Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental section
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(10):2593-2605. doi:10.7150/thno.19894 This issue Cite
Research Paper
Self-Assembly DNA Polyplex Vaccine inside Dissolving Microneedles for High-Potency Intradermal Vaccination
Jing-Fong Liao1,#, Jin-Ching Lee2,#, Chun-Kuang Lin3, Kuo-Chen Wei4, Pin-Yuan Chen5, Hung-Wei Yang1 ![]()
1. Institute of Medical Science and Technology, National Sun Yat-sen University, Kaohsiung 80424, Taiwan;
2. Department of Biotechnology, Kaohsiung Medical University, Kaohsiung 80708, Taiwan;
3. Doctoral Degree Program in Marine Biotechnology, National Sun Yat-sen University, Kaohsiung 80424, Taiwan;
4. Department of Neurosurgery, Linkou Chang Gung Memorial Hospital, Taoyuan 33305, Taiwan. School of Medicine, Chang Gung University, Taoyuan 33302, Taiwan;
5. Department of Neurosurgery, Chang Gung Memorial Hospital, Keelung 20401, Taiwan
School of Medicine, Chang Gung University, Taoyuan 33302, Taiwan.
# These authors contributed equally.
Received 2017-3-3; Accepted 2017-4-13; Published 2017-6-25
Abstract

The strong immunogenicity induction is the powerful weapon to prevent the virus infections. This study demonstrated that one-step synthesis of DNA polyplex vaccine in microneedle (MN) patches can induce high immunogenicity through intradermal vaccination and increase the vaccine stability for storage outside the cold chain. More negative charged DNA vaccine was entrapped into the needle region of MNs followed by DNA polyplex formation with branched polyethylenimine (bPEI) pre-coated in the cavities of polydimethylsiloxane (PDMS) molds that can deliver more DNA vaccine to immune-cell rich epidermis with high transfection efficiency. Our data in this study support the safety and immunogenicity of the MN-based vaccine; the MN patch delivery system induced an immune response 3.5-fold as strong as seen with conventional intramuscular administration; the DNA polyplex formulation provided excellent vaccine stability at high temperature (could be stored at 45ºC for at least 4 months); the DNA vaccine is expected to be manufactured at low cost and not generate sharps waste. We think this study is significant to public health because there is a pressing need for an effective vaccination in developing countries.
Keywords: dissolvable microneedles, self-assembly, thermostability, DNA vaccine, intradermal delivery.
Introduction
There are limitations to the manufacturing capacity and production time of conventional vaccines. Currently, almost all vaccines are generated in liquid form, and must be refrigerated to retain their activity [1]. Conventional vaccines are prepared using viruses grown in eggs or cell cultures that are often broken apart by detergents into a subunit vaccine [2]. DNA vaccines are considered an attractive alternative to conventional vaccines because they are relatively easy and inexpensive to produce, stable at room temperature, and able to stimulate potent cellular and humoral immune responses for pAPCrotection against infection.3 Whereas DNA vaccines are safe and well tolerated, the antibody titers induced by first-generation DNA vaccines are low or nonexistent [3, 4]. Thus, second-generation DNA vaccines were designed that more broadly activate CD8+ cytotoxic T cells in larger animal models [5].
DNA vaccinations rely mainly on the cellular delivery of plasmid DNA (pDNA) to overexpress an encoded antigen; however, the poor transfection ability of DNA vaccines is considered one of the most critical reasons for the low level of immunity they generate [6]. Viral vectors are often used to improve vaccine immunogenicity by increasing DNA delivery into cells, but they can induce undesirable immune responses, exhibit oncogenic properties, and have unknown long-term effects [7]. Non-viral vectors for DNA delivery, including cationic polymers and lipids that can form polyplex with DNA vaccines, are efficiently protected from enzymatic degradation and exhibit better cell-transfection efficiency [8, 9]. Nevertheless, polyplex-based DNA vaccines still require a trained health-care professional to perform their administration with a syringe. As an alternative, electroporation and gene gun-based gene-delivery systems have been used to improve immunization results; however, these methods often cause pain or discomfort to the patient and possibly tissue damage, and also require special expertise to operate the equipment [10, 11]. Thus, the development of more convenient and painless DNA vaccine delivery systems is urgently required to overcome these shortcomings.
Skin vaccination is an alternate route of immunization that increases vaccine immunogenicity [12]. The skin's layers are known to have abundant professional antigen-presenting cells (APCs; i.e., macrophages, Langerhans cells, and dendritic cells), which play an important role in inducing immune responses [13, 14]. Currently, injection with a hypodermic needle is the most commonly used method of administering vaccines, but this technique is painful, has low patient acceptance, and cannot deliver the vaccines to the epidermis. Recently, an alternative vaccination method has been developed: an easy-to-apply microneedle (MN) patch, which addresses these concerns by offering a more patient-compliant and safer method of administering vaccines into the epidermis and superficial dermis by minimally trained personnel [15, 16], enabling increased vaccine thermostability and generating no sharps waste [17-19]. In previous studies, MNs coated with naked DNA vaccines have been used to significantly increase the antibody- and cell-mediated immune responses. However, the repeated dip-coating and drying of highly concentrated and viscous DNA vaccine solutions necessary for MN manufacture cause initial activity loss [20, 21]. Another approach is the layer-by-layer deposition of polyelectrolytes and DNA vaccines on the surface of MNs, which precisely controls the DNA loading amount and avoids initial activity loss [22-24]. However, the coated DNA vaccines can be scraped away from the MN surface by skin tissues (especially the stratum corneum) during insertion, reducing the amount of vaccine in the epidermis. To overcome this problem, dissolving MNs were designed to protect the vaccines by their entrapment inside the MN using casting procedures (vacuum or centrifugation). These MNs only dissolve in the interstitial fluid of the skin upon application. However, expensive vaccines are always wasted during procedures that significantly increase the cost of manufacturing [25, 26].
The challenges of dissolvable MN-based DNA vaccine delivery are:
- how to concentrate the DNA vaccines in the needle region of MNs;
- how to retain the potency of DNA vaccines during MN fabrication and after storage for a long time at an elevated temperature; and
- how to improve the low transfection ability of DNA vaccines.
To overcome these challenges, we developed a new fabrication procedure for one-step synthesis of DNA polyplex vaccine in MNs and also entrapment of more DNA vaccines in the needle region of dissolving MNs, which is different with the multilayer coated MNs [24]. The benefits of this new procedure include a reduction in the cost manufacturing as a result of low DNA vaccine waste, and improved delivery of DNA vaccine into the epidermis with consequent stronger immunogenicity. Our preclinical data support the safety and high immunogenicity of the MNs vaccination; the MNs delivery system enables vaccine administration by minimally trained personnel; and the formulation provides remarkable vaccine stability without refrigeration.
Experimental section
Fabrication of the microneedle array mold
The biodegradable polymer polylactide (PLA; 1.1 dL g-1; Lakeshore Biomaterials, Birmingham, AL, USA) was used to fabricate the master MN array. A stainless-steel micropillar was micromachined to yield a 10 × 10 array form; each micropillar had a diameter of 640 μm. The master MN array was fabricated using a spatially discrete thermal drawing system as previously reported [27]. The fabrication of the MN arrays was performed in four steps. First, PLA film on a heating substrate was heated to 190°C, then the micropillar was gently lowered to make contact with the heated PLA film. PLA in the contact area was drawn upward by 100 μm by the micropillar, which was heated to 120°C. After a short pause, the drawn PLA was further lifted to 860 μm by the micropillar at 160°C. To disconnect the neck of the drawn PLA, the temperature of the substrate was decreased to 60°C while the temperature of the micropillar was increased to 190°C. This high temperature at the micropillar ensured that the neck of the drawn polymer structure broke without deforming to form the MN master, with a base 640 μm wide and a total height of 860 μm [including needle (560 μm) and tapered base (300 μm)].
Next, an MN mold made from PDMS (Sylgard 184; Dow Corning Corp., Midland, MI, USA) was created using the PLA master. A reusable inverse female PDMS mold was obtained by pouring PDMS over the male master structure, allowing it to cure for 2.5 h at 70°C, and carefully peeling the resulting mold off the master structure.
Construction of porcine circovirus Type 2-open reading frame 2 plasmid DNA
The backbone of the PCV2-ORF2 expression plasmid was pcDNA4/myc-His. PCV2-ORF2 was amplified by polymerase chain reaction from PCV2 genomic DNA kindly provided by Dr. Cheng-Yao Yang (Animal Technology Laboratories at Agricultural Techonolgy Research Institute, Taiwan), and cloned into pcDNA/myc-His using the BglII restriction enzyme sites under the control of the cytomegalovirus (CMV) immediate-early gene promoter. The final expression construct was named pCMV-PCV2.
Dissolving MN patch fabrication with SRB loading
Flexible and dissolvable MN patches were fabricated by casting two solutions in PDMS molds using scraping and pressing procedures. The first casting with 30 μL of 10% PVA/2.5% bPEI aqueous solution (molecular weight [Mw] 6000 Da for PVA; Mw 10000 Da for bPEI; Sigma-Aldrich Corp., St. Louis, MO, USA) containing SRB was followed by centrifugation (3,500 rpm, 10 min) and vacuumization at room temperature for 0.5 h to dry the solution into the mold. The second casting with 30 μL of a mixture of 30% PVA and 10% polyvinylpyrrolidone (PVP; Mw 10000 Da; Sigma-Aldrich Corp., Louis, MO, USA) was followed by vacuumization at room temperature for 0.5 h, then pressing the semi-dry PVA/PVP on to the mold to push more PVA/PVP into the needles using a roller and vacuumization for another 1 h. To obtain flexible and stronger MN patches, we repeated the second step again, then scraped the surplus PVA/PVP from the mold and vacuum-treated for 2 h. After drying overnight at room temperature, the MN patches were carefully removed from the PDMS molds using PU film to obtain SRB-loaded MN patches, and stored in a desiccator.
Self-entrapment of GFP-pDNA or the DNA vaccine for porcine circovirus Type 2 in MN patches
The fabrication procedures were similar to those described in the preceding section. Briefly, the first casting with 30 μL of 10% PVA/2.5% bPEI aqueous solution was followed by centrifugation (3,500 rpm, 10 min) and vacuumization at room temperature for 0.5 h to fill the cavities and the solution in the cavities was dried under vacuum after removal of redundancy, forming a thin film with a positive charge in need region of the PDMS mold. Then the thin layers on the surface of the PDMS mold was brushed by moist swab to make sure the thin layers with positive charge was only existed inside the cavities. Next, 30 μL of different concentrations of negatively charged GFP-pDNA or pcDNA4-PCV2 vaccine solution was mixed with 1% tripolyphosphate (TPP; Sigma-Aldrich Corp., St. Louis, MO, USA) then dropped on the molds to fill the cavities, followed by vacuumization at room temperature for 1 h, the vaccine in the cavities will be attracted to needle region by pre-coated bPEI film then formed DNA vaccine polyplex. The second casting was conducted with a mixture of 30 μL of 30% PVA and 10% PVP, followed by vacuumization at room temperature for 0.5 h, then the semi-dry PVA/PVP was pressed on to the mold to push more PVA/PVP into the needles using a roller and the mold was vacuum-treated again for another 1 h. To obtain flexible and stronger MN patches, we repeated the second step again followed by scraping the surplus PVA/PVP from the mold and vacuumization for 2 h. After drying overnight at room temperature, the MN patches were was carefully peeled off directly with an adhesive plate (PU film) on the base to obtain GFP-pDNA or pcDNA4-PCV2 vaccine-loaded MN patches, and stored in a desiccator.
Quantification of DNA vaccines loaded into MN patches
GFP-pDNA or pcDNA4-PCV2 were loaded into MN patches, then the MN patches were solidified after drying in lyophilizer for 24 h. Subsequently, the needles were cut into a centrifuge tube, then 200 μL of DI water was added to solve the needles. A QIAquick® Gel Extraction Kit (Qiagen, Hilden, Germany) was used to purify the DNA vaccines from the polymer solution. The concentration of DNA vaccines was measured by NanoDrop (NanoDrop One, Thermo Scientific, Wilmington, DE, USA).
Tests of mechanical properties
Tests of mechanical properties were performed using a displacement-force test station (Model 921A, Tricor Systems Inc., Elgin, IL, USA), as described previously. A 10 × 10 array of MNs was attached to the mount of a moving sensor, and an axial force (10 N) was applied to move the mount at a speed of 8 µm/s. The mount pressed the MNs against a flat, rigid surface of stainless steel oriented perpendicularly to the axis of mount movement. The test station recorded the force required to move the mount as a function of distance.
Agarose gel electrophoresis of the DNA vaccines to test their stability
The stability of GFP-pDNA or pcDNA4-PCV2 in the MN patches was identified by the electric mobility of pDNA into a 1% agarose gel. The patches were stored for 1-121 days at 45°C, and then dissolved in DI water. A QIAquick® Gel Extraction Kit was used to purify the DNA vaccines from the polymer solution. Electrophoresis was performed at a constant voltage of 100 V (Bio-Rad Laboratories, Inc., Hercules, CA, USA) for 30 min in TAE buffer (40 mM Tris-base, 1 mM sodium ethylenediaminetetraacetic acid [EDTA], and 20 mM acetic acid [pH 8.5]), and then stained with GelStar™ Nucleic Acid Gel Stain (Lonza Rockland, Inc., Rockland, ME, USA). The gels were visualized under an ultraviolet transluminator (Gel Doc 2000; Bio-Rad Laboratories, Inc.).
In-vitro gene transfection and expression study
HEK293 cells were added to 24-well plates at a density of about 1 × 105 cells per well in 0.5 mL of Dulbecco's modified Eagle's medium (DMEM) containing 2.2 mg/mL sodium carbonate, 10% fetal bovine serum (FBS), 50 μg/mL gentamicin, and 50 μg/mL penicillin, then incubated overnight before transfection; all incubations were performed at 37°C in 5% CO2. Next, the cells were transfected with GFP-pDNA (2 μg/well) obtained by dissolving MNs in a total volume of 500 μL of culture medium using a PolySci transfection reagent (Q001; Qida Biomedical, Taiwan). Eight hours later, the medium was replaced with fresh medium, and the cells were incubated overnight. Cells were washed three times with serum-free DMEM, then their nuclei were stained using Hoechst. Fluorescence images of GFP expression in cells were captured using an inverted fluorescence microscope (Eclipse Ti-S; Nikon Corp., Tokyo, Japan). All images were collected using the same imaging parameters to facilitate direct comparison between figures.
MNs skin insertion
To determine whether the MN patches could penetrate skin, the patches were inserted into full-thickness, shaved pig cadaver skin with the subcutaneous fat removed. SRB-loaded MN patches were inserted into the skin by pressing against the backside of an MN patch with a thumb using a force of approximately 1.5 N, and then removed after MNs insertion (1, 3, 5, 8 min). Next, the inserted area of skin was visualized, and images of the microneedle-punctured skin were collected using fluorescence stereomicroscopy (SZX7; Olympus Corp., Tokyo, Japan). The ImageJ software was used to analyze the area of residual needle region then calculated how many percentage of needle region was dissolved in skin compared with the original one.
To prepare histological specimens, MNs insertion sites were cut from bulk skin using a scalpel. Each skin section was embedded in Optimum Cutting Temperature (OCT) compound in a cryostat mold and frozen in liquid nitrogen immediately. The frozen OCT-skin samples were subsequently sliced into 50-μm thick sections using a cryotome and collected by silane coated glass slides. The skin sections were finally viewed using an inverted fluorescence microscope (Eclipse Ti-S; Nikon Corp., Tokyo, Japan).
Stability tests for the DNA vaccine for porcine circovirus Type 2 in MN patches
MN patches containing pcDNA4-PCV2 were stored at 45°C for 1-121 days, and then dissolved in DI water. A QIAquick® Gel Extraction Kit was used to purify the pcDNA4-PCV2 from the polymer solution. The stability of pcDNA4-PCV2 in the MN patches was determined using western blotting to analyze the ORF2 protein expression in cells. Huh-7 cells were maintained in DMEM containing 10% heat-inactivated FBS, 1% antibiotic-antimycotic, and 1% non-essential amino acids, and incubated at 37°C with a 5% CO2 supplement. Huh-7 cells were seeded on a 24-well plate at a density of 5 × 104 cells per well. The next day, the Huh-7 cells were transfected with pcDNA4-PCV2 using T-pro P-Fect Transfection Reagent (Ji-Feng Biotechnology Co., Ltd., Taipei, Taiwan) following the manufacturer's instructions. After 6 h of transfection, the medium was changed with fresh medium, followed by incubation for another 2 days.
The transfected Huh-7 cells were collected with RIPA lysis buffer (50 mM Tric-HCl 150 mM NaCl, 5 mM EDTA, 2% sodium dodecyl sulfate [SDS], and 1% NP-40) from the plate. Each sample was clarified by centrifugation at 13000 revolutions per minute for 60 min at 4°C. Ten micrograms of total protein from each sample was resolved by 8% SDS-polyacrylamide gel electrophoresis and subsequently transferred to a polyvinylidene difluoride membrane (Pall Corp., Pensacola, FL, USA). The protein-transferred membranes were blocked with 5% skim milk in phosphate-buffered saline (PBS) containing 0.05% Tween-20 (PBST) for 4 h at room temperature, and then probed with rabbit polyclonal anti-PCV2 (Abomics Co., Ltd., New Taipei City, Taiwan) and anti-GAPDH (GeneTex, Inc., Irvine, CA, USA) antibodies. The blotting signals were developed using an ECL Detection Kit (PerkinElmer, Norwalk, CT, USA). The protein expression levels were quantified using the software Quantity One® (Bio-Rad Laboratories, Inc.).
Anti-active Caspase-3 staining
The skin tissues of mice with (specimen collection after 12 h of insertion) or without the insertion of MN patches were placed in 4% PFA and paraffin embedded were cut into sections of 4 μm thickness. The immunohistochemistry was performed with the Quanto Detection System HRP (Thermo Fisher Scientific, Inc., Fremont, CA, USA); the sections were incubated with antigen retrieval solution pH 6.0, then blocked with endogenous peroxidase Ultra V Block for 10 min. (Thermo Fisher Scientific). Next, the sections were incubated with UltraVision protein block (Thermo Fisher Scientific) for 5 min to reduce nonspecific background. Apoptosis staining was detected using anti-active caspase 3 antibody (1:500, BD #559565) incubated at 4˚C overnight; and then the signaling was amplified using the Primary Antibody Amplifier Quanto (Thermo Fisher Scientific) for 10 minutes. Then, the secondary antibody (HRP Polymer Quanto) was added for 10 minutes. Finally, the DAB chromogen was added for 5 min and counterstained with Mayers' hematoxylin (DAKO) then examined under a light microscope.
Immunization of mice
Mice were obtained from National Laboratory Animal Center, Taiwan, and housed at the Kaohsiung Medical University Division of Animal Resources. All mice were immunized twice at 2-week intervals with 20 μg of pcDNA4-PCV2, either by IM injection or MN patches. Mice were anesthetized with 2.5% isoflurane using XGI-8 Gas Anesthesia System, and their hair was removed using Nair depilatory cream (Church & Dwight Co., Inc., Princeton, NJ, USA). The skin was dried using cotton balls with ethanol, and MN patches were manually inserted into the skin and left in place for 10 min.
Mice were randomly divided into six groups with six mice per group. Mice were immunized with:
pcDNA4 vehicle via IM injection;
pcDNA4 vehicle-loaded bPEI-precoated MN patches;
naked pcDNA4-PCV2 vaccine via IM injection;
pcDNA4-PCV2 vaccine-loaded MN patches
pcDNA4-PCV2 vaccine-loaded bPEI-precoated MN patches; or
pcDNA4-PCV2 vaccine-loaded bPEI-precoated MN patches stored for 1 month.
All animal studies were approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University.
For intradermal administration, vaccine was delivered to the shaved posterior-abdominal skin of mice under isoflurane anesthesia. Blood samples were collected 1 week after each immunization, and serum samples were collected and stored at -80ºC until further analysis.
Detection of antigen-specific antibodies
The levels of PCV2-specific antibodies were measured in individual mouse serum samples by ELISA to determine the induced immune response. Microplates (Corning Costar Corp., Cambridge, MA, USA) were coated overnight at 4ºC with purified PCV2-ORF2 protein as a coating antigen at a concentration of 2 μg/mL. A standard curve was generated by coating control wells with known concentrations of mouse IgG. The microplates were then washed with PBST prior to blocking with 200 μL per well of 2% bovine serum Albumin in PBST for 1 h at 37ºC. Serial dilute serum samples (1:500 in dilution; 200 μL) were added into the wells and incubated at room temperature for 2 h. The microplates were washed again with PBST, and incubated with horseradish peroxidase-labeled goat anti-mouse secondary antibody (1:5000 dilution in PBST). After a final wash, the plates were developed with 3,39,5,59-tetramethylbenzidine (Sigma-Aldrich Corp., Louis, MO, USA). The color reaction was stopped with 0.2 N hydrochloric acid. The microplates were read at 450 nm in a microplate reader (SpectraMax M2, Molecular Devices, Sunnyvale, CA, USA), and the concentrations of PCV2-specific antibodies in the serum samples were calculated using the standard curves obtained and expressed as the amount of antigen-specific antibody in 1 mL of serum sample (ng/mL).
Statistics
The statistical significance was calculated for selected groups by two-tailed unpaired Students t test, and p≤0.05 was considered significant. Average values of PCV2-ORF2 expression in Western blotting and antibodies titers were analyzed using one-way ANOVA with Dunnett's multiple comparison post-test.
Results and Discussion
Characterization of dissolving MNs
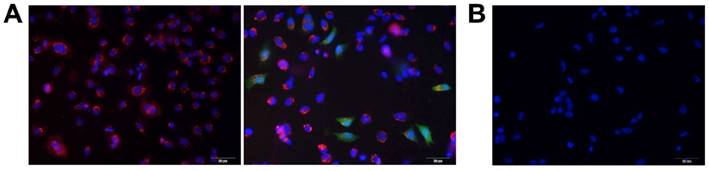
To meet the design goals, polydimethylsiloxane (PDMS) molds were copied from the MN master (needles and supporting bases), then treated with oxygen plasma to produce hydrophilic PDMS molds (contact angle changed to 72º from 128º; SI: Figure S1) with a rich hydroxyl groups on surface for easily facilitate further casting of water-soluble vaccine or polymer solution. The thin layers with positive charge were generated in needle region of MNs by filling branched polyethylenimine (bPEI) solution in the cavities under centrifugation and vaccum that enabled rapid entrapment of the negatively charged DNA vaccine into the needle region of MNs followed by DNA polyplex vaccine formation with the size of 56.1 ± 2.7 nm (SI: Figure S2) between the pre-coated bPEI and the DNA vaccines through electrical interactions (Figure 1A, B), which can significantly enhance the cell transfection efficiency. The entrapped GFP-pDNA polyplex in MNs were stained with GelRed then incubated with HEK293 cells, the results showed the stained GFP-pDNA polyplex could be taken up into cytoplasm by cells (red color) within 6 h (Figure 2A, left). Following 12 h of incubation, the GFP-pDNA expressed the green fluorescence protein inside cells showing green fluorescence (Figure 2A, right). Conversely, no signal of red or green fluorescence was observed in cells when incubating the GelRed stained GFP-pDNA extracted from MNs with HEK293 cells for 18 h (Figure 2B), indicating that self-assembly DNA polyplex could enter the cells and effectively express encoding protein in the absence of transfection reagent assistance. This one-step synthesis of self-assembly DNA polyplex vaccine inside MNs is simpler than those reports that must prepare polyplex first then loaded in MNs [22, 28, 29]. This purposed MN patch can protect the DNA vaccine well inside MNs, which is quite different from the surface DNA vaccine coated MNs [24]. The produced MN patch is a simple-to-administer vaccination strategy for everyone, we just need to apply and slightly press the MN patch on the skin, the MNs are strong enough to penetrate the skin and dissolved in the epidermis to release the DNA polyplex vaccine within 6 min for the induction of immunogenicity (Figure S3).
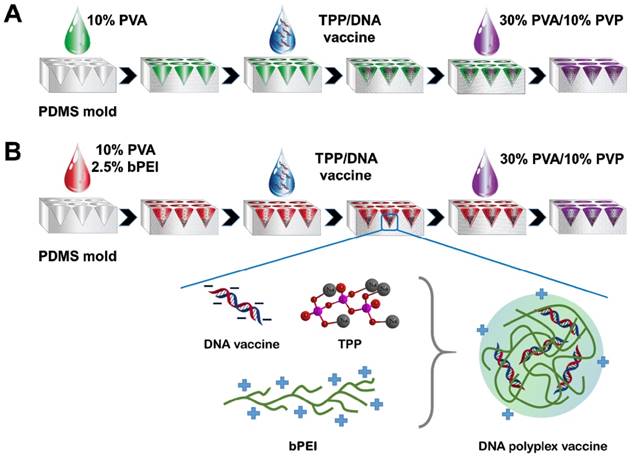
Schematic illustrating the fabrication procedure for microneedle (MN) DNA vaccine patches (A) without branched polyethylenimine (bPEI) pre-coating and (B) with bPEI pre-coating (inset: the formation of self-assembly DNA polyplex vaccine in MNs).
Fluorescence micrographs of HEK293 cells after incubation with (A) GelRed stained GFP-pDNA polyplex extracted from MNs for 6 h (left) and 18 h (right); (B) GelRed stained GFP-pDNA extracted from MNs for 18 h (scale bar: 50 μm). Red color: GelRed stained DNA; Green color: green fluorescence protein; Blue color: nuclei.
Compared to conventional needle-and-syringe vaccination, the dissolving MN patch is designed to be smaller and painless, generate no sharps waste and be simple to administer by minimally trained personnel (Figure 3A). Scanning electron microscopy revealed that the resulting MNs were 860 μm high [including needle (560 μm) and tapered base (300 μm)] and 640 μm wide at the base, the width of pinpoint was only 10.4 μm that was sharp enough to penetrate stratum corneum (Figure 3B). The MN patch was arranged in a 10 × 10 array covering an area of 0.64 cm2 (SI: Figure S4). To verify whether the MNs were strong enough to penetrate the skin for the efficient intradermal delivery of DNA vaccines, sulforhodamine B (SRB) was loaded into MNs made of highly water-soluble PVA/bPEI, which dissolves in the skin and, thus, generates no sharps waste. After insertion of the MNs into porcine cadaver skin, 100 holes were generated, and all of them filled with SRB (red color) that could not be wiped from the skin's surface, indicating that the SRB was located within skin and that the MNs were mechanically robust enough to withstand insertion into porcine cadaver skin (Figure 3C). The strength of the MNs was also confirmed by compression test: the failure forces for MNs encapsulating pcDNA4-PCV2 were 0.17 ± 0.02 N/needle and 27.7 ± 3.4 N/patch (SI: Figure S5), which is much larger than the force reportedly required to puncture human skin (~0.058 N/needle or 16 N/patch) [30, 31], demonstrating that the fabrication procedure creates strong MN patches.
(A) Magnified image of a MN patch containing Trypan blue placed adjacent to a 22-gauge needle for scale. (B) Scanning electron microscopy image of the MNs. (C) Skin-insertion capability of sulforhodamine B (SRB)-loaded MN patches and bright-field micrograph of porcine cadaver skin after insertion of the MN patch (inset: magnification of part of the micrograph). (D) Fluorescence micrographs showing dissolution kinetics of microneedle (MN) patches encapsulating SRB after their insertion into porcine cadaver skin for 0, 3, and 5 min, and the histogram of residual needle region on the patch. The ImageJ was used to analyze the area of residual needle region then calculated how many percentage of needle region was dissolved in skin compared with the original one. The white region was defined as needle region.
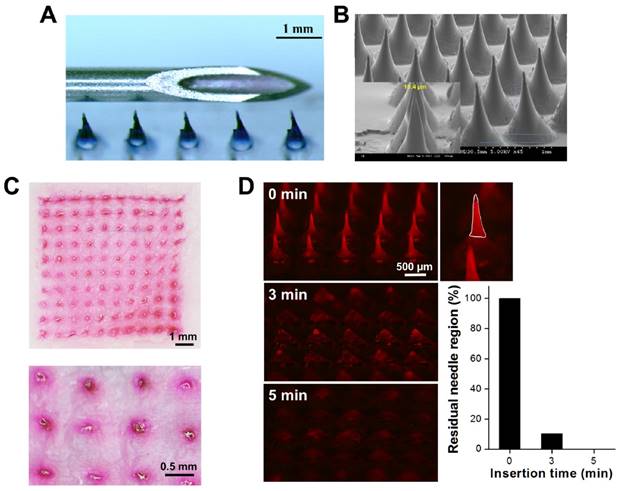
Dynamic dissolution of MNs in the skin
In this study, we used highly water-soluble PVA/PVP to fabricate MN patches that can rapidly dissolve in the skin to release DNA polyplex vaccine. To meet our goal, it was necessary to determine the dissolution kinetics of the MNs in the skin. We imaged the dissolution of MNs containing SRB after insertion into the skin over time (Figure 3D). We found that the MNs penetrated and dissolved in the skin: the tips of the MNs dissolved within 0.5 min, the needle region was almost dissolved (89.7 ± 3.3%) after 3 min of insertion and only the bases remained on the patch after more 2 min of insertion (all needles were dissolved in skin), indicating the most of vaccine in the MNs could be released into epidermis. The results demonstrated that the patients can remove the MN patches and throw away in 6 min after insertion. Therefore, the MN-based intradermal vaccination is fast, safe, and environmentally friendly.
DNA vaccine encapsulation in MNs
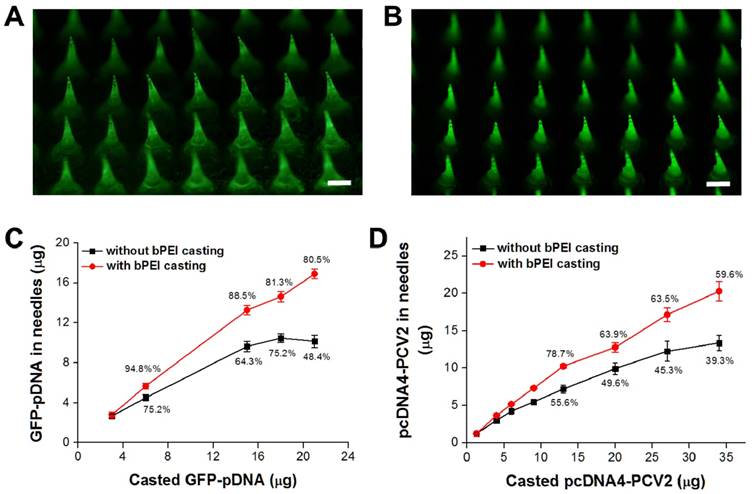
The focus of this study is the simple method we developed to load more DNA vaccine into the needle region of MN patch compared with vacuum or centrifugation self-assembly DNA polyplex vaccine formation. Although there are studies presenting effective tip-loaded MN fabrication approaches already [32, 33], no one likes our approach which can concentrate the encapsulated DNA vaccine to the needle region of MNs and also form DNA polyplex vaccine in the MNs using one-step fabrication process. To clarify the improvement, we stained the loaded DNA vaccine for porcine circovirus Type 2 (pcDNA4-PCV2) using GelStar™ Nucleic Acid Gel Stain (Lonza Rockland, Inc., Rockland, ME, USA), during which green fluorescence was used to monitor the distribution of DNA vaccine in the MNs. Without bPEI pre-coating, the pcDNA4-PCV2 was loaded into the MNs with the assistance of gravity alone, resulting in the distribution of pcDNA4-PCV2 throughout whole needle, from base to top (Figure 4A). Conversely, with bPEI pre-coating, most pcDNA4-PCV2 was concentrated into the needle region of the MNs as a result of electrical interactions between the bPEI and pcDNA4-PCV2, and no green fluorescence was evident in the base (Figure 4B). Guided by these results, we were able to encapsulate 16.9 ± 0.5 μg Green fluorescent protein (GFP)-pDNA or 20.3 ± 1.3 μg pcDNA4-PCV2 in the needle region of MNs per patch when bPEI pre-coating was used before DNA-vaccine loading, which was 1.7-fold (GFP-pDNA) or 1.5-fold (pcDNA4-PCV2) higher than the encapsulation possible without bPEI pre-coating (10.2 ± 0.6 μg for GFP-pDNA; 13.4 ± 1.1 μg for pcDNA4-PCV2; Figure 4C, D). This difference may be explained by the strong guiding force of electrical interactions between bPEI and the DNA vaccines, which effectively concentrate DNA vaccines into the needle region of MNs compared with gravity alone at the same DNA vaccine concentration, and allow the delivery of more DNA vaccine into the epidermis because only the top two thirds of MNs are inside the skin and dissolved to release the DNA vaccine after insertion of an MN patch. This approach highlights the use of electrostatic charges to load more DNA vaccine in the needle region of MNs that is significantly different from the layer-by-layer deposition of polyelectrolytes and DNA vaccines on the surface of MNs [24], also protect the vaccines would not scrap away from the MNs surface by skin tissues (especially the stratum corneum) during insertion.
The distribution of the DNA vaccine for porcine circovirus Type 2 (pcDNA4-PCV2) stained using GelStar™ Nucleic Acid Gel Stain (Lonza Rockland, Inc., Rockland, ME, USA) in microneedles (MNs) (A) without branched polyethylenimine (bPEI) pre-coating and (B) with bPEI pre-coating on the polydimethylsiloxane mold (scale bar: 500 μm). Amount of DNA vaccine in the needle region of the MNs after loading (C) Green fluorescent protein-plasmid DNA and (D) pcDNA4-PCV2 at different concentrations. The percentages show the loading efficiency in the needle region of the MNs (i.e., not in the base). Data represent average ± standard deviation (n = 3 replicates).
Stability of DNA vaccine in MNs
The concern for conventional vaccines is the stability during storage, usually must be stored at 4 ºC to maintain its activity that would significantly increase the storage cost. The cold chain would cost hundreds of million dollars per vaccine program, thus a vaccine is stable at room temperature or higher temperature could be of significant financial benefit. Thus, we investigated whether the integrity of DNA vaccines is maintained after their encapsulation, and tested their long-term thermostability in MN patches stored at 45ºC. We used GFP-pDNA as the DNA vaccine model for the convenient observation of protein expression in cells. Gel electrophoresis showed that GFP-pDNA reconstituted from MN patches after storage at 45ºC for 1 (Figure 5A, Lane 3), 7 (Figure 5A, Lane 5), and 60 (Figure 5A, Lane 6) days retained its supercoiled structure and was partially transformed into the nicked-circle form compared to naked GFP-pDNA (Figure 5A, Lane 2), but its protein expression ability remained the same as that of naked GFP-pDNA from stock solution (Figure 5B). In contrast, naked GFP-pDNA was completely degraded to fragments after storage at 45ºC for 7 days (Figure 5A, Lane 4). These results were also confirmed by the transfection of GFP-pDNA into HEK293 cells. Interestingly, the encapsulated GFP-pDNA mixed with pre-coated bPEI to form polyplex, which can self-transfect cells without the use of a transfection reagent (i.e., PolySci transfection reagent) that would effectively improve the low transfection ability of DNA vaccine in epidermis. Assessment of transfection efficiency showed that the GFP-pDNA reconstituted from MN patches after storage at 45ºC for 7 [expression level: 41.5 ± 4.7%; Figure 5B(iii)] and 60 [expression level: 38.9 ± 5.3%; Figure 5B(iv)] days was similar to naked GFP-pDNA from stock solution [expression level: 39.6 ± 6.4%; Figure 5B(i)], indicating that the GFP-pDNA in MNs was protected and showed high thermostability, and also confirmed the formation of DNA polyplex vaccine in MNs. In contrast, naked GFP-pDNA was damaged and lost its protein-expression activity when stored at 45ºC for 7 days [no green fluorescence; Figure 5B(ii)].
(A) Agarose gel retardation analysis to identify structural changes in Green fluorescent protein-plasmid DNA (GFP-pDNA). Lane 1: DNA ladder, Lane 2: GFP-pDNA stock, Lane 3: GFP-pDNA from reconstituted microneedles (MNs) after storage at 45ºC for 1 day, Lane 4: Naked GFP-pDNA stored at 45ºC for 7 days, Lane 5: GFP-pDNA from reconstituted MNs after storage at 45ºC for 7 days, Lane 6: GFP-pDNA from reconstituted MNs after storage at 45ºC for 60 days. (B) Fluorescence micrographs of HEK293 cells after different GFP-pDNA transfection protocols. Cells were incubated with: (i) GFP-pDNA stock transfected to cells in the presence of PolySci transfection reagent, (ii) Naked GFP-pDNA stored at 45ºC for 7 days then transfected to cells in the presence of PolySci transfection reagent, (iii) GFP-pDNA from reconstituted MNs after storage at 45ºC for 7 days in the absence of PolySci transfection reagent, (iv) GFP-pDNA from reconstituted MNs after storage at 45ºC for 60 days in the absence of PolySci transfection reagent. Blue color: nuclei; green color: GFP (scale bar: 200 μm). (C) Agarose gel retardation analysis to identify structural changes in the DNA vaccine for porcine circovirus Type 2 (pcDNA4-PCV2). Lane 1: Naked pcDNA4-PCV2 stored at 45ºC for 7 days, Lane 2: pcDNA4-PCV2 from reconstituted MNs after storage at 45ºC for 7 days, Lane 3: Naked pcDNA4-PCV2 stored at 45ºC for 15 days, Lanes 4-20: pcDNA4-PCV2 from reconstituted MNs after storage at 45ºC for 15, 17, 20, 23, 26, 29, 32, 37, 39, 44, 54, 58, 61, 75, 90, 107, and 121 days. (D) Western-blotting analysis of PCV2-open reading frame 2 (ORF2) protein expressions in Huh-7 cells after treatment with different samples (pcDNA4, pcDNA4-PCV2 stock, or pcDNA4-PCV2 from reconstituted MNs). (E) Western-blotting analysis of PCV2-ORF2 protein expression in Huh-7 cells after treatment with pcDNA4-PCV2 stored at 45ºC for 0, 14, and 28 days. Values are expressed as means ± standard deviation (SD; n = 3). Asterisk indicates a significant difference (Student's t-test, *p < 0.001). (F) Western-blotting analysis of PCV2-ORF2 protein expression in Huh-7 cells after treatment with pcDNA4-PCV2 from reconstituted MNs stored at 45ºC for 0, 14, and 28 days. Values are expressed as means ± SD (n = 3).
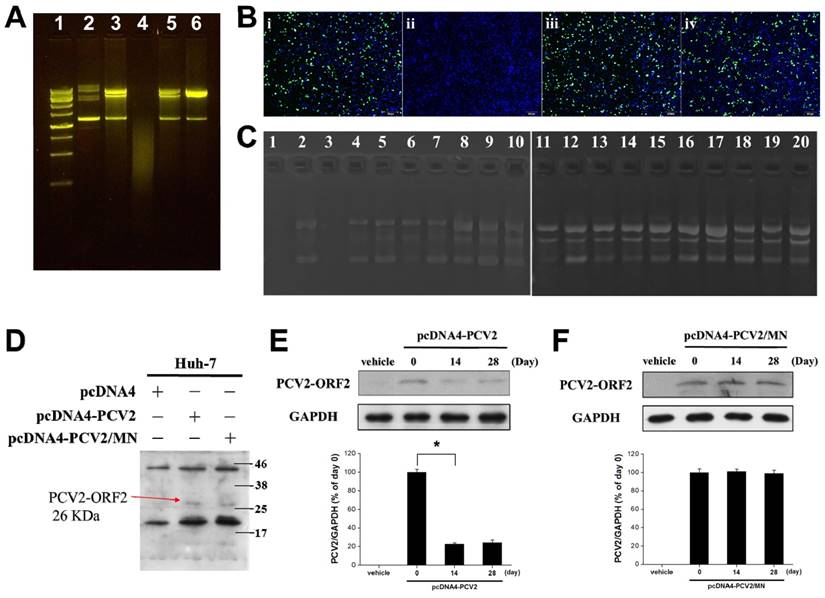
To achieve our long-term vision of DNA vaccination using MN patches that do not require refrigeration, MN patches containing pcDNA4-PCV2 were stored at 45ºC for up to 121 days. The pcDNA4-PCV2 stock solution was completely degraded to fragments on Days 7 and 15 (Figure 5C, Lanes 1 and 3). The pcDNA4-PCV2 in MN patches was more thermostable than that in a liquid formulation, and remained in a supercoiled form until 121 days of storage at 45ºC; 36.8 ± 4.6% transformation of pcDNA4-PCV2 from the supercoiled form to the nicked-circle form occurred (Figure 5C, Lanes 2 and 4-20) but still remained the protein expression ability, showing that pcDNA4-PCV2 is protected by loading in MNs, possibly due to electrostatic interactions that prevent degradation during the MN-fabrication process [34]. These results suggest that MN patches containing DNA vaccines (i.e., pcDNA4-PCV2) can be stably stored scorching environment without refrigeration.
To assess the effect of pcDNA4-PCV2 strand-breaking due to vaccine instability on protein expression, we transfected Huh-7 cells with pcDNA4 vehicle, naked pcDNA4-PCV2, or pcDNA4-PCV2 reconstituted from MNs, and found that pcDNA4-PCV2 extracted from reconstituted MNs suffered no significant loss in PCV2-open reading frame 2 (PCV2-ORF2) expression levels compared with naked pcDNA4-PCV2 stock solution: both exhibited a signal band of PCV2-ORF2 at 26 kDa (Figure 5D). After long-term storage at 45ºC, the naked pcDNA4-PCV2 lost its initial PCV2-ORF2 expression activity: only 24.4 ± 2.8% of the expression level remained at Day 28 (Figure 5E). Conversely, the pcDNA4-PCV2 in MNs remained intact, with high PCV2-ORF2 expression similarly to stock pcDNA4-PCV2 even at 28 days of storage (Figure 5F). These results indicate that the pcDNA4-PCV2 was protected from damage by MNs and exhibits high PCV2-ORF2 expression that may induce high immunogenicity.
In vivo intradermal delivery of pcDNA4-PCV2 to mouse
We finally vaccinated mice using MN patches containing pcDNA4-PCV2. After their insertion into the skin for 8 min, the MNs had completely dissolved, and 100 holes in μm-scale were evident on the skin's surface in the shape of the MN arrays. The procedure was well tolerated by the mice with no significant erythema, edema, or other effects, and 50 min later, the skin returned to normal appearance (Figure 6A). We also determined the release kinetics of DNA polyplex vaccine from MNs after insertion in mouse skin by measurement of residual DNA in the undissolved MN patch using NanoDrop. The release ratio of DNA polyplex vaccine from bPEI-precoated MNs to epidermis was 22.4 ± 3.9% after 1 min insertion, then increased to 93.4 ± 2.2% at 5 min, which was higher than that of MNs in the absence of bPEI (75.8 ± 3.9%; Figure S6). Comparison with the data of dynamic MNs dissolution in skin (only the bases remained after 5 min of insertion) confirmed most of DNA was located in the needle region of MNs in the presence of bPEI. To further investigate MNs insertion and the penetration depth, the MNs inserted skin of the mice was sliced away for histologic observation after 8 min of insertion, and showed that the SRB released from the MNs was deposited at the puncture sites. The encapsulated SRB was released and diffused to the dermal layer where it exhibited red fluorescence inside the skin tissue, reaching a penetration depth of 318 μm most likely due to its superior mechanical strength and sharp pinpoint (Figure 6B) and confirming the results of the in-vitro porcine skin-insertion study. We also investigated the cell apoptosis in skin after MNs insertion to evaluate the safety of MN patch-based vaccination. We did not find the significant active Caspase-3 in MNs inserted skin tissue after 12 h of MNs insertion compared to no treated skin tissue, indicating that the formulation of MNs and minimal blemishes induced by MNs would not cause any toxicity and side effects (Figure 6C).
(A) Representative photographs of a mouse after insertion of an MN patch encapsulating the DNA vaccine for porcine circovirus Type 2 (pcDNA4-PCV2) to determine the restoration state. (B) Fluorescence micrograph of histologic sections of the skin pierced by the MN patch encapsulating SRB. The arrow shows the insertion depth of the MN patch. (C) IHC on paraffin sections of mouse skin for apoptosis staining by detection of active Caspase-3 normal mouse skin without (top) and with (bottom) MNs insertion. The specimen was collected after 12 h of MNs insertion. (D) PCV2 total virus-specific serum Immunoglobulin G responses after the vaccination of mice with pcDNA4 by intramuscular (IM) injection (pcDNA4 IM), pcDNA4 by bPEI-precoated MNs (pcDNA4 bPEI-precoated MNs), pcDNA4-PCV2 by IM injection (pcDNA4-PCV2 IM), pcDNA4-PCV2 by MNs (pcDNA4-PCV2 MNs), pcDNA4-PCV2 by bPEI-precoated MNs (pcDNA4-PCV2 bPEI-precoated MNs), or pcDNA4-PCV2 by bPEI-precoated MNs after storage at 45ºC for 1 month (pcDNA4-PCV2 bPEI-precoated MNs/1 month). Values are expressed as means ± standard deviation (n = 6). Asterisk indicates a significant difference (Student's t-test, *p < 0.05 compared with pcDNA4-PCV2 IM, **p < 0.001 compared with pcDNA4-PCV2 bPEI-precoated MNs).
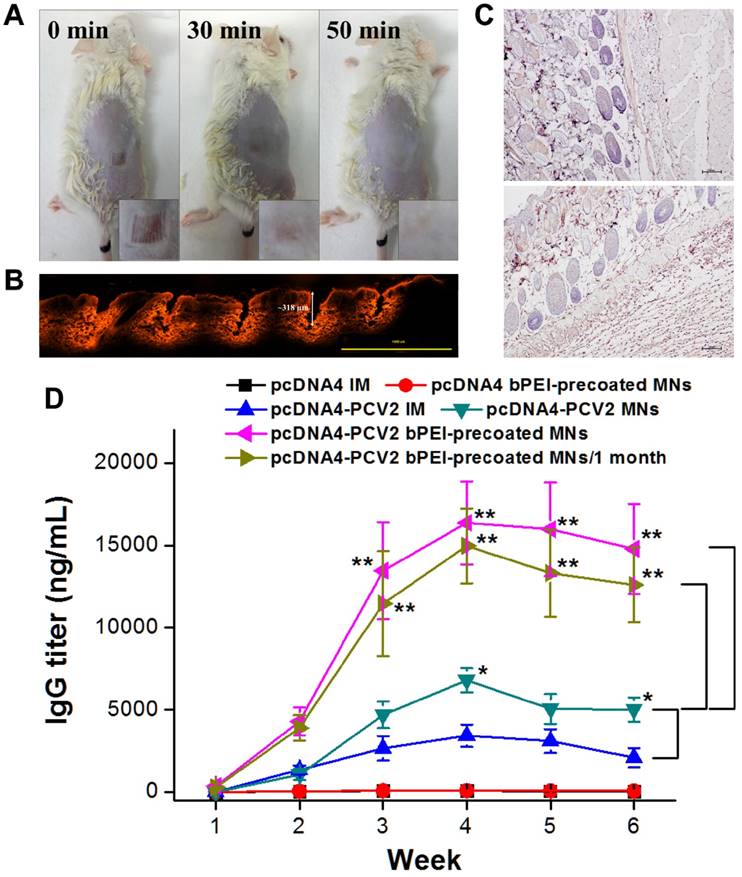
As a final and most important assessment of vaccine thermostability, mice were divided into six groups (pcDNA4 intramuscular [IM], pcDNA4 MNs, pcDNA4-PCV2 IM, pcDNA4-PCV2 MNs, pcDNA4-PCV2 bPEI-precoated MNs, and pcDNA4-PCV2 bPEI-precoated MNs/1 month; six mice per group) and immunized twice at 2-week intervals with 20 μg of pcDNA4-PCV2 vaccine, either with naked pcDNA4-PCV2 liquid formulation or pcDNA4-PCV2 in MNs administered either by IM injection or MN patch. Serum samples were collected once weekly and tested for antibodies to PCV2: the MN-administered group was compared with the positive control, i.e., the IM injection of naked pcDNA4-PCV2 in a liquid formulation.
To monitor the progress of the immune response, serum samples were tested for the presence of PCV2-specific Immunoglobulin G (IgG) by enzyme-linked immunosorbent assay (ELISA) (Figure 6D). Total antigen-specific IgG titers after vaccination with pcDNA4 vehicle administered either by IM injection (black line) or bPEI-precoated MN patches (red line) showed no significant induction of an immune response, indicating the pcDNA4 vehicle and bPEI would not induce significant immune response. However, the specific antibodies were first detected at 2 weeks post-vaccination for pcDNA4-PCV2 administered by IM injection, and increased to a peak in Week 4 of approximately 3421.9 ± 683.9 ng/mL (blue line), the titers slightly increased to 6789.8 ± 735.5 ng/mL administered by MNs (dark cyan line), approximately 1.9-fold higher than the maximum triggered by pcDNA4-PCV2 delivered by IM injection. This may be because the pcDNA4-PCV2 was precisely delivered to the Langerhans cell-rich epidermis (approximately 500-1000 cells/mm2) by MN patches [35, 36]. Likewise, antibodies were significantly induced and increased to a maximum of 16363.7 ± 2539.5 ng/mL in Week 4 in the pcDNA4-PCV2-loaded bPEI-precoated MNs group (magenta line), approximately 2.4-fold higher than the maximum triggered by pcDNA4-PCV2 delivered by without bPEI-precoated MNs and 3.5-fold higher than the maximum triggered by pcDNA4-PCV2 delivered by IM injection, indicating that the polyplex formation can enhance the transfection of DNA vaccine to cells and delivery of pcDNA4-PCV2 using MN patches induced a stronger immune response. Additionally, whereas MN patches stored at 45ºC for 4 weeks could still induce a high concentration of antibodies (14963.7 ± 2274.4 ng/mL) after administration to the skin (dark yellow line), there was no significant difference in antibody induction compared with MN patches stored for 1 day at 25ºC. This finding demonstrates that this fabrication procedure with bPEI precoating is not only increasing the transfection by self-formation of polyplex in MNs but also entrapping more DNA vaccine in the needle region of MNs. Moreover, DNA vaccines were protected in MN patches, allowing them to induce a strong immune response, and can be conveniently stored for a long period of time at 25ºC and 45ºC.
Conclusion
In conclusion, we demonstrated a new procedure to fabricate MN patches that can entrap more DNA vaccines or other negatively charged biomolecules in their needle region and one-step synthesis of DNA polyplex vaccine in MNs. In this study, the concentration of the model vaccine, pcDNA4-PCV2, in the needle region of the MNs was increased 1.5-fold compared with that loaded by gravity alone, and induced a 3.5-fold higher immunogenicity than IM administration. The MNs developed are sufficiently strong and easily inserted into the skin with thumb pressure, and dissolved in the skin within 5 min to release the DNA vaccine. In addition, DNA vaccines in MN patches retain potency after storage for a long time at an elevated temperature (45ºC). These findings provide a promising approach to intradermal DNA-based vaccination that facilitates increased vaccination coverage and also meets the needs of developing countries, where serious infectious diseases are most prevalent.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was financially supported by the Ministry of Science and Technology and National Health Research Institutes, Taiwan (R.O.C.), for the financial assistance provided (MOST105-2314-B-110-001, MOST105-2119-M-182-001 and NHRI-EX106-10502NI). We would also like to thank the Chang Gung Memorial Hospital Microscopy Core Laboratory for the assistance of SEM.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chu LY, Ye L, Dong K. et al. Enhanced stability of inactivated influenza vaccine encapsulated in dissolving microneedle patches. Pharm. Res. 2016;33:868-78
2. Ellebedy AH, Webby RJ. Influenza vaccines. Vaccine. 2009;27:D65-D68
3. Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat. Rev. Genet. 2008;9:776-88
4. Jones S, Evans K, McElwaine-Johnn H. et al. DNA vaccination protects against an influenza challenge in a double-blind randomised placebo-controlled phase 1b clinical trial. Vaccines. 2009;27:2506-12
5. Ferraro B, Morrow MP, Hutnick NA. et al. Clinical applications of DNA vaccines: current progress. Clin. Infect. Dis. 2011;53:296-302
6. Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol. Rev. 2011;239:62-84
7. Ura T, Okuda K, Shimada M. Developments in viral vector-based vaccines. Vaccine. 2014;2:624-41
8. Kim NW, Lee MS, Kim KR. et al. Polyplex-releasing microneedles for enhanced cutaneous delivery of DNA vaccine. J. Control. Release. 2014;179:11-7
9. Endmann A, Klunder K, Kapp K. et al. Cationic lipid-formulated DNA vaccine against hepatitis B virus: immunogenicity of MIDGE-Th1 vectors encoding small and large surface antigen in comparison to a licensed protein vaccine. Plos One. 2014;9:e101715
10. Weiland O, Ahlén G, Diepolder H. et al. Therapeutic DNA vaccination using in vivo electroporation followed by standard of care therapy in patients with genotype 1 chronic hepatitis C. Mol. Ther. 2013;21:1796-805
11. Prausnitz MR, Langer R. Transdermal drug delivery. Nat. Biotechnol. 2008;26:1261-8
12. Glenn GM, Kenney RT, Ellingsworth LR. et al. Transcutaneous immunization and immunostimulant strategies: capitalizing on the immunocompetence of the skin. Expert Review Of Vaccines. 2003;2:253-67
13. Nagao K, Ginhoux F, Leitner WW. et al. Murine epidermal Langerhans cells and langerin-expressing dermal dendritic cells are unrelated and exhibit distinct functions. Proc. Natl. Acad. Sci. USA. 2009;106:3312-7
14. Pearton M, Pirri D, Kang SM. et al. Host responses in human skin after conventional intradermal injection or microneedle administration of virus-like-particle influenza vaccine. Adv. Healthc. Mater. 2013;2:1401-10
15. Donnelly R, Douroumis D. Microneedles for drug and vaccine delivery and patient monitoring. Drug Deliv. Transl. Res. 2015;5:311-2
16. Koutsonanos DG, Compans RW, Skountzou I. Targeting the skin for microneedle delivery of influenza vaccine. Adv. Exp. Med. Biol. 2013;785:121-32
17. Kim YC, Yoo DG, Compans RW. et al. Cross-protection by co-immunization with influenza hemagglutinin DNA and inactivated virus vaccine using coated microneedles. J. Control. Release. 2013;172:579-88
18. Song JM, Kim YC, O E. et al. DNA vaccination in the skin using microneedles improves protection against influenza. Mol. Ther. 2012;20:1472-80
19. Yan G, Arelly N, Farhan N. et al. Enhancing DNA delivery into the skin with a motorized microneedle device. Eur. J. Pharm. Sci. 2014;52:215-22
20. Kim YC, Song JM, Lipatov AS. et al. Increased immunogenicity of avian influenza DNA vaccine delivered to the skin using a microneedle patch. Eur. J. Pharm. Biopharm. 2012;81:239-47
21. Kim YC, Quan FS, Compans RW. et al. Stability kinetics of influenza vaccine coated onto microneedles during drying and storage. Pharm. Res. 2011;28:135-44
22. DeMuth PC, Su XF, Samuel RE. et al. Nano-layered microneedles for transcutaneous delivery of polymer nanoparticles and plasmid DNA. Adv. Mater. 2010;22:4851-6
23. Saurer EM, Flessner RM, Sullivan SP. et al. Layer-by-layer assembly of DNA-and protein-containing films on microneedles for drug delivery to the skin. Biomacromolecules. 2010;11:3136-43
24. DeMuth PC, Min Y, Huang B. et al. Polymer multilayer tattooing for enhanced DNA vaccination. Nat. Mater. 2013;12:367-76
25. Chen MC, Ling MH, Lai KY. et al. Chitosan microneedle patches for sustained transdermal delivery of macromolecules. Biomacromolecules. 2012;13:4022-31
26. Bediz B, Korkmaz E, Khilwani R. et al. Dissolvable microneedle arrays for intradermal delivery of biologics: fabrication and application. Pharm. Res. 2014;31:117-35
27. Choi CK, Lee KJ, Youn YN. et al. Spatially discrete thermal drawing of biodegradable microneedles for vascular drug delivery. Eur. J. Pharm. Biopharm. 2013;83:224-33
28. Yang HW, Ye L, Guo XD. et al. Ebola vaccination using a DNA vaccine coated on PLGA-PLL/γPGA nanoparticles administered using a microneedle patch. Adv. Healthc. Mater. 2017;6:1600750
29. Kumar A, Wonganan P, Sandoval MA. et al. Microneedle-mediated transcutaneous immunization with plasmid DNA coated on cationic PLGA nanoparticles. J. Control. Release. 2012;163:230-9
30. Park JH, Allen MG, Prausnitz MR. Biodegradable polymer microneedles: Fabrication, mechanics and transdermal drug delivery. J. Control. Release. 2005;104:51-66
31. Xiang Z, Wang H, Pant A. et al. Development of vertical SU-8 microneedles for transdermal drug delivery by double drawing lithography technology. Biomicrofluidics. 2013;7:066501
32. Kim JY, Han MR, Kim YH. et al. Tip-loaded dissolving microneedles for transdermal delivery of donepezil hydrochloride for treatment of Alzheimer's disease. Eur. J. Pharm. Biopharm. 2016;105:148-55
33. Wang QL, Zhu DD, Liu XB. et al. Microneedles with controlled bubble sizes and drug distributions for efficient transdermal drug delivery. Sci. Rep. 2016;6:38755
34. Nawwab Al-Deen F, Ma C, Xiang SD. et al. On the efficacy of malaria DNA vaccination with magnetic gene vectors. J. Control. Release. 2013;168:10-7
35. Chen D, Payne LG. Targeting epidermal Langerhans cells by epidermal powder immunization. Cell Res. 2002;12:97-104
36. Seneschal J, Clark RA, Gehad A. et al. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36:873-84
Author contact
![]() Corresponding author: Tel.: +886-7-5252000x5842; fax: +886-7-5250151 E-mail address: howardyangnsysu.edu.tw
Corresponding author: Tel.: +886-7-5252000x5842; fax: +886-7-5250151 E-mail address: howardyangnsysu.edu.tw