Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(7):1875-1889. doi:10.7150/thno.18985 This issue Cite
Research Paper
Self-Assembly of Gold Nanoparticles Shows Microenvironment-Mediated Dynamic Switching and Enhanced Brain Tumor Targeting
Qishuai Feng1, 2, Yajing Shen2, Yingjie Fu2, 3, Megan E. Muroski4, Peng Zhang4, Qiaoyue Wang2, Chang Xu2, Maciej S. Lesniak4 ![]() , Gang Li1
, Gang Li1 ![]() , Yu Cheng2
, Yu Cheng2 ![]()
1. Department of Neurology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, 200120, China;
2. The Institute for Translational Nanomedicine, Shanghai East Hospital
The Institute for Biomedical Engineering & Nano Science, Tongji University School of Medicine, Shanghai, 200120, China
3. College of Chemistry and Molecular Science, The Institute for Advanced Studies, Wuhan University, Wuhan 430072, P.R. China;
4. Northwestern University Feinberg School of Medicine, 676 North Saint Clair Street, Suite 2210, Chicago, Illinois 60611, United States.
Received 2016-12-30; Accepted 2017-1-26; Published 2017-4-10
Abstract

Inorganic nanoparticles with unique physical properties have been explored as nanomedicines for brain tumor treatment. However, the clinical applications of the inorganic formulations are often hindered by the biological barriers and failure to be bioeliminated. The size of the nanoparticle is an essential design parameter which plays a significant role to affect the tumor targeting and biodistribution. Here, we report a feasible approach for the assembly of gold nanoparticles into ~80 nm nanospheres as a drug delivery platform for enhanced retention in brain tumors with the ability to be dynamically switched into the single formulation for excretion. These nanoassemblies can target epidermal growth factor receptors on cancer cells and are responsive to tumor microenvironmental characteristics, including high vascular permeability and acidic and redox conditions. Anticancer drug release was controlled by a pH-responsive mechanism. Intracellular L-glutathione (GSH) triggered the complete breakdown of nanoassemblies to single gold nanoparticles. Furthermore, in vivo studies have shown that nanospheres display enhanced tumor-targeting efficiency and therapeutic effects relative to single-nanoparticle formulations. Hence, gold nanoassemblies present an effective targeting strategy for brain tumor treatment.
Keywords: Self-Assembly, Gold nanoparticles, Tumor microenvironment, L-glutathione (GSH), Blood-brain barrier (BBB).
Introduction
Nanoparticles of <100 nm are generally suitable for delivering anticancer drugs due to their preferential and selective accumulation at tumor sites due to the enhanced permeability and retention (EPR) effect [1-4]. Because of the aberrant vasculature, elevated interstitial fluid pressure, and dense extracellular matrices in tumor microenvironments, nanoparticles must overcome considerable interstitial transport barriers to achieve deep and uniform tumor penetration [3, 5]. Versatile inorganic nanoparticle systems with unique physical properties, such as gold nanoparticles, magnetic nanoparticles, and mesoporous silica nanoparticles, have been explored for targeted tumor delivery [6-8]. Nanoparticle size is an important factor in cellular uptake and tumor-targeting efficiency [9, 10]. Previous work has shown that nanoparticles in the range of 40-60 nm are favorable for receptor-mediated endocytosis and exhibit higher tumor accumulation rates than smaller nanoparticles in the 15-nm range [9]. In terms of tumor permeability, smaller nanoparticles of 20 nm rapidly migrate throughout tumor tissues, whereas larger nanoparticles primarily accumulate near vascular tissues [10]. Furthermore, smaller nanoparticles have hydrodynamic diameters that facilitate renal clearance [11, 12]. Thus, the design of a renal-clearable nanoparticle delivery system with high tumor accumulation and strong tumor-mass penetration is challenging.
Brain tumors are life-threating diseases of the central nervous system [13-15]. Although the blood-brain barrier (BBB) is significantly compromised in tumors, high intratumoral interstitial pressure prevents a majority of anticancer drugs from accessing the brain [16-19]. Nanoparticles with tunable sizes and surfaces accumulate in brain tumors via the EPR effect and by active targeting [20]. Inorganic nanoparticles modified with antibodies and peptides have been applied as drug delivery platforms for brain tumor treatments [17, 21, 22]. These nanoparticles are often in the <20-nm range and are able to pass the BBB and be excreted [17, 23]. However, due to the low tumor-targeting ability of these nanoparticles, effective therapeutic doses have not been achieved.
Self-assembled nanostructures have emerged as efficient platforms for designing tumor-targeting epitopes [24, 25]. Efforts have been made to create size-tunable and biodegradable self-assembled nanostructures that are responsive to endogenous stimuli (e.g., enzymes and changes in pH) [26-29]. For example, amphiphilic small molecules have been self-assembled into 95.4-nm-sized spherical nanoparticles that are then decomposed via ester hydrolysis that is catalyzed by cytoplasmic esterase [24]. Nanoassemblies of iron oxide nanoclusters and gold nanoshells with gelatin in the size range of 205 nm were sensitive to pH and matrix metalloproteinases in tumor microenvironments and were bioeliminated from the body [25]. Although assembling approaches of gold nanoparticles with different size and shapes have been reported [30-34], their ability to cross the BBB and brain tumor targeting mechanism studies have not yet been explored and are important to advance the field.
In this study, we rationally designed a simple approach to prepare self-assembled gold nanospheres via cross-linking with dithiol-polyethylene glycol (HS-PEG-SH) as a targeted drug delivery platform for enhanced brain tumor treatment (Scheme 1). We hypothesized that such nanoassemblies of single gold nanoparticles could take the advantage of disrupted BBB in the brain tumor and increase the nanoparticle retention in the tumor tissue via passive targeting effect. In order to further increase the tumor targeting efficiency, the nanospheres were functionalized with epidermal growth factor (EGF) peptide for active tumor targeting. In tumor microenvironments, which are characterized by high vascular permeability and acidic and redox environments, anticancer drug release is controllable. Under these conditions, the nanoassemblies can be broken down into single gold nanoparticles. We thus present an effective and feasible targeting strategy for brain tumor treatment.
Schematic design of the self-assembly of gold nanoparticles. A) Gold nanoparticles self-assemble into nanospheres via cross-linking with dithiol-polyethylene glycol (HS-PEG-SH) that are further modified with drug and EGF peptide. B) The nanoassemblies show the EPR effect and active targeting in brain tumors. Acidic and redox environments in tumors can mediate dynamic structural switching and controlled drug release.
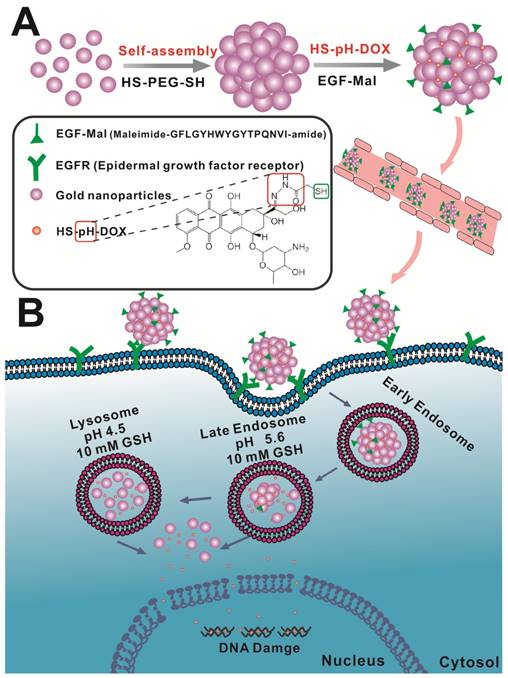
Materials and Methods
Materials
Tetra-n-octylammonium bromide (TOAB) and reduced L-glutathione (GSH) were purchased from Sigma-Aldrich (St Louis, MO, USA). Dodecylamine (DDA) and sodiumborohydride (NaBH4) were purchased from Fluka (Shanghai, China). Chloroauric acid (HAuCl4) was purchased from Civi-Chem (Suzhou, China). mPEG-SH (MW=5,000) and HS-PEG-SH (MW=5,000) were purchased from Pengshuo Biotech Co., Ltd.(Shanghai, China). Cy5.5 was purchased from Xibao Biotech Co., Ltd. (Shanghai, China). MTT kits were purchased from Roche (Shanghai, China). Cleaved caspase-3 antibody (Alexa Fluor®647 conjugate) was purchased from Cell Signaling Technology, Inc. (Lexington, MA, USA).
Preparation
Gold nanoparticles (AuNPs) were synthesized based on procedures described in the literature. Briefly, TOAB (0.25 mmol, 136.7 mg) and DDA (0.6 mmol, 111.21 mg) were dissolved in 5 mL toluene. Next, 0.53 mmol (180.09 mg) HAuCl4 was added. Subsequently, 2 mmol (75.66 mg) NaBH4 in 1 mL refrigerated deionized water was slowly added under 2 h of vigorous mixing at room temperature. Afterward, the mixture was poured into 40 mL ethanol. The resulting precipitate was collected by centrifugation at 4,000 rpm for 10 min and redispersed in 4 mL of chloroform.
Synthesis of self-assembled AuNPs (SA-AuNPs)
The bifunctional reagent HS-PEG-SH (MW 5,000 Daltons, 2.24×10-5 mol) and the monofunctional reagent CH3O-PEG-SH (MW 5,000 Daltons, 2.24×10-5 mol) were uniformly mixed in chloroform. Next, 4×10-8 mol AuNPs were added, and the mixture was stirred for 24 h at room temperature. The solution was purified by centrifuging 3-4 times at 4,000 rpm for 20 min to remove unreacted PEG. Subsequently, the -SH groups on the AuNPs were coupled with the maleimide groups on the EGF peptide to synthesize EGF-SA-AuNPs.
To synthesize single AuNPs (AuNPs), AuNPs in chloroform (4×10-8 mol) were treated with a mixture of the bifunctional reagent HOOC-PEG-SH (MW 5,000 Daltons, 2.24×10-5mol) and CH3O-PEG-SH (MW 5,000 Daltons, 2.24×10-5mol) for 24 h. After purifying by centrifugation, the -COOH groups on the AuNPs were activated with EDC and sulfo-NHS for 2 h in MES buffer and coupled with the -NH2 groups on the EGF peptide to obtain EGF-AuNPs.
DOX loading into AuNPs
Briefly, 100 μL of HS-pH-DOX in DMF was added to 900 μL of the gold solution in PBS (pH 7.4) at a molar ratio of 1:200 and was stirred for 24 h. The mixed solution was then filtered using centrifuge tubes (10-kDa membrane cutoff) to remove unbound, free DOX.
Synthesis of Cy5.5-labeled AuNPs
Thirty microliters of Cy5.5 in DMF solution was added to 970 μL of gold solution in PBS (pH 7.4) at a molar ratio of 1:100 and was stirred for 24 h. After free Cy5.5 was extracted with toluene, the solution was filtered using centrifuge tubes (10-kDa membrane cutoff) to remove the organic solvent.
Characterization
Sample morphology was observed via TEM. The concentrations of all samples were determined by UV-Vis spectroscopy (Cary 60 UV-Vis, Agilent Technologies). To assess GSH-responsive behavior, 2 μL of DOX-EGF-SA-AuNPs were dispersed in 1 mL of PBS (pH 7.4) containing 10 mM GSH. Changes in the morphology of the DOX-EGF-SA-AuNPs were observed at different time intervals, from 5 min to 72 h.
To determine DOX release behavior, 5 μL of DOX-EGF-SA-AuNPs were dispersed in 1 mL of deionized water or PBS at different pH values (7.4 or 4.5) with or without 10 mM GSH in sample cuvettes. Next, 1 mL of toluene was carefully added to the aqueous phase, and the cuvettes were incubated under gentle shaking. The fluorescence spectra of DOX released from the DOX-EGF-SA-AuNPs were measured using a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies, USA) at 488 nm excitation at predetermined time intervals from 0 h to 72 h. All procedures, including incubation and fluorescence detection, were performed at 37°C in the dark.
In vitro cytotoxicity determination
U87, GBM43 and U251 cell lines were cultured in DMEM containing 10% fetal bovine serum (Hyclone, Thermo Scientific, USA) and 1% penicillin-streptomycin (Hyclone, Thermo Scientific, USA). All cell lines were incubated at 37°C under 5% CO2 atmosphere in an incubator (Thermo Scientific). To determine the efficiency of brain tumor cell growth inhibition, U87, GBM43 and U251 cells were seeded into 96-well cell culture plates at a density of 4×104 cells per well. After incubating overnight, the cells were treated with DOX, DOX-SA-AuNPs, DOX-EGF-AuNPs or DOX-EGF-SA-AuNPs at a series of DOX concentrations (i.e., 10 μM, 5 μM, 1 μM, 500 nM, 100 nM, 50 nM and 10 nM) and incubated for 72 h. The cell killing efficiency was determined by MTT assays, according to the instructions provided in the Cell Proliferation Kit I (MTT; Roche Applied Sciences, Indianapolis). Briefly, a mixed solution (0.5 mg/mL) of MTT and fresh culture medium were added to each well and incubated for 4 h at 37°C and 5% CO2. Absorbances were measured at a test wavelength of 570 nm and a reference wavelength of 630 nm using a microplate reader (ELx808, BioTek). The fluorescence intensity of caspase-3 activity was detected by labeling with a cleaved caspase-3 antibody (Alexa Fluor®647 conjugate) followed by flow cytometry (Attune®NxT flow cytometer, Life Technologies, Thermo Fisher, USA).
Intracellular trafficking
U87 cells were seeded onto a confocal microscopy dish (NEST) at a density of 5×104 cells per well. After culturing for 24 h, the cultures were treated with DOX-EGF-SA-AuNPs (1 μM DOX) for 4 h. The cultures were washed twice using PBS (pH 7.4), stained with LysoTracker Red (1:13,000 dilution; Life Technologies, USA) and stored in an incubator at 37°C for 30 min. The cultures were then washed twice with PBS on ice and immediately observed using confocal laser scanning microscopy (LeicaTCS SP5, Germany). To determine the intracellular GSH responsiveness and the DOX release from DOX-EGF-SA-AuNPs, the U87 cultures were pre-incubated with or without 10 mM GSH for 2 h and subsequently treated with 1 μM DOX-EGF-SA-AuNPs at 37°C for 24 h. The cultures were then stained with 4',6- diamidino-2-phenylindole (DAPI) for 10 min at room temperature. Finally, the cultures were washed with 1 mL PBS. The fluorescence emission spectra of DOX (Ex/Em=488/570 nm) and DAPI (Ex/Em=350/461 nm) were immediately captured using a confocal fluorescence microscope (LeicaTCS SP5, Germany).
To assess subcellular localization, U87 cells were seeded into 6-well plates and incubated with 100 nM DOX-EGF-SA-AuNPs for 24 h. Next, the cells were fixed in glutaraldehyde and post-fixed in 1% osmium tetroxide for 1 h. After subsequent dehydration in a graded ethanol series, the cells were embedded in Epon. The samples were then maintained at 60°C for 48 h. Subsequently, resin blocks were cut using an ultramicrotome to approximately 100-nm thickness and transferred onto copper TEM grids. TEM images were obtained on an FEI Tecnai F30 scanning transmission electron microscope using a high-performance CCD camera.
Animal Experiments
Animals and in situ brain tumor models
All animals were treated in accordance with the guidelines of the animal care and use committee at Tongji University. Male nude mice were purchased and bred at the Center of Experimental Animals at Tongji University. To prepare the U87 brain tumor model, male nude mice were anesthetized by intraperitoneal injection with 10% chloralhydrate and fixed using a brain stereotactic fixation device with a mouse adapter. Subsequently, U87 cells (5×105 cells suspended in 5 μL PBS) were implanted into the right striatum (3-mm depth) of each mouse.
In vivo and ex vivo imaging for tumor brain accumulation and each tissue biodistribution
For the in vivo fluorescence assay, the U87-bearing nude mice were intravenously injected with Cy5.5-EGF-SA-AuNPs or Cy5.5-EGF-10%SA-AuNPs at gold doses of 500 nmol/kg (Cy5.5 concentration). The whole-body fluorescence distributions were observed 24 h after injection using a Berthold NightOWL LB 983 In Vivo Imaging System (Bad Wildbad, Germany). The mice were then sacrificed and dissected to obtain ex vivo fluorescence images of the brain and main organs (i.e., heart, liver, spleen, lungs, and kidneys) using standard operating procedures for a routine animal blood draw. The fluorescence intensities in regions of interest (ROI) were calculated using the Indigo software that accompanied the In Vivo Imaging System. All organs were sampled and fixed with 4% paraformaldehyde; hematoxylin and eosin (H&E) staining and silver staining were then applied. To confirm BBB permeability of assembled AuNPs, EGF-SA-AuNPs and EGF-AuNPs were injected via the tail vein in the normal mouse model. The mice were sacrificed at 24 h post-injection and brains were collected. The Au contents in the normal brain and tumor brain were performed by measuring the Au content through ICP-MS.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 5 (GraphPad Software Inc.,San Diego CA). Presented data are reported as means±SEM. P-values of less than 0.05 were considered significant.
Results
Synthesis and characterization of DOX-EGF-SA-AuNPs
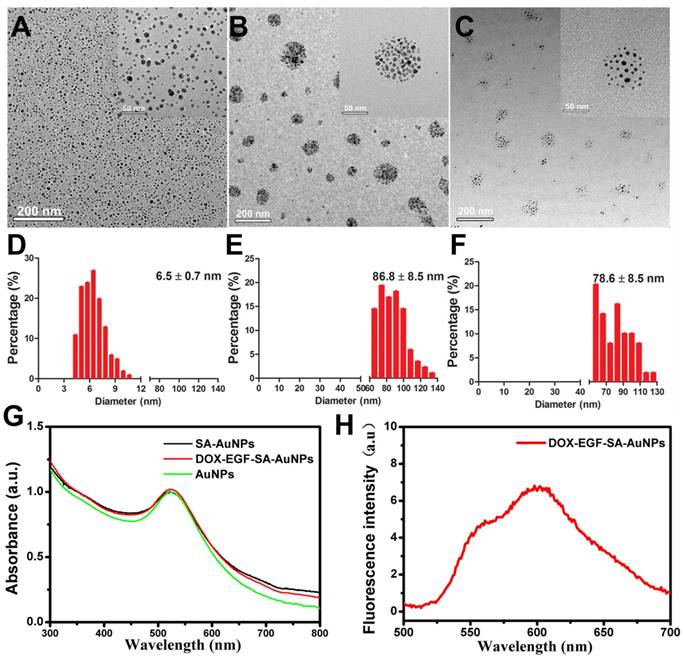
To obtain self-assembling gold nanospheres (SA-AuNPs), gold nanoparticles (AuNPs) were modified by mixing solutions of HS-PEG-SH (MW 5,000 Da) and mPEG-SH (MW 5,000 Da) at a molar ratio of 1:1 for 24 h at room temperature. After purification, AuNP nanoassemblies were obtained. Transmission electron microscopy (TEM) showed that the average size of the monodispersed AuNPs was 6.5±0.7 nm (Figure 1A). The average size of the SA-AuNPs was 86.8±8.5 nm due to the self-assembly of HS-PEG-SH on the AuNPs due to S-S bonding (Figure 1B). Next, the SA-AuNPs were modified with EGF peptides to produce EGF-SA-AuNPs by coupling nanoparticle sulfhydryl groups to EGF peptide maleimide groups. A pH-sensitive, thiol-modified prodrug, HS-pH-DOX, was derived from the anticancer drug doxorubicin (DOX) and verified by nuclear magnetic resonance spectroscopy and mass spectroscopy (Figures S1 and S2). DOX-EGF-SA-AuNPs was produced by mixing HS-pH-DOX with EGF-SA-AuNPs for 24 h. The mixture was washed by centrifugation to remove excess DOX molecules. The recovered DOX-EGF-SA-AuNPs appeared as self-assembled nanospheres with an average diameter of 78.6±8.5 nm (Figure 1C and 1F) with the plasmon resonance peak at 525 nm (Figure 1G) and fluorescence at 590 nm (Figure 1H). Compared with the single AuNP, the nanoassemblies exhibited a 5 nm red-shift in the UV-Vis spectra (Figure 1G). Notably, the HS-pH-DOX prodrug could potentially dissociate the nanoassemblies. Therefore, the ratio of HS-pH-DOX to AuNPs in the final mixture was optimized at 200:1 to maintain the assembled structure. The DOX-loading capacity (i.e., the molar ratio of DOX to nanoparticles) of the nanoassemblies was 16%. It should also be noted that the average interparticle distance of SA-AuNPs is 3.61 ± 0.9 nm, and DOX-EGF-SA-AuNPs is 8.38 ± 1.9 nm (Figure S3).
Characterization of self-assembled AuNPs. (A-C) Representative TEM images of AuNPs, SA-AuNPs and DOX-EGF-SA-AuNPs, respectively. (D-F) Histograms of the particle size distribution through 100 particles statistics from TEM images by measuring the diameter of the particles. (G) UV-Vis spectra of AuNPs (black line), SA-AuNPs (red line) and DOX-EGF-SA-AuNPs (green line). (f) Fluorescence spectrum of DOX-EGF-SA-AuNPs solution under 488 nm excitation.
Responsive behaviors of DOX-EGF-SA-AuNPs to tumor microenvironments
To study the response of DOX-EGF-SA-AuNPs to redox environments, the DOX-EGF-SA-AuNPs were treated with 10 mM GSH. Morphological changes to the nanoassemblies were determined by TEM imaging at different time points (i.e., at 5 min, 30 min, 4 h, 24 h, 48 h and 72 h; Figure 2A). GSH-triggered disassembly of the nanosphere assemblies was observed to begin after 5 min. Lose spherical shapes and decreased numbers of AuNPs in the nanosphere assemblies were observed at 30 min. After 4 h, the structures of the nanosphere assemblies had partially changed into more-linear shapes. At 24 h, a majority of the nanosphere assemblies had dissociated. Finally, they degraded into single gold nanoparticles at 48 h and complete dissociation was observed at 72 h. These results agreed well with size distribution measurements by dynamic laser scattering (DLS; Figure S4A). The polydispersity index (PDI) of the nanosphere assemblies immediately increased upon treatment with GSH and reached a peak value at 4 h. During the treatment period, the absorption of nanoassemblies did not change (Figure S4B). This result indicated that GSH was able to dissociate the nanoassemblies and that the properties of the monodispersed AuNPs were maintained. The disulfide bonds that were formed by the bifunctional HS-PEG-SH in the nanosphere assemblies were cleaved by the GSH molecules. The thiol groups on the GSH molecule competed with the Au-S bonds and replaced the PEG-SH bonds on the nanosphere assemblies. These two reactions together contributed to the GSH-triggered disassembly of the nanospheres and sequentially degraded the assemblies into smaller clusters. This doubly responsive structural disruption may be highly desirable for the in vitro and in vivo excretion of the nanoassemblies.
Dual-stimulus-responsive biodegradation and drug release of DOX-EGF-SA-AuNPs in acidic and redox environments. (A) Representative TEM images of DOX-EGF-SA-AuNPs with 10 mM GSH at different time points (5 min, 30 min, 4 h, 24 h, 48 h and 72 h). Scale bars, 200 nm. (B) Fluorescence intensities of DOX released from DOX-EGF-SA-AuNPs at different pH levels (4.5 and 7.4) at different time intervals. (C) Fluorescence intensities of DOX released from DOX-EGF-SA-AuNPs with GSH (10 mM) at pH 4.5 and 7.4 at different time points.
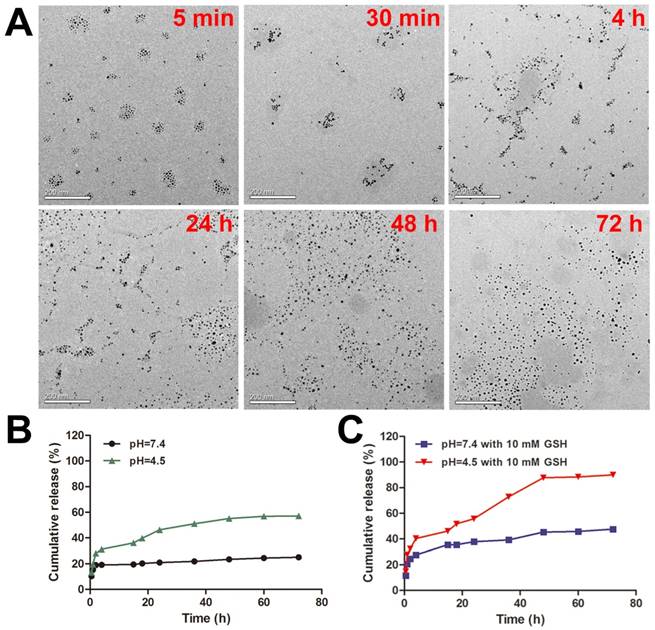
To assess the acid-triggered release of DOX, the DOX release profiles of the DOX-EGF-SA-AuNPs were investigated in physiological (pH 7.4) and acidic (pH 4.5) solutions (Figure 2B). Only 20% of the drug was released after 72 h at pH 7.4, whereas a faster release of DOX was observed at pH 4.5, with over 50% of the drug was released after 72 h. The weakly acidic environment likely facilitated drug release by hydrolyzing the hydrazone groups of the prodrug. Furthermore, GSH-mediated drug release was observed (Figure 2C). Interestingly, DOX released from DOX-EGF-SA-AuNPs with GSH (10 mM) at pH 4.5 displayed two-stage release behavior (Figure 2C). The first-stage release behavior for 4 h was initial burst release. The drug release from AuNP assemblies featured a burst release associated with smaller quantities of drug attached to the surface of the AuNP assemblies, which happened in a very short time compared to the entire release process. The second stage behavior from 4 h to 48 h was the sustained release, which was associated with degradation of AuNP assemblies and release of the encapsulated drug. In the second stage, the internal of the AuNP assemblies was destroyed by GSH-triggered disulfide bond cleavage and then the free DOX was further released. Besides, HS-pH-DOX bond to the surface of AuNP assemblies could release the drug by pH-triggered hydrazone bond cleavage. Finally, the drug release reached a plateau after 48 h. In the acidic buffer with 10 mM GSH, 88% of the loaded drug was released within 48 h, which was significantly higher than that released (23%) at pH 7.4 with 10 mM GSH. This phenomenon may be attributed to the ligand exchange reaction of the Au-S bond on the AuNPs via GSH. Together, the acidic and redox environments were able to synergistically control the release of drugs from the DOX-EGF-SA-AuNPs.
Enhanced in vitro therapeutic effect of DOX-EGF-SA-AuNPs for brain tumor therapy
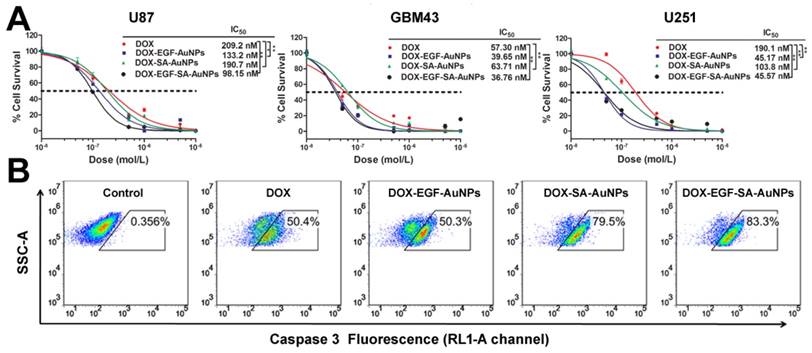
To evaluate the inhibition efficiency of DOX-EGF-SA-AuNPs for brain tumors, three brain tumor cell lines were tested and treated with DOX, DOX-SA-AuNPs, DOX-EGF-AuNPs or DOX-EGF-SA-AuNPs for 72 h. The DOX-EGF-SA-AuNPs-treated cells showed significant morphological changes (Figure S5) and enhanced cytotoxicity (Figure 3A). For U87 cells, the IC50 of the DOX-EGF-SA-AuNPs was 98.2 nM, which was 2-fold lower than that of free DOX (209.2 nM). Compared with the untargeted DOX-SA-AuNPs (IC50 of 190.7 nM), the nanoassemblies showed an enhanced cytotoxicity for cancer cell inhibition (Figure 3A). Enhanced cytotoxicity of the DOX-EGF-SA-AuNPs was also observed for the U251 and GBM43 glioma cell lines. Interestingly, the cytotoxicity of unassembled DOX-EGF-AuNPs was similar to that of the nanoassemblies. Cleaved caspase-3, an important mediator of cell apoptosis, was examined by flow cytometry to determine cell apoptosis. Approximately 83.3% of U87 cells treated with DOX-EGF-SA-AuNPs showed cleaved caspase-3; this value was significantly higher than that of the DOX and DOX-SA-AuNPs-treated groups (Figure 3B) and was consistent with the cytotoxicity assays.
In vitro cytotoxicity in brain tumor cell lines. (A) Cytotoxicity assays of U87, GBM43 and U251 cells treated with DOX, DOX-SA-AuNPs, DOX-EGF-AuNPs and DOX-EGF-SA-AuNPs for 72 h. Error bars indicate s.e. (n=6). *P<0.05, **P<0.01 and ***P<0.001 compared with the free-DOX group. (B) Flow cytometric analysis of cleaved caspase-3 in U87 cells treated with 1 μM DOX, DOX-SA-AuNPs, DOX-EGF-AuNPs or DOX-EGF-SA-AuNPs for 72 h; the RL1-A channel is shown.
The AuNP assemblies with plasmonic coupling could be utilized for laser irradiation for near infrared photothermal therapy (PTT) [30,31, 33]. In our study, the AuNPs assemblies exhibited a red-shift from 520 nm to 525 nm in the UV-Vis spectrum compared to the monodispersed AuNPs. According to the TEM, the interparticle distance in the AuNPs was 3-8 nm, which led to weak coupling between the adjacent NPs. We investigated the PTT effects of AuNPs assemblies in the U87 cells with an 808 nm laser irradiation (2 W cm-2) (Figure S6). There was no significant PTT effect at the current parameters, which could be due to the low absorption of the nanoassemblies in the 600-800 nm range. The results suggest that the optical properties of the AuNP assemblies in the current form should be further optimized in order to achieve good photothermal efficiency.
According to the previous report, gold nanoparticles with size smaller than 20 nm can increase intracellular reactive oxygen species (ROS) production, which is associated with cell apoptosis [34]. Thus, the disassembled AuNPs itself may also play an important role in cytotoxicity. To exclude the possibility of the disassembled AuNPs itself for inducing cell apoptosis, we investigated intracellular ROS level of U87 cells incubated with 12.5 nM of EGF-AuNPs and EGF-SA-AuNPs for 24 h. The ROS expression level is very low in EGF-AuNPs and EGF-SA-AuNPs treated cells (Figure S7). In addition, we found EGF-AuNPs and EGF-SA-AuNPs at less than 25 nM Au concentration had no inhibitive effects on U87 cells after 72 h incubation, while DOX-EGF-AuNPs and DOX-EGF-SA-AuNPs exhibited more than half inhibition rate of cell viability at 25 and 12.5 nM Au concentration (Figure S8). Taken together, these results confirm that EGF-AuNPs and EGF-SA-AuNPs do not induce the intracellular ROS generation and will not lead to the cell apoptosis and could be utilized as a delivery system for brain cancer treatment.
Intracellular GSH-induced disruption of nanoassemblies and DOX release
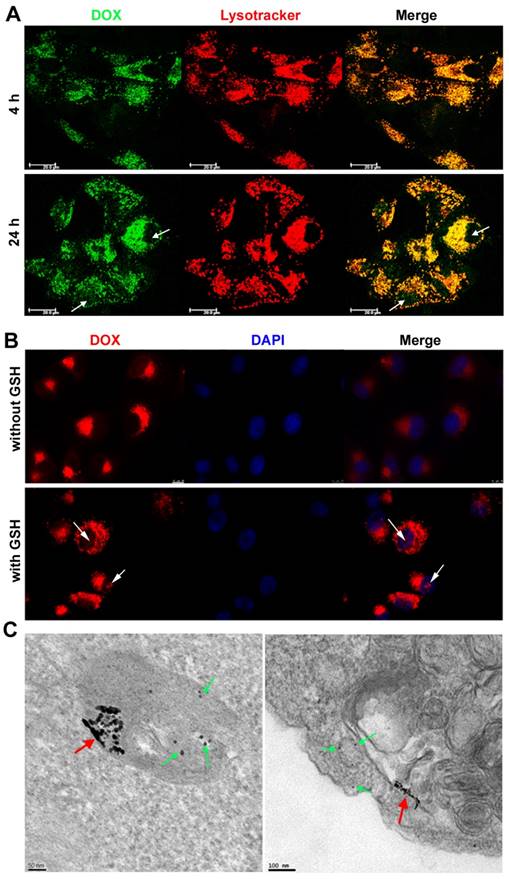
We then further explored the intracellular delivery of DOX-EGF-SA-AuNPs into U87 cells using confocal laser scanning microscopy (Figure 4A). DOX fluorescence was clearly observed in lysosomes after 4 h of incubation with the DOX-EGF-SA-AuNPs, indicating the cellular internalization of the nanospheres. When the incubation time was prolonged to 24 h, DOX was released from the lysosomes and localized within the nuclei of the U87 cells, as indicated by red fluorescence (Figure 4B). The nuclear localization of DOX in U87 cells that were pre-treated with 10 mM GSH was much stronger than in non-GSH treated cells. This difference showed that DOX release was dependent on the acidic environment and the GSH concentration. Next, the endocytic process of the DOX-EGF-SA-AuNPs was visualized by TEM imaging (Figure 4C) of U87 cells that were treated with DOX-EGF-SA-AuNPs for 24 h. A few self-assembled structures were observed in the lysosomes. A majority of the nanospheres had disassembled into linear shapes and single AuNPs, in accord with the in vitro results discussed above.
Intracellular distribution and drug release in brain cancer cells. (A) Confocal analysis for cellular accumulation and distribution of DOX-EGF-SA-AuNPs in U87 cells at DOX concentration of 2.5 μM for 4 h and 24 h. (B) Confocal analysis of DOX release and distribution in U87 cells incubated with or without 10 mM GSH before treatment with 1 μM DOX-EGF-SA-AuNPs for 24 h. (C) Representative TEM images of U87 cells incubated with DOX-EGF-SA-AuNPs for 24 h in the endosome (left) and cytosol (right). Red arrows show fused nanoassemblies; green arrows show single gold nanoparticles dispersed from nanoassemblies.
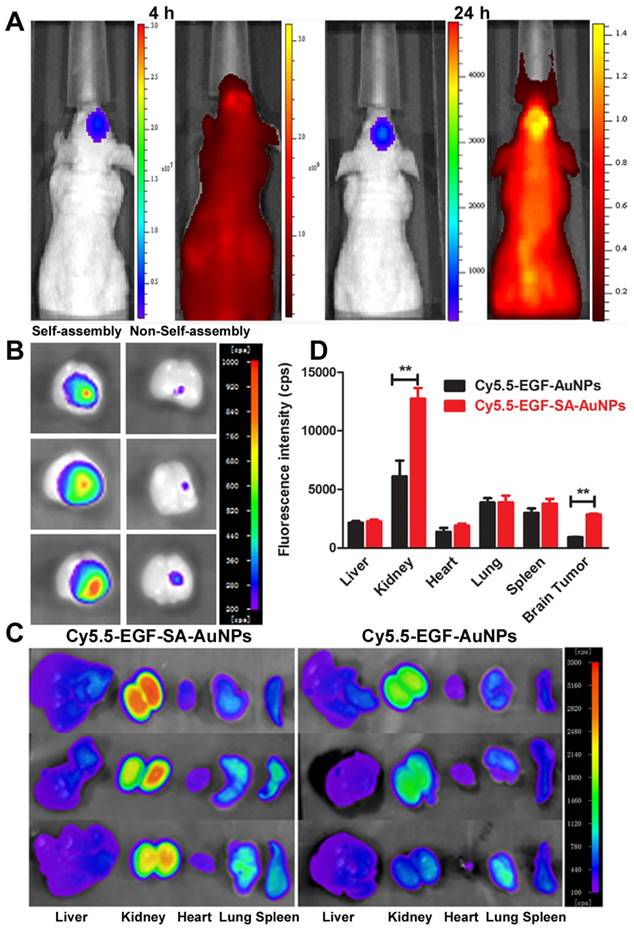
Brain tumor targeting and biodistribution of gold nanoassemblies
The brain-tumor-targeting capability of nanoassemblies was evaluated on intracranial brain tumor xenografts. U87 brain tumor-bearing mice were intravenously administered Cy5.5-labeled EGF-SA-AuNPs. Cy5.5 fluorescence signals were observed in the mouse brain and overlapped with tumor luciferase signals at 4 and 24 h after injection (Figure 5A). This result clearly showed that the EGF-SA-AuNPs accumulated in the tumor. At 24 h post-injection, the mouse brains and organs were collected for ex vivo imaging (Figures. 5B and 5C). The fluorescence intensity of the Cy5.5-EGF-SA-AuNPs at the brain tumor site was significantly higher than that for unassembled Cy5.5-EGF-AuNPs (Figure 5B). Based on a quantitative region-of-interest analysis, the fluorescence intensity of the Cy5.5-EGF-SA-AuNPs at the tumor site was approximately 3-fold higher than that of the Cy5.5-EGF-AuNPs (Figure 5D). The nanoassemblies preferentially targeted the brain tumor to a greater extent than did the single-nanoparticle formulations. To further verify the targeting capability of the nanoassemblies, brain tissue slices were stained with silver enhancing agents to visualize the AuNP distribution (Figure 5A). The EGF-SA-AuNPs and the EGF-AuNPs penetrated the tumors and selectively accumulated in the tumor areas, as shown by black dots in the images (Figure 5B). A majority of the AuNPs were found in the tumor areas, with few particles in the normal brain tissues. The targeting effects of the delivery systems were further verified via confocal microscopy. The fluorescence signals of Cy5.5-EGF-SA-AuNPs were higher in the brain tumor area than those of the Cy5.5-EGF-AuNPs (Figure 5B).
In vivo and ex vivo fluorescence imaging for tumor targeting and biodistribution. (A) Bioluminescent and fluorescent images after 4 h and 24 h of a mouse with a U87 brain tumor that was intravenously injected with Cy5.5-EGF-SA-AuNPs. U87 cells are labeled with luciferase. (B) Ex vivo fluorescence imaging of brains collected from mice treated with Cy5.5-EGF-SA-AuNPs (left) or Cy5.5-EGF-AuNPs (right) at 24 h post-injection. (C) Ex vivo fluorescence imaging of organs from mice treated with Cy5.5-EGF-SA-AuNPs or Cy5.5-EGF-AuNPs. The organs from left to right are as follows: liver, kidney, heart, lung and spleen. (D) Region-of-interest analyses of fluorescent signals from the tumor and normal tissues. Error bars indicate s.d. (n=3).
As the tumor targeting is highly dependent on the circulation behavior of the nanoassemblies, the time-dependent study of Au content in the blood was investigated. EGF-SA-AuNPs and EGF-AuNPs were injected into the mice respectively and the blood content was collected and analyzed by ICP-MS. The results showed that EGF-SA-AuNPs consistently exhibit enhanced blood retention over a 24 h period when compared with the EGF-AuNPs (Figure 6A). For example, at 4 and 8 h after injection, EGF-SA-AuNPs exhibited blood retentions of 19.3% ID/g and 15.7% ID/g, respectively, as compared to only 10.8 ID/g and 9.4% ID/g by the EGF-AuNPs (p<0.01) (Figure 6A). It should be noted that both EGF-SA-AuNPs and EGF-AuNPs showed similar blood retention (<5% ID/g) after 24 h post injection. Using a two compartment model to fit the circulation results, the circulation half-life of the EGF-SA-AuNPs was ~11.7 h, as compared to 5.0 h for the EGF-AuNPs. The observed long circulation lifetime of EGF-SA-AuNPs indicates the robustness of the AuNP assemblies in the blood.
Brian tumor-targeting efficiency and biodistribution of nanoassemblies. (A) Blood retentions of EGF-SA-AuNPs and EGF-AuNPs in normal mice over 24 h post injection. (B) Biodistribution of EGF-SA-AuNPs and EGF-AuNPs in the brain of U87-bearing mice or normal mice after i.v. injection for 24 h. (C) Distributions of Cy5.5-EGF-SA-AuNPs and Cy5.5-EGF-AuNPs by H&E and silver staining in normal and tumor brain sections. Slices were visualized under an inverted optical microscope with a 100× objective. (D) Confocal images of Cy5.5-EGF-SA-AuNPs and Cy5.5-EGF-AuNPs distributions in brains. Bright field and fluorescence images and their overlays are presented. (E) Silver and H&E staining results of heart, liver, spleen, lung and kidneys. Slices were visualized under an inverted optical microscope with a 200× objective.
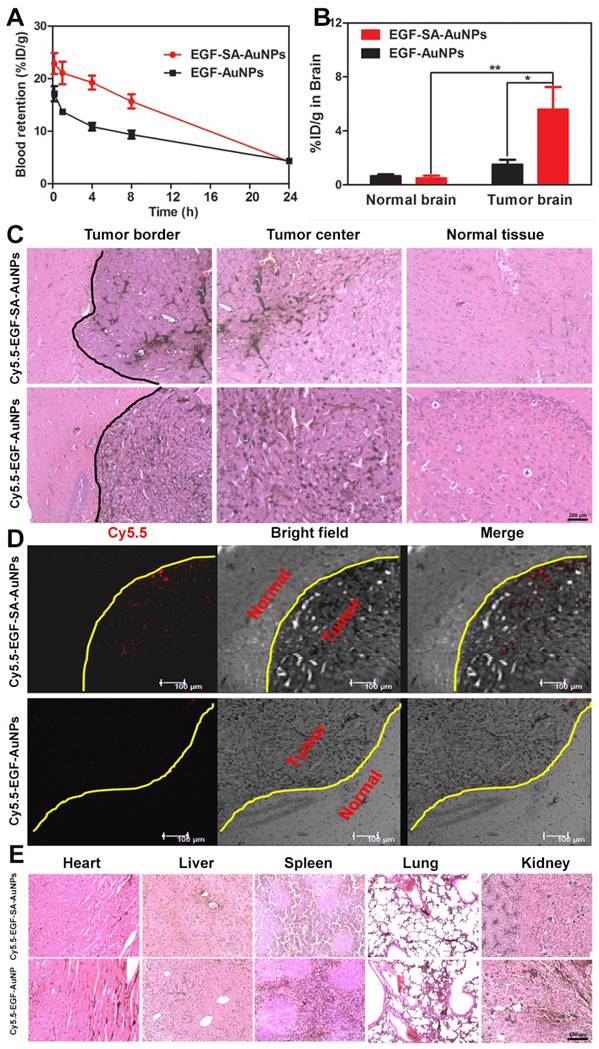
BBB permeability is another parameter to affect the tumor targeting efficiency. EGF-SA-AuNPs and EGF-AuNPs were injected via the tail vein in the normal mouse model, respectively. The mice were sacrificed at 24 h post-injection and brains were collected for analysis. After 24 h, EGF-SA-AuNPs treated mice showed 0.51±0.17% ID/g gold content in the normal brains. In the EGF-AuNPs treated mice, the gold content was 0.68±0.11% ID/g (Figure 6B). These results suggest that EGF-SA-AuNPs and EGF-AuNPs could cross the BBB with similar permeability.
It should be noted that BBB in the tumor area are significantly compromised [19]. In the intracranial U87 xenograft mouse model, we found that the gold content in the brains of assembled EGF-SA-AuNPs treated mice displayed 3.7 fold enhancement compared to EGF-AuNPs (5.6±2.55% ID/g vs. 1.5±0.42% ID/g; p < 0.05) (Figure 6 B), which was consistent with the fluorescence quantification results (Figure 5D). Compared with the normal mouse brain, the AuNP assemblies show 11 fold enhancement in the brain of the U87 glioma bearing mouse. These results suggest that the assembled AuNPs with sub-80 nm in size could accumulate selectively in the tumor because of the EPR effect.
To further verify the targeting capability of the nanoassemblies, brain tissue slices were stained with silver enhancing agents to visualize the AuNPs distribution (Figure 6C). The EGF-SA-AuNPs and the EGF-AuNPs penetrated the tumors and selectively accumulated in the tumor areas, as shown by black dots in the images (Figure 6C). A majority of the AuNPs were found in the tumor areas, with few particles in the normal brain tissues. The targeting effects of the delivery systems were further verified via confocal microscopy. The fluorescence signals of Cy5.5-EGF-SA-AuNPs were higher in the brain tumor area than those of the Cy5.5-EGF-AuNPs (Figure 6D). This potent tumor-targeting capability of the SA-AuNPs can be attributed to the combined EPR effect and active targeting mechanisms. Both of the fluorescence imaging and histology studies showed that the AuNPs significantly accumulated in the kidney (Figure 5C and 6E). The AuNPs clearly accumulated in the glomeruli and kidney tubules. The gold content was also detected in the urine, strongly suggesting that the nanoparticles could be dissociated and excreted via renal clearance. The nanoparticle distributions in the heart, liver, lung and spleen were at lower levels relative to the kidneys, in accord with the tissue fluorescence (Figure 5C and 5D).
Anti-tumor efficacy of nanoassemblies
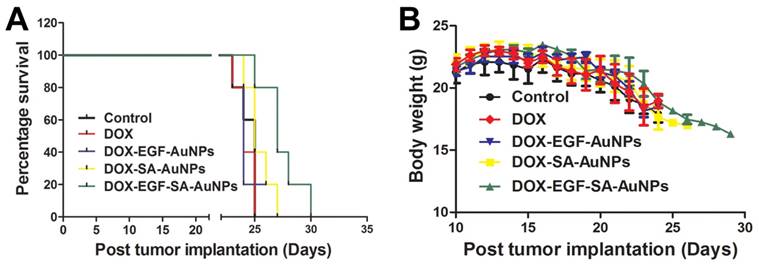
In order to mimic the disease environment including the BBB and tumor microenvironment, the anti-tumor efficacy of the nanoassemblies were evaluated in the intracranial implanted brain tumor model. U87 tumor-bearing mice were intravenously administered with free DOX, DOX-EGF-AuNPs, DOX-SA-AuNPs or DOX-EGF-SA-AuNPs at DOX doses of 1.5 mg/kg. When tumor progression was evaluated, the DOX-EGF-SA-AuNPs group showed improved survival rates relative to the control group (Figure 7A), indicating that the DOX-EGF-SA-AuNPs had a therapeutic effect for brain tumors. There were no significant differences observed among the free DOX, DOX-EGF-AuNPs and DOX-SA-AuNPs treatment groups (Figure 7A). Additionally, no significant differences in body weight were observed among the groups (Figure 7B).
In vivo antitumor activity and toxicology evaluation in situ brain tumor model. (A) Survival curve of U87 brain tumor-bearing mice. Mice were treated at a DOX dose of 1.5 mg/kg via intravenous administration on days 8 and 15 after tumor inoculation. Error bars indicate s.d. (n=6). (B) The body weight variations of U87 tumor-bearing mice during treatment (n=6).
Discussion
Inorganic nanoparticle-based delivery systems typically show low targeting efficacy and systemic toxicity, which prevent optimal therapeutic outcomes [35, 36]. To adequately address these problems while achieving effective therapeutic doses, nanoparticles must be rationally designed to substantially accumulate via the EPR effect and to biodegrade in response to endogenous stimuli in tumor microenvironments [26, 29, 37-40]. A stimulus-responsive, self-assembled system based on physicochemical properties may improve the therapeutic efficacy of nanoparticle drug delivery vehicles by increasing blood circulation times, tumor vascular extravasation rates, tumor penetration, cell internalization, and intracellular drug release [39, 41, 42].
One key design of DOX-EGF-SA-AuNPs is the responsibility to biological stimuli such as the acidic condition and elevated intracellular GSH level. The self-assembled nanospheres were able to fuse to promote drug release and were also degradable in mild acidic and GSH-containing environments [41, 43]. The drug release curve indicated that DOX-EGF-SA-AuNPs could release DOX in the acidic cellular compartments at pH 4.5 and observed that GSH had the synergistic effect on drug release (Figure 2B and 2C). When the nanoassemblies distributed into tissue sites or taken up into cancer cells, DOX could be rapidly released from AuNPs and diffuse into the cytosol and later into the nucleus [44]. Furthermore, the morphological change of nanospheres was found due to 10 mM GSH triggered-disulfide bond breaking (Figure 3). It has been validated that the penetration of nanoparticles in tumor sites depended heavily on the particle size, with the consensus that smaller particles have improved renal clearance [12, 45]. In vivo experiments, our results showed an approximately 80-nm diameter SA-AuNPs showed increased a 3-fold accumulation in brain tumors when compared with single gold nanoparticles (Figure 5B and 5D). Moreover, DOX prodrugs chemically bound to pH-responsive SA-AuNPs were released in acidic environments to effectively kill cancer cells; this delivery vehicle overcomes the inability of free DOX to penetrate the BBB [46].
Developing dynamic switching drug delivery systems has attached significant attention for cancer therapy [47-50]. Interesting, our findings have shown that these self-assembled nanoplatforms can accumulate in tumors and are quickly eliminated from the body through renal clearance. It was noted that previous delivery systems mainly focused on single nanoparticle based platforms with small sizes to facilitate brain tumor penetration. Compared with those studies, our strategy has several unique features to overcome vascular extravasation and BBB for enhanced tumor targeting and efficient body clearance. Achieving this goal is vitally important because these barriers are interconnected, and simply overcoming one individual barrier is not adequate to produce proper therapeutic outcomes [51, 52].
The size of the assembled AuNPs plays an important role for enhanced tumor accumulation. Compared with monodispersed AuNPs, AuNP assemblies show significant enhancement in terms of long-term circulation half-life. Moreover, similar BBB permeability is observed in the normal mouse brain in both assembled and monodispersed forms. Compared with the normal mouse brain, the AuNP assemblies show 11 fold enhancement in the brain of the U87 glioma bearing mouse. All the results indicate that the enhanced tumor targeting efficiency is mainly attributed to the synergistic effects of improved circulation half-life, enhanced accumulation via ERP effect and active targeting.
In this study, our nanoassemblies system enables its basic physicochemical properties to adaptively change in response to the endogenous stimuli of the tumor microenvironment to accomplish improved therapeutic efficacy. We have developed novel, self-assembled gold nanospheres as a drug delivery platform for targeted brain tumor therapy. The platform was easily developed and tailored via ligand-mediated self-assembly using S-S and Au-S bonds. Different types and ratios of polymers were used as cross-linking agents to control the biological delivery and clearance of nanoparticles. Our results indicate that the described platform utilizes a very promising approach for the simple production of self-assembled metal nanospheres.
Conclusions
In this study, the dynamic switching gold nanospheres self-assembled from the use of HS-PEG-SH have been developed successfully for brain tumor targeting. Our delivery systems hold several features, including a nanoscaled material for crossing BBB, enhanced accumulation at the brain tumor site, tumor microenvironment triggered drug release and the self-assembled structure for dynamic switching and bioelimination. Moreover, the flexible nanoassemblies using molecular ligands could overcome the rapid clearance of small particles and the slow clearance of stable nanostructures. Thus, such dynamic switching nanoassemblies have the promising potential as a favorable platform for constructing excellent drug delivery systems for brain tumor therapy. Overall, our work offers a simple and feasible strategy for inorganic-based nanomedicines to target brain tumors and minimize the potential toxicity. It can be of great interest for researchers to rational design nanoparticles for brain tumor imaging and therapy.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was financed by National Science Foundation of China (No.81571803 to Y.C.) and NIH (R35CA197725, R01NS077388 to M.S.L.). Y.C. thanks the Thousand Talents Plan, Shanghai Pujiang Program (No.15PJ1407800) and Shanghai Science and International Cooperation Program (No. 16410724300) for support. This work was also supported by grants obtained from the Ministry of Science and Technology (No.2016YFA0101301), the National Nature Science Foundation of China (No.81271289), Shanghai Science and Technology Commission (No. 16511105000-16511105002) and Academic Leader Training Program of Pudong Health Bureau of Shanghai, China (No. PWRd2012-06).
Author contributions
Q.F. initiated the project, designed the research, synthesized and characterized the nanoassemblies. Q.F. and Y.S. performed the in vitro studies. Y.F. designed and characterized the prodrug. Q.F., M.E.M., P.Z., Q.W., and X.C carried out the animal and histology studies. Q.F. analyzed the data and wrote the manuscript. Y.C., G.L. and M.S.L. supervised the project.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Luo Z, Zhang S. Designer nanomaterials using chiral self-assembling peptide systems and their emerging benefit for society. Chem Soc Rev. 2012;41:4736-54
2. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
3. Li HJ, Du JZ, Du XJ, Xu CF, Sun CY, Wang HX. et al. Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy. Proc Natl Acad Sci U S A. 2016;113:4164-9
4. Lu Y, Hu Q, Lin Y, Pacardo DB, Wang C, Sun W. et al. Transformable liquid-metal nanomedicine. Nat Commun. 2015;6:10066
5. Peller M, Willerding L, Limmer S, Hossann M, Dietrich O, Ingrisch M. et al. Surrogate MRI markers for hyperthermia-induced release of doxorubicin from thermosensitive liposomes in tumors. J Control Release. 2016;237:138-46
6. Xiao D, Jia HZ, Zhang J, Liu CW, Zhuo RX, Zhang XZ. A dual-responsive mesoporous silica nanoparticle for tumor-triggered targeting drug delivery. Small. 2014;10:591-8
7. Huang X, Peng X, Wang Y, Wang Y, Shin DM, El-Sayed MA. et al. A reexamination of active and passive tumor targeting by using rod-shaped gold nanocrystals and covalently conjugated peptide ligands. ACS Nano. 2010;4:5887-96
8. Kievit FM, Stephen ZR, Veiseh O, Arami H, Wang T, Lai VP. et al. Targeting of primary breast cancers and metastases in a transgenic mouse model using rationally designed multifunctional SPIONs. ACS Nano. 2012;6:2591-601
9. Sykes EA, Chen J, Zheng G, Chan WC. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8:5696-706
10. Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9:1909-15
11. Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG. et al. Design considerations for tumour-targeted nanoparticles. Nat Nanotechnol. 2010;5:42-7
12. Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B. et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25:1165-70
13. Conrad CA. Chemotherapy for metastatic tumors to the central nervous system. Curr Oncol Rep. 2001;3:490-4
14. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96
15. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology. 2009;10:459-66
16. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610-22
17. Cheng Y, Dai Q, Morshed RA, Fan X, Wegscheid ML, Wainwright DA. et al. Blood-brain barrier permeable gold nanoparticles: an efficient delivery platform for enhanced malignant glioma therapy and imaging. Small. 2014;10:5137-50
18. del Burgo LS, Hernandez RM, Orive G, Pedraz JL. Nanotherapeutic approaches for brain cancer management. Nanomedicine. 2014;10:905-19
19. Lesniak MS, Brem H. Targeted therapy for brain tumours. Nat Rev Drug Discov. 2004;3:499-508
20. Cheng Y, Morshed RA, Auffinger B, Tobias AL, Lesniak MS. Multifunctional nanoparticles for brain tumor imaging and therapy. Adv Drug Deliv Rev. 2014;66:42-57
21. Jensen SA, Day ES, Ko CH, Hurley LA, Luciano JP, Kouri FM. et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci Transl Med. 2013;5:209ra152
22. Dixit S, Novak T, Miller K, Zhu Y, Kenney ME, Broome AM. Transferrin receptor-targeted theranostic gold nanoparticles for photosensitizer delivery in brain tumors. Nanoscale. 2015;7:1782-90
23. Cheng Y, Meyers JD, Agnes RS, Doane TL, Kenney ME, Broome AM, Burda C, Basilion JP. Addressing brain tumors with targeted gold nanoparticles: a new gold standard for hydrophobic drug delivery? Small. 2011;7:2301-6
24. Ma Y, Mou Q, Sun M, Yu C, Li J, Huang X. et al. Cancer Theranostic Nanoparticles Self-Assembled from Amphiphilic Small Molecules with Equilibrium Shift-Induced Renal Clearance. Theranostics. 2016;6:1703-16
25. Li L, Fu S, Chen C, Wang X, Fu C, Wang S. et al. Microenvironment-Driven Bioelimination of Magnetoplasmonic Nanoassemblies and Their Multimodal Imaging-Guided Tumor Photothermal Therapy. ACS Nano. 2016;10:7094-105
26. Thambi T, You DG, Han HS, Deepagan VG, Jeon SM, Suh YD. et al. Bioreducible carboxymethyl dextran nanoparticles for tumor-targeted drug delivery. Adv Healthc Mater. 2014;3:1829-38
27. Yao C, Liu J, Wu X, Tai Z, Gao Y, Zhu Q. et al. Reducible self-assembling cationic polypeptide-based micelles mediate co-delivery of doxorubicin and microRNA-34a for androgen-independent prostate cancer therapy. Journal Control Release. 2016;232:203-14
28. Ruan S, Cao X, Cun X, Hu G, Zhou Y, Zhang Y. et al. Matrix metalloproteinase-sensitive size-shrinkable nanoparticles for deep tumor penetration and pH triggered doxorubicin release. Biomaterials. 2015;60:100-10
29. Ruan S, Yuan M, Zhang L, Hu G, Chen J, Cun X. et al. Tumor microenvironment sensitive doxorubicin delivery and release to glioma using angiopep-2 decorated gold nanoparticles. Biomaterials. 2015;37:425-35
30. Tam JM, Tam JO, Murthy A, Ingram DR, Ma LL, Travis K. et al. Controlled assembly of biodegradable plasmonic nanoclusters for near-infrared imaging and therapeutic applications. ACS Nano. 2010;4:2178-84
31. Liu Y, He J, Yang K, Yi C, Liu Y, Nie L. et al. Folding up of gold nanoparticle strings into plasmonic vesicles for enhanced photoacoustic imaging. Angew Chem Int Ed Engl. 2015;54:15809-12
32. Niikura K, Iyo N, Matsuo Y, Mitomo H, Ijiro K. Sub-100 nm gold nanoparticle vesicles as a drug delivery carrier enabling rapid drug release upon light irradiation. ACS Appl Mater Interfaces. 2013;5:3900-7
33. Deng H, Dai F, Ma G, Zhang X. Theranostic gold nanomicelles made from biocompatible comb-like polymers for thermochemotherapy and multifunctional imaging with rapid clearance. Adv Mater. 2015;27:3645-53
34. Dong L, Li M, Zhang S, Li J, Shen G, Tu Y. et al. Cytotoxicity of BSA-stabilized gold nanoclusters: In vitro and in vivo study. Small. 2015;11:2571-81
35. Choi HS. Nanoparticle assembly: building blocks for tumour delivery. Nat Nanotechnol. 2014;9:93-4
36. Orr G, Panther DJ, Phillips JL, Tarasevich BJ, Dohnalkova A, Hu D. et al. Submicrometer and nanoscale inorganic particles exploit the actin machinery to be propelled along microvilli-like structures into alveolar cells. ACS Nano. 2007;1:463-75
37. Chou LY, Zagorovsky K, Chan WC. DNA assembly of nanoparticle superstructures for controlled biological delivery and elimination. Nat Nanotechnol. 2014;9:148-55
38. Wang L, Xu L, Kuang H, Xu C, Kotov NA. Dynamic nanoparticle assemblies. Acc Chem Res. 2012;45:1916-26
39. Melemenidis S, Jefferson A, Ruparelia N, Akhtar AM, Xie J, Allen D. et al. Molecular magnetic resonance imaging of angiogenesis in vivo using polyvalent cyclic RGD-iron oxide microparticle conjugates. Theranostics. 2015;5:515-29
40. Wang J, Sun X, Mao W, Sun W, Tang J, Sui M. et al. Tumor redox heterogeneity-responsive prodrug nanocapsules for cancer chemotherapy. Adv Mater. 2013;25:3670-6
41. Cui Y, Dong H, Cai X, Wang D, Li Y. Mesoporous silica nanoparticles capped with disulfide-linked PEG gatekeepers for glutathione-mediated controlled release. ACS Appl Mater Interfaces. 2012;4:3177-83
42. Tong X, Wang Z, Sun X, Song J, Jacobson O, Niu G. et al. Size Dependent Kinetics of Gold Nanorods in EPR Mediated Tumor Delivery. Theranostics. 2016;6:2039-51
43. Gao C, Liu T, Dang Y, Yu Z, Wang W, Guo J. et al. pH/redox responsive core cross-linked nanoparticles from thiolated carboxymethyl chitosan for in vitro release study of methotrexate. Carbohydr Polym. 2014;111:964-70
44. Lee CC, Cramer AT, Szoka FC Jr, Frechet JM. An intramolecular cyclization reaction is responsible for the in vivo inefficacy and apparent pH insensitive hydrolysis kinetics of hydrazone carboxylate derivatives of doxorubicin. Bioconjug Chem. 2006;17:1364-8
45. Liu J, Yu M, Zhou C, Yang S, Ning X, Zheng J. Passive tumor targeting of renal-clearable luminescent gold nanoparticles: long tumor retention and fast normal tissue clearance. J Am Chem Soc. 2013;135:4978-81
46. Rousselle C, Clair P, Lefauconnier JM, Kaczorek M, Scherrmann JM, Temsamani J. New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol Pharmacol. 2000;57:679-86
47. Ling D, Park W, Park SJ, Lu Y, Kim KS, Hackett MJ. et al. Multifunctional tumor pH-sensitive self-assembled nanoparticles for bimodal imaging and treatment of resistant heterogeneous tumors. J Am Chem Soc. 2014;136:5647-55
48. Jeong H, Huh M, Lee SJ, Koo H, Kwon IC, Jeong SY. et al. Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy. Theranostics. 2011;1:230-9
49. Hou W, Zhao X, Qian X, Pan F, Zhang C, Yang Y. et al. pH-Sensitive self-assembling nanoparticles for tumor near-infrared fluorescence imaging and chemo-photodynamic combination therapy. Nanoscale. 2016;8:104-16
50. Kopecka J, Porto S, Lusa S, Gazzano E, Salzano G, Giordano A. et al. Self-assembling nanoparticles encapsulating zoledronic acid revert multidrug resistance in cancer cells. Oncotarget. 2015;6:31461-78
51. Sun Q, Sun X, Ma X, Zhou Z, Jin E, Zhang B. et al. Integration of nanoassembly functions for an effective delivery cascade for cancer drugs. Adv Mater. 2014;26:7615-21
52. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941-51
Author contact
![]() Corresponding author: maciej.lesniakedu; ligangedu.cn; yuchengedu.cn
Corresponding author: maciej.lesniakedu; ligangedu.cn; yuchengedu.cn