Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(13):3260-3275. doi:10.7150/thno.19979 This issue Cite
Research Paper
Direct Macromolecular Drug Delivery to Cerebral Ischemia Area using Neutrophil-Mediated Nanoparticles
Chun Zhang1, Cheng-li Ling1, 3, Liang Pang1, Qi Wang2, Jing-xin Liu1, Bing-shan Wang1, Jian-ming Liang1, Yi-zhen Guo1, Jing Qin1, 4 ![]() , Jian-xin Wang1, 2
, Jian-xin Wang1, 2 ![]()
1. Department of Pharmaceutics, School of Pharmacy, Fudan University & Key Laboratory of Smart Drug Delivery, Ministry of Education, Shanghai 201203, PR China;
2. Institute of Clinical Pharmacology, Guangzhou University of Traditional Chinese Medicine, Guangzhou 510006, PR China;
3. School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 610072, PR China;
4. Key Laboratory of Drug Targeting and Drug Delivery System, Ministry of Education (Sichuan University).
Received 2017-3-8; Accepted 2017-5-10; Published 2017-7-23
Abstract

Delivery of macromolecular drugs to the brain is impeded by the blood brain barrier. The recruitment of leukocytes to lesions in the brain, a typical feature of neuroinflammation response which occurs in cerebral ischemia, offers a unique opportunity to deliver drugs to inflammation sites in the brain. In the present study, cross-linked dendrigraft poly-L-lysine (DGL) nanoparticles containing cis-aconitic anhydride-modified catalase and modified with PGP, an endogenous tripeptide that acts as a ligand with high affinity to neutrophils, were developed to form the cl PGP-PEG-DGL/CAT-Aco system. Significant binding efficiency to neutrophils, efficient protection of catalase enzymatic activity from degradation and effective transport to receiver cells were revealed in the delivery system. Delivery of catalase to ischemic subregions and cerebral neurocytes in MCAO mice was significantly enhanced, which obviously reducing infarct volume in MCAO mice. Thus, the therapeutic outcome of cerebral ischemia was greatly improved. The underlying mechanism was found to be related to the inhibition of ROS-mediated apoptosis. Considering that neuroinflammation occurs in many neurological disorders, the strategy developed here is not only promising for treatment of cerebral ischemia but also an effective approach for various CNS diseases related to inflammation.
Keywords: brain targeting, catalase, neutrophils, PGP, ischemic stroke.
Introduction
Macromolecular drugs, including peptides and proteins, have been shown to be one of the promising treatments of neurological illness [1]. However, the inherent instability in plasma and the weak penetration through the blood brain barrier (BBB) of macromolecular drugs have hindered their further success [2]. Therefore, restricted entry of macromolecular neurotherapeutics to the brain has been a major bottleneck in the treatment of brain-related diseases, especially ischemic stroke (IS) [3]. Even though BBB integrity can be partially compromised during stroke, the reduction in blood supply to the ischemic area and subsequent “no-reflow” seriously impedes drug transport [4, 5]. Hence, an efficient macromolecular drug delivery system with brain targeting ability is highly desired to treat IS.
It has been reported that an ischemic cascade which involves overproduction of reactive oxygen species (ROS), failure of endogenous antioxidants to detoxify these entities, abnormal recruitment of inflammatory cells, and initiation of remarkable levels of cell apoptosis leads to severe brain damage and infarction [6]. Antioxidant enzymes can protect neurocytes from ROS-induced injury and have the potential to improve therapeutic outcomes for IS patients. As a promising neuroprotectant, catalase (CAT) can convert hydrogen peroxide, a representative ROS, to water and molecular oxygen with high turnover rates. It is an alternate source of oxygen and assists in neuroprotection under hypoxic conditions [7]. However, clinical application of CAT is limited by its short half-life, proteolytic degradation, immunogenicity and inability to be delivered into the brain [8].
To address these issues, an effective approach has been reported in which macrophages combined with nanoparticles (NPs) not only protected CAT from degradation but also delivered it across the BBB to the inflammation site [9]. However, the use of carrier cells is limited by invasive procedures, complicated fabrication and unique suitability for chronic diseases [10]. To avoid these disadvantages, endogenous circulatory leukocytes-mediated delivery of nanotherapeutics has gained increasing attention [11]. Detailed reviews have been provided by Dong and Agrahari, respectively, to unveil the current research status and the promising future of leukocyte-mediated delivery of nanotherapeutics in inflammatory diseases [12, 13]. Based on these considerations, it is promising to deliver drugs to disease sites by loading them into NPs, administering intravenously and then specifically uptake by endogenous circulating leukocytes.
We previously developed drug-loaded liposomes modified with a cRGD peptide, which is specifically recognized by αvβ1 integrins that are highly expressed on the surface of monocytes [14, 15]. Although the results confirmed that inflammatory monocytes could be recruited and could transport drugs to injury sites [16], the number of blood monocytes (approximately 5 % of leukocytes) is not sufficient for delivery of drug throughout the entire pathogenesis process during cerebral ischemia. Compared with monocytes, neutrophils, working as secretory and phagocytic cells of the innate immune system, are the most abundant leukocytes in the human body (approximately 50 % of leukocytes). Neutrophils are the first blood-borne cells found in ischemic areas [17]; they reach peak numbers at days 2-4 after transient ischemia and decline thereafter [18]. More importantly, a systemic immune response to ischemic insult engages an increased number of circulating neutrophils in stroke patients [19], which are then recruited and migrate to the brain [20]. It has been reported employing neutrophils as vehicle to deliver NPs into inflammatory site. For instance, Chu et al. revealed that albumin NPs can be internalized and then delivered across blood barrier into inflammatory lungs and tumors by activated neutrophils [21,]. They also demonstrated that the tumor accumulation of albumin NPs could be mediated by neutrophils when co-injected with TA99 antibody [22]. Therefore, a circulatory neutrophil-NP combination carrier may be an ideal platform to achieve brain targeting drug delivery at sequential time points following IS.
CXCR2, a classic chemoattractant receptor, was chosen as a targeting receptor in this study because it is prominently expressed on phagocytic cells, especially neutrophils [23]. As an endogenous degradation product of extracellular collagen, the tripeptide agonist N-acetyl Pro-Gly-Pro (Ac-PGP) exhibits high binding affinity and specificity to the CXCR2 receptor and a low risk of immunogenicity [24]. Ac-PGP has been recognized to be a key chemoattractant in inflammatory diseases and plays an important role in neutrophil influx in inflammatory conditions [25]. Hence, PGP can probably serve as a neutrophil anchoring peptide for brain targeting drug delivery.
Polymeric NPs are the most widely studied protein encapsulating system, which promote the stabilization and delivery efficiency of biopharmaceutical agents [26]. To improve drug loading and targeting of phagocytic cells in the circulatory system, complexation and assembly of proteins with oppositely charged polymers is very important [27]. In the present study, dendrigraft poly-L-lysine (DGL) generation 2 was used as the vector due to its excellent biodegradability and synthetic controllability as well as its excess of positive charge [28]. DGL was coupled with the targeting ligand PGP and with N-succinimidyl 3-(2-pyridyldithio)-propionate SPDP for active neutrophil targeting and improved stability [29-32], yielding a PGP-PEG-DGL-PDP polymer. CAT was modified with cis-aconitic anhydride to increase the charge density for better complexation with PGP-PEG-DGL-PDP through electronic interactions [33-35]. The system, cl PGP-PEG-DGL/CAT-Aco NPs, is illustrated in Figure. 2A.
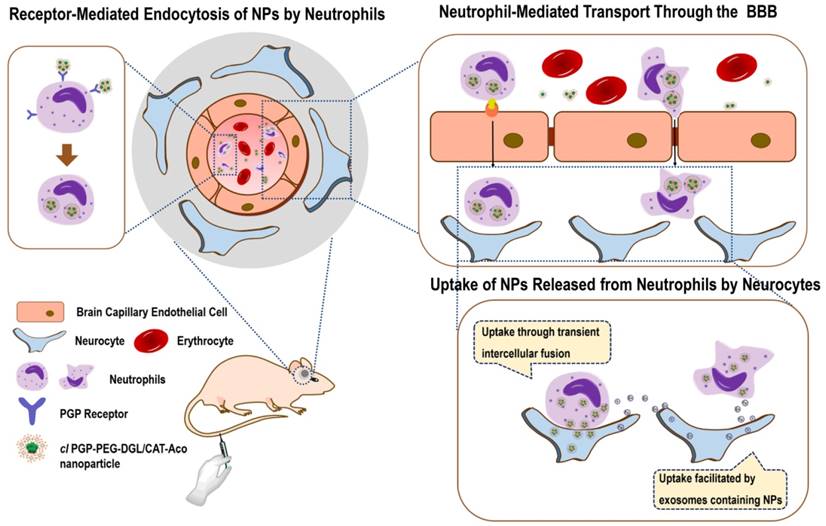
In the system, cross-linkage of the polymer fixes the structure of the NP, suppresses dissociation and increases drug stability [29]. Modification with PGP ligands can endow the NPs with effective and selective susceptibility to phagocytosis in the circulation because of their high affinity to the receptors expressed on the surface of neutrophils. In principle, it is expected that proteins will be released from the carrier cells in the brain if they are not degraded by cellular proteases. The targeting ability of the constructed protein delivery system can be realized in three steps: first, arriving at the brain subregion by crossing the BBB; second, targeting to ischemic brain by neutrophil-mediated inflammatory migration; third, entering the cerebral neurocytes by transient intercellular connections and exosomes (Scheme 1). Thus, the targeting efficiency and the therapeutic efficacy of protein-based drugs for IS could be significantly improved. Considering that neuroinflammation occurs in many neurological disorders, the strategy presented here offers broad potential applications for the treatment of brain diseases.
In vivo processes of thecirculatory neutrophil-mediated multi-brain targeting drug delivery system during brain ischemia.
Methods
Materials and Animals
All reagents were purchased from Sigma unless otherwise indicated. Recombinant Interleukin 8 Receptor Beta (IL8Rb) was from Cloud-Clone Corp (Product no. RPC006Hu01, Wuhan, China). DGL containing 48 primary amino groups (generation 2) was purchased from COLCOM (Montpellier Cedex, France). a-Malemidyl-u-N-hydroxysuccinimidyl polyethylene glycol (NHS-PEG-MAL, MW 3500) was obtained from JenKem Technology (Beijing, China). PGP peptide (Ac-Pro-Gly-Pro-His-His-His-Lys-Cys) was synthesized by China Peptides Co., Ltd. (Suzhou, Jiangsu, China). SPDP was purchased from ProteoChem, Inc. CAT from bovine liver was purchased from Calbiochem (CA, USA). BCA Protein Assay Kit was purchased from Beyotime Co. (Nantong, China). Dihydrorhodamine 123 (DHR) was purchased from AAT Bioquest Inc. (Sunnyvale, USA). TUNEL Assay Kit was obtained from Roche Diagnostics Corp.
The human promyelocytic leukemia cell line HL-60 was purchased from the Cell Bank at the Chinese Academy of Sciences (Shanghai, China). Adult male Sprague-Dawley rats (250-300 g), male C57BL/6 mice (25-30 g) and male BALB/c nude mice (25-30 g) were purchased from Sino-British SIPPR/BK Lab. Animal Co., Ltd. (Shanghai, China). The animal experiment protocol was approved by the Animal Experimentation Ethics Committee of Fudan University.
Synthesis and Characterization of PGP-PEG-DGL-PDP
The primary amino groups on the surface of DGL were specifically reacted with the NHS groups of the bifunctional PEG. NHS-PEG3.5K-MAL was dissolved in PBS 7.4 and then added drop-by-drop to stirred DGL (5 mg/ml) in PBS 7.4 solution and reacted for 1 h at a 5:1 molar ratio at room temperature. The PEG-DGL conjugate was obtained by purification using an ultrafiltration membrane (cutoff = 5 kDa). The MAL groups in PEG-DGL could specifically react with the PGP thiol groups. Thus, the PEG-DGL was mixed with PGP peptide at a 1:2 molar ratio (PEG-DGL: PGP) in PBS 7.4 for 24 h at room temperature, and the reaction product was purified with an ultrafiltration membrane (cutoff = 5 kDa) to obtain PGP-PEG-DGL. A two-fold molar excess of SPDP dissolved in DMF was added to the PGP-PEG-DGL and allowed to react for an additional 1 h under gentle stirring in PBS with EDTA (100 mM Na3PO4, 1 mM EDTA, PH 7.4) at room temperature. A portion of the amino groups of DGL were substituted with pyridyldithiopropionyl (PDP) groups and purified with an ultrafiltration membrane (cutoff = 5 kDa) to obtain PGP-PEG-DGL-PDP polymer in a 10nM pH 7.4 Tris buffer. The above-purified polymer was freeze-dried for preparation of NPs. Then, 3 mg of polymer was redissolved in D2O and analyzed in a 600 MHz spectrometer.
Characterization of the PGP/CXCR2 Interaction Using SPR
Binding was monitored with surface plasmon resonance (SPR) using a BIACORE XTM system. Anti-GST was deposited on a CM5 sensor chip (GE Healthcare) through amide-linkages. Recombinant IL8Rb/CXCR2 with two N-terminal GST tags (10 μg/ml in running buffer composed of 50 mM HEPES, pH 7.0, 0.01% CHS, 0.1% CHAPS, and 0.33 mM synthetic phospholipid blend [dioleoyl] DOPC:DOPS [7:3 wt/wt; Avanti polar lipids])[36] was then applied to the chip coated with anti-GST Ab at a flow rate of 5 L/min. Free PGP and cl PGP-modified NPs were dissolved in running buffer at a defined concentration and flowed over the CXCR2-immobilized CM5 chip to record resonance changes to assess binding affinity.
Preparation of Dendrimer/CAT-Aco NPs
cis-Aconitic anhydride (cis-Aco) was selected to react with free amino groups on the surface of CAT to enhance the negative charge on the protein surface. Briefly, cis-aconitic anhydride was dissolved in DMSO and added to CAT (5 mg/ml) in a pH 8.5 Na2CO3 solution at a 1:5 weight ratio (cis-Aco:CAT). N,N-diisopropylethylamine was added to the mixture, and the mixture was mechanically stirred at room temperature for 2 h. CAT-cis-aconitic anhydride (CAT-Aco) was purified from low molecular weight residuals using gel filtration with a Sephadex G-25 column in 10 mM Tris pH 7.4 and then lyophilized. CAT-Aco was mixed with different DGL derivatives (PEG-DGL, PGP-PEG-DGL, PEG-DGL-PDP, PGP-PEG-DGL-PDP) to electrostatically form NPs. CAT-Aco and the DGL derivative were dissolved in 10 mM pH 7.4 Tris at room temperature [37]. DTT (2 mg/ml) was added to the SPDP-modified DGL derivative (DGL 2.5 mg/ml) solution at a 1:40 volume ratio (DTT:DGL) and immediately vortexed for 15 min to reduce the disulfide bond of PDP to a thiol and then added dropwise to the stirred enzyme solution (1 mg/ml) to achieve a 2.5:1 weight ratio (DGL:CAT-Aco). The mixture was mechanically stirred at room temperature for 20 min. Then, DTT and released 2-thiopyridone were removed with dialysis against 5 L of Tris for 1 h. The SPDP-modified dendrimer/CAT-ACo NPs were cross-linked by oxidation of thiols introduced in the side chains of the lysine units of DGL to obtain cl (cross-linked) NPs during the dialysis process. Non-cl NPs were prepared using the same method described above, except the DGL derivative solution (without PDP groups) was not mixed with DTT.
Characterization of Dendrimer/CAT-Aco NPs
The encapsulation of CAT-Aco in DGL NPs was examined using an acrylamide gel shift assay, with naked CAT-Aco as the control. NPs composed of different weight ratios of PGP-PEG-DGL were mixed with appropriate amounts of 2× loading buffer and then electrophoresed on an 8.0% acrylamide gel containing 5 mM Tris and 50 mM glycine, at pH 8.3, under non-denaturing conditions (in the absence of SDS) to preserve the binding. The protein bands were stained with rabbit polyclonal anti-CAT antibody (Ab 6573, Abcam; 1:1000) and visualized with Immobilon Western Chemiluminescent HRP substrate (Millipore).
cl PGP-PEG-DGL/CAT-Aco NPs were freshly prepared with a specified ratio (2.5:1, DGL to CAT-Aco, w/w). The mean diameter and zeta potential of the NPs were determined with a Zeta Potential/Particle Sizer Nicomp 380 ZLS system (PPS Nicomp Particle Size System, USA). The morphology of the cl PGP-PEG-DGL/CAT-Aco NPs was examined under a high-resolution transmission electron microscope (TEM) (JEM-2010HT, Japan).
Cell Culture
HL-60 cells (a human promyelocytic leukemia cell line) were cultured in Iscove's modified Dulbecco's minimum essential medium (IDMEM; Gibco Ltd, Paisley, UK) with 20% FBS and 1% penicillin-streptomycin (PS), and differentiated into PMN-like cells by adding 1.3% DMSO for 96 h as previously reported [24]. PC12 (a neuron-like rat pheochromocytoma cell line) neuronal cells were maintained in RPMI 1640 medium (Corning Cellgro, Mediatech, USA) supplemented with 10% FBS and 1% PS and 5% nonessential amino acids. Cells were maintained in an incubator in a controlled atmosphere of 37 °C, 95% relative humidity and 5% CO2.
Cellular Uptake and Mechanism Studies In Vitro
HL-60 cells were added into each well of a 24-well plate (2.0x105 cells/well) and then were co-incubated with FITC-labeled CAT alone or with different NPs at a CAT concentration of 20 μg/ml for 30 min at 37 °C in medium containing 10% FBS. After being washed with PBS twice, the cells were dispersed in DMEM and analyzed using flow cytometry (FACSCalibur, BD, USA).
The inhibition effect of different inhibitors on the cellular uptake of NPs by HL-60 cells was also measured. The cells were treated as described for the above procedures. After checking the morphology under a microscope, free PGP (0.5 mM), SB225002 (2 μg/ml), phenylarsine oxide (1 µM), filipin III (6 μg/ml) or colchicine (2 μg/ml) was added to each group of wells. HL-60 cells were incubated at 37 °C for 10 min. At the end of the incubation, the cells were rinsed with PBS twice and cultured in fresh medium. Subsequently, FITC-labeled cl PGP-PEG-DGL/CAT-Aco NPs with different inhibitors (the concentration of inhibitors was the same as that used for pre-incubation) was added to the corresponding group of wells. After 30 min of incubation with a DGL concentration of 100 μg/ml at 37 °C, the cell suspension was collected. The cells were washed three times with PBS and analyzed using flow cytometry. At least 10,000 events (cells) were analyzed for each sample.
Intracellular Trafficking Studies
To investigate the integrity of the NPs in the cells, first, DGL derivatives were labeled with BODIPY and CAT was labeled with FITC, and then, cl PGP-PEG-DGL/CAT-Aco NPs were prepared with the labeled molecules. To investigate the subcellular co-localization of NPs in the cells, BODIPY-labeled CAT was applied to complex with DGL, while the acidic endosomes were labeled with LysoTracker Green.
HL-60 cells were plated in 2-cm2 dishes at a density of 2.5 × 106 cells/dish and were then pretreated with LysoTracker Green (100 nM) for 60 min to label the acid compartments, followed by incubation with DAPI (0.05 mg/ml) for 5 min. After the nuclei were stained, the cells were incubated with BODIPY-labeled cl PGP-PEG-DGL/CAT-Aco NPs at a CAT concentration of 100 µg/ml for 5 min, 15 min 30 min, or 60 min. The mixture was harvested and then centrifuged at 500 g for 5 min at 4 °C. After being washed with PBS twice, cells were dispersed in PBS and fixed using Glycerol Jelly Mounting Medium. In addition, cells were treated with double dye-labeled cl PGP-PEG-DGL/CAT-Aco complexes as described previously.
Cell-to-Cell Contact for NP Transfer
HL-60 cells were plated in 6-well plates (3 × 107 cells/well) and incubated with cl PGP-PEG-DGL/CAT-Aco NPs labeled with CAT-BODIPY (0.25 mg/ml DGL) for 1 h and were then cultured in 2 ml of DMEM supplemented with 4 µl of DiO (10 mg/ml) for 30 min at 37 °C. After being washed with HBSS twice, cells were dispersed in HBSS and added to PC12 neuronal cells. The appearance of NP-laden lipids from carrier cells (HL-60s) in receiver cells (PC12 cells) was monitored with confocal microscopy at various time points.
Exosome-facilitated NP Transfer
HL-60 cells labeled with DiO were loaded with BODIPY 650/665-labeled NPs in the same manner as described above. Then, cells were supplemented with 20 μl of IFN-γ (20 ng/ml) and 1 μl of LPS (1 mg/ml) in 2 ml of HBSS solution for 2 h at 37 °C. Meanwhile, the cells were observed with confocal microscopy. Subsequently, the suspension was collected and filtered twice through low protein-binding 0.45 μm filters and then purified by gel filtration on a Sephadex G-25 column in 10 mM PH 7.4 Tris buffer to remove IFN and LPS. Isolated exosomes with encapsulated NPs were added to PC12 neuronal cells, and the dynamics of NP accumulation in receiver cells were observed with confocal microscopy.
Model of Focal Cerebral Ischemia
Transient MCAO mouse models were constructed according to a previously reported protocol, with minor modifications. Briefly, after 12 h of fasting, adult male BALB/c nude or C57BL/6 mice (25-30 g) were anesthetized with 10% chloral hydrate (6 ml/kg) via intraperitoneal (i.p.) injection. The left external carotid artery was incised under an operating microscope, and a monofilament nylon thread (Sunbio Biotech, China) was inserted through this incision into the internal carotid artery and further until it reached the bifurcation of the middle cerebral artery (MCA), a distance of up to 9-10 mm. A rounded tip coated with poly-L-lysine was used to occlude the entrance to the MCA and block the blood supply to the ipsilateral hemisphere of the mouse brain. After occlusion for 1.5 h, the filament was slowly withdrawn, and 1 ml of saline was administered via i.p. injection.
Adult male Sprague-Dawley rat (250-300 g) MCAO models were also constructed according to the protocol described above.
Uptake of NPs by Neutrophils in vivo
To quantify the uptake of NPs by granulocytes, NPs labeled with CAT-BODIPY 650/665 (7.5 mg/kg DGL) were injected into rats subjected to MCAO. Whole blood specimens were taken from rat eyes after 5 min, 15 min and 30 min. The red blood cells were lysed using RBC Lysis Buffer (eBioscience, San Diego, CA), and the cell suspension was washed with PBS. Then neutrophils were separated from total white blood cells by flow cytometry based on cellular size and granularity in the dot plot of forward scatter versus side scatter as previously described [38, 39]. The combination efficiency of the neutrophils with fluorescent NPs were measured by calculating BODIPY650/665 positive percentage of neutrophils with flow cytometry.
Qualitative assessment of NP internalization by neutrophils was carried out in vivo. Fluorescence-labeled NPs were injected into rats. At 15 min post-injection, granulocytes were isolated from abdominal aortic blood samples using OptiPrep™ Density Gradient Medium (Axis-Shield PoC AS, Oslo, Norway). Neutrophils with NPs were isolated with fluorescence activated cell sorting (FACS). The uptake of NPs was observed and recorded with confocal microscopy and TEM.
In Vivo Imaging
Male nude mice with IS were used for brain targeting evaluation. cl Dendrimer/CAT-Aco NPs were labeled with BODIPY 650/665 (Molecular Probes). and were then injected to the nude mice at a dose of 200 μg DGL/mouse 24 h post-establishment of MCAO model. At defined time points, fluorescence images of the brain were acquired using an in vivo imaging system (IVIS Spectrum, Caliper, USA). Mice were sacrificed, and the brains were dissected 3 h after injection to check the relative accumulation. In addition, the IS nude mice were administrated with BODIPY 650/665-labeled NPs as descripted above. After 1 h, the main organs were dissected for ex vivo fluorescence imaging.
Transport across the BBB In Vivo
To understand the targeting mechanism of cl PGP-PEG-DGL/CAT-Aco NPs, an immunofluorescence assay was performed. BODIPY 650/665-labeled NPs were intravenously administered to IS mice 24 h after reperfusion. Approximately 1 h later, mice were anesthetized, and their hearts were perfused with PBS followed by 4% formaldehyde in PBS for further study. Then, the brains were removed, fixed overnight in 4% formaldehyde, dehydrated in 15% sucrose solution until subsidence (12 h), and then in 30% sucrose until subsidence (24 h). Brains were frozen with O.C.T (Sakura, Torrance, CA, USA) at 80 °C and cut into 20-μm slices. Sections were blocked with 5% goat serum at 37 °C for 1 h. Microvessels were labeled with anti-CD31 antibody, neurons were stained with anti-MAP2 antibody, and DAPI was used for nuclear counterstaining. The distribution of fluorescence was visualized with a confocal microscope.
In Vivo MRI Studies
In vivo MRI images were acquired in C57 mice at 3 h, 24 h and 72 h post-establishment of the stroke model. Animals were anesthetized by inhalation of 2% isoflurane and then positioned in the magnet of a 7.0 T small animal MR scanner (Bruker PharmaScan, Germany). The mice were kept warm with a heating pad, and their respiration rates were monitored. Axial fast spin-echo T2-weighted magnetic resonance (T2W-MR) (repetition time ms /echo time ms: 3000/36, 1 average) images were acquired to show the ischemic territories. Mice were injected i.v. with NPs, PBS, or CAT-Aco alone (250 U/mouse) immediately following MRI, and MR examinations were repeated 48 h after treatment. The infarct volume visualized in the T2W images was quantified using the NIH Image J program and normalized to the volume of half the contralateral hemisphere.
Statistical Analysis
The data are presented as the mean ± standard deviation unless otherwise indicated. Student's t-test and one-way analysis of variance (ANOVA) were applied to determine statistical significance between two groups and among more than two groups, respectively. A value of P < 0.05 was considered to be significant.
Results and Discussion
Preparation and Optimization of CAT-Aco NPs
Characterization of PGP-PEG-DGL-PDP
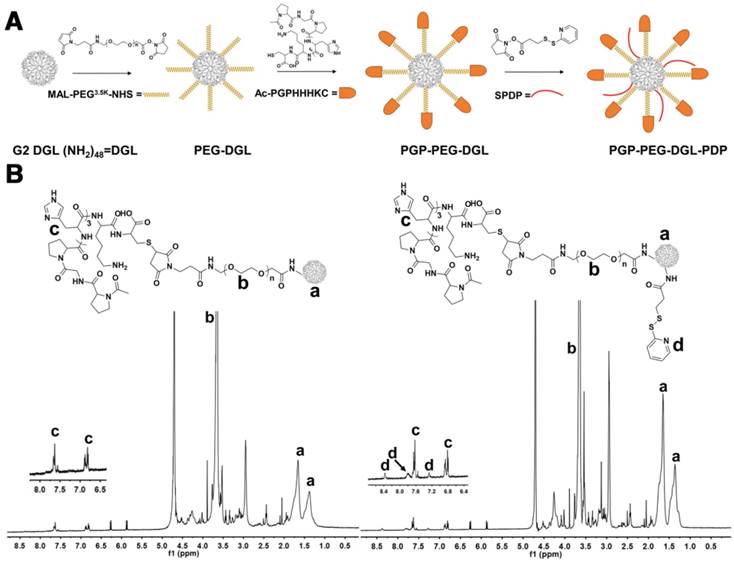
PGP-PEG-DGL-PDP was synthesized as illustrated in Fig. 1A. In 1H NMR spectra, the peaks between 1 and 2 ppm represented the protons of the three CH2 units in lysine (Figure 1B, a). Peaks near 3.6 ppm indicated repeat PEG units (Figure 1B, b). The disappearance of the characteristic peak of the MAL group at 6.7 ppm resulted from the reaction between the MAL group in PEG and the thiol group in the PGP peptide. The peak at approximately 7.6 ppm and 6.8 ppm represented the protons in the PGP histidine units, indicating the presence of PGP peptide (Figure 1B, c). The short peaks between 7.0 and 8.5 ppm in the 1H NMR spectra of PGP-PEG-DGL-PDP represented the pyridine ring in PDP (Figure 1B, d). Thus, the 1H NMR spectra confirmed the successful synthesis of PGP-PEG-DGL-PDP.
(A) Synthetic route for PGP-PEG-DGL-PDP. (B) The 1H-NMR spectrum of PGP-PEG-DGL-PDP in D2O at 600 MHz.
Interaction of the PGP Peptide with Receptor
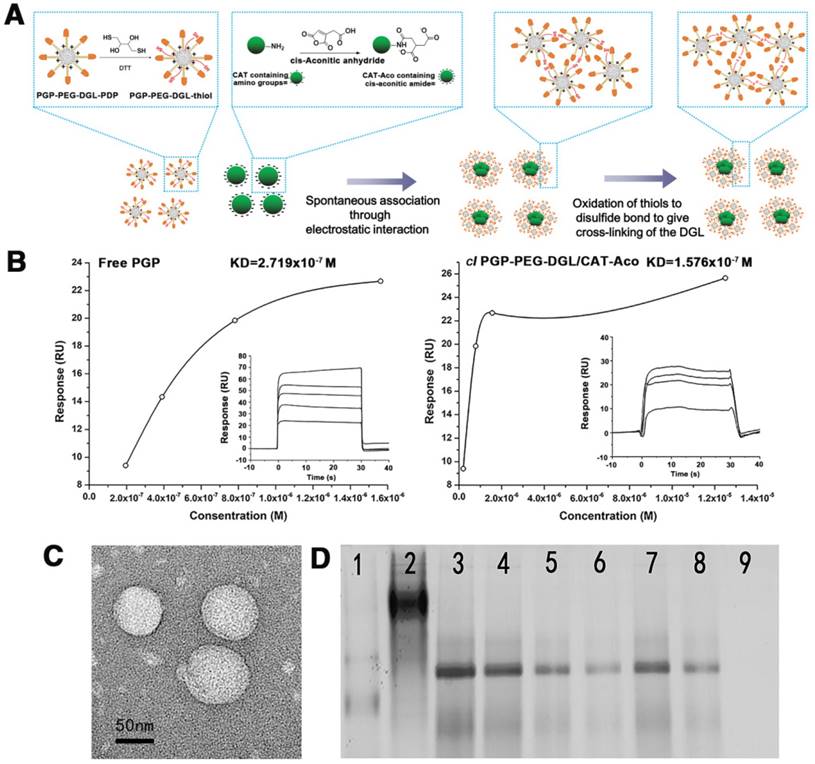
The preparation process of cl PGP-PEG-DGL/CAT-Aco NPs is illustrated in Figure 2A. The binding affinity of PGP peptides with the targeted receptors is crucial in this innovative design. SPR was conducted to assess the capacity of PGP peptide to bind CXCR2 immobilized on a BIAcore sensor chip coated with a lipid bilayer. As shown in Figure 2B (insert), the interaction between PGP and CXCR2 was dose dependent, which indicated that the PGP peptide was capable of binding to CXCR2 immobilized on the sensor chip. Free PGP and cl PGP-PEG-DGL/CAT-Aco NPs displayed similar binding affinity to CXCR2, and their equilibrium dissociation constants (KD) were 2.719x10-7 and 1.576x10-7 M, respectively. The SPR results indicated that conjugation of the PGP peptide on the surface of the NP did not alter its binding affinity with CXCR2.
(A) The construction of the cl PGP-PEG-DGL/CAT-Aco NP system. (B) The interaction between PGP/CXCR2 was assessed with SPR (main graph). The saturation curve indicated plots of RU vs. analyte concentration from which an average dissociation constant (KD) can be obtained in both (left panel) and (right panel) (inset). (C) Transmission electron micrograph of the cl PGP-PEG-DGL/CAT-Aco NPs. (D) CAT-Aco mobility shift assay of dendrimer/CAT-Aco NPs. 1: Protein marker; 2: native CAT; 3: naked CAT-Aco; 4-6: non-cl PGP-PEG-DGL/CAT-Aco NPs; 7-9: cl PGP-PEG-DGL/CAT-Aco NPs at weight ratios (DGL:CAT-Aco) of 0.3:1, 1.25:1 and 2.5:1, respectively.
Characterization of Dendrimer/CAT-Aco NPs
As shown in Table S1, the size and zeta potential of the NPs were approximately 100 nm and 5.0 mV, respectively. Conjugation of the peptide ligand did not significantly affect the properties of the NPs. The particle size of the cl NPs was approximately 100 nm, with a positive charge, and favorable for adsorption to the negatively charged membrane of phagocytic cells and subsequent internalization [40]. The transmission electron micrograph (Figure 2C) showed that cl PGP-PEG-DGL/CAT-Aco NPs had a well-formed spherical shape.
The results of a gel shift assay shown in Figure 2D indicate that modified CAT could be partially encapsulated by a non-cl vector but completely encapsulated by the cl vector. Compared with naked CAT-Aco, there was no electrophoretic shift when the weight ratio of DGL to CAT-Aco was greater than 2.5, because the mobility of the CAT-Aco was significantly slowed.
Besides the qualitative assessment by acrylamide gel shift assay, we also measured the amount of protein entrapped by the dendritic polymers by CAT-Aco complexation efficiency [41]. Ultracentrifugation was used to separate CAT-loaded nanoparticles from the aqueous suspension medium. Absorbance of free CAT-Aco (at 404 nm) in the supernatant was tested and the corresponding concentration of free CAT-Aco was determined with a standard curve. The complexation efficiency of was calculated using the formula as reported previously [1]. In the present study, the complexation efficiency of CAT-Aco in cl PGP-PEG-DGL/CAT-Aco NPs was 70.69±0.46%, which was much higher than that in non-cl PGP-PEG-DGL/CAT-Aco NPs of 58.83±2.28%.
Cytotoxicity of NPs in Neutrophil-like Differentiated HL-60 cells
HL-60 neutrophils were used as an in vitro model to evaluate the cytotoxicity of non-cl and cl PGP-PEG-DGL/CAT NPs (Figure S1A). The concentration of DGL ranged from 20 to 500 μg/ml. The cells treated with cl NPs showed higher viability (lower cytotoxicity) than those treated with non-cl NPs. No apparent decrease in cell viability was observed when the concentration of cl NPs was higher than 500 μg/ml. The percentage of viable cells after treatment with cl NPs remained higher than 70% at NP concentrations lower than 200 μg/ml. The cytotoxicity of non-cl NPs increased when the concentration was lower than 80 μg/ml but decreased when the concentration of DGL was higher than 80 μg/ml. This might be caused by the release of DGL, which can interact with negatively charged cellular membranes and other large-molecules through its polycation, as documented in the literature [42].
Preservation of Catalase Activity Against Degradation in Carrier Cells
The ability of different NPs to protect the enzymatic activity of CAT inside neutrophils was assayed (Figure S1B&C). HL-60 cells were incubated with CAT alone or PGP-PEG-DGL/CAT-Aco complex for 1 h, washed with PBS, and then incubated with CAT-free media for different lengths of time. The activity of CAT released into the media and retained in the cells was determined by examining the rate of hydrogen peroxide decomposition. The results clearly indicated that cross-linked NPs showed significant protection of CAT inside neutrophils compared to free CAT and non-cl NPs. CAT loaded in non-cl NPs was degraded in cells to a lesser extent than the “naked” enzyme (Free CAT-Aco) before 4 h. In medium, the enzyme activity of both cl NPs and non-cl NPs increased with time. Preservation of CAT enzymatic activity in neutrophils by the NPs was primarily due to Protection from the acid in endocytic comopartmentsin the cells by the “proton sponge effect” of the amino groups on the surface of the NPs [43].
Targeting Mechanism Investigation In Vitro
Comparison of the Uptake of Different CAT-Aco NPs by HL-60 cells
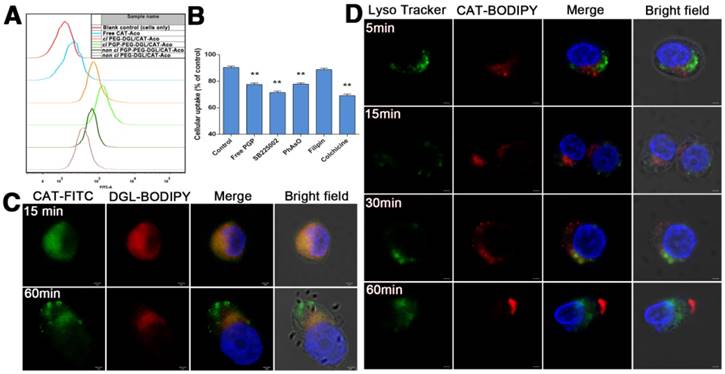
The cellular uptake of FITC-labeled “naked” CAT and various DGL vector/CAT-Aco NPs were compared in HL-60 cells using a flow cytometric analysis. From the results shown in Figure 3A & S2, we can find that the uptake of CAT was enhanced after encapsulating into NPs. The effect was further facilitated by the modification of PGP and the cross-linking of NPs. Thus, the cl NPs were selected and studied in further experiments.
(A) Flow cytometric determination of fluorescence intensity within HL-60 cells treated with FITC-labeled catalase NPs with or without modification of PGP ligand. (B) Inhibition of the cellular uptake of cl PGP-PEG-DGL/CAT-Aco NPs. The data represent the mean ± SD, n=3. **P<0.01, compared with control group. (C) Confocal images of HL-60 cells incubated with cl PGP-PEG-DGL/CAT-Aco NPs for 15 min and 60 min. Green: FITC-labeled catalase. Red: BODIPY 650/665-labeled DGL. The scale bar represents 2 μm. (D) Confocal images of HL-60 cells exposed to cl PGP-PEG-DGL/CAT-Aco NPs for 5 min, 15 min, 30 min and 60 min. Green: LysoTracker Green used to label acidic endosomes. Red: BODIPY 650/665-labeled catalase. Bars represent 2 μm.
Mechanism of Cell Uptake of NPs
HL-60 cells have been intensively applied as a reliable cell line for evaluation of the functional regulation of neutrophils [44]. To clarify the endocytic mechanism of cl PGP-PEG-DGL/CAT-Aco NP uptake, HL-60 cells were pretreated with different endocytic pathway inhibitors. As shown in Figure 3B, the results of the competition assay revealed that the addition of free PGP peptide [25] and SB225002, a CXCR2 receptor antagonist [45], significantly inhibited the cellular uptake of cl PGP-PEG-DGL/CAT-Aco NPs, which indicated that the PGP modification is crucial for effective internalization of NPs by neutrophils through specific binding to CXCR2 receptors on HL-60 cells. Other inhibitors involved in various endocytic processes were also studied. PhAso, an inhibitor of clathrin-dependent endocytosis [46], also remarkably decreased the cellular uptake of cl PGP-PEG-DGL/CAT-Aco NPs. In contrast, filipin III [47], an inhibitor of caveolin-mediated endocytosis, which is relevant to the surface charge of the NPs, had no effect on the uptake of cl PGP-PEG-DGL/CAT-Aco NPs, Meanwhile, treatment with colchicine, which could signify particle size-related micropinocytosis, inhibited the uptake of NPs. Hence, the results show that clathrin-mediated endocytosis and micropinocytosis were primarily responsible for the internalization of cl PGP-modified NPs by neutrophils.
Intracellular Trafficking of NPs
The integrity of drug-loaded NPs in carrier cells is very important for delivery of the enzyme to the brain. To investigate the structure of cl PGP-PEG-DGL/CAT-Aco NPs in HL-60 cells, we labeled CAT with FITC and DGL with BODIPY 650/665 and observed their intracellular localization in HL-60 cells with confocal microscopy (Figure 3C). As expected, with the cl NPs, total co-localization of the polymer and CAT was observed inside HL-60 cells after 15 min of incubation. Interestingly, when the NPs were incubated with cells for 60 min, a portion of them were dissociated and separation of the CAT and DGL vector fluorescence could be observed, which was presumably due to cleavage of NP disulfide bonds while they were in the cells [29, 33].
Co-localization of the NPs and acidified endosomes was detected using confocal microscopy (Figure 3D). Fluorescently labeled CAT was used in cl PGP-PEG-DGL/CAT-Aco NPs and acidic endosomes were stained with LysoTracker Green. Initially, cl NPs were found to be mostly distributed in the cytoplasm (5 min), and then, they partially co-localized with acidic endosomes at 15 min and 30-60 min while mainly presented in the cytoplasm. The results indicated that cl PGP-PEG-DGL/CAT-Aco NPs might not be primarily taken up by HL-60 cells via endosome-lysosome pathways. This intracellular process might shield most proteins from degradation by cellular proteases. Endosomal escape of cl NPs was in agreement with the results reported by many other investigators concerning the “proton sponge” effect induced by amino groups on the surface of NPs [9, 48]. In addition, the acidified endosomes might to a certain degree induce charge conversion of modified CAT (CAT-Aco) to the original protein (CAT) [33].
Brain-targeting Effect In Vitro
Transwell migration assay was used to mimic the fate of neutrophils containing nanoparticles migrate across a monolayer of endothelial cells (HUVECs) and then deliver the nanoparticles to neuron cells [49, 50]. HUVECs were grown to confluency on porous membranes for 9 days followed by co-cultured with PC12 neuronal cells for 3 days. PC12 neuronal cells were labeled with cell tracer (CFSE) in the lower chamber. Meanwhile, BODIPY 650/665-labeled NPs were loaded with neutrophil-like differential HL-60s stained with HOECHST. Then the HL-60 cells were collected and added to the upper chamber above confluent HUVECs. IL-8 was added into the lower chamber to establish a chemotactic gradient for neutrophil migration across a monolayer of endothelial cells (HUVECs). Quantitative measurement of BODIPY-positive cells by flow cytometry was shown in Figure S3. It was found that the number of neutrophils and neuron cells containing NPs significantly increased in lower chamber with the stimulation of IL-8. The results demonstrated that inflammation promotes the transendothelial migration of NPs via neutrophils, which were contributable to the upregulation of nanoparticle uptake by neuron cells. Confocal microscopy images also clearly showed that NPs (red) released from neutrophils (blue) were co-localized with neuron cells (green) when IL-8 was added to the lower chamber. (Figure S4).
Cell-to-Cell Interaction for the Transfer of NPs
When neutrophils carrying NPs are recruited to the sites of inflammation in the brain, it is of interest to know how the enzyme-loaded NPs are delivered into neuronal cells. A time course of NPs labeled with CAT-BODIPY 650/665 transferred from HL-60 cells to PC12 neuronal cells was observed using confocal microscopy. HL-60 cells stained with DiO dye (green) were loaded with BODIPY-labeled NPs (red), collected and then placed on top of PC 12 neuronal cells (contact). Accumulation and co-localization of HL-60 lipids (green) with NPs (red) in non-stained PC12 neuronal cells were manifested in yellow (Figure 4A, shown with arrows). The data revealed that cell-to-cell contact facilitated the transfer of NPs along with lipid compartments from HL-60 cells to PC12 neuronal cells.
(A) Transfer of NPs from HL-60 cells to PC12 neuronal cells through intercellular fusion. Bar represents 10 μm. (B) Facilitated release of NPs from activated HL-60 cells. Bar represents 10 μm. (C) Kinetic transport of NPs via exosomes from HL-60 cells to PC12 neuronal cells. Bar represents 20 μm.
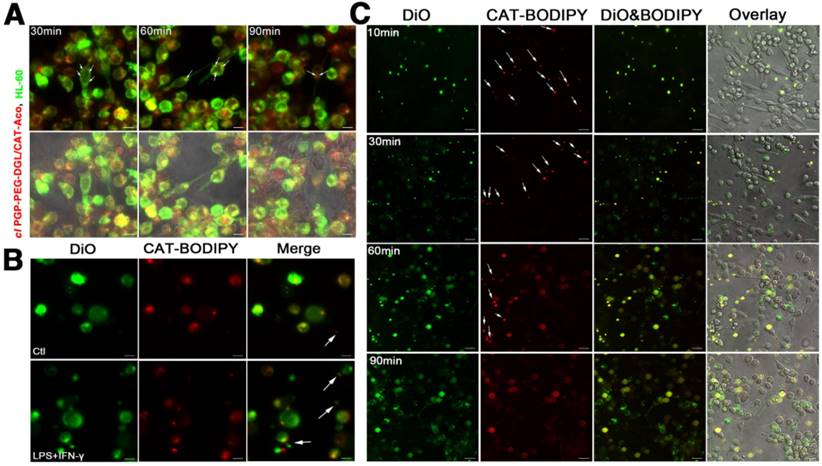
Effect of Exosomes during the Transfer of NPs
To investigate whether exosomes facilitate the transfer of NPs from neutrophils to neuronal cells, DiO (green)-labeled HL-60 cells were pre-incubated with NPs labeled with CAT-BODIPY 650/665 (red) and were visualized using confocal microscopy after two hours of incubation with or without stimulation of lipopolysaccharide (LPS) and interferon gamma (IFN-γ). Few exosomes were observed in non-activated neutrophils; meanwhile, in LPS- and IFN-γ-activated neutrophils, exosomes were clearly secreted from the cells, suggesting release of NPs from the carrier cells at the inflammation site (Figure 4B). The exosomes with encapsulated NPs released from neutrophils into the extracellular medium were harvested and purified. Then, the isolated exosomes were added to neuron-like differentiated PC12 neuronal cells. The dynamics of NP accumulation within receiver cells were visualized with confocal microscopy (Figure 4C). Images clearly showed that the exosomes could be adsorbed to the surface of the receiver cells and fuse with the cellular membrane. Discharge of NPs into the cytoplasm of neuronal cells resulted in diffuse fluorescence. The phenomenon could be explained by NP incorporation into exosomes, which can increase binding of the drug to lipid membranes of receiver cells.
Evaluation of the In Vivo Targeting Effect
Uptake of NPs by Neutrophils In Vivo
Flow cytometry, transmission electron microscopy and confocal microscopy were utilized to elucidate the role of circulating leukocytes, mainly neutrophils, in transporting NPs to the brain. Circulating neutrophils were isolated using RBC Lysis Buffer from the blood of rats given cl PEG-DGL/CAT-Aco and cl PGP-PEG-DGL/CAT-Aco NPs. The extent of fluorescence-labeled NP endocytosis was detected with flow cytometry over time after injection of the NPs (Figure 5A & S5). Cellular uptake results reflected that cl PGP-PEG-DGL/CAT-Aco NPs were rapidly internalized by neutrophils 5 min postinjection followed by overtime elimination. It was evident that cl PGP-PEG-DGL/CAT-Aco NPs displayed significantly higher cellular uptake than unmodified cl PEG-DGL/CAT-Aco NPs, especially at shorter time points.
(A) Flow cytometry assay of NP endocytosis by neutrophils isolated from MCAO rat plasma at different times. The data shown represents the mean ± SD, n=5. P<0.05, compared with the cl PEG-DGL/CAT-Aco group. (B) TEM images of NPs inside the circulatory neutrophils of MCAO rats after injection of cl PGP-PEG-DGL/CAT-Aco. Some typical NPs are indicated by arrows. (C) Fluorescence microscopy images of NPs taken up by neutrophils in rat blood after i.v. injection of double dye-labeled cl PGP-PEG-DGL/CAT-Aco NPs. Bars represent 5 μm. (D) Distribution of BODIPY 650/665-labeled CAT in the brain subregion after administration of different cl dendrimer/CAT-Aco NPs. Blood vessels were stained with anti-CD31 (green). (E) Uptake of fluorescence-modified CAT into neurons of MCAO mice after injection of cl NPs. Neurons were labeled with MAP-2 (green). Red represents the drug. I.H., ischemic hemisphere; C.H., control hemisphere. Bar represents 10 μm.
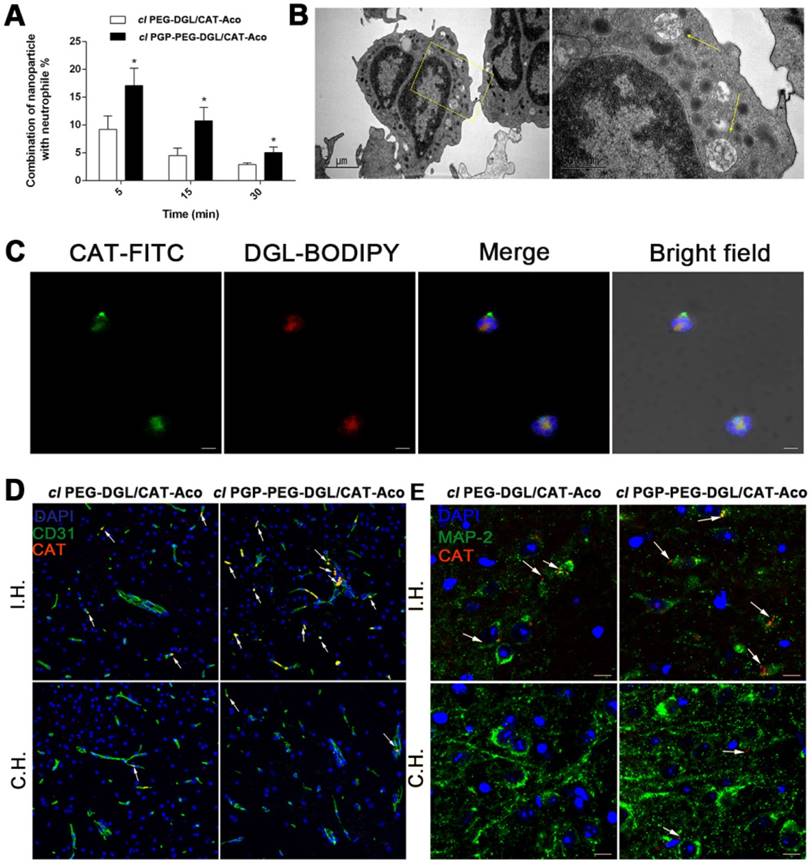
To obtain further evidence to confirm endocytosis of NPs by neutrophils, we isolated neutrophils from rat blood 15 min post-administration of NPs labeled with CAT-BODIPY using OptiPrep™ Density Gradient Medium. The neutrophils with NPs were isolated using FACS. The presence of NPs in the endocytic compartments of cells was identified by TEM (Figure 5B). Confocal microscopy images illustrated that the components of NPs, both DGL vector and CAT, could be simultaneously detected in neutrophils, indicating that NPs were effectively taken up by neutrophils (Figure 5C).
Evaluation of NP Brain targeting effect
The brain targeting efficiency of cl PGP-PEG-DGL/CAT-Aco NPs and cl PEG-DGL/CAT-Aco NPs was compared using two parallel experiments in nude mice pre-treated with transient MCAO. Extravasation of CAT in vesicular structures was observed mostly in ischemic regions 1 h after treatment with cl PGP-PEG-DGL/CAT-Aco NPs (Figure 5D). The distribution of cl PGP-PEG-DGL NPs was also found to a relatively greater extent in healthy brain capillaries in stroke mice, which could be explained by the fact that CXCR2, as a PGP-specific receptor, is also expressed on brain microvascular endothelial cells [51]. It is worth noting that a high combination of cl PGP-PEG-DGL/CAT-Aco NPs with endothelial cells is beneficial for treatment of cerebrovascular disorders, including IS, due to the protection of the BBB from oxidative damage.
Furthermore, the fluorescence of cl PGP-PEG-DGL/CAT-Aco NPs, which was co-localized with neutrophils, was higher than that of cl PEG-DGL/CAT-Aco NPs in brain ischemic area (shown as arrows). It depicted that PGP modified NPs, which was effectively taken up by neutrophils, showed higher drugs accumulation in ischemic subregion. (Figure S6). The phenomenon presumably attributes to the transendothelial migration of neutrophils with NPs. Co-localization of the fluorescence-labeled CAT in cl PGP-PEG-DGL/CAT-Aco group and MAP2 antibody-labeled neurons was observed, indicating entry of translocated vesicular structures into recipient cells in MCAO mice (Figure 5E). Unsurprisingly, the amount of labeled enzyme in cl PGP-PEG-DGL/CAT-Aco group was much more than that in cl PEG-DGL/CAT-Aco group.
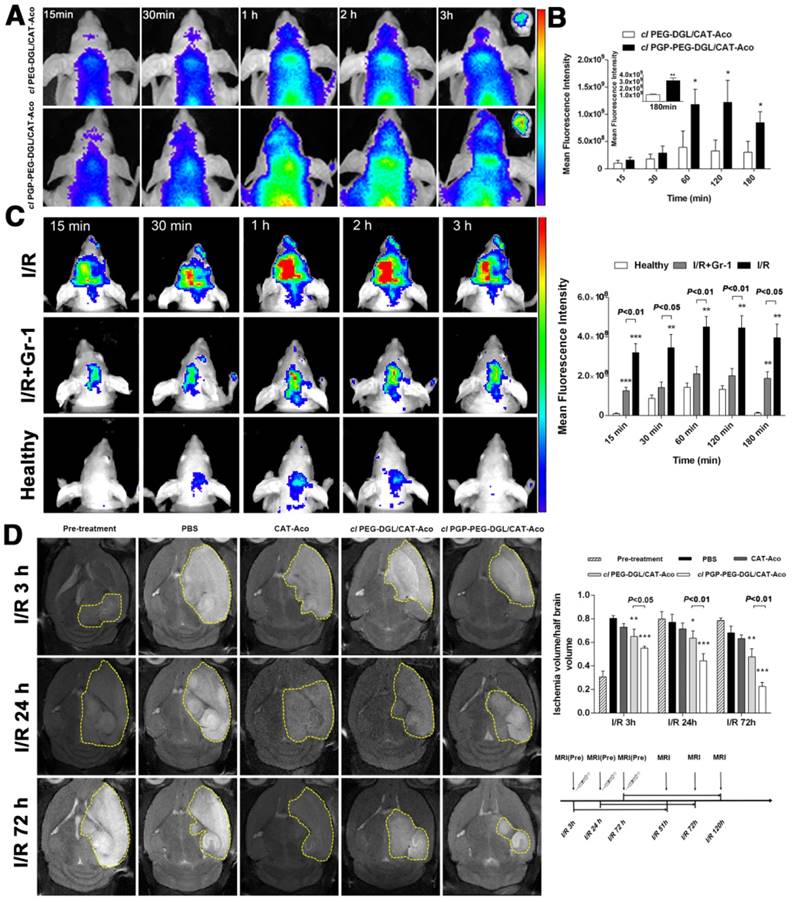
Images of NPs loaded with BODIPY 650/665-labeled CAT are shown in Figure 6A. The in vivo imaging results reflected that the protein drug, loaded in cl PGP-PEG-DGL/CAT-Aco NPs, accumulated in brain in 2 h followed by a slow elimination (Figure 6A). Semi-quantitative ROI analysis revealed that an obviously enhanced brain targeting efficiency of cl PGP-PEG-DGL/CAT-Aco NPs compared with NPs without PGP decoration (Figure 6B). The ex vivo images of the distribution of NPs in main organs were acquired 1 h after i.v. injection. Compared with cl PEG-DGL/CAT-Aco NPs, cl PGP-PEG-DGL/CAT-Aco NPs showed increased accumulation in the brain. There was no obvious difference in other organs between two groups (Figure S6). The result confirmed that the modification of PGP and its rapid binding with the neutrophils in the blood accounts for the targeting specificity of the NPs to the brain. In order to further elucidate the effect of neutrophils on the delivery of drug-loaded NPs into the ischemic brain, we depleted neutrophils using anti-Gr-1 antibody as reported previously [21, 52]. MCAO models were constructed two days after neutrophils depletion via i.p. injection with anti-Gr-1 antibody in nude mice. BODIPY 650/665-labeled cl PGP-PEG-DGL/CAT-Aco NPs were intravenously administered to IS mice 24 hours after reperfusion. The in vivo brain fluorescence images (Figure 6C) demonstrated that the injection of anti-Gr-1 antibody could significantly reduce the brain distribution of NPs in MCAO mice, which confirmed the importance of neutrophils during the delivery of NPs to the ischemic brain. Based on the above results, we concluded that cl PGP-PEG-DGL/CAT-Aco NPs not only enhanced enzyme accumulation in the brain but in the ischemic subregion and cerebral neurocytes in response to stroke.
(A) In vivo fluorescence images of the brains of nude mice subjected to MCAO after administration of cl dendrimer/CAT-Aco NPs labeled with CAT-BODIPY; ex vivo NIR fluorescence image of harvested brain 3 h post-treatment. (B) Semi-quantitative ROI analysis of the average fluorescence intensity from the BODIPY 650/665-labeled NPs in ischemic brain (mean ± SD, n=3), *P<0.05, **P<0.01 vs. cl PEG-DGL/CAT-Aco. (C) In vivo fluorescence images of nude mice brain after cl PGP-PEG-DGL/CAT-Aco NPs administration with or without the injection of anti-Gr-1 antibody (mean ± SD, n=3). NPs were labeled with CAT-BODIPY. **P<0.01, ***P<0.001 vs. the healthy (D) In vivo T2W-MRI images of ischemic mouse brain at 48 h post-injection of PBS, CAT-Aco alone (250 U/mouse), cl PEG-DGL/CAT-Aco and cl PGP-PEG-DGL/CAT-Aco (mean ± SD, n=6). *P<0.05, **P<0.01, ***P<0.001 vs. the PBS group.
Therapeutic Efficacy of NPs in MCAO Mice with Ischemia/Reperfusion Injury
The therapeutic efficiency of the neutrophil-mediated neuroprotectant delivery system was evaluated in a mouse MCAO model of IS. To assess the efficacy of cl PGP-PEG-DGL/CAT-Aco NPs in treating brain injury, we used in vivo T2W-MRI as a non-invasive imaging technology to delineate the ischemic volume 48 h after different treatments. The images in Figure 6D depict the ischemic territory as a hyper-intensive signal corresponding to cerebral edema after transient focal cerebral ischemia. In the mice that received cl PGP-PEG-DGL/CAT-Aco NPs at 3 h, 24 h and 72 h after reperfusion, the infarct volume was significantly reduced to approximately 68.5%, 57.7% and 33.1% of the PBS group (P<0.001), respectively. The result was closely related to the general neurological score acquired before MRI, which showed significant reduction to approximately 68.1%, 55.8% and 48.3% of the PBS group (P<0.001) at the same time point after cl PGP-PEG-DGL/CAT-Aco NPs administration (Figure S8). Both evaluation experiments jointly referred to the point that the effect of cl PGP-PEG-DGL/CAT-Aco NPs was much better than that of the cl PEG-DGL/CAT-Aco NPs (P<0.05 at 3 h and P<0.01 at 24 h and 72 h). Furthermore, we presumed that the therapeutic efficiency of cl PGP-PEG-DGL/CAT-Aco NPs improved with time, primarily due to the increased amount of CAT delivered by the neutrophils recruited to the ischemic area via inflammatory response. This is of considerable importance for extension of therapeutic time window in acute cerebral ischemia.
To verify that the significant neuroprotective effect of NPs was a result of CAT-mediated ROS scavenging, the ROS levels in ischemic brains were detected by injection of the hydrogen peroxide-sensitive mitochondrial probe DHR, which can be oxidized by ROS to fluorescent rhodamine 123 (RH123). The level of ROS in ischemic brains 24 h post-treatment was evaluated by visualizing the RH123 fluorescence. The therapeutic effect was further evaluated by quantifying the apoptotic cells in the ischemic area via TUNEL staining. The RH123 and TUNEL signals were distributed widely in the ischemic area of the mice injected with PBS or CAT-Aco alone, while fewer and less intense signals were found in mice injected with NPs, especially the NPs decorated with PGP. There was a significant difference in the percentage of RH123-positive cells between the mice treated with cl PGP-PEG-DGL/Cat-Aco NPs and those treated with cl PEG-DGL/Cat-Aco NPs as well as in the percentage of TUNEL-positive cells. The reduced percentage of RH123-positive and TUNEL-positive cells was interpreted to indicate that prohibition of the ROS-dependent apoptosis pathway played an important role in the efficacy. Hematoxylin and eosin (HE) staining indicated that nearly all of the neurocyte nuclei were pyknotic in the ischemic area (I). Above all, high fluorescence intensity from RH123 and TUNEL was localized in the ischemic area (I) marked with HE staining (Figure S9).
Conclusion
In this study, we successfully constructed novel cl PGP-PEG-DGL/CAT-Aco NPs for delivery of macromolecular protein-based drugs, and the NPs exhibited an excellent multi-brain targeting effect. The effect was attributed to the strong binding efficiency of PGP ligands on the surface of the NPs with neutrophils in the blood, the ability of neutrophils to migrate through the BBB, and the tropism of leucocytes to the inflammation site and the cells in the brain. Realization of the purpose of these NPs depends on high protein drug loading and encapsulation efficiency, specific and high internalization of NPs by phagocytic cells in the circulation, sufficient protection of the protein-based drug from degradation within carrier cells, targeted delivery to the diseased sites in the brain and triggered release at the site of inflammation. The process and mechanisms were comprehensively illustrated in vitro and in vivo in our study. Administration of cl PGP-PEG-DGL/CAT-Aco NPs significantly reduced the infarct volume of MCAO mice and enhanced the therapeutic outcome of cerebral ischemia. Considering that neuroinflammation occurs in many neurological disorders, the strategy reported here offers broad potential applications for treatment of brain diseases. We believe that the developed delivery system is not only promising for efficient treatment of IS but may also be an effective approach for various CNS diseases related to inflammation.
Abbreviations
cross-linked, cl; catalase, CAT; cis-aconitic anhydride, cis-Aco; CAT-cis-Aco, CAT-Aco; nanoparticle, NP.
Acknowledgements
We are thankful for financial support from the National Natural Science Foundation of China (81690263, 81673372, 81361140344), the National Basic Research Program of China (2013CB 932500), the Opening Project of Key Laboratory of Drug Targeting and Drug Delivery System, Ministry of Education (Sichuan University) and the Development Project of Shanghai Peak Disciplines - Integrated Medicine (No. 20150407).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brasnjevic I, Steinbusch HW, Schmitz C, Martinez-Martinez P, European NanoBioPharmaceutics Research I. Delivery of peptide and protein drugs over the blood-brain barrier. Prog Neurobiol. 2009;87:212-51
2. Tomlinson IM. Next-generation protein drugs. Nat Biotechnol. 2004;22:521-2
3. Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T. et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008;7:84-96
4. Suzuki Y, Nagai N, Umemura K. A Review of the mechanisms of blood-brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front Cell Neurosci. 2016;10:2
5. Mori E, Delzoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712-8
6. Pandya RS, Mao L, Zhou H, Zhou S, Zeng J, Popp AJ. et al. Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem. 2011;11:81-97
7. Singhal A, Morris VB, Labhasetwar V, Ghorpade A. Nanoparticle-mediated catalase delivery protects human neurons from oxidative stress. Cell Death Dis. 2013;4:e903
8. Jaffer H, Morris VB, Stewart D, Labhasetwar V. Advances in stroke therapy. Drug Deliv Transl Res. 2011;1:409-19
9. Klyachko NL, Haney MJ, Zhao Y, Manickam DS, Mahajan V, Suresh P. et al. Macrophages offer a paradigm switch for CNS delivery of therapeutic proteins. Nanomedicine. 2014;9:1403-22
10. Batrakova EV, Gendelman HE, Kabanov AV. Cell-mediated drug delivery. Expert Opin Drug Deliv. 2011;8:415-33
11. Qin J, Yang X, Mi J, Wang J, Hou J, Shen T. et al. Enhanced antidepressant-like effects of the macromolecule trefoil factor 3 by loading into negatively charged liposomes. Int J nanomedicine. 2014;9:5247-57
12. Dong X, Chu D, Wang Z. Leukocyte-mediated Delivery of Nanotherapeutics in Inflammatory and Tumor Sites. Theranostics. 2017;7:751-63
13. Agrahari V, Agrahari V, Mitra AK. Next generation drug delivery: circulatory cells-mediated nanotherapeutic approaches. Expert Opin Drug Deliv. 2017;14:285-9
14. Qin J, Yang X, Zhang RX, Luo YX, Li JL, Hou J. et al. Monocyte mediated brain targeting delivery of macromolecular drug for the therapy of depression. Nanomedicine. 2015;11:391-400
15. Qin J, Zhang RX, Li JL, Wang JX, Hou J, Yang X. et al. cRGD mediated liposomes enhanced antidepressant-like effects of edaravone in rats. Eur J Pharm Sci. 2014;58:63-71
16. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796-808
17. Qiu Y, Palankar R, Echeverria M, Medvedev N, Moya SE, Delcea M. Design of hybrid multimodal poly(lactic-co-glycolic acid) polymer nanoparticles for neutrophil labeling, imaging and tracking. Nanoscale. 2013;5:12624-32
18. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA. et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849-57
19. Courties G, Herisson F, Sager HB, Heidt T, Ye Y, Wei Y. et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res. 2015;116:407-17
20. Gronberg NV, Johansen FF, Kristiansen U, Hasseldam H. Leukocyte infiltration in experimental stroke. J Neuroinflammation. 2013;10:9
21. Chu D, Gao J, Wang Z. Neutrophil-mediated delivery of therapeutic nanoparticles across blood vessel barrier for treatment of inflammation and infection. ACS nano. 2015;9:11800-11
22. Chu D, Zhao Q, Yu J, Zhang F, Zhang H, Wang Z. Nanoparticle Targeting of Neutrophils for Improved Cancer Immunotherapy. Adv Healthc Mater. 2016;5:1088-93
23. Tazzyman S, Barry ST, Ashton S, Wood P, Blakey D, Lewis CE. et al. Inhibition of neutrophil infiltration into A549 lung tumors in vitro and in vivo using a CXCR2-specific antagonist is associated with reduced tumor growth. Int J Cancer. 2011;129:847-58
24. Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS. et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317-23
25. Kim SD, Lee HY, Shim JW, Kim HJ, Yoo YH, Park JS. et al. Activation of CXCR2 by extracellular matrix degradation product acetylated Pro-Gly-Pro has therapeutic effects against sepsis. Am J Respir Crit Care. 2011;184:243-51
26. Patel A, Patel M, Yang X, Mitra AK. Recent advances in protein and Peptide drug delivery: a special emphasis on polymeric nanoparticles. Protein Pept Lett. 2014;21:1102-20
27. Sisavath N, Leclercq L, Le Saux T, Oukacine F, Cottet H. Study of interactions between oppositely charged dendrigraft poly-L-lysine and human serum albumin by continuous frontal analysis capillary electrophoresis and fluorescence spectroscopy. J Chromatogr A. 2013;1289:127-32
28. Han L, Guo Y, Ma H, He X, Kuang Y, Zhang N. et al. Acid active receptor-specific peptide ligand for in vivo tumor-targeted delivery. Small. 2013;9:3647-58
29. Kakizawa Y, Harada A, Kataoka K. Environment-sensitive stabilization of core-shell structured polyion complex micelle by reversible cross-linking of the core through disulfide bond. J Am Chem Soc. 1999;121:11247-8
30. Zhou Z, Shen Y, Tang J, Fan M, Van Kirk EA, Murdoch WJ. et al. Charge-reversal drug conjugate for targeted cancer cell nuclear drug delivery. Adv Funct Mater. 2009;19:3580-9
31. Nam K, Park JW, Bark H, Han J, Nah JW, Jang MK. et al. Enhanced gene delivery system using disulfide-linked chitosan immobilized with polyamidoamine. Macromol Res. 2014;22:370-6
32. Hu Y, Tang G, Liu J, Cheng W, Yue Y, Li J. et al. FGF receptor-mediated gene delivery using ligands coupled to PEI-beta-CyD. J Biomed Biotechnol. 2012;2012:989235
33. Lee Y, Ishii T, Cabral H, Kim HJ, Seo JH, Nishiyama N. et al. Charge-conversional polyionic complex micelles-efficient nanocarriers for protein delivery into cytoplasm. Angewandte Chemie Int Ed Engl. 2009;48:5309-12
34. Miyata K, Christie RJ, Kataoka K. Polymeric micelles for nano-scale drug delivery. React Funct Polym. 2011;71:227-34
35. Crombez M, Chevallier P, Gaudreault RC, Petitclerc E, Mantovani D, Laroche G. Improving arterial prosthesis neo-endothelialization: application of a proactive VEGF construct onto PTFE surfaces. Biomaterials. 2005;26:7402-9
36. Giagulli C, Magiera AK, Bugatti A, Caccuri F, Marsico S, Rusnati M. et al. HIV-1 matrix protein p17 binds to the IL-8 receptor CXCR1 and shows IL-8-like chemokine activity on monocytes through Rho/ROCK activation. Blood. 2012;119:2274-83
37. Batrakova EV, Li S, Reynolds AD, Mosley RL, Bronich TK, Kabanov AV. et al. A macrophage-nanozyme delivery system for Parkinson's disease. Bioconjug chem. 2007;18:1498-506
38. Mendelson AA, Guan QN, Chafeeva I, Da Roza GA, Kizhakkedathu JN, Du CG. Hyperbranched polyglycerol is an efficacious and biocompatible novel osmotic agent in a rodent model of peritoneal dialysis. Perit Dial Int. 2013;33:15-27
39. Li W, Chung S. Flow cytometric evaluation of leukocyte function in rat whole blood. In Vitro Cell Dev Biol Anim. 2003;39:413-9
40. Li Y, Lian Y, Zhang LT, Aldousari SM, Hedia HS, Asiri SA. et al. Cell and nanoparticle transport in tumour microvasculature: the role of size, shape and surface functionality of nanoparticles. Interface focus. 2016;6:20150086
41. Sideratou Z, Sterioti N, Tsiourvas D, Tziveleka LA, Thanassoulas A, Nounesis G. et al. Arginine end-functionalized poly(L-lysine) dendrigrafts for the stabilization and controlled release of insulin. J Colloid Interface Sci. 2010;351:433-41
42. Tsogas I, Theodossiou T, Sideratou Z, Paleos CM, Collet H, Rossi JC. et al. Interaction and transport of poly(L-lysine) dendrigrafts through liposomal and cellular membranes: the role of generation and surface functionalization. Biomacromolecules. 2007;8:3263-70
43. Behr JP. The proton sponge: A trick to enter cells the viruses did not exploit. Chimia (Aarau). 1997;51:34-6
44. Klinker JF, Wenzel-Seifert K, Seifert R. G-protein-coupled receptors in HL-60 human leukemia cells. Gen Pharmacol. 1996;27:33-54
45. Manjavachi MN, Quintao NL, Campos MM, Deschamps IK, Yunes RA, Nunes RJ. et al. The effects of the selective and non-peptide CXCR2 receptor antagonist SB225002 on acute and long-lasting models of nociception in mice. Eur J Pain. 2010;14:23-31
46. Hillaireau H, Couvreur P. Nanocarriers' entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66:2873-96
47. Sharma DK, Choudhury A, Singh RD, Wheatley CL, Marks DL, Pagano RE. Glycosphingolipids internalized via caveolar-related endocytosis rapidly merge with the clathrin pathway in early endosomes and form microdomains for recycling. J Biol Chem. 2003;278:7564-72
48. Huang R, Liu S, Shao K, Han L, Ke W, Liu Y. et al. Evaluation and mechanism studies of PEGylated dendrigraft poly-L-lysines as novel gene delivery vectors. Nanotechnology. 2010;21:265101
49. Dorovinizis K, Bowman PD, Prameya R. Adhesion and migration of human polymorphonuclear leukocytes across cultured bovine brain microvessel endothelial cells. J Neuropathol Exp Neurol. 1992;51:194-205
50. Sans E, Delachanal E, Duperray A. Analysis of the roles of ICAM-1 in neutrophil transmigration using a reconstituted mammalian cell expression model: Implication of ICAM-1 cytoplasmic domain and Rho-dependent signaling pathway. J Immunol. 2001;166:544-51
51. Dwyer J, Hebda JK, Le Guelte A, Galan-Moya EM, Smith SS, Azzi S. et al. Glioblastoma cell-secreted interleukin-8 induces brain endothelial cell permeability via CXCR2. PloS one. 2012;7:e45562
52. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64-70
Author contact
![]() Corresponding authors: Jing Qin, E-mail: Qinjingedu.cn (J.Q.), Tel.: 86-21-51980087, Fax: 86-21-51980087; Jianxin Wang, E-mail: jxwangedu.cn (J.W.), Tel.: 86-21-51980088, Fax: 86-21-51980088.
Corresponding authors: Jing Qin, E-mail: Qinjingedu.cn (J.Q.), Tel.: 86-21-51980087, Fax: 86-21-51980087; Jianxin Wang, E-mail: jxwangedu.cn (J.W.), Tel.: 86-21-51980088, Fax: 86-21-51980088.