Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(6):1633-1649. doi:10.7150/thno.17588 This issue Cite
Research Paper
Multifunctional Nucleus-targeting Nanoparticles with Ultra-high Gene Transfection Efficiency for In Vivo Gene Therapy
Ling Li1, Xia Li1, Yuzhe Wu2, Linjiang Song1, Xi Yang1, Tao He1, Ning Wang1, Suleixin Yang1, Yan Zeng1, Qinjie Wu1, Zhiyong Qian1, Yuquan Wei1, Changyang Gong1 ![]()
1. State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, 610041, P. R. China;
2. College of Materials, Xiamen University, Xiamen 361005, P.R. China.
Received 2016-9-15; Accepted 2017-2-14; Published 2017-4-10
Abstract

Cancer stem cell-like cells (CSCL) are responsible for tumor recurrence associated with conventional therapy (e.g. surgery, radiation, and chemotherapy). Here, we developed a novel multifunctional nucleus-targeting nanoparticle-based gene delivery system which is capable of targeting and eradicating CSCL. These nanoparticles can facilitate efficient endosomal escape and spontaneously penetrate into nucleus without additional nuclear localization signal. They also induced extremely high gene transfection efficiency (>95%) even in culture medium containing 30% serum, which significantly surpassed that of some commercial transfection reagents, such as Lipofectamine 2000 and Lipofectamine 3000 etc. Especially, when loaded with the TRAIL gene, this system mediated remarkable depletion of CSCL. Upon systemic administration, the nanoparticles accumulated in tumor sites while sparing the non-cancer tissues and significantly inhibited the growth of tumors with no evident systemic toxicity. Taken together, our results suggest that these novel multifunctional, nucleus-targeting nanoparticles are a very promising in vivo gene delivery system capable of targeting CSCL and represent a new treatment candidate for improving the survival of cancer patients.
Keywords: gene delivery, nuclear targeting, hyaluronic acid, TRAIL, cancer stem cells, colon cancer.
Introduction
Colorectal cancer is one of the most common causes of cancer-associated deaths worldwide [1]. Despite the treatment modalities of surgery, radiation, and chemotherapy used in the management of colon carcinoma, the survival of patients has not improved significantly in the past decade, making it a major public health problem for the foreseeable future. Major impediments of these treatments include uncertain efficacy and undesirable side effects [2-4]. Importantly, the incapability to eradicate cancer stem cell-like cells (CSCL) in tumor tissues which leads to the recurrence of colorectal cancer, also limits the application of the conventional treatment modalities [5-6]. Thus, there is an urgent need for developing new therapies. Gene-based therapy, which has a great potential for treating inherited and acquired diseases not amenable to conventional strategies, may be an alternative strategy [7-11]. This method encompasses targeting the diseased tissues with therapeutic genes using a variety of delivery carriers followed by increasing or reducing the transcription and translation of the targeted genes. There are several impediments to this approach including the membrane permeability inhibition due to the negative charges of the gene, rapid degradation by plasma enzymes, and induction of an immune response following systemic administration. Therefore, safe and effective gene delivery systems are needed for gene-based therapies [12-13].
Although some clinical trials reported promising outcomes with viral gene carriers, the fundamental drawbacks of severe immune/inflammatory reactions, recombination with wild-type viruses, limited DNA packaging capacity, and difficulty of vector production severely limit their application [14-18]. In the past decades, non-viral gene delivery vectors had attracted much attention due to their safety, limited immunogenicity, and ability to load and deliver large-size genetic payloads [19-22]. Nevertheless, low transfection potency, cytotoxicity caused by the high positive charge density, and poor formulation stability had largely limited their application [23-25]. Although various strategies have been developed to eliminate or minimize limitations associated with non-viral gene carriers, efficient targeted gene delivery with low toxicity remains a challenge [26-28].
Herein, we have designed a novel ternary multifunctional nanoparticle with multi-stage, multi-targeting, and depth penetration capability for in vivo gene delivery (Fig. 1). The system consists of a spontaneous nucleus-targeting core (PF33/pDNA) with extremely high gene transfection efficiency and a negatively charged capsid-like shell (RGD-R8-PEG-HA, RRPH) with multi-stage active targeting capability. PF33 is a fluorinated polymer with very high transfection efficiency to form the binary complexes (PF33/pDNA) with the plasmid (pDNA). The positively charged surface of PF33/pDNA may result in aggregation in blood and nonspecific phagocytosis by the reticuloendothelial system (RES). We, therefore, coated the binary complexes with RRPH, a hyaluronan polymer grafted with PEG side chains, which are further conjugated with a R8-RGD (Cys-RRRRRRRR-c(RGDfK)) tandem peptide. Hyaluronan (HA) can specifically bind CD44, which is an important marker of a subset population of CSCL in many tumors [29-30]. HA could be partially degraded by hyaluronidase (HAase) overexpressed in tumor tissues and tumor intracellular compartments promoting the release of the payloads [31]. To further promote the active targeting, we modified some of PEG side chains with R8-RGD. cRGD peptide can be used as a specific ligand for integrin αvβ3 receptors while R8 can mediate the depth penetration in solid tumors [32].
Schematic representation of RRPHC nanoparticles for gene delivery.
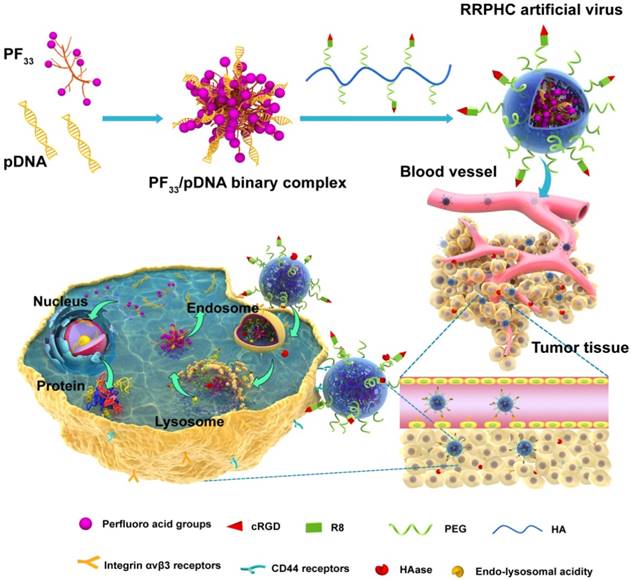
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is shown to be a promising anti-cancer therapeutic, which triggers apoptosis through interaction with the death receptors DR4 and DR5 [33-34]. It has been widely considered an optimal candidate for cancer treatment owing to its tumor cell specificity and minimal side effects [35-36]. Previous reports had demonstrated that human colon carcinoma cells, such as HCT 116 and SW 480, are sensitive to the apoptosis induced by TRAIL. More importantly, some reports also proved that the sub-population (SP) of SW 480, representing CSCL, was also sensitive to TRAIL [37]. Depletion of SP was observed after treatment with TRAIL, suggesting that TRAIL could be a candidate for the eradicating CSCL.
In our previous work, we applied RRPHC to deliver CRISPR-Cas9 genome editing system to explore the application of this platform in gene editing field. This system mediated efficient delivery of large-size CRISPR-Cas9 plasmid in vivo [38]. In this study, we chose TRAIL as a therapeutic gene for colon cancer. We used the novel multifunctional, nucleus-targeting nanoparticles for in vivo TRAIL delivery to target and eradicate CSCL in cancer gene therapy. First, the cellular uptake, multiple targeting and penetration capability, endosomal escape, the ability to deplete CSCL, and gene transfection were carefully examined in vitro. We then further evaluated the in vivo distribution, tumor growth inhibition, and the underlying mechanisms in xenografted tumors of human colon carcinoma cells in mice.
Materials and Methods
Materials
Hyaluronic acid (HA, 35 kDa) was purchased from Shandong Freda Biochem Co., Ltd. (Shandong, China). Maleimide-PEG2000-NH2 was obtained from JenKem Technology Co., Ltd. (Beijing, China). R8-RGD peptides with a terminal cysteine [Cys-RRRRRRRR-c(RGDfK), cysteine modified octa-arginine conjugated to the branch of lysine] were synthesized by Chinapeptides Co. Ltd. (Shanghai, China). PEI 25K and PEI 1.8K were purchased from Sigma-aldrich and Alfa Aesar, respectively. All other chemicals were purchased from Sigma-Aldrich (China) and used without further purification.
YOYO-1, TOTO-3, Lyso-Tracker red, Hoechst 33342, and Lipofectamine 2000 and 3000 transfection reagents were purchased from Invitrogen (USA). pUNO1-hTRAILa (hTRAIL) and pUNO1-MCS (MCS) plasmid (InvivoGen, San Diego, CA, USA) were purified with QIAGEN Plasmid Mega Kit (Qiagen GmbH, Hilden, Germany). Annexin V-FITC /PI apoptosis detection kit was purchased from Nanjing KeyGen Biotech. Co., Ltd (Nanjing,China). Antibodies used for western blot were acquired from Cell Signaling Technology (CST, Beverly, Mass), while anti-TRAIL, anti-DR4, anti-DR5 antibodies for IHC were obtained from Abcam (Cambridge, MA).
Preparation and Characterization of Binary and Ternary Complexes
The PF33/pDNA binary complex was prepared by gently mixing 2 μg of pDNA and PF33 (20 μg) solution in DNase-free water and incubated at room temperature for 30 min. RRPHC/pDNA ternary complex was obtained by adding RRPH (60 μg) solution to PF33/pDNA binary complex by incubation for another 20 min. The HAC/pDNA ternary complexs was prepared by a procedure similar to the synthesis of RRPHC/pDNA ternary complex by replacing the RRPH with HA. The size and zeta potentials of the prepared binary and ternary complexes were evaluated using dynamic light scattering (DLS) measurements (Malvern, Zetasizer NanoZS ZEN 3600, UK). The condensation ability of the binary and ternary complexes was assessed by gel retardation assays. Briefly, the PF33/pDNA complex was freshly prepared and loaded onto 2% (w/v) agarose gel with tris-acetate (TAE) running buffering at 120 V for 30 min. DNA retardation was examined by a UV illuminator using a Gel Doc System (Bio-Rad Laboratories, Hercules, CA, USA).
CD44 Expression Level and Cellular Uptake Analysis
FITC-conjugated anti-CD44 antibody was applied to examine the CD44 expression level on the surface of HCT116 cells. Briefly, cells were detached, washed twice with PBS, and incubated with 0.5 µl of FITC-conjugated anti-CD44 antibody for 30 min. Subsequently, cells were washed and analyzed by flow cytometry. FITC-conjugated IgG 2a antibody was used as an isotype control to exclude the nonspecific binding.
To analyze the cellular uptake, pDNA was labeled with YOYO-1 according to manufacturer's protocol. HCT116 cells were plated into 6-well plates at a density of 3×105 cells/well and allowed to attach overnight. The complex loaded with YOYO-1 labeled pDNA was then incubated with the cells for another 2 h at 37 oC. The cells were harvested, rinsed with cold PBS, and re-suspended in PBS for the flow cytometry analysis after incubation. In the competitive receptor study, cells were pre-incubated with excess free HA or (and) RGD peptide for 1 h before for blocking the CD44 or (and) integrin αvβ3 receptors of HCT 116 cells.
Penetration into 3D Tumor Spheroids
Since HCT 116 cells cannot form 3D tumor spheroids in conventional culture conditions, 3D tumor spheroids of U-87 (human glioblastoma cells) were used for this purpose. Briefly, the 96-well plate was precoated with 60 μL of 2% (w/v) low melting point agarose. U-87 cells were seeded into the wells at a density of 5×103 cells/well and incubated for 5 days. The uniform and compact spheroids were selected and incubated with the binary and ternary complexes loaded with YOYO-1 labeled pDNA for another 4 h. Subsequently, the spheroids were rinsed three times with cold PBS and fixed in 4% paraformaldehyde for 30 min before subjecting to CLSM (Leica, Germany).
In Vitro Distribution and Nucleus-Targeting Analysis
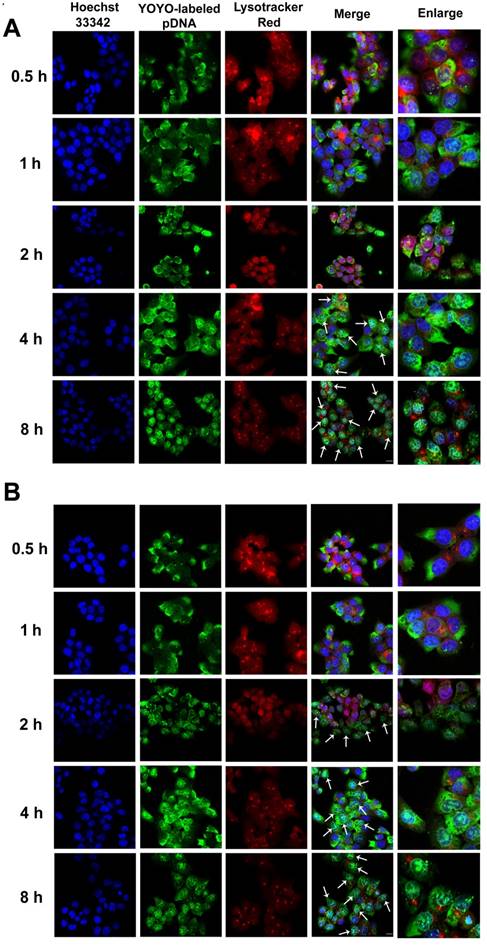
To examine the in vitro distribution of different complexes, HCT116 cells (2×105 cells/well) were seeded into 6-well plates over glass cover slips the day before use. The complexes loaded with YOYO-1 labeled pDNA were then incubated with the cells in serum-free medium. The cells were pretreated with Lyso-Tracker Red dye for 1 h to visualize lysosomes and endosomes before stopping the experiment. At determined time intervals (0.5, 1, 2, 4, 8 h), cells were washed with PBS and incubated with Hoechst 33342 for 10 min to visualize nuclei. Subsequently, cells were washed three times with cold PBS, fixed in 4% paraformaldehyde and subjected to CLSM (ZEISS, LSM 880, Germany).
In vitro Gene Transfection Analysis
To evaluate the transfection efficiency of the binary and ternary complexes in HCT116 cells, pGFP (Green Fluorescent Protein plasmid) was used as a reporter gene. Briefly, HCT116 cells were seeded into 6-well plates before transfection. The medium was replaced with 800 μL serum-free medium or medium containing 10% ~ 30% serum, 100 μL of PF33/pGFP and RRPHC/pGFP complexes were added to each well and incubated for 4 h. The medium was replaced with 2 mL fresh medium containing 10% serum and further incubated at 37oC for another 24 h. PEI 25K/pGFP and PEI 1.8K/pGFP polyplexes, Lipofectamine 2000/pGFP and Lipofectamine 3000/ pGFP lipoplexes at their optimal condition were used as controls. After incubation, the cells were rinsed twice with PBS. The GFP expression was analyzed by a fluorescence microscope (Olympus, Japan). The transfection efficiency and the fluorescence intensity of GFP were quantified by flow cytometry (Calibur, BD, USA).
In vitro Apoptosis and CSCL Analysis PostTransfection with hTRAIL
The anticancer activity of the PF33/hTRAIL, HAC/hTRAIL, and RRPHC/hTRAIL complexes on HCT116 cells was studied. HCT116 cells were seeded into 6-well plates and treated with PF33/hTRAIL, HAC/hTRAIL, and RRPHC/hTRAIL, respectively. After 24 h or 48 h treatment, cells were trypsinized, washed, and resuspended in binding buffer. FITC-conjugated annexin-V and PI were added and incubated with cells for 10 min. Then the samples were immediately analyzed by FACS. PF33/MCS, HAC/MCS, and RRPHC/MCS were used as controls.
For Western blotting analysis, the cells treated with different complexes were harvested, washed, and resuspended in RIPA lysis buffer supplemented with Complete Protease Inhibitor Cocktail. After incubation on ice for 1 h, the cell lysates were centrifugated for 15 min at 12, 000 g. The protein concentration was quantified. Total protein (~30μg) was separated with 15% SDS-PAGE gel and transferred to a PVDF membrane. The PVDF membranes were blocked in 5% non-fat milk for 2 h, followed by incubation with antibodies against TRAIL, cleaved caspase 3, and cleaved caspase 9 at 4 oC overnight. Subsequently, the PVDF membranes were washed with TBST and incubated with HRP-conjugated secondary antibodies in 5% non-fat milk for 45 min at 37 oC. Then PVDF membranes were washed three times with TBST and the protein bands were visualized using a chemiluminescence (ECL) detection system.
For the CSCL analysis, the procedures were similar to the apoptosis analysis described above. After treatment, cells were trypsinized, washed and incubated with PE mouse anti-human CD44 antibody and PerCP-cy 5.5 mouse anti-human CD24 antibody (BD Biosciences) for 30 min at 4 oC. Subsequently, the samples were immediately analyzed by FACS.
Animals and Tumor Xenograft Models
The female BALB/C nude mice (16-18 g) were provided by the Vital Laboratory Animal Center (Beijing, China). All animals were treated in accordance with the Guide for Care and Use of Laboratory Animals, approved by the Ethics Committee of Sichuan University (Chengdu, P.R. China). The xenograft tumor model was generated by subcutaneous injection of HCT 116 cells (1×107 cells for each mouse) in the flank of the BALB/C nude mice. The tumor size was monitored and the tumor volume (V) was calculated as V=L×W2 /2, where L and W indicated the length and width of the tumor, respectively.
In vivo Transfection and Tumor Targeting
For in vivo transfection, when the tumor volume was about 200 mm3, mice were divided into three groups and intratumorally injected with either PF33/pGFP, HAC/pGFP, or RRPHC/pGFP loaded with 5 μg pGFP. Two days after injection, the tumor tissues were collected and rinsed with cold PBS, the tumor tissues were embedded in OCT glue, frozen and cut into 6 μm thick cryosections for fluorescence microscopic analysis (Leica, Germany).
The in vivo imaging study was performed to investigate the tumor targeting capability of RRPHC/pDNA comple. TOTO-3 with far-red fluorescence was used to label the pDNA. When the tumor volume reached 400-500 mm3, 200 μL of RRPHC/pDNA or HAC/pDNA complexes loaded with 5 μg of TOTO-3 labeled pDNA were intravenously administered. Images were taken on IVIS Lumina imaging system (Caliper, USA) at 2, 6, 12 and 24 h post injection.
IHC of Human Colon Cancer Tissues
Human colon cancer tissues were obtained from patients undergoing tumor resection surgery. The study was approved by the Ethics Committee of the West China Hospital, Sichuan University, China (project no. 2015-263). IHC was performed according to established procedures. Briefly, slides were deparaffinized and then rehydrated with downgraded ethanol. Endogenous peroxidase was blocked with fresh 0.3% hydrogen peroxide in methanol for 15 min following which the slides were washed with PBS and heated to 95 oC in 10 mM citrate buffer (pH 6.0) for 30 min. Sections were blocked in goat serum, incubated overnight at 4 oC in primary antibodies, washed with TBST (Tris buffered saline with Tween 20), and incubated with secondary antibodies (60 min; RT). The slides were visualized with Betazoid DAB Chromogen Kit (Biocare) and followed by hematoxylin counterstaining. The number of positive cells was visually evaluated in each core by a pathologist from West China Hospital. The staining intensity of DR4, DR5, and TRAIL was classified based on an improved semi-quantitative system developed by Allred et al [39] which assesses the proportion of positive cells and intensity of staining (none~weak = 1; intermediate = 2; moderate~strong = 3; strong=4).
In vivo Anti-tumor Efficacy and Safety Evaluation
The tumor model with HCT 116 xenograft was established as described above. When the tumor volumes were around 100 mm3 after cell implantation, the mice were divided into six groups (n=6), and treated with PBS, RRPH, HAC/MCS, HAC/hTRAIL, RRPHC/MCS and RRPHC/hTRAIL once every other day by intravenous injections. The dosage of hTRAIL or MCS of each injection was 5μg per mouse. The tumor volumes and body weights of mice were monitored every three days. Statistical analysis of the tumor volume was performed with two-way ANOVA (time and treatment) followed by post-hoc Tukey test for treatment. At the end of the experiment, mice were euthanized; the tumors as well as the major organs were collected, photographed, weighed, and then fixed in 4% paraformaldehyde for hematoxylin and eosin (H&E) staining and IHC analysis. The blood of mice was also collected for chemistry profile and complete blood count (CBC) test. The in vivo experiments were performed in a blinded fashion with two independent experienced investigators.
Statistical Analysis
Statistical analysis was performed by one-way ANOVA unless otherwise specified. Significant differences between groups were indicated by *p < 0.05, **p < 0.01 and ***p < 0.001.
Results and Discussion
Preparation and Characterization of the Binary and Ternary Complexes
In the past decades, fluorine was widely applied to modulate the properties of drugs and bio-macromolecules [40-41]. Fluorinated compounds exhibite many unique characteristics, such as both hydrophobic and lipophobic properties and high phase-separation tendency [42]. More recently, some reports demonstrated that fluorination strategy can effectively improve the gene transfection efficiency of cationic polymers. In this preliminary work, we synthesized a series of fluorinated polymers (PFs) (Fig. 2) [43]. The PF33 polymers exhibited excellent gene transfection efficacy in HEK-293 cells (almost 100%) with low cytotoxicity (data not shown). Based on our preliminary experiment, PF33 showed excellent gene transfection efficacy. The PF33/pDNA binary complexes showed an average size of 81.3 ± 2.2 nm and a moderately positive zeta potential of +21.8 ± 2.2 mV (Fig. S1A and S1B). The condensation ability of these fluorinated polymers was further assessed by the gel retardation assay. The DNA mobility in PF33/pDNA complexes was completely retarded at a low mass ratio of 1:1 (PF33: pDNA) (Fig. S1C). This may be attributed to the low surface energy of fluorinated polymers that preferably self-associate at low concentrations [44-45]. Coating with RRPH or HA (control, without PEG or R8-RGD) showed moderate increase in particle size to 117.0 ± 4.2 nm (HAC/pDNA) or 132.8 ± 3.8 nm (RRPHC/pDNA). The surface became negatively charged with values of -24.1 ± 1.8 mV and -20.1 ± 2.0 mV for HAC/pDNA,and RRPHC/pDNA, respectively. After coating PF33/pDNA complexes with synthetic RRPH polymers, no DNA detachment was observed, indicating the negatively charged RRPH did not interfere with DNA condensation (Fig. S1C).
Synthesis scheme of PF33 and RGD-R8-PEG-HA (RRPH).
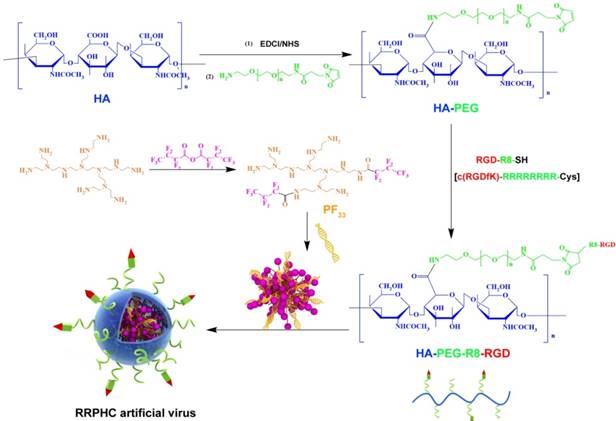
Cellular Uptake Evaluation
The expression levels of receptors that are are involved in internalization of polymer/DNA complexes are critical for cellular entry. Thus prior to evaluating the cellular uptake efficiency of the above complexes, the CD44 expression levels of HCT 116 were assessed by flow cytometry. The results revealed that HCT 116 cell lines had high CD44 expression levels (almost 100%) (Fig. S2) as reported for most malignant tumors derived from epithelial tissues.
We subsequently measured the cellular uptake efficiency of the above binary and ternary complexes. As shown in Fig. 3A and 3B, PF33/pDNA complexes exhibited the highest cellular uptake efficiency (95%). This was consistent with the notion that the positively charged nanoparticles are easily taken up by cells through electrostatically adsorptive endocytosis. Compared with the polyplexes of PEI 25K (81.4%), the higher efficiency of cellular uptake may be attributed to the hydrophobic feature of fluorinated compounds with improved affinity for the cell membrane. Generally, anionic shielding might reduce the electrostatic interaction between the negatively charged cell membrane and cationic complexes. However, in our experiments, the negatively charged HAC/pDNA complexes showed cellular uptake efficiency (84%) comparable to that of PEI 25K, indicating that the CD44-mediated endocytosis played an important role in promoting the intracellular accumulation of pDNA. Notably, the negatively charged RRPHC/pDNA complexes displayed Mean Fluorescence Intensity (MFI) (~ 80) higher than that of HAC/pDNA complexes (~ 40).
Intracellular uptake of complexes loaded with YOYO-1 pDNA in HCT 116 cells after incubation for 2 h at 37oC. (A, C) Flow cytometry analysis of the cellular uptake of various complexes. (B, D) Quantitative analysis of the cellular uptake efficiency after treatment with various complexes. *p < 0.05, **p < 0.01 and ***p < 0.001.
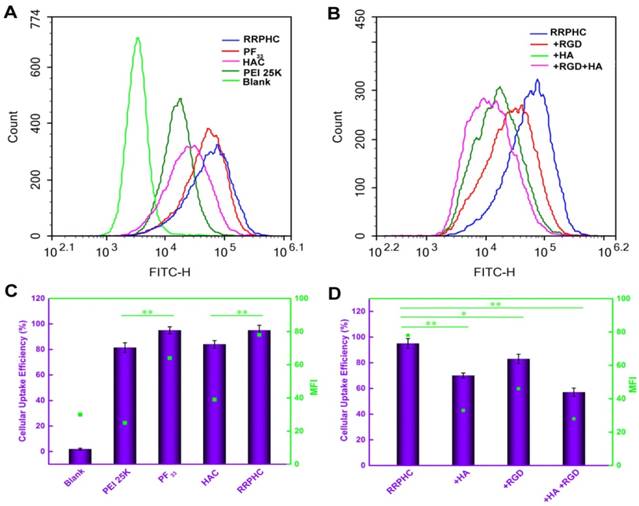
To evaluate whether cellular uptake of RRPHC ternary complexes was associated with CD44 and integrin αvβ3 receptors, the receptor competitive assay was performed. Pre-blocking the CD44 and/or integrin αvβ3 receptors significantly decreased the cellular uptake efficiency of RRPHC/pDNA complexes (Fig. 3C and 3D). These results confirmed that the enhanced intracellular uptake of RRPHC/pDNA complexes was relevant to the dual receptor-mediated endocytosis.
The highly desirable features of an ideal gene carrier are efficiency in cellular uptake and high permeability in targeted tissues [46]. 3D tumor spheroids have been used as an ideal format for better understanding of the influence of the cellular microenvironment in tumor biology [47-49]. We prepared three dimensional tumor spheroids to evaluate the penetration ability of the PF33/pDNA and RRPHC/pDNA complexes. Compared to all other groups, RRPHC/pDNA complexes displayed much stronger green fluorescence at different depths (Fig. 4) validating that R8-RGD possessed stronger penetration ability.
Penetration ability of various formulations (PEI 25K/pDNA (PEI 25K), PF33/pDNA (PF33), HAC/pDNA (HAC), RRPHC/pDNA (RRPHC)) into 3D tumor spheroids.
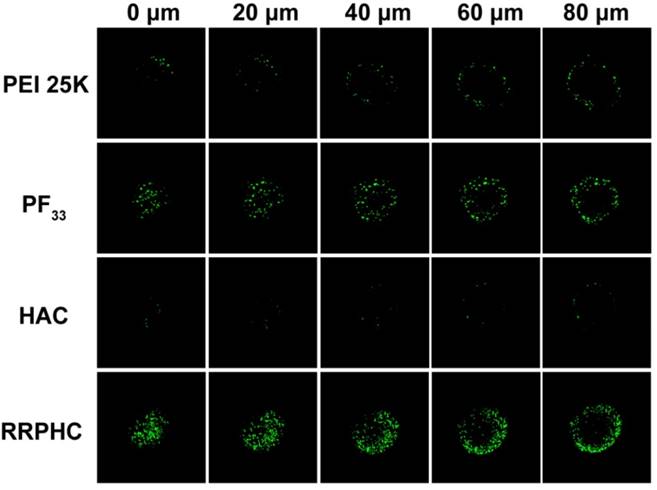
In Vitro Distribution and Nucleus-Targeting Analysis
Efficient accumulation into nuclei of targeted cells is critical for effective gene transfer. Cationic polymers such as PEI can effectively escape from endosomes and accumulate into nuclei [50]. It has been reported fluorination can facilitate endosomal escape of the polymers by fusion with endosome/lysosome membrane [51-52]. We, therefore, investigated whether RRPHC/pDNA complexes could promote endosomal escape of the pDNA.
The intracellular distribution of YOYO-1-labeled complexes occurred in a time-dependent manner. As shown in Fig. 5A, after 4 h of incubation, large amounts of RRPHC/pDNA complexes effectively escaped from the endosomes and penetrated into the nuclei. We investigated the enhanced endosomal escape ability of PF33/pDNA complex. Interestingly, some of the YOYO-1-lableded complexes were already associated with the nuclei in the first 2 h and there was an almost complete overlap between all nuclei and the green dots within 4 h of incubation (Fig. 5B). The excellent endosomal escape ability of RRPHC/pDNA complexes may be largely attributed to PF33/pDNA core. The RRPHC/pDNA complexes showed slightly delayed endosomal escape in the first 2 h. This delay could be because the RRPH coating had to be degraded by HAase in the lysosome exposing PF33/pDNA complexes which then facilitated the endosomal escape.
Confocal images of HCT 116 cells treated with (A) RRPHC/pDNA and (B) PF33/pDNA for 0.5, 1, 2, 4 and 8 h. pDNA was labeled with YOYO-1, the endosomes and lysosomes were stained with Lyso-Tracker Red while the nuclei were stained with Hoechst 33342. The arrows indicate co-localization of YOYO-1-labeled pDNA and the nuclei.
In vitro Gene Transfection Study
We evaluated the in vitro transfection efficiency of the PF33/pDNA and RRPHC/pDNA complexes in HCT 116 cells using pGFP as a reporter gene. PEI 25K and PEI 1.8K were used as controls and the transfection experiment was first performed in the serum-free culture medium. As shown in Fig. 6A and B, the PF33/pGFP complexes achieved extremely high transfection efficiency (nearly 100%) at a low mass ratio (indicating low N/P ratio) which significantly higher than that of the commonly used transfection reagents (PEI 25K, lower than 30%) in HCT 116 cells. Enhanced cellular uptake and accelerated endosomal escape of the PF33/pGFP complexes may contribute to the high transfection efficiency. In comparison, the polyplexes of the PEI 1.8K showed the lowest transfection efficiency (less than 10%).
Transfection efficiencies of various complexes in HCT 116 cells at 24 h. (A) Comparison of various complexes in serum-free medium, PEI 1.8K/pGFP (a), PEI 25K/pGFP (b), PF33/pGFP at mass ratio of 5:1 (c), 10:1(d), HAC/pGFP (e), and RRPHC/pGFP (f). (B) Quantitative analysis of transfection efficiency in serum-free medium by flow cytometry. (C) Comparison of PF33/pGFP (PF33), RRPHC/pGFP (RRPHC) and Lipofectamine 2000/pGFP (Lipo 2000) in medium containing 10% - 30% serum. (D) Quantitative analysis of transfection efficiency in medium containing 10% - 30% serum by flow cytometry. *p < 0.05, **p < 0.01 and ***p < 0.001. pGFP indicates GFP pDNA. The scale bar indicates 100 μm.
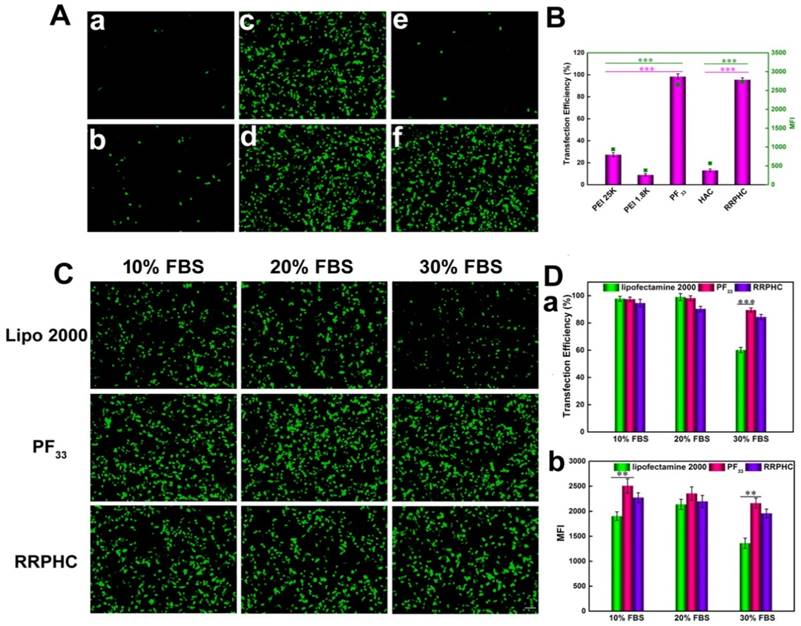
These results implied that introducing perfluoro acid groups to cationic polymers greatly change the properties of the polymer significantly improving the transfection efficiency. An excellent gene transfection efficiency using the fluorinated PAMAM dendrimers was reported by Cheng et al [50]. Especially, RRPHC/pGFP complexes exhibited high gene transfection efficiency (~95%) comparable to the PF33/pGFP complexes which was significantly higher than HAC/pGFP complexes. This could be explained by the fact that RRPHC/pGFP complexes were efficiently taken up by the cells and quickly penetrated into the nuclei while the cellular uptake of HAC/pGFP complexes was relatively low even with the CD44-mediated endocytosis. On the other hand, the HAC/pGFP complexes showed lower zeta potential than RRPHC/pGFP complexes, possibly due to the tighter interaction of HA with the core than RRPH. Also, the steric hindrance with PEG resulted in difficult release of pGFP and poor transfection efficiency which was consistent with a previous study [53].
Serum concentration is an important parameter for gene transfection. Thus we evaluated the transfection efficacy of the PF33/pGFP and RRPHC/pGFP complexes in medium containing 0~30% serum. Lipofectamine 2000, a commercial transfection reagent with high gene transfection efficacy in serum-containing medium, was used as a control. The PF33/pGFP complexes and RRPHC/pGFP complexes maintained their high efficiency in the presence of 10% ~ 30% serum which surpassed the efficiency of Lipofectamine 2000. Even in the medium containing 30% serum, the PF33/pGFP and RRPHC/pGFP complexes showed high transfection efficiency (> 90% positive GFP cells). In contrast, Lipofectamine 2000 showed a decrease in gene transfection to 60% after transfection in medium with 30% serum (Fig. 6C and D). Furthermore, we noted that many cells exhibited round shape indicative of cytotoxicity after transfection with the nanoplexes using Lipofectamine 2000. Inspired by the above results, we further compared the transfection efficiency of the PF33/pGFP and RRPHC/pGFP complexes with that of Lipofectamine 3000, which is a more efficient commercial transfection reagent than Lipofectamine 2000. Notably, the PF33/pGFP and RRPHC/pGFP complexes showed significantly higher transfection efficacy than Lipofectamine 3000 in the presence of 0~30% serum (Fig. 7).
Comparison of the transfection efficiency of PF33/pGFP (PF33), RRPHC/pGFP (RRPHC) and Lipofectamine 3000/pGFP (Lipo 3000) in medium containing 0% - 30% serum in HCT 116 cell. (A) Fluorescence microscopy images. (B) Analysis of transfection efficiency in serum-free medium by flow cytometry. (C) Analysis of the transfection efficiency in medium containing 30% serum by flow cytometry. (D, E) Quantitative analysis of GFP-positive cells (%) and Mean Fluorescence Intensity (MFI) by flow cytometry. pGFP indicates GFP pDNA. The scale bar indicates 200 μm.
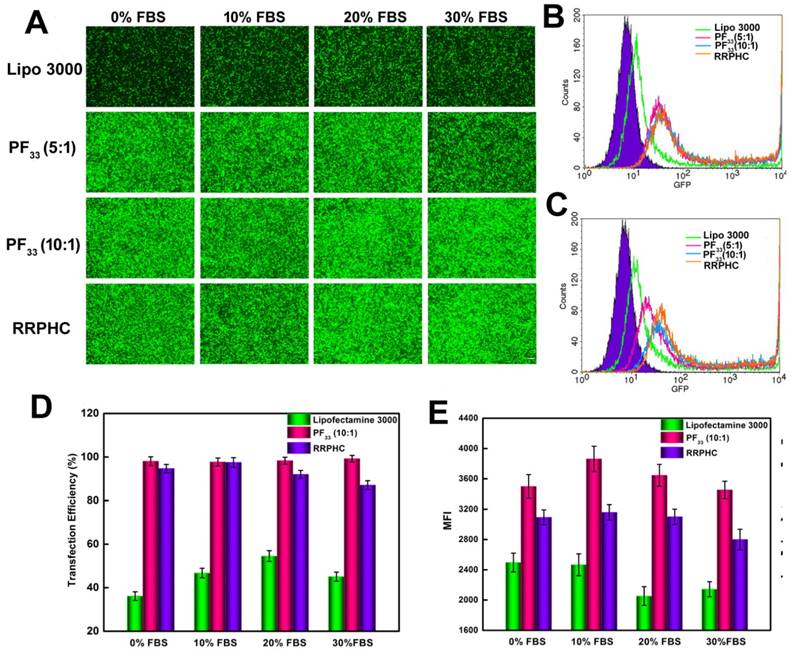
All of these results demonstrated that fluorination significantly improved the in vitro transfection efficacy and both the PF33/pGFP and RRPHC/pGFP complexes are efficient transfection reagents in vitro. Coating with RRPH did not decrease the transfection efficiency of the PF33/pGFP complexes.
Apoptosis-inducing Ability of hTRAIL
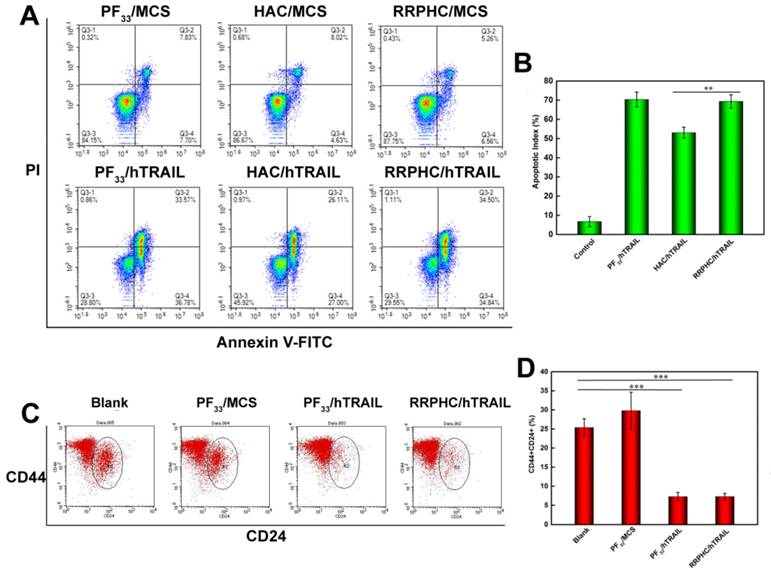
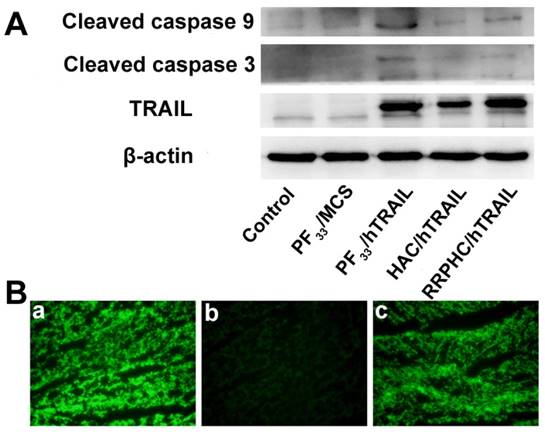
We evaluated the apoptosis-inducing effect of the PF33/pDNA and RRPHC/pDNA complexes loaded with hTRAIL (human TRAIL isoform) gene. As shown in Fig. 8A, the total apoptotic ratio of PF33/hTRAIL and RRPHC/hTRAIL treated- HCT116 cells was 70.35% and 69.34%, respectively, at 24h, which was much higher than that of HAC/hTRAIL (43.11%). As expected, PF33, RRPH, and all other formulations loaded with MCS (controlled pDNA without target gene) showed minimal apoptosis-inducing effect (Fig. S3). We also tested TRAIL protein expression transfected by the above complexes using a Western blotting assay. The expression levels of the TRAIL protein correlated with the apoptotic ratios. PF33/hTRAIL- and RRPHC/hTRAIL-treated groups showed significantly higher TRAIL protein expression than that of HAC/hTRAIL-treated group (Fig. 9A). Similar results were observed in the expression of the cleaved caspase 9 and cleaved caspase 3 proteins, both of which were classical pro-apoptotic proteins. These results confirmed that the apoptosis effect was indeed induced by the protein coded by the hTRAIL gene.
(A, B) In vitro apoptosis-inducing effect in HCT 116 cells following transfection with various formulations loaded with hTRAIL plasmid at 24 h. Early apoptotic cells are located in the lower right quadrant (Q2-4) while the late apoptotic cells are in the upper right quadrant (Q2-2). (C, D) Change in CSCL (CD44+CD24+) after transfection with various formulations detected by flow cytometry. *p < 0.05, **p < 0.01 and ***p < 0.001.
(A) Protein expression levels of TRAIL, cleaved caspase 9, and cleaved caspase 3 in HCT 116 cells following transfection with various formulations. (B) Representative fluorescence micrographs of tumor frozen sections 2 days after injection with (a) PF33/pGFP, (b) HAC/pGFP, and (c) RRPHC/pGFP. pGFP indicates GFP pDNA.
Based on the reports suggesting that CSCL cells are sensitive to TRAIL, we evaluated the change in the number of CSCL in HCT 116 cell line after treatment with PF33/hTRAIL complexes. Though controversial, many reports have shown enrichment of CD44+CD24+ cancer stem cell population in colorectal cancer cell lines. The CD44+CD24+ subpopulation was clonogenic and retained the ability to self-renew and differentiate into all four CD44/CD24 subpopulations [54]. We labeled the cells after treatment with PE-CD44 and Percp-cy 5.5-CD24 antibodies. As shown in Fig. 8B, the ratio of CD44+CD24+ subpopulation decreased significantly in HCT 116 after treatment with PF33/hTRAIL and RRPHC/hTRAIL complexes (from 25.39% to 7.27% and 7.28%, respectively) indicating depletion of CSCL cells in cultured colorectal cancer cells.
In vivo Gene Transfection and Imaging
Since the PF33/pGFP and RRPHC/pGFP have shown considerable gene transfection and penetration abilities in vitro, we examined their in vivo effect in BALB/C nude mice with xenografted tumors of colon carcinoma HCT 116 cells. As seen in the in vitro transfection study, the gene expression of PF33/pGFP and RRPHC/pGFP complexes was comparable in vivo and much higher than that of the HAC/pGFP (Fig. 9B).
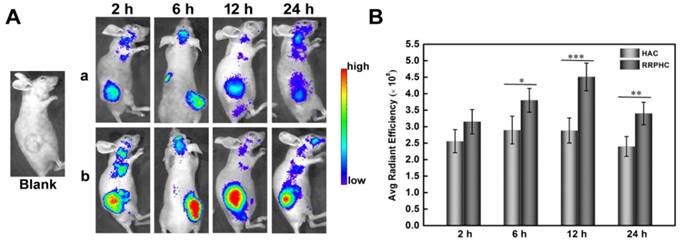
The tumor targeting capability of RRPHC/pDNA and HAC/pDNA complexes was assessed in vivo following systemic administration. We examined the accumulation of TOTO-3-labeled pDNA in the tumor at pre-determined time points after intravenous injection of RRPHC/pDNA and HAC/pDNA complexes by IVI Spectrum system. RRPHC/pDNA complexes rendered a significantly stronger fluorescence signal in the tumor region over a long time (from 6 h to 24 h) compared with HAC/pDNA complexes, further confirming a more effective tumor targeting effect of RRPHC/pDNA complexes (Fig. 10A and B).
(A) In vivo fluorescence imaging of the HCT 116 tumor-bearing nude mice at 2, 6, 12, and 24 h after intravenous injection of HAC/pDNA (HAC) and RRPHC/pDNA (RRPHC). The pDNA was labeled with TOTO-3. (B) Region-of-interest analysis of fluorescent signals from the tumors. *p < 0.05, **p < 0.01 and ***p < 0.001.
Demonstration of DR4, DR5 and TRAIL Expression in Colon Cancer Samples from Patients
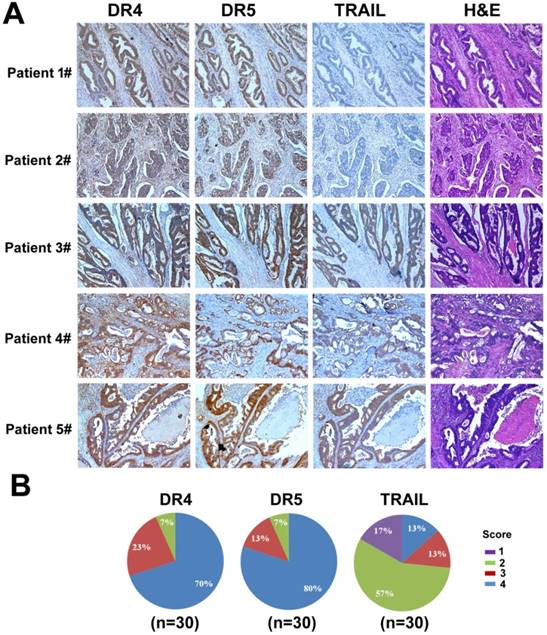
We examined the expression of DR4, DR5, and TRAIL by immunohistochemistry (IHC) in tumor samples of colorectal cancer patients. Samples were collected from 30 colon cancer cases that underwent resection at West China Hospital from January 1, 2015 to October 1, 2015. High expression of DR4 and DR5 (score 3 to 4) was detected in 93% of colon cancer cases (28 cases). In contrast, only 26% of cases (8 cases) had high TRAIL expression (score 3 to 4), whereas most cases (22 cases) displayed little TRAIL expression (score 1 to 2) (Fig. 11). Interestingly, we also observed that the expression of DR4 and DR5 in normal intestine tissues was low (data not shown). Therefore, TRAIL could be a promising candidate for colon cancer treatment as it showed efficacy in targeting cancer cells while sparing the non-cancer tissues based on the differential expression levels of its receptors. However, although many studies have shown the antitumor activity of recombinant TRAIL protein, the in vivo efficacy was limited due to its short half-life in plasma [55-56]. Thus, a delivery system for TRAIL gene is urgently needed as TRAIL gene therapy can serve as a potential strategy to treat colon cancer.
Analysis of DR4, DR5, and TRAIL expression in colon cancer. (A) Examples of IHC staining for DR4, DR5, and TRAIL. Positive DR4 staining was cytoplasmic, positive DR5 staining was nuclear, and positive TRAIL staining was cytoplasmic. (B) Differences in proportion of positive cells and intensity of staining were noted in positively stained cases and formed the basis of the grading system.
Anti-tumor Effect of RRPHC Carrying hTRAIL Gene Following Intravenous Injection
The therapeutic anti-tumor potential of the RRPHC/hTRAIL and HAC/hTRAIL complexes was then evaluated in tumor xenografts of HCT 116 cells in vivo. The tumor growth was remarkably inhibited after treatment with RRPHC/hTRAIL or HAC/hTRAIL compared to other control groups, such as PBS, RRPH, RRPHC/MCS and HAC/MCS, indicating that the antitumor effect was hTRAIL-specific. In particular, RRPHC/hTRAIL treatment demonstrated significantly better anti-tumor efficacy than HAC/hTRAIL (*p < 0.05, two-way ANOVA followed by post-hoc Tukey test). It is of note that the HAC/hTRAIL treatment exhibited strong effects on inhibiting the tumor growth due to the degradation of HA shell by HAase overexpressed in the tumor stroma and subsequent HA-CD44 receptor-mediated tumor-targeting (Fig. 12A and 12C). The body weights of the mice did not change much during the period of treatment in all groups (Fig. 12B). At the end of experiment, the tumors were harvested and mass ratio determined. Compared with all other groups, the mice treated with RRPHC/hTRAIL exhibited much lower mass ratio of the tumor followed by the HAC/hTRAIL treatment (Fig. 12D).
In vivo anti-tumor activity evaluation. (A) Inhibition of tumor growth in the tumor model with the HCT 116 xenograft after treatment with different formulations. (B) The body weight variation of HCT 116 tumor-bearing mice during treatment. (C, D) Images and tumor weights of HCT 116 tumors after treatment with different formulations at Day 31. (E) Ex vivo IHC analyses of the expression of hTRAIL, DR4, DR5, Ki-67 and TUNEL in tumor sections after treatment with different formulations. *p < 0.05, **p < 0.01 and ***p < 0.001.
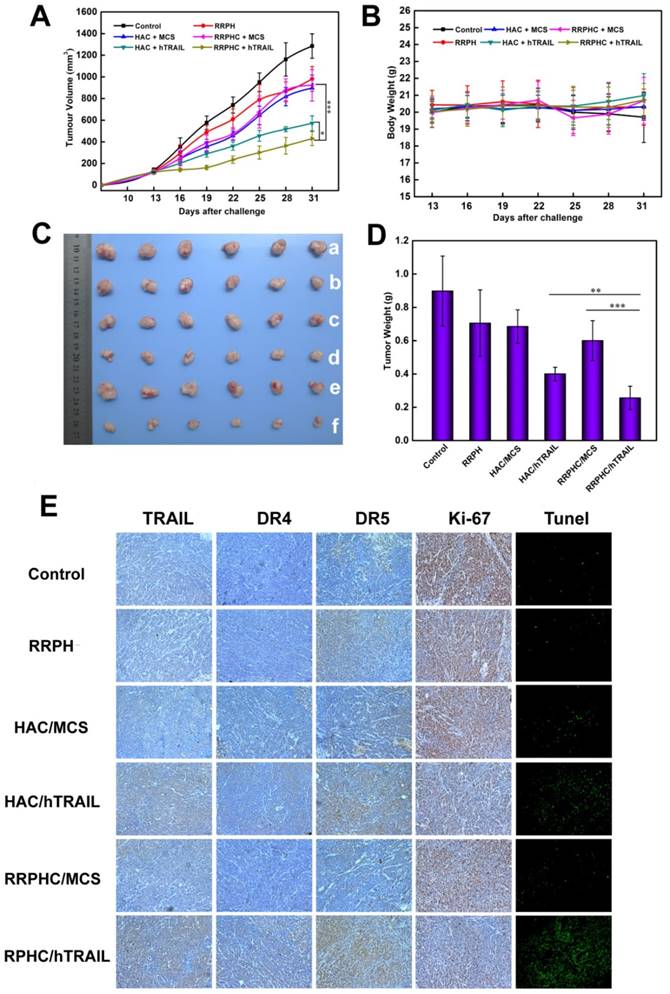
To further examine whether inhibited tumor growth by RRPHC/hTRAIL was associated with hTRAIL gene expression in tumor cells, the tumors were excised for ex vivo IHC analysis after the completion of experiment. The IHC staining of tumor tissues showed significant amount of TRAIL protein after treatment with RRPHC/hTRAIL. Interestingly, we also noted the upregulated expression of DR4 and DR5 indicating that the increased expression of TRAIL in tumor tissues may in turn promote the expression of its receptors (DR4, DR5) (Fig. 12E). These results demonstrated that the retarded tumor growth achieved after treatment with RRPHC/hTRAIL was largely attributed to the expression of TRAIL protein in tumor tissues.
The cell proliferation in tumor tissues post treatment was also assessed. A decrease of Ki-67-positive tumor cells (brown) in tumor tissues was seen after treatment with RRPHC/hTRAIL and HAC/hTRAIL. Furthermore, in comparison with HAC/hTRAIL treatment, RRPHC/hTRAIL was more effective in preventing tumor cell proliferation with less Ki-67-positive tumor cells. Also, treatment with RRPHC/hTRAIL induced the strongest apoptosis in tumor cells consistent with the tumor growth inhibition efficiency. The control groups including PBS, RRPH materials, RRPHC/MCS and HAC/MCS did not significantly affect the numbers of the proliferating Ki-67-positive tumor cells and TUNEL-positive tumor cells as expected (Fig. 12E).
Safety Evaluation
For the safety evaluation, complete blood count (CBC) test was carried out after treatment. WBC, RBC, HGB, and PLT counts were all in normal range and no significant differences were observed after treatment with RRPHC/hTRAIL and HAC/hTRAIL. Because of the reported liver toxicity of TRAIL, the blood chemistry profile analysis was performed to assess the liver toxicity after treatment [57]. No significant difference in the amount of ALB, ALT, AST, and TP between RRPHC/hTRAIL and PBS treatments was observed (Fig. S4A, Fig. S5). Histological analysis of the normal organs including heart, liver, spleen, lung, and kidney showed no evidence of abnormal and inflammatory cell infiltration in these organs after treatment with RRPHC/hTRAIL (Fig. S5B). Collectively, these results indicated that RRPHC/hTRAIL treatment did not cause toxicity to normal tissues and organs of the mice and RRPHC was a safe and potential nanocarrier for in vivo cancer gene therapy.
Conclusion
We have synthesized a library of fluorinated polymers (PFs) via simple chemical reactions. These PFs showed extremely high transfection efficiency (nearly 100%) compared to various commercial reagents. Furthermore, their transfection efficiency was serum-independent, i.e. >90% in medium containing 30% serum. To promote the stability in physiological conditions and to confer tumor-targeting capability, RRPH polymers were designed to coat the binary complexes, formulating “core-shell” nanoparticles. RRPH coating endowed the ternary complexes with multi-stage, multi-targeting effects and depth penetration ability. The “core-shell” nanoparticles improved the 2D and 3D cellular uptake of the binary complexes due to the multi-targeting and effective penetration ability. In particular, the “core-shell” nanoparticles also maintained the excellent transfection efficiency of the binary complexes in vitro. When used as delivery vehicles for the pro-apototic hTRAIL plasmid in vitro, both PF33/hTRAIL and RRPHC/hTRAIL significantly inhibited the growth of tumor cells, enhanced apoptosis, and decreased the percentage of CSCL cells. RRPH coating significantly improved gene delivery into tumor sites while reducing accumulation in other organs such as liver and kidney after intravenous injection. We, therefore, employed RRPHC ternary complexes to deliver hTRAIL gene for in vivo applications. The tumor growth was greatly inhibited after treatment with RRPHC/hTRAIL withno systemic toxicity. In the future, this system has the potentioal to become a promising platform for effective therapeutic gene delivery and to advance the treatment of colon cancer.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was financially supported by National Young Top-notch Talent Program and Distinguished Young Scholars of Sichuan University (2015SCU04A42).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S. et al. Colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;5:619-25
2. Manchun S, Dass CR, Cheewatanakornkool K, Sriamornsak P. Enhanced anti-tumor effect of pH-responsive dextrin nanogels delivering doxorubicin on colorectal cancer. Carbohyd Polym. 2015;126:222-30
3. Ubel PA, Abernethy AP, Zafar SY. Full disclosure—out-of-pocket costs as side effects. N Engl J Med. 2013;369:1484-6
4. Mayer C, Popanda O, Greve B, Fritz E, Illig T, Eckardt-Schupp F. et al. A radiation-induced gene expression signature as a tool to predict acute radiotherapy-induced adverse side effects. Cancer Lett. 2011;302:20-8
5. Vermeulen L, Melo FS, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 2012;13:e83-9
6. Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D. et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011;8:511-24
7. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P. et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669-72
8. Centlivre M, Legrand N, Klamer S, Liu YP, von Eije K, Bohne M. et al. Preclinical in vivo evaluation of the safety of a multi-shRNA-based gene therapy against HIV-1. Mol Ther-Nucl Acids. 2013;2:e120
9. Guerrero-Cázares H, Tzeng SY, Young NP, Abutaleb AO, Quiñones-Hinojosa A, Green JJ. Biodegradable polymeric nanoparticles show high efficacy and specificity at DNA delivery to human glioblastoma in vitro and in vivo. ACS Nano. 2014;8:5141-53
10. Hayakawa K, Uchida S, Ogata T, Tanaka S, Kataoka K, Itaka K. Intrathecal injection of a therapeutic gene-containing polyplex to treat spinal cord injury. J Control Release. 2015;197:1-9
11. Won YW, Bull DA, Kim SW. Functional polymers of gene delivery for treatment of myocardial infarct. J Control Release. 2014;195:110-9
12. Ashley CE, Carnes EC, Epler KE, Padilla DP, Phillips GK, Castillo RE. et al. Delivery of small interfering RNA by peptide-targeted mesoporous silica nanoparticle-supported lipid bilayers. ACS Nano. 2012;6:2174-88
13. Li J, Zheng C, Cansiz S, Wu C, Xu J, Cui C. et al. Self-assembly of DNA nanohydrogels with controllable size and stimuli-responsive property for targeted gene regulation therapy. J Am Chem Soc. 2015;137:1412-5
14. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541-55
15. Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. 2006;17:253-63
16. Bessis N, Cozar G, Boissier M. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004;11:S10-7
17. Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573-87
18. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346-58
19. Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4:581-93
20. Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev. 2008;109:259-302
21. Guo X, Huang L. Recent advances in nonviral vectors for gene delivery. Accounts Chem Res. 2011;45:971-9
22. Mangraviti A, Tzeng SY, Kozielski KL, Wang Y, Jin Y, Gullotti D. et al. Polymeric Nanoparticles for Nonviral Gene Therapy Extend Brain Tumor Survival in Vivo. ACS Nano. 2015;9:1236-49
23. Woodrow KA, Cu Y, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzma WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater. 2009;8:526-33
24. Zhou J, Liu J, Cheng CJ, Patel TR, Weller CE, Piepmeier JM. et al. Biodegradable poly (amine-co-ester) terpolymers for targeted gene delivery. Nat Mater. 2012;11:82-90
25. Liu H, Wang H, Yang W, Cheng Y. Disulfide cross-linked low generation dendrimers with high gene transfection efficacy, low cytotoxicity, and low cost. J Am Chem Soc. 2012;134:17680-7
26. Nam JP, Nam K, Jung S, Nah JW, Kim SW. Evaluation of dendrimer type bio-reducible polymer as a siRNA delivery carrier for cancer therapy. J Control Release. 2015;209:179-85
27. Han L, Zhao J, Liu J, Duan XL, Li LH, Wei XF. et al. A universal gene carrier platform for treatment of human prostatic carcinoma by p53 transfection. Biomaterials. 2014;35:3110-20
28. Eltoukhy AA, Chen D, Alabi CA, Langer R, Anderson DG. Degradable terpolymers with alkyl side chains demonstrate enhanced gene delivery potency and nanoparticle stability. Adv Mater. 2013;25:1487-93
29. Du L, Wang H, He L, Zhang J, Ni B, Wang X. et al. CD44 is of functional importance for colorectal cancer stem cells. Clin Cancer Res. 2008;14:6751-60
30. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. P Natl Acad Sci. 2003;100:3983-8
31. Sun Q, Kang Z, Xue L, Shang Y, Su Z, Sun H. et al. A Collaborative Assembly Strategy for Tumor-Targeted siRNA Delivery. J Am Chem Soc. 2015;137:6000-10
32. Yamada Y, Hashida M, Harashima H. Hyaluronic acid controls the uptake pathway and intracellular trafficking of an octaarginine-modified gene vector in CD44 positive-and CD44 negative-cells. Biomaterials. 2015;52:189-98
33. Edagawa M, Kawauchi J, Hirata M, Goshima H, Inoue M, Okamoto T. et al. Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum (ER) stress-induced sensitization of p53-deficient human colon cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib. J Biol Chem. 2014;289:21544-61
34. Pietro RD, Zauli G. Emerging non-apoptotic functions of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)/Apo2L. J Cell Physiol. 2004;201:331-40
35. Park SJ, Kim MJ, Kim HB, Sohn HY, Bae JH, Kang CD. et al. Cotreatment with apicidin overcomes TRAIL resistance via inhibition of Bcr-Abl signaling pathway in K562 leukemia cells. Exp Cell Res. 2009;315:1809-18
36. Hu YL, Huang B, Zhang TY, Miao PH, Tang GP, Tabata Y. et al. Mesenchymal stem cells as a novel carrier for targeted delivery of gene in cancer therapy based on nonviral transfection. Mol Pharm. 2012;9:2698-709
37. Sussman RT, Ricci MS, Hart LS, Sun SY, El-Deiry WS. Chemo-therapy- resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL-receptor DR4. Cancer Biol Ther. 2007;6:1486-91
38. Li L, Song L, Liu X, Yang X, Li X, He T. et al. Artificial virus delivers CRISPR-Cas9 system for genome editing of cells in mice. ACS Nano. 2017;11:95-111
39. Allred D, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Modern Pathol. 1998;11:155-68
40. Müller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881-6
41. Buer BC, Meagher JL, Stuckey JA, Marsh ENG. Structural basis for the enhanced stability of highly fluorinated proteins. P Natl Acad Sci. 2012;109:4810-5
42. Xiong SD, Li L, Jiang J, Tong LP, Wu S, Xu ZS. et al. Cationic fluorine-containing amphiphilic graft copolymers as DNA carriers. Biomaterials. 2010;31:2673-85
43. Li L, Song L, Yang X, Li X, Wu Y, He T. et al. Multifunctional nucleus-targeting “core-shell” nanoparticles of gene delivery for treatment of aggressive melanoma. Biomaterials. 2016;111:124-37
44. Brittain SM, Ficarro SB, Brock A, Peters EC. Enrichment and analysis of peptide subsets using fluorous affinity tags and mass spectrometry. Nat Biotechnol. 2005;23:463-9
45. Percec V, Imam MR, Peterca M, Leowanawat P. Self-organizable vesicular columns assembled from polymers dendronized with semifluorinated Janus dendrimers act as reverse thermal actuators. J Am Chem Soc. 2012;134:4408-20
46. Wang H, Wang Y, Wang Y, Hu J, Li T, Liu H. et al. Self-Assembled Fluorodendrimers Combine the Features of Lipid and Polymeric Vectors in Gene Delivery. Angew Chem Int Edit. 2015;54:11647-51
47. Haycock JW. 3D cell culture: a review of current approaches and techniques. 3D Cell Culture, Springer. 2011:1-15
48. Kunz-Schughart LA, Freyer JP, Hofstaedter F, Ebner R. The use of 3-D cultures for high-throughput screening: the multicellular spheroid model. J Biomol Screen. 2004;9:273-85
49. Gao W, Xiang B, Meng TT, Liu F, Qi XR. Chemotherapeutic drug delivery to cancer cells using a combination of folate targeting and tumor microenvironment-sensitive polypeptides. Biomaterials. 2013;34:4137-49
50. Kim J, Kim H, Kim WJ. Single-Layered MoS2-PEI-PEG Nanocomposite-Mediated Gene Delivery Controlled by Photo and Redox Stimuli. Small. 2016;12:1184-92
51. Neil E, Marsh G. Towards the nonstick egg: designing fluorous proteins. Chem Biol. 2000;7:R153-7
52. Wang M, Liu H, Li L, Cheng Y. A fluorinated dendrimer achieves excellent gene transfection efficacy at extremely low nitrogen to phosphorus ratios. Nat Commun. 2014;5:3053
53. He Y, Nie Y, Xie L, Song H, Gu Z. P53 mediated apoptosis by reduction sensitive shielding ternary complexes based on disulfide linked PEI ternary complexes. Biomaterials. 2014;35:1657-66
54. Yeung TM, Gandhi SC, Wilding JL, Muschel R, Bodmer WF. Cancer stem cells from colorectal cancer-derived cell lines. P Natl Acad Sci. 2010;107:3722-7
55. Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA. et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155-62
56. Kontermann RE. Strategies for extended serum half-life of protein therapeu- tics. Curr Opin Biotechnol. 2011;22:868-76
57. Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR. et al. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat Med. 2000;6:564-7
Author contact
![]() Corresponding author: (C Gong) E-mail: chygong14com or gongchangyangedu.cn
Corresponding author: (C Gong) E-mail: chygong14com or gongchangyangedu.cn