Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(14):3461-3477. doi:10.7150/thno.20657 This issue Cite
Research Paper
Sulforaphane-Induced Cell Cycle Arrest and Senescence are accompanied by DNA Hypomethylation and Changes in microRNA Profile in Breast Cancer Cells
Anna Lewinska1 ![]() , Jagoda Adamczyk-Grochala1, Anna Deregowska2, 3, Maciej Wnuk2
, Jagoda Adamczyk-Grochala1, Anna Deregowska2, 3, Maciej Wnuk2
1. Laboratory of Cell Biology, University of Rzeszow, Werynia 502, 36-100 Kolbuszowa, Poland;
2. Department of Genetics, University of Rzeszow, Kolbuszowa, Poland;
3. Postgraduate School of Molecular Medicine, Medical University of Warsaw, Warsaw, Poland.
Received 2017-4-19; Accepted 2017-5-29; Published 2017-8-15
Abstract

Cancer cells are characterized by genetic and epigenetic alterations and phytochemicals, epigenetic modulators, are considered as promising candidates for epigenetic therapy of cancer. In the present study, we have investigated cancer cell fates upon stimulation of breast cancer cells (MCF-7, MDA-MB-231, SK-BR-3) with low doses of sulforaphane (SFN), an isothiocyanate. SFN (5-10 µM) promoted cell cycle arrest, elevation in the levels of p21 and p27 and cellular senescence, whereas at the concentration of 20 µM, apoptosis was induced. The effects were accompanied by nitro-oxidative stress, genotoxicity and diminished AKT signaling. Moreover, SFN stimulated energy stress as judged by decreased pools of ATP and AMPK activation, and autophagy induction. Anticancer effects of SFN were mediated by global DNA hypomethylation, decreased levels of DNA methyltransferases (DNMT1, DNMT3B) and diminished pools of N6-methyladenosine (m6A) RNA methylation. SFN (10 µM) also affected microRNA profiles, namely SFN caused upregulation of sixty microRNAs and downregulation of thirty two microRNAs, and SFN promoted statistically significant decrease in the levels of miR-23b, miR-92b, miR-381 and miR-382 in three breast cancer cells. Taken together, we show for the first time that SFN is an epigenetic modulator in breast cancer cells that results in cell cycle arrest and senescence, and SFN may be considered to be used in epigenome-focused anticancer therapy.
Keywords: sulforaphane, breast cancer cells, DNA methylation, microRNA.
Introduction
The maintenance of tissue-specific gene expression patterns in normal mammalian cells is achieved by epigenetic mechanisms such as DNA methylation, histone post-transcriptional modifications, nucleosome remodeling and positioning, and non-coding RNAs, specifically microRNA expression and globally affected epigenetic landscape is considered to be a hallmark of cancer [1-4]. Cancer epigenome is characterized by promoter specific DNA hypermethylation (e.g. tumor suppressor genes), global DNA hypomethylation (e.g. tissue-specific genes, oncogenes, repetitive regions) and altered status of histone methylation and acetylation (e.g. H3K4ac, H3K9ac, H3K18ac, H4K16ac, H3K4me2, H3K4me3, H3K27me3, H4K20me3) that may in turn lead to chromosome instability and fragility, aberrant gene silencing and abnormal global microRNA expression (e.g. onco-microRNAs and tumor suppressor microRNAs) promoting activation of oncogenes and silencing of tumor suppressor genes [1-6]. As epigenetic modifications/alterations are implicated in virtually every step of tumorigenesis, the cancer epigenome is considered as a prevention/therapy target [7-10]. Targeted epigenetic therapy of cancer is based on the use of epigenetic drugs (epi-drugs) that have been developed for the specific inhibition of epi-enzymes, e.g., DNA methyltransferase inhibitors (DNMTi) and histone deacetylase inhibitors (HDACi) [7-10]. Treatment with epi-drugs results in changes in gene expression in multiple pathways, including cell cycle and apoptosis that may in turn induce tumor regression or sensitize to chemotherapy [7-10].
More recently, plant-derived bioactive compounds with numerous anticancer activities, such as curcumin (turmeric), genistein (soybean), tea polyphenols (green tea), resveratrol (grapes) and sulforaphane (cruciferous vegetables) [11] have been also reported to modulate epigenetic marks in cancer cells (e.g., DNA methylation, histone modifications and microRNA expression) that may be a basis for new phytochemical-mediated therapeutic or preventive approaches [12-17].
In the present study, we have investigated the mechanisms of cytostatic action of low doses of sulforaphane (SFN), an isothiocyanate, against phenotypically distinct breast cancer cells MCF-7 (ER+, PR+/-, HER2-), MDA-MB-231 (ER-, PR-, HER2-) and SK-BR-3 (ER-, PR-, HER2+). We found that SFN-induced cell cycle arrest, nitro-oxidative stress and genotoxicity were accompanied by global DNA hypomethylation, decreased levels of DNMT1 and DNMT3B, diminished pools of m6A RNA methylation and changes in microRNA profile. SFN treatment resulted in statistically significant decrease in the levels of miR-23b, miR-92b, miR-381 and miR-382 in three breast cancer cells used.
Materials and Methods
Reagents
The reagents used were purchased from Sigma-Aldrich (Poland). Sulforaphane (1-isothiocyanato-4-(methylsulfinyl)-butane, SFN) was dissolved in dimethyl sulfoxide (DMSO). DMSO concentrations used (up to 0.1%) did not affect cell vitality and viability.
Cell lines
Human breast cancer cells MCF-7, MDA-MB-231 and SK-BR-3 were obtained from ATCC (Manassas, VA, USA) and normal human mammary epithelial cells (HMEC) were obtained from Lonza (Basel, Switzerland). Cells were cultured as described comprehensively elsewhere [18].
MTT test
SFN cytotoxicity was estimated using an MTT assay [19]. Briefly, 5000 cells per a well of 96-well plate were stimulated with 2.5, 5, 10, 20, 30 and 50 µM SFN for 24 h and metabolic activity (MTT test) was then analyzed. The concentrations of 5, 10 and 20 µM SFN and 24 h treatment were selected for further experiments.
Cell cycle
After SFN treatment, DNA content-based analysis of cell cycle was assessed using flow cytometry (Muse™ Cell Analyzer, Merck Millipore) and Muse™ Cell Cycle Kit according to the manufacturer's instructions.
Senescence-associated β-galactosidase activity (SA-β-gal)
Cells were stimulated with SFN for 24 h and 7 days after SFN removal SA-β-gal activity was conducted [20].
Apoptosis
After SFN treatment, apoptosis was assessed using flow cytometry and Muse™ Annexin V and Dead Cell Assay Kit and Muse™ Multi-caspase Assay Kit (Merck Millipore) as described elsewhere [21]. 30 min treatment with 10 mM hydrogen peroxide (HP) was considered as an apoptotic stimulus (positive control) [22].
Autophagy
SFN-induced autophagy was measured using flow cytometry and Muse™ Autophagy LC3-antibody based Kit (Merck Millipore). Cells starved in Earle's Balanced Salt Solution (EBSS) for 6 h served as a positive control [23].
Nitro-oxidative stress
After SFN treatment, several oxidative and nitrosative stress parameters were analyzed. The production of reactive oxygen species (ROS), total and mitochondrial superoxide was evaluated using the fluorogenic probes a chloromethyl derivative of H2DCF-DA (CM-H2DCF-DA), dihydroethidium and MitoSOX™ Red reagent, respectively [19]. SFN-mediated protein carbonylation was analyzed using an OxyBlot™ Protein Oxidation Detection Kit (Merck Millipore). SFN-induced changes in the levels of nitric oxide were assessed using flow cytometry and Muse™ Nitric Oxide Kit (Merck Millipore). Cells treated for 5 min with a nitric oxide donor MAHMA NONOate (1 mM) (Cayman Chemical Company, Ann Arbor, Michigan, USA) served as a positive control [23].
Genotoxicity and DNA damage response
DNA double strand breaks (DSBs) and DNA single strand breaks (SSBs) were evaluated using neutral and alkaline comet assay, respectively [24]. The percentage of tail DNA was used as a parameter of DNA damage. The activation of ATM and H2AX was analyzed using flow cytometry and Muse™ Multi-Color DNA Damage kit (Merck Millipore) [23]. Cells treated for 24 h with 20 µM etoposide served as a positive control.
Immunostaining
An immunostaining protocol used was comprehensively described elsewhere [21]. Briefly, SFN-treated and fixed cells were subjected to incubation with the primary antibody anti-53BP1 (1:200) (Novus Biologicals) and a secondary antibody conjugated to Texas Red (1:1000) (Thermo Fisher Scientific). 53BP1 foci per nucleus were quantified using In Cell Analyzer software (GE Healthcare).
Western blotting
Western blotting protocol was comprehensively described elsewhere [20]. The following primary and secondary antibodies were used: anti-p21 (1:100), anti-p53 (1:500), anti-p27 (1:200), anti-GLUT1 (1:1000), anti-HK2 (1:200), anti-PKM2 (1:1000), anti-LDHA (1:1000), anti-phospho-AMPKα (Thr172) (1:750), anti-AMPKα (1:1000), anti-phospho-AKT (Ser473) (1:1750), anti-AKT (1:1000), anti-DNMT1 (1:200), anti-DNMT2 (1:500), anti-DNMT3A (1:200), anti-DNMT3B (1:200), anti-β-actin (1:1000) and secondary antibodies conjugated to HRP (1:50000) (Thermo Fisher Scientific, Santa Cruz, Abcam, Cell Signaling Technology, Sigma-Aldrich). Densitometry analysis was conducted using GelQuantNET software (http://biochemlabsolutions.com/GelQuantNET.html). The data represent the relative density normalized to β-actin. Phospho-AMPK and phospho-AKT signals were also normalized to AMPK and AKT signals, respectively.
ATP and lactate assays
ATP and lactate levels were evaluated in SFN-treated cells using ATP assay kit and lactate assay kit (Sigma-Aldrich), respectively [23]. ATP and lactate concentrations were calculated on the basis of a standard curve obtained for ATP and lactate solutions, respectively, and are presented in ng/µl.
ERK1/2 activity
SFN-mediated extracellular signal-regulated kinase 1/2 (ERK1/2) activity was analyzed using flow cytometry and Muse™ MAPK Activation Dual Detection Kit (Merck Millipore) [23].
Real-Time PCR using TaqMan® Arrays
After 10 µM SFN treatment, RNA was extracted using GenElute™ Mammalian Total RNA Miniprep Kit (Sigma-Aldrich) and cDNA was synthesized using 2 µg of RNA as a template and Transcriptor First Strand cDNA Synthesis Kit (Roche) according to the manufacturer's instructions. The expression of cyclin and cell cycle regulation-associated genes and DNA methylation and transcriptional repression-associated genes was evaluated using Applied Biosystems StepOnePlus™ Real-Time PCR System and dedicated Real-Time PCR TaqMan® Array Plates, namely TaqMan® Array 96-Well FAST Plate Human Cyclins and Cell Cycle Regulation (4418768, Applied Biosystems™) and TaqMan® Array 96-Well FAST Plate Human DNA Methylation and Transcriptional Repression (4418772, Applied Biosystems™), respectively, according to the manufacturer's instructions. GAPDH gene was used as a housekeeping gene. The qRT-PCR products that were amplified after 35 cycles were discarded. The expression profiles were created using Genesis 1.7.7 software [25] (https://genome.tugraz.at/genesisclient/genesisclient_download.shtml) on the basis of ΔCt values and log10 to log2, log2 transform ratio and hierarchical clustering functions.
Global DNA methylation
DNA methylation was evaluated as a 5-methyl-2'-deoxycytidine (5-mdC) level using ELISA-based assay, namely MethylFlash™ Methylated DNA Quantification Kit (Epigentek, Farmingdale, NY, USA) [21]. Cells treated for 24 h with an inhibitor of DNA methylation 5-aza-2'-deoxycytidine (5-aza-dC) (5 µM) served as a negative control [21].
N6-methyladenosine (m6A) RNA methylation
N6-methyladenosine (m6A) in RNA was assayed using ELISA-based EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric) (Epigentek) [26].
microRNA profiling
After 10 µM SFN treatment, RNA was extracted using GenElute™ Mammalian Total RNA Miniprep Kit (Sigma-Aldrich) and cDNA was synthesized using 100 ng of RNA as a template and EPIK miRNA Panel Assays (Bioline Ltd.) containing RT Primer Pool, EPIK RT Enzyme, EPIK RT Buffer and RNA Spike according to the manufacturer's instructions. The expression of 352 microRNAs (http://www.bioline.com/list-of-cancer-panel-plates#tab2) was evaluated using Applied Biosystems StepOnePlus™ Real-Time PCR System and dedicated EPIK Cancer miRNA Panel (Hi-ROX Plate 0.1Y, BIO-66032, Bioline Ltd.) according to the manufacturer's instructions. The qRT-PCR products that were amplified after 35 cycles were discarded. The expression profiles were created using Genesis 1.7.7 software [25] (https://genome.tugraz.at/genesisclient/genesisclient_download.shtml) on the basis of global normalized Ct values and log10 to log2, log2 transform ratio and hierarchical clustering functions. The interactions between four selected microRNAs, namely miR-23b, miR-92b, miR-381, miR-382 and cell cycle signaling pathways were analyzed using iBioGuide (Advaita Bio, Plymouth, USA; https://www.advaitabio.com/ibioguide.html) on the basis of KEGG PATHWAY Database.
Statistical analysis
The mean values ± SD were calculated on the basis of at least three independent experiments. Box and whisker plots were also considered. Statistical significance was evaluated using GraphPad Prism 5 using 1-way ANOVA and Dunnett's test. The differences in microRNA expression between control vs treated samples were assessed using Student's t-test.
Results
SFN treatment results in different and concentration-dependent cell fates in breast cancer cells - cell cycle arrest, senescence, apoptosis and autophagy
As cancer cell populations are heterogeneous, we decided to evaluate if sulforaphane (SFN) would promote different cell fates in breast cancer cells. According to the initial screen based on MTT assay that involved twenty eight phytochemicals, namely ellagic acid, eugenol, silibinin, allyl sulfide, emodin, vanillin, morin, baicalein, genistein, quercetin, rutin, naringin, hesperidin, phloretin, taxifolin, myricetin, luteolin, galangin, pelargonidin, epigallocatechin gallate, catechin, curcumin, capsaicin, sulforaphane, 6-gingerol, indole-3-carbinol, fisetin and apigenin (data not shown), we have selected SFN because it significantly affected breast cancer cell metabolic activity when used at low micromolar range, namely at the concentration as low as 2.5 µM and 24 h treatment (Fig. 1A).
SFN-induced cytotoxicity (A), changes in the cell cycle and cell cycle regulators (B, C, D, E) and stress-induced premature senescence (SIPS) (F) in breast cancer cells. (A) MTT test. Metabolic activity at control conditions is considered as 100%. The effect of solvent used (0.1% DMSO) is also shown. Bars indicate SD, n = 5, ***p < 0.001, **p < 0.01, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). (B) DNA content-based analysis of cell cycle was conducted using flow cytometry and Muse™ Cell Cycle Kit. Bars indicate SD, n = 3, ***p < 0.001, **p < 0.01, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). Representative histograms are also presented. (C, D) The expression profile of selected genes involved in the regulation of cell cycle. (C) A heat map generated from qRT-PCR data is shown. Hierarchical clustering was created using Genesis 1.7.7 software. (D) SFN-mediated upregulation (red) and downregulation (blue) of cell cycle genes. ΔΔCt values are shown. (E) Western blot analysis of the levels of p21, p27 and p53 cell cycle inhibitors. Anti-β-actin antibody was used as a loading control. The data represent the relative density normalized to β-actin. Bars indicate SD, n = 3, ***p < 0.001, **p < 0.01, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). (F) Senescence-associated β-galactosidase (SA-β-gal) activity. Bars indicate SD, n=3, ***p < 0.001 compared to the control (ANOVA and Dunnett's a posteriori test). SFN, sulforaphane.
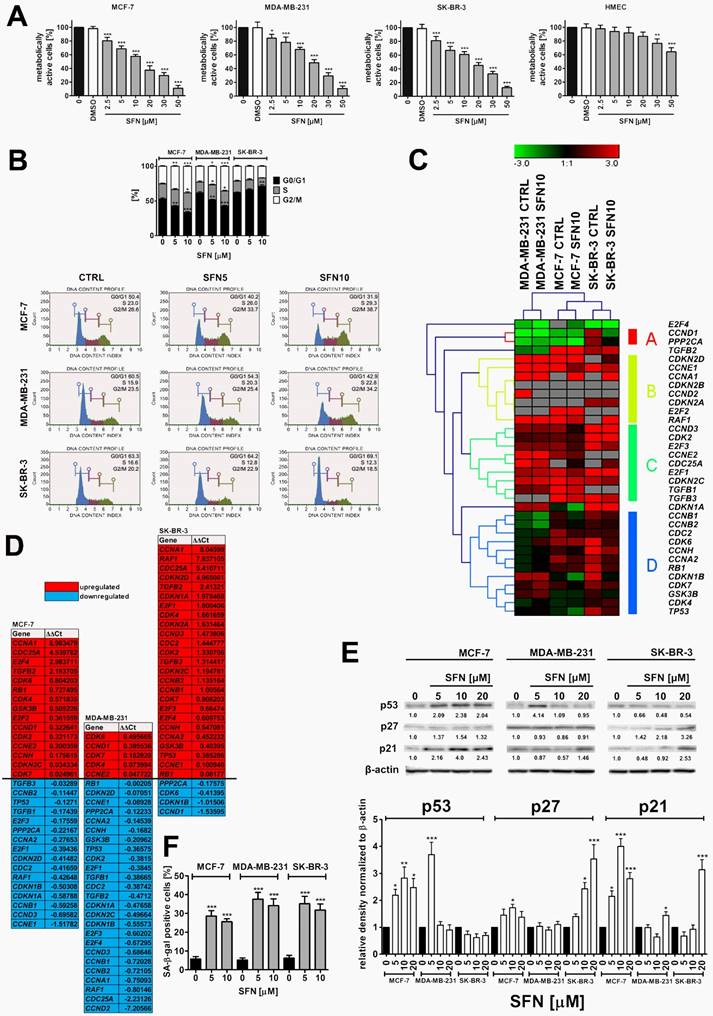
Calculated IC50 values were 14.05 µM, 19.35 µM and 16.64 µM for MCF-7, MDA-MB-231 and SK-BR-3 cells, respectively (Fig. 1A). In contrast, normal human mammary epithelial cells (HMEC) were not sensitive to SFN when used at low micromolar range (IC50 = 81.24 µM, Fig. 1A). SFN significantly affected HMEC metabolic activity when used at higher concentrations (30 to 50 µM) (Fig. 1A). Three concentrations of SFN, namely 5 µM, 10 µM and 20 µM were selected for further analysis (Fig. 1A). SFN (5 and 10 µM) caused the accumulation of cells in the G2/M phase of the cell cycle of MCF-7 and MDA-MB-231 cells, whereas SFN (5 and 10 µM) promoted the accumulation of cells in the G0/G1 phase of the cell cycle of SK-BR-3 cells (Fig. 1B). An increase of 12.1 and 10.7% in the levels of MCF-7 and MDA-MB-231 cells in the G2/M phase of the cell cycle and an increase of 5.8% in the levels of SK-BR-3 cells in the G0/G1 phase of the cell cycle were observed after 10 µM SFN treatment (Fig. 1B). The effect of 10 µM SFN on the expression of forty four genes involved in the regulation of cell cycle was then studied (Fig. 1C and D). Cancer cell line-dependent expression profile was revealed as control and treated categories of each cancer cell line analyzed were grouped together and MCF-7 and SK-BR-3 expression profiles were more similar to each other than MDA-MB-231 expression profile with its own category (Fig. 1C). SFN treatment resulted in a decrease in mRNA levels of CCNA2 (cyclin A2), CCNB1 (cyclin B1), CCNB2 (cyclin B2), CCND3 (cyclin D3) and CCNE1 (cyclin E1) in MCF-7 and MDA-MB-231 cells, CCND1 (cyclin D1) in SK-BR-3 cells, and CCND2 (cyclin D2) and CCNH (cyclin H) in MDA-MB-231 cells (Fig. 1D). SFN also caused an increase in TGFB2 (transforming growth factor beta 2) mRNA levels in MCF-7 and SK-BR-3 cells and TGFB3 (transforming growth factor beta 3) mRNA levels in SK-BR-3 cells (Fig. 1D). As increased mRNA levels of TP53 (p53) and CDKN1A (p21) were observed in SFN-treated SK-BR-3 cells, we decided to evaluate then the corresponding protein levels (Fig. 1E). SFN caused an increase in p53 levels in MCF-7 cells (wild type p53) (Fig. 1E). Except of 5 µM SFN-treated MDA-MB-231 cells, similar effects were not observed in MDA-MB-231 and SK-BR-3 cells (mutant p53) (Fig. 1E). SFN treatment also resulted in an increase in p21 levels in three breast cancer cell lines used (Fig. 1E). Moreover, an increase in p27 levels was noticed in SFN-treated MCF-7 and SK-BR-3 cells (Fig. 1E). In general, upregulation of p53, p21 and p27 at the protein levels did not correspond to TP53, CDKN1A and CDKN1B mRNA levels that may suggest that p53, p21 and p27 are stabilized in SFN-treated breast cancer cells (Fig. 1D and E).
We have then studied if SFN-induced cell cycle arrest was a transient or a permanent phenomenon in breast cancer cells (Fig. 1F). After 7 days of SFN removal (5 and 10 µM), an increase in senescence-associated beta-galactosidase (SA-β-gal)-positive cells was observed in three breast cancer cells considered (Fig. 1F). The effect was slightly more accented after treatment with 5 µM SFN than after treatment with 10 µM SFN that indicated that this is not a concentration-dependent phenomenon (Fig. 1F). Pro-senescent activity of SFN was the most accented in 5 µM SFN-treated MDA-MB-231 cells (Fig. 1F).
Cytotoxic action (apoptosis induction) of SFN was exclusively observed when SFN was used at the concentration of 20 µM as judged by phosphatidylserine externalization (Supplementary Material, Fig. S1A) and multicaspase activity (Supplementary Material, Fig. S1B). Pro-apoptotic activity of SFN (20 µM) was slightly more evident in MDA-MD-231 cells (20.29% of Annexin V-positive cells, 35.13% of cells with multicaspase activity) than in MCF-7 cells (18.94% of Annexin V-positive cells, 22.91% of cells with multicaspase activity) and SK-BR-3 cells (11.25% of Annexin V-positive cells, 22.11% of cells with multicaspase activity) (Fig. S1A and B). In contrast, SFN (5 to 20 µM) did not promote phosphatidylserine externalization in normal human mammary epithelial cells (HMEC) that may suggest that pro-apoptotic action of SFN is specific to breast cancer cells (Fig. S1A).
We have then evaluated if 20 µM SFN-induced apoptosis is accompanied by diminished pro-survival signal of phospho-ERK1/2 (Supplementary Material, Fig. S2). In general, breast cancer cell lines used are characterized by different steady state levels of phosphorylated form of ERK1/2 in the control conditions, namely MCF-7 cells are 91.2% phospho-ERK1/2-negative, MDA-MB-231 cells are 89.6% phospho-ERK1/2-positive and SK-BR-3 cells are 49.6% phospho-ERK1/2-positive in the control conditions (Fig. S2). After 20 µM SFN stimulation, the levels of phospho-ERK1/2-negative cells were increased of 30% in MDA-MB-231 cells but were unaffected in SK-BR-3 cells (Fig. S2). In contrast, an increase of 17% in phospho-ERK1/2-positive cells was observed in MCF-7 cells after 20 µM SFN treatment (Fig. S2). Taken together, this suggests that SFN-induced apoptosis may be mediated by decreased phospho-ERK1/2 levels in MDA-MB-231 cells but not in MCF-7 and SK-BR-3 cells (Fig. S2).
SFN induces nitro-oxidative stress
In general, SFN treatment (5 to 20 µM) promoted the production of reactive oxygen species (ROS) (total ROS, total superoxide and mitochondrial superoxide) at comparable levels in three breast cancer cells (Fig. 2A).
SFN-mediated oxidative (A, B) and nitrosative (C) stress in breast cancer cells. (A) Total reactive oxygen species (ROS) production, total superoxide production and mitochondrial superoxide production were assessed using CM-H2DCF-DA, DHE and MitoSOX fluorogenic probes, respectively. Bars indicate SD, n=5, ***p < 0.001, **p < 0.01, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). (B) Protein carbonylation was revealed using 2,4-dinitrophenylhydrazine (DNPH) derivatization and anti-DNP antibody (OxyBlot™ Protein Oxidation Detection Kit). A positive control with a mixture of standard proteins with attached DNP residues (lane M) is also shown. (C) Nitric oxide levels were measured using flow cytometry and Muse™ Nitric Oxide Kit. As a positive control, cells were treated for 5 min with a nitric oxide donor 1 mM MAHMA NONOate. Bars indicate SD, n = 3, ***p < 0.001, **p < 0.01, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). SFN, sulforaphane; C+, MAHMA NONOate positive control.
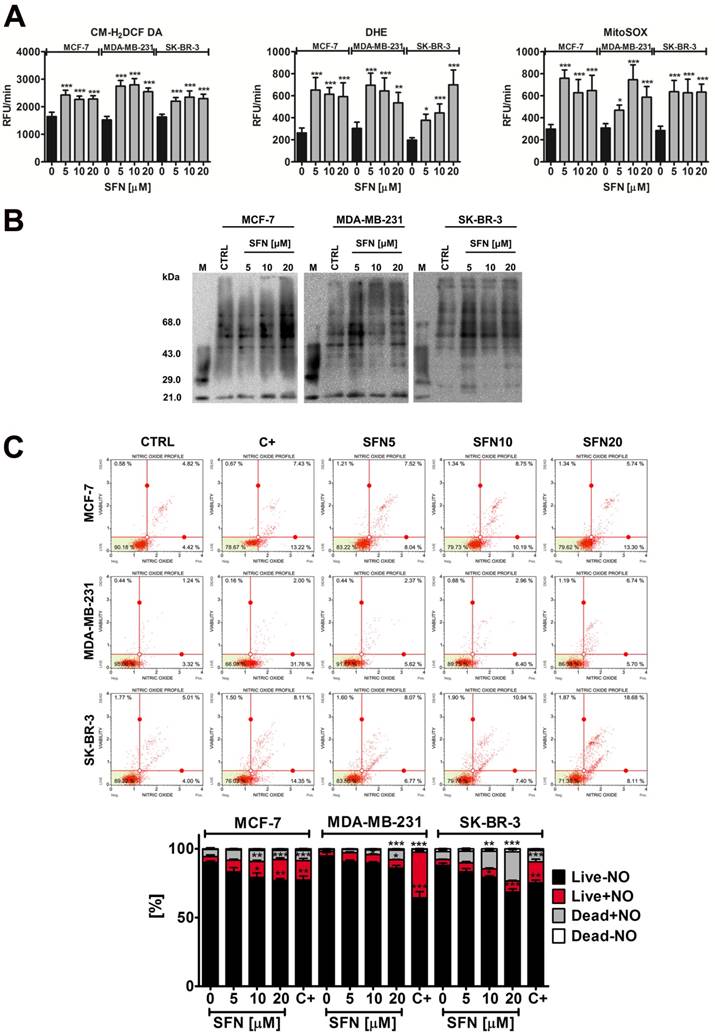
However, MDA-MB-231 and SK-BR-3 cells were more susceptible to protein carbonylation than MCF-7 cells when SFN was used at the concentration of 5 µM (Fig. 2B). Nitric oxide production was the most evident after 20 µM SFN treatment and the highest levels of nitric oxide were observed in SK-BR-3 cells (Fig. 2C).
SFN-mediated genotoxicity
SFN induced both DNA double strand breaks (DSBs) and single strand breaks (SSBs) in breast cancer cells (Fig. 3A). However, DSBs were much more accented than SSBs after SFN stimulation (Fig. 3A). Genotoxic potential of SFN was the most pronounced in MDA-MB-231 cells (Fig. 3A).
SFN-induced DNA damage (A) and DNA damage response (B, C) in breast cancer cells. (A) Comet assay. DNA double strand breaks (DSBs) (neutral comet assay) and DNA single strand breaks (SSBs) (alkaline comet assay) are presented. The percentage of tail DNA was used as a parameter of DNA damage. Bars indicate SD, n = 150, *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control (ANOVA and Dunnett's a posteriori test). (B) pATM and pH2AX were measured using flow cytometry and Muse™ Multi-Color DNA Damage Kit. As a positive control for DNA damage, 24 h treatment with 20 µM etoposide was used (C+). Bars indicate SD, n = 3, ***p < 0.001, **p < 0.01, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). (C) 53BP1 foci formation was revealed using 53BP1 immunostaining and calculated per nucleus. Box and whisker plots are shown, n = 100, *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control (ANOVA and Dunnett's a posteriori test). SFN, sulforaphane.
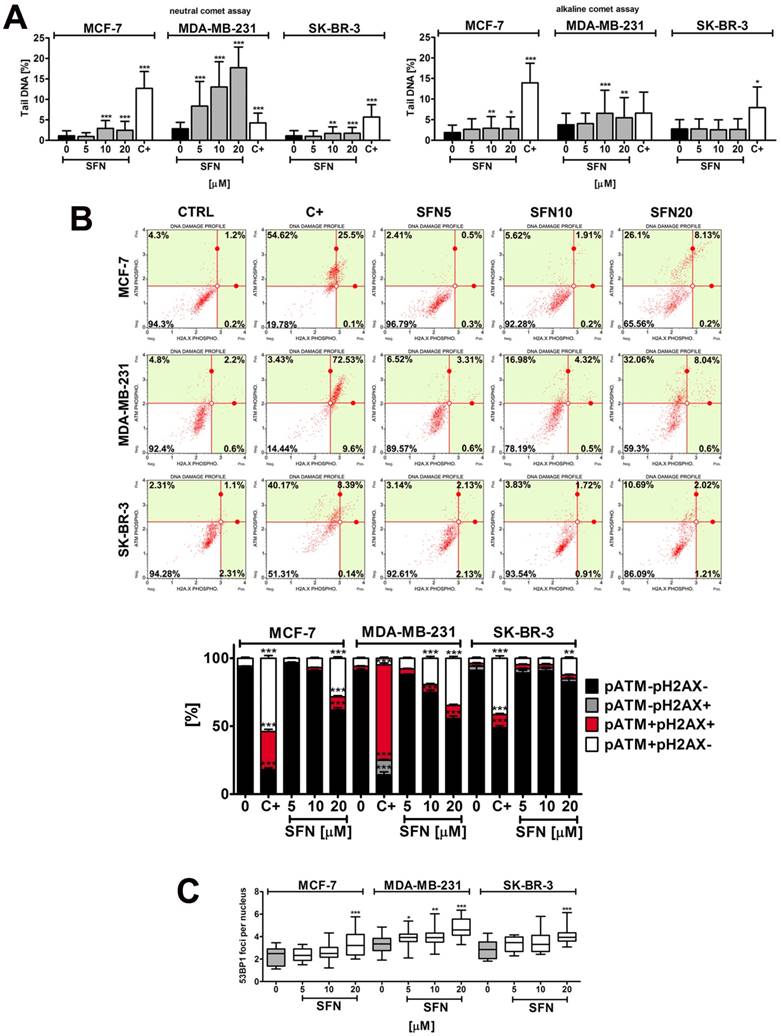
SFN-induced genotoxicity resulted in increased levels of ATM phosphorylation but not by H2AX phosphorylation (Fig. 3B). 53BP1 foci formation was also noticed (Fig. 3C). DNA damage response (DDR) was the most accented in MDA-MB-231 cells that were the most sensitive to SFN-induced genotoxicity (Fig. 3).
SFN-associated changes in energy metabolism and autophagy
We have then investigated if SFN may modulate glycolytic pathway (Warburg effect) that potentially may lead to cell cycle arrest and cytotoxicity in breast cancer cells (Fig. 4).
SFN-mediated changes in glycolysis and related signaling pathways, and autophagy induction. (A) ATP levels. Bars indicate SD, n=3, *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control (ANOVA and Dunnett's a posteriori test). (B) Lactate levels. Bars indicate SD, n=3, **p < 0.01, ***p < 0.001 compared to the control (ANOVA and Dunnett's a posteriori test). (C) Western blot analysis of GLUT1, HK2, PKM2, LDHA, AMPK, pAMPK, AKT and pAKT levels. Anti-β-actin antibody was used as a loading control. The data represent the relative density normalized to β-actin. For pAMPK and pAKT quantification, normalization to AMPK and AKT was also considered, respectively. Bars indicate SD, n = 3, ***p < 0.001, **p < 0.01, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). (D) Autophagy was measured using flow cytometry and Muse™ Autophagy LC3-antibody based Kit. As a positive control, cells were incubated in EBSS at 37°C for 6 h. Autophagy induction ratio (test sample fluorescence, red histogram, versus control sample fluorescence, gray histogram) is presented. Bars indicate SD, n = 3, ***p < 0.001, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). SFN, sulforaphane.
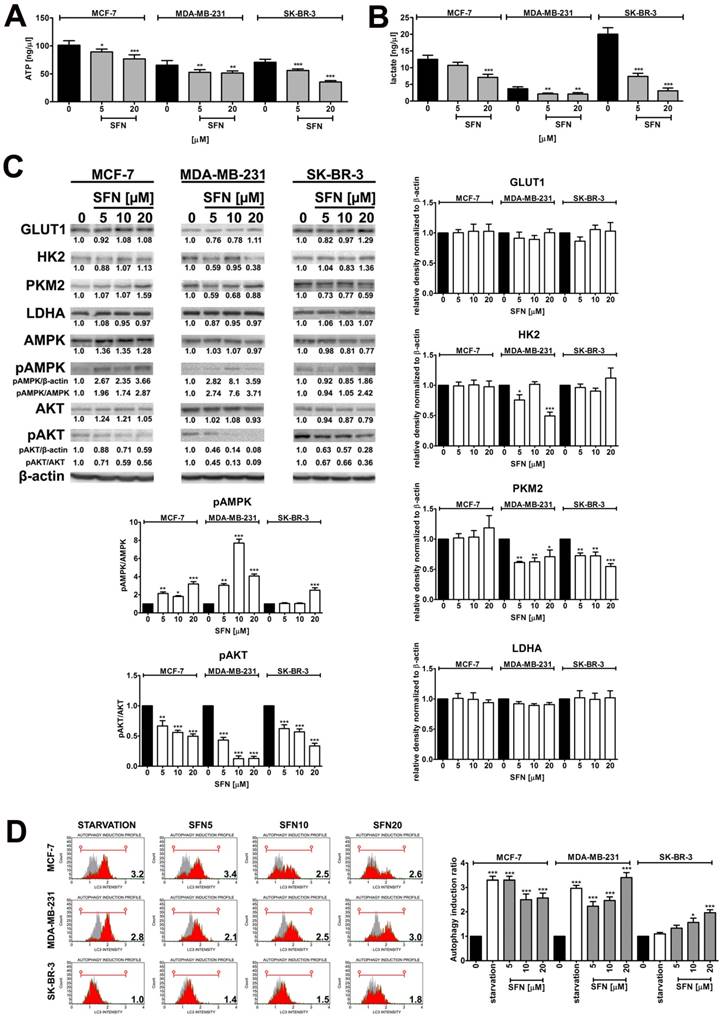
Treatment with SFN, even when used at the non-toxic concentration of 5 µM, resulted in a decrease in intracellular ATP levels in three breast cancer cell lines used (Fig. 4A). Similar results were obtained for lactate levels (Fig. 4B). SFN-mediated diminution in ATP pools induced energy stress as judged by AMPK activation (Fig. 4C). SFN (5 to 20 µM) also resulted in decreased levels of phospho-AKT, the “Warburg kinase”, in three breast cancer cell lines used (Fig. 4C). However, this did not dramatically affect the protein levels of glucose transporter GLUT1 and glycolytic enzymes such as hexokinase 2 (HK2), pyruvate kinase isoform 2 (PKM2) and lactate dehydrogenase (LDHA) in three breast cancer cell lines used (Fig. 4C). A decrease in HK2 levels was observed in SFN-treated MDA-MB-231 cells and a decrease in PKM2 levels was noticed in SFN-treated MDA-MB-231 and SK-BR-3 cells (Fig. 4C). As SFN (5 to 20 µM) induced energy stress (decreased levels of ATP and AMPK activation, Fig. 4A and C), we have then evaluated if SFN may also promote autophagy in breast cancer cells (Fig. 4D). Indeed, SFN treatment that resulted in both cell cycle arrest and senescence (5 and 10 µM SFN, Fig. 1) and apoptosis (20 µM SFN, Fig. S1) also promoted autophagy in breast cancer cells (Fig. 4D) that may be considered as both cytostatic autophagy (5 and 10 µM SFN) and cytotoxic autophagy (20 µM SFN).
SFN is an epigenetic modulator in breast cancer cells
SFN was found to be a DNA hypomethylating agent in three breast cancer cells used as judged by decreased 5-methyl-2′-deoxycytidine (5-mdC) levels after SFN treatment (Fig. 5A).
SFN-mediated changes in global DNA methylation (A), the expression profile and mRNA levels of selected genes involved in DNA methylation and transcriptional repression (B, C), the levels of DNA methyltransferase proteins, namely DNMT1, DNMT2, DNMT3A and DNMT3B (D) and the levels of m6A RNA methylation (E) in breast cancer cells. (A) DNA methylation was estimated as a 5-methyl-2′-deoxycytidine (5-mdC) level using ELISA-based assay (Epigentek). Bars indicate SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control (ANOVA and Dunnett's a posteriori test). Treatment with an inhibitor of DNA methylation 5-aza-2'-deoxycytidine (5-aza-dC) (5 µM, 24 h treatment) served as a negative control. (B, C) The expression profile of selected genes involved in DNA methylation and transcriptional repression. (B) A heat map generated from qRT-PCR data is shown. Hierarchical clustering was created using Genesis 1.7.7 software. (C) SFN-mediated upregulation (red) and downregulation (blue) of DNA methylation and transcriptional repression genes. ΔΔCt values are shown. (D) Western blot analysis of DNMT1, DNMT2, DNMT3A and DNMT3B levels. Anti-β-actin antibody was used as a loading control. The data represent the relative density normalized to β-actin. Bars indicate SD, n = 3, ***p < 0.001, **p < 0.01, *p < 0.05 compared to the control (ANOVA and Dunnett's a posteriori test). (E) The levels of N6-methyladenosine (m6A) in RNA samples were measured using EpiQuik m6A RNA Methylation Quantification Kit. Bars indicate SD, n=3, ***p < 0.001 compared to the control (ANOVA and Dunnett's a posteriori test). SFN, sulforaphane.
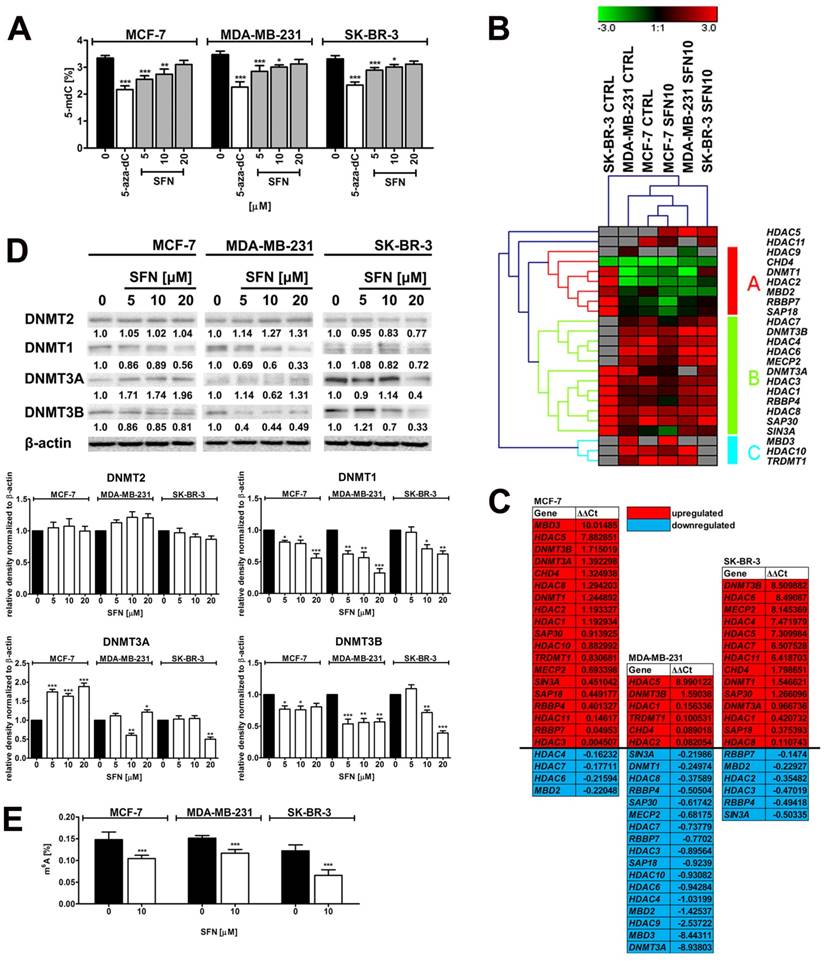
The effect of cytostatic concentration of SFN (10 µM) on the expression profiles of twenty eight genes involved in DNA methylation and transcriptional repression was then considered (Fig. 5B and C). The gene expression profiles of SFN-treated breast cancer cells were grouped together that suggest that three breast cancer cell lines responded to SFN treatment in a similar manner (Fig. 5B). Surprisingly, SFN upregulated HDAC5 mRNA levels (histone deacetylase 5) in three breast cancer cells (Fig. 5C). In contrast, HDAC3 (histone deacetylase 3), HDAC4 (histone deacetylase 4), HDAC6 (histone deacetylase 6), HDAC7 (histone deacetylase 7), HDAC8 (histone deacetylase 8), HDAC9 (histone deacetylase 9) and HDAC10 (histone deacetylase 10) mRNA levels were decreased in SFN-treated MDA-MB-231 cells (Fig. 5C). HDAC2 and HDAC3 mRNA levels were slightly decreased in SFN-treated SK-BR-3 cells and HDAC4, HDAC6 and HDAC7 mRNA levels were also slightly diminished in SFN-treated MCF-7 cells (Fig. 5C). Moreover, SIN3A (transcriptional co-repressor and histone deacetylase complex subunit Sin3A) mRNA levels were downregulated in SFN-treated MDA-MB-231 and SK-BR-3 cells, and SAP18 (Sin3A-associated protein 18 and histone deacetylase complex subunit SAP18) and SAP30 (Sin3A-associated protein 30 and histone deacetylase complex subunit SAP30) mRNA levels were downregulated in SFN-treated MDA-MB-231 cells (Fig. 5C). RBBP4 (RB binding protein 4, chromatin remodeling factor), RBBP7 (RB binding protein 7, chromatin remodeling factor), MECP2 (methyl-CpG binding protein 2), MBD2 (methyl-CpG binding domain protein 2) and MBD3 (methyl-CpG binding domain protein 3) mRNA levels were decreased in SFN-treated MDA-MB-231 cells (Fig. 5C). RBBP4 and RBBP7 mRNA levels were also diminished in SFN-treated SK-BR-3 cells and MBD2 mRNA levels were also decreased in SFN-treated MCF-7 and SK-BR-3 cells (Fig. 5C). Changes in global DNA methylation patterns did not correlate with corresponding changes in mRNA levels of DNA methyltransferases (DNMTs) (Fig. 5C). However, a decrease in protein levels of DNMT1 and DNMT3B was observed in three breast cancer cells analyzed (Fig. 5D). Diminished DNMT3A mRNA levels were also accompanied by decreased protein levels of DNMT3A in 10 µM SFN-treated MDA-MB-231 cells (Fig. 5C and D).
SFN, when used at cytostatic concentration of 10 µM, also promoted a decrease in the levels of N6-methyladenosine (m6A) in RNA pools in three breast cancer cells used (Fig. 5E).
10 µM SFN-mediated changes in the profile of 352 microRNAs (microRNAs that levels are suggested to be altered in cancer) in three breast cancer cell lines were also considered (Fig. 6).
SFN-mediated changes in microRNA profile in breast cancer cells. The expression of 352 microRNAs (http://www.bioline.com/list-of-cancer-panel-plates#tab2) was evaluated using EPIK Cancer miRNA Panel. (A) A heat map generated from qRT-PCR data is shown. Hierarchical clustering was created using Genesis 1.7.7 software. (B) SFN-mediated upregulation (red) and downregulation (blue) of selected microRNAs in three breast cancer cells. Log2 values are shown; *p < 0.05, **p < 0.01 compared to the control (Student's t-test). SFN, sulforaphane.

According to hierarchical clustering, 10 µM SFN treatment affected microRNA profile of MDA-MB-231 and SK-BR-3 cells more than microRNA profile of MCF-7 cells as control and treated categories of MCF-7 cells were grouped together (Fig. 6A). In general, SFN treatment promoted changes in the levels of ninety two microRNAs (Fig. 6). We have also considered a joined analysis of microRNA profiles in three breast cancer cells and according to log2 values, thirty two microRNAs were downregulated and sixty microRNAs were upregulated in three breast cancer cells used upon SFN stimulation (Fig. 6B). Four microRNAs, namely miR-23b-3p, miR-92b-3p, miR-381-3p and miR-382-5p were significantly decreased in SFN-treated MCF-7, MDA-MB-231 and SK-BR-3 cells (Fig. 6B).
We have then considered the interactions between four selected microRNAs (miR-23b-3p, miR-92b-3p, miR-381-3p and miR-382-5p) and cell cycle pathways (Fig. 7).
Selected microRNA-target (cell cycle pathways) interaction data. Four microRNAs, namely miR-23b, miR-92b, miR-381 and miR-382, that levels were significantly decreased upon SFN stimulation were subjected to target analysis based on KEGG PATHWAY database and iBioGuide (https://www.advaitabio.com/ibioguide.html). Red circles indicate miR-23b targets, blue circles indicate miR-92b targets, yellow circles indicate miR-381 targets and gray circles indicate miR-382 targets. TGF-β was recognized as a target of miR-23b, miR-92b and miR-381.
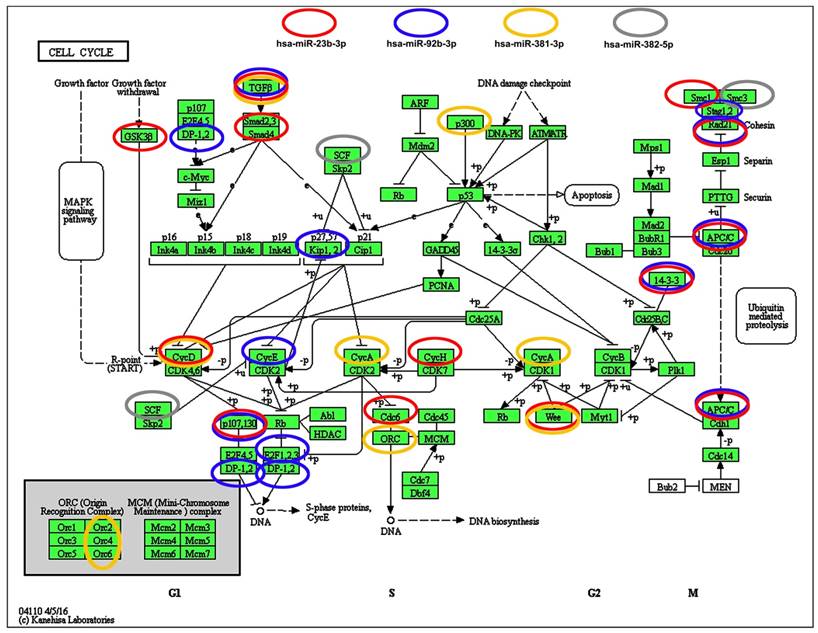
According to KEGG PATHWAY database and iBioGuide, three microRNAs, namely miR-23b-3p, miR-92b-3p and miR-381-3p may affect TGFβ/Smad signaling pathway (Fig. 7). Indeed, TGFB2 mRNA levels were elevated in SFN-treated MCF-7 and SK-BR-3 cells and TGFB3 mRNA levels were increased in SK-BR-3 cells that may in turn promote an increase in the mRNA levels of cell cycle inhibitor CDKN1A (p21) in SK-BR-3 cells (Fig. 1D).
Discussion
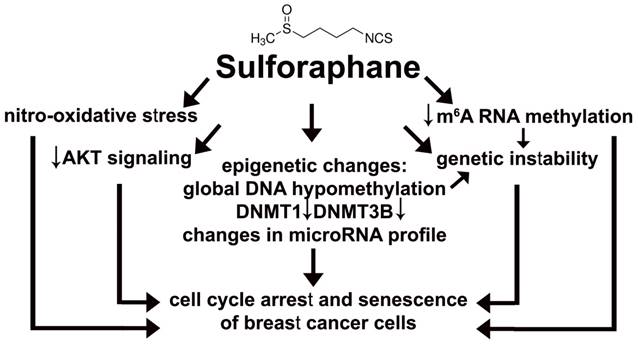
Sulforaphane (SFN), a dietary phytochemical, was found to be an epigenetic modulator, namely SFN promoted global DNA hypomethylation and changes in microRNA profile that resulted in cell cycle arrest and cellular senescence in three breast cancer cell lines (Fig. 8).
SFN-provoked cell cycle arrest and senescence are mediated by epigenetic changes, namely global DNA hypomethylation, decreased levels of DNMT1 and DNMT3B and changes in microRNA profile in breast cancer cells. Moreover, SFN induced a decrease in m6A RNA methylation that is also considered as an epigenetic regulation at the RNA level. SFN may promote genetic instability directly or indirectly by SFN-mediated DNA hypomethylation and/or diminution in m6A RNA methylation pools. SFN-associated cytostatic action may also result from SFN-induced nitro-oxidative stress and decreased AKT signaling.
SFN (5 to 10 µM) induced G2/M cell cycle arrest in MCF-7 and MDA-MB-231 cells, whereas in SFN-treated SK-BR-3 cells, G0/G1 cell cycle arrest was observed. SFN (15 µM) has already been shown to promote G2/M cell cycle arrest and affect the polymerization of mitotic microtubules in MCF-7 cells [27]. More recently, we have also reported that SFN inhibited cell proliferation of MCF-7, MDA-MB-231 and SK-BR-3 breast cancer cells when used at low micromolar range (5 to 10 µM) [18]. Interestingly, SFN also prevented the growth of breast cancer stem cells both in vitro and in vivo and downregulated the Wnt/β-catenin self-renewal pathway [28]. Moreover, mRNA levels of selected cyclins were decreased, namely CCNA2 (cyclin A2), CCNB1 (cyclin B1), CCNB2 (cyclin B2), CCND3 (cyclin D3) and CCNE1 (cyclin E1) in SFN-treated MCF-7 and MDA-MB-231 cells, CCND1 (cyclin D1) in SFN-treated SK-BR-3 cells, and CCND2 (cyclin D2) and CCNH (cyclin H) in SFN-treated MDA-MB-231 cells (this study) that is in agreement with previous findings on SFN-mediated decrease in cyclin D1 levels [28] and cyclin A and cyclin B1 levels [29] in breast cancer cells. SFN-mediated G2/M cell cycle arrest was also accompanied by elevation in the levels of p53 and p21 in MCF-7 cells (wild type p53 [30]) (this study). As p21 upregulation was also observed in SFN-treated MDA-MB-231 and SK-BR-3 cells (mutant p53 [30]), one can conclude that active p53 is not required for activation of p21 in breast cancer cells.
SFN-induced cell cycle arrest was permanent in three breast cancer cells as judged by increased senescence-associated beta-galactosidase staining after 7 days of SFN removal. To the best of our knowledge, this is the first report on SFN-induced cellular senescence in cancer cells. SFN also promoted cellular senescence in mesenchymal stem cells (MSCs), but the potent pro-senescent activity of SFN was observed at cytotoxic concentration of 20 µM [31]. In contrast, the pro-senescent activity of SFN in breast cancer cells was limited to the concentrations ranging from 5 to 10 µM and at the concentration of 20 µM, apoptosis was induced. Despite the fact that MCF-7 cells are caspase 3-deficient (a deletion mutation in exon 3 of the CASP3 gene [32]), we did not observe decreased susceptibility of MCF-7 cells to SFN-induced apoptosis compared to MDA-MB-231 and SK-BR-3 cells. Our data are in agreement with previous results on SFN-induced cell type-specific apoptosis in MDA-MB-231, MDA-MB-468, MCF-7 and T47D breast cancer cell lines [33]. It has been reported that tumor necrosis factor (TNF) and staurosporine [32], and Bax [34] may induce apoptosis in MCF-7 cells, and Bax-induced apoptosis is associated with executioner caspase 6 activation in MCF-7 cells [34]. Thus, executioner caspase 3 may not be required for triggering apoptosis in MCF-7 cells. As Ras/Raf/extracellular signal-regulated kinase (ERK) signaling pathway may be implicated in the regulation of cell death [35], we have then analyzed the phosphorylation status of ERK1/2 and found that SFN-mediated apoptotic cell death in MDA-MB-231 cells was accompanied by decreased phospho-ERK1/2 signals, whereas similar effect was not observed during SFN-induced apoptosis in MCF-7 and SK-BR-3 cells. Thus, decreased ERK signaling is not a common phenomenon during SFN-stimulated apoptosis in breast cancer cells.
In contrast, SFN-induced cell cycle arrest and apoptosis were accompanied by nitro-oxidative stress, genotoxicity and decreased AKT signaling in three breast cancer cells. Isothiocyanates (ITCs) including SFN may possess both antioxidant and pro-oxidant properties in biological systems in vitro and in vivo [36-41]. SFN increased the production of reactive oxygen species (ROS), inhibited ROS-scavenging enzymes and impaired glutathione recycling as judged by the inhibition of glutathione reductase (GR) activity and combined inhibition of glutathione peroxidase (GPx) gene expression and enzyme activity that resulted in decreased cell viability and apoptosis in the p53-null MG-63 osteosarcoma cells [38]. SFN-mediated glutathione depletion and ROS production were also correlated with apoptosis in pancreatic cancer cell lines MIA PaCa-2 and PANC-1 [40]. Modulation of oxidative stress by targeting the antioxidant capacity of cancer cells may be considered as an anticancer strategy [42]. However, one should remember that ROS may play a dual role in cancer biology as cancer-suppressing and cancer-promoting effects of ROS depend on their levels and cell context [43]. SFN also promoted DNA damage and ataxia telangiectasia mutated (ATM) kinase activation and 53BP1 foci formation in breast cancer cells, however increased phosphorylation of H2AX was not observed (this study). ATM, DNA damage response kinase [44], may also be activated directly by reactive oxygen species [45] and reactive nitrogen species [46] being a part of cytotoxic response in MCF-7 cells [46]. Genotoxic potential of SFN has been already documented in MG-63 osteosarcoma [47] and HeLa cervical carcinoma cells [48].
SFN also decreased AKT signaling as judged by diminished phospho-AKT levels in three breast cancer cells (this study). AKT signaling pathway is frequently hiperactivated in cancer cells that may result in resistance to apoptosis, increased cell growth, cell proliferation, metastasis, angiogenesis and cellular energy metabolism [49-53]. Decreased AKT signaling was accompanied by cell cycle arrest and apoptosis, but its effect on the levels of glucose transporter GLUT1 and selected glycolytic enzymes was limited (this study). However, SFN induced energy stress as judged by diminished ATP pools that promoted AMPK activation and autophagy in three breast cancer cells both at cytostatic and cytotoxic concentrations of SFN. Data on interplay between SFN-induced autophagy and apoptosis are rather contradictory [29, 54-57]. Autophagy may be considered as a protective mechanism against SFN-induced apoptosis because autophagy inhibition promoted apoptosis in prostate, colon and breast cancer cells [29, 54, 55]. Autophagy inhibition may also promote apoptosis in SFN-treated tumor vascular endothelial cells that potentiated the anti-angiogenic effect of SFN [56]. Autophagy and cell death signaling may also act independently of each other in SFN-treated pancreatic carcinoma cells [57].
Finally, SFN modulated breast cancer cell epigenome as evidenced by SFN-mediated global DNA hypomethylation, decreased levels of DNMT1 and DNMT3B and changes in microRNA profile of three breast cancer cells. As global DNA hypomethylation is considered to be a hallmark of aging [58, 59], a decrease in 5-methyl-2′-deoxycytidine (5-mdC) levels may also be associated with cellular senescence and genetic instability in SFN-treated breast cancer cells. SFN has been already shown to reduce the levels of DNMT1 in CaCo-2 colon cancer cells [60] and DNMT1 and DNMT3A in MCF-7 and MDA-MB-231 cells [61]. SFN promoted epigenetic repression of telomerase reverse transcriptase (hTERT) expression that provoked apoptosis in breast cancer cells [61]. This epigenetic mechanism is based on SFN-induced hyperacetylation that facilitated the binding of many hTERT repressor proteins such as MAD1 and CTCF to the hTERT regulatory region [61]. SFN has been also found to be a potent HDAC inhibitor both in vitro and in vivo cancer models [62-65]. SFN-mediated HDAC inhibitory activity resulted in histone acetylation at the p21WAF1/CIP1 promoter to enhance its expression in HCT116 colorectal cancer cells [62] and BPH-1, LNCaP and PC-3 prostate cancer cells [63] and mice [64] that in turn promoted p21-associated cell cycle arrest and apoptosis in prostate cancer cells [63] and suppressed tumorigenesis in Apcmin mice [64]. SFN also retarded the growth of prostate cancer PC-3 tumor xenografts and inhibited HDAC activity in human subjects [65]. In human subjects, a single dose of 68 g broccoli sprouts (approximately 105 mg SFN; equivalent to approximately 570 g of mature broccoli) inhibited HDAC activity significantly in peripheral blood mononuclear cells (PBMC) at 3 and 6 hrs following consumption [65]. Importantly, following oral dosing, SFN metabolites were readily measurable in human breast tissue enriched for epithelial cells [66]. Except of SFN-mediated increase in mRNA levels of HDAC5 in three breast cancer cells, mRNA levels of selected histone deacetylases were decreased upon SFN stimulation that was especially seen in MDA-MB-231 cells (this study). Moreover, in SFN-treated MDA-MB-231 cells, mRNA levels of SIN3A (transcriptional co-repressor and histone deacetylase complex subunit Sin3A), SAP18 (Sin3A-associated protein 18 and histone deacetylase complex subunit SAP18) and SAP30 (Sin3A-associated protein 30 and histone deacetylase complex subunit SAP30) were downregulated. It has been shown that the targeted disruption of Sin3 function by introduction of a Sin3 interaction domain (SID) decoy that interferes with Sin3A/B-paired amphipathic alpha-helices (PAH2) binding to SID-containing partner proteins reverted the silencing of genes involved in cell growth and differentiation in breast cancer cells [67]. The use of SID decoy restored the sensitivity to 17beta-estradiol, tamoxifen and retinoids in MDA-MB-231 cells [67]. Moreover, Sin3A repressed expression of key apoptotic genes, including TRAIL, TRAILR1, CASP10 and APAF1 in ERα-positive breast cancer cells and knockdown of Sin3A inhibited breast cancer cell growth by increasing apoptosis [68]. Thus, Sin3A expression promoted maximum growth and survival of ERα-positive breast cancer cells [68].
SFN also diminished m6A RNA methylation in three breast cancer cells (this study). N6-methyladenosine (m6A) is the most abundant mRNA post-transcriptional modification in eukaryotes that has broad roles in the regulation of adipogenesis, spermatogenesis, development, carcinogenesis and stem cell renewal that highlighted the existence of another layer of epigenetic regulation at the RNA level [69, 70]. More recently, the importance of m6A in the ultraviolet-responsive DNA damage response has been revealed [71]. Methylation at the 6 position of adenosine in RNA is rapidly (within 2 min) and transiently induced at DNA damage sites and m6A RNA serves as a beacon for the recruitment of DNA polymerase kappa to damage sites to facilitate repair and cell survival [71]. In the absence of methyltransferase METTL3, cells displayed delayed repair of ultraviolet-induced cyclobutane pyrimidine adducts and elevated sensitivity to ultraviolet [71]. Perhaps, SFN-mediated decrease in m6A RNA methylation may also promote genetic instability in three breast cancer cells (this study).
Finally, SFN affected the expression of ninety two microRNAs and statistically significant decrease in the levels of miR-23b, miR-92b, miR-381 and miR-382 was revealed in three breast cancer cells. Pro-oncogenic factors miR-23b and miR-27b are highly upregulated in breast cancer cells that is achieved by elevated HER2/neu (ERBB2), EGF and TNF-α through the AKT/NF-κB signaling cascade that correlates with poor outcome in breast cancer [72]. Moreover, engineered knockdown of miR-23b and miR-27b substantially repressed breast cancer growth [72]. It has been suggested that the upregulation of miR-92b and deletions of PTEN could coordinately regulate the cell cycle pathway and hence contribute to the progression of breast cancer [73]. MiR-92b is considered to be an oncogene and its levels are upregulated in A549 lung cancer cells that resulted in PTEN downregulation and growth promotion [74]. MiR-92b is also significantly increased in U87, U251 and SHG66 glioblastoma cells and samples, and silencing of miR-92b inhibited the viability of glioblastoma cells through the upregulation of TGF-β/Smad3/p21 signaling pathway [75]. According to microRNA-target (cell cycle pathways) interaction data, miR-23b, miR-92b and miR-381 may target TGF-β/Smad signaling pathway. Indeed, SFN-mediated decrease in miR-23b, miR-92b and miR-381 was correlated with elevated mRNA levels of TGFB2 and CDKN1A in SK-BR-3 cells that may account for p21-mediated cell cycle arrest (this study). MiR-382 is also considered to be an onco-microRNA in breast cancer cells as it promoted breast cancer cell viability, clonogenicity, survival, migration, invasion and in vivo tumorigenesis/metastasis by regulating the RERG/Ras/ERK signaling axis and its expression is negatively correlated with tumor suppressor RERG expression that is associated with higher incidence and poorer prognosis of breast cancer [76].
In summary, we have shown for the first time that SFN-mediated cell cycle arrest and senescence in breast cancer cells may be mediated by epigenetic changes, namely global DNA hypomethylation, decreased m6A RNA methylation and changes in microRNA profile (Fig. 8). Thus, SFN may be considered to be used in epigenome-focused anticancer therapy. SFN also promoted nitro-oxidative stress, genetic instability and decreased AKT signaling that may also contribute to its cytostatic action in breast cancer cells (Fig. 8).
Acknowledgements
This study was supported by Grant from the National Science Center, 2013/11/D/NZ7/00939.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683-92
2. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057-68
3. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27-36
4. Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22(1):50-5
5. Elsheikh SE, Green AR, Rakha EA. et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009;69(9):3802-9
6. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259-69
7. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630-41
8. Ahuja N, Sharma AR, Baylin SB. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu Rev Med. 2016;67:73-89
9. Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest. 2014;124(1):56-63
10. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12-27
11. Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol. 2006;71(10):1397-421
12. Meeran SM, Ahmed A, Tollefsbol TO. Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin Epigenetics. 2010;1(3-4):101-16
13. Khan SI, Aumsuwan P, Khan IA. et al. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem Res Toxicol. 2012;25(1):61-73
14. Schneider-Stock R, Ghantous A, Bajbouj K. et al. Epigenetic mechanisms of plant-derived anticancer drugs. Front Biosci (Landmark Ed). 2012;17:129-73
15. Myzak MC, Ho E, Dashwood RH. Dietary agents as histone deacetylase inhibitors. Mol Carcinog. 2006;45(6):443-6
16. Ratovitski EA. Anticancer Natural Compounds as Epigenetic Modulators of Gene Expression. Curr Genomics. 2017;18(2):175-205
17. Singh BN, Singh HB, Singh A. et al. Dietary phytochemicals alter epigenetic events and signaling pathways for inhibition of metastasis cascade: phytoblockers of metastasis cascade. Cancer Metastasis Rev. 2014;33(1):41-85
18. Lewinska A, Bednarz D, Adamczyk-Grochala J. et al. Phytochemical-induced nucleolar stress results in the inhibition of breast cancer cell proliferation. Redox Biol. 2017;12:469-82
19. Lewinska A, Siwak J, Rzeszutek I. et al. Diosmin induces genotoxicity and apoptosis in DU145 prostate cancer cell line. Toxicol In Vitro. 2015;29(3):417-25
20. Lewinska A, Adamczyk-Grochala J, Kwasniewicz E. et al. Diosmin-induced senescence, apoptosis and autophagy in breast cancer cells of different p53 status and ERK activity. Toxicol Lett. 2017;265:117-30
21. Lewinska A, Jarosz P, Czech J. et al. Capsaicin-induced genotoxic stress does not promote apoptosis in A549 human lung and DU145 prostate cancer cells. Mutat Res Genet Toxicol Environ Mutagen. 2015;779:23-34
22. Lewinska A, Chochrek P, Smolag K. et al. Oxidant-based anticancer activity of a novel synthetic analogue of capsaicin, capsaicin epoxide. Redox Rep. 2015;20(3):116-25
23. Lewinska A, Adamczyk-Grochala J, Kwasniewicz E. et al. Ursolic acid-mediated changes in glycolytic pathway promote cytotoxic autophagy and apoptosis in phenotypically different breast cancer cells. Apoptosis. 2017;22(6):800-15
24. Dworak N, Wnuk M, Zebrowski J. et al. Genotoxic and mutagenic activity of diamond nanoparticles in human peripheral lymphocytes in vitro. Carbon. 2014;68:763-76
25. Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18(1):207-8
26. Lewinska A, Adamczyk-Grochala J, Kwasniewicz E. et al. Downregulation of methyltransferase Dnmt2 results in condition-dependent telomere shortening and senescence or apoptosis in mouse fibroblasts. J Cell Physiol. 2017 doi: 10.1002/jcp.25848
27. Jackson SJ, Singletary KW. Sulforaphane inhibits human MCF-7 mammary cancer cell mitotic progression and tubulin polymerization. J Nutr. 2004;134(9):2229-36
28. Li Y, Zhang T, Korkaya H. et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res. 2010;16(9):2580-90
29. Kanematsu S, Uehara N, Miki H. et al. Autophagy inhibition enhances sulforaphane-induced apoptosis in human breast cancer cells. Anticancer Res. 2010;30(9):3381-90
30. Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer. 2006;13(2):293-325
31. Zanichelli F, Capasso S, Cipollaro M. et al. Dose-dependent effects of R-sulforaphane isothiocyanate on the biology of human mesenchymal stem cells, at dietary amounts, it promotes cell proliferation and reduces senescence and apoptosis, while at anti-cancer drug doses, it has a cytotoxic effect. Age (Dordr). 2012;34(2):281-93
32. Janicke RU, Sprengart ML, Wati MR. et al. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273(16):9357-60
33. Pledgie-Tracy A, Sobolewski MD, Davidson NE. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther. 2007;6(3):1013-21
34. Kagawa S, Gu J, Honda T. et al. Deficiency of caspase-3 in MCF7 cells blocks Bax-mediated nuclear fragmentation but not cell death. Clin Cancer Res. 2001;7(5):1474-80
35. Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2010;277(1):2-21
36. Valgimigli L, Iori R. Antioxidant and pro-oxidant capacities of ITCs. Environ Mol Mutagen. 2009;50(3):222-37
37. Singh P, Sharma R, McElhanon K. et al. Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic Biol Med. 2015;86:90-101
38. Ferreira de Oliveira JM, Costa M, Pedrosa T. et al. Sulforaphane induces oxidative stress and death by p53-independent mechanism: implication of impaired glutathione recycling. PLoS One. 2014;9(3):e92980
39. Zanichelli F, Capasso S, Di Bernardo G. et al. Low concentrations of isothiocyanates protect mesenchymal stem cells from oxidative injuries, while high concentrations exacerbate DNA damage. Apoptosis. 2012;17(9):964-74
40. Pham NA, Jacobberger JW, Schimmer AD. et al. The dietary isothiocyanate sulforaphane targets pathways of apoptosis, cell cycle arrest, and oxidative stress in human pancreatic cancer cells and inhibits tumor growth in severe combined immunodeficient mice. Mol Cancer Ther. 2004;3(10):1239-48
41. Negrette-Guzman M, Huerta-Yepez S, Tapia E. et al. Modulation of mitochondrial functions by the indirect antioxidant sulforaphane: a seemingly contradictory dual role and an integrative hypothesis. Free Radic Biol Med. 2013;65:1078-89
42. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931-47
43. Gupta SC, Hevia D, Patchva S. et al. Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal. 2012;16(11):1295-322
44. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197-210
45. Alexander A, Cai SL, Kim J. et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107(9):4153-8
46. Tripathi DN, Chowdhury R, Trudel LJ. et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci U S A. 2013;110(32):E2950-7
47. Ferreira de Oliveira JM, Remedios C, Oliveira H. et al. Sulforaphane induces DNA damage and mitotic abnormalities in human osteosarcoma MG-63 cells: correlation with cell cycle arrest and apoptosis. Nutr Cancer. 2014;66(2):325-34
48. Sekine-Suzuki E, Yu D, Kubota N. et al. Sulforaphane induces DNA double strand breaks predominantly repaired by homologous recombination pathway in human cancer cells. Biochem Biophys Res Commun. 2008;377(2):341-5
49. Cicenas J, Urban P, Vuaroqueaux V. et al. Increased level of phosphorylated akt measured by chemiluminescence-linked immunosorbent assay is a predictor of poor prognosis in primary breast cancer overexpressing ErbB-2. Breast Cancer Res. 2005;7(4):R394-401
50. Semba S, Moriya T, Kimura W. et al. Phosphorylated Akt/PKB controls cell growth and apoptosis in intraductal papillary-mucinous tumor and invasive ductal adenocarcinoma of the pancreas. Pancreas. 2003;26(3):250-7
51. Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15(2):177-82
52. Elstrom RL, Bauer DE, Buzzai M. et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892-9
53. Robey RB, Hay N. Is Akt the "Warburg kinase"?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19(1):25-31
54. Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006;66(11):5828-35
55. Nishikawa T, Tsuno NH, Okaji Y. et al. Inhibition of autophagy potentiates sulforaphane-induced apoptosis in human colon cancer cells. Ann Surg Oncol. 2010;17(2):592-602
56. Nishikawa T, Tsuno NH, Okaji Y. et al. The inhibition of autophagy potentiates anti-angiogenic effects of sulforaphane by inducing apoptosis. Angiogenesis. 2010;13(3):227-38
57. Naumann P, Fortunato F, Zentgraf H. et al. Autophagy and cell death signaling following dietary sulforaphane act independently of each other and require oxidative stress in pancreatic cancer. Int J Oncol. 2011;39(1):101-9
58. Johnson AA, Akman K, Calimport SR. et al. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012;15(5):483-94
59. Lopez-Otin C, Blasco MA, Partridge L. et al. The hallmarks of aging. Cell. 2013;153(6):1194-217
60. Traka M, Gasper AV, Smith JA. et al. Transcriptome analysis of human colon Caco-2 cells exposed to sulforaphane. J Nutr. 2005;135(8):1865-72
61. Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One. 2010;5(7):e11457
62. Myzak MC, Karplus PA, Chung FL. et al. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64(16):5767-74
63. Myzak MC, Hardin K, Wang R. et al. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. 2006;27(4):811-9
64. Myzak MC, Dashwood WM, Orner GA. et al. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006;20(3):506-8
65. Myzak MC, Tong P, Dashwood WM. et al. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood). 2007;232(2):227-34
66. Cornblatt BS, Ye L, Dinkova-Kostova AT. et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis. 2007;28(7):1485-90
67. Farias EF, Petrie K, Leibovitch B. et al. Interference with Sin3 function induces epigenetic reprogramming and differentiation in breast cancer cells. Proc Natl Acad Sci U S A. 2010;107(26):11811-6
68. Ellison-Zelski SJ, Alarid ET. Maximum growth and survival of estrogen receptor-alpha positive breast cancer cells requires the Sin3A transcriptional repressor. Mol Cancer. 2010;9:263
69. Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29(13):1343-55
70. Cao G, Li HB, Yin Z. et al. Recent advances in dynamic m6A RNA modification. Open Biol. 2016;6(4):160003
71. Xiang Y, Laurent B, Hsu CH. et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543(7646):573-6
72. Jin L, Wessely O, Marcusson EG. et al. Prooncogenic factors miR-23b and miR-27b are regulated by Her2/Neu, EGF, and TNF-alpha in breast cancer. Cancer Res. 2013;73(9):2884-96
73. Peng F, Zhang Y, Wang R. et al. Identification of differentially expressed miRNAs in individual breast cancer patient and application in personalized medicine. Oncogenesis. 2016;5:e194
74. Li Y, Li L, Guan Y. et al. MiR-92b regulates the cell growth, cisplatin chemosensitivity of A549 non small cell lung cancer cell line and target PTEN. Biochem Biophys Res Commun. 2013;440(4):604-10
75. Wu ZB, Cai L, Lin SJ. et al. The miR-92b functions as a potential oncogene by targeting on Smad3 in glioblastomas. Brain Res. 2013;1529:16-25
76. Ho JY, Hsu RJ, Liu JM. et al. MicroRNA-382-5p aggravates breast cancer progression by regulating the RERG/Ras/ERK signaling axis. Oncotarget. 2017;8(14):22443-59
Author contact
![]() Corresponding author: Anna Lewinska, Laboratory of Cell Biology, University of Rzeszow, Werynia 502, 36-100 Kolbuszowa, Poland. Tel.: +48 17 872 37 11. Fax: +48 17 872 37 11. E-mail: lewinskaedu.pl
Corresponding author: Anna Lewinska, Laboratory of Cell Biology, University of Rzeszow, Werynia 502, 36-100 Kolbuszowa, Poland. Tel.: +48 17 872 37 11. Fax: +48 17 872 37 11. E-mail: lewinskaedu.pl