Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(7):2003-2014. doi:10.7150/thno.19404 This issue Cite
Research Paper
Drug-Bearing Supramolecular Filament Hydrogels as Anti-Inflammatory Agents
Zhipeng Chen1,2,4*, Lei Xing3*, Qin Fan1,2*, Andrew G. Cheetham4, Ran Lin4, Barbara Holt4, Liwen Chen4, Yanyu Xiao3 ![]() , Honggang Cui4,5,6
, Honggang Cui4,5,6 ![]()
1. Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization, Nanjing University of Chinese Medicine, Nanjing 210023, China;
2. Department of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210023, China;
3. State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, China;
4. Department of Chemical and Biomolecular Engineering and Institute for NanoBioTechnology, The Johns Hopkins University, Baltimore, MD 21218, USA;
5. Department of Oncology and Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA;
6. Center for Nanomedicine, The Wilmer Eye Institute, Johns Hopkins University School of Medicine, 400 North Broadway, Baltimore, MD 21231, USA.
* These authors contributed equally to this work
Received 2017-1-30; Accepted 2017-3-3; Published 2017-5-12
Abstract

We report here on the covalent conversion of the anti-inflammatory agent ketoprofen into self-assembling prodrugs that enable the effective purification of ketoprofen enantiomers, the improved selectivity and potency of ketoprofen, as well as the formation of one-component drug-bearing supramolecular hydrogels. We found that the ketoprofen hydrogelator could exhibit much-enhanced selectivity for cyclooxygenase 2 (COX-2) over COX-1, reduce the concentration of inflammatory cytokines (IL-1 and TNFα), and induce apoptosis in fibroblast-like synoviocytes while maintaining biocompatibility with healthy chondrocytes. In addition, these anti-inflammatory agent-containing hydrogels demonstrated the ability to retain the therapeutic within a joint cavity after intra-articular injection, exhibiting a slow, steady release into the plasma. We believe that upon further optimization these drug-based injectable supramolecular hydrogels could provide the basis for a local treatment strategy for rheumatoid arthritis and similar conditions.
Keywords: supramolecular hydrogel, self-assembly, drug delivery, peptide-drug conjugate, rheumatoid arthritis.
Introduction
The chemical modification of drugs to bestow additional beneficial properties is a widely explored bioconjugation strategy, offering the potential for improved targeting, greater bioavailability, enhanced efficacy, and reduced side-effects1-4. Peptide conjugation is one adopted approach that takes advantage of the multitude of peptides' biological roles, providing highly specific ligands that target particular cell receptors and sequences that can improve cellular uptake and avoid drug resistance mechanisms5-8. Furthermore, the conjugation of peptides that can lead to potential supramolecular assembly behavior has become a notable extension of this over the last decade9-17. In doing so, the drug becomes part of a larger nanostructure with its own distinct physicochemical properties, one that can be tailored toward a particular route of drug administration—local or systemic4.
For drugs that can act directly at disease sites, local administration is the preferred mode of delivery since it can offer several distinct advantages over systemic delivery18-22. These benefits include increased bioavailability, reduced systemic exposure that limits off-target effects and associated adverse events, and a lower total drug cost23-27. Anti-inflammatory drugs are well placed to benefit from local delivery strategies since their continued use can lead to significant side effects, including gastrointestinal and kidney problems28, 29. While their use may be an acceptable short-term risk for acute conditions, the long-term relief of localized chronic diseases would benefit greatly from alternative approaches. One method is the use of in situ hydrogels, either polymer or supramolecular-based, as localized drug depots30-38. The study of these systems has attracted increasing attention due to their abundant advantages in drug administration, including ease of preparation, biocompatibility, convenience and improved patient compliance, high regional drug concentration and low systemic toxicity34, 39-44. Furthermore, hydrogels can be designed with the ability to gel in response to physiological stimuli45-47 such as temperature48-50, pH43, 51-53, physical changes in biomaterials54, 55, enzymatic modification33, 56-59 or chemical reactions60-62.
Herein, we report on the design and preparation of a series of self-assembling prodrugs (SAPD) derived from anti-inflammatory agents that are capable of forming hydrogels under the physiological conditions. This concept of SAPD design has attracted much recent attention for medical applications due to their inherent biocompatibility and biodegradability, as well as the simplicity of the system design2-4, 57, 63-71. We, therefore, hypothesized that a hydrogel-forming SAPD that incorporates a non-steroidal anti-inflammatory drug (NSAID) into its structure could have great potential for the localized delivery of the NSAID (Figure 1a). The studied amphiphilic peptide-drug conjugates were synthesized through the conjugation of the hydrophobic NSAID (R/S)-ketoprofen (Ket) to a short peptide with overall hydrophilicity, separating the resulting epimers via HPLC. The chirality of the Ket molecule was found to have a substantial impact on the assembled filament morphology, and their efficacy as anti-inflammatory agents, showing excellent selectivity for the inhibition of cyclooxygenase-2 (COX-2) and inducing apoptosis in fibroblast-like synoviocytes with no associated chondrotoxicity. Furthermore, their potential utility as a localized NSAID depot for rheumatoid arthritis treatment was assessed through injection of the (S)-Ket-conjugate hydrogels into the articular cavity of a knee joint (rat), demonstrating the slow and sustained release of the drug through diffusion and degradation of the SAPD.
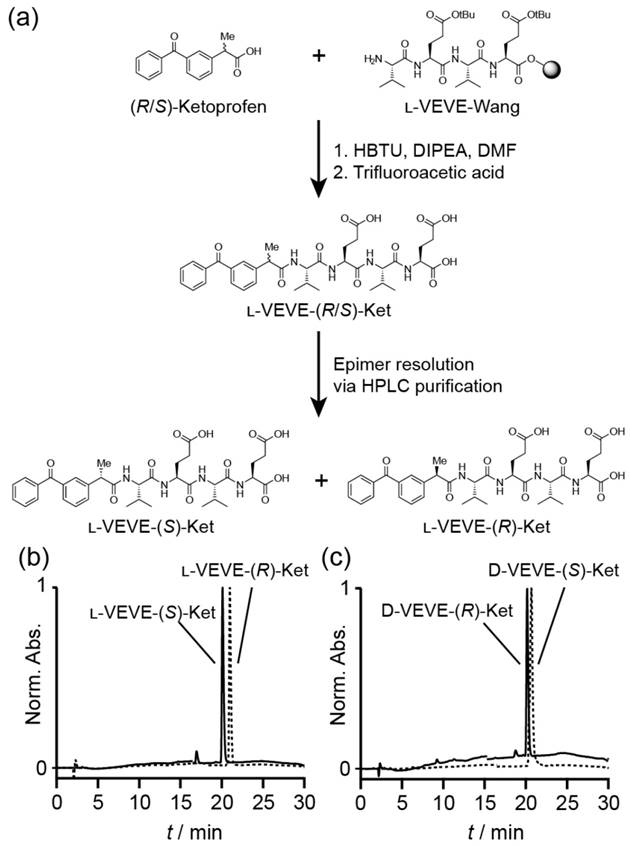
Synthesis of Ketoprofen-peptide conjugates. (a) Representative example of the synthetic procedure, showing the separation of the epimers l-VEVE-(S)-Ket and l-VEVE-(R)-Ket via HPLC purification. (b) and (c) HPLC chromatograms of the separated epimers of the l-VEVE and d-VEVE Ketoprofen conjugates, respectively.
Materials and Methods
Materials
Amino acids were purchased from Ampebiochem Co. Ltd (Chengdu, China) and Ketoprofen was obtained from energy-chemical Co. Ltd (Shang'hai, China). Uranyl acetate (>99.9%) was obtained from Xi'an Dingtian Chemical Co. Ltd (Xi'an, China). All other chemical reagents and solvents were used as received from commercial sources without further purification.
3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma (St. Louis, MO, USA). TNF-α detection kit was purchased from Nanjing built Biological Technology Co., Ltd. (Nanjing, China). BCG vaccine was purchased from Shanghai Institute of Biological Products Co., Ltd. (Shanghai, China). Trypsin, fetal calf serum and DMEM (Gibco®) were purchased from Thermo Fisher Scientific (USA). Annexin V-FITC/PI Cell Cycle and Apoptosis Analysis Kit was purchased from Nanjing Keygen Co., Ltd. (Nanjing, China). Deionized distilled water was used for the preparation of all solutions, and the other solvents were HPLC grade.
Cells and animals
Fibroblast-like synovial (FLS) cells were separated from the joint synovial tissues in rats with adjuvant-induced arthritis. New Zealand white rabbits were obtained from the Experimental Animal Center (Nanjing University of Chinese Medicine, China) and Sprague Dawley rats were purchased from the Animal Center of Nanjing University of Chinese Medicine (Nanjing, China). All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. The procedures were approved by the Ethical and Research Committee of the Nanjing University of Chinese Medicine.
Preparation of Ketoprofen-N-hydroxysuccinimide ester (Ket-NHS)
N-Hydroxysuccinimide (115 mg, 1.0 mmol) and Ketoprofen (230 mg, 1.0 mmol) were dissolved in chloroform (10 mL), and N,N′-diisopropyl-carbodiimide (DIC, 152 mg, 1.2 mmol) was added. After stirring for 2 h at room temperature, the mixture was filtered to remove precipitated urea, and the solvent evaporated under reduced pressure. Recrystallization from ethanol was performed to give the pure Ket-NHS ester.
Solid-phase peptide synthesis (SPPS) of Ket-conjugates
All conjugates were prepared by solid-phase peptide synthesis (SPPS) using a 2-chlorotrityl chloride resin (100-200 mesh and 0.3-0.8 mmol/g) and Fmoc-protected L- or D-amino acids with appropriate side chain protection—tert-butyl group for acids (e.g. Fmoc-L-Glu(tBu)-OH). The resin was initially bubbled with N2 in dry dichloromethane (DCM) for 20 min, after which the swollen resin was washed with dry N,N-dimethylformamide (DMF) (3 × 3 mL). Next, the first amino acid was loaded onto resin via its C-terminal carboxylate group by bubbling the resin in a DMF solution of Fmoc-protected amino acid (2 equiv.) and N,N-diisopropylethylamine (DIPEA) for 0.5 h. After washing with DMF (3 × 3 mL), the resin was bubbled with the blocking solution (16:3:1 of DCM/MeOH/DIPEA) for 0.5 h to deactivate the unreacted sites. The resins were then treated with 20 % piperidine (in DMF) for 0.5 h to remove the protecting group, followed by coupling of the next Fmoc-protected amino acid (2 equiv.) in the sequence to the free amino group on the resin. A mixture of DIPEA and O-benzotriazole-N,N,N',N'-tetramethyluronium hexafluorophosphate (HBTU) (2 equiv.) was used to effect coupling. These two steps were repeated to elongate the peptide chain until the final sequence was obtained. The resin was washed with DMF for 3-5 times after each step. Next, the terminal Fmoc group of the peptide was removed, and Ketoprofen attached through reaction with the Ket-NHS ester. Finally, the drug-peptide conjugate was cleaved with TFA (10 mL) for 2 h, followed by trituration into diethyl ether. The resulting crude products were purified by reverse phase HPLC, using a slow gradient (see below for conditions) and careful fractionation. Collected fractions were analyzed for epimeric purity, with appropriate fractions combined and lyophilized. The purity was confirmed by HPLC (Figure 1(b) and (c)) and the identity verified by LC-MS analysis (Figure S1).
Reverse phase HPLC protocols
Preparative HPLC was performed using a Varian ProStar Model 325 HPLC (Agilent Technologies, Santa Clara, CA) equipped with a fraction collector. Preparative separations utilized a Varian PLRP-S column (150 × 25 mm, 100 Å, 10 µm), with a mobile phase comprising 0.1% (v/v) aqueous NH4OH and acetonitrile (20:80 0.01 min, 45:55 25.00 min, 20:80 26.00 min, stop 30 min) at a flow rate of 25 mL/min.
Analytical HPLC was performed on a C18 analytical column (4.6 mm × 250 mm, 5 μm; Hanbon Sci. & Tech. Huanan, China) coupled with a C18 guard cartridge (4.6 mm × 10 mm, 5 μm; Hedera), maintained at 30 °C. The mobile phase comprised acetonitrile and 0.1 % aqueous TFA, and a gradient method was employed for the analysis (34:66 0.01 min, 41:59 25.00 min, 34:66 25.01 min, stop 30 min). The mobile phase was filtered through a 0.45 μm filter and delivered at a flow rate of 1.0 mL/min. The Ket absorbance was monitored at 254 nm.
General procedure for hydrogel preparation
To prepare hydrogels of L- or D-VEVE-(S)-Ket, 4 mg of the appropriate Ket-conjugate was dissolved in 0.5 mL of alkaline water (pH 9.0, room temperature) with the aid of sonication. Careful adjustment of the solution pH to 7.0 with 1 M HCl afforded a stable hydrogel within seconds.
Transmission electron microscopy
All produced nanostructures were visualized by transmission electron microscopy (TEM) using the following representative procedure. The sample to be imaged was deposited onto a copper grid (300 mesh) coated with a carbon membrane and then stained with 2 % uranyl acetate for 1 min. A piece of filter paper was used to blot most of the solution way, resulting in a thin liquid film. The grid was then allowed to dry before characterization. After air drying, the sample was observed using a TEM/H-7650 (Hitachi, Japan) operating at 80 kV. Images were captured with a CCD/MT280B camera.
COX inhibitor screening assay
COX inhibition by (S)-Ket and (S)-Ket-conjugate hydrogelators was determined using a COX Fluorescent Inhibitor Screening Assay Kit. Each compound was tested against COX-1 (ovine) and COX-2 (human recombinant) enzymes separately in 96-well black assay plates. COX activity was monitored through the action of its peroxidase component on the reaction between PGG2 and ADHP (10-acetyl-3,7-dihydroxyphenoxazine), which produces the highly fluorescent compound resorufin. The resorufin production was quantified using a multimode detector with an excitation wavelength in the 530-540 nm range and an emission range of 585-595 nm. All compounds were assayed in triplicate. After combining 10 μL of inhibitor, 10 μL of Heme solution, 10 μL of the fluorometric substrate, 10 μL of enzyme (either COX-1 or COX-2), and 150 μL of assay buffer, 10 μL of arachidonic acid solution was added to initiate the reaction. The plate was analyzed exactly two minutes after incubation at room temperature. In this experiment, the blank data without enzyme and inhibitors, and the control data without inhibitors were also measured.
In vitro FLS cell toxicity assay
The cytotoxicity of the (S)-Ket-conjugates towards FLS cells was quantified by an MTT assay using FLS cells obtained directly from arthritis-inflicted joints of a rat animal model. FLS cells were incubated at 37 °C in MEM media containing 10 % fetal calf serum under an atmosphere of 5 % CO2 in air. For the cell toxicity assay, FLS Cells were pre-cultured in 96-well cell culture plates at 1.0×105/well in 100 µl medium and grown overnight. The cells were then incubated with various concentrations of L-VEVE-(S)-Ket, D-VEVE-(S)-Ket and (S)-Ket for 48 h. After the incubation times, 20 µl of 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution in PBS (pH 7.4) was added to each well. After the cells had been incubated for another 4 h, the medium was withdrawn and 150 µl DMSO was added to each well to dissolve the MTT formazan crystals. The absorbance of each well was then measured at 490 nm in a microplate reader. Cell viability was calculated as a percentage of viable cells after treated with drugs compared with the untreated cells. The IC50 values of these hydrogelators were read from their activity curves, which were measured with six different concentrations of the hydrogelators.
Determination of IL-1 and TNF-α
FLS cells were pre-cultured in 96-well cell culture plates at 1×105/well in 100 µl in MEM media containing 10 % fetal calf serum at 37 °C under an atmosphere of 5 % CO2 in air and grown overnight. The cells were then incubated with L-VEVE-(S)-Ket, D-VEVE-(S)-Ket, and (S)-Ket (0.355 mg/ml) for 48 h. Cell supernatants were analyzed by ELISA to evaluate the levels of IL-1 and IFN-γ. Cells were acquired on a flow cytometer, and data were analyzed using the FlowJo software. Production of IFN-γ was analyzed in CD11cþ gated population.
Apoptosis detection
FLS cells were seeded at a density of 1 × 106 on each well of a 24-well plate and allowed to grow overnight. Cells were treated with L-VEVE-(S)-Ket, D-VEVE-(S)-Ket, and (S)-Ket at the concentration of 0.016-0.71 mg/ml for 48 h, followed by trypsinization. The cells were repeatedly washed and centrifuged at 1800 rpm for 3 min, and the supernatants were discarded. Cell density was determined. The cells were then re-suspended in annexin-binding buffer at ∼1×106 cells/mL, preparing a sufficient volume for 100 μL per assay. To 100 μL of cell suspension were added 5 μL of Alexa Fluor 488 annexin V and 5 μL of 100 μg/mL PI working solution, after which the solution was incubated for 15 min at room temperature. After the incubation period, 400 μL of Annexin binding buffer was added to each sample, followed by gentle mixing in an ice-bath. The samples were immediately analyzed using flow cytometry.
Chondrocyte isolation and culture
Normal articular cartilage harvested from two New Zealand White rabbits was washed three times with phosphate-buffered solution (PBS) containing 1 % penicillin and streptomycin. The cartilage slices were digested with 0.25 % trypsin at 37 °C for 30 min, thoroughly washed, and treated with 0.2 % type II collagenase for 4 hours in an incubator at 37 °C with 5 % CO2. The isolated chondrocytes were cultured to confluence in 75 cm3 flasks at 37 °C in a humidified atmosphere containing 5 % CO2 for 5 days. The culture medium was Dulbecco's Modified Eagle's Medium supplemented with 10 % fetal calf serum.
Chondrocyte biocompatibility test
Chondrocytes were seeded onto a 96-well plate (1 × 104 cells/well) in 100 μL of DMEM medium with 10 % FBS. After 18 hours of incubation at 37 °C and 5 % CO2, the chondrocytes were attached to the bottom of the 96-well plate. The medium was then replaced by another 100 μL of growth medium that contained serial diluents of Ket and hydrogelators at concentrations in the 0.016-0.71 mg/ml range and the chondrocytes were incubated at 37 °C and 5 % CO2 for an additional 72 h. Chondrocyte proliferation was determined by the addition of 20 μL of MTT (5 mg/mL) and incubating at 37 °C for another 4 h in the dark, the media were withdrawn and 150 µl DMSO was added to each well to dissolve the MTT formazan crystals. The absorbance of each well was then measured at 490 nm using a Microplate Reader (TECAN Austria GmbH). Chondrocytes without treatment were used as a control. All experiments were conducted in triplicate.
Determination of the in vitro release profile
200 μL of (S)-Ket-conjugate hydrogels and (S)-Ket-conjugate solution and (S)-Ket were prepared, all with concentrations of 4 mg/ml of (S)-Ket or (S)-Ket-conjugate. Solutions were dialyzed (Spectra/Por® membrane: MWCO = 800-1000) for 3 days against PBS buffer solution. With the addition of PBS buffer (200 μL, pH 7.4) onto the surface of hydrogels, the gels were incubated at 37 °C for 24 hours. The release solutions (200 μL) were taken and refreshed at a predetermined time, which were analyzed by analytical HPLC at 260 nm to determine the quantities of released Ket-conjugate hydrogelators.
In vivo hydrogel release experimental protocol
Eighteen male Sprague Dawley rats (230-240 g) were randomly divided into 3 groups, and then the rats were administered 200 μL of (S)-Ket solution, L-VEVE-(S)-Ket hydrogel and D-VEVE-(S)-Ket hydrogel via IA injection.
The dynamic behavior of the Ket was monitored at predetermined intervals (0.5, 1, 2, 3, 4, 6, 8 and 24 hours, and 2, 4, 8, 16, 24 and 32 days) post-injection by LC-MS/MS. The rats were anesthetized with ether. A heparinized capillary was then inserted into eyeground veins to obtain 0.3 mL blood in a plastic centrifuge tube. The plasma samples were collected after centrifugation at 4,000 rpm for 10 min and immediately stored at -20 °C until analysis. Next, 200 μl methanol containing an internal standard (chlorzoxazone, 50 ng/ml) was added to the 50 μl plasma samples to precipitate protein, centrifuged at 12,000 rpm for 10 min, and 20 μl of supernatant was assayed by LC-MS/MS.
LC-MS/MS quantitation of Ket plasma concentrations
Plasma samples collected from the rats were analyzed by quantitative LC-MS/MS. The LC-MS/MS equipment consisted of two LC-20AD pumps, an SIL-20ACHT auto-sampler, a CTO-20AC column oven (Shimadzu Corporation, Japan) and a 4000 Qtrap® mass spectrometer (AB SCIEX, USA), equipped with a Turbo ion source. Analyst software (Version 1.5.1) and Multi Quant software (Version 1.5.1) were used for data acquisition and analysis, respectively. LC separation was performed on a Agilent Extend C18 column (30 mm × 4.6 mm, 1.8 μm) using gradient elution with a mobile phase of methanol and 5 mM aqueous ammonium acetate (10/90 0.01 min, 10/90 0.5 min, 90/10 2 min, 90/10 4.5 min, 10/90 5.0 min, stop 6 min, v/v) at a flow rate of 1.0 mL/min. The column temperature was maintained at 40 °C and the auto-sampler temperature at 4 °C. The mass spectrometer was operated in the negative ionization mode. The injection volume was 10 μL. The ion spray voltage was set to -4500 V, and the ion source temperature was set at 450 °C. The curtain gas and collision gas were 35.0 psi and set at medium level, respectively. Ion source gas 1 and ion source gas 2 were both set at 40 psi. The declustering potential (DP), entrance potential (EP), collision energy (CE) and collision cell exit potential (CXP) for ketoprofen were -20, -10, -11 and -15 V, respectively. Detection of the ions was performed in the multiple reaction monitoring (MRM) mode, monitoring the transition of the m/z 253 precursor ion to the m/z 209 product ion for ketoprofen, and the m/z 709 precursor ion to the m/z 132 product ion for the Ket-conjugates. The standard, chlorzoxazone, was monitored using the m/z 285 precursor ion to the m/z 193 product ion transition.
Results and Discussion
Ket-conjugate synthesis
The NSAID-based hydrogelators utilized in this study were created through the connection of racemic (R/S)-Ketoprofen to the backbone of small tetrapeptides using standard solid-phase Fmoc peptide synthesis protocols72 (Figure 1a). To provide the necessary intermolecular interactions for one-dimensional nanostructure formation, the tetrapeptide sequence Val-Glu-Val-Glu (VEVE) was chosen as this possesses alternating hydrophobic-hydrophilic side-chain functionalities. This particular peptide sequence can, when conjugated to a larger hydrophobic moiety, form nanobelt structures and even undergo hydrogel formation73-76. L- or D-amino acids were utilized to fully probe the influence that the stereochemistry has on the self-assembly behavior and to evaluate any correlation between the structure and activity of NSAID hydrogelators77. By conjugating the racemic (R/S)-Ket to the enantiomerically pure L- or D-VEVE peptides, two sets of epimeric conjugates are formed: L-VEVE-(S)-Ket and L-VEVE-(R)-Ket (Figure 1a), and D-VEVE-(S)-Ket and D-VEVE-(R)-Ket. Surprisingly, after conjugation, both sets can be separated by HPLC purification, as indicated by a difference in retention time in their respective HPLC traces (Figure 1b-c). In contrast, the racemic mixtures of (R/S)-Ketoprofen have identical retention times (data not shown) and thus cannot be purified by the HPLC techniques. The separation of the racemic mixture after peptide conjugation is noteworthy, likely due to a difference in binding affinity to the HPLC column's stationary phase as a result of the peptide conjugation.
Self-assembly characterization
The self-assembly of all the Ket-conjugates was initiated by directly dissolving each molecule into an aqueous solution to reach a predetermined concentration. After aging for at least 24 h, transmission electron microscopy (TEM) imaging was conducted to examine the morphological characteristics of the respective Ket-conjugate assemblies. We found that assembly of L-VEVE-(S)-Ket afforded large, rigid, multi-strand nanoribbons with average widths of 49 ± 10 nm, with the constituent strands averaging 4.6 ± 0.5 nm in width (Figure 2a). These filamentous structures also possessed a tendency to align up to form parallel bundles. In contrast, the associated epimer L-VEVE-(R)-Ket gave a more filament-like morphology with average widths of 12 ± 1.7 nm (Figure 2b). A similar difference was observed for the D-peptide conjugates. D-VEVE-(R)-Ket, the enantiomer of L-VEVE-(S)-Ket, formed multi-strand intertwined nanoribbons (ribbon widths of 22 ± 5 nm and strand widths of 4.8 ± 0.9 nm) (Figure 2d). D-VEVE-(S)-Ket, the enantiomer of L-VEVE-(R)-Ket, formed the more filament-like structures with average widths of 11 ± 1.7 nm (Figure 2c).
Self-assembly of the ketoprofen-peptide conjugates. (a)-(d) Representative transmission electron micrographs of l-VEVE-(S)-Ket (a), l-VEVE-(R)-Ket (b), d-VEVE-(S)-Ket (c), and d-VEVE-(R)-Ket (d). All scale bars are 200 nm. The stereochemical relationships between the four conjugates are also indicated. (e) Newman projections of the four conjugates showing the Ketoprofen methyl alignment relative to the hydrophobic Val and hydrophilic Glu side chains. View shown is along the peptide axis from the Ketoprofen α-carbon, extending to the first Glu residue.
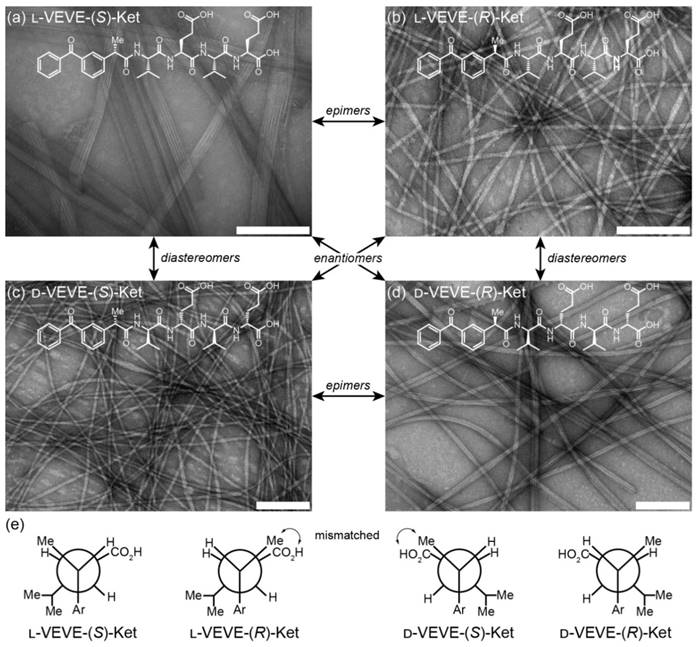
This difference in morphology—ribbons/bundles vs. individual filaments—may arise from the inherent chirality of the Ketoprofen molecule matching with the chirality of the chosen amino acids. The projection of the methyl group at the chiral carbon relative to the amino acid side chains likely has a substantial impact on the molecular assembly pathway and internal packing. For the enantiomeric L-VEVE-(S)- Ket and D-VEVE-(R)-Ket conjugates, the methyl group of Ket is projected into the same plane as the hydrophobic isopropyl groups of the Val side-chains and so would contribute to the hydrophobic interactions (Figure 2e). For the corresponding epimers L-VEVE-(R)-Ket and D-VEVE-(S)-Ket, however, the methyl group projects into the same plane as the hydrophilic carboxylic acid groups of the Glu side chains. This hydrophobic group in the predominantly hydrophilic plane may disrupt the packing that would prevent extensive lateral associations and thus give a narrower filament-like appearance.
Inhibition of COX enzymes
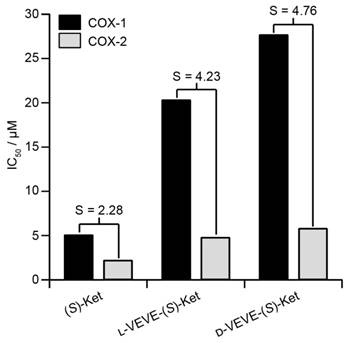
(S)-Ketoprofen is a commonly used drug for the management of arthritic conditions such as rheumatoid arthritis (RA), with its (R) form possessing little bioactivity. One mode of action for (S)-Ket is the inhibition of the cyclooxygenase enzymes COX-1 and COX-2. However, it is the inhibition of COX-1 that can lead to gastrointestinal ulceration, and so it is advantageous if the conjugate can show greater selectivity for inhibition of COX-2 over COX-1. We, therefore, performed in vitro inhibition assays for both COX-1 and COX-2 enzymes to evaluate the efficacies of the NSAID-containing SAPDs (Figure 3). The calculated IC50 values of L-VEVE-(S)-Ket and D-VEVE-(S)-Ket against COX-2 were 4.8 and 5.8 μM, respectively. Since the IC50 of (S)-Ket against COX-2 was found to be 2.2 μM, these IC50 values for the two conjugates are reasonable for the inhibition of COX-2, especially for the intended local delivery application. The IC50 values of L-VEVE(S)-Ket and D-VEVE-(S)-Ket against COX-1 enzyme were found to be 20.3 and 27.7 μM, respectively, both much higher than the IC50 value of 5.1 μM for (S)-Ket. However, the reduction in Ketoprofen efficacy against both COX-1 and COX-2 is compensated by the much-improved selectivities against COX-2. In both designs of employing natural and non-natural amino acids, L-VEVE-(S)-Ket and D-VEVE-(S)-Ket exhibited excellent selectivities of S = 4.2 and 4.8, respectively, toward COX-2, similar to those observed for other NSAID hydrogelators78-80. Undoubtedly, the attachment of the small L- or D-peptides to Ket greatly impedes its binding to COX-1, which should reduce any associated adverse gastrointestinal and renal effects.
Inhibition (IC50) of COX-1 and COX-2 by (S)-Ket, l-VEVE-(S)-Ket and d-VEVE-(S)-Ket. The selectivity, S, is defined as the ratio of the IC50 values towards COX-1 and COX-2.
Inhibitory activity against fibroblast-like-synoviocytes
In RA, fibroblast-like synoviocytes (FLS) play a significant role in the inflammation response and subsequent damage to the cartilage and bone of the joint 81, 82, and constitute a major component of a synovial pannus that forms within the joint. In this pannus, the FLS have transitioned to a more aggressive phenotype in response to the initial inflammatory conditions and no longer undergo apoptosis 83. These FLS cells produce pro-inflammatory cytokines, chemokines and proteases 84, that invade and destroy cartilage and bone 85. Inflammatory cytokines have been implicated as important mediators of joint destruction in RA, among which interleukin-1 (IL-1) and tumor necrosis factor α (TNFα) play central roles 86. Addressing the issue of FLS-mediated tissue damage in RA is, therefore, a viable strategy for the prevention or limitation of further joint degradation.
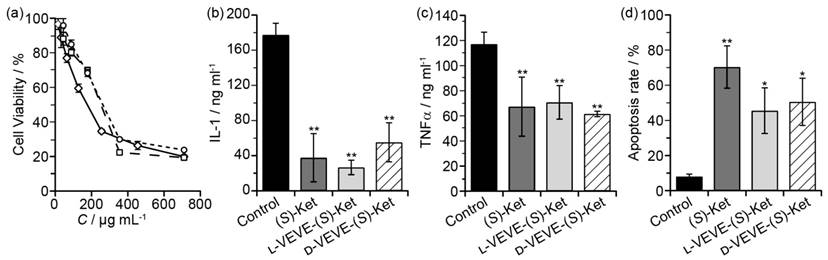
To evaluate if our (S)-Ket conjugates can affect the proliferation of FLS cells, we incubated FLS cells obtained from rabbits with arthritic joints with the conjugates and assessed their proliferation after 48 h. Surprisingly, we found little significant difference in the extent of FLS proliferation between those treated with L-VEVE-(S)-Ket or D-VEVE-(S)-Ket and those of the control group (Ket) (Figure 4a), with all three groups exhibiting IC50 values in the 0.1-0.3 mg/ml range. Additionally, compared with the untreated control group, the FLS cells produced trace amounts of IL-1 (Figure 4b) and a comparatively lower amount of TNFα (Figure 4c) after treatment with Ket, L-VEVE-(S)-Ket, or D-VEVE-(S)-Ket. Furthermore, an assay to determine the extent of apoptosis in the FLS cells upon treatment with Ket, L-VEVE-(S)-Ket, and D-VEVE-Ket (all at 0.4 mg/ml Ket), showed that all three induced a greater degree of cell death than untreated groups (Figure 4d). A notable aspect is that the concentration of (S)-Ket required to induce significant apoptosis is in the millimolar range, a concentration that would not only be difficult to attain within the articular cavity through systemic delivery but would also result in severe side-effects due to the dosage required to do so. By converting the drug into a hydrogelator, it thus becomes possible to deliver the drug at a concentration that would be able to reduce the FLS population within the synovial pannus and limit further damage, without the risk of significant side-effects.
In vitro studies of FLS toxicity. (a) Cytotoxicity assay of FLS cells treated with (S)-Ket (™, IC50 = 159 μg/mL), l-VEVE-(S)-Ket (£, IC50 = 223 μg/mL), and d-VEVE-(S)-Ket (¯, IC50 = 278 μg/mL) for 48 h. The effect on IL-1 (b) and TNFα (c) production by FLS cells treated with (S)-Ket, l-VEVE-(S)-Ket, and d-VEVE-Ket. (d) Induction of apoptosis on FLS cells treated with Ket, l-VEVE-(S)-Ket, and d-VEVE-(S)-Ket. All experiments were performed in triplicate (n = 3). Asterisks (*) denote statistically significant differences calculated by one-way ANOVA test, *p < 0.05, **p < 0.01.
Due to potential chondrotoxicity, the two Ket-conjugates were further investigated for biocompatibility through incubation with primary rabbit chondrocytes for 72 h at 37 °C (Figure S2). Encouragingly, no toxic effect could be seen, with the chondrocytes maintaining an almost 100% viability in the presence of either conjugate, even at the higher concentration of 0.71 mg/mL. This result indicates that they are chondrocyte compatible and would be suitable as locally administrable therapeutics.
In vitro and in vivo drug release
We next evaluated the potential of using the conjugates as a local delivery platform. Gelation tests of L-VEVE-(S)-Ket and D-VEVE-(S)-Ket clearly suggested that the two conjugates were indeed suitable hydrogelators, forming stable, slightly opaque hydrogels in water at a concentration of 5 mM or higher (Figure 5a). The cloudiness of the hydrogels could stem from the bundling and alignment of the filaments which scatter light in a different manner87. The hydrogels were then assessed for their ability to act as a long-term drug depot. Accordingly, we incubated 200 μL of both L- and D-VEVE-(S)-Ket-conjugate hydrogels (4 mg/ml) at 37 °C for 40 d with 200 μL of PBS buffer solution (pH 7.4) in separate dialysis bags, which were refreshed and monitored at predetermined time points (Figure 5b). (S)-Ket solution and L-VEVE-(S)-Ket-conjugate solution (assembled but not gelled) were used as controls. As expected, the hydrogels exhibited the slowest release kinetics with 100% of the (S)-Ket-conjugate released only after 30 d had elapsed. Dynamic light scattering of the released hydrogel solution exhibits little difference with that of the non-gelled (S)-Ket-conjugate solution (data not shown), suggesting the conjugates are released as a mixture of nanostructures and small aggregates. In sharp contrast, drug release from the (S)-Ket solution and L-VEVE-(S)-Ket-conjugate solution releases within a few hours.
Study of the (S)-Ketoprofen-containing conjugates. (a) Images of the self-supporting hydrogels formed by l-VEVE-(S)-Ket and d-VEVE-(S)-Ket (both 5 mM in water). (b) In vitro release profiles of Ket-based formulations: (S)-Ket solution (£), d-VEVE-(S)-Ket prodrug (¸), l-VEVE-(S)-Ket hydrogel (™) and d-VEVE-(S)-Ket hydrogel (¯) (Data points are slightly offset in the x-axis for clarity). (c) Illustration of the proposed intra-articular delivery of an NSAID-containing hydrogel to an RA-afflicted joint. The hydrogel forms from the entanglement of self-assembled NSAID-peptide conjugate nanostructures. (d) In vivo pharmacokinetic study of intra-articularly administered (S)-Ket solution (£), l-VEVE-(S)-Ket hydrogel (™) and d-VEVE-(S)-Ket hydrogel (¯).
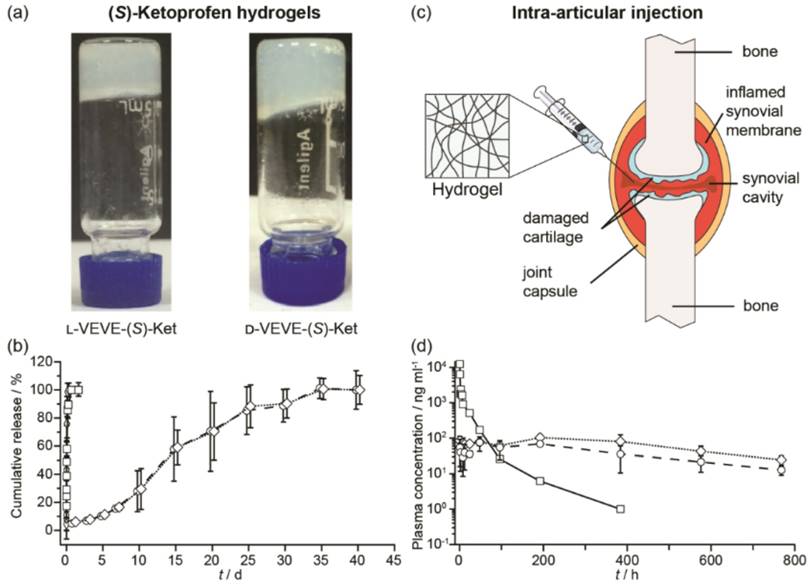
To evaluate the potential of the hydrogels as a local drug depot in an in vivo environment, six male rats were administered 200 μL of L-VEVE-(S)-Ket and D-VEVE-(S)-Ket hydrogels (containing 400 μg Ket-conjugate) via intra-articular injection (Figure 5c). Supramolecular filament hydrogels of similar types are known to be shear thinning and injectable and can be delivered using a needle mounted on a syringe. Following intra-articular injection with a needle, the dynamic behavior of the hydrogels was monitored at predetermined intervals post-injection using LC-MS/MS analysis to quantify the Ketoprofen plasma concentration. It can be clearly seen that both hydrogels slowly release the (S)-Ket-conjugate over an extended period, maintaining a near constant plasma concentration close to 100 ng/mL with no burst release characteristics (Figure 5d). In contrast, (S)-Ket solution exhibited rapid release with the classic burst release profile (Cmax = 10 μg/mL). These results suggest that these Ket-conjugate hydrogels are capable of retaining the bulk of the administered therapeutic within the arterial cavity, thus maintaining a high local concentration. That Ket-containing species are released indicates that the hydrogel is undergoing slow decay within the cavity, either through diffusional loss or biological activity (e.g. proteolysis), releasing the Ket-conjugate into the surrounding environment at a steady rate. Although many experimental parameters remain to be tested and optimized, these release experiments clearly demonstrate the clinical potential of using these supramolecular filament hydrogels as local anti-inflammatory agents to treat rheumatoid arthritis.
Conclusions
In conclusion, a novel supramolecular anti-inflammatory hydrogel made entirely of a peptide-NSAID conjugate was designed for the potential treatment of arthritic diseases via intra-articular administration, constituting a new platform for the delivery of therapeutic agents by biocompatible, target specific and long-acting hydrogels. Additionally, the incorporation of L- or D-amino acids make the Ket-conjugates highly selective for inhibition of COX-2 over COX-1, thereby minimizing adverse effects associated with inhibition of the latter. In vitro studies revealed that the Ket-conjugate supramolecular hydrogels could benefit RA-afflicted joints by suppressing the levels of IL-1 and TNF-α through induction of apoptosis in FLS, and thus may protect RA damaged joints from further degradation. Furthermore, an in vivo release study demonstrated that this platform is capable of a steady and sustained drug release profile that would favor long term retention at the injection site. Overall, the presented Ket-peptide hydrogelators represent a highly promising platform upon which to base a new local therapy for rheumatoid arthritis, and will be explored through further optimization and in vivo experiments to assess their efficacy against an RA model. The ability to deliver a high concentration of Ketoprofen may not only aid with pain relief and minimize gastrointestinal side-effects but may also prevent further progression of the disease through their inhibitory action against FLS cell proliferation. This approach could also provide a useful strategy for reducing the adverse drug reactions of other therapeutic agents that would benefit from local administration.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Science Foundation (DMR 1255281), and by the National Natural Science Foundation of China (No: 81473147, 81274100, 21301191), 2011 Collaborative Innovation Center of Chinese Medicinal Resources Industrialization (No: ZDXM-2-3), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Six talent peaks project in Jiangsu Province, Outstanding talent training program of Nanjing University of Chinese Medicine of Pharmacy.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12128-33
2. Cheetham AG, Zhang P, Lin Y-A, Lock LL, Cui H. Supramolecular nanostructures formed by anticancer drug assembly. Journal of the American Chemical Society. 2013;135:2907-10
3. Ma W, Cheetham AG, Cui HG. Building nanostructures with drugs. Nano Today. 2016;11:13-30
4. Su H, Koo JM, Cui HG. One-component nanomedicine. Journal of Controlled Release. 2015;219:383-95
5. Geng LL, Wang ZH, Jia XQ, Han QJ, Xiang ZC, Li D. et al. HER2 Targeting Peptides Screening and Applications in Tumor Imaging and Drug Delivery. Theranostics. 2016;6:1261-73
6. Park SH, Zheng JH, Nguyen VH, Jiang SN, Kim DY, Szardenings M. et al. RGD Peptide Cell-Surface Display Enhances the Targeting and Therapeutic Efficacy of Attenuated Salmonella-mediated Cancer Therapy. Theranostics. 2016;6:1672-82
7. Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Advanced Drug Delivery Reviews. 2016 http://dx.doi.org/10.1016/j.addr.2016.06.015
8. Chen Z, Zhang P, Cheetham AG, Moon JH, Moxley JW, Lin Y-a. et al. Controlled release of free doxorubicin from peptide-drug conjugates by drug loading. Journal of Controlled Release. 2014;191:123-30
9. Shu JY, Panganiban B, Xu T. Peptide-polymer conjugates: from fundamental science to application. Annual review of physical chemistry. 2013;64:631-57
10. Fleming S, Ulijn RV. Design of nanostructures based on aromatic peptide amphiphiles. Chem Soc Rev. 2014;43:8150-77
11. Hamley IW. Lipopeptides: from self-assembly to bioactivity. Chem Commun. 2015;51:8574-83
12. Du X, Zhou J, Guvench O, Sangiorgi FO, Li X, Zhou N. et al. Supramolecular assemblies of a conjugate of nucleobase, amino acids, and saccharide act as agonists for proliferation of embryonic stem cells and development of zygotes. Bioconjugate chemistry. 2014;25:1031-5
13. Hartgerink JD, Beniash E, Stupp SI. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science. 2001;294:1684-8
14. Stupp SI, Zha RH, Palmer LC, Cui HG, Bitton R. Self-assembly of biomolecular soft matter. Faraday Discussions. 2013;166:9-30
15. Mlinar LB, Chung EJ, Wonder EA, Tirrell M. Active targeting of early and mid-stage atherosclerotic plaques using self-assembled peptide amphiphile micelles. Biomaterials. 2014;35:8678-86
16. Trent A, Marullo R, Lin B, Black M, Tirrell M. Structural properties of soluble peptide amphiphile micelles. Soft Matter. 2011;7:9572-82
17. Li Y, Wang F, Cui H. Peptide-Based Supramolecular Hydrogels for Delivery of Biologics. Bioengineering & Translational Medicine. 2016;1:306-322
18. Wolinsky JB, Colson YL, Grinstaff MW. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafers. Journal of Controlled Release. 2012;159:14-26
19. Xi HL, Cun DM, Xiang RW, Guan YL, Zhang YX, Li YR. et al. Intra-articular drug delivery from an optimized topical patch containing teriflunomide and lornoxicam for rheumatoid arthritis treatment: Does the topical patch really enhance a local treatment? Journal of Controlled Release. 2013;169:73-81
20. Lin R, Cui H. Supramolecular nanostructures as drug carriers. Current Opinion in Chemical Engineering. 2015;7:75-83
21. Sun JEP, Stewart B, Litan A, Lee SJ, Schneider JP, Langhans SA. et al. Sustained release of active chemotherapeutics from injectable-solid beta-hairpin peptide hydrogel. Biomaterials Science. 2016;4:839-48
22. Tatman PD, Muhonen EG, Wickers ST, Gee AO, Kim ES, Kim DH. Self-assembling peptides for stem cell and tissue engineering. Biomaterials Science. 2016;4:543-54
23. Thakkar H, Sharma RK, Mishra A, Chuttani K, Murthy R. Efficacy of chitosan microspheres for controlled intra-articular delivery of celecoxib in inflamed joints. Journal of Pharmacy and Pharmacology. 2004;56:1091-9
24. Bakker AC, ARNTZ J. Prevention of murine collagen-induced arthritis in the knee and ipsilateral paw by local expression of human interleukin-l receptor antagonist protein in the knee. Arthritis Rheum. 1997;40:893-900
25. Larsen C, Østergaard J, Larsen SW, Jensen H, Jacobsen S, Lindegaard C. et al. Intra-articular depot formulation principles: Role in the management of postoperative pain and arthritic disorders. Journal of Pharmaceutical Sciences. 2008;97:4622-54
26. Burt HM, Tsallas A, Gilchrist S, Liang LS. Intra-articular drug delivery systems: overcoming the shortcomings of joint disease therapy. Expert Opin Drug Deliv. 2009;6:17-26
27. Betre H, Liu W, Zalutsky MR, Chilkoti A, Kraus VB, Setton LA. A thermally responsive biopolymer for intra-articular drug delivery. Journal of Controlled Release. 2006;115:175-82
28. Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A. et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis - The CLASS study: A randomized controlled trial. Jama-Journal of the American Medical Association. 2000;284:1247-55
29. Wallace JL. Nonsteroidal anti-inflammatory drugs and gastroenteropathy: The second hundred years. Gastroenterology. 1997;112:1000-16
30. Zhao F, Ma ML, Xu B. Molecular hydrogels of therapeutic agents. Chem Soc Rev. 2009;38:883-91
31. Chen CY, Ke CJ, Yen KC, Hsieh HC, Sun JS, Lin FH. 3D Porous Calcium-Alginate Scaffolds Cell Culture System Improved Human Osteoblast Cell Clusters for Cell Therapy. Theranostics. 2015;5:643-55
32. Tondera C, Hauser S, Kruger-Genge A, Jung F, Neffe AT, Lendlein A. et al. Gelatin-based Hydrogel Degradation and Tissue Interaction in vivo: Insights from Multimodal Preclinical Imaging in Immunocompetent Nude Mice. Theranostics. 2016;6:2114-28
33. Zelzer M, Todd SJ, Hirst AR, McDonald TO, Ulijn RV. Enzyme responsive materials: design strategies and future developments. Biomaterials Science. 2013;1:11-39
34. Du X, Zhou J, Shi J, Xu B. Supramolecular hydrogelators and hydrogels: from soft matter to molecular biomaterials. Chem Rev. 2015;115:13165-307
35. Giano MC, Pochan DJ, Schneider JP. Controlled biodegradation of Self-assembling beta-hairpin Peptide hydrogels by proteolysis with matrix metalloproteinase-13. Biomaterials. 2011;32:6471-7
36. Boekhoven J, Stupp SI. 25th anniversary article: supramolecular materials for regenerative medicine. Advanced materials. 2014;26:1642-59
37. Tansik G, Kilic E, Beter M, Demiralp B, Sendur GK, Can N. et al. A glycosaminoglycan mimetic peptide nanofiber gel as an osteoinductive scaffold. Biomaterials Science. 2016;4:1328-39
38. Yu ZQ, Cai Z, Chen QL, Liu MH, Ye L, Ren JY. et al. Engineering beta-sheet peptide assemblies for biomedical applications. Biomaterials Science. 2016;4:365-74
39. Hoffman AS. Hydrogels for biomedical applications. Advanced Drug Delivery Reviews. 2012;64:18-23
40. Lee KY, Mooney DJ. Hydrogels for tissue engineering. Chemical Reviews. 2001;101:1869-80
41. Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Advanced materials. 2006;18:1345-60
42. Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Reversible hydrogels from self-assembling artificial proteins. Science. 1998;281:389-92
43. Dong L-C, Hoffman AS. A novel approach for preparation of pH-sensitive hydrogels for enteric drug delivery. Journal of Controlled Release. 1991;15:141-52
44. Haraguchi K, Takehisa T. Nanocomposite hydrogels: a unique organic-inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties. Advanced materials. 2002;14:1120-4
45. Draper ER, Adams DJ. Chapter 12 Responsive Materials by the Self-assembly of Low Molecular Weight Gelators. Chemoresponsive Materials: Stimulation by Chemical and Biological Signals: The Royal Society of Chemistry. 2015:332-63
46. Hu Y, Lin R, Patel K, Cheetham AG, Kan CY, Cui HG. Spatiotemporal control of the creation and immolation of peptide assemblies. Coordination Chemistry Reviews. 2016;320:2-17
47. Frederix P, Scott GG, Abul-Haija YM, Kalafatovic D, Pappas CG, Javid N. et al. Exploring the sequence space for (tri-) peptide self-assembly to design and discover. Nature Chemistry. 2015;7:30-7
48. Qiu Y, Park K. Environment-sensitive hydrogels for drug delivery. Advanced Drug Delivery Reviews. 2012;64:49-60
49. Yoshida R, Uchida K, Kaneko Y, Sakai K, Kikuchi A, Sakurai Y. et al. Comb-type grafted hydrogels with rapid deswelling response to temperature changes. Nature. 1995;374:240-2
50. Rodríguez-Llansola F, Hermida-Merino D, Nieto-Ortega B, Ramírez FJ, Navarrete JTL, Casado J. et al. Self-Assembly Studies of a Chiral Bisurea-Based Superhydrogelator. Chem Eur J. 2012;18:14725-31
51. Matson JB, Newcomb CJ, Bitton R, Stupp SI. Nanostructure-templated control of drug release from peptide amphiphile nanofiber gels. Soft Matter. 2012;8:3586-95
52. Matson JB, Stupp SI. Drug release from hydrazone-containing peptide amphiphiles. Chem Commun. 2011;47:7962-4
53. Schmaljohann D. Thermo-and pH-responsive polymers in drug delivery. Advanced Drug Delivery Reviews. 2006;58:1655-70
54. Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science. 2009;324:59-63
55. Foo CTWP, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. Two-component protein-engineered physical hydrogels for cell encapsulation. Proceedings of the National Academy of Sciences. 2009;106:22067-72
56. Kalafatovic D, Nobis M, Son JY, Anderson KI, Ulijn RV. MMP-9 triggered self-assembly of doxorubicin nanofiber depots halts tumor growth. Biomaterials. 2016;98:192-202
57. Gao Y, Yang Z, Kuang Y, Ma M-L, Li J, Zhao F. et al. Enzyme-instructed self-assembly of peptide derivatives to form nanofibers and hydrogels. Pept Sci. 2010;94:19-31
58. Yang Z, Gu H, Fu D, Gao P, Lam JK, Xu B. Enzymatic formation of supramolecular hydrogels. Adv Mater. 2004;16:1440-4
59. Zhou J, Du X, Yamagata N, Xu B. Enzyme-instructed self-assembly of small D-peptides as a multiple-step process for selectively killing cancer cells. J Am Chem Soc. 2016;138:3813-23
60. Serizawa T, Nanameki K, Yamamoto K, Akashi M. Thermoresponsive ultrathin hydrogels prepared by sequential chemical reactions. Macromolecules. 2002;35:2184-9
61. Hennink W, Van Nostrum CF. Novel crosslinking methods to design hydrogels. Advanced Drug Delivery Reviews. 2012;64:223-36
62. Sargeant TD, Aparicio C, Goldberger JE, Cui H, Stupp SI. Mineralization of peptide amphiphile nanofibers and its effect on the differentiation of human mesenchymal stem cells. Acta biomaterialia. 2012;8:2456-65
63. Cheetham AG, Ou YC, Zhang PC, Cui HG. Linker-determined drug release mechanism of free camptothecin from self-assembling drug amphiphiles. Chemical Communications. 2014;50:6039-42
64. Su H, Zhang P, Cheetham AG, Koo JM, Lin R, Masood A. et al. Supramolecular Crafting of Self-Assembling Camptothecin Prodrugs with Enhanced Efficacy against Primary Cancer Cells. Theranostics. 2016;6:1065-74
65. MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nature Materials. 2009;8:993-9
66. Keyes-Baig C, Duhamel J, Fung S-Y, Bezaire J, Chen P. Self-assembling peptide as a potential carrier of hydrophobic compounds. Journal of the American Chemical Society. 2004;126:7522-32
67. Zhang P, Cheetham AG, Lin Y-A, Cui H. Self-assembled Tat nanofibers as effective drug carrier and transporter. Acs Nano. 2013;7:5965-77
68. Carter JM, Qian Y, Foster JC, Matson JB. Peptide-based hydrogen sulphide-releasing gels. Chemical Communications. 2015;51:13131-4
69. Lin R, Cheetham AG, Zhang P, Lin Y-a, Cui H. Supramolecular filaments containing a fixed 41% paclitaxel loading. Chemical Communications. 2013;49:4968-70
70. Lin Y-A, Cheetham AG, Zhang P, Ou Y-C, Li Y, Liu G. et al. Multiwalled nanotubes formed by catanionic mixtures of drug amphiphiles. ACS nano. 2014;8:12690-700
71. Ma W, Su H, Cheetham AG, Zhang W, Wang Y, Kan Q. et al. Synergistic antitumor activity of a self-assembling camptothecin and capecitabine hybrid prodrug for improved efficacy. Journal of Controlled Release. 2017 http://dx.doi.org/10.1016/j.jconrel.2017.01.015
72. Ottinger EA, Shekels LL, Bernlohr DA, Barany G. Synthesis of phosphotyrosine-containing peptides and their use as substrates for protein tyrosine phosphatases. Biochemistry. 1993;32:4354-61
73. Moyer TJ, Cui HG, Stupp SI. Tuning Nanostructure Dimensions with Supramolecular Twisting. Journal of Physical Chemistry B. 2013;117:4604-10
74. Cui HG, Cheetham AG, Pashuck ET, Stupp SI. Amino Acid Sequence in Constitutionally Isomeric Tetrapeptide Amphiphiles Dictates Architecture of One-Dimensional Nanostructures. Journal of the American Chemical Society. 2014;136:12461-8
75. Cui H, Muraoka T, Cheetham AG, Stupp SI. Self-assembly of giant peptide nanobelts. Nano Letters. 2009;9:945-51
76. Hu Y, Lin R, Zhang P, Fern J, Cheetham AG, Patel K. et al. Electrostatic-Driven Lamination and Untwisting of β-Sheet Assemblies. Acs Nano. 2016;10:880-8
77. Shi J, Du X, Yuan D, Zhou J, Zhou N, Huang Y. et al. D-amino acids modulate the cellular response of enzymatic-instructed supramolecular nanofibers of small peptides. Biomacromolecules. 2014;15:3559-68
78. Li JY, Kuang Y, Gao Y, Du XW, Shi JF, Xu B. D-Amino Acids Boost the Selectivity and Confer Supramolecular Hydrogels of a Nonsteroidal Anti-Inflammatory Drug (NSAID). Journal of the American Chemical Society. 2013;135:542-5
79. Li JY, Kuang Y, Shi JF, Gao YA, Zhou J, Xu B. The conjugation of nonsteroidal anti-inflammatory drugs (NSAID) to small peptides for generating multifunctional supramolecular nanofibers/hydrogels. Beilstein Journal of Organic Chemistry. 2013;9:908-17
80. Li XM, Li JY, Gao YA, Kuang Y, Shi JF, Xu B. Molecular Nanofibers of Olsalazine Form Supramolecular Hydrogels for Reductive Release of an Anti-inflammatory Agent. Journal of the American Chemical Society. 2010;132:17707-9
81. Mor A, Abramson SB, Pillinger MH. The fibroblast-like synovial cell in rheumatoid arthritis: a key player in inflammation and joint destruction. Clinical Immunology. 2005;115:118-28
82. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunological Reviews. 2010;233:233-55
83. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nature Reviews Rheumatology. 2013;9:24-33
84. Dayer J-M, Demczuk S. Cytokines and other mediators in rheumatoid arthritis. Springer Seminars in Immunopathology. 1984
85. Müller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE. et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. The American journal of pathology. 1996;149:1607
86. Arend WP, Dayer JM. Inhibition of the production and effects of interleukins-1 and tumor necrosis factor α in rheumatoid arthritis. Arthritis & Rheumatism. 1995;38:151-60
87. Cui HG, Pashuck ET, Velichko YS, Weigand SJ, Cheetham AG, Newcomb CJ. et al. Spontaneous and X-ray-Triggered Crystallization at Long Range in Self-Assembling Filament Networks. Science. 2010;327:555-9
Author contact
![]() Corresponding authors: cpuyanyuxiaocom, hcui6edu
Corresponding authors: cpuyanyuxiaocom, hcui6edu