels and deep penetration of SPIONs for cancer cell binding, while most control-SPIONs were washed out and thus rarely detected in tumor. Yellow arrows indicate blood vessels of tumor tissue. (C) Quantification of Prussian blue reaction products from the representative tumor sections. **, P<0.01; * * *, P<0.001 compared with Ctrl P-Dex-Fe3O4 group.
Three-dimensional-reconstructed MR images can reveal the details of tumor structure, vascular location, and the spatial distribution pattern of each contrast nonoprobe. In the supplementary materials, we show a 3D rotational MRI video of an HSP1-SPION-contrasted tumor (Video S1). In summary, all of our MRI data are consistent with the optical imaging data for phage homing (Figure 2B-C). HSP1 exhibits the highest tumor-binding efficiency and longest tumor retention time of the tested peptides, making it promising for development into multimodal imaging probes for lung cancer diagnostics.
HSP1, HSP2, and HSP4 improve liposomal drug binding, intracellular delivery, and cytotoxicity
Since receptor-mediated endocytosis greatly enhances targeted drug delivery by improving drug penetration, release, and efficacy, we proceeded to examine whether HSP1, HSP2, or HSP4 are able to promote liposomal drug internalization by human lung cancer cells. Unlike chemotherapeutic drugs, fluorescence dye SRB does not cause cell death, even at high concentrations, making it ideal for measuring the uptake efficiency of living cells. Through time-course experiments in which we washed out surface-bound liposomes with acidic buffer, we found that targeting peptides (HSP1, HSP2, or HSP4) conjugated to LSRB enhanced liposome endocytosis in H460 (Figure S8) and H1993 (Figure S9A) cells, as compared to non-targeting LSRB. Interestingly, we also observed that at low concentrations, HSP2 enhanced intracellular delivery to H460 cells as compared to non-targeting LSRB. However, at higher concentrations, this did not occur, which may reflect receptor saturation (Figure S8). On the contrary, HSP4 clearly enhanced uptake at higher doses. This phenomenon suggests that HSP1, HSP2, and HSP4 may target different receptors on the cell surface, with different receptor densities.
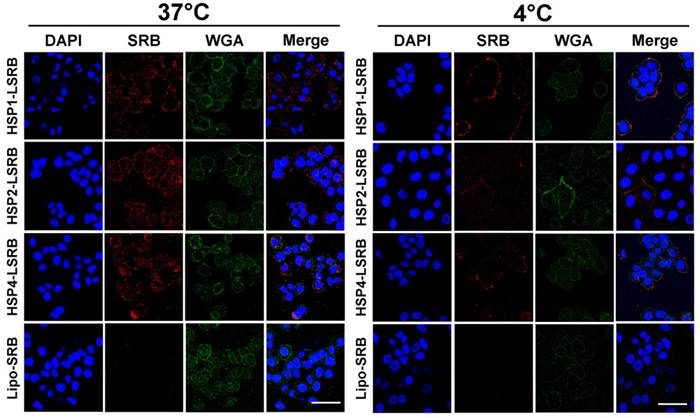
We also examined targeting peptide-conjugated LSRB in lung cancer cells using confocal microscopy (Figure 5). We observed large amounts of LSRB in the cytoplasm of H460 cells incubated with targeting LSRB at 37°C. This was true for both HSP2-LSRB and HSP4-LSRB, whereas minimal SRB fluorescence was detectable in cells incubated with non-targeting LSRB. If cells were incubated at 4°C, targeting peptide-conjugated LSRB bound to the outer membrane of H460 cells, with HSP1-LSRB showing maximal surface binding (Figure 5).
Endocytosis of HSP1, HSP2, and HSP4-LSRB by H460 cells, as examined by confocal microscopy. H460 cells were separately incubated with 10 μM each of HSP1, HSP2, or HSP4 peptide-conjugated LSRB at 37°C (A) or 4°C (B) for 30 min. After removal of non-bound liposomes by washing, confocal microscopy was used to examine liposomal fluorescence. Non-targeting LSRB (10 μM) was used as a control. The nuclei were stained with DAPI. Scale bar, 50 μm.
Subsequently, we examined whether HSP1, HSP2, and HSP4 enhance the therapeutic efficacy of liposomal drugs through their observed targeting and endocytosis abilities. We performed in vitro cytotoxicity assays using HSP1-, HSP2-, or HSP4-conjugated LD in H460 cells (Figure S10). All three targeting peptides significantly enhanced the cytotoxicity of cancer cells to LD. At their optimal peptide ratios, HSP1, HSP2, and HSP4 decreased the IC50 in H460 cells by 12.5-, 13-, and 9.4-fold, respectively.
In brief, HSP1, HSP2, and HSP4 not only bind to lung cancer cells with high specificity, but also trigger liposomal drug internalization and enhance therapeutic efficacy in vitro.
HSP1-, HSP2-, and HSP4-mediated drug delivery systems enhance biodistribution and therapeutic efficacy in vivo
To determine whether HSP1, HSP2, and HSP4 can improve the chemotherapeutic efficacy of anticancer drugs in vivo, we formulated targeting drug delivery systems by coupling the peptides with PEGylated LD. We examined the therapeutic effect of HSP-1, HSP2-, and HSP4-LD in H460 human LCC (Figure 6A-B) and H1993 human lung adenocarcinoma xenograft models (Figure 6C). Mice bearing H460 small tumors (average tumor size of ~75 mm3) were treated with 1 mg/kg of doxorubicin once a week for 4 weeks. The tumor volume decreased significantly in targeting-LD groups (Figure 6A). HSP1, HSP2, and HSP4 significantly improved the therapeutic efficacy of LD in mice bearing H460 large tumors (average tumor size of ~500 mm3). In particular, HSP2- and HSP4-LD treatment decreased tumor volume by half as compared to tumors treated with LD (Figure 6B). The in vivo biodistribution and pharmacodynamic results are consistent with the finding that HSP2 and HSP4 enhance the drug delivery efficacy of LD to H460 tumor tissues by up to 2-fold (Figure 6D). Mice bearing large tumors with size matched H1993 were intravenously injected with 1 mg/kg of HSP1-LD, HSP2-LD, HSP4-LD, LD, FD, or equivalent volumes of PBS twice a week for three weeks (Figure 6C). HSP4-LD resulted in the greatest therapeutic effect, as measured by tumor volume, which was significantly decreased as early as 10.5 days after treatment (after 3 injections).
Targeting peptides HSP1, HSP2, and HSP4 improve the therapeutic efficacy of LD in vivo by increasing drug bioavailability. Mice bearing human LCC H460-derived xenografts with an average tumor size of (A) ~75 mm3 (n=8 in each group) or (B) ~500 mm3 (n=7 in each group) were intravenously injected with FD, LD, targeting liposomes (HSP1-LD, HSP2-LD, or HSP4-LD), or an equal volume of PBS. The administration regimens for each experiment are shown under their respective abscissae. Data points, mean tumor volumes. Error bars, SE. *, P<0.05 compared with non-targeting LD group; ***, P<0.001 compared with FD group. (C) Mice bearing human lung adenocarcinoma H1993 xenografts with an average tumor size of ~300 mm3 were intravenously injected with FD, LD, HSP1-LD, HSP2-LD, HSP4-LD (1 mg/kg, twice a week for three weeks), or an equal volume of PBS. n=7 in each group. Data points, mean tumor volumes. Error bars, SE. *, P<0.05; **, P<0.01; ***, P<0.001. All significant P values arise from comparison with the LD group. (D-E) Pharmacokinetic and pharmacodynamic analyses of LD, HSP1-LD, HSP2-LD, and HSP4-LD in H460 xenografts. At selected time points (1 hr and 24 hr) after a single dose injection (2 mg/kg), the biodistribution patterns of free form, liposomal, and targeting liposomal drug in serum (D, upper panel), tumor (D, middle panel), and normal tissues (E) were estimated by measuring doxorubicin auto-fluorescence signals. n=3 mice in each group. Auto-fluorescence signals were also detected in the nuclei of tumors (D, lower panel). *, P<0.05; **, P<0.01; ***, P<0.001; NS, no significance; Bars, mean; error bars, SD. HSP2 and HSP4 peptides selectively and significantly enhanced drug delivery to tumors and tumor nuclei. (E) Liposomal drugs have similar biodistribution patterns in normal tissues.
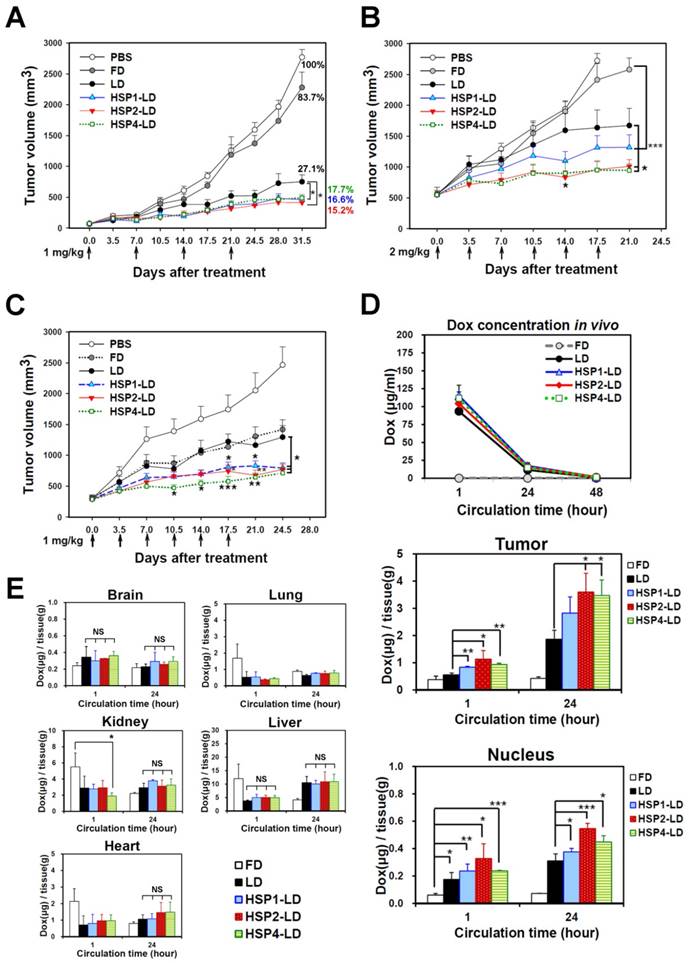
To explore the mechanisms underlying the enhanced anticancer effects of HSP1-, HSP2-, or HSP4-conjugated liposomal drugs in vivo, we performed pharmacodynamic and biodistribution experiments to measure drug accumulation in tumor tissues. Mice bearing H460 xenograft tumors were intravenously injected with a single dose of 2 mg/kg FD, LD, HSP1-LD, HSP2-LD, or HSP4-LD. Either 1 or 24 hr after systemic administration, doxorubicin concentrations in serum (Figure 6D, upper panel), tumors (Figure 6D, middle and lower panels), and normal organs (Figure 6E) were estimated by measuring doxorubicin fluorescence signals after purification. The mean intra-tumor doxorubicin concentrations in the HSP1-, HSP2-, and HSP4-LD groups were about 1.5-, 2-, and 2-fold higher than that in the LD group, respectively (Figure 6D, middle panel). The in vitro peptide stabilities were measured by MALDI-TOF mass spectrometry (Figure S11), and the HSP peptides were predicted to be stable enough to reach the tumor sites within 24 hr while in circulation in vivo (Figure 6D). Because doxorubicin functions by intercalation into DNA, the accumulation of drugs in cancer nuclei was apparent (Figure 6D, lower panel). Liposomal formulation drugs (LD, HSP1-LD, HSP2-LD, and HSP4-LD) displayed similar biodistribution profiles in plasma and normal organs, whereas free doxorubicin had a much shorter half-life in plasma (Figure 6D, upper panel). Moreover, these targeting peptides did not increase doxorubicin accumulation in normal organs, such as brain, heart, lungs, liver, or kidney, in animal models (Figure 6E). Liver toxicity was not observed in any group by hematoxylin and eosin staining (Figure S12). The above experiments demonstrate that targeting peptides, especially HSP2 and HSP4, showed prominent intratumoral delivery in vivo, thereby increasing the therapeutic index of these treatments.
Characterization and comparison of HSP4-conjugated LD and sLV
The difficulty in loading and retention of vinorelbine in liposomes is related to its hydrophobicity; more lipophilic drugs have higher membrane permeability and are released more rapidly [55]. In the present work, vinorelbine was loaded into the designated lipid composition of vesicle at a suitable drug-to-lipid ratio (350 mg/mol) using the ammonium 5-sulfosalicylate gradient method. This optimal construction of sLV increased the drug retention and resulted in high encapsulation efficiency (>95%).
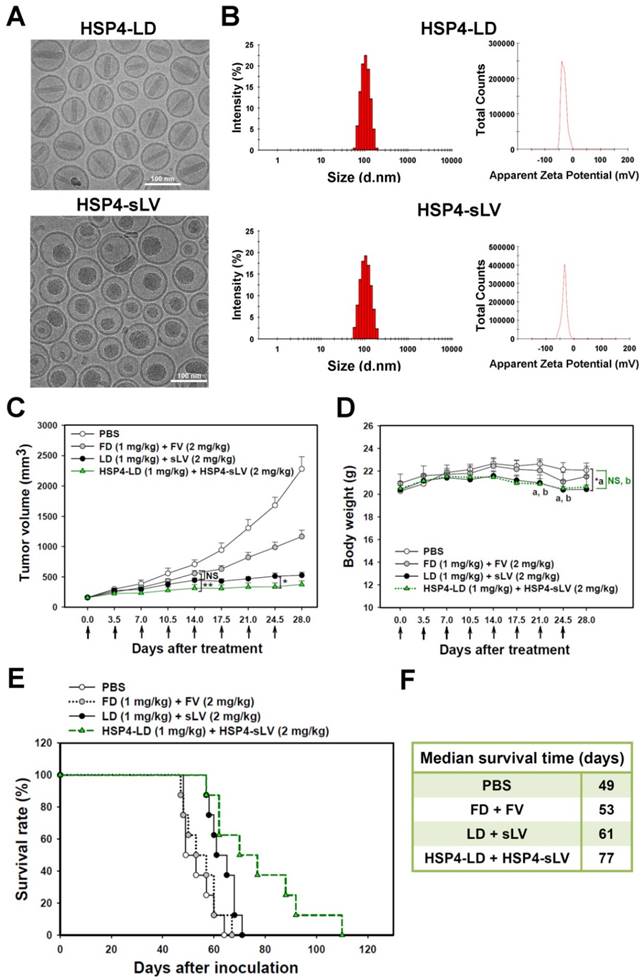
For the purpose of combinatory drug delivery, HSP4 peptide was incorporated into both LD and sLV for NSCLC-targeted therapy. The microstructure of HSP4-conjugated LD and sLV were visualized with a cryo-TEM, which revealed that coupling to targeting peptides did not cause any significant morphological changes (Figure 7A). For detailed characterization of non-targeting LD and sLV, please refer to our related publication [60]. The precipitate inside HSP4-LD exhibited crystalline morphology due to the ammonium sulfate loading technique, while the precipitate of HSP4-sLV appeared amorphous (Figure 7A). A DLS analyzer was used to show that the average particle size of targeting liposomes was 102.3 ± 5 nm for HSP4-LD and 100.6 ± 10 nm for HSP4-sLV (Figure 7B). The zeta-potential of the targeting liposomes was -32 mV for HSP4-LD and -34.6 mV for HSP4-sLV, respectively (Figure 7B). Thus, while the influence of targeting peptide insertion on size distribution was negligible, targeted vesicles became more negatively charged as DSPE-PEG increased, as compared to nontargeting liposomes (zeta potential = -20 ~ -30 mV).
Combination therapy with HSP4-LD and HSP4-sLV in human lung LCC xenografts. (A) Cryo-TEM micrographs of HSP4-LD and HSP4-sLV. Scale bar, 100 nm. (B) Particle size distribution and zeta potential of HSP4-LD and HSP4-sLV. (C) Mice bearing H460-derived lung cancer xenografts with an average tumor size of ~200 mm3 were intravenously injected with FD/FV, LD/sLV, or HSP4-LD/HSP4-sLV at combination doses of 1 mg/kg vinorelbine and 2 mg/kg doxorubicin twice a week for four weeks, or with an equivalent volume of PBS. n=8 in each group. Data points, mean tumor volumes. Error bars, SE. *, P<0.05; **, P<0.01. (D) Body weight during treatment. a, LD+sLV compared to PBS group with P<0.05 (*). b, HSP4-LD+HSP4-sLV compared to PBS group with no significance (NS). (E) Kaplan-Meier survival curve, revealing markedly longer lifespan in mice treated with HSP4-targeted liposomal drugs as compared to other groups. (F) Median survival (in days) was significantly prolonged by HSP4-targeted LD and sLV as compared to non-targeting liposomes (*, P=0.0254).
Targeting liposome-based combination therapy further improved overall survival
Due to genomic instability and genetic heterogeneity of cancerous cells, single-drug monotherapy often strengthens redundant signaling pathways, and accelerates the development of chemoresistant mutations and recurrence. The combined use of multiple chemotherapeutics with different mechanisms of actions has become the primary strategy to treat drug-resistant cancers. Based on this principle, we co-delivered HSP4-LD (a DNA damaging agent) and HSP4-sLV (a microtubule inhibitor) at a 1:2 ratio, to H460 xenografts (Figure 7C-F). The administration regimen of these two drugs was tested and optimized in vivo. The use of targeting peptides significantly decreased tumor volume at 14 and 24.5 days after treatment, as compared to LD + sLV control (Figure 7C). Importantly, targeting peptides did not affect body weight (Figure 7D), and mice treated with HSP4-targeted combinatorial liposomes exhibited longer overall survival than mice treated with non-targeting liposome or free drug (Figure 7E-F). Combinatorial targeting of liposomes prolonged median survival by up to 16 days as compared to non-targeting liposomes (77 vs. 61 days; P = 0.0254).
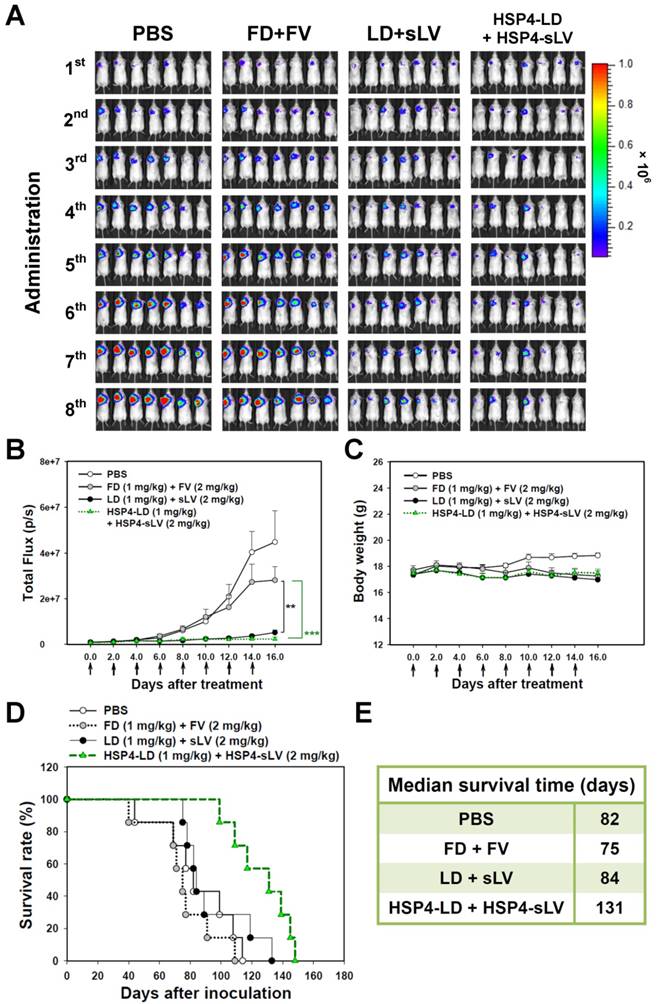
We also investigated the 1:2 LD and sLV combinatorial regimen in H460 LCC (Figure S13) and A549 adenocarcinoma (Figure 8) orthotopic models, which successfully recapitulate tumor-microenvironment interactions. In the H460 orthotopic model, luciferase-expressing tumor mass was significantly decreased by HSP4-targeting liposome treatment, but not by non-targeting liposome treatment, as compared to free drug treatment (Figure S13A-B). The highly aggressive nature of the H460 orthotopic model caused all mice to experience severe body weight loss due to cancer cachexia syndrome (Figure S13C). However, the median survival time was prolonged by 6 days with targeting liposome as compared to non-targeting liposome (76 vs. 70 days; Figure S13D-E). In the A549 orthotopic model, treatment with HSP4-liposomes significantly prolonged overall survival rate and increased median survival times by up to 47 days, as compared to non-targeting liposome treatment (131 vs. 84 days; P = 0.0248) (Figure 8D-E). Our findings demonstrate that HSP4 improves the therapeutic efficacy of nanodrugs (Figure 8A-B), without causing overt adverse effects (Figure 8C).
HSP4-LD and HSP4-sLV combination therapy in an orthotopic model of A549 adenocarcinoma. (A) Imaging drug response of mice with luciferase-expressing A549 cell transplants to combination therapy with FD/FV, LD/sLV, or HSP4-LD/HSP4-sLV at doses of 1 mg/kg vinorelbine and 2 mg/kg doxorubicin, delivered by i.v. injection. Controls were treated with an equal volume of PBS. A total of 5 × 105 cells were transplanted with Matrigel, and treatment started 5 days after cancer cell transplantation (once every two days for 16 days). n=7 in each group. (B) Luminescence signals in tumor were quantified using IVIS200 software. **, P<0.01; ***, P<0.001. (C) Body weight during the course of treatment. The overall survival rate (D) and median survival days (E) were significantly prolonged by HSP4-targeted LD and sLV, as compared to non-targeting liposomes (*, P=0.0248).
Discussion
Although LCC comprises only 10-15% of all lung cancers, it tends to grow and spread quickly, making it harder to treat [70]. On the other hand, its heterogeneity and lack of specific genomic features make it difficult to identify druggable cell-surface markers. Large-scale systematic analyses of lung cancer genomes have reclassified LLC as an undifferentiated type with genetic alterations dispersed across all other subtypes, including adenocarcinoma, SCC, or neuro-endocrine (similar to SCLC) designations [3, 4]. In order to develop diagnostics and tailored TDDS for both SCLC and NSCLC, we have identified three novel peptides, HSP1, HSP2, and HSP4, by phage display-mediated selection of an undifferentiated subtype, LCC. These peptides could also selectively bind to adenocarcinoma, SCC, or SCLC, but not to normal cells in vitro, in vivo, or within clinical samples. Further research is necessary to identify the target molecules of HSP peptides. LCC lacks specific gene expression patterns, mutations, and somatic copy number alteration (SCNA) patterns [3]. Thus, compared to alterations that may be discovered through genomic and proteomic profiling, aberrant posttranslational modifications (PTM) on proteins or lipids, such as glycosylation, may be more likely to act as cell-surface signatures for HSP binding. In the future, appropriate cross-linkers adjusted to different kinds of molecules should be used for peptides to “bait” their target molecules. Once cross-linked, the targets can be immunoprecipitated and identified. Since the targets of HSP peptides have not yet been identified, we used membrane components extracted from H460 cells as the target mixture for HSP binding to measure the association rates (ka), dissociation rates (kd), and equilibrium dissociation constants (KD) (Figure S14). HSP1, HSP2, and HSP4 have KD values of ~5.72, ~12.5, and ~5.52 μM, respectively (Figure S14C). While HSP1 exhibits the fastest association rate among the three peptides, HSP4 shows the slowest dissociation rate. Therefore, these three peptides exhibit distinct target interaction characteristics. Peptide ligands tend to have lower binding affinity than antibodies do, and most of them have typical KD values in the micromolar range [71-73]. Multivalent interactions occur between the surface of nanoparticles with numerous targeting ligands (in this study, 500 peptides per liposome) and cells with high receptor density (for example, overexpressed tumor-associated antigens). This multivalent targeting strategy can provide sufficient avidity and enhanced effective association to overcome the low affinity of each monovalent peptide [16, 74, 75]. The localization of HSP1, HSP2, and HSP4 target molecules in the cell membrane and cytosol of membrane-permeabilized H460 cells were examined by confocal microscopy of FITC-labeled peptides (Figure S15).
The shorter serum half-lives and higher biodegradability of peptides compared to those of antibodies make them ideal targeting probes for many imaging modalities, especially those that involve radioisotopes (e.g., PET or SPECT) [76]. HSP1 exhibits the most favorable surface binding to cancer cells in vivo and displays sustained signal reduction in T2 contrast MR imaging as well as the highest tumor fluorescent intensity in optical phage homing imaging. Moreover, HSP1 significantly prolonged the tumor retention time of SPIONs to 6-24 hr. The extended steady-state acquisition window enables wider applications of HSP1 to other multimodal or theranostic platforms, including novel T1-T2 dual-modal MRI contrast agents [33, 34, 37], magnetic hyperthermia therapy [15, 77], NIR-absorbing agents for photothermal therapy [78], 89Zr-labeled iron oxide nanoparticles for PET/MRI [79], or 111In-labeled iron oxide nanoparticles for SPECT/MRI [27, 80]. Lung MRI is challenging due to the high air occupancy and abundant air-tissue interfaces of the alveolar wall, which generate weak intrinsic MR signals [81, 82]. Therefore, SPIONs contrasted with hyperpolarized (HP) 3He or 129Xe MRI have been studied for detecting pulmonary micrometastases [83]. Our peptide targeted probe provides a powerful molecular imaging tool for lung cancer diagnosis and detection. Furthermore, it does not require radiation, and may preferentially label malignant nodules.
HSP1-, HSP2-, and HSP4 specifically bind to lung cancer cells, which in turn trigger receptor-mediated endocytosis to discharge payloads intracellularly, resulting in about a 10-fold reduction of IC50 in vitro at their optimal peptide ratios. HSP2 and HSP4 enhanced LD delivery by up to 2-fold when compared to non-targeting LD in the H460 model, and significantly improved drug bioavailability in vivo, thereby increasing the therapeutic index. In this study, multiple-subtype targeting peptides have been evaluated for preclinical enhancement of chemotherapeutic efficacy in animal models of LCC and adenocarcinoma (the most predominant lung cancer subtype, ~40%). HSP4-targeting of combinatorial liposomes (LD and sLV) enhanced overall survival, suggesting that this therapeutic strategy may help overcome clinical chemoresistance and delay cancer relapse. The above results also support the use of these targeting peptides for designing tailored therapeutic regimens against SCC and SCLC. SCLC is the most aggressive form of neuroendocrine tumor, accounting for about 15% of all diagnosed cases of lung cancer, and up to 25% of lung cancer deaths annually [11, 84]. It is worth noting that patients diagnosed as having LCC with neuroendocrine features (genetic similarity) have the second poorest survival rate among all lung cancer subtype patients, which is similar to overall survival in SCLC [3]. At present, the treatment of SCLC remains a challenge. Chemotherapeutics, such as cisplatin and etoposide, with or without radiation therapy, remain the sole first-line treatment for SCLC [12, 85]. However, the effect is usually transient, with a high rate of early recurrence. Topotecan is the only FDA-approved agent for second-line treatment of SCLC, and yet it may not provide any survival benefit [86]. Our findings introduce a multivalent targeted approach that is guided by targeting peptides and has many options for drug payload to their indicated lung cancer subtype. HSP1 and HSP4 showed prominent SCLC binding activities in cell lines and clinical specimens and may be suitable for future development into tailored TDDS, which combines liposomal cisplatin and liposomal topotecan for SCLC treatment. Besides lung cancer, HPC1 and HPC4 also exhibit certain degree of binding affinities to SAS (tongue SCC), SKOV-3 (ovarian adenocarcinoma), and MIA PaCa-2 (pancreas ductal adenocarcinoma) cell lines (Figure S16A), showing broader cancer indications.
An overall scheme of this research is depicted in the Graphical Abstract. In summary, our research indicates that HSP1, HSP2, and HSP4 have significant potential to be incorporated into effective theranostic strategies, such as tailored targeting therapy, companion diagnostics, and non-invasive imaging, and hence, are meaningful for translational and precision medicine.
Supplementary Material
Additional File 1Supplementary Tables and Figures.
Video S1.
Abbreviations
ALK: anaplastic lymphoma kinase;
BAC: bronchioloalveolar carcinoma;
Cryo-TEM: cryogenic transmission electron microscope;
CT: computed tomography;
Ctrl P: control peptide;
DDR2: discoidin domain receptor 2;
DDS: drug delivery systems;
DNA: deoxyribonucleic acid;
EGFR: epidermal growth factor receptor;
ELISA: enzyme-linked immunosorbent assay;
EML4: echinoderm microtubule associated protein like 4;
EPR effect: enhanced permeability and retention effect;
FACS: fluorescence-activated cell sorting;
FDA: Food and Drug Administration;
FGFR: fibroblast growth factor receptor;
FITC: fluorescein isothiocyanate;
HL750: HiLyte Fluor 750;
IC50: half maximal inhibitory concentration;
IFA: immunofluorescence assays;
IFP: interstitial fluid pressure;
IHC: immunohistochemistry;
i.v.: intravenous;
LCC: large cell carcinoma;
LD: liposomal doxorubicin;
MRI: Magnetic Resonance Imaging;
NSCLC: non-small-cell lung carcinoma;
NSF: nephrogenic systemic fibrosis;
PEG: polyethylene glycol;
PET: positron emission tomography;
SCC: squamous cell carcinoma;
SCID: severe combined immunodeficiency;
SCLC: small-cell lung carcinoma;
sLV: stable formulation of liposomal vinorelbine;
SOX2: SRY (sex determining region Y)-box 2;
SPECT: single-photon emission computed tomography;
SPIONs: super-paramagnetic iron oxide nanoparticles;
SRB: sulforhodamine B;
TDDS: targeted drug delivery systems;
TKIs: tyrosine kinase inhibitors.
Acknowledgements
The authors thank Ms. Ching-Chun Lin and the Core Facility of the Institute of Cellular and Organismic Biology (ICOB), Academia Sinica, for their assistance in peptide synthesis and FITC conjugation. We are also grateful to Dr. Yuan-Chih Chang and Ms. Hui-Ju Huang for the assistance in Cryo-EM imaging and for use of the Tecnai F20 in the Cryo-EM Core Facility, Department of Academic Affairs and Instrument Service at Academia Sinica. We would also like to acknowledge the financial support received from the ChemBank and High-Throughput Screening Resource Center of the National Research Program for Biopharmaceuticals Project (ChemBank and High-Throughput ScreeningVI, NSC-105-2325-B-001-012) in the form of grants to support the sensorgrams and KD values of each peptide measured by FortéBIO Octet® HTX system, and the technical support provided by Dr. Ying-Ta Wu's lab. Finally, we thank the Taiwan Mouse Clinic (MOST 104-2325-B-001-011), which is funded by the National Research Program for Biopharmaceuticals (NRPB) at the Ministry of Science and Technology (MOST) of Taiwan, for technical support in MRI experiments, especially the great service from Dr. Chih-Cheng Chen and Mr. Ming-Huang Lin.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
2. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535-46
3. Clinical Lung Cancer Genome Project (CLCGP), Network Genomic Medicine (NGM). A genomics-based classification of human lung tumors. Sci Transl Med. 2013;5:209ra153
4. Bunn PA Jr, Franklin W, Doebele RC. The evolution of tumor classification: a role for genomics? Cancer cell. 2013;24:693-4
5. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S. et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123-32
6. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S. et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561-6
7. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519-25
8. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B. et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4:120ra17
9. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786-92
10. Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C. et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3543-51
11. Joshi M, Ayoola A, Belani CP. Small-cell lung cancer: an update on targeted therapies. Adv Exp Med Biol. 2013;779:385-404
12. William WN Jr, Glisson BS. Novel strategies for the treatment of small-cell lung carcinoma. Nat Rev Clin Oncol. 2011;8:611-9
13. American Cancer Society. Cancer Facts & Figures 2016. Atlanta: American Cancer Society. 2016
14. Cheng WW, Allen TM. The use of single chain Fv as targeting agents for immunoliposomes: an update on immunoliposomal drugs for cancer treatment. Expert Opin Drug Deliv. 2010;7:461-78
15. Bakhtiary Z, Saei AA, Hajipour MJ, Raoufi M, Vermesh O, Mahmoudi M. Targeted superparamagnetic iron oxide nanoparticles for early detection of cancer: Possibilities and challenges. Nanomedicine. 2016;12:287-307
16. Choi SK. Synthetic multivalent molecules: concepts and biomedical applications. Hoboken, USA: John Wiley & Sons, Inc. 2004
17. The National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD. et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365:395-409
18. Sodickson A, Baeyens PF, Andriole KP, Prevedello LM, Nawfel RD, Hanson R. et al. Recurrent CT, cumulative radiation exposure, and associated radiation-induced cancer risks from CT of adults. Radiology. 2009;251:175-84
19. Frush DP, Applegate K. Computed tomography and radiation: understanding the issues. J Am Coll Radiol. 2004;1:113-9
20. Ost D, Fein AM, Feinsilver SH. Clinical practice. The solitary pulmonary nodule. N Engl J Med. 2003;348:2535-42
21. Winer-Muram HT. The solitary pulmonary nodule. Radiology. 2006;239:34-49
22. Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer. 2002;2:683-93
23. Harris RS, Schuster DP. Visualizing lung function with positron emission tomography. J Appl Physiol (1985). 2007;102:448-58
24. Kozlowska D, Foran P, MacMahon P, Shelly MJ, Eustace S, O'Kennedy R. Molecular and magnetic resonance imaging: The value of immunoliposomes. Adv Drug Deliv Rev. 2009;61:1402-11
25. Degen CL, Poggio M, Mamin HJ, Rettner CT, Rugar D. Nanoscale magnetic resonance imaging. Proc Natl Acad Sci U S A. 2009;106:1313-7
26. Buxton RB. Introduction to functional magnetic resonance imaging: Principles and techniques. 2nd ed. Cambridge, UK: Cambridge University Press. 2009
27. Gao Z, Ma T, Zhao E, Docter D, Yang W, Stauber RH. et al. Small is smarter: Nano MRI contrast agents - advantages and recent achievements. Small. 2016;12:556-76
28. Kobayashi H, Brechbiel MW. Nano-sized MRI contrast agents with dendrimer cores. Adv Drug Deliv Rev. 2005;57:2271-86
29. Penfield JG, Reilly RF Jr. What nephrologists need to know about gadolinium. Nat Clin Pract Nephrol. 2007;3:654-68
30. Marckmann P, Skov L, Rossen K, Dupont A, Damholt MB, Heaf JG. et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17:2359-62
31. Pouliquen D, Le Jeune JJ, Perdrisot R, Ermias A, Jallet P. Iron oxide nanoparticles for use as an MRI contrast agent: pharmacokinetics and metabolism. Magn Reson Imaging. 1991;9:275-83
32. Weinstein JS, Varallyay CG, Dosa E, Gahramanov S, Hamilton B, Rooney WD. et al. Superparamagnetic iron oxide nanoparticles: diagnostic magnetic resonance imaging and potential therapeutic applications in neurooncology and central nervous system inflammatory pathologies, a review. J Cereb Blood Flow Metab. 2010;30:15-35
33. Kim BH, Lee N, Kim H, An K, Park YI, Choi Y. et al. Large-scale synthesis of uniform and extremely small-sized iron oxide nanoparticles for high-resolution T1 magnetic resonance imaging contrast agents. J Am Chem Soc. 2011;133:12624-31
34. Huang G, Li H, Chen J, Zhao Z, Yang L, Chi X. et al. Tunable T1 and T2 contrast abilities of manganese-engineered iron oxide nanoparticles through size control. Nanoscale. 2014;6:10404-12
35. Zhao Z, Zhou Z, Bao J, Wang Z, Hu J, Chi X. et al. Octapod iron oxide nanoparticles as high-performance T2 contrast agents for magnetic resonance imaging. Nat Commun. 2013;4:2266
36. Lee N, Choi Y, Lee Y, Park M, Moon WK, Choi SH. et al. Water-dispersible ferrimagnetic iron oxide nanocubes with extremely high r2 relaxivity for highly sensitive in vivo MRI of tumors. Nano Lett. 2012;12:3127-31
37. Zhou Z, Wu C, Liu H, Zhu X, Zhao Z, Wang L. et al. Surface and interfacial engineering of iron oxide nanoplates for highly efficient magnetic resonance angiography. ACS Nano. 2015;9:3012-22
38. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145-60
39. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271-84
40. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387-92
41. Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136-51
42. Kohlschutter J, Michelfelder S, Trepel M. Drug delivery in acute myeloid leukemia. Expert Opin Drug Deliv. 2008;5:653-63
43. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65:36-48
44. Tiwari G, Tiwari R, Sriwastawa B, Bhati L, Pandey S, Pandey P. et al. Drug delivery systems: An updated review. Int J Pharm Investig. 2012;2:2-11
45. Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA. et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10:389-97
46. Lu RM, Chang YL, Chen MS, Wu HC. Single chain anti-c-Met antibody conjugated nanoparticles for in vivo tumor-targeted imaging and drug delivery. Biomaterials. 2011;32:3265-74
47. Svensen N, Walton JGA, Bradley M. Peptides for cell-selective drug delivery. Trends Pharmacol Sci. 2012;33:186-92
48. Torchilin V. Antibody-modified liposomes for cancer chemotherapy. Expert Opin Drug Deliv. 2008;5:1003-25
49. Lo A, Lin CT, Wu HC. Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Mol Cancer Ther. 2008;7:579-89
50. Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1:297-315
51. Wu HC, Chi YH, Wu CH. Targeting liposomes for drug delivery in cancer therapy. In: (ed.) Souto EB. Lipid Nanocarriers in Cancer Diagnosis and Therapy, 1st ed. United Kingdom: iSmithers Press. 2011:85-136
52. Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211-7
53. Depierre A, Lemarie E, Dabouis G, Garnier G, Jacoulet P, Dalphin JC. A phase II study of Navelbine (vinorelbine) in the treatment of non-small-cell lung cancer. Am J Clin Oncol. 1991;14:115-9
54. Faller BA, Pandit TN. Safety and efficacy of vinorelbine in the treatment of non-small cell lung cancer. Clin Med Insights Oncol. 2011;5:131-44
55. Zhigaltsev IV, Maurer N, Akhong QF, Leone R, Leng E, Wang J. et al. Liposome-encapsulated vincristine, vinblastine and vinorelbine: a comparative study of drug loading and retention. J Control Release. 2005;104:103-11
56. Yang SH, Lin CC, Lin ZZ, Tseng YL, Hong RL. A phase I and pharmacokinetic study of liposomal vinorelbine in patients with advanced solid tumor. Invest New Drugs. 2012;30:282-9
57. Drummond DC, Noble CO, Guo Z, Hayes ME, Park JW, Ou CJ. et al. Improved pharmacokinetics and efficacy of a highly stable nanoliposomal vinorelbine. J Pharmacol Exp Ther. 2009;328:321-30
58. Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496-504
59. Li CL, Cui JX, Wang CX, Zhang L, Li YH, Zhang L. et al. Development of pegylated liposomal vinorelbine formulation using "post-insertion" technology. Int J Pharm. 2010;391:230-6
60. Wu CH, Kuo YH, Hong RL, Wu HC. α-Enolase-binding peptide enhances drug delivery efficiency and therapeutic efficacy against colorectal cancer. Sci Transl Med. 2015;7:290ra91
61. Lau D, Guo L, Liu R, Marik J, Lam K. Peptide ligands targeting integrin alpha3beta1 in non-small cell lung cancer. Lung Cancer. 2006;52:291-7
62. Chang DK, Lin CT, Wu CH, Wu HC. A novel peptide enhances therapeutic efficacy of liposomal anti-cancer drugs in mice models of human lung cancer. PLoS One. 2009;4:e4171
63. McGuire MJ, Gray BP, Li S, Cupka D, Byers LA, Wu L. et al. Identification and characterization of a suite of tumor targeting peptides for non-small cell lung cancer. Sci Rep. 2014;4:4480
64. Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, Wu R. et al. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am J Respir Cell Mol Biol. 1997;17:353-60
65. Lee TY, Wu HC, Tseng YL, Lin CT. A novel peptide specifically binding to nasopharyngeal carcinoma for targeted drug delivery. Cancer Res. 2004;64:8002-8
66. Yang SY, Jian ZF, Horng HE, Hong C-Y, Yang HC, Wu CC. et al. Dual immobilization and magnetic manipulation of magnetic nanoparticles. J Magn Magn Mater. 2008;320:2688-91
67. Huang KW, Chieh JJ, Horng HE, Hong CY, Yang HC. Characteristics of magnetic labeling on liver tumors with anti-alpha-fetoprotein-mediated Fe3O4 magnetic nanoparticles. Int J Nanomedicine. 2012;7:2987-96
68. Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466-8
69. Lee TY, Lin CT, Kuo SY, Chang DK, Wu HC. Peptide-mediated targeting to tumor blood vessels of lung cancer for drug delivery. Cancer Res. 2007;67:10958-65
70. American Cancer Society. Lung Cancer (Non-Small Cell). 2016. 16 May. 2016 Available: http://www.cancer.org/cancer/lungcancer-non-smallcell/detailedguide/non-small-cell-lung-cancer-what-is-non-small-cell-lung-cancer
71. Ruzza P, Rubini C, Siligardi G, Hussain R, Calderan A, Guiotto A. et al. Effect of 4-fluoro-L-proline on the SH3 binding affinity. Adv Exp Med Biol. 2009;611:499-500
72. Yokoyama K, Sakai T, Ishikawa H, Morita Y, Tamiya E. Screening and design of hybrid peptide that binds with glucose oxidase. In: (ed.) Lebl M, Houghten RA. Peptides: The wave of the future. California: Springer Netherlands. 2001:202-3
73. Causey LD, Dwyer DS. Detection of low affinity interactions between peptides and heat shock proteins by chemiluminescence of enhanced avidity reactions (CLEAR). Nat Biotechnol. 1996;14:348-51
74. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
75. Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker Jr JR, Banaszak Holl MM. The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem Biol. 2007;14:107-15
76. Chen X. Molecular imaging probes for cancer research. Singapore: World Scientific Publishing. 2012
77. Laurent S, Dutz S, Häfeli UO, Mahmoudi M. Magnetic fluid hyperthermia: focus on superparamagnetic iron oxide nanoparticles. Adv Colloid Interface Sci. 2011;166:8-23
78. Yu J, Yang C, Li J, Ding Y, Zhang L, Yousaf MZ. et al. Multifunctional Fe5C2 nanoparticles: a targeted theranostic platform for magnetic resonance imaging and photoacoustic tomography-guided photothermal therapy. Adv Mater. 2014;26:4114-20
79. Thorek DL, Ulmert D, Diop NF, Lupu ME, Doran MG, Huang R. et al. Non-invasive mapping of deep-tissue lymph nodes in live animals using a multimodal PET/MRI nanoparticle. Nat Commun. 2014;5:3097
80. Zeng J, Jia B, Qiao R, Wang C, Jing L, Wang F. et al. In situ 111In-doping for achieving biocompatible and non-leachable 111In-labeled Fe3O4 nanoparticles. Chem Commun (Camb). 2014;50:2170-2
81. Hatabu H, Alsop DC, Listerud J, Bonnet M, Gefter WB. T2* and proton density measurement of normal human lung parenchyma using submillisecond echo time gradient echo magnetic resonance imaging. Eur J Radiol. 1999;29:245-52
82. Kuethe DO, Adolphi NL, Fukushima E. Short data-acquisition times improve projection images of lung tissue. Magn Reson Med. 2007;57:1058-64
83. Branca RT, Cleveland ZI, Fubara B, Kumar CS, Maronpot RR, Leuschner C. et al. Molecular MRI for sensitive and specific detection of lung metastases. Proc Natl Acad Sci U S A. 2010;107:3693-7
84. Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A. et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol. 2006;24:4539-44
85. Thatcher N, Eckardt J, Green M. Options for first- and second-line therapy in small cell lung cancer-a workshop discussion. Lung Cancer. 2003;41(Suppl 4):S37-41
86. Ardizzoni A, Hansen H, Dombernowsky P, Gamucci T, Kaplan S, Postmus P. et al. Topotecan, a new active drug in the second-line treatment of small-cell lung cancer: a phase II study in patients with refractory and sensitive disease. The European Organization for Research and Treatment of Cancer Early Clinical Studies Group and New Drug Development Office, and the Lung Cancer Cooperative Group. J Clin Oncol. 1997;15:2090-6
Author contact
![]() Corresponding author: Dr. Han-Chung Wu, Institute of Cellular and Organismic Biology, Academia Sinica, 128 Academia Road, Section 2, Nankang, Taipei 11529, Taiwan E-mail: hcw0928sinica.edu.tw
Corresponding author: Dr. Han-Chung Wu, Institute of Cellular and Organismic Biology, Academia Sinica, 128 Academia Road, Section 2, Nankang, Taipei 11529, Taiwan E-mail: hcw0928sinica.edu.tw