Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(7):1914-1927. doi:10.7150/thno.17852 This issue Cite
Research Paper
Triptolide Inhibits the AR Signaling Pathway to Suppress the Proliferation of Enzalutamide Resistant Prostate Cancer Cells
Yangyang Han1#, Weiwei Huang1#, Jiakuan Liu2, Dandan Liu3,4, Yangyan Cui2, Ruimin Huang3,4, Jun Yan2 ![]() , Ming Lei1,5
, Ming Lei1,5 ![]()
1. College of Life Sciences, Northwest A&F University, Yangling, Shaanxi, China
2. State Key Laboratory of Pharmaceutical Biotechnology and MOE Key Laboratory of Model Animals for Disease Study, Model Animal Research Center of Nanjing University, Nanjing, Jiangsu, China
3. Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
4. University of Chinese Academy of Sciences, Beijing 100049, China
5. Institute of Biophysics, Chinese Academy of Sciences, Chaoyang District, Beijing, China
# These authors contributed equally to this work.
Received 2016-10-8; Accepted 2017-2-21; Published 2017-4-20
Abstract

Enzalutamide is a second-generation androgen receptor (AR) antagonist for the treatment of metastatic castration-resistant prostate cancer (mCRPC). Unfortunately, AR dysfunction means that resistance to enzalutamide will eventually develop. Thus, novel agents are urgently needed to treat this devastating disease. Triptolide (TPL), a key active compound extracted from the Chinese herb Thunder God Vine (Tripterygium wilfordii Hook F.), possesses anti-cancer activity in human prostate cancer cells. However, the effects of TPL against CRPC cells and the underlying mechanism of any such effect are unknown. In this study, we found that TPL at low dose inhibits the transactivation activity of both full-length and truncated AR without changing their protein levels. Interestingly, TPL inhibits phosphorylation of AR and its CRPC-associated variant AR-V7 at Ser515 through XPB/CDK7. As a result, TPL suppresses the binding of AR to promoter regions in AR target genes along with reduced TFIIH and RNA Pol II recruitment. Moreover, TPL at low dose reduces the viability of prostate cancer cells expressing AR or AR-Vs. Low-dose TPL also shows a synergistic effect with enzalutamide to inhibit CRPC cell survival in vitro, and enhances the anti-cancer effect of enzalutamide on CRPC xenografts with minimal side effects. Taken together, our data demonstrate that TPL targets the transactivation activity of both full-length and truncated ARs. Our results also suggest that TPL is a potential drug for CRPC, and can be used in combination with enzalutamide to treat CRPC.
Keywords: Triptolide, Enzalutamide, Castration-resistant prostate cancer, Androgen receptor, Phosphorylation.
Introduction
Prostate cancer (PCa) is among the most common adult malignancies and is the second leading cause of cancer death in men. In 2016, there were an estimated 180,890 diagnoses and almost 26,120 deaths in American men.[1] In past decades, surgical or medical androgen deprivation therapy (ADT) has been the primary treatment paradigm for PCa patients.[2] However, almost all ADT treatments eventually fail due to the development of metastatic castration-resistant prostate cancer (mCRPC). Recently, 'second-generation' drugs, such as enzalutamide (also named MDV3100), abiraterone acetate, and cabazitaxel, have been administered to patients who develop mCRPC.[3-7] Enzalutamide is an AR antagonist which has an 8-fold higher affinity for AR than bicalutamide, and inhibits the ability of AR to translocate into the nucleus and bind DNA.[8] Although these new drugs have shown improved efficacies in the clinic, nearly all treated mCRPC patients eventually develop resistance to these agents, possibly due to amplification or gain-of-function somatic mutation of the AR gene, aberrant posttranslational modification of the AR protein, alternative splicing events that result in hyperactive receptors, and cofactor dysregulation and/or intracrine androgen synthesis.[8]
Compared to hormone-naïve cancers, most CRPCs contain AR splice variants (AR-Vs), which are often up-regulated. Such selection for AR-Vs in CRPC has also been demonstrated in several preclinical models.[9,10] These AR-Vs lack the ligand binding domain (LBD), and thus are insensitive to drugs such as enzalutamide that target the LBD. AR-Vs remain constitutively active as transcription factors in a ligand-independent manner.[11,12] Two major AR-Vs, AR-V7 (also called AR3, which contains exons 1-3 and CE3, a small expressed tag after exon 3) and AR-V567es (also known as AR-V12, which contains exons 1-4 and exon 8), are capable of regulating gene expression in the absence of the full-length AR protein (AR-FL).[13] Patients with high AR-V7 or detectable AR-V567es expression levels have significantly shorter cancer-specific survival than other CRPC patients.[14] In CRPC xenografts, resistance to both abiraterone and enzalutamide is associated with increased expression of AR truncated variants.[15,16] Furthermore, it has been reported recently that in CRPC cells expressing both endogenous AR-FL and AR-Vs, AR-Vs drive resistance to enzalutamide by functioning as independent drivers of the AR transcriptional program.[14,17,18] Thus, combining enzalutamide with an AR-V-targeting agent may be a viable approach to overcome resistance to enzalutamide.
Based on the important roles of AR dysfunction, including overexpression, mutation and the presence of AR truncated variants, in the development of mCRPC and resistance to clinical drugs, the search for novel compounds that target AR signaling has become a hotspot in PCa research. Natural compounds provide an excellent resource for finding novel antiandrogens. Several natural products were recently reported to show an anti-PCa effect by targeting AR or AR-Vs.[19-21] Maytenus royleanus extract had potent growth inhibition and apoptosis induction effects on PCa in vitro and in vivo.[19] Urolithins from walnut polyphenol metabolites suppressed the proliferation of PCa cells by repressing AR expression.[20] The marine compound Rhizochalinin (Rhiz) re-sensitized AR-V7-positive PCa cells to enzalutamide and had a pronounced anti-cancer effect on enzalutamide- and abiraterone-resistant AR-V7-positive cells, indicating that Rhiz is a potential drug for treating PCa patients with AR-V7 expression and enzalutamide or abiraterone resistance.[21] Sintokamide A (SINT 1) inhibited the growth of enzalutamide-resistant PCa cells with AR-Vs and induced regression of CRPC xenografts by binding to the activation function-1 (AF-1) region in the N-terminal domain of AR and suppressing the transactivation of both AR and AR-Vs.[22] We have focused our attention on Triptolide (TPL), a major active compound extracted from the Chinese medicine "Thunder God Vine" (Tripterygium wilfordii Hook F.), which possesses potent anti-cancer, anti-fertility, anti-inflammatory and immunosuppressive properties.[23] Our previous study has proved that TPL has potent anti-PCa effects.[24] However, the severe toxicity of TPL and our poor understanding of the mechanism of its anti-cancer effect on CRPC has limited its clinical use. In this study, we found that TPL inhibits the transactivation activity of both AR and AR-Vs, and reduces phosphorylation of AR at Ser515 through XPB/CDK7. A low dose of TPL inhibits the growth of CRPC cells and has a synergistic effect with enzalutamide in vitro. TPL also enhances the anti-cancer effect of enzalutamide on CRPC xenografts. In all, our data indicate that the combination of TPL with enzalutamide is a potential therapeutic treatment for CRPC.
Materials and Methods
Cell culture and reagents
LNCaP, 22Rv1, PC3, DU145, and 293T cells were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and maintained in RPMI-1640 supplemented with 10% FBS and 100 units/ml of penicillin and streptomycin. All the cell lines were recently authenticated by short tandem repeat analysis at the Cell Bank of Type Culture Collection of Chinese Academy of Sciences and the Genetic Testing Biotechnology Company (Suzhou, China, April 2016). C4-2 cells were provided and authenticated by Dr S. Li. C4-2/Luci and C4-2/AR-V7 cells were generated by infecting C4-2 cells with either control virus pLVX-Luci or virus expressing AR-V7 (pLVX-AR-V7). C4-2 shCTL and C4-2 shXPB were generated by infecting C4-2 cells with either a scrambled shRNA or pLKO.1 encoding shXPB. Infected cells were maintained in RPMI 1640 medium containing 1 mg/ml puromycin. To generate enzalutamide-resistant C4-2 (C4-2R) cells, C4-2 cells were chronically exposed to increasing concentrations of enzalutamide (5-20 μM) by passage for more than 6 months and maintained with 20 μM enzalutamide. C4-2 parental cells were passaged as a control. TPL (> 98% purity) was purchased from π-π Technologies, Inc (Guangzhou, China), and dissolved in DMSO at a stock concentration of 100 mM. Enzalutamide (MDV3100, Cat #: S1250) was from Selleck Chemicals (Houston, TX, USA). BS-181 (Cat #: HY-13266) was from MedChem Express (USA). Plasmids are detailed in the Supplementary Materials and Methods.
Transcriptional reporter assay
LNCaP cells (1×105 cells/well) were transfected with 300 ng of pGL3-PSA-Luc reporter plasmid or the control plasmid along with 300 ng AR-FL or AR-V7. Transactivation of the AR NTD (AR 1-558) was measured by co-transfecting LNCaP cells with 5×UAS-TATA-luciferase and AR-(1-558)-Gal4DBD or Gal4DBD for 24 h prior to treatment with TPL (6.25 nM) for 1 h. Cells were then incubated with forskolin (Sigma; 50 µM) or IL-6 (Peprotech, Rocky Hill, NJ; 50 ng/ml) or vehicle for an additional 24 h. Transfected cells were incubated for 24 h in the absence and presence of 1 nM R1881 with or without inhibitors. Luciferase activity was determined using a dual luciferase reporter assay system (Promega). Luciferase activities were normalized to the protein concentrations of the samples.
RNA isolation, reverse transcription, and quantitative real-time PCR
The procedures are described in the Supplementary Materials and Methods, and the qPCR primers are listed in Table S1.
Western blotting
Western blotting was carried out following the standard method. The following antibodies were used: phosphor-AR (S515) (ab128250, Abcam), AR (N-20, Santa Cruz), PSA (L106, Bioworld), XPB (10580-1-AP, Proteintech), RPB1 (RLT4173, Ruiyingbio), CDK7 (RLT0838, Ruiyingbio), VEGFA (19003-1-AP, Proteintech) and Tubulin (4D9, Bioworld). Tubulin was used as the loading control.
Chromatin immunoprecipitation (ChIP) assay
Cells were plated in 150 mm dishes (7×106 cells) in RPMI 1640 supplemented with 8% CSS (charcoal-stripped FBS) for 72 h. Cells were pretreated with TPL or DMSO as vehicle for 1 h before treatment with 1 nM R1881 or vehicle (ethanol) for 6 h. ChIP assays were performed using a ChIP kit (Millipore, 17-295) according to the manufacturer's instructions. Antibodies against AR (C-19, sc-815, Santa Cruz), CDK7 (sc-856, Santa Cruz), XPB (sc-293, Santa Cruz), FLAG (Sigma) or RNA pol II S5 (ab5408, Abcam) were used. IgG antibody was used as negative control. The bound DNA was amplified by qPCR using the primers listed in Table S1.
Viability assay
The MTT assay or SRB assay was used to quantify the cell viability. The procedures are described in the Supplementary Materials and Methods.
Clonogenic assay
PCa cells were seeded in triplicate at a density of 1000 cells/well into 12-well plates. After two days, cells were treated with different concentrations of TPL for 14 days. Colonies were fixed with methanol and stained with 0.5% crystal violet (Sigma). Colonies with 50 cells or greater were counted in each well.
Calculation of Combination Index by the Chou-Talalay Method
The type of interaction between Triptolide and enzalutamide was evaluated by comparing the cytotoxic effects obtained after simultaneous exposure to the drugs. The combination index (CI) was calculated using Calcusyn 2.0 software based on the Chou-Talalay method [25,26]: CI < 1 indicates a synergistic effect, CI = 1 indicates an additive effect, and CI > 1 indicates an antagonistic effect.
Animal study
The animal study (Project number: YJ32) was approved by the Institutional Animal Care and Use Committee of the Model Animal Research Center at Nanjing University and strictly followed ethical regulatory standards. Six-week-old male nude mice (STOCK-Foxn1nu/Nju) were inoculated subcutaneously with 5×106 22Rv1 cells suspended in 50% Matrigel in both dorsal flanks. The day following inoculation, mice were randomly divided into four groups (n = 10) and treated every other day for total 3 weeks as follows: (1) vehicle control [1% CMC (sodium salt of carboxymethyl cellulose, Sangon Biotech, Shanghai, China), 0.1% Tween-80, and 5% DMSO, oral gavage], (2) enzalutamide (25 mg/kg, oral gavage), (3) TPL (75 μg/kg, i.p.), and (4) enzalutamide (25 mg/kg, oral gavage) + TPL (75 μg/kg, i.p.). Body weights and tumor dimensions were monitored three times a week. Tumor volumes were calculated using the equation V= 0.524 × width2 × length/10.[15] The level of serum prostate specific antigen (PSA) in mouse blood was determined using quantitative ELISA (Cusabio Biotech. Wuhan, China)
Immunohistochemistry (IHC)
For IHC staining, sections were cut from paraffin blocks. Staining was carried out using anti-Ki-67 (Cat #: M7240; 1:1000; DAKO, Inc., City, CA, USA), anti-Cleaved-Caspase-3 (Cat #: 9661; 1:3000; Cell Signal Technologies, Inc), and anti-AR (N-20, Cat #: sc-27136; 1:1000; Santa Cruz) as primary antibodies following the standard protocol.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (version 6.01; GraphPad Software). Except where specified, comparisons between groups were performed with 2-tailed Student's t test, and differences were considered statistically significant at p < 0.05.
Results
TPL inhibits AR transcription activity
Persistent AR activity through mutation of the AR gene in CRPC cells accounts for the failure of enzalutamide therapy. Hence, we assessed whether TPL attenuates the ligand-dependent and ligand-independent transactivation activities of AR. We found that the expression of luciferase, drive by the PSA promoter, was inhibited in the presence of enzalutamide (10 μM) or TPL (6.25 nM), suggesting that a low dose of TPL can inhibit R1881-induced AR transactivation activity without affecting the levels of endogenous AR (Figure 1A and 1B). Next, we co-transfected LNCaP cells with a reporter vector containing the Gal4 binding site and an expression vector encoding a chimeric protein consisting of the N-terminal domain (NTD) of human AR (AR-NTD; amino acids 1-558) fused to the Gal4-DNA binding domain (Gal4DBD), which can be activated by forskolin (FSK) or IL-6 in the absence of androgen and serum to stimulate PKA and STAT3 signaling, respectively. The results showed that TPL reduces both FSK- and IL6-induced transactivation activity of AR-NTD (Figure 1B and 1C) without affecting the levels of the fusion protein AR-NTD-Gal4DBD (Figure 1E and 1F). TPL treatment also significantly inhibited endogenous AR activation, induced by FSK or IL6, without changing the AR protein level (Figure S1A and B). These data indicate that TPL possesses the ability to inhibit both ligand-dependent and ligand-independent transactivation of AR, and the effect is not due to a reduction in the AR protein level. Next, to directly examine whether TPL can inhibit the transactivation activity of the AR variants AR-V567es and AR-V7, we co-transfected plasmids expressing AR-FL and AR variants (pcDNA3-AR-FL/-AR-V567es/-AR-V7) and a reporter plasmid (pGL3-PSA-Luc) into 293T cells, which do not express endogenous AR. The results showed that TPL inhibits the transactivation activity of AR-FL and the two AR variants without affecting their protein levels (Figure S1C and D). Similar results were also observed in AR-negative PC3 cells (Figure S1E and F). Therefore, we demonstrated that TPL can inhibit the ligand-dependent and ligand-independent transactivation activity of AR-FL and AR variants.
TPL inhibits the transactivation activity of AR. (A) TPL inhibits ligand-dependent transactivation activity of AR. LNCaP cells were transfected with a PSA-luciferase reporter and treated with TPL and R1881 under serum-free conditions for 24 h. (B) and (C) Transactivation assays of the AR-NTD were performed in LNCaP cells cotransfected with p5x3 Gal4UAS-TATA-luciferase and AR-NTD-Gal4 DBD. Enzalutamide (Enza) or TPL was added 1 h before incubation with FSK or IL-6 for 24 h. (D), (E) and (F) Western blots showing the levels of AR and AR-NTD in cell extracts from (A), (B) and (C), respectively. (G) and (H) TPL inhibits expression of endogenous target genes of AR and AR-Vs. (G) LNCaP cells were pretreated with TPL then incubated for 24 h with R1881. (H) 22Rv1 cells were treated with TPL for 24 h. mRNA levels of AR and AR-V target genes were measured by qRT-PCR and normalized to β-actin mRNA. Bars represent the mean ± SD. (I) and (J) TPL reduces the protein levels of PSA in LNCaP (I) and 22Rv1 (J) cells. The results are represented as means ±SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
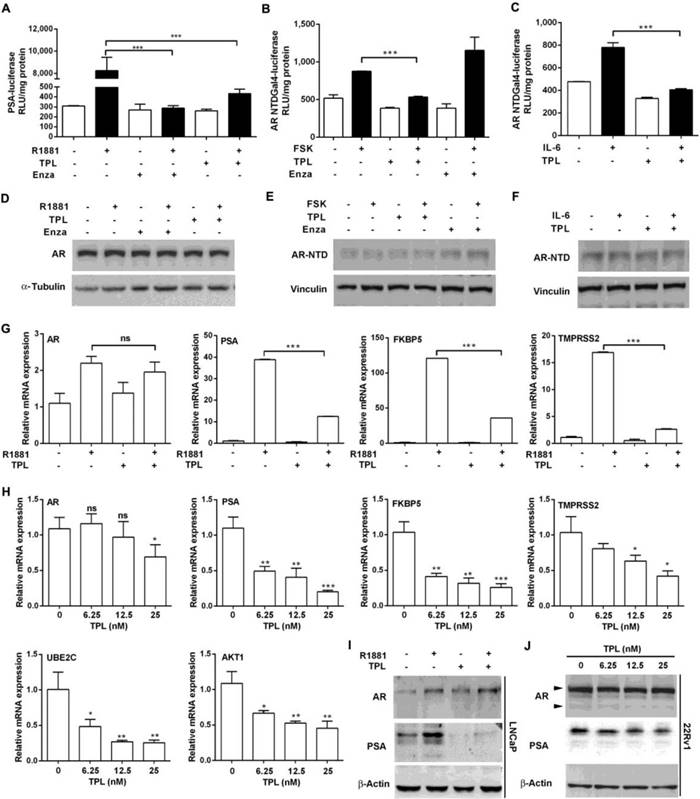
To further examine the effects of TPL on AR transactivation activity, we examined the mRNA levels of endogenous target genes of AR-FL (PSA, FKBP5, TMPRSS2) and AR-V (UBE2C and AKT1). In LNCaP cells, which express only AR-FL, TPL (6.25 nM) effectively repressed R1881-induced transcription of AR target genes (Figure 1G). TPL also suppressed the expression of AR-FL and AR-Vs target genes in 22Rv1 cells in a dose-dependent manner, without changing the AR mRNA level (Figure 1H). The results were validated by western blotting for AR and PSA protein levels (Figure 1I and 1J). Taken together, these results show that TPL inhibits the transactivation activity of AR-FL and AR variants at a low concentration (6.25 nM) without affecting their protein level.
TPL inhibits the transactivation activity of AR through CDK7 and XPB
Since we observed that the activity of AR is suppressed by TPL in PCa cells with no effects on its expression, we hypothesized that TPL may affect AR at the post-translational level. Interestingly, we found that 6.25 nM TPL decreased the level of AR phosphorylated at Ser515 (pAR S515) in LNCaP cells and the CRPC cell line C4-2/AR-V7 (Figure 2A), which stably expresses the AR-V7 variant (Figure S2A) and is more resistant to enzalutamide than control C4-2/Luci cells (Figure S2B). Given that CDK7 in the TFIIH complex is responsible for phosphorylating AR at Ser515,[27] we examined CDK7 as well as XPB and RPB1, two other proteins in the TFIIH complex. We found that 6.25 nM TPL did not change their expression levels (Figure 2A). To further examine whether CDK7 is involved in the effect of TPL on pAR S515, we treated PCa cells expressing AR-FL or the AR-V7 variant with the selective CDK7 inhibitor BS-181. We found that BS-181 has a similar effect to TPL on the level of pAR S515 (Figure 2B), which indicates that TPL may decrease the level of pAR S515 by indirectly affecting the activity of CDK7. Furthermore, to determine whether TPL inhibits AR transcriptional activity by reducing the level of pAR S515, we co-transfected 293T cells with the reporter plasmid PSA-Luc and plasmids expressing AR with different phosphorylation statuses, either AR/WT (wild-type AR) or AR/S515E (constitutively activated phosphorylation mutant). Transfected cells were pretreated with TPL prior to incubation with R1881 for 24 h.
TPL reduces the recruitment of AR to the PSA promoter by inhibiting phosphorylation of AR at S515. (A) Effect of TPL on the levels of pAR S515 and related proteins in PCa cells. LNCaP and C4-2/AR-V7 cells were pretreated with TPL or BS-181 for 1 h, and then incubated with R1881 for 4 h. Cell lysates were subjected to western blotting analysis with the indicated antibodies and the protein levels of AR and pAR S515 were quantified (B). (C) Effect of the phosphorylation status of AR on TPL activity. 293T cells were transiently co-transfected with pGL3.PSA-Luc and pcDNA3.1-AR/WT, or -AR/S515E, and then pretreated with TPL for 1 h prior to incubation with R1881 for 24 h. Luciferase activity was then measured. (D) and (E) Effect of TPL on the binding of AR or AR-V7 to the promoter of PSA. LNCaP (D) and C4-2/AR-V7 (E) cells were pretreated with TPL prior to the addition of R1881 for 6 h. ChIP assays were performed with rabbit or mouse IgG, anti-AR antibody, anti-CDK7 antibody, anti-XPB antibody, anti-Flag antibody or anti-RNA pol II S5 antibody. The results are represented as means ±SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
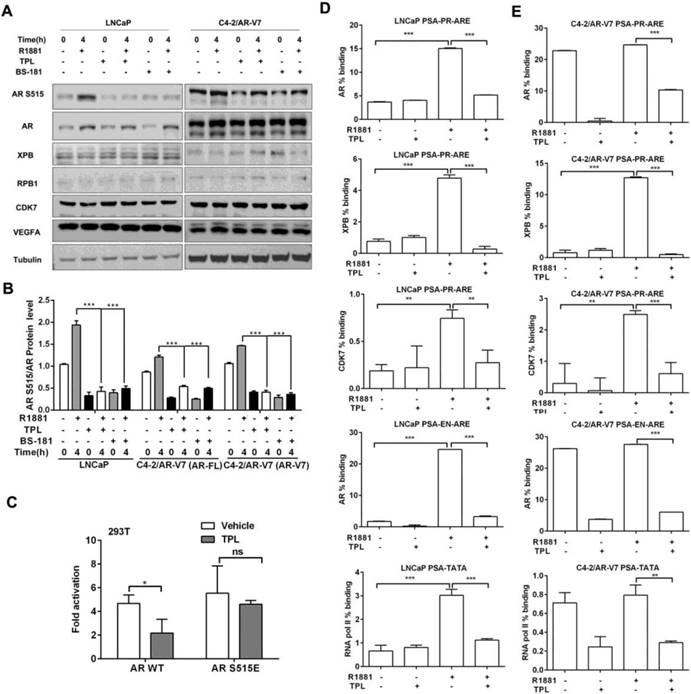
As shown in Figure 2C, TPL treatment significantly inhibits AR/WT transactivation activity, whereas AR/S515E abrogates the effect of TPL. This indicated that the phosphorylation of AR at Ser515 is essential for the suppression of AR activity by TPL. Next, to examine the effects of TPL on the DNA binding activity of AR, we performed ChIP assays and found that TPL significantly suppressed R1881-mediated AR binding to the androgen response element (ARE) and enhancer of the PSA gene. The binding of XPB and CDK7 to the promoter of the PSA gene was also decreased (Figure 2D). Consequently, less recruitment of RNA pol II (pS5) was found, which is consistent with the observation that TPL induces disrupted binding of AR and TFIIH. Similar experiments were also performed in C4-2/AR-V7 cells, which stably express AR-V7. TPL significantly inhibited the recruitment of these proteins to the PSA promoter, independent of the presence of R1881 (Figure 2E). Together, these data indicate that TPL also suppresses AR-mediated transcriptional activation by inhibiting AR binding and RNA pol II recruitment to target gene promoters.
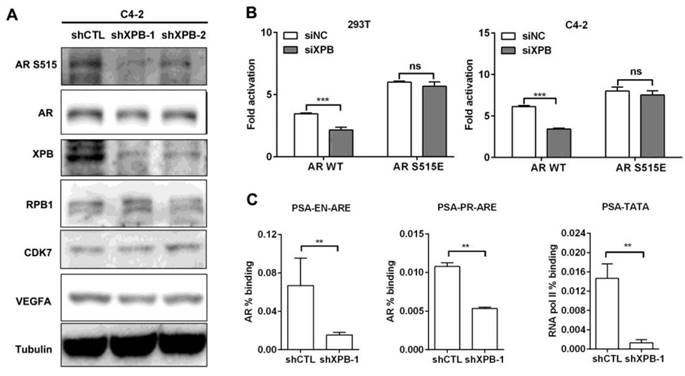
TPL was found to directly bind to XPB protein within the TFIIH complex,[28] which also contains CDK7 and XPD. Since XPD mutation was reported to suppress CDK7-mediated phosphorylation of AR at Ser515,[27] we hypothesized that TPL may function through XPB to suppress CDK7-mediated phosphorylation of AR at Ser515. Depletion of XPB by RNA interference reduced the level of pAR S515 while the CDK7 protein level was unaffected (Figure 3A). Accordingly, knockdown of XPB suppressed the transactivation activity of AR/WT, and had no effect on AR/S515E (Figure 3B). These results indicate that TPL suppresses CDK7-mediated phosphorylation of AR at Ser515 through XPB. Moreover, knockdown of XPB suppressed the occupancy of AR on the PSA promoter, and the recruitment of RNA pol II (pS5) to the PSA transcription start site (Figure 3C). These results demonstrate that XPB is the TPL-sensitive mediator in the AR signaling pathway.
Knockdown of XPB mimics the effect of TPL on AR activity in PCa cells. (A) Western blotting analysis of AR phosphorylation and several related proteins in C4-2 cells with XPB depletion. C4-2 cells were infected with lentivirus carrying control shRNA or XPB shRNA. The transduced cells were lysed and subjected to western blotting analysis with the indicated antibodies. (B) Effect of XPB depletion on the AR transactivation activity. 293T or C4-2 cells with stable knockdown of XPB were transiently co-transfected with pGL3.PSA-Luc and pcDNA3.1-AR/WT or -AR/S515E plasmids for 24 h. Luciferase activity was then measured. (C) Effect of XPB depletion on the DNA-binding activity of AR and RNA pol II. C4-2 cells with stable knockdown of XPB were lysed and subjected to ChIP assays with control IgG, anti-AR antibody and anti-RNA pol II S5 antibody. The results are represented as means ±SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Low concentrations of TPL inhibit PCa cell growth
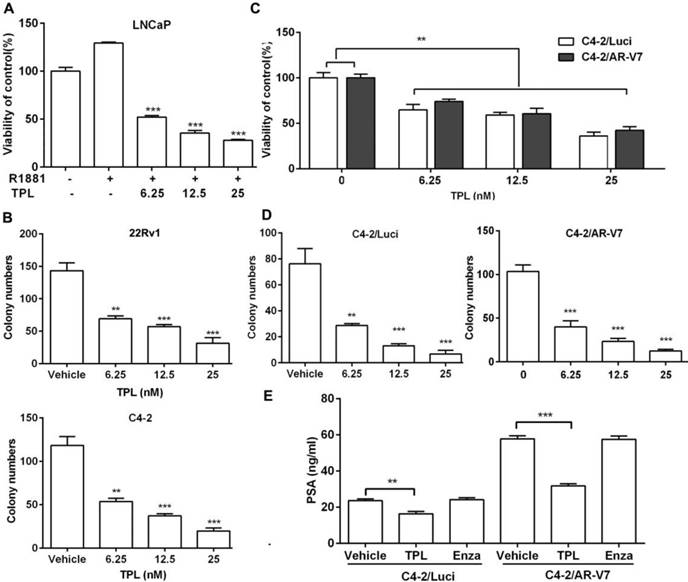
We and others have proved that TPL has effective anti-cancer activity against PCa at high concentrations (micromolar).[24,29,30] Based on the aforementioned results on AR transactivation with TPL in the nanomolar range, we were interested to examine whether low TPL concentrations (which are likely to be less toxic in vivo) still possess anti-PCa effects, particularly on CRPC cells. As shown in Figure 4A, low concentrations of TPL significantly reduced the R1881-stimulated growth of LNCaP cells over a period of 4 days with an IC50 of 6.25 nM. Meanwhile, low concentrations of TPL significantly suppressed the colony-forming ability of C4-2 and 22Rv1 cells in a dose-dependent manner (Figure 4B). These results confirm that low concentrations of TPL also inhibit the growth of PCa cells. We examined whether low TPL concentrations also show anti-PCa effects on the C4-2/AR-V7 cell line, which stably expresses AR-V7. The results showed that TPL suppresses the viability and colony-forming ability of both C4-2/Luci and C4-2/AR-V7 cells in a dose-dependent manner (Figure 4C and 4D). Moreover, TPL significantly reduced the R1881-induced expression of PSA in C4-2/AR-V7 cells (Figure 4E). These results indicate that low concentrations of TPL have similar cytotoxic effects on PCa cells expressing AR variants. In addition, we also examined the ability of low concentrations of TPL to induce apoptosis, as this is considered to be the major mechanism underlying the anti-cancer activity of TPL. The FACS data showed that TPL at a dose of 6.25 nM has a moderate apoptosis induction effect on LNCaP cells and a weak effect on C4-2/Luci and C4-2/AR-V7 cells, while higher concentrations of TPL have a marked apoptosis induction effect on all three cell lines (Figure S3). These results indicated that at low concentrations, such as 6.25 nM, TPL may exert its anti-PCa activity mainly through cell growth inhibition, while higher concentrations of TPL act through both cell growth inhibition and apoptosis induction. Collectively, our data reveal that low concentrations of TPL also have effective anti-cancer activity on PCa cells expressing AR-FL or AR variants.
Low doses of TPL block the proliferation of CRPC cells in vitro. (A) LNCaP cells were treated with 6.25 nM TPL for 1 h before the addition of R1881 (1 nM) for 3 days. Cell viability was measured by MTT. (B) Number of colonies formed by CRPC cells (22Rv1 and C4-2) following incubation with different concentrations of TPL. (C) C4-2/Luci and C4-2/AR-V7 cells were treated with different concentrations of TPL for 4 days. Cell viability was measured by MTT. (D) Number of colonies formed by C4-2/Luci and C4-2/AR-V7 cells after treatment with TPL. (E) C4-2/Luci and C4-2/AR-V7 cells were cultured in 8% CSS 1640 medium for 24 h, followed by treatment with 6.25 nM TPL or 20 μM enzalutamide for 72 h. PSA was then detected by ELISA. The results are represented as means ±SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Co-treatment with TPL and enzalutamide suppresses cell growth and induces apoptosis in CRPC cells
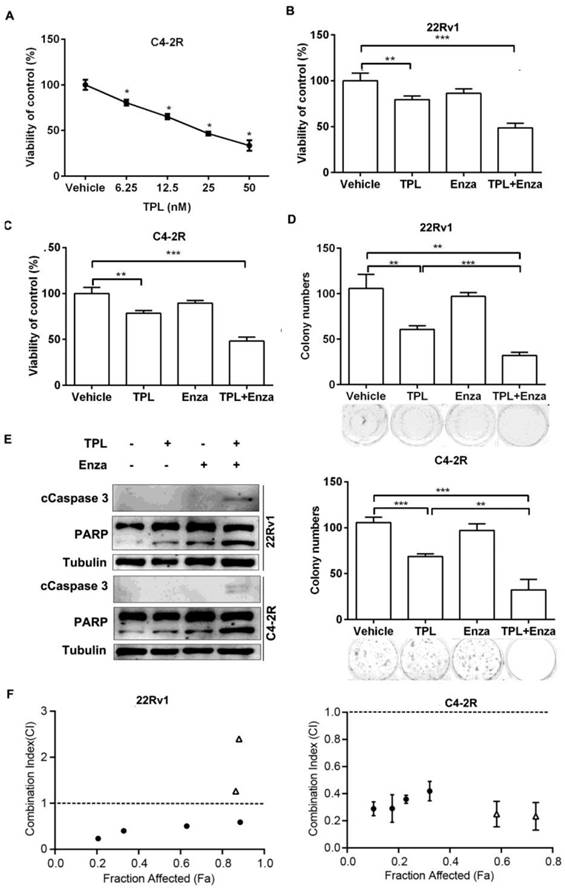
To further examine the effects of TPL on CRPC cells, we generated an enzalutamide-resistant PCa cell line C4-2R (C4-2 enzalutamide resistant) by continuous culture of C4-2 cells in medium containing enzalutamide for 7 months. The C4-2R cells showed more resistance to enzalutamide than the parental C4-2 cells (Figure S4A), and expressed higher protein levels of nuclear AR-V7 (Figure S4B-E). Interestingly, we found that the viability of C4-2R cells was significantly reduced by TPL in a dose-dependent manner (Figure 5A). This indicates that TPL can overcome the enzalutamide resistance of PCa cells expressing AR-V, and further suggests that TPL may be combined with enzalutamide against CRPC. To examine the combined effects of the two agents, we treated 22Rv1 cells with TPL (6.25 nM) in the presence or absence of enzalutamide for 4 days. The results showed that the co-treatment with TPL and enzalutamide has a stronger inhibitory effect on the viability of 22Rv1 cells than TPL or enzalutamide alone (Figure 5B). Similar results were also observed in the enzalutamide-resistant cell line C4-2R (Figure 5C). Co-treatment with TPL and enzalutamide had a stronger inhibitory effect on the colony-forming ability of 22Rv1 and C4-2R cells (Figure 5D). Co-treatment also induced higher levels of the cleaved products of two markers of apoptosis, Caspase-3 and PARP, in 22Rv1 and C4-2R cells (Figure 5E), indicating that the combined-treatment is also more cytotoxic to CRPC cells. These data suggest that low concentrations of TPL may have a synergistic effect with enzalutamide against PCa cells in vitro. We then examined the viability of 22Rv1 and C4-2R cells that were exposed to individual or combined TPL and enzalutamide at various concentrations, and calculated the combination index (CI) between TPL and enzalutamide using the Chou-Talalay method. As shown in Figure 5F, the results demonstrated that the combined TPL and enzalutamide treatment at low concentrations (<60 nM for TPL and <200 μM for enzalutamide) had a synergistic effect on the growth inhibition of both 22Rv1 and C4-2R cells. Taken together, our data demonstrate that low concentrations of TPL have a synergistic anti-PCa effect with enzalutamide in vitro. Co-treatment with TPL and enzalutamide may therefore be a potential therapeutic program for CRPC.
TPL shows a synergistic anti-cancer effect with enzalutamide on PCa cells in vitro. (A) C4-2R cells were cultured in medium containing 8% FBS and treated with different concentrations of TPL for 72 h. MTT assays were then carried out. (B) 22Rv1 or (C) C4-2R cells were treated with 6.25 nM TPL with or without 20 μM enzalutamide. MTT assays were carried out after 96 h. (D) Number of colonies formed by 22Rv1 or C4-2R cells treated with individual or combined TPL and enzalutamide. (E) Western blotting was performed to detect apoptotic marker proteins in 22Rv1 or C4-2R cells treated with individual or combined TPL and enzalutamide. (F) 22Rv1 and C4-2R cells were treated with individual or combined TPL and enzalutamide at various concentrations for 48 h and then subjected to SRB assay. The Combination Index of TPL and enzalutamide in the two cell lines was calculated by the Chou-Talalay method. Open triangles indicate high combination concentrations of TPL and enzalutamide (≥60 nM for TPL and ≥200 μM for enzalutamide); black dots indicate low combination concentrations of TPL and enzalutamide (<60 nM for TPL and <200 μM for enzalutamide). The results are represented as means ±SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Co-treatment with TPL and enzalutamide inhibits CRPC tumor progression in vivo
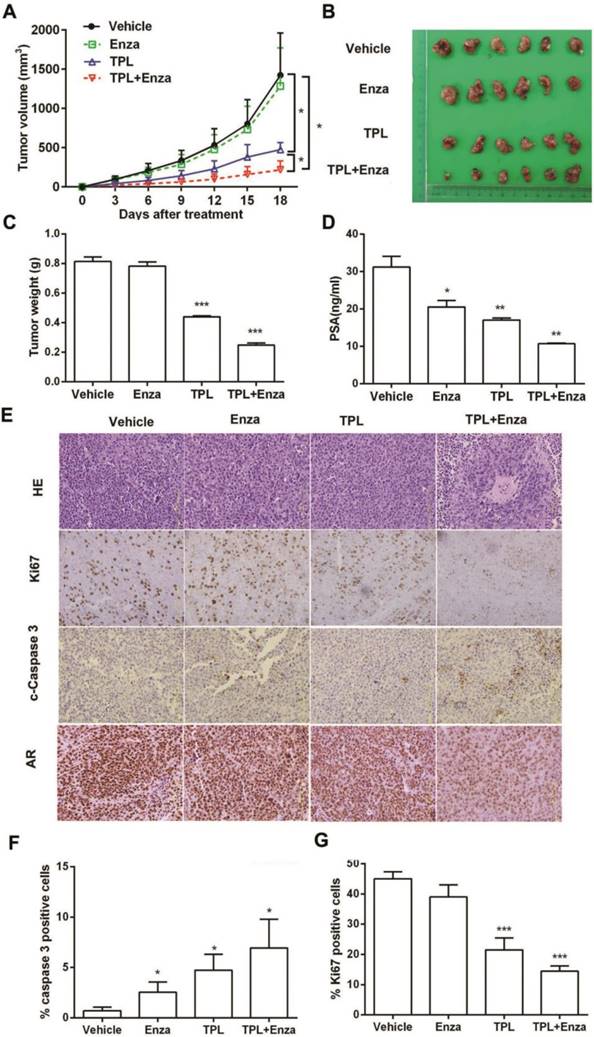
To evaluate the anti-PCa effect of co-treatment with TPL and enzalutamide in vivo, we made a CRPC xenograft model in mice using 22Rv1 cells. Male nude mice bearing 22Rv1 xenografts were randomized and treated with either TPL (75 μg/kg), enzalutamide (25 mg/kg), or both for 3 weeks. At the end of 3 weeks, the mice were euthanized and xenografts were collected for further analysis. As shown in Figure 6, co-treatment with TPL and enzalutamide is highly effective at reducing the volumes of xenograft tumors, compared with the moderate effect of TPL and the slight effect of enzalutamide (Figure 6A). Accordingly, the xenograft tumors in the co-treatment group appeared smaller than those from the other groups (Figure 6B). The weight of xenograft tumors from the co-treatment group was also lower than those from the other groups (Figure 6C). Moreover, the PSA level in mice in the co-treatment group was the lowest among the four treatment groups (Figure 6D). Histologic analysis showed that the cells in the xenograft tumors from the co-treatment group were packed together less tightly than those in the other groups (Figure 6E). IHC analysis revealed that the xenograft tumors from the co-treatment group showed less Ki-67 and AR staining, and more cleaved-Caspase-3 staining, than the other groups (Figure 6E-G), which indicated that the xenograft tumors undergo less proliferation and more apoptosis under co-treatment with TPL and enzalutamide. These results demonstrate that TPL and enzalutamide together show more effective anti-CRPC activity than either drug alone, which indicates that TPL enhances the anti-cancer effect of enzalutamide on CRPC in vivo. We also examined the body weights of the mice during treatment and found that none of the three treatments had an obvious effect on growth (Figure S5A). No obvious changes were detected in the weight and tissue structure of essential organs, such as lung, heart, liver, spleen, and kidney (Figure S5B and C). These data also demonstrated that a low dose of TPL, either alone or in combination with enzalutamide, has no significant toxicity to mice. Taken together, our results indicate that co-treatment with TPL and enzalutamide may be a potential treatment for CRPC.
TPL enhances the anti-cancer effects of enzalutamide in vivo. Mice bearing 22Rv1 xenografts were treated with vehicle control, enzalutamide, TPL, or enzalutamide + TPL for 3 weeks. (A) Tumor volumes in the different groups (volumes were measured twice every week). (B) Photographs of xenograft tumors harvested at day 21. (C) Weight of the xenograft tumors in the different groups. (D) Serum PSA level of the different groups. (E) Hematoxylin and eosin (H&E) staining and IHC staining of Ki67, cleaved-Caspase-3 (c-Caspase-3) and AR of representative sections of xenograft tumors. (F) and (G) Quantification of c-Caspase-3 staining (apoptotic index) and Ki-67 staining (proliferative index) in tumor sections. The results are represented as means ±SD. Scale bars represent 125 μm in all micrographs. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
As a representative of 'second generation' drugs and as an AR antagonist, enzalutamide has many favorable effects on mCRPC patients based on data from some phase I/II/III trials. However, patients who respond to enzalutamide usually relapse within 1 to 2 years, which is mainly due to the dysfunction of AR or AR variants.[31] Therefore, there is an urgent need for new agents that can overcome enzalutamide resistance. Our study revealed that TPL at low concentrations reduced the level of pAR S515 via XPB/CDK7, and reduced the binding of AR and recruitment of phosphorylated RNA Pol II (S5) to the promoters of AR target genes, resulting in the inhibition of AR transactivation activity. Low concentrations of TPL also inhibited the growth of PCa cells that express AR-FL or functional AR variants. Furthermore, TPL and enzalutamide together showed synergistic and efficient anti-CRPC effects in vitro and in vivo, including inhibition of CRPC cell proliferation and survival, without noticeable side-effects. Our data indicate that the combination of TPL with enzalutamide is a potential therapeutic treatment for CRPC.
The role of AR variants in the mechanism underlying enzalutamide resistance is supported by clinical data.[31] The most common AR-V isoforms associated with CRPC and subsequent metastases are AR-V7 and AR-V567es. In addition to regulating AR-FL downstream targets, AR-Vs also regulate a unique set of genes, most of which are associated with mitotic and anti-apoptotic functions, e.g. UBE2C.[32] Overexpression of UBE2C has been correlated with the presence of AR-V7 in clinical samples.[18,33] Hence, several drugs, such as EPI-001 and its analogs, which disrupt the transactivation of AR and AR-V in particular are more effective at preventing the growth of CRPCs.[34] Some compounds, such as 20(S)-protopanaxadiol-aglycone, Galeterone and its analog VNPT55 exert their anti-CRPC effect by depleting the AR-FL and AR-V protein levels or inducing AR-FL and AR-V degradation.[35-38] In our study, we found that TPL at low concentration effectively suppresses the transactivation activity of both AR-FL and AR-Vs, without changing their expression levels. However, such inhibition is mediated by XPB/CDK7 in the TFIIH complex, not by direct binding of TPL to AR, as we did not detect direct interaction between TPL and AR-FL or AR-NTD (AR-AF1) using Biomolecular Interaction Analysis (BIA, data not shown).
Herein, we found that TPL inhibits the transactivation activity of AR-FL and AR-Vs by reducing the level of pAR S515. Phosphorylation plays an important role in modulating the functional activity of AR. More than 15 distinct phosphorylation sites have been found within AR, most of which localize in the AR-NTD.[39] The phosphorylation of AR has been implicated in various AR activities, including cellular localization, expression, DNA binding, transcriptional activity and stability/degradation.[40] Phosphorylation of AR at Ser515 was proved to be a key step for accurate transcriptional activation by AR, including the cyclic recruitment of the transcription machinery and turnover of AR itself.[27] Unlike phosphorylation at other sites, phosphorylation of AR at Ser515 is not required for translocation of AR into the nucleus, but specifically affects the ability of AR to bind to its responsive elements. Impaired phosphorylation of AR at Ser515 disrupts AR-mediated transcription.[27] Phosphorylation of AR at Ser515 was also reported to be involved in the epidermal growth factor (EGF)-induced increase in AR transcriptional activity.[41] These studies demonstrated that pAR S515 is important for AR transactivation. In this study, we found that TPL effectively decreases the level of pAR S515, suggesting that TPL suppresses AR transactivation activity by inhibiting the phosphorylation of AR. In addition, the level of pAR S515 was related with EGF-induced PCa cell growth.[41] pAR S515 was also proved to be a potential predictive marker for relapse in PCa.[42] These studies indicated that pAR S515 is related to PCa progression. Hence, the decreased level of pAR S515 may also contribute to the anti-CRPC activity of TPL.
The phosphorylation of AR at Ser515 was proved to be mediated by the CDK7 kinase in the TFIIH complex, which is one of the general transcription factors that participates in transcription initiation.[27] TFIIH is composed of two subcomplexes, the core complex which consists of 7 subunits (XPD, XPB, p62, p52, p44 p34 and TTDA) and the cyclin activating kinase-subcomplex (CDK7, MAT1, and cyclin H).[43] Among these subunits, the helicases XPD and XPB uncouple the promoter while CDK7 phosphorylates RNA polymerase II (RNAPII) and transcription factors to initiate transcription.[43] AR interacts strongly with XPB and XPD, and weakly with CDK7, which eventually enhances AR transactivation by phosphorylating AR at S515.[27,43] However, we found that TPL attenuates the level of pAR S515 without impact on CDK7 expression. TPL was reported to activate CDK7, which led to phosphorylation of Rbp1 at S1878, the largest subunit of RNAPII, followed by degradation of Rbp1.[44,45] We found that inhibition of CDK7 with a selective inhibitor BS-181 had a similar effect to TPL on the phosphorylation of AR at Ser515 in PCa cells. This confirms that CDK7 is involved in TPL-induced attenuation of pAR S515. The next question is how TPL decreases the phosphorylation of AR at Ser515 through CDK7. Loss-of-function mutation of XPD has been proved to reduce the level of pAR S515 through CDK7,[27] which suggests that XPD may play a bridging role in CDK7-mediated AR phosphorylation. XPD and XPB are both helicase subunits in TFIIH, so it is possible that XPB may play a similar role to XPD in CDK7-mediated phosphorylation of AR, since XPB also binds directly with CDK7, like XPD.[27] TPL was reported to directly target XPB, leading to inhibition of XPB ATPase activity.[28] We found that knockdown of XPB by RNA interference had a similar effect to TPL, including a decreased level of pAR S515, inhibition of AR transactivation activity and reduced RNA pol II recruitment. These results demonstrated that TPL attenuates AR activity through inhibition of the XPB/CDK7-mediated phosphorylation of AR at Ser515.
Nowadays, the second-generation drug enzalutamide is widely used to treat PCa in the clinic with potent treatment effects. However, the development of enzalutamide resistance is a newly encountered problem, which is mainly induced by AR dysfunction. Therapeutic strategies have been investigated in which enzalutamide is used in combination with other drugs.[46] Several compounds were proved to enhance the anti-PCa effect of enzalutamide in vitro or in vivo, such as the RSK inhibitor SL0101,[47] the 5α-reductase inhibitor dutasteride,[48] and AZD5363,[49] which targets the PI3K/Akt pathway. Our data demonstrated that TPL is a potential drug for CRPC therapy. TPL showed effective anti-CRPC activity in PCa cells expressing either AR-FL or AR-Vs by abrogating the XPB/CDK7-mediated phosphorylation of AR at Ser515, which inhibited the transactivation activity of both AR-FL and AR-Vs. Furthermore, TPL showed a synergistic anti-PCa effect with enzalutamide in vitro. TPL also enhanced the anti-PCa effect of enzalutamide in vivo. These data demonstrated that the combined TPL/enzalutamide treatment strategy has potential for CRPC therapy.
TPL has been proved to be a promising anti-cancer drug, based on its potent and broad spectrum anti-cancer effects in vitro and in vivo. However, several barriers prevent the clinical application of TPL. Firstly, TPL is water-insoluble, which limits its clinical use. Several water-soluble derivatives of TPL have been developed, such as PG490-88 [50] and Minnelide [51], which can convert to TPL in vivo and retain most of the bioactivity of TPL. PG490-88 and Minnelide showed potent anti-cancer effects and were approved for entry into Phase I clinical trials for prostate cancer [52] and pancreatic cancer,[51] respectively. Secondly, TPL is highly cytotoxic. In traditional Chinese medicine, the "Thunder God Vine", from which TPL is extracted, is thought to be very toxic and has been used as a pesticide although it is mainly used to treat autoimmune and inflammatory diseases. Furthermore, the anti-cancer effect of TPL is attributed to its cytotoxicity. We found that TPL is cytotoxic to both cancer cells and normal cells in vitro, although the cytotoxic effect of TPL on normal cells is less than on cancer cells.[53] The toxicity of TPL is dose-dependent. The distinction between the safe dose and the toxic dose of TPL is very marginal,[54] which leads to a narrow therapeutic window. Therefore, development of TPL derivatives with moderate toxicity is an effective solution. The cytotoxicity of the novel TPL derivative LLDT-8, which was developed to treat rheumatoid arthritis, is 122-fold lower than TPL in cells,[55] yet it still has effective anti-cancer activity. In this study, we found that TPL, even at a concentration as low as 6.25 nM, shows effective anti-CRPC activity and has a synergistic effect with enzalutamide. The low dose of TPL did not cause any obvious adverse effects in the xenograft mouse model. Using a low dose of TPL in clinical applications is therefore another approach to reducing its toxicity. Thirdly, TPL has severe side-effects in animal models and patients, including gastrointestinal disturbances, kidney dysfunction, leucopenia, aplastic anemia and infertility, which limits its clinical application.[23] The adverse reactions of TPL are mainly due to its diverse biological activities. Besides anti-cancer activity, TPL also has anti-inflammatory, immunosuppressive, anti-fertility and anti-cystogenic properties. The relationship between the anti-cancer activity and immunosuppressive activity of TPL is very interesting and needs to be elucidated. Recently, glutriptolide, a new glucose-conjugated TPL derivative which selectively targets cancer cells overexpressing the glucose transporter, was developed. It has greater water solubility than TPL and higher cytotoxicity towards cancer cells than normal cells.[56] Glutriptolide provides a novel example of TPL modification and application. Fourth, the molecular mechanism underlying the anti-cancer effect of TPL is still unclear. Several TPL-binding proteins have been identified, including XPB, polycystin-2 (PC-2), disintegrin and metalloprotease 10 (ADAM10), dCTP pyrophosphatase 1 (DCTPP1) and TAB1.[28,57-60] Of these, XPB is thought to be the primary target of TPL and this is consistent with the phenotype of TPL-induced transcription inhibition. However, some genes are up-regulated with TPL treatment.[61] Overexpression of XPB does not totally neutralize the anti-cancer effect of TPL.[24] TPL-induced Rpb1 degradation was proved to be more important than XPB for the anti-cancer activity of TPL.[45] Glutriptolide has no inhibition effect on the activity of XPB in vitro.[56] We have found several new potential TPL-binding proteins using protein chips (data not shown). The anticancer mechanism of TPL should therefore be extensively explored, as this may be helpful for uncovering the anti-cancer mechanism of TPL and for developing potent new TPL derivatives or clinical applications with high therapeutic effect and low side-effects. In summary, although many difficult problems must be solved before TPL can be used for clinical applications, TPL shows potent anti-cancer effects and is a promising anti-cancer drug.
Conclusion
In summary, our study shows that TPL significantly inhibits the transactivation activity of AR-FL and AR-Vs by disrupting the phosphorylation of AR at Ser515 through XPB/CDK7. Moreover, TPL shows effective anti-PCa activities even at a low dose. TPL also synergizes with enzalutamide in attenuating PCa cell survival in vitro, and enhances the anti-PCa effect of enzalutamide on CRPC xenografts growth in vivo. These results provide a strong rationale for further evaluation of this combination in the clinic.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31571194 to ML; 81672873 and 81372168 to JY), the National Forestry Public Welfare Industry Research Project (201204603 to ML), the Research Innovation Team Project of Shaanxi (2013KCT-27 to ML), the Natural Science Foundation for Universities in Jiangsu Province (BK20151396 to JY), the National Key Research and Development Plan of China (2016YFC0902202 to JY), One Hundred Talent Program of Chinese Academy of Sciences (to RH), and the Research Fund of Institutes for Drug Discovery and Development, Chinese Academy of Sciences (CASIMM0120164001 to RH).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
2. Tannock IF, de Wit R, Berry WR. et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502-12
3. Morris MJ, Rathkopf DE, Novotny W. et al. Phase Ib Study of Enzalutamide in Combination with Docetaxel in Men with Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2016;22:3774-81
4. Beer TM, Armstrong AJ, Rathkopf DE. et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424-33
5. Scher HI, Fizazi K, Saad F. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187-97
6. Ryan CJ, Shah S, Efstathiou E. et al. Phase II study of abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer displaying bone flare discordant with serologic response. Clin Cancer Res. 2011;17:4854-61
7. Meisel A, von Felten S, Vogt DR. et al. Severe neutropenia during cabazitaxel treatment is associated with survival benefit in men with metastatic castration-resistant prostate cancer (mCRPC): A post-hoc analysis of the TROPIC phase III trial. Eur J Cancer. 2016;56:93-100
8. Crona DJ, Milowsky MI, Whang YE. Androgen receptor targeting drugs in castration-resistant prostate cancer and mechanisms of resistance. Clin Pharmacol Ther. 2015;98:582-9
9. Qu Y, Dai B, Ye D. et al. Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci Rep. 2015;5:7654
10. Ware KE, Garcia-Blanco MA, Armstrong AJ. et al. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr Relat Cancer. 2014;21:T87-T103
11. Hu R, Dunn TA, Wei S. et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16-22
12. Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287:19736-49
13. Xu D, Zhan Y, Qi Y. et al. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res. 2015;75:3663-71
14. Antonarakis ES, Lu C, Wang H. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028-38
15. Cao B, Qi Y, Zhang G. et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5:1646-56
16. Li Y, Chan SC, Brand LJ. et al. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483-9
17. Stone L. Prostate cancer: predicting resistance-AR-V7 is a potential biomarker. Nat Rev Urol. 2014;11:606
18. Hu R, Lu C, Mostaghel EA. et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457-62
19. Shabbir M, Syed DN, Lall RK. et al. Potent anti-proliferative, pro-apoptotic activity of the Maytenus royleanus extract against prostate cancer cells: evidence in in-vitro and in-vivo models. PLoS One. 2015;10:e0119859
20. Sanchez-Gonzalez C, Ciudad CJ, Noe V. et al. Walnut polyphenol metabolites, urolithins A and B, inhibit the expression of the prostate-specific antigen and the androgen receptor in prostate cancer cells. Food Funct. 2014;5:2922-30
21. Dyshlovoy SA, Otte K, Alsdorf WH. et al. Marine compound rhizochalinin shows high in vitro and in vivo efficacy in castration resistant prostate cancer. Oncotarget. 2016;7:69703-17
22. Banuelos CA, Tavakoli I, Tien AH. et al. Sintokamide A Is a Novel Antagonist of Androgen Receptor That Uniquely Binds Activation Function-1 in Its Amino-terminal Domain. J Biol Chem. 2016;291:22231-43
23. Liu Q. Triptolide and its expanding multiple pharmacological functions. Int Immunopharmacol. 2011;11:377-83
24. Huang W, He T, Chai C. et al. Triptolide inhibits the proliferation of prostate cancer cells and down-regulates SUMO-specific protease 1 expression. PLoS One. 2012;7:e37693
25. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621-81
26. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440-6
27. Chymkowitch P, Le May N, Charneau P. et al. The phosphorylation of the androgen receptor by TFIIH directs the ubiquitin/proteasome process. EMBO J. 2011;30:468-79
28. Titov DV, Gilman B, He QL. et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol. 2011;7:182-8
29. Hu XW, Sun Y. Triptolide sensitizes TRAIL-induced apoptosis in prostate cancer cells via p53-mediated DR5 up-regulation. Mol Biol Rep. 2012;39:8763-70
30. Tamgue O, Chai CS, Hao L. et al. Triptolide inhibits histone methyltransferase EZH2 and modulates the expression of its target genes in prostate cancer cells. Asian Pac J Cancer Prev. 2013;14:5663-9
31. Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18:R183-196
32. Hao Z, Zhang H, Cowell J. Ubiquitin-conjugating enzyme UBE2C: molecular biology, role in tumorigenesis, and potential as a biomarker. Tumour Biol. 2012;33:723-30
33. Stockley J, Markert E, Zhou Y. et al. The RNA-binding protein Sam68 regulates expression and transcription function of the androgen receptor splice variant AR-V7. Sci Rep. 2015;5:13426
34. Myung JK, Banuelos CA, Fernandez JG. et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948-60
35. Cao B, Liu X, Li J. et al. 20(S)-protopanaxadiol-aglycone downregulation of the full-length and splice variants of androgen receptor. Int J Cancer. 2013;132:1277-87
36. Montgomery B, Eisenberger MA, Rettig MB. et al. Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clin Cancer Res. 2016;22:1356-63
37. Kwegyir-Afful AK, Ramalingam S, Purushottamachar P. et al. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer. Oncotarget. 2015;6:27440-60
38. Yu Z, Cai C, Gao S. et al. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin Cancer Res. 2014;20:4075-85
39. Daniels G, Pei ZH, Logan S. et al. Mini-review: androgen receptor phosphorylation in prostate cancer. Am J Clin Exp Urol. 2013;1:25-9
40. Gao YF, Chen SY. Proline-Directed Androgen Receptor Phosphorylation. J Mol Genet Med. 2013;7:75
41. Ponguta LA, Gregory CW, French FS. et al. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J Biol Chem. 2008;283:20989-1001
42. Willder JM, Heng SJ, McCall P. et al. Androgen receptor phosphorylation at serine 515 by Cdk1 predicts biochemical relapse in prostate cancer patients. Br J Cancer. 2013;108:139-48
43. Lee DK, Duan HO, Chang C. From androgen receptor to the general transcription factor TFIIH. Identification of cdk activating kinase (CAK) as an androgen receptor NH(2)-terminal associated coactivator. J Biol Chem. 2000;275:9308-13
44. Manzo SG, Zhou ZL, Wang YQ. et al. Natural product triptolide mediates cancer cell death by triggering CDK7-dependent degradation of RNA polymerase II. Cancer Res. 2012;72:5363-73
45. Yi JM, Huan XJ, Song SS. et al. Triptolide Induces Cell Killing in Multidrug-Resistant Tumor Cells via CDK7/RPB1 Rather than XPB or p44. Mol Cancer Ther. 2016;15:1495-503
46. Drake CG, Sharma P, Gerritsen W. Metastatic castration-resistant prostate cancer: new therapies, novel combination strategies and implications for immunotherapy. Oncogene. 2014;33:5053-64
47. Shiota M, Yokomizo A, Takeuchi A. et al. Inhibition of RSK/YB-1 signaling enhances the anti-cancer effect of enzalutamide in prostate cancer. Prostate. 2014;74:959-69
48. Hamid AR, Verhaegh GW, Smit FP. et al. Dutasteride and enzalutamide synergistically suppress prostate tumor cell proliferation. J Urol. 2015;193:1023-9
49. Toren P, Kim S, Cordonnier T. et al. Combination AZD5363 with Enzalutamide Significantly Delays Enzalutamide-resistant Prostate Cancer in Preclinical Models. Eur Urol. 2015;67:986-90
50. Fidler JM, Li K, Chung C. et al. PG490-88, a derivative of triptolide, causes tumor regression and sensitizes tumors to chemotherapy. Mol Cancer Ther. 2003;2:855-62
51. Chugh R, Sangwan V, Patil SP. et al. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci Transl Med. 2012;4:156ra139
52. Kiviharju TM, Lecane PS, Sellers RG. et al. Antiproliferative and proapoptotic activities of triptolide (PG490), a natural product entering clinical trials, on primary cultures of human prostatic epithelial cells. Clin Cancer Res. 2002;8:2666-74
53. Zhao F, Huang W, Ousman T. et al. Triptolide induces growth inhibition and apoptosis of human laryngocarcinoma cells by enhancing p53 activities and suppressing E6-mediated p53 degradation. PLoS One. 2013;8:e80784
54. Li Z, Zhou ZL, Miao ZH. et al. Design and synthesis of novel C14-hydroxyl substituted triptolide derivatives as potential selective antitumor agents. J Med Chem. 2009;52:5115-23
55. Zhou R, Zhang F, He PL. et al. (5R)-5-hydroxytriptolide (LLDT-8), a novel triptolide analog mediates immunosuppressive effects in vitro and in vivo. Int Immunopharmacol. 2005;5:1895-903
56. He QL, Minn I, Wang Q. et al. Targeted Delivery and Sustained Antitumor Activity of Triptolide through Glucose Conjugation. Angew Chem Int Ed Engl. 2016;55:12035-9
57. Leuenroth SJ, Okuhara D, Shotwell JD. et al. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci U S A. 2007;104:4389-94
58. Soundararajan R, Sayat R, Robertson GS. et al. Triptolide: An inhibitor of a disintegrin and metalloproteinase 10 (ADAM10) in cancer cells. Cancer Biol Ther. 2009;8:2054-62
59. Corson TW, Cavga H, Aberle N. et al. Triptolide directly inhibits dCTP pyrophosphatase. Chembiochem. 2011;12:1767-73
60. Lu Y, Zhang Y, Li L. et al. TAB1: a target of triptolide in macrophages. Chem Biol. 2014;21:246-56
61. Zhou ZL, Yang YX, Ding J. et al. Triptolide: structural modifications, structure-activity relationships, bioactivities, clinical development and mechanisms. Nat Prod Rep. 2012;29:457-75
Author contact
![]() Corresponding authors: Dr Jun Yan, State Key Laboratory of Pharmaceutical Biotechnology and MOE Key Laboratory of Model Animals for Disease Study, Model Animal Research Center of Nanjing University, 12 Xuefu Road, Nanjing, Jiangsu, 210061, China. Tel: 86-025-58641535; Email: yanjunedu.cn; Dr Ming Lei, College of Life Sciences, Northwest A&F University, Taicheng Road, Yangling, Shaanxi, China. Tel: 86-029-87080160; Fax: 86-029-87080160; Email: leimingac.cn.
Corresponding authors: Dr Jun Yan, State Key Laboratory of Pharmaceutical Biotechnology and MOE Key Laboratory of Model Animals for Disease Study, Model Animal Research Center of Nanjing University, 12 Xuefu Road, Nanjing, Jiangsu, 210061, China. Tel: 86-025-58641535; Email: yanjunedu.cn; Dr Ming Lei, College of Life Sciences, Northwest A&F University, Taicheng Road, Yangling, Shaanxi, China. Tel: 86-029-87080160; Fax: 86-029-87080160; Email: leimingac.cn.