Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(14):3398-3414. doi:10.7150/thno.20919 This issue Cite
Research Paper
Towards Translational ImmunoPET/MR Imaging of Invasive Pulmonary Aspergillosis: The Humanised Monoclonal Antibody JF5 Detects Aspergillus Lung Infections In Vivo
Genna Davies1*, Anna-Maria Rolle2*, Andreas Maurer2, Philipp R. Spycher3, Claudia Schillinger2, Djamschid Solouk-Saran4, Mike Hasenberg4, Juliane Weski4, Jesper Fonslet5, 6, Adrien Dubois7, Frederic Boschetti7, Franck Denat8, Matthias Gunzer4, Martin Eichner9, Lauren S Ryder1, Mikael Jensen5, 6, Roger Schibli3, Bernd J. Pichler2, Stefan Wiehr2, Christopher R. Thornton1 ![]()
1. ISCA Diagnostics Ltd. and Biosciences, College of Life & Environmental Sciences, University of Exeter, Exeter, United Kingdom;
2. Werner Siemens Imaging Center, Department of Preclinical Imaging and Radiopharmacy, Eberhard Karls University Tübingen, Germany;
3. Paul Scherrer Institute, Research Department Biology and Chemistry, Center for Radiopharmaceutical Sciences (CRS), Switzerland;
4. Institute for Experimental Immunology and Imaging, University Hospital, University of Duisburg-Essen, Essen, Germany;
5. The Hevesy Laboratory, DTU Nutech, Technical University of Denmark;
6. Center for Nanomedicine and Theranostics, Technical University of Denmark;
7. CheMatech, Faculté des Sciences Mirande, Dijon, France;
8. Institut de Chimie Moléculaire, Université de Bourgogne, Dijon, France;
9. Institute for Clinical Epidemiology and Applied Biometry, Eberhard Karls University Tübingen, Germany.
* GD and AMR contributed equally to this work.
Received 2017-5-8; Accepted 2017-7-4; Published 2017-8-11
Abstract

Invasive pulmonary aspergillosis (IPA) is a life-threatening lung disease of hematological malignancy or bone marrow transplant patients caused by the ubiquitous environmental fungus Aspergillus fumigatus. Current diagnostic tests for the disease lack sensitivity as well as specificity, and culture of the fungus from invasive lung biopsy, considered the gold standard for IPA detection, is slow and often not possible in critically ill patients. In a previous study, we reported the development of a novel non-invasive procedure for IPA diagnosis based on antibody-guided positron emission tomography and magnetic resonance imaging (immunoPET/MRI) using a [64Cu]DOTA-labeled mouse monoclonal antibody (mAb), mJF5, specific to Aspergillus. To enable translation of the tracer to the clinical setting, we report here the development of a humanised version of the antibody (hJF5), and pre-clinical imaging of lung infection using a [64Cu]NODAGA-hJF5 tracer. The humanised antibody tracer shows a significant increase in in vivo biodistribution in A. fumigatus infected lungs compared to its radiolabeled murine counterpart [64Cu]NODAGA-mJF5. Using reverse genetics of the pathogen, we show that the antibody binds to the antigenic determinant β1,5-galactofuranose (Galf) present in a diagnostic mannoprotein antigen released by the pathogen during invasive growth in the lung. The absence of the epitope Galf in mammalian carbohydrates, coupled with the enhanced imaging capabilities of the hJF5 antibody, means that the [64Cu]NODAGA-hJF5 tracer developed here represents an ideal candidate for the diagnosis of IPA and translation to the clinical setting.
Keywords: Infectious Diseases, Aspergillus, Aspergillosis, Monoclonal Antibody, JF5, ImmunoPET/MRI.
Introduction
Infectious diseases caused by viruses, bacteria, and fungi remain a leading cause of death worldwide [1], with fungi responsible for an estimated 2 million life-threatening infections each year [2]. At present, there is a paucity of diagnostic tests that can accurately differentiate these infectious etiologies, resulting in the incorrect prescription of antimicrobial drugs and emergence of widespread resistance to antibiotics and antifungals [3]. Both rapid point-of-care tests [4] and more sophisticated diagnostic modalities [5] compatible with contemporary medical facilities are urgently needed to allow pathogen discrimination and implementation of targeted treatments [3]. Here, molecular imaging has huge potential to deliver accurate, non-invasive, and rapid detection of infectious diseases with the prospect of earlier diagnosis, efficient therapy and improved survival rates. While still in their infancy, highly specific antibody-guided imaging technologies have recently been described for the in vivo diagnosis of viral [6], bacterial [7] and fungal diseases [8] in animal models, with genuine potential for translation to human disease diagnosis.
Invasive pulmonary aspergillosis (IPA) is a life-threatening lung disease of immunocompromised individuals with the majority of infections caused by the fungus Aspergillus fumigatus. Conservative estimates show that the opportunistic pathogen is responsible for >200,000 infections of hematological malignancy, hematopoietic stem cell transplant (HSCT) and solid organ transplant patients worldwide each year, with an associated mortality rate of between 30 and 90 % [2]. While the disease is typically associated with prolonged neutropenia, it is being increasingly reported in non-neutropenic patients, such as those with underlying lung diseases including steroid-treated chronic obstructive pulmonary disease (COPD), asthma, lung cancer, or autoimmune diseases with pulmonary involvement [4]. In addition, it has been estimated that chronic pulmonary aspergillosis (CPA) and allergic bronchopulmonary aspergillosis (ABPA) affect >15 million people globally each year [2]. Taken together, diseases caused by Aspergillus spp. constitute a serious but often hidden threat to human health, adding to patient morbidities and hospital treatment costs. Arguably, much of this burden derives from the lack of accurate tests for disease diagnosis.
Diagnosis of IPA is a major clinical challenge since manifestations of the disease (fevers and chills, hemoptysis, shortness of breath, chest pains, headaches) are non-specific, and assays that are presently used to detect Aspergillus cell wall galactomannans and fungal β-D-glucan in serum or bronchoalveolar lavage fluids lack either sensitivity or specificity [9]. High-resolution CT is increasingly used to identify IPA, but lung abnormalities suggestive of the disease such as nodules and “halo sign” can be caused by other angio-invasive fungal and bacterial pathogens or even non-infectious pathologies, and so are unable to differentiate IPA from other pulmonary diseases [9].
Positron emission tomography and magnetic resonance imaging (PET/MRI) has proven to be an extremely powerful tool for the diagnosis of cancer [5], but has yet to be fully exploited in the diagnosis of microbial infections [10, 11]. However, we recently demonstrated the enormous potential of antibody-guided PET/MRI (immunoPET/MRI) for the molecular imaging of both bacterial [7] and fungal [8] infections in vivo. Importantly, we showed that a mouse monoclonal antibody (mJF5), which specifically binds to a diagnostic Aspergillus mannoprotein antigen [12, 13], when conjugated to [64Cu]DOTA, can be used successfully in a neutropenic mouse model of IPA to image Aspergillus lung infections in vivo [8]. The highly specific [64Cu]DOTA-mJF5 tracer allows repeated imaging of A. fumigatus lung infections and differentiation of IPA from pulmonary inflammation and from infections caused by bacteria.
In this study, we report the development of a humanized version of JF5 (hJF5), generated using CDR grafting, and investigate its accuracy in detecting A. fumigatus lung infections in vivo compared to the mouse antibody. We show that conjugation of mJF5 to [64Cu]DOTAGA or [64Cu]NODAGA allows specific detection of lung infections, while [64Cu]NODAGA-labeled hJF5 enables additional improvements in disease detection compared to the mouse tracers. Furthermore, using reverse genetics to produce mutants of A. fumigatus deficient in UDP-galactopyranose mutase, a key enzyme in the biosynthesis of galactofuranose containing glycoconjugates in A. fumigatus [14-16], we show that the antibodies bind to the antigenic determinant β1,5-galactofuranose (Galf) present in the diagnostic mannoprotein antigen. The absence of the epitope Galf in mammalian carbohydrates [17], coupled with the improved imaging capabilities of the hJF5 antibody, means that the [64Cu]NODAGA-hJF5 tracer represents an ideal candidate for the diagnosis of IPA in humans and translation to the clinical setting.
Material and Methods
Design of humanised JF5 antibody
Full-length antibody constructs were generated from the sequenced variable regions of the Aspergillus-specific mouse monoclonal antibody JF5 (mJF5) [12]. The full-length humanised antibody was created using a human IgG1 framework and mJF5 variable regions, by grafting the murine complementary determining regions (CDR) sequences onto human antibody donor sequences.
Cell culture
All culture media was supplied by Invitrogen (UK) and supplemented with 8 mM L-glutamine (25030-024, Invitrogen) unless otherwise stated. CD DG44 medium (12610-010, Invitrogen) was supplemented with 1.8 % Pluronic F68 (24040-032, Invitrogen) prior to use. All cells were incubated at 37 °C with 8 % CO2 in vented Erlenmeyer flasks (431143, 431144, 431145, Corning, Netherlands), with shaking on a Labnet orbital 1000 shaking platform (ST2060, Appleton Woods Ltd., UK) rotating at 135 rpm unless otherwise stated. Cells were passaged by centrifugation at 300 g for 5 minutes and re-suspension in appropriate medium, and cell numbers and viabilities were determined using a hemocytometer and Trypan blue exclusion, respectively.
Expression vectors
The gene coding for dihydrofolate reductase (DHFR) expression was cloned into the expression vector pVitro1-neo-mcs (Autogen-Bioclear UK Ltd., UK) following the internal ribosome entry sites (IRES) sequence, to generate pVitro DHFR3. DNA encoding the full-length 150 kDa antibody (hereafter referred to as Ab633) was cloned into the pVitro DHFR3 mammalian expression vector.
Transient transfection
Suspension adapted Chinese Hamster Ovary (CHO) K1 cells, were routinely cultivated at 2.0 - 3.0 x 105 cells/ml at 150 rpm, in Pro CHO 4 serum free medium (BE12-029Q, Lonza, UK) plus 100 µM hypoxanthine/16 µM thymidine (H/T) (11607-030, Invitrogen). 1 x 108 cells were transfected with 1.25 µg/ml of DNA (Ab633 pVitro DHFR3) in Pro CHO 5 medium (BE12-766Q, Lonza) supplemented with H/T. Cultures were incubated for 11 days at 150 rpm. Medium scale transient transfection expression analysis was carried out to determine expression yields from CHO cells, with cell line Ab633-HC3-LC2 taken forward for stable transfection.
Stable transfection
Linearisation of Ab633 pVitro DHFR3 vector
One hundred and fifty µg of Ab633-HC3-LC2 pVitro DHFR3 was linearised in a restriction reaction consisting of 176 μl of endonuclease free water, 30 µl of NE buffer 2 (R0547S, New England Biolabs, UK), 3 µl of 100X BSA (R0547S, New England Biolabs) and 5 µl of Pac1 (R0547S, New England Biolabs). The digest was incubated at 37 °C overnight and then purified.
Purification of Ab633 plasmid
Six hundred µl of cold ethanol was added to 300 µl of linearisation mix and incubated on ice for 5 minutes. Following centrifugation at 16,200 g at room temperature for 30 min the pellet was washed in 1 ml of 70 % ethanol and centrifuged for 10 min at 16,200 g. Following removal of the supernatant, the pellet was air dried for 30 minutes and 100 µl of endotoxin-free water was added to the pellet and dissolved overnight at 4 °C. One µl of the linearised plasmid was run on a 1.5 % agarose gel and visualized on a Kodak gel imager. The DNA concentration was calculated using a spectrophotometer (SmartSpec reader, Bio-Rad, US).
Transfection
Prior to transfection, cGMP CHO DG44 cells (A11000-01, Invitrogen) were routinely cultured in CD DG44 medium. Cells were diluted to a concentration of 3.0 x 105 cells/ml, incubated with shaking at 165 rpm, and passaged every 3-4 days. Twenty-four hours prior to transfection, DG44 cells were seeded, at a density of 7.0 x 105 cells/ml, into 80 ml of CD DG44 medium and shaken at 165 rpm. On the day of transfection, 1.5 x 107 DG44 cells were seeded into 30 ml CD DG44 medium in a T175 flask (431080, Corning, US), and allowed to attach for 2 hours in static culture. Fifteen µl of Freestyle Max transfection reagent (16447-100, Invitrogen) was added to 18 µg total of linearised Ab633 pVitro DHFR3 vector diluted in 600 µl OptiPRO serum free medium (12309-019, Invitrogen), and incubated for 10 min at room temperature. The transfection mix was added drop wise to the T175 flask of cells and incubated, as a static culture, for 48 h. Following transfection, stably transfected cells were selected by culture in CD OptiCHO medium (12681029, Invitrogen) in T175 flasks, at a concentration of 1.0 x 106 cells/ml. Cells were passaged until the viability was seen to begin to recover, and the culture was transferred to shaking cultures at 3.0 x 105 cells/ml and incubated for 3-4 days. Cells were then seeded in CD OptiCHO selection medium supplemented with 500 µg/ml G418 sulfate (ant-gn-5, Autogen-Bioclear). Cells were routinely passaged by dilution in fresh selection medium at a density of 3.0 x 105 cells/ml every 3-4 days until the viability was seen to increase to >90 % and a stable pool of cells was generated which expressed Ab633-HC3-LC2. Cell line T1 was selected for genomic amplification as it had the highest antibody production in picograms/cell/day (PCD) of 0.9.
Genomic amplification and sub-cloning
A culture of Ab633-HC3-LC2-T1 with a viability of >90 % was subjected to a single round of amplification in amplification medium (CD OptiCHO medium supplemented with 500 µg/ml G418 and 500 nM Methotrexate (A6770, Sigma, UK)). Cultures were seeded at 5.0 x 105 cells/ml and passaged every 4-5 days in amplification medium until the culture was observed to recover viability to >90 %. Cells were then routinely passaged at 3.0 x 105 cells/ml with incubation for 3-4 days. Cells were subsequently sub-cloned by limiting dilution to ensure monoclonality. The cloning medium consisted of 60 % (v/v) conditioned medium (no L-glutamine), 35.5 % CD OptiCHO medium, 3 % 200 mM L glutamine, 1 % 100X H/T, 0.3 % Phenol red (P0290, Sigma), and 500 nM Methotrexate. The sub-clone Ab633-HC3-LC2-T1-1B3 with a PCD of 17.1 was selected and, after a second round of sub-cloning by limiting dilution, sub-clone 1G10 with a PCD of 33.5 was selected.
Culture and preservation of Ab633-HC3-LC2-T1-1B3-1G10 cell line
Following adaptation to shaking culture, the cell line Ab633-HC3-LC2-T1-1B3-1G10 was routinely cultured in CD OptiCHO culture medium supplemented with 500 nM MTX and 1.0 % (v/v) dextran sulphate (D49110, Sigma) by seeding at 3.0 x 105 cells/ml. Once cell viability had dropped to approximately 50 %, cultures were harvested. Cell cultures with viabilities >90 % were frozen at -80 °C in freezing medium (46.25 % conditioned medium, 46.25 % fresh medium, 7.5 % sterile DMSO (D2650, Sigma)) prior to transfer to the vapour phase of liquid nitrogen for long-term storage. The cell line Ab633-HC3-LC2-T1-1B3-1G10 was deposited in the European Collection of Cell Cultures at Public Health England (Porton Down, UK; Patent Deposit Reference Number 15032401).
Production and purification of antibodies
For the production and purification of mJF5, the procedures described previously [8] were used. For purification of the humanised JF5 antibody (hereafter referred to as hJF5) from the cell line Ab633-HC3-LC2-T1-1B3-1G10, culture supernatants were harvested by centrifugation at 2147 g for 40 min at 4 °C, followed by filtration through a 0.8 μM cellulose acetate filter (10462240, GE Healthcare Life Sciences, UK). Culture supernatant was loaded onto a HiTrap Protein A column (17-0402-01, GE Healthcare Life Sciences) using a peristaltic pump P-1 (18-1110-91, GE Healthcare Life Sciences) with a low pulsation flow of 1 ml/minute. Columns were equilibrated with 10 ml of phosphate-buffered saline (PBS), and column-bound antibody was eluted with 5 ml of 0.1 M glycine-HCL buffer (pH 2.5) with a flow rate of 0.5 ml/min. The buffer of the purified antibody was exchanged to PBS using a disposable PD-10 desalting column (17-0851-01, GE Healthcare Life Sciences). Following purification, the antibody was sterile filtered with a 0.24 µm syringe filter (85037-574-44, Sartorius UK, Ltd., UK) and stored at 4 °C. Protein concentration was determined using a spectrophotometer (Nanodrop 1000, Thermo Fisher Scientific, USA), and purity was confirmed by sodium-dodecyl-sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and gel staining using Coomassie Brilliant Blue R-250 dye (Thermo Fisher Scientific).
Mouse strains
Eight to 14-week-old female C57BL/6 OlaHsd mice were purchased from Envigo (Huntingdon, Cambridgeshire, UK). The animals were kept under standardized and sterile environmental conditions (20 ± 1 °C room temperature, 50 ± 10 % relative humidity, 12 h light-dark cycle) and received food and water ad libitum. All experiments were performed according to the German Animal Protection Law with permission from the responsible local authorities (Tübingen, Magdeburg, Essen; Germany).
Production of A. fumigatus inoculum
For intratracheal infection of animals, the wild-type Aspergillus fumigatus strain ATCC 46645 was used. The fungus was grown on Aspergillus minimal medium (AMM) containing 56 mM D(+)glucose, 70 mM sodium nitrate, 7 mM potassium chloride, 11 mM potassium dihydrogen orthophosphate, 2 mM magnesium sulfate, 1x Hutner's trace elements and 30 g agar per liter for spore production. Conidia were harvested after 3 days of incubation at 37 °C by flushing with PBS and subsequent filtering through a cell strainer (40 μm pore size).
Generation of A. fumigatus UDP-galactopyranose mutase-deficient mutants
Targeted replacement of the A. fumigatus UDP-galactopyranose mutase-encoding gene glfA, with the hygromycin B phosphotransferase-encoding gene (hph), was performed using the split marker recombination method [18-20]. The glfA gene and flanking sequences were obtained from the Aspergillus Genome Database (AspGD, http://www.aspergillusgenome.org/) and used to design primers accordingly (Table S1). Primer pairs glf50.1F/glf50.1R and glf30.1F/glf30.1R were used to amplify the 5' (LF, 1.0-kb) and 3' (RF, 1.0-kb) flanking regions of the glfA gene, respectively, from A. fumigatus Af293 genomic DNA. Simultaneously, split hph templates were amplified to create the 5' amplicon (HY, 1.2-kb) using primers HY split/M13F, and 3' amplicon (YG, 0.8-kb) using primers YG split/M13R. Fusion PCR resulted in two products, LFHY (2.1-kb) and RFYG (1.8-kb), using primer pairs glf50.4F/HY split and glf30.1R/nested YG split, respectively. The amplicons were gel-purified and transformed into protoplasts of Af293, replacing glfA with the assembled hph gene and conferring resistance to hygromycin B. Putative ΔglfA::hph transformants were selected in the presence of hygromycin B (600µg/mL) and gene replacement was confirmed by Southern blot. The restriction enzyme Xba1 was used for digestion of genomic DNA prior to gel electrophoresis and blotting onto Hybond-NX membrane. The membrane was probed a 1.0-kb fragment of the glfA ORF, amplified from A. fumigatus genomic DNA using the primer pair PROBEF/PROBER. The probe was predicted to hybridise to a 3.6-kb fragment present in Af293 that, if successfully deleted, would not be present in the mutant strains. Therefore, absence of the 3.6-kb band in Southern blots of digested genomic DNA would show successful deletion of the glfA gene.
Phenotypic characterisation of mutants
For each strain, a 20 µl drop containing spores at a concentration of 103 spores/ml, was used to inoculate the centre of a malt extract agar (MEA) plate and then allowed to air dry. Plates were incubated at 30 °C and 37 °C for 3 days, after which plates were scanned and the area of growth (mm2) was measured using Image J software (United States National Institutes of Health, Bethesda, Maryland, USA). This data was further analysed using SPSS to perform an ANOVA with post hoc Tukey-Kramer analysis. Ten-ml flasks of malt extract broth (MEB) were inoculated with spores to a final concentration of 104 spores/ml and incubated at 30 °C and agitation at 60 rpm. At 24 hour intervals, replicates of each strain were removed and culture fluids separated from mycelia by centrifugation at 4000 rpm for 10 min. Mycelial biomass was dried for 2 days at 80 °C, weighed, and culture filtrates assayed with mAbs mJF5 and hJF5 by using ELISA and western blotting.
Immunoassays
Recognition of ΔglfA::hph mutant strains and wild-type strain Af293 by mAbs mJF5 and hJF5 was determined in ELISA, western blotting and immunofluorescence studies. For ELISA tests, 50 µl volumes of MEB culture fluids (or MEB only as a negative control), were used to coat wells of microtiter plates and incubated at 4 °C for 16 hours to allow antigen immobilisation. Wells were washed three times with PBST (PBS (0.8 % NaCl; 0.02 % KCl; 0.115 % Na2HPO4; 0.02 % KH2PO4; pH 7.2) containing 0.05 % (v/v) Tween-20), once with PBS and once with dH2O before being air-dried at 23 °C in a laminar flow hood. Wells were blocked for 15 min using 150 µl PBS containing 1 % (w/v) bovine serum albumin (BSA) and rinsed with PBS prior to adding PBST containing 4 µg/ml of Protein A-purified mJF5 or hJF5 antibody to each well. The plates were incubated for 1 hour and then washed 3 times with PBST (5 min each) before secondary antibody was added for 1 hour. For hJF5, goat anti-human polyvalent immunoglobulins (G, A, M) peroxidase conjugate (A8400, Sigma) was used at a concentration of 1:1000 in PBST, while a 1:1000 PBST dilution of goat anti-mouse polyvalent immunoglobulins (G, A, M) peroxidase conjugate (A0412, Sigma) was used for mJF5. After washing wells 3 times with PBST and once with PBS, tetramethyl benzidine substrate solution [21] was added to the wells and incubated for 30 min. Reactions were stopped with 3 M H2SO4 and absorbance measured at 450 nm using a microplate reader (Tecan GENios, Tecan Austria GmbH). Working volumes were 50 µl and all incubation steps were performed at 23 °C in sealed plastic bags.
Having shown in ELISA tests that strongest recognition of the mannoprotein antigen by mAbs mJF5 and hJF5 was achieved with day 3 culture fluids of Af293, 3-day-old culture fluids from the wild-type and mutant strains were subjected to sodium-dodecyl-sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting. For SDS-PAGE, culture filtrates were mixed 1:1 (v/v) with Laemmli buffer [22] and were denatured by heating at 95 oC for 10 min. Antigens were separated in 4-20 % (w/v) polyacrylamide gradient gels (161-1159, Bio-Rad) for 1.5 hours at 23 oC (165V) under denaturing conditions, and pre-stained broad range markers (161-0318, Bio-Rad) were used for molecular weight determinations. For westerns, separated antigens were transferred electrophoretically to a PVDF membrane (162-0175, Bio-Rad). The membranes were blocked for 16 hours at 4 oC with PBS containing 1 % (w/v) BSA and incubated with Protein A-purified mJF5 or hJF5 (both at 2 μg protein/ml) in PBS containing 0.5 % (w/v) BSA (PBSA) for 2 h at 23 oC. After washing three times with PBS, membranes were incubated for 1 h with goat anti-mouse polyvalent immunoglobulins (G, A, M) peroxidase conjugate (A0412, Sigma) or goat anti-human polyvalent immunoglobulins (G, A, M) peroxidase conjugate (A8400, Sigma), both diluted 1:1000 in PBSA. After washing twice with PBS and once with PBST, the membranes were incubated for 5 min in Amersham ECL western blotting detection reagent (RPN2106, GE Healthcare Lifesciences) and chemiluminescence was measured at 428 nm using a gel doc system (Syngene G, Box, Syngene UK).
For immunofluorescence (IF), sterilised glass slides were coated with washed spore suspensions containing 1 % (w/v) glucose solution and incubated at 30 °C for 16 hours to allow spore germination and formation of germ tubes. After air-drying, the cells were fixed to the slides [21] and incubated with Protein A-purified mJF5 or hJF5 diluted to 2 µg protein/ml in PBS for 1 h, followed by three 5 min PBS washes. Slides were then incubated with goat anti-mouse polyvalent fluorescein isothiocyanate (FITC) conjugate (F1010, Sigma) or goat anti-human IgG (Fc specific) FITC conjugate (F9512, Sigma), each diluted 1 in 40 in PBS, for 30 min. Slides were given three 5 min washes with PBS and mounted in PBS-glycerol mounting medium (F4680, Sigma) before overlaying with coverslips. All incubation steps were performed at 23 °C in a humid environment to prevent evaporation and slides were stored in the dark, at 4 °C, prior to examination using an epifluorescence microscope (Olympus IX81) fitted with 495 nm (excitation) and 518 nm (emission) filters for FITC.
A. fumigatus infection procedure
To induce neutropenia, mice received an intraperitoneal (i.p.) injection of 100 µg of a 1 mg/ml solution of anti-Ly-6G/anti-Ly6C antibody (clone RB6-8C5, BioXCell, West Lebanon, NH, USA) 24 hours prior to the infection procedure. For pulmonary infection, animals were anesthetized with an i.p. injection of 100 µl Ketamin/Rompun solution (Ketamin: [80 mg/kg], Ratiopharm GmbH, Ulm, Germany; Rompun: [15 mg/kg], Bayer HealthCare, Leverkusen, Germany). After reaching deep narcosis, mice were intubated using a 22-gauge indwelling venous catheter (Vasofix Braunüle, B. Braun AG, Melsungen, Germany) and 100 μl of the freshly prepared A. fumigatus spore suspension (4 x 106/ml) was applied. Animals were ventilated for 1 min with a small animal respirator (MiniVent, Hugo Sachs, March-Hugstetten, Germany) at a rate of 250 breaths per minute and an inhalation volume of 300 μl per breath to achieve a better distribution of the spores. Control animals received an equivalent volume of phosphate-buffered saline only (DPBS, Life Technologies, Carlsbad, CA, USA).
Fluorescence microscopy of infected lungs
For fluorescence microscopy, purified hJF5 IgG1 antibody or human IgG1 isotype control antibody (clone ET901, Biolegend, USA) were coupled to DyLight650 (Thermo Fisher Scientific) and were used in in situ and in vivo studies of A. fumigatus lung infections as described previously [8].
Chelator conjugation of antibodies
hJF5 antibody and human IgG1 isotype control antibody
For conjugation to chelators, purified mAb hJF5 was first incubated with EDTA (0.5 mM) for 45 min at room temperature with gentle shaking to remove metal contamination. Trace-metal basis 0.1M boric acid (Sigma-Aldrich, Switzerland), pH 9.1, equilibrated with PD-10 (GE Healthcare, Switzerland), was used for buffer exchange and to remove excess EDTA from the hJF5 antibody. Eluted antibody was collected in Amicon Ultra-15 centrifugal filter units with a molecular weight cut off of 10 kDa (Merck Millipore, Switzerland). Chelators p-NCS-Bz-DOTAGA or p-NCS-Bz-NODAGA (Chematech, France) were dissolved in DMSO at 33.3 mM and were applied at a 20-fold molar excess versus the antibody (33.3 µM). The conjugation reaction was carried out at 4 °C for 16 hours and, after purification with a PD-10 column, the antibody was equilibrated in Chelex-treated PBS, pH 7.4 (Bio-Rad, Switzerland). The eluted conjugate was collected in an Amicon Ultra-15 centrifugal filter unit (10 kDa molecular weight cut off) and was purified two more times with Chelex-treated PBS (pH 7.4). NODAGA-NCS conjugation of the isotype IgG1 control antibody ET901 was performed in the same manner as above, but with a 25 molar excess compared to the antibody (33.3 µM).
mJF5
Protein A-purified mAb mJF5 (IgG3) was buffer exchanged using PBS (pH 7.4, Chelex-treated) equilibrated PD-10. Eluted antibody was collected in Amicon Ultra-15 centrifugal filter units (10 kDa molecular weight cut-off). p-NCS-Bz-DOTA-GA or p-NCS-Bz-NODA-GA chelators were dissolved in DMSO at 33.33 mM and were applied at a 13-fold molar excess versus the antibody (13.33 µM). The conjugation reaction was carried out for 16 hours at 37 °C, with subsequent antibody purification via a PD-10 column. The antibody was equilibrated in Chelex-treated PBS (pH 7.4). The eluted conjugate was collected in an Amicon Ultra-15 centrifugal filter unit (molecular weight cut off: 10 kDa) and was purified two more times with Chelex-treated PBS (pH 7.4).
Determination of antibody-to-chelator ratio
Antibody-to-chelator ratios were determined with a LC-MS-ESI (Waters LCT Premier mass spectrometer). Antibody samples were de-glycosylated overnight using PNGase F (Roche, Switzerland), and reduced with 50 mM DTT containing 0.5 mM EDTA for 30 min at 37 °C. The samples were chromatographed on an Aeris WIDEPORE XB-C18 column (3.6 μm, 100 mm x 2.1 mm; Phenomenex, USA) at 80 °C. The following gradient was applied: 0-3min with 20 % A, 75 % B, and 5 % C; 3-18 min with 22 % A, 73 % B, and 5 % C; 18-20 min with 85 % A, 10 % B, and 5 % C (where A = acetonitrile containing 0.1% formic acid, B = water containing 0.1 % formic acid, and C = isopropanol) using a constant flow of 0.5 mL/min. MassLynxV4.1 was applied for data collection, and MaxEnt1 for data deconvolution.
Immuno-reactivities and serum stabilities of the mJF5 and hJF5 antibodies and their DOTAGA and NODAGA chelator derivatives
Immuno-reactivities of the antibodies and their chelator derivatives were determined in ELISA, western blotting and IF studies as described, with modifications. For ELISA and western blotting studies, purified Aspergillus mannoprotein antigen [8] was used in place of Aspergillus culture filtrates, with microtiter plates coated with saturating concentrations of antigen (1 mg antigen/mL PBS). For western blotting studies, purified antigen was denatured in Laemmli buffer at 0.5 mg/mL buffer, and 40 μL volumes subjected to SDS-PAGE, electrophoretic transfer to PVDF and processing with antibodies as described.
For serum stability tests, one volume of the respective chelator-conjugated (DOTAGA, NODAGA), 64Cu-labeled JF5 antibodies (mJF5, hJF5) in their final formulations were cleaned with a Bio-Spin 6 column and each was incubated with three volumes of C57BL/6 serum at 37 °C. The samples were removed after 1h, 24h, and 48 h and immediately analyzed by radio high-performance size exclusion chromatography (HPSEC; Phenomenex Biosep SEC-s3000, 1.5 mL/min saline sodium citrate). In addition, the samples were run on iTLC-SG paper with 0.1 M sodium citrate (pH 5.0) and analyzed using autoradiography.
PET tracer production and radiolabeling of antibodies
64Cu was produced at the in-house radiopharmacy of the Department of Preclinical Imaging and Radiopharmacy in Tübingen, using a PETtrace cyclotron (General Electric Medical Systems (GEMS). 64Cu was generated by 12.5 MeV proton irradiation of enriched 64Ni metal (35-75 mg; Isoflex, >98 % enrichment) electroplated on a platinum/iridium disc via the 64Ni(p,n)64Cu nuclear reaction as described previously [8]. In brief, 64Cu was separated from the bulk nickel target and other metallic impurities by acid dissolution followed by anion exchange chromatography (AG1 × 8, Biorad) in aqueous hydrochloric acid media (Trace Select, Sigma Aldrich). The fractions containing the 64Cu product were dried under argon at 120 °C.
For antibody radiolabeling, the dry 64CuCl2 was re-dissolved in 0.1 M HCl and the pH was adjusted to 6-7 using 0.5 M ammonium acetate. Two µg of p-NCS-Bz-DOTAGA or p-NCS-benzyl-NODAGA conjugated antibodies (NODAGA-mJF5, DOTAGA-mJF5, NODAGA-hJF5, NODAGA-isotype IgG1 control ET901) was added per MBq of 64Cu and incubated at 42 °C for 60 min. Thin layer chromatography (Polygram SIL G/UV254, Macherey-Nagel; mobile phase: 0.1 M sodium citrate, pH 5.0) and HPSEC (Phenomenex BioSep SEC-s3000 300 x 4.6 mm, 1.5 ml/min saline sodium citrate) were conducted for quality control of the radiolabeled antibodies.
PET/MR imaging, ex vivo biodistribution and autoradiography
The animal infection and imaging protocol (Fig. S2) included simultaneous PET/MR imaging of A. fumigatus-infected and non-infected control mice at three consecutive time points (3 h, 24 h, and 48 h post-infection (p.i.)) in groups of n=4-5. In vivo biodistributions of the respective antibody-based PET tracers were assessed using a small animal PET insert (Bruker Biospin GmbH, Ettlingen, Germany) yielding a spatial resolution of approximately 1.3 mm in the reconstructed images [23]. For administration of tracers, animals were anaesthetised briefly with 1.5 % isoflurane and 0.8 L/min 100 % oxygen, and injected intravenously (i.v.) with 20 µg of [64Cu]DOTAGA-labeled or [64Cu]NODAGA-labeled mJF5 antibody, [64Cu]NODAGA-labeled hJF5 antibody, or [64Cu]NODAGA-labeled isotype control antibody ET901, corresponding to 12-14 MBq for immunoPET imaging. Static PET scans (10 min) were acquired 3 h, 24 h and 48 h after the injection of the respective tracer. During PET and MR imaging, the animals were anesthetized with 1.5 % isoflurane mixed with 100 % oxygen. PET data were acquired in list-mode, histogrammed in one 10 min time frame and reconstructed using a 2D iterative ordered subset expectation maximization (OSEM2D) algorithm. MR imaging was performed on a 7 T, 300 MHz small animal MR tomograph (Bruker Biospin GmbH) for the acquisition of anatomical information. The images were acquired using a T2 fat saturated 3D sequence with a TE/TR of 90.51/1800.000 ms. Additionally, a T1 3D fast low angle shot (FLASH) sequence with a TE/TR of 6.000/30.000 ms was performed. PET images were normalized to each other, subsequently fused to the respective MR images and analyzed using Inveon Research Workplace software (Siemens Preclinical Solutions, Knoxville, TN, USA). Regions of interest (ROIs) were drawn around the respective tissues based on the anatomical information from the MR images. Absolute quantification of the PET data is expressed as percentage of the injected dose per cubic centimeter (%ID/cc). After the final PET scan, all animals were sacrificed by cervical dislocation under deep anesthesia and dissected. Organs were removed and radioactivity was quantified with an aliquot of the injected radiotracer in a γ-counter (Wallac 2480 WIZARD 3”, Perkin-Elmer, Waltham, MA, USA) using an energy window between 350 and 650 keV. The results are expressed as % injected dose per g (%ID/g) of tissue. Autoradiography was also performed after the last PET scan. Animals were sacrificed, the lungs were carefully dissected, embedded in optimal cutting temperature compound (TissueTek, Sakura, Zoeterwonde, The Netherlands) and snap frozen. Twenty µm cryosections of tissue were exposed for 24 h to phosphor screens and autoradiograms were acquired with a storage phosphor imager (445SI, Molecular Dynamics, Sunnyvale, CA, USA). The spatial resolution of the phosphor imager was set to 50 µm. The autoradiography data were analyzed using ImageJ software and normalized to the injected dose. The same tissue slices used for the autoradiography were stained with hematoxylin and eosin (H&E) following standard procedures. Subsequently, the slides were scanned using the digital slide scanner NanoZoomer 2.0-HT (Hamamatsu Photonics K.K., Hamamatsu, Japan).
Statistical analysis
For experiments with more than two investigated groups, p-values were calculated using one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test conducted with Origin 8 software (OriginLab Corporation, Northampton, MA, USA). Means were considered statistically significant at *P < 0.05. Unless otherwise stated, all quantitative data are shown as the mean ± 1 standard deviation (SD).
Results
Characterisation of the hJF5 antibody
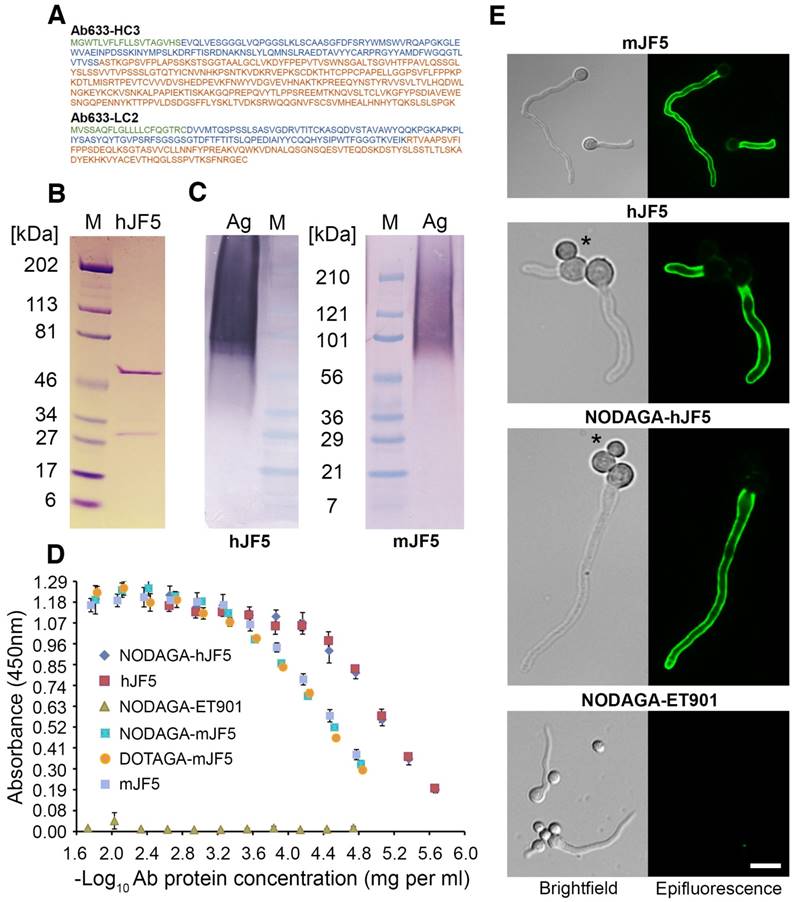
The amino acid sequences of the heavy and light chains of the humanised antibody Ab633-HC3-LC2-T1-1B3-1G10 (hJF5) are shown in Fig. 1A. The full length humanized antibody hJF5 comprising heavy and light chains (Fig. 1B) showed characteristic binding in western blots to purified Aspergillus mannoprotein, with molecular weights of ~36 kDa to >210 kDa (Fig. 1C). The pattern of staining was consistent with that of the mouse antibody mJF5 (Fig. 1C). To further test the immuno-reactivities of the unlabeled and chelator-labeled antibodies after modification, additional in vitro tests were performed. Immunoreactivity of the mJF5 antibody following labeling with the chelators DOTAGA and NODAGA was investigated in ELISA tests. There was no significant loss in mJF5 immunoreactivity in ELISA, using a saturating concentration of purified mannoprotein antigen, as a consequence of labeling with the two chelators (Fig. 1D). Similarly, there was no notable loss in hJF5 immunoreactivity following labeling with NODAGA. Indeed, both the unlabeled and NODAGA-labeled hJF5 antibodies showed increased binding to the mannoprotein target, compared to mJF5 and its chelator derivatives, at equivalent antibody protein concentrations. The human IgG1 isotype control antibody ET901 conjugated to NODAGA showed no binding to the target antigen in ELISA tests (Fig. 1D). Labeling of the hJF5 antibody with NODAGA did not alter the hyphal-specific nature of the antibody to its target mannoprotein antigen (Fig. 1E). Intense immunofluorescence of hyphae, but lack of staining of un-germinated spores of the fungus (marked by asterisks), was observed with unconjugated mJF5, unconjugated hJF5, and NODAGA-labeled hJF5. No fluorescence was found using the IgG1 isotype control antibody ET901 conjugated to NODAGA, further demonstrating the specificities of the mJF5 and hJF5 antibodies.
Immuno-reactivities of the mJF5 and hJF5 antibodies and their DOTAGA and NODAGA chelator-labeled derivatives. A. Amino acid sequences of the heavy and light chains of the humanised antibody Ab633-HC3-LC2-T1-1B3-1G10 (hJF5); the signal peptides (cleaved off in the mature antibody) are highlighted in green, the variable domains are highlighted in blue, and the constant domains are highlighted in orange. B. Purity of the Protein A-purified antibody hJF5, with heavy and light chains of ~50kDa and ~31kDa respectively, determined by using SDS-PAGE under denaturing conditions followed by Coomassie staining. C. Western immunoblots using the purified hJF5 antibody (left) and mJF5 antibody (right) tested against purified Aspergillus mannoprotein antigen. Wells were loaded with 40 μl of a 0.5 mg/mL solution of antigen denatured in Laemmli buffer. Both antibodies show a characteristic smearing pattern, binding to glycoprotein antigen with molecular weights of between ~36 kDa and >210 kDa. D. ELISA of mJF5, hJF5, human IgG1 isotype control antibody ET901, and non-radiolabeled DOTAGA- or NODAGA-conjugated antibody derivatives, using microtiter plates coated with 1 mg/mL of purified Aspergillus mannoprotein antigen. No notable reductions in binding of the mJF5 and hJF5 antibodies are apparent as a result of conjugation to the chelators DOTAGA or NODAGA. Note, however, the increase in binding of the hJF5 and NODAGA-hJF5 antibodies compared to the mJF5 and NODAGA-mJF5 counterparts at equivalent antibody protein concentrations. The lack of binding of the NODAGA-conjugated human IgG1 isotype control ET901, demonstrates the specific binding of the mJF5 and hJF5 antibodies to the target antigen, and is confirmed by the molecular imaging studies in vivo. E. Immunofluorescence of germinating spores of Af293 using mJF5, hJF5, NODAGA-hJF5, and NODAGA-ET901. Brightfield images of cells are shown alongside their corresponding epifluorescence images following staining of the cells with the antibodies and FITC conjugates. Under epifluorescence, mJF5, hJF5 and NODAGA-hJF5 can be seen to bind to extracellular cell-wall bound antigen displayed on the surface of Af293 germ tubes, while the NODAGA conjugated human IgG1 isotype control antibody shows no fluorescence, consistent with hyphal-specific binding of the mJF5 and hJF5 antibodies. The scale bar that applies to all of the images = 6 μm.
mJF5 and hJF5 bind to the epitope β1,5-galactofuranose (Galf)
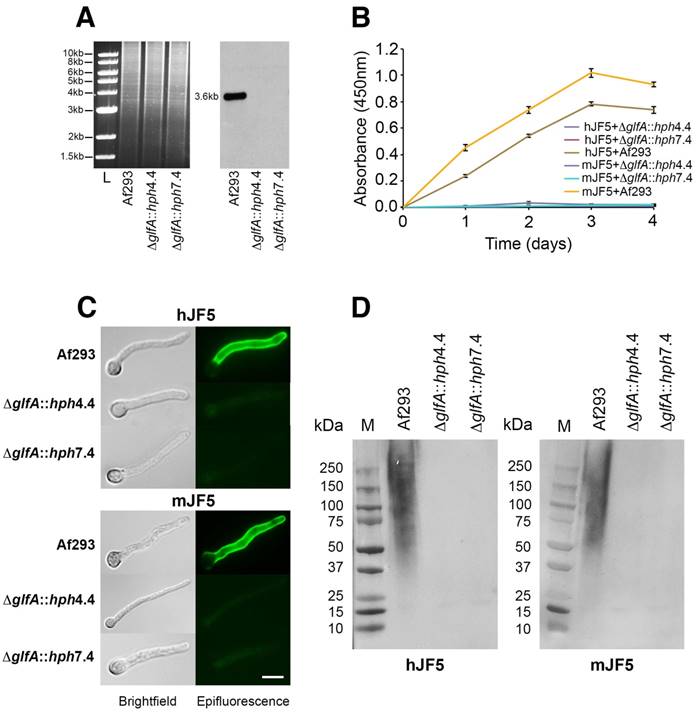
Aspergillus fumigatus mutants deficient in the epitope β1,5-galactofuranose (Galf) were successfully generated by targeted deletion of the UDP-galactopyranose mutase-encoding gene glfA (Fig. 2A). Two independent mutant strains (ΔglfA::hph4.4 and ΔglfA::hph7.4) showed characteristic reductions in growth at 30 °C and 37 °C compared to the wild-type strain Af293 (Fig. S1A), which is consistent with growth defects of A. fumigatus Galf-deficient mutants across a range of temperatures [16]. The mutants and Af293 were grown for 4 days in liquid shake cultures (Fig. S1B), and the corresponding fluids tested by ELISA using mAbs mJF5 and hJF5 (Fig. 2B). While the mAbs reacted strongly with extracellular antigen produced by Af293, there was complete loss of binding to antigen produced by the mutants over the 4-day sampling period (Fig. 2B). The mutants were also shown to be deficient in the Galf epitope in IF and western blotting studies, with complete lack of binding of the antibodies to cell-wall bound and secreted antigens (Figs. 2C and 2D, respectively). Taken together, these results indicate that the mAbs bind to the epitope Galf present in secreted and cell wall bound (galacto)mannoproteins of A. fumigatus.
Characterisation of the epitopes bound by mAbs mJF5 and hJF5. A. Genomic DNA of the Aspergillus fumigatus wild-type strain Af293 and the putative UDP-galactopyranose mutase-deficient mutants ΔglfA::hph4.4 and ΔglfA::hph7.4 (left panel), was digested with the restriction enzymes Xba1, fractionated by gel electrophoresis and blotted onto Hybond-NX membrane; the Southern blot (right panel) was probed with a 1.0-kb fragment of the glfA ORF. Note the presence of the predicted 3.6-kb band in the wild type strain, but absence in ΔglfA::hph4.4 and ΔglfA::hph7.4, showing successful deletion of the glfA gene in the mutant strains. B. ELISA of extracellular antigens in fluids from shake cultures of Af293 and mutants ΔglfA::hph4.4 and ΔglfA::hph7.4. While mAbs mJF5 and hJF5 showed strong recognition of secreted antigens in Af293 culture fluids, there was no recognition of antigens in the culture fluids of the mutant strains over the 4-day sampling period. C. Immunofluorescence of germinating spores of Af293 and the two mutant strains using mAbs mJF5 and hJF5. Brightfield images of cells are shown alongside their corresponding epifluorescence images following staining of the cells with the two antibodies and FITC conjugates. Under epifluorescence, both antibodies can be seen to bind to extracellular cell-wall bound antigen displayed on the surface of Af293 germ tubes, but there is no recognition of germinated spores of the wild-type strain, consistent with the hyphal-specific binding characteristics of the two antibodies. In contrast, neither antibody binds to the germ tubes of the two mutant strains. The scale bar that applies to all of the images = 4 µm. D. Western immunoblots using fluids from 3-day-old shake cultures of Af293 and the two mutant strains, shown in ELISA to contain greatest amounts antibody-reactive extracellular antigen from Af293 (Fig. 1B). Both mJF5 and hJF5 show a characteristic smearing pattern, binding to glycoproteins with molecular weights of between ~36 kDa and >210 kDa. In contrast, there is complete loss of antibody binding to extracellular antigens from corresponding 3-day-old shake culture fluids of ΔglfA::hph4.4 and ΔglfA::hph7.4.
hJF5 binds to A. fumigatus in vivo
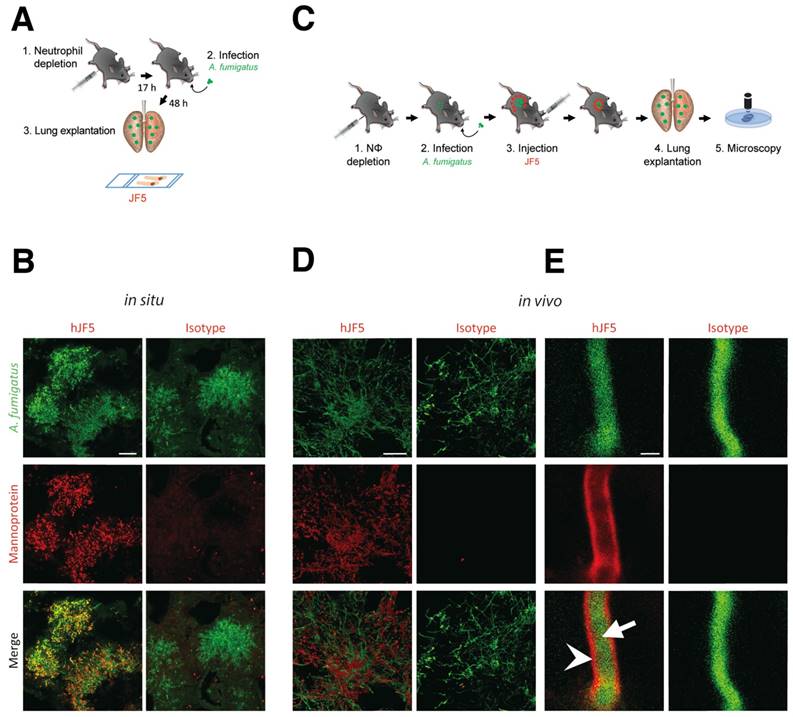
The suitability of mJF5 as an intravital whole-body diagnostic antibody, when equipped with a suitable label, was demonstrated previously [8]. To determine the diagnostic accuracy of the newly developed hJF5 antibody, we first investigated whether fluorochrome-labeled hJF5 was able to detect A. fumigatus in infected lungs of experimental animals. Neutrophil-depleted mice were intra-tracheally (i.t.) infected with conidia of a genetically-modified strain of the fungus expressing the fluorescent protein tdTomato (A. fumigatustdTomato) and, 48 h later, the lungs were excised and tissue sections were probed with hJF5 or the IgG1 isotype control antibody ET901 both labeled with the fluorochrome DyLight650 (Fig. 3A). Large fluorescent (false colour green) masses of A. fumigatustdTomato mycelium were detected in both cases (Fig. 3B). However, while hJF5 gave an intense red fluorescence of fungal elements, the isotype control was negative (Fig. 3B). In a second set of experiments, the labeled hJF5 or control antibodies were injected 24 h after infection into the circulation of mice (Fig. 3C). After an additional 24 h, lung lobes were dissected and were examined under confocal microscopy (Fig. 3D). This also showed red fluorescence of the hJF5-DyLight650 tracer which co-localised with green fluorescing fungal elements, with no fluorescence exhibited by the control. Individual hyphae showed green fluorescence of the A. fumigatustdTomato cytoplasm and specific binding of hJF5-DyLight650 (red) to the hyphal cell wall (Fig. 3E). The isotype controls were negative and showed green fluorescence of the hyphal cytoplasm only (Fig. 3E).
Binding specificity of hJF5 to A. fumigatus in infected mouse lungs. A. Experimental workflow for the in situ binding of mAb hJF5: (1) Neutrophil depletion by i.p. injection of 100 μg of the anti-Gr-1 antibody 17 h before injection with the pathogen; (2) Intra-tracheal infection with A. fumigatustdTomato; (3) After 48 h, the lungs were explanted, fixed, and fungal biomass stained in situ with DyLight650 conjugates of hJF5 or the human IgG1 isotype control antibody ET901. B. Micrographs of infected lungs showing cytoplasmic fluorescence (false colour green) of the genetically modified strain of the pathogen A. fumigatustdTomato, and co-localisation of cell-wall-bound mannoprotein antigen with the probes hJF5-DyLight650 (red fluorescence) or ET901-DyLight650 control (red fluorescence). Note red fluorescence of tissue probed with hJF5-DyLight650, but lack of fluorescence with the control antibody. Scale bar = 50 μm. C. Experimental workflow for hJF5 binding in vivo: (1) Neutrophil depletion by i.p. injection of 100 μg anti-Gr-1 antibody (clone RB6-8C5) 17 h before injection with the pathogen; (2) Intra-tracheal infection with A. fumigatustdTomato; (3) i.v. injection of hJF5-DyLight650 or ET901-DyLight650 antibodies 24 h after fungal infection; (4) Another 24 h later, the lungs were explanted and (5) longitudinal sections of explanted lung tissues were examined under confocal microscopy. D. Micrographs of A. fumigatustdTomato infected lungs (shown in false colour green) following i.v. injection with hJF5-DyLight650 (red) or ET901-DyLight650 (red). Note red fluorescence of lungs with hJF5-DyLight650, but lack of fluorescence with the control. Scale bar = 50 μm. E. An individual hypha is shown at higher magnification, with green fluorescence of A. fumigatustdTomato cytoplasm (white arrow) and binding of hJF5-DyLight650 (red) to the target mannoprotein antigen in the hyphal cell wall (white arrowhead). Note the lack of red fluorescence of the outer cell wall with the isotype control, further demonstrating the specificity of the hJF5 antibody. Scale bar = 2 μm.
PET/MR imaging of invasive pulmonary aspergillosis using [64Cu]DOTAGA-mJF5, [64Cu]NODAGA-mJF5 and [64Cu]NODAGA-hJF5
For immunoPET imaging, mJF5 was conjugated with the chelators DOTAGA and NODAGA, and hJF5 with the chelator NODAGA. The chelated antibodies were then labeled with the radionuclide 64Cu, which allows consecutive imaging for up to 3 d. The specific activities of the tracers used in the in vivo biodistribution studies was determined to be in the region of 500-625 MBq/mg. Radiolabeling efficiencies were determined by TLC with the 64Cu-labeled, chelator-conjugated antibodies [64Cu]DOTAGA-mJF5, [64Cu]NODAGA-mJF5 and [64Cu]NODAGA-hJF5 having labeling efficiencies of 92%, 98% and 97%, respectively. The serum stabilities of the chelator-conjugated antibodies were determined by radio-HPSEC, with the analysis showing no signs of proteolytic degradation, protein aggregation or copper transchelation to serum proteins over a 48 h time period (Fig. S3).
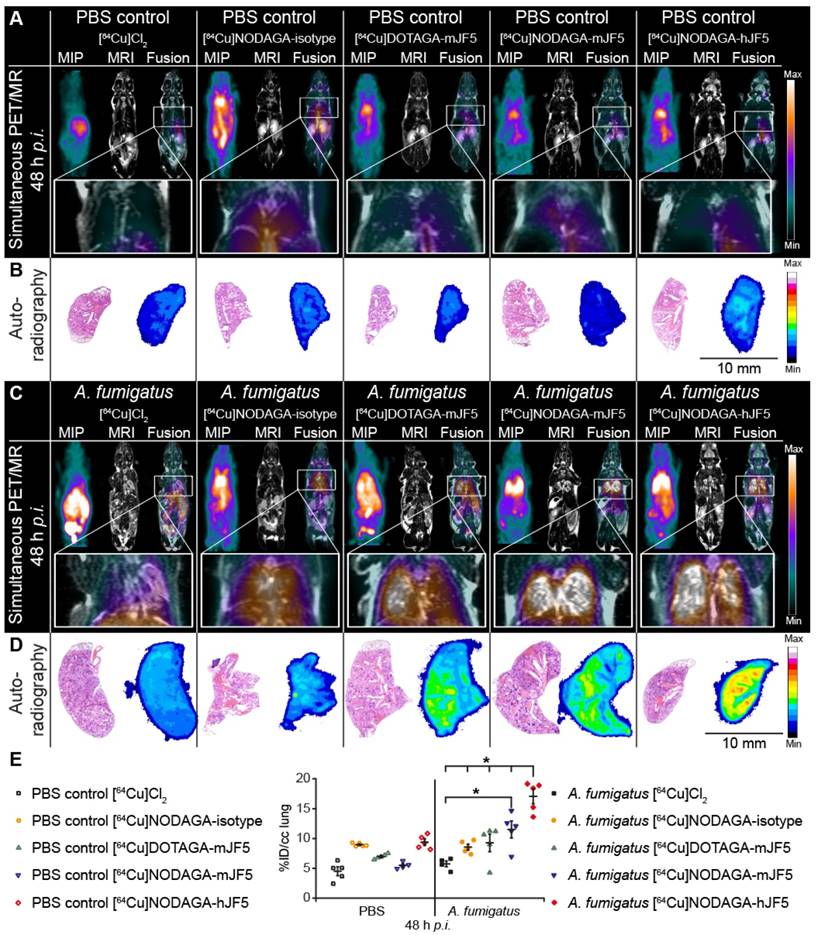
Neutropenic mice infected with A. fumigatus were injected with [64Cu]Cl2 only, [64Cu]NODAGA-isotype control antibody ET901, [64Cu]DOTAGA-mJF5, [64Cu]NODAGA-mJF5 or [64Cu]NODAGA-hJF5 and their biodistributions evaluated by PET/MRI at 3h (Fig. S4), 24 h (Fig. S5), and 48 h post infection (Fig. 4). Control mice were i.t. treated with PBS inducing sterile inflammation in the lung, but were otherwise treated the same.
In vivo biodistribution of [64Cu]Cl2, [64Cu]NODAGA-isotype control antibody ET901, [64Cu]DOTAGA-mJF5, [64Cu]NODAGA-mJF5 and [64Cu]NODAGA-hJF5 in PET/MR imaging at 48 h p.i. A and C. Coronal MIP, MR and fused PET/MR images of PBS treated mice and A. fumigatus infected mice injected with the respective tracers. The MR images of infected animals clearly reveal pulmonary infection and hyphal growth of A. fumigatus (hyperintense regions). The PET insert images of infected mice display specific lung uptake of the radiolabeled JF5 antibody-tracers compared to infected animals receiving [64Cu]Cl2 only or the 64Cu-labeled isotype control antibody ET901. Additionally, the high liver uptake of [64Cu]Cl2 and [64Cu]DOTAGA-mJF5 in infected animals can be observed. B and D. Representative autoradiographic images with subsequent H&E staining of the lung tissue 48 h p.i. Autoradiographs of lung sections (right) with the corresponding H&E staining (left). Ex vivo autoradiography shows higher tracer accumulation in the lungs of infected animals receiving the JF5 antibody tracers compared to the respective PBS controls. Infected animals receiving the JF5 antibody tracers show higher signal intensities in the lung compared to the A. fumigatus infected mice receiving [64Cu]Cl2 or the control tracer [64Cu]NODAGA-isotype. Furthermore, higher uptake of [64Cu]NODAGA-hJF5 in the lungs of infected animals compared to the other infected groups can be observed. (E) Quantification of the PET images 48 h p.i. for lung tissues. Uptake of the various tracers in the lungs of infected animals compared to PBS controls at 48 h p.i. is shown in groups of n=4-5 mice. Significantly higher uptake of [64Cu]NODAGA-hJF5 in the lungs of A. fumigatus infected animals was obtained compared to all other tracers. Significantly higher lung uptake was also observed in infected animals injected with the tracer [64Cu]NODAGA-mJF5 compared to [64Cu]Cl2 . Data are expressed as the mean ± SD %ID/cc. Group differences were examined using one-way ANOVA, followed by post hoc Tukey-Kramer, *P < 0.05.
[64Cu]NODAGA-hJF5 improves the in vivo detection of A. fumigatus lung infections compared to [64Cu]DOTAGA-mJF5 and [64Cu]NODAGA-mJF5
PET images showed increased uptake of the antibody-based PET tracers [64Cu]DOTAGA-mJF5, [64Cu]NODAGA-mJF5 and [64Cu]NODAGA-hJF5 in the lungs of A. fumigatus-infected animals 48 h p.i., compared to the lungs of PBS-treated control mice or lungs of PBS control and A. fumigatus-infected mice injected with [64Cu]Cl2 only (Fig. 4A and 4C). In vivo quantification (Fig. 4E) showed higher uptake of the antibody-based tracers in the lungs of infected animals compared to the PBS-treated controls or infected and control animals injected with [64Cu]Cl2 only. Results from in vivo PET imaging were confirmed by the ex vivo biodistribution data at 48 h p.i. (Table 1). The in vivo uptake of [64Cu]NODAGA-hJF5 48 h p.i. in the lungs of infected animals (17.1 ± 2.5 %ID/cc) was higher than in the lungs of the corresponding PBS-treated control animals (9.4 ± 1.1 %ID/cc; Fig. 4E). The ex vivo biodistribution results similarly showed higher uptake of [64Cu]NODAGA-hJF5 (33.8 ± 4.0 %ID/g) in the lungs of infected animals compared to the corresponding PBS-treated control animals (10.9 ± 1.8%ID/g). A similar trend in in vivo lung uptake in infected animals was found with [64Cu]NODAGA-mJF5 (11.5 ± 2.8 %ID/cc) compared to the corresponding PBS-treated control animals (5.5 ± 0.6 %ID/cc), with ex vivo biodistribution values of 31.6 ± 5.1 %ID/g and 7.7 ± 1.2 %ID/g, respectively. While ex vivo biodistribution results (Table 1) revealed a higher uptake of [64Cu]DOTAGA-mJF5 in the lungs of infected animals (26.0 ± 5.4 %ID/g) compared to the corresponding PBS-treated controls (10.1 ± 1.9 %ID/g), uptake of this tracer in the A. fumigatus-infected lung was less compared to the tracer [64Cu]NODAGA-mJF5. Taken together, the results show that the strength of uptake of the tracers in the A. fumigatus-infected lung followed the sequence [64Cu]NODAGA-hJF5 > [64Cu]NODAGA-mJF5 > [64Cu]DOTAGA-mJF5 > [64Cu]NODAGA-ET901 > [64Cu]Cl2 only, thereby showing that the uptake of [64Cu]NODAGA-hJF5 in the lungs of A. fumigatus infected mice was significantly higher compared to all other tested tracers 48 h p.i. Also the uptake of [64Cu]NODAGA-mJF5 in the lungs of A. fumigatus infected mice was significantly higher compared to [64Cu]Cl2 (Fig. 4E). This trend in uptake was further evidenced by ex vivo autoradiography of the lungs of infected and control animals (Fig. 4B and 4D).
Ex vivo biodistribution at 48 h p.i. The data confirm the results of the in vivo biodistribution obtained 48 h p.i. Significantly higher uptake of the [64Cu]DOTAGA-mJF5, [64Cu]NODAGA-mJF5 and [64Cu]NODAGA-hJF5 tracers in the lungs of infected animals compared to the corresponding PBS control animals confirm the specificities of the radiolabeled JF5 antibody tracers for the detection of IPA in vivo.
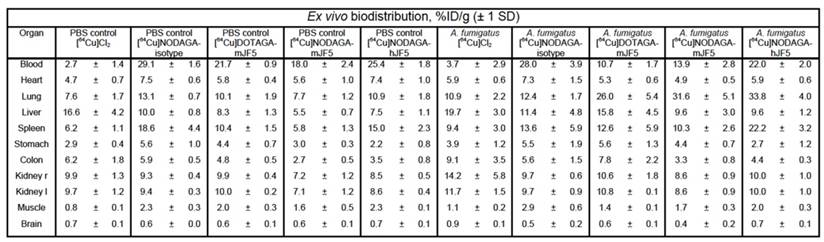
Data are results of n=4-5 animals per group and expressed as the mean ± SD %ID/g.
Conjugation of mJF5 and hJF5 antibodies to [64Cu]NODAGA gives non-specific liver enrichment similar to [64Cu]DOTAGA
In a previous study [8], we showed that while [64Cu]DOTA-mJF5 allowed specific detection of A. fumigatus lung infections in vivo, uptake by the liver was also evident. We hypothesized that this was due to non-specific hepatic clearance of the radiolabeled antibody or transchelation of 64Cu upon unstable binding to its chelator as seen by others [24] and that liver uptake might be reduced by using the chelators DOTAGA or NODAGA in place of DOTA. In vivo biodistribution of the tracers [64Cu]NODAGA-ET901, [64Cu]DOTAGA-mJF5, [64Cu]NODAGA-mJF5 and [64Cu]NODAGA-hJF5 showed no significant difference in uptake between DOTAGA- and NODAGA-labeled JF5 antibodies in the liver 3 h, 24 h and 48 h p.i. (Fig. S6). However, [64Cu]DOTAGA-mJF5 was highest in the liver of A. fumigatus infected animals 48 h p.i. with 12.9 ± 4.1 %ID/cc compared to [64Cu]NODAGA-mJF5 (7.3 ± 1.8 %ID/cc) and [64Cu]NODAGA-hJF5 (10.0 ± 0.8 %ID/cc). [64Cu]Cl2 was injected for control purposes to preclude unspecific perfusion effects of the antibody-based tracer and presented with significantly higher uptake in the liver of A. fumigatus infected animals with 26.0 ± 5.0 %ID/cc (3 h p.i.) compared to all other investigated groups. A. fumigatus infected mice presented with higher liver uptake of [64Cu]Cl2 and [64Cu]DOTAGA-mJF5 compared to the radiolabeled ET901 isotype control antibody and [64Cu]NODAGA-labeled JF5-based tracers (Fig. S6, Table 1).
Discussion
Early detection of invasive pulmonary aspergillosis (IPA) and treatment with appropriate antifungal drugs is critical to patient survival, but clinical diagnosis remains extremely difficult. Mycological culture from sterile biopsy, still regarded as the gold standard for IPA diagnosis, is limited by poor sensitivity and lengthy turnaround time, and assays used to detect biomarkers of infection (Aspergillus cell wall galactomannan and 'pan-fungal' β-D-glucan) in serum or bronchoalveolar lavage fluid (BALf), lack either sensitivity or specificity and, in the case of BALf, involve invasive procedures [9]. While a point-of-care lateral-flow assay for IPA diagnosis has been developed [12] and has the potential to increase the speed and accuracy of detection [4], the present limitations of IPA diagnostics has led to the empirical or 'fever-driven' use of antifungals, resulting in the sometimes unnecessary exposure of patients to toxic drugs, increased likelihood of severe drug-drug interactions, inflated treatment costs, and contributes to the emergence of resistance to mould-active triazoles in Aspergillus species. Empirical antifungal therapy also reduces the sensitivity of culture-based and non-culture-based diagnostic tests, which are needed to establish a conclusive diagnosis, to determine drug sensitivities of cultured pathogens, and to monitor responsiveness to treatments.
Anti-fungal therapy guided by diagnostic-driven approaches has been shown to be more effective than empirical treatment both in terms of cost and improved patient outcomes [9]. Such a diagnostic-driven approach to IPA treatment typically relies on radiographic imaging, especially computed tomography (CT), coupled with frequent testing for fungal biomarkers. A chest CT that reveals dense, well-delineated, infiltrates in the lung with or without a 'halo sign' in a patient with prolonged neutropenia and persistent or recurrent fever of unknown cause that is unresponsive to empirical antibacterial agents, is suggestive of IPA. However, these radiological signs are not pathognomonic since other mould pathogens and bacterial pathogens capable of angio-invasion can produce a similar appearance in CT.
Despite the limited specificity of standard CT for diagnosing IPA, or invasive fungal diseases (IFDs) in general, CT remains a trigger for commencing antifungal treatment in many hematology units. Attempts have been made to improve the accuracy of radiographic imaging for early diagnosis and monitoring of IPA, by combining measures of metabolic activity with positron emission tomography (PET), but these methods fail to discriminate between infection and non-infectious pathologies [10, 11]. A number of studies have suggested that [18F]fluorodeoxyglucose ([18F]FDG), a diagnostic tracer that accumulates in metabolically-active inflammatory and cancer cells, might be useful for differentiating between non-invasive and invasive aspergillosis and for monitoring IPA response to antifungal treatment [25]. However, a recent study conducted by our group, using PET and magnetic resonance imaging (PET/MRI) to detect A. fumigatus lung infections in vivo, showed that increased uptake of [18F]FDG in IPA was indistinguishable from the uptake seen during inflammatory reactions due to sterile triggers, or during infections by the bacterial pathogens Streptococcus pneumonia and Yersinia enterocolitica [8].
Improvements in the accuracy of PET-based imaging for infectious disease diagnosis have been met through the use of small molecules such as 18F-labelled maltohexaose, fluorodeoxysorbitol, or ciprofloxacin, but the majority of these target bacterial infections [26]. Few tracers have been developed that specifically target IFDs, but some success has been achieved in visualizing IPA by combining microPET/CT with 68Ga-radiolabeled siderophores [27]. The use of monoclonal antibodies in the molecular imaging of IFDs has yet to be exploited, but we recently demonstrated the enormous potential of antibody-guided PET/MRI (immunoPET/MRI) for accurate diagnosis of IPA [8]. Our diagnostic probe, which allows non-invasive detection of A. fumigatus lung infections in vivo, employs the Aspergillus-specific mouse monoclonal antibody (mAb) mJF5. The mAb, which forms the basis of the Aspergillus LFA [4, 12], binds to a diagnostic mannoprotein antigen which is secreted during active growth of the pathogen only, and so is able to discriminate between inactive spores present in inhaled air and invasive hyphae that infect the lung of the neutropenic host. When coupled to [64Cu]DOTA, the long half-life of the [64Cu]DOTA-mJF5 tracer enables sequential imaging of IPA and so represents an ideal candidate for repeated imaging of patients following a single injection of the radioactive tracer. Furthermore, the hyphal-specific nature of the immuno-PET tracer may prove useful in monitoring infection in response to antifungal treatment.
In this study, we have engineered the JF5 antibody for use in humans by grafting of the complementarity determining regions of the mouse antibody into a human IgG1 antibody framework, to make a humanised JF5 antibody (hJF5). The pre-clinical studies conducted here using the 64Cu-labeled hJF5 antibody in a neutropenic mouse model of IPA, have demonstrated improved uptake of the hJF5-based PET tracer in infected lung tissues compared to its murine counterpart. Increased binding of hJF5 and hJF5-NODAGA compared to mJF5 and mJF5-NODAGA, although not significant, was further demonstrated in vitro during ELISA tests with the purified mannoprotein antigen. The reasons for this increased affinity have yet to be established, but there is increasing evidence to suggest that exchanging murine and human immunoglobulin constant chains can have profound effects on the kinetics of antigen binding [28, 29]. Furthermore, while there is extensive anecdotal and unpublished evidence that attempts to generate humanised mAbs based on mouse V regions have failed to produce useful antibodies because of loss of affinity and specificity, our work shows successful generation of a higher affinity (humanised) antibody for in vivo imaging of IPA, and retention of specificity for the invasive hyphal growth phase of the pathogen.
In our previous work [8], specific uptake of [64Cu]DOTA-mJF5 was shown in the lungs of A. fumigatus infected animals compared to PBS-treated control mice, but was also accompanied by high liver uptake in all animals. Uptake of [64Cu]DOTA-mJF5 was increased in the livers of A. fumigatus infected animals (27.35 ± 2.72 %ID/g), and in PBS control mice (15.77 ± 3.05 %ID/g) in ex vivo biodistribution studies at 48 h p.i. [8]. Possible reasons for this were considered to be (1) non-specific hepatic clearance of the radiolabeled antibody, (2) binding of the tracer to mannoprotein in the liver following release of soluble antigen from infection foci in the lungs, and (3) trans-chelation of 64Cu to abundant liver proteins due to insufficiently strong binding of 64Cu to its chelator. The chelator DOTA is known to have poor in vivo stability, which results in loss of the radio-metal and its non-specific accumulation in non-target tissue. In recent years, alternative chelators such as DOTAGA and NODAGA have been reported which have increased in vivo stability [30, 31, 32]. Here, we found that NODAGA provided the lowest liver uptake of both the mJF5 and hJF5-labeled antibodies in A. fumigatus infected animals (9.6 ± 3.0 %ID/g and 9.6 ± 1.2 %ID/g, respectively), and in PBS-treated mice (5.5 ± 0.7 %ID/g and 7.5 ± 1.1 %ID/g, respectively), while maintaining the highest level of uptake in the infected lungs (31.6 ± 5.1 %ID/g and 33.8 ± 4.0 %ID/g, respectively). Serum stability showed no signs of proteolytic degradation, protein aggregation or transchelation of 64Cu to serum proteins. Thus, the improved biodistribution of the DOTAGA- and NODAGA-labeled antibodies is consistent with the improved in vivo stability of these chelators, resulting in decreased uptake of radiolabeled immuno-conjugates in the liver [30, 31, 32].
In conclusion, we have shown that the humanised JF5 antibody tracer has enhanced diagnostic performance for non-invasive imaging of A. fumigatus lung infections in vivo compared to its mouse counterpart. It is essentially translatable to the clinical setting since the mAb targets a signature molecule of Aspergillus infection that has been clinically authenticated for IPA diagnosis in humans based on LFA tests of serum and bronchoalveolar lavage fluids [4, 12]. Furthermore, we have shown that the mJF5 and hJF5 antibodies bind to the antigenic determinant β1,5-galactofuranose (Galf) present in the target mannoprotein antigen. The absence of the epitope Galf in mammalian carbohydrates [17], coupled with the improved imaging capabilities of the hJF5 antibody, means that the [64Cu]NODAGA-hJF5 tracer should not bind to human structures non-specifically. This is very promising for the specificity of the imaging approach. At the same time, it reduces substantially the likelihood of adverse events when applying this antibody to humans. The hJF5 antibody therefore represents an excellent candidate for clinical studies of radiological IPA detection.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We are grateful to Walter Ehrlichmann for the 64Cu production and Maren Harant, Sandro Aidone and Ramona Stumm for expert technical assistance. This work was supported by the European Union Seventh Framework Programme FP7/2007-2013 under Grant 602820, the Deutsche Forschungsgemeinschaft (Grant WI3777/1-2 to SW), and the Werner Siemens Foundation. We thank Sven Krappman for use of the A. fumigatustdTomato strain, and acknowledge the Imaging Centre Essen (IMCES) for assistance with optical imaging of lungs.
Authors' Contributions
Conception and design: G.D., A.-M.R., M.H., F.B., M.G., M.J., R.S., B.J.P., S.W., and C.R.T. Development of methodology: G.D., A.-M.R., A.M., P.R.S., C.S., D.S.-S., M.H., J.W., J.F., A.D., F.B., F.D., M.G., L.S.R., M.J., R.S., B.J.P., S.W., and C.R.T. Statistical analysis: A.-M.R., M.E., S.W. and C.R.T. Writing of the manuscript: G.D., A.-M.R., A.M., P.R.S., M.H., J.F., F.B., M.G., M.E. M.J., R.S., B.J.P., S.W., and C.R.T.
Competing Interests
B.J.P. receives grant/research support from: Bayer Healthcare, Boehringer-Ingelheim, and Siemens; however, none of the grants are directly related to this work. C.R.T. is director of ISCA Diagnostics Ltd.
References
1. WHO: Geneva, Switzerland. World Health Statistics 2014. http://www.who.int/mediacentre/news/releases/2014/world-health-statistics-2014/en/
2. Brown GD, Denning DW, Gow NAR. et al. Hidden killers: human fungal infections. Sci Transl Med. 2012;4(165):165rv13
3. HM Government: London, United Kingdom. Review on antimicrobial resistance. Tackling drug-resistant infections globally: final report and recommendations. 2016. https://amr-review.org/
4. Prattes J, Prueller F, Koidl C. et al. Novel tests for diagnosis of invasive pulmonary aspergillosis in patients with underlying respiratory diseases. Am J Resp Crit Care Med. 2014;190(8):922-929
5. Pichler BJ, Kolb A, Nägele T. et al. PET/MRI: paving the way for the next generation of clinical multimodality imaging applications. J Nucl Med. 2010;51(3):333-336
6. Santangelo PJ, Rogers KA, Zurla C. et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiteroviral therapy-treated macaques. Nat Methods. 2015;12(5):427-432
7. Wiehr S, Warnke P, Rolle A-M. et al. New pathogen-specific immunoPET/MR tracer for molecular imaging of a systemic bacterial infection. Oncotarget. 2016;7:10990-11001
8. Rolle A-M, Hasenberg M, Thornton CR. et al. ImmunoPET/MR imaging allows specific detection of Aspergillus fumigatus lung infection in vivo. Proc Nat Acad Sci USA. 2016;113(8):1026-1033
9. Segal BM. Aspergillosis. New Engl J Med. 2009;360:1870-1884
10. Glaudemans AW, Quintero AM, Signore A. PET/MRI in infectious and inflammatory diseases: will it be a useful improvement? Eur J Nucl Med Mol Imaging. 2012;39(5):745-749
11. Glaudemans AW, Slart RH, van Dijl DM. et al. Molecular imaging of infectious and inflammatory diseases: a terra incognita. J Nucl Med. 2015;56(5):659-661
12. Thornton CR. Development of an immunochromatographic lateral flow device for rapid serodiagnosis of invasive aspergillosis. Clin Vacc Immunol. 2008;15(7):1095-1105
13. Thornton CR. Breaking the mould - novel diagnostic and therapeutic strategies for invasive pulmonary aspergillosis in the immune deficient patient. Expert Rev Clin Immunol. 2014;6(10):771-780
14. Komachi Y, Hatakeyama S, Motomatsu H. et al. gfsA encodes a novel galactofuranosyltransferase involved in biosynthesis of galactofuranose antigen of O-glycan in Aspergillus nidulans and Aspergillus fumigatus. Mol Microbiol. 2013;90(5):1054-1073
15. Latgé JP. Galactofuranose containing molecules in Aspergillus fumigatus. Med Mycol. 2009;47:S104-S109
16. Schmalhorst PS, Krappman S, Vervecken W. et al. Contribution of galactofuranose to the virulence of the opportunistic pathogen Aspergillus fumigatus. Euk Cell. 2008;7(8):1268-1277
17. Tefsen B, Ram AFJ, van Die I. et al. Galactofuranose in eukaryotes: aspects of biosynthesis and functional impact. Glycobiology. 2012;22(4):456-469
18. Catlett NL, Lee BE, Yoder OC. et al. Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet Newsl. 2003;50:9-11
19. Kershaw MJ, Talbot NJ. Genome-wide functional analysis reveals that infection-related fungal autophagy is necessary for rice blast disease. Proc Natl Acad Sci USA. 2009;106:15967-15972
20. Thornton CR, Ryder LS, Le Cocq K. et al. Identifying the emerging human pathogen Scedosporium prolificans by using a species-specific monoclonal antibody that binds to the melanin biosynthetic enzyme tetrahydroxynaphthalene reductase. Env Microbiol. 2015;17:1023-1048
21. Thornton CR. Immunological methods for fungi. In Molecular and Cellular Biology of Filamentous Fungi, A Practical Approach. Talbot, N.J. (ed.). Oxford, UK: University Press. 2001 pp. 227-257
22. Laemmli UK. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature. 1970;227:680-695
23. Wehrl HF, Hossain M, Lankes K. et al. Simultaneous PET-MRI brain function in activated and resting state on metabolic, hemodynamic and multiple temporal scales. Nat Med. 2013;19:1184-1189
24. Wu AM, Olafsen T. Antibodies for molecular imaging of cancer. Cancer J. 2008;14(3):191-197
25. Hot A, Maunoury C, Poiree S. et al. Diagnostic contribution of positron emission tomography with [18F]fluorodeoxyglucose for invasive fungal infections. Clin Microbiol Infect. 2011;17(3):409-417
26. Van Oosten M, Hahn M, Crane LMA. et al. Targeted imaging of bacterial infections: advances, hurdles and hopes. FEMS Microbiol Rev. 2015;39:892-916
27. Petrik M, Haas H, Dobrozemsky G. et al. 68Ga-siderophores for PET imaging of invasive pulmonary aspergillosis: Proof of principle. J Nucl Med. 2010;51(4):639-645
28. Dillon MJ, Loban RA, Reed DE. et al. Contribution of murine IgG Fc regions to antibody binding to the capsule of Burkolderia pseudomallei. Virulence. 2016;7(6):691-701
29. Torres M, Fernandez-Fuentes N, Fiser A. et al. Exchanging murine and human immunoglobulin constant chains affects the kinetics and thermodynamics of antigen binding and chimeric antibody autoreactivity. PLoS ONE. 2007;12:e1310
30. Boswell CA, Sun XK, Niu WJ. et al. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J Med Chem. 2004;47:1465-1474
31. Ghosh SC, Pinkston KL, Robinson H. et al. Comparison of DOTA and NODAGA as chelators for 64Cu-labeled immunoconjugates. Nucl Med Biol. 2015;42(2):177-183
32. Westerlund K, Honarvar H, Norrtsrom E. et al. Increasing the net negative charge by replacement of DOTA chelator with DOTAGA improves the biodistribution of radiolabeled second-generation synthetic affibody molecules. Mol Pharmaceutics. 2016;13(5):1668-1678
Author contact
![]() Corresponding author: Christopher R. Thornton, ISCA Diagnostics Ltd. and Biosciences, College of Life & Environmental Sciences, University of Exeter, Stocker Road, EX4 4QD, Exeter, United Kingdom. Phone 44-1392-725172; Fax: 44-1392-723434; E-mail: c.r.thorntonac.uk
Corresponding author: Christopher R. Thornton, ISCA Diagnostics Ltd. and Biosciences, College of Life & Environmental Sciences, University of Exeter, Stocker Road, EX4 4QD, Exeter, United Kingdom. Phone 44-1392-725172; Fax: 44-1392-723434; E-mail: c.r.thorntonac.uk