Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2017; 7(7):2067-2077. doi:10.7150/thno.19427 This issue Cite
Review
Current Strategies and Challenges for Purification of Cardiomyocytes Derived from Human Pluripotent Stem Cells
Kiwon Ban1, Seongho Bae2, Young-sup Yoon2, 3 ![]()
1. Department of Biomedical Sciences, City University of Hong Kong, Hong Kong;
2. Department of Medicine, Division of Cardiology, Emory University, Atlanta, Georgia, USA;
3. Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Korea.
Received 2017-1-31; Accepted 2017-3-22; Published 2017-5-17
Abstract

Cardiomyocytes (CMs) derived from human pluripotent stem cells (hPSCs) are considered a most promising option for cell-based cardiac repair. Hence, various protocols have been developed for differentiating hPSCs into CMs. Despite remarkable improvement in the generation of hPSC-CMs, without purification, these protocols can only generate mixed cell populations including undifferentiated hPSCs or non-CMs, which may elicit adverse outcomes. Therefore, one of the major challenges for clinical use of hPSC-CMs is the development of efficient isolation techniques that allow enrichment of hPSC-CMs. In this review, we will discuss diverse strategies that have been developed to enrich hPSC-CMs. We will describe major characteristics of individual hPSC-CM purification methods including their scientific principles, advantages, limitations, and needed improvements. Development of a comprehensive system which can enrich hPSC-CMs will be ultimately useful for cell therapy for diseased hearts, human cardiac disease modeling, cardiac toxicity screening, and cardiac tissue engineering.
Keywords: Cardiomyocytes, hPSCs
Introduction
Heart failure is the leading cause of death worldwide [1]. Approximately 6 million people suffer from heart failure in the United States every year [1]. Despite this high incidence, existing surgical and pharmacological interventions for treating heart failure are limited because these approaches only delay the progression of the disease; they cannot directly repair the damaged hearts [2]. In the case of large myocardial infarction (MI), patients progress to heart failure and die within short time from the onset of symptoms [3].
The adult human heart has minimal regenerative capacity, because during mammalian development, the proliferative capacity of cardiomyocytes (CMs) progressively diminishes and becomes terminally differentiated shortly after birth [4].Therefore, once CMs are damaged, they are rarely restored [5]. When MI occurs, the infarcted area is easily converted to non-contractile scar tissue due to loss of CMs and replacement by fibrosis [6]. Development of a fibroblastic scar initiates a series of events that lead to adverse remodeling, hypertrophy, and eventual heart failure [2, 3, 7].
While heart transplantation is considered the most viable option for treating advanced heart failure, the number of available donor hearts is always less than needed [6]. Therefore, more realistic therapeutic options have been required [2]. Accordingly, over the past two decades, cell-based cardiac repair has been intensively pursued [2, 7]. Several different cell types have been tested and varied outcomes were obtained. Indeed, the key factor for successful cell-based cardiac repair is to find the optimal cell type that can restore normal heart function. Naturally, CMs have been considered the best cell type to repair a damaged heart [8]. In fact, many scientists hypothesized that implanted CMs would survive in damaged hearts and form junctions with host CMs and synchronously contract with the host myocardium [9]. In fact, animal studies with primary fetal or neonatal CMs demonstrated that transplanted CMs could survive in infarcted hearts [9-11]. These primary CMs reduced scar size, increased wall thickness, and improved cardiac contractile function with signs of electro-mechanical integration [9-11]. These studies strongly suggest that CMs can be a promising source to repair the heart. However, the short supply and ethical concerns disallow using primary human CMs. In a patient with ischemic cardiomyopathy, about 40-50% of the CMs are lost in 40 to 60 grams of heart tissue [7]. Even if we seek to regenerate a fairly small portion of the damaged myocardium, a large number of human primary CMs would be required, which is impossible.
Accordingly, CMs differentiated from human pluripotent stem cells (hPSCs) including both embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) have emerged as a promising option for candidate CMs for cell therapy [12, 13]. hPSCs have many advantages as a source for CMs. First, hPSCs have obvious cardiomyogenic potential. hPSC derived-CMs (hPSC-CMs) possess a clear cardiac phenotype, displaying spontaneous contraction, cardiac excitation-contraction (EC) coupling, and expression of cardiac transcription factors, cardiac ion channels, and cardiac structural proteins [14, 15]. Second, undifferentiated hPSCs and their differentiated cardiac progeny display significant proliferation capacity, allowing generation of a large number of hPSC-CMs. Lastly, many pre-clinical studies demonstrated that implantation of hPSC-CMs can repair injured hearts and improve cardiac function [16-19]. Histologically, implanted hPSC-CMs are engrafted, aligned and coupled with the host CMs in a synchronized manner [16-19].
In the last two decades, various protocols for differentiating hPSCs into CMs have been developed to improve the efficiency, purity and clinical compatibility [20] [18]. The reported differentiation methods include, but are not limited to: differentiation via embryoid body (EB) formation [20], co-culture with END-2 cells [18], and monolayer culture [15, 21, 22]. The EB-mediated CM differentiation protocol is one of the most widely employed methods due to its simple procedure and low cost. However, it often becomes labor-intensive to produce scalable EBs for further differentiation, which makes it difficult for therapeutic applications. EB-mediated differentiation also produces inconsistent results, showing beating CMs from 5% to 70% of EBs. Recently, researchers developed monolayer methods to complement the problems of EB-based methods [15, 21, 22]. In one representative protocol, hPSCs are cultured at a high density (up to 80%) and treated with a high concentration of Activin A (100 ng/ml) for 1 day and BMP4 (10 ng/ml) for 4 days followed by continuous culture on regular RPMI media with B27 [15]. This protocol induces spontaneous beating at approximately 12 days and produces approximately 40% CMs after 3 weeks. These hPSC-CMs can be further cultured in RPMI-B27 medium for another 2-3 weeks without significant cell damage [15]. However, these protocols use media with proprietary formulations, which complicates clinical application. As shown, most monolayer-based methods employ B27, which is a complex mix of 21 components. Some of the components of B27, including bovine serum albumin (BSA), are animal-derived products, and the effects of B27 components on differentiation, maturation or subtype specification processes are poorly defined. In 2014, Burridge and his colleagues developed an advanced protocol that is defined, cost-effective and efficient [22]. By subtracting one component from B27 at a time and proceeding with cardiac differentiation, the researchers reported that BSA and L-ascorbic acid 2-phosphate are essential components in cardiac differentiation. Subsequently, by replacing BSA with rice-derived recombinant human albumin, the chemically defined medium with 3 components (CDM3) was produced. The application of a GSK-inhibitor, CHIR99021, for the first 2 days followed by 2 days of the Wnt-inhibitor Wnt-59 to cells is an optimal culture condition in CDM3 resulting in similar levels of live-cell yields and CM differentiation [22].
Despite remarkable improvement in the generation of hPSC-CMs, obtaining pure populations of hPSC-CMs still remains challenging. Currently available methods can only generate a mixture of cells which include not only CMs but other cell types. This is one of the most critical barriers for applications of hPSC-CMs in regenerative therapy, drug discovery, and disease investigation. For Instance, cardiac transplantation of non-pure hPSC-CMs mixed with undifferentiated hPSCs or other cell types may produce tumors or unwanted cell types in hearts [23-28]. Accordingly, a pure or enriched population of hPSC-CMs would be required, particularly for cardiac cell therapy. Enriched hPSC-CMs would also be more beneficial for myocardial repair due to improved electric and mechanical properties [29]. A pure, homogeneous population of hPSC-CMs would pose less arrhythmic risk and have enhanced contractile performance, and would be more useful in disease modeling as they better reflect native CM physiology. Finally, purified hPSC-CMs would better serve for testing drug efficacy and toxicity. Therefore, many researchers have tried to develop methods to purify CMs from cardiomyogenically differentiated hPSCs.
There are three important topics that are not addressed in this review. First is the beneficial role of other cell types such as endothelial cells and fibroblasts in the integration, survival, and function of CMs [30-32]. We did not discuss this issue because it would need a separate review due to the volume of material. While the roles of such cells are important, the value of having purified hPSC-CMs is not diminished. Although cell mixtures or tissue engineered products can be used, unless purified CMs are employed, they would form tumors or other cells/tissues when implanted in vivo. Our point here is that even if cardiomyocytes are mixed with non-CMs, all cells should be clearly defined and purified as well. If the mixture is made in a non-purified or non-defined manner (for example, an unsophisticated top-down approach), there would be undefined cells that are neither CMs, ECs, nor fibroblasts and these unidentified cells will make aberrant tissues or tumors. Second, we did not deal with maturation of hPSC-CMs because of its broad scope and depth [33, 34]. Third is direct reprogramming or conversion of somatic cells into CMs. There has been another advancement in the generation of CMs by directly reprogramming or converting somatic cells into CM-like cells by introducing a combination of cardiac transcription factors (TFs) or muscle-specific microRNAs (miRNAs) both in vitro and in vivo [35-41]. These cells are referred to as induced CMs (iCMs) or cardiac-like myocytes (iCLMs). While this is an important advancement, we did not cover this topic either due to its size. Accordingly, this review will focus on the various strategies for purifying or enriching hPSC-CMs reported to date (Figure 1).
Currently available strategies for enriching cardiomyocytes derived from human pluripotent stem cells.
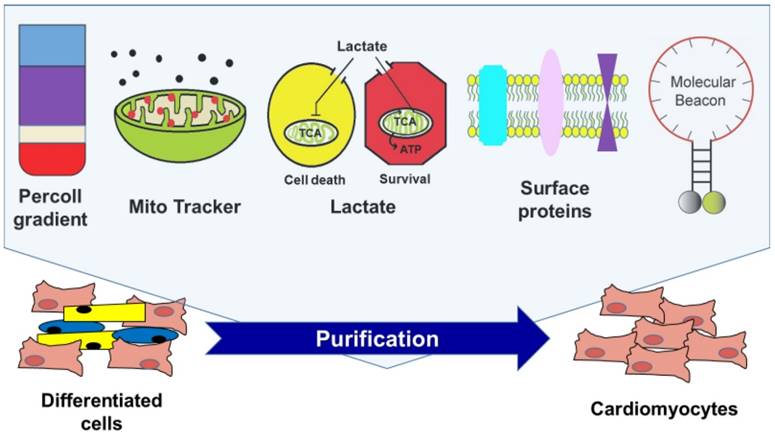
Strategies for Enriching Cardiomyocytes Derived From hPSCs
Manual enrichment
Early on, researchers isolated hPSC-CMs manually under microscopy by mechanically separating out the beating areas from myogenically differentiating hPSC cultures [18, 20, 42]. This method usually generates 5-70% hPSC-CMs. Although generally crude, it can enrich even higher percentages of CMs with further culture. This manual isolation method has the advantage of being easy, but while it can be useful for small-scale research, it is very labor intensive and not scalable, precluding large scale research or clinical application.
Physical enrichment
Xu et el. reported that hPSC-CMs, due to their physical and structural properties, can be enriched by Percoll density gradient centrifugation [43]. Percoll was first formulated by Pertoft et al [44] and it was originally developed for the isolation of cells, organelles, or viruses by density centrifugation. The Percoll-based method has several advantages. The procedure for Percoll-based separation is very simple and easy, it is inexpensive, and its low viscosity allows more rapid sedimentation and lower centrifugal forces compared to a sucrose density gradient. Lastly, it can be prepared and kept for a long time in an isotonic solution to maintain osmolarity. Although Percoll separation has resulted in major improvements in hPSC-CM isolation procedures, it has clear limitations with regard to purity and scalability. Previous studies found that Percoll separation is only able to enrich 40 -70% of hPSC-CMs. It is also not compatible with large-scale enrichment of hPSC-CMs.
Genetic modification based CM Isolation
Another traditional method for purifying hPSC-CMs is based on the expression of a drug resistant gene or a fluorescent reporter gene such as eGFP or DsRed, which is driven by a cardiac specific promoter in genetically modified hPSC lines [45, 46]. Here, enrichment of hPSC-CMs can be achieved by either drug treatment to eliminate cells that do not express the drug resistant gene or with FACS to isolate fluorescent cells [47, 48].
Briefly, enrichment of PSC-CMs by genetically based selection was first reported by Klug et al [49]. The authors generated murine ES cell lines via permanent gene transfection of the aminoglycoside phosphotransferase gene driven by the βMHC (MYH7) promoter. With this approach, highly purified murine ESC-CMs up to 99% were achieved. Next, several studies reported the use of various CM-specific promoters to enrich ESC-CMs such as αMhc (Myh6), Myh7, Ncx (Sodium Calcium exchanger) and Mlc2v (Myl2) [46, 50, 51]. In the case of hESCs, αMHC/EGFP hESCs were generated by permanent transfection of the EGFP-tagged αMHC promoter [52]. Similarly, an NKX2.5/eGFP hESC line was generated to enrich GFP positive CMs [53]. However, since αMHC and NKX2.5 are expressed in general CMs, the resulting CMs contain a mixture of the three subtypes of CMs, nodal-, atrial-, and ventricular-like CMs. To enrich only ventricular-like CMs, Huber et al. generated MLC2v/GFP ESCs to be able to isolate MLC2v/GFP positive ventricular-like cells by FACS [52] [54-57]. In addition, the cGATA6 gene was used to purify nodal-like hESC-CMs [58]. Future studies should focus on testing new types of cardiac specific promoters and devising advanced selection procedures to improve this strategy.
While fluorescence-based cell sorting is more widely used, the drug selection method may be a better approach to enrich high purity of hPSC-CMs during differentiation/culture as it does not require FACS. The advantage is its capability for high-purity cell enrichment due to specific gene-based cell sorting. These highly pure cells can allow more precise mechanistic studies and disease modeling. Despite its many advantages, the primary weakness of genetic selection is genetic manipulation, which disallows its use for therapeutic application. Insertion of reporter genes into the host genome requires viral or nonviral transfection/transduction methods, which can induce mutagenesis and tumor formation [50, 59-61].
Surface Protein-Based Enrichment
Practically, antibody-based cell enrichment is the best method for cell purification to date. When cell type-specific surface proteins or “marker” proteins are known, one can tag cells with antibodies against the proteins and sort the target cells by FACS or magnetic-activated cell sorting (MACS). The main advantage is its specificity and sensitivity, and its utility is well demonstrated in research and even in clinical therapy with hematopoietic cells [62]. Another advantage is that multiple surface markers can be used at the same time to isolate target cells when one marker is not sufficient. However, no studies have reported surface markers that are specific for CMs, even after many years. Recently, though, several researchers demonstrated that certain proteins can be useful for isolating hPSC-CMs.
In earlier studies, KDR (FLK1 or VEGFR2) and PDGFR-α were used to isolate cardiac progenitor cells [63]. However, since these markers are also expressed on hematopoietic cells, endothelial cells, and smooth muscle cells, they could not enrich only hPSC-CMs. Next, two independent studies reported two surface proteins, SIRPA [64] and VCAM-1 [65], which it was claimed could specifically identify hPSC-CMs. Dubois et al. screened a panel of 370 known antibodies against CMs differentiated from hESCs and identified SIRPA as a specific surface protein expressed on hPSC-CMs [64]. FACS with anti-SIRPA antibody enabled the purification of CMs and cardiac precursors from cardiomyogenically differentiating hPSC cultures, producing cardiac troponin T (TNNT2, also known as cTNT)-positive cells, which are generally considered hPSC-CMs, with up to 98% purity. In addition, a study performed by Elliot and colleagues identified another cell surface marker, VCAM1 [53]. In this study, the authors used NKX2.5/eGFP hESCs to generate hPSC-CMs, allowing the cells to be sorted by their NKX2.5 expression. NKX2.5 is a well-known cardiac transcription factor and a specific marker for cardiac progenitor cells [66, 67]. To identify CM-specific surface proteins, the authors performed expression profiling analyses and found that expression levels of both VCAM1 and SIRPA were significantly upregulated in NKX2.5/eGFP+ cells. Flow cytometry results showed that both proteins were expressed on the cell surface of NKX2.5/eGFP+ cells. Differentiation day 14 NKX2.5/eGFP+ cells expressed VCAM1 (71 %) or SIRPA (85%) or both VCAM1 and SIRPA (37%). When the FACS-sorted SIRPA-VCAM1-, SIRPA+ or SIRPA+VCAM1+ cells were further cultured, only SIRPA+ or SIRPA+VCAM1+ cells showed NKX2.5/eGFP+ contracting portion. Of note, NKX2.5/eGFP and SIRPA positive cells showed higher expression of smooth muscle cell and endothelial cell markers indicating that cells sorted solely based on SIRPA expression may not be of pure cardiac lineage. Hence, the authors concluded that a more purified population of hPSC-CMs could be isolated by sorting with both cell surface markers. Despite significant improvements, it appears that these surface markers are not exclusively specific for CMs as these antibodies also mark other cell types including smooth muscle cells and endothelial cells. Furthermore, they are also known to be expressed in the brain and the lung, which raises concerns whether these surface proteins can be used as sole markers for the purification of hPSC-CMs compatible for clinical applications.
More recently, Protze et al. reported successful differentiation and enrichment of sinoatrial node-like pacemaker cells (SANLPCs) from differentiating hPSCs by using cell surface markers and an NKX2-5-reporter hPSC line [68]. They found that BMP signaling specified cardiac mesoderm toward the SANLPC fate and retinoic acid signaling enhanced the pacemaker phenotype. Furthermore, they showed that later inhibition of the FGF pathway, the TFGβ pathway, and the WNT pathway shifted cell fate into SANLPCs, and final cell sorting for SIRPA-positive and CD90-negative cells resulted in enrichment of SANLPCs up to ~83%. These SIRPA+CD90- cells showed the molecular, cellular and electrophysiological characteristics of SANLPCs [68]. While this study makes important progress in enriching SANLPCs by modulating signaling pathways, no specific surface markers for SANLPCs were identified and the yield was still short of what is usually expected for cells purified via FACS.
MITO tracker-based enrichment
Hattori et al. developed a highly efficient non-genetic method for purifying hPSC-derived CMs, in which they employed a red fluorescent dye, tetramethylrhodamine methyl ester perchlorate (TMRM), that can label active mitochondria. Since CMs contain a large number of mitochondria, CMs from mice and marmosets (monkey) could be strongly stained with TMRM [69]. They further found that primary CMs from several different types of animals and CMs derived from both mESCs and hESCs were successfully purified by FACS up to 99% based on the TMRM signals. In addition to its efficiency for CM enrichment, TMRM did not affect cell viability and disappeared completely from the cells within 24 hrs. Importantly, injected hPSC-CMs purified in this way did not form teratoma in the heart tissues. However, since TMRM only functions in CMs with high mitochondrial density, this method cannot purify entire populations of hPSC-CMs [64]. While originally TMRM was claimed to be able to isolate mature hPSC-CMs, mounting evidence indicates that hPSC-CMs are similar to immature human CMs at embryonic or fetal stages. Therefore, both the exact phenotype of the cells isolated by TMRM and its utility are rather questionable [33, 34]. Two subsequent studies demonstrated that TMRM failed to accurately distinguish hPSC-CMs due to the insufficient amounts of mitochondria [64].
Lactate-based hPSC-CM enrichment
Employing the unique metabolic properties of CMs, Tohyama et al. developed an elegant purification method to enrich PSC-CMs [70]. This approach is based on the remarkable biochemical differences in lactate and glucose metabolism between CMs and non-CMs, including undifferentiated cells. Mammalian cells use glucose as their main energy source [71]. However, CMs are capable of energy production from different sources such as lactate or fatty acids [71]. A comparative transcriptome analysis was performed to detect metabolism-related genes which have different expression patterns between newborn mouse CMs and undifferentiated mouse ESCs. These results showed that CMs expressed genes encoding tricarboxylic acid (TCA) cycle enzymes more than genes related to lipid and amino acid synthesis and the pentose phosphate cycle compared to undifferentiated ESCs. To further prove this observation, they compared the metabolites of these pathways using fluxome analysis between CMs and other cell types such as ESCs, hepatocytes and skeletal muscle cells, and found that CMs have lower levels of metabolites related to lipid and amino acid synthesis and pentose phosphate. Subsequently, authors cultured newborn rat CMs and mouse ESCs in media with lactate, forcing the cells to use the TCA cycle instead of glucose, and they observed that CMs were the only cells to survive this condition for even 96 hrs. They further found that when PSC derivatives were cultured in lactate-supplemented and glucose-depleted culture medium, only CMs survived. Their yield of CM population was up to 99% and no tumors were formed when these CMs were transplanted into hearts. This lactate-based method has many advantages: its simple procedures, ease of application, no use of FACS for cell sorting, and relatively low cost. More recently, this method was applied to large-scale CM aggregates to ensure scalability. As a follow-up study, the same group recently reported a more refined lactate-based enrichment method which further depletes glutamine in addition to glucose [72]. The authors found that glutamine is essential for the survival of hPSCs since hPSCs are highly dependent on glycolysis for energy production rather than oxidative phosphorylation. The use of glutamine- and glucose-depleted lactate-containing media resulted in more highly purified hPSC-CMs with less than 0.001% of residual PSCs [72]. One concern of this lactate-based enrichment method is the health of the purified hPSC-CMs, because physiological and functional characteristics of hPSC-CMs cultured in glucose- and glutamine-depleted media for a long time may have functional impairment since CMs with mature mitochondria were not able to survive without glucose and glutamine, although they were able to use lactate to synthesize pyruvate and glutamate [72]. In addition, this lactate-based strategy can only be applied to hPSC- CMs, but not other hPSC derived cells such as neuron or β-cells.
mRNA-based method (Molecular Beacon)
Our group also recently reported a new method to isolate hPSC-CMs by directly labelling cardiac specific mRNAs using nano-sized probes called molecular beacons (MBs) [29, 73, 74]. Designed to detect intracellular mRNA targets, MBs are dual-labeled antisense oligonucleotide (ODN) nano-scale probes with a DNA or RNA backbone, a Cy3 fluorophore at the 5' end, and a Black Hole quencher 2 (BHQ2) at the 3' end [75, 76]. They form a stem-loop (hairpin) structure in the absence of a complementary target, quenching the fluorescence of the reporter. Hybridization with the target mRNA opens the hairpin and physically separates the reporter from the quencher, allowing a fluorescence signal to be emitted upon excitation. The MB-based method can be applied to the purification of any cell type that has known specific gene(s) [77].
In one study [29], we designed five MBs targeting unique sites in TNNT2 or MYH6/7 mRNA in both mouse and human. To determine the most efficient transfection method to deliver MBs into living cells, various methods were tested and nucleofection was found to have the highest efficiency. Next, we tested the sensitivity and specificity of MBs using an immortalized mouse CM cell line, HL-1, and other cell types. Finally, we narrowed it down to one MB, MHC-MB, which showed >98% sensitivity and > 95% specificity. This MHC-MB was applied to cardiomyogenically differentiated mouse and human PSCs and FACS sorting was performed. The resultant MHC-MB-positive cells expressed cardiac proteins at ~97% when measured by flow cytometry. These sorted cells also demonstrated spontaneous contraction and all the molecular and electrophysiological signatures of human CMs. Importantly, when these purified CMs were injected into the mouse infarcted myocardium, they were well integrated into the myocardium without forming any tumors, and they improved cardiac function.
In a subsequent study [74], we refined a method to enrich ventricular CMs from differentiating PSCs (vCMs) by targeting a transcription factor which is not robustly expressed in cells. Since vCMs are the main source for generating cardiac contractile forces and the most frequently damaged in the heart, there has been great demand to develop a method that can obtain a pure population of vCMs for cardiac repair. Despite this critical unmet need, no studies have demonstrated the feasibility of isolating ventricular CMs without permanently altering their genome. Accordingly, we first designed MBs targeting the Iroquois homeobox protein 4 (Irx4) mRNA, a vCM specific transcription factor [78, 79]. After testing sensitivity and specificity, one IRX4-MB was selected and applied to myogenically differentiated mPSCs. The FACS-sorted IRX4-MB-positive cells exhibited vCM-like action potentials in more than 98% of cells when measured by several electrophysiological analyses including patch clamp and Ca2+ transient analyses. Furthermore, these cells maintained spontaneous contraction and expression of vCM-specific proteins.
The MB-based cell purification method is theoretically the most broadly applicable technology among the purification methods because it can isolate any target cells expressing any specific gene. Thus, the MB-based sorting technique can be applied to the isolation of other cell types such as neural-lineage cells or islet cells, which are critical elements in regenerative medicine but do not have specific surface proteins identified to date. In addition, theoretically, this technology may have the highest efficiency when MBs are designed to have the maximum sensitivity and specificity for the cells of interest, but not others. These characteristics are particularly important for cell therapy. Despite these advantages, the delivery method of MB into the cells needs to be improved. So far, nucleofection is the best delivery method, but caused some cell damage with < 70% cell viability. Thus, development of a safer delivery method will enable wider application of MB-based cell enrichment.
MicroRNA-based enrichment
Recently, Miki and colleagues reported a novel method for purifying cells of interest based on endogenous miRNA activity [80]. Miki et al. employed several synthetic mRNA switches (= miRNA switch), which consist of synthetic mRNA sequences that include a recognition sequence for miRNA and an open reading frame that codes a desired gene, such as a regulatory protein that emits fluorescence or promotes cell death. If the miRNA recognition sequence binds to miRNA expressed in the desired cells, the expression of the regulatory protein is suppressed, thus distinguishing the cell type from others that do not contain the miRNA and express the protein.
Briefly, the authors first identified 109 miRNA candidates differentially expressed in distinct stages of hPSC-CMs (differentiation day 8 and 20). Next, they found that 14 miRNAs were co-expressed in hPSC-CMs at day 8 and day 20 and generated synthetic mRNAs that recognize these 14 miRNA, called miRNA switches. Among those miRNA switches, miR-1-, miR-208a-, and miR-499a-5p-switches successfully enriched hPSC-CMs with purity of sorted cells up to 96% determined by TNNT2 intracellular flow cytometry. Particularly, hPSC-CMs enriched by the miR-1-switch showed substantially higher expression of several cardiac specific genes/proteins and lower expression of non-CM genes/proteins compared with control cells. Patch clamp confirmed that these purified hPSC-CMs possessed both ventricular-like and atrial-like action potentials.
One of the major advantages of this technology is its wider applicability to other cell types. miRNA switches have the flexibility to design the open reading frame in the mRNA sequence such that any desired transgene can be incorporated into the miRNA switches to regulate the cell phenotype based on miRNA activity. The authors tested this possibility by incorporating BIM sequence, an apoptosis inducer, into the cardiac specific miR-1- and miR-208a switches and tested whether they could selectively induce apoptosis in non-CMs. They found that miR-1- and miR-208a-Bim-switches successfully enriched cTNT-positive hPSC-CMs without cell sorting. Enriched hPSC-CMs by 208a-Bim-switch were injected into the hearts of mice with acute MI and they engrafted, survived, expressed both cTNT and CX43, and formed gap junctions with the host myocardium. No teratoma was detected. In addition, other miRNA switches such as miR-126-, miR-122-5p-, and miR-375-switches targeting endothelial cells, hepatocytes, and β-cells, respectively, successfully enriched these cell types differentiated from hPSCs. However, identification of specific miRNAs expressed only in the specific cell type of interest and verification of their specificity in target cells will be key issues for continuing to use this miRNA-based cell enrichment method.
Microfluidic systems
Recent advances in biomedical engineering have contributed to developing systems that can isolate target cells using physicochemical properties of the cells. Microfluidic systems have been intensively applied for cell separation due to recent improvements in miniaturizing a cell culture system [81-83]. These advances made possible the design of automated microfluidic devices with cellular microenvironments and controlled fluid flows that save time and cost in experiments. Thus, there have been an increasing number of studies seeking to apply the microfluidic system for cell separation. Among the first, Singh et al. tested the possibility of using a microfluidic system for the separation of hPSC [84] by preparative detachment of hPSCs from differentiating cultures based on differences in the adhesion properties of different cell types. Distinct streams of buffer that generated varying levels of shear stress further allowed selective enrichment of hPSC colonies from mixed populations of adherent non-hPSCs, achieving up to 95% purity. Of note, this strategy produced hPSC survival rates almost two times higher than FACS, reaching 80%.
Subsequently, for hPSC-CMs purification, Xin et al. developed a microfluidic system with integrated ridge-like flow derivations and fishnet-like microcolumns for the enrichment of hiPSC-CMs [85]. This device is composed of a 250 mm-long microfluidic channel, which has two integrated parallel microcolumns with surfaces functionalized with anti-human TRA-1 antibody for undifferentiated hiPSC trapping. Aided by the ridge-like surface patterns on the upper wall of the channel, micro-streams are generated so that the cell suspension of mixed undifferentiated hiPSCs and hiPSC-CMs are forced to cross the functionalized fishnet-like microcolumns, resulting in trapping of undifferentiated hiPSCs due to the interaction between the hiPSCs and the columns, and the untrapped hiPSC-CMs are eventually separated. By modulating flow and coating with anti-human TRA-1 antibody, they were able to enrich CMs to more than 80% purity with 70% viability. While this study demonstrated that a microfluidic device could be used for purifying hPSC-CMs, it was not realistic because the authors used a mixture of only undifferentiated hiPSCs and hiPSC-CMs. In real cardiomyogenically differentiated hiPSCs, undifferentiated hiPSCs are rare and many intermediate stage cells or other cell types are present, so the idea that this simple device can select only hiPSC-CMs from a complex mixture is uncertain.
Overall, the advantages of microfluidic system based cell isolation include fast speed, improved cell viability and low cost owing to the automated microfluidic devices that can control cellular microenvironments and fluid flows [86-88]. However, microfluidic-based cell purification methods have limitations in terms of low purity and scalability [89-92]. In fact, there have been only a few studies demonstrating the feasibility that microfluidic device-based cell separation could achieve higher than 80% purity of target cells. Furthermore, currently available microfluidic devices allow only separation of a small number of cells (< 1011). To employ microfluidic devices for large-scale cell production, we need to develop a next generation of microfluidic devices that can achieve a throughput greater than 1011 sorted cells per hour with > 95% purity.
Conclusion
Having available a large quantity of a homogeneous population of cells of interest is an important factor in advancing biomedical research and clinical medicine, and is especially true for hPSC-CMs. While remarkable progress has been made in the methods for differentiating hPSCs into CMs, technologies to enrich hPSC-CMs, particularly those which are clinically applicable, have been emerging only over the last few years. Contamination with other cell types and even the heterogeneous nature of hPSC-CMs significantly hinder their use for several future applications such as cardiac drug toxicology screening, human cardiac disease modeling, and cell-based cardiac repair. For instance, cardiac drug-screening assays require pure populations of hPSC-CMs, so that the observed signals can be attributed to effects on human CMs. Studies of human cardiac diseases can also be more adequately interpreted with purified populations of patient derived hiPSC-CMs. Clinical applications with hPSC-CMs will need to be free of other PSC derivatives to minimize the risk of teratoma formation and other adverse outcomes.
Summary of representative methods for hPSC-CM purification
| Purification methods | Purity (%) | Yield or Viability (%) | Tumorigenicity | Scalability | Clinical applicability | Application to other type of cells |
|---|---|---|---|---|---|---|
| Manual enrichment | 5 - 70% | > 70% | Not reported | Difficult | No | No |
| Physical enrichment by percoll gradient | 40 - 70% | < 70% | No tumor detected | Difficult | Yes | No |
| Genetic modification- based method | > 95% | ~ 70% | No tumor detected | Yes | No | Yes |
| Surface protein-based method | > 95% | ~ 70% | No tumor detected | Yes | Yes | Yes |
| MITO tracker-based enrichment | Up to 99% | ~ 70% | No tumor detected | Yes | Yes | No |
| Lactate-based enrichment | Up to 99% | > 80% | No tumor detected | Yes | Yes | No |
| mRNA-based method (Molecular Beacon) | Up to 97% | < 70% | No tumor detected | Yes | Yes | Yes |
| MicroRNA-based method | Up to 96% | ~ 90% | No tumor detected | Yes | Yes | Yes |
| Microfluidic systems | < 80% | > 70% | Not tested | Difficult | Yes | Yes |
Schematic pictures of microfluidic device for enriching hiPSC-CMs. (A) The part of the device designed for trapping undifferentiated hiPSCs. (B) (Left) Illustration of the overall microfluidic device assembled with peristaltic pump, cell suspension reservoirs, and a serpentine channel. (Right) Magnified image showing a channel combining microcolumns and ridge-like flow derivation structures. Modified from Li et al. On chip purification of hiPSC-derived cardiomyocytes using a fishnet-like microstructure. Biofabrication. 2016 Sep 8;8(3): 035017

Therefore, development of reproducible, effective, non-mutagenic, scalable, and economical technologies for purifying hPSC-CMs, independent of hPSC lines or differentiation protocols, is a fundamental requirement for the success of hPSC-CM applications. Fortunately, new technologies based on the biological specificity of CMs such as MITO-tracker, molecular beacons, lactate-enriched-glucose depleted-media, and microRNA switches have been developed. In addition, technologies based on engineering principles have recently yielded a promising platform using microfluidic technology. While due to the short history of this field, more studies are needed to verify the utility of these technologies, the growing attention toward this research is a welcome move.
Another important question raised recently is how to non-genetically purify chamber-specific subtypes of CMs such as ventricular-like, atrial-like and nodal-like hPSC-CMs. So far, only a few studies have addressed this potential with human PSCs. We also showed that a molecular beacon-based strategy could enrich ventricular CMs differentiated from PSCs [74]. Another study demonstrated generation of SA-node like pacemaker cells by using a stepwise treatment of various morphogens and small molecules followed by cell sorting with several sub-specific surface markers. However, the yield of both studies was relatively low (<85%). Given the growing clinical importance of chamber-specific CMs, the strategies for purifying specific subtypes of CM that are independent of hPSC lines or differentiation protocols should be continuously developed. A recently reported cell surface capture-technology [93, 94] may facilitate identification of chamber specific CM proteins that will be useful for target CM isolation.
In summary, technological advances in the purification of hPSC-CMs have opened an avenue for realistic application of hPSC-CMs. Although initial success was achieved for purification of CMs from differentiating hPSC cultures, questions such as scalability, clinical compatibility, and cellular damage remain to be answered and isolation of human subtype CMs has yet to be demonstrated. While there are other challenges such as maturity, in vivo integration, and arrhythmogenecity, this development of purification technology represents major progress in the field and will provide unprecedented opportunities for cell-based therapy, disease modeling, drug discovery, and precision medicine. Furthermore, the availability of chamber-specific CMs with single cell analyses will facilitate more sophisticated investigation of human cardiac development and cardiac pathophysiology.
Acknowledgements
This work was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIP) (No 2015M3A9C6031514), the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI15C2782, HI16C2211) and grants from NHLBI (R01HL127759, R01HL129511), NIDDK (DP3-DK108245). This work was also supported by a CityU Start-up Grant (No 7200492), a CityU Research Project (No 9610355), and a Georgia Immuno Engineering Consortium through funding from Georgia Institute of Technology, Emory University, and the Georgia Research Alliance.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M. et al. Heart disease and stroke statistics—2016 update. Circulation. 2016;133:e38-e360
2. Ptaszek LM, Mansour M, Ruskin JN, Chien KR. Towards regenerative therapy for cardiac disease. Lancet. 2012;379:933-42
3. Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007-18
4. Pasumarthi KBS, Field LJ. Cardiomyocyte cell cycle regulation. Circ Res. 2002;90:1044-54
5. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S. et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98-102
6. Passier R, van Laake LW, Mummery CL. Stem-cell-based therapy and lessons from the heart. Nature. 2008;453:322-9
7. Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326-35
8. Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD. et al. Growth of engineered human myocardium with mechanical loading and vascular coculture / Novelty and significance. Circ Res. 2011;109:47-59
9. Reinecke H, Zhang M, Bartosek T, Murry CE. Survival, integration, and differentiation of cardiomyocyte grafts: A study in normal and injured rat hearts. Circulation. 1999;100:193-202
10. Leor J, Patterson M, Quinones MJ, Kedes LH, Kloner RA. Transplantation of fetal myocardial tissue into the infarcted myocardium of rat. A potential method for repair of infarcted myocardium? Circulation. 1996;94:II332-6
11. Li R-K, Mickle DAG, Weisel RD, Zhang J, Mohabeer MK. In vivo survival and function of transplanted rat cardiomyocytes. Circ Res. 1996;78:283-8
12. Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J. et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012;489:322-5
13. Shigeru M, Satsuki F, Yukiko I, Takuji K, Noriko Mochizuki O, Shigeo M. et al. Building a new treatment for heart failure-transplantation of induced pluripotent stem cell-derived cells into the heart. Curr Gene Ther. 2016;16:5-13
14. Mignone JL, Kreutziger KL, Paige SL, Murry CE. Cardiogenesis from human embryonic stem cells - Mechanisms and applications. Circulation J. 2010;74:2517-26
15. Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK. et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015-24
16. Yuasa S, Itabashi Y, Koshimizu U, Tanaka T, Sugimura K, Kinoshita M. et al. Transient inhibition of BMP signaling by Noggin induces cardiomyocyte differentiation of mouse embryonic stem cells. Nat Biotechnol. 2005;23:607-11
17. Nemir M, Croquelois A, Pedrazzini T, Radtke F. Induction of cardiogenesis in embryonic stem cells via downregulation of Notch1 signaling. Circ Res. 2006;98:1471-8
18. Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R. et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733-40
19. Passier R, Oostwaard DW, Snapper J, Kloots J, Hassink RJ, Kuijk E. et al. Increased cardiomyocyte differentiation from human embryonic stem cells in serum-free cultures. Stem Cells. 2005;23:772-80
20. Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A. et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407-14
21. Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M. et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524-8
22. Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD. et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855-60
23. Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A. et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407-14
24. Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP. et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30-41
25. Nussbaum J, Minami E, Laflamme MA, Virag JAI, Ware CB, Masino A. et al. Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB J. 2007;21:1345-57
26. Lee AS, Tang C, Rao MS, Weissman IL, Wu JC. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med. 2013;19:998-1004
27. Masuda S, Miyagawa S, Fukushima S, Sougawa N, Ito E, Takeda M. et al. Emerging innovation towards safety in the clinical application of ESCs and iPSCs. Nat Rev Cardiol. 2014;11:553-4
28. Masuda S, Miyagawa S, Fukushima S, Sougawa N, Okimoto K, Tada C. et al. Eliminating residual iPS cells for safety in clinical application. Protein Cell. 2015;6:469-71
29. Ban K, Wile B, Kim S, Park H-J, Byun J, Cho K-W. et al. Purification of cardiomyocytes from differentiating pluripotent stem cells using molecular beacons that target cardiomyocyte-specific mRNA. Circulation. 2013;128:1897-909
30. Masumoto H, Ikuno T, Takeda M, Fukushima H, Marui A, Katayama S. et al. Human iPS cell-engineered cardiac tissue sheets with cardiomyocytes and vascular cells for cardiac regeneration. Sci Rep. 2014;4:6716
31. Kensah G, Roa Lara A, Dahlmann J, Zweigerdt R, Schwanke K, Hegermann J. et al. Murine and human pluripotent stem cell-derived cardiac bodies form contractile myocardial tissue in vitro. Eur Heart J. 2013;34:1134-46
32. Thavandiran N, Dubois N, Mikryukov A, Massé S, Beca B, Simmons CA. et al. Design and formulation of functional pluripotent stem cell-derived cardiac microtissues. Proc Natl Acad Sci U S A. 2013;110:E4698-E707
33. Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res. 2014;114:511-23
34. Robertson C, Tran DD, George SC. Concise review: maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem Cells. 2013;31:829-37
35. Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375-86
36. Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L. et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593-8
37. Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K. et al. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res. 2012;110:1465-73
38. Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN. et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599-604
39. Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K. et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci U S A. 2013;110:5588-93
40. Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson CP, Pratt RE. et al. MicroRNA induced cardiac reprogramming in vivo: Evidence for mature cardiac myocytes and improved cardiac function. Circ Res. 2015;116:418-24
41. Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T. et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc Natl Acad Sci U S A. 2013;110:12667-72
42. Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A. et al. Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol. 2007;50:1884-93
43. Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91:501-8
44. Pertoft H, Laurent TC, Låås T, Kågedal L. Density gradients prepared from colloidal silica particles coated by polyvinylpyrrolidone (Percoll). Anal Biochem. 1978;88:271-82
45. Doevendans PA, Becker KD, An RH, Kass RS. The utility of fluorescentin vivoreporter genes in molecular cardiology. Biochem Biophys Res Commun. 1996;222:352-8
46. Ritner C, Wong SSY, King FW, Mihardja SS, Liszewski W, Erle DJ. et al. An engineered cardiac reporter cell line identifies human embryonic stem cell-derived myocardial precursors. PLoS One. 2011;6:e16004
47. Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ. et al. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol. 2011;301:H2006-H17
48. Xu XQ, Zweigerdt R, Soo SY, Ngoh ZX, Tham SC, Wang ST. et al. Highly enriched cardiomyocytes from human embryonic stem cells. Cytotherapy. 2008;10:376-89
49. Klug MG, Soonpaa MH, Koh GY, Field LJ. Genetically selected cardiomyocytes from differentiating embronic stem cells form stable intracardiac grafts. J Clin Invest. 1996;98:216-24
50. Anderson D, Self T, Mellor IR, Goh G, Hill SJ, Denning C. Transgenic enrichment of cardiomyocytes from human embryonic stem cells. Mol Ther. 2007;15:2027-36
51. Fu J-D, Jiang P, Rushing S, Liu J, Chiamvimonvat N, Li RA. Na+/Ca2+ exchanger is a determinant of excitation-contraction coupling in human embryonic stem cell-derived ventricular cardiomyocytes. Stem Cells Dev. 2009;19:773-82
52. Huber I, Itzhaki I, Caspi O, Arbel G, Tzukerman M, Gepstein A. et al. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. FASEB J. 2007;21:2551-63
53. Elliott DA, Braam SR, Koutsis K, Ng ES, Jenny R, Lagerqvist EL. et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat Methods. 2011;8:1037-40
54. Bizy A, Guerrero-Serna G, Hu B, Ponce-Balbuena D, Willis BC, Zarzoso M. et al. Myosin light chain 2-based selection of human iPSC-derived early ventricular cardiac myocytes. Stem Cell Res. 2013;11:1335-47
55. Lee MY, Sun B, Schliffke S, Yue Z, Ye M, Paavola J. et al. Derivation of functional ventricular cardiomyocytes using endogenous promoter sequence from murine embryonic stem cells. Stem Cell Res. 2012;8:49-57
56. MÜLLER M, FLEISCHMANN BK, SELBERT S, JI GJ, ENDL E, MIDDELER G. et al. Selection of ventricular-like cardiomyocytes from ES cells in vitro. FASEB J. 2000;14:2540-8
57. Zhang Q, Jiang J, Han P, Yuan Q, Zhang J, Zhang X. et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011;21:579-87
58. Zhu W-Z, Xie Y, Moyes KW, Gold JD, Askari B, Laflamme MA. Neuregulin/ErbB signaling regulates cardiac subtype specification in differentiating human embryonic stem cells; Novelty and significance. Circ Res. 2010;107:776-86
59. Recchia A, Bonini C, Magnani Z, Urbinati F, Sartori D, Muraro S. et al. Retroviral vector integration deregulates gene expression but has no consequence on the biology and function of transplanted T cells. Proc Natl Acad Sci U S A. 2006;103:1457-62
60. Tsukahara T, Agawa H, Matsumoto S, Matsuda M, Ueno S, Yamashita Y. et al. Murine leukemia virus vector integration favors promoter regions and regional hot spots in a human T-cell line. Biochem Biophys Res Commun. 2006;345:1099-107
61. Woods N-B, Muessig A, Schmidt M, Flygare J, Olsson K, Salmon P. et al. Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood. 2003;101:1284-9
62. Thomas TE, Miller CL, Eaves CJ. Purification of hematopoietic stem cells for further biological study. Methods. 1999;17:202-18
63. Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A. et al. Stage-specific optimization of Activin/Nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228-40
64. Dubois NC, Craft AM, Sharma P, Elliott DA, Stanley EG, Elefanty AG. et al. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotechnol. 2011;29:1011-8
65. Uosaki H, Fukushima H, Takeuchi A, Matsuoka S, Nakatsuji N, Yamanaka S. et al. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLoS One. 2011;6:e23657
66. Komuro I, Izumo S. Csx: a murine homeobox-containing gene specifically expressed in the developing heart. Proc Natl Acad Sci U S A. 1993;90:8145-9
67. Domian IJ, Chiravuri M, van der Meer P, Feinberg AW, Shi X, Shao Y. et al. Generation of functional ventricular heart muscle from mouse ventricular progenitor cells. Science. 2009;326:426-9
68. Protze SI, Liu J, Nussinovitch U, Ohana L, Backx PH, Gepstein L. et al. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat Biotechnol. 2017;35:56-68
69. Hattori F, Chen H, Yamashita H, Tohyama S, Satoh Y-s, Yuasa S. et al. Nongenetic method for purifying stem cell-derived cardiomyocytes. Nat Methods. 2010;7:61-6
70. Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T. et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127-37
71. Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56:130-40
72. Tohyama S, Fujita J, Hishiki T, Matsuura T, Hattori F, Ohno R. et al. Glutamine oxidation is indispensable for survival of human pluripotent stem cells. Cell Metab. 2016;23:663-74
73. Wile BM, Ban K, Yoon Y-S, Bao G. Molecular beacon-enabled purification of living cells by targeting cell type-specific mRNAs. Nat Protoc. 2014;9:2411-24
74. Ban K, Wile B, Cho K-W, Kim S, Song M-K, Kim Sang Y. et al. Non-genetic purification of ventricular cardiomyocytes from differentiating embryonic stem cells through molecular beacons targeting IRX-4. Stem Cell Reports. 2015;5:1239-49
75. Heyduk T, Heyduk E. Molecular beacons for detecting DNA binding proteins. Nat Biotechnol. 2002;20:171-6
76. Santangelo P, Nitin N, Bao G. Nanostructured probes for RNA detection in living cells. Ann Biomed Eng. 2006;34:39-50
77. Rhee WJ, Bao G. Simultaneous detection of mRNA and protein stem cell markers in live cells. BMC Biotechnol. 2009;9:30
78. Wang GF, Nikovits W, Bao Z-Z, Stockdale FE. Irx4 forms an inhibitory complex with the vitamin D and retinoic X receptors to regulate cardiac chamber-specific slow MyHC3 expression. J Biol Chem. 2001;276:28835-41
79. Bruneau BG, Bao Z-Z, Tanaka M, Schott J-J, Izumo S, Cepko CL. et al. Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by Nkx2-5 and dHand. Dev Biol. 2000;217:266-77
80. Miki K, Endo K, Takahashi S, Funakoshi S, Takei I, Katayama S. et al. Efficient detection and purification of cell populations using synthetic microRNA switches. Cell Stem Cell. 2015;16:699-711
81. Khademhosseini A, Langer R, Borenstein J, Vacanti JP. Microscale technologies for tissue engineering and biology. Proc Natl Acad Sci U S A. 2006;103:2480-7
82. Cimetta E, Sirabella D, Yeager K, Davidson K, Simon J, Moon RT. et al. Microfluidic bioreactor for dynamic regulation of early mesodermal commitment in human pluripotent stem cells. Lab Chip. 2013;13:355-64
83. Yoshimitsu R, Hattori K, Sugiura S, Kondo Y, Yamada R, Tachikawa S. et al. Microfluidic perfusion culture of human induced pluripotent stem cells under fully defined culture conditions. Biotechnol Bioeng. 2014;111:937-47
84. Singh A, Suri S, Lee T, Chilton JM, Cooke MT, Chen W. et al. Adhesion strength-based, label-free isolation of human pluripotent stem cells. Nat Methods. 2013;10:438-44
85. Xin L, Leqian Y, Junjun L, Itsunari M, Minako N, Yuichiro N. et al. On chip purification of hiPSC-derived cardiomyocytes using a fishnet-like microstructure. Biofabrication. 2016;8:035017
86. Guo K-T, SchÄfer R, Paul A, Gerber A, Ziemer G, Wendel HP. A new technique for the isolation and surface immobilization of mesenchymal stem cells from whole bone marrow using high-specific DNA aptamers. Stem Cells. 2006;24:2220-31
87. Vykoukal J, Vykoukal DM, Freyberg S, Alt EU, Gascoyne PRC. Enrichment of putative stem cells from adipose tissue using dielectrophoretic field-flow fractionation. Lab Chip. 2008;8:1386-93
88. Didar TF, Li K, Veres T, Tabrizian M. Separation of rare oligodendrocyte progenitor cells from brain using a high-throughput multilayer thermoplastic-based microfluidic device. Biomaterials. 2013;34:5588-93
89. Myers FB, Zarins CK, Abilez OJ, Lee LP. Label-free electrophysiological cytometry for stem cell-derived cardiomyocyte clusters. Lab Chip. 2013;13:220-8
90. Sofla A, Cirkovic B, Hsieh A, Miklas JW, Filipovic N, Radisic M. Enrichment of live unlabelled cardiomyocytes from heterogeneous cell populations using manipulation of cell settling velocity by magnetic field. Biomicrofluidics. 2013;7:014110
91. Zhang B, Green JV, Murthy SK, Radisic M. Label-free enrichment of functional cardiomyocytes using microfluidic deterministic lateral flow displacement. PLoS One. 2012;7:e37619
92. Murthy SK, Sethu P, Vunjak-Novakovic G, Toner M, Radisic M. Size-based microfluidic enrichment of neonatal rat cardiac cell populations. Biomedical Microdevices. 2006;8:231-7
93. Doerr A. Snapshots of the cell surface. Nat Methods. 2009;6:401
94. Bausch-Fluck D, Hofmann A, Bock T, Frei AP, Cerciello F, Jacobs A. et al. A mass spectrometric-derived cell surface protein atlas. PLoS One. 2015;10:e0121314
Author contact
![]() Corresponding author: Young-sup Yoon, M.D., Ph.D., Department of Medicine, Division of Cardiology, Emory University, 101 Woodruff Dr. WMB 3009, Atlanta, GA, 30322, USA; Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Korea, E-mail: yyoon5edu.
Corresponding author: Young-sup Yoon, M.D., Ph.D., Department of Medicine, Division of Cardiology, Emory University, 101 Woodruff Dr. WMB 3009, Atlanta, GA, 30322, USA; Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Korea, E-mail: yyoon5edu.