Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2017; 7(14):3504-3516. doi:10.7150/thno.19017 This issue Cite
Review
Bioengineering of Artificial Antigen Presenting Cells and Lymphoid Organs
Chao Wang1, 2, Wujin Sun1, 2, Yanqi Ye1, 2, Hunter N. Bomba1, Zhen Gu1, 2, 3 ![]()
1. Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA.
2. Division of Pharmacoengineering and Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
3. Department of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Received 2017-1-2; Accepted 2017-3-24; Published 2017-8-17
Abstract

The immune system protects the body against a wide range of infectious diseases and cancer by leveraging the efficiency of immune cells and lymphoid organs. Over the past decade, immune cell/organ therapies based on the manipulation, infusion, and implantation of autologous or allogeneic immune cells/organs into patients have been widely tested and have made great progress in clinical applications. Despite these advances, therapy with natural immune cells or lymphoid organs is relatively expensive and time-consuming. Alternatively, biomimetic materials and strategies have been applied to develop artificial immune cells and lymphoid organs, which have attracted considerable attentions. In this review, we survey the latest studies on engineering biomimetic materials for immunotherapy, focusing on the perspectives of bioengineering artificial antigen presenting cells and lymphoid organs. The opportunities and challenges of this field are also discussed.
Keywords: artificial immune cells, lymphoid organs
Introduction
The immune system is made up of lymphoid organs (such as the thymus, bone marrow, spleen, and lymph nodes), specialized cells (such as T cells and B cells), and secreted proteins (such as cytokines).[1] This network collaborates to protect the body against many diseases.[1] However, immune surveillance can be evaded by a variety of mechanisms.[2-5] For example, cancer cells can produce a variety of ligands, such as programmed death ligand 1 (PD-L1) to inhibit T cell attacking, resulting in PD-L1-mediated T cell apoptosis.[6, 7] Immunotherapy aims to re-boost effective immune responses to destroy harmful cells[8-12] or to protect normal tissues from immune attack in autoimmune disease and rejection of transplanted organs.[11-14] Among them, immunotherapy based on the manipulation and transfusion of engineered immune cells or tissues has recently made great progresses in clinical applications.[15, 16] However, therapy with natural immune cells or lymphoid organs is often costly, time-consuming, and may have a limited effectiveness in clinical trials.[17, 18] The development of artificial immune cells and artificial lymphoid organs using biomimetic strategies has become an emerging field as an alternative.[19-21] Bioengineering technologies, especially immune engineering technologies leveraging biomaterials, such as micro- or nanoparticles and scaffolds, have attracted tremendous attentions.[22-34] Among them, artificial biomimetic materials, inspired by nature, have frequently been studied as immunotherapeutics to treat infectious diseases and cancer.[18-20, 35-39] Here we survey the latest studies using biomaterials-incorporated strategies to mimic immune cells, with a focus on artificial antigen presenting cells (aAPCs) and lymphoid organs. Emerging trends and clinical challenges in this field are also discussed.
Artificial antigen presenting cell system
Antigen-presenting cells (APCs) act as a link between the innate and adaptive immune responses.[40] Upon internalization of an antigen, the APCs can display antigen-class I and II major histocompatibility complex (MHC) on the membrane together with co-stimulatory signals to activate antigen-specific T cells, which play a key role in the adaptive immune response.[41] Generally, antigen-specific T cells can be primed and amplified ex vivo before they are transferred back to the patient. For example, in adoptive cell transfer (ACT), tumor-specific T cells are isolated then expanded ex vivo to obtain a large number of cells for transfusion.[16] As one of the APCs, dendritic cells (DCs) are usually used to maximize T cell stimulation ex vivo.[42] Particularly, the initiation of MHC-I-restricted CD8 cytotoxic lymphocytes (CTL) is primarily carried out by cross-presentation of specific DC subsets.[43] However, the preparation of DCs for clinical use is challenging.[20] In addition to the need for autologous cells, the preparation of appropriately differentiated and activated DCs is a delicate task requiring high level of skills. Moreover, the isolation and culture of DCs is a time-consuming process that obstructs the translation of T cell based therapy.[44] Therefore, technologies for efficient expansion of the therapeutic lymphocytes are highly desired. [13, 25, 32, 34, 35, 45]
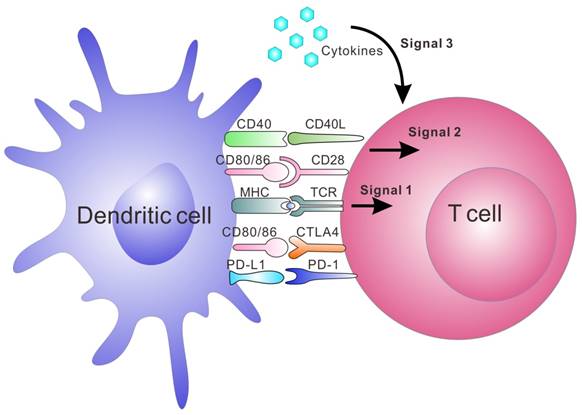
Artificial APCs (aAPCs) are engineered platforms for T cell activation and expansion that aim to bypass the aforementioned obstacles by mimicking the interaction between DCs and T cells.[20, 35-38, 46] They include multiple systems that utilize engineered cells or synthesized biomaterials. Since these aAPCs are ready-to-use to the patients, this strategy is becoming a common tool in immunology and clinical applications.[47, 48] Synthetic aAPCs based on biomaterials have shown great success in generating anti-tumor immune responses in vitro and in vivo.[20, 36, 37, 44, 49] They are often conjugated or encapsulated with three signals required for T cell activation (Figure 1). Signal 1 is the binding of peptide-MHC complexes to provide T cell receptor (TCR) specificity. TCR agonists, such as recombinant peptide-MHC complexes or antibodies directed towards CD3, lead to ligation of the TCR, which triggers the activation of the T cell. Signal 2 is provided by co-stimulatory molecules, which are upregulated on APCs; this leads to the complete activation of T cell. Co-stimulatory agonists, such as the anti-CD28 monoclonal antibody (mAb), are known to provide the necessary co-stimulatory signals to T cells. Lastly, signal 3 involves cytokines produced by either APCs or T cells, which are essential for T cell expansion and differentiation.[50-53] IL-2 is one of the most known cytokines for CD8+ T cell survival. Other cytokines such as IL-7, IL-15 and IL-21 have been investigated and may promote better expansion or differentiation into more optimal T cell phenotypes.[50-53] In addition to the activation of T cells by aAPC, tolerogenic aAPCs coupling with negative signals, such as apoptosis-inducing molecules, deplete targeted antigen-specific T cells. It is another strategy to treat autoimmune diseases and allograft rejection.[54-56] Here, we summarize recent advances in aAPC systems with an emphasis on synthetic and biomimetic biomaterials for both ex vivo and in vivo applications in immunotherapy.
The major interactions between T cells and DCs and the three signals leading to activation and expansion of T cells. Signal 1 is antigen presentation by interaction between the peptide-MHC complex and TCR; Signal 2 is co-stimulation by co-stimulatory molecule interaction. The binding of CD80/CD86 on DCs and CD28 on T cells is one of the co-stimulatory signals. Negative co-stimulatory interactions such as PD-L1/PD-1 and CD80/CTLA-4 are also shown in this figure. Signal 3 is release of cytokines, which are essential for T cell expansion and differentiation.[153]
Lipid based aAPC
The dynamic lipid bilayer is essential for the molecular interactions in the natural systems.[57] To mimic natural interactions between natural APCs and T cells, lipid bilayer-based particles with a fluid membrane have been developed as aAPCs.[58-62] For instance, MHC-containing liposomes developed by Prakken et al. [58] have been reported to activate CD4+ T cells in vitro, leading to T cell proliferation as well as IL-2 secretion. It indicated the role of the lipid membrane as a scaffold supporting MHC-restricted antigen presentation. In addition, preclustering of MHC-peptide complexes on the APC membrane microdomains enhanced the efficiency of T cell activation.[63] In fact, natural APCs have been shown to precluster antigens even in the absence of T cells. Therefore, Ding et al. [59] designed reconstituted liposomes with membrane microdomains containing enriched epitope/MHC complexes, also known as RAFTsomes, to stimulate CD4+ T cell proliferation. Similarly, Giannoni et al. [60] described an aAPC system for ex vivo stimulation of human polyclonal T cells. The described aAPCs were based on artificial membrane bilayers containing T cell ligands membrane microdomains. They showed that preclustering of MHC molecules triggered a higher degree of T cell activation than soluble tetramers and aAPCs with MHC molecules uniformly distributed in artificial bilayer membranes. In a subsequent study by the same group, anti-LFA-1 (an adhesion molecule to allow for an efficient aAPC-T-cell interaction) together with anti-CD3 and anti-CD28 were preclustered in microdomains as before which resulted in an increased expansion of polyclonal T cells or antigen-specific T cells (lymphocytes from tumor-invaded lymph nodes cultured with the cognate antigen before) compared to commercially available systems (Dynabeads® CD3/CD28 T Cell Expander).[61]
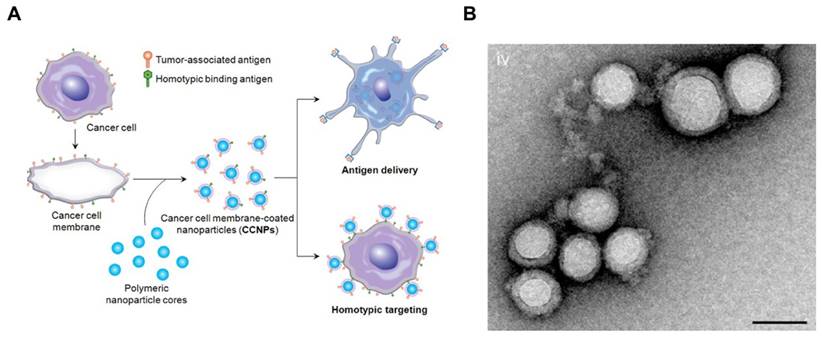
To enhance the stability of the liposomes, researchers also used solid particles as a scaffold or core for the lipid bilayer, also called supported lipid bilayers (SLBs).[64] Different SLB systems have been developed recently,[65-68] which offered improved stability to standard liposomal formulations.[69] For example, Ashley et al. [70] developed a protocell that consists of a nanoporous silica core and a SLB, which enabled increased stability and drug loading capacity. The authors found that the nanoporous silica particles showed increased membrane fluidity compared to the protocells formed from nonporous solid silica nanoparticles or unsupported liposomes. In another example, using silica beads coated with a lipid bilayer was shown to be more efficient than liposomes to boost CTL responses.[71] In addition, various types of natural cell membranes were extracted for particle coating.[65, 68, 72-75] For example, plasma membrane of tumor cells vesicles have been coated on poly (lactic-co-glycolic acid) (PLGA) particles[65], silica microbeads, or latex microbeads[76] for immune stimulation, leading to increased immune activity (Figure 2). This technology presents the potential to synthesize particles that are coated with natural DC membranes for aAPCs preparation.
Cancer Cell Membrane-Coated Nanoparticles. (A) Schematic of CCNP synthesis. The cancer cell membrane along with its associated antigens were coated onto PLGA polymeric nanoparticle cores. The resulting CCNPs was used to deliver tumor-associated antigens to antigen presenting cells or to homotypically target the source cancer cells. (B) Transmission electron micrographs (TEM) of cancer cell membrane-coated nanoparticles. Reprinted with permission from ref. [65]. Copyright 2014 American Chemical Society.
Polymeric aAPC
Various polymers, such as biodegradable PLGA[77-83] and non-biodegradable sepharose or polystyrene beads[84-95], have been incorporated into aAPC systems. Immunomodulatory compounds, such as anti-CD3, pMHC and anti-CD28 mAbs can be anchored on the surface of polymeric particles, while IL-2 or other soluble molecules can be gradually released from within the aAPC.
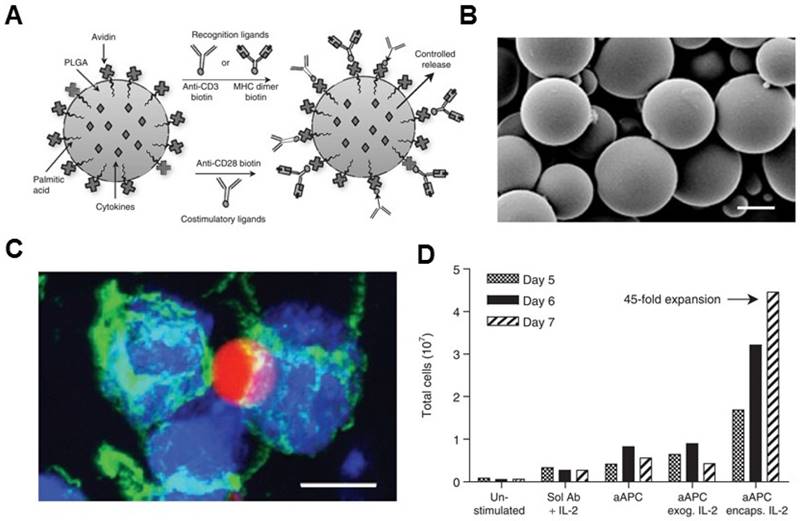
Non-biodegradable sepharose or polystyrene beads were the first synthetic bead-based platforms used to activate T cells.[96, 97] It was reported that the optimal size of microbeads was between 4 to 5 μm, which is one important parameter for T cell activation.[84] The polymer bead-based aAPCs have been used as a tool to study T cell biology.[98, 99] Additionally, aAPC systems can be synthesized from a variety of biodegradable polymers, such as PLGA. Steenblock and Fahmy developed a PLGA-based biodegradable aAPC that presented both recognition ligands and co-stimulatory ligands (anti-CD3, anti-CD28 mAbs and peptide-MHC complexes) on its surface and slowly released the encapsulated cytokines (IL-2) from particles (Figure 3).[79] Surface ligand presentation was stable about 20 days, leading to a significantly secretion of interferon gamma (IFNγ) by the murine and human T cells ex vivo. The PLGA particles of 6-10 μm in size were found to be the most effective for T cell activation and expansion ex vivo.[79] In another study, Kosmides et al. [81] combined an antigen-specific PLGA-based aAPC and a checkpoint blockade molecule, anti-PD-1 monoclonal antibody, for cancer immunotherapy. The PLGA particle was functionalized by antigen specific MHC-I and anti-CD28 mAbs. Combinatorial treatment with aAPCs and anti-PD-1 enhanced CD8+ T cells in vitro, significantly delayed tumor growth in vivo, and increased median survival time in mice after systemic administration. It was associated with reduced PD-1 expression and enhanced antigen-specific proliferation of CD8+ T cells within the tumor microenvironment and spleen.
Biodegradable polymeric artificial antigen-presenting cells. (A) Schematic of a biodegradable PLGA aAPC. Anti-CD3, anti-CD28 mAbs, and pMHC complexes were loaded onto aAPCs through biotin-avidin conjugation. Encapsulated cytokines were released from particles in a time-dependent manner. (B) SEM imaging of the microparticle. (C) Fluorescence imaging of aAPC-T-cell binding. (D) Expansion of T cells after various treatment as indicated. Reprinted with permission from ref. [79]. Copyright 2008 American Society of Gene & Cell Therapy.
It is known that shape of aAPC appears to be an important factor for T cell activation as the natural APCs are not spherical.[80, 100] Therefore, non-spherical PLGA-based microparticles have been designed to mimic the natural situation by increasing the contact area of particles. The film-stretching method was used for controlling the shape of PLGA microparticles to generate ellipsoidal aAPCs with varying long axis lengths and aspect ratios.[101] It was reported that elongated PLGA particles with a more ellipsoidal shape were more efficient as aAPCs compared to spherical particles, leading to a higher T cell proliferation in vitro.[80] In a subsequent study by the same group, Meyer et al. [102] synthesized nanoellipsoidal PLGA aAPCs as before and they activated T cells in vivo following systemic administration. After systemic administration, these nanoellipsoidal aAPCs stimulated stronger in vivo immune cell responses comparable to previously reported spherical aAPCs at a reduced overall protein dose. Moreover, the authors found that these nanoellipsoidal aAPCs had enhanced pharmacokinetic properties, properly due to their resistance to hepatic and splenic elimination.
In addition to PLGA, a new class of semi-flexible and filamentous polymers comprising of poly (isocyano dipeptide) and oligo (ethylene oxide) side chains were developed by Rowan and coworkers.[103] Anti-CD3 mAbs were decorated to these highly controlled, semi-stiff polymers to mimic dendritic cells. These anti-CD3 polymers induced a more robust T cell response than PLGA microparticles. This enhanced activity can be attributed to the structural flexibility and multivalency of these polymers, assisting in the formation of TCR nanoclusters on the T cell surface. In addition, these semi-flexible and filamentous polymers were biocompatible and non-toxic, highlighting their promise for the induction of both ex vivo and in vivo T cell responses.
Inorganic aAPCS
Synthetic aAPCs may also contain superparamagnetic parts for further separation from cells by the magnetic field before transfusion into patients. Magnetic particles are of particular interest for ex vivo T cell expansion.[86, 104-108] Levine et al.[106] used magnetic beads covalently linked to anti-CD3 and anti-CD28 mAbs to first demonstrate expansion of purified CD4+ T cells in vitro. Today, T cell expansion ex vivo using anti-CD3/anti-CD28-coated magnetic beads has been applied in clinical trials of ACT to treat various types of cancer[109-113]. In addition to magnetic beads, magnetic nano-aAPCs were recently developed. Perica et al. [107] developed a strategy using magnetic nano-aAPCs and an externally applied magnetic field to enhance the T cell receptor clustering. After binding to the TCR, the magnetic field was used to drive aggregation of these magnetic nano-aAPCs, resulting in TCR clustering and increased T cell expansion in vitro and after adoptive transfer in vivo. Magnetic field-enhanced nano-aAPC stimulation is a novel approach to drive receptor clustering. In another example, a magnetic field was used for the enrichment of rare tumor-specific T cells and activate them to induce proliferation.[108] This enrichment and expansion strategy using paramagnetic nano-aAPC could effectively expand tumor-specific T cells, resulting in a greater than 1000-fold expansion of tumor-specific T cells in one week.[108]
Janus particles, named in reference to the Roman god, Janus, who had two faces, are particles that have two distinct faces.[114] Double-sided Janus particles could prove useful in the development of aAPCs. Yu and co-workers[115] designed a magnetic Janus microparticle to control T cell activation remotely. One side of these particles was decorated with a thin film of magnetically responsive materials, and on the other side coated with stimulatory ligand (anti CD3) for T cell activation. By simultaneously controlling the rotation and locomotion of the Janus particles, the authors demonstrated the initiation of T cell activation in single-cell precision.
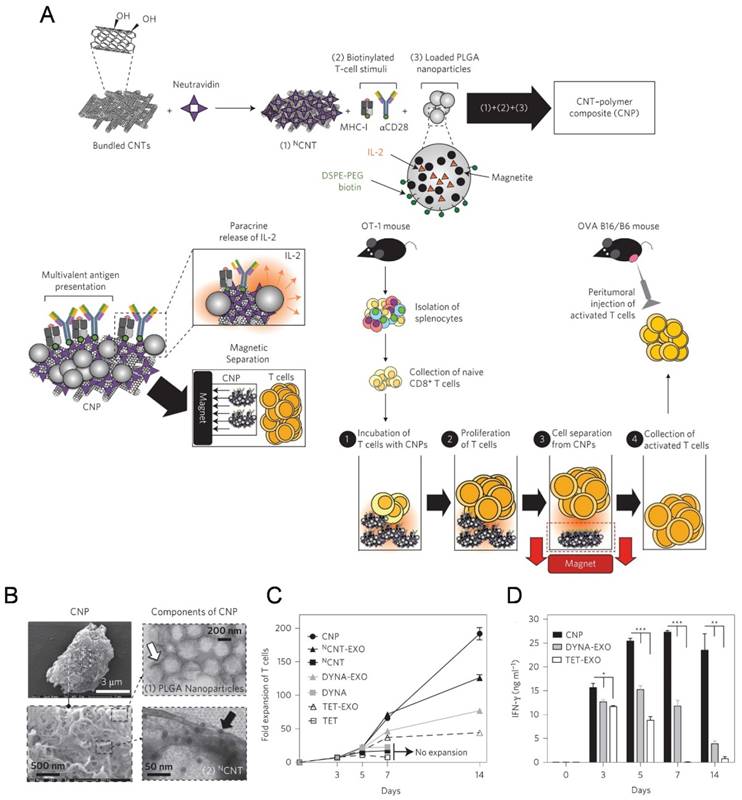
The high-surface-area carbon nanotubes have been widely used as in aAPCs for ex vivo T cell expansion[116-119]. Fadel et al. [116] demonstrated that anti-CD3-coated tubes induced a higher specific T cell activation and IL-2 production than other high surface area materials (activated carbon, polystyrene, and C60 nanoparticles). A potential mechanism for the enhanced stimulation response with treated single-walled nanotubes (SWNTs) surface may be due to the local clustering of the antibody stimuli in defect regions combined with the chemical nature of the environment surrounding these clusters. In the following work, a carbon nanotube-polymer composite (CNP) was developed to expand T cells ex vivo (Figure 4).[118] The CNP was made of carbon nanotubes with magnetite and PLG (polylactate-co-glycolate) nanoparticles loaded with IL-2. The release of IL-2 from the PLG nanoparticles facilitated antigen presentation to T cells and proliferative capacity of T cells. These CNPs could remarkably activate the T cells ex vivo. Meanwhile, the magnetic properties of the PLG nanoparticles allowed for separation of CNPs from the expanded T cells after activation ex vivo. After adoptively transferred into the tumor bearing mice, the expanded T cells induced a significant delay in tumor growth in a murine melanoma model and increased T cell infiltration in tumor sites.
A carbon nanotube-polymer composite for T-cell therapy. (A) Schematic of bundled CNTs as aAPCs to activate T cells. (B) SEM images of a CNP at low (top) and high (bottom) magnifications. Right: TEM images of PLGA nanoparticles (top) and CNT (bottom). (C) Expansion ability of OT-1 CD8+ T cells by CNTs aAPCs. (D) Level of IFN-γ release from CD8+ T cells after various treatment as indicated. Reprinted with permission from ref. [118]. Copyright 2014 Nature Publishing Group.
Artificial Lymphoid Organs
Lymphoid organs such as the thymus, spleen, and lymph nodes are essential for the immune response.[1] Bioengineering artificial lymphoid tissues aims to use these organs for treatment of various diseases is an emerging field.[19] Successfully bioengineered artificial lymphoid organs are able to mimic the natural organs by recruiting and organizing lymphocytes into such structures and ensuring their survival, interaction, activation and function.[18, 19, 39, 120, 121] Biomaterial-based three-dimensional (3D) scaffolds have been widely studied for engineering of lymphoid organs. Various scaffold materials, including natural proteins such as fibroin[122], spidroin[123], alginate[124] and collagen[125], synthetic polymers including PLG (polylactate-co-glycolate), PLA (polylactate), and PGA (polyglycolate),[126] and inorganic mesoporous silica rods[127] have been applied for constructing scaffolds.
Lymph node
The artificial lymph nodes that imitate the structure of a lymph node organ have been developed. First, several in vitro models of lymph node were created. In one approach, the researchers developed a bioreactor that imitated human cell microenvironment and homeostasis of primary follicles.[120] It was developed using macroporous matrix sheets with dendritic cells or a suspension of lymphocytes wherein the soluble factors and cells could communicate with each other. Both the T and B lymphocytes and dendritic cells formed clusters within the matrix, indicating their potential functionality. Additionally, this system represented some of the processes in a lymph node, for example, the migration and interaction of lymphocytes with dendritic cells. In another example, Matloubian et al. constructed an in vitro lymph node model to study the local inflammation in lymph nodes.[128] The model consisted of a matrix populated with fibroblast reticular cells under the controlled flow of lymphatic fluid. It was found that lymph flow affected not only the expression of the chemokines but also the rate of cell division, indicating that increased lymph flow may act as an early inflammatory cue to enhance efficient immune response. Recently, Purwada et al. examined the ex vivo generation of artificial lymphoid follicles with active germinal center (GC) reactions.[129] Here, the authors developed an artificial B cell follicle organoid made of a RGD-loaded hydrogel scaffold which reinforced with silicate nanoparticles (SiNP). The scaffold mimicked the anatomical microenvironment of a lymphoid tissue and could accelerate development and differentiation of primary naïve B cells into the GC phenotype ex vivo by providing extracellular matrix and signals to co-cultured naïve B cells. The described approach is promising for artificial production of antigen specific antibodies.
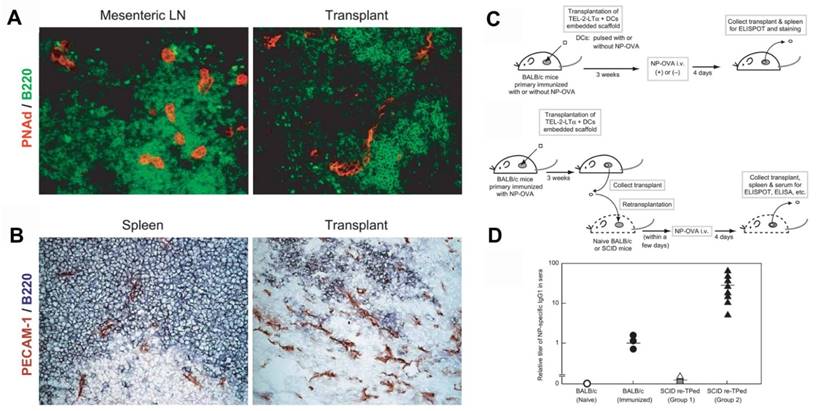
Furthermore, in vivo models of artificial lymph nodes have been demonstrated. Irvine and co-workers[130] designed injectable DCs-carrying alginate gels. An in vivo self-gelling formulation of alginate was designed, which was obtained by mixing calcium-loaded alginate microspheres with soluble alginate solution and dendritic cells. When subcutaneously injected into mice, these DCs' alginate 'nodes' attracted both host DCs and T cells to the injection sites over a week in vivo. Meanwhile, some of the inoculated DCs moved to the draining lymph nodes. This gel/DC system showed great promise in mimicking antigen specific lymph nodes to treat tumors or infections. Watanabe and co-authors[131, 132] developed a system based on a collagen matrix. The thymus-derived stromal cell line TEL-2 expressing lymphotoxin β receptor (LTbR) and vascular cell adhesion molecule-1 (VCAM-1) were embedded in 3D structure sponge-like collagenous scaffold (Figure 5). Murine lymphotoxin alpha (LTα) was also introduced into TEL-2 cells to establish a LTα-expressing TEL-2 cell line (TEL-2-LTα). The authors transplanted the collagenous scaffold with TEL-2-LTa cells and activated DCs to the renal subcapsular space of mice. It has been found that the transplants, which were infiltrated frequently, contained many T cell and B cell clusters. Moreover, these T cells and B cells had similar organization to that of the normal lymph node. They also demonstrated that several high endothelial venule (HEV) markers of secondary lymphoid organs and blood vessel-like structures were detected in the matrix, suggesting they had become organized tissues in mice. After transplantation of the matrix into BALB/c mice immunized with alum-precipitated NP-OVA, the mice displayed secretion of antigen-specific IgG antibodies in the transplants, indicating lymphocyte infiltration in the matrix organoids. Similar results were also demonstrated in immunodeficient (SCID) mice.
Generation of a synthetic lymphoid tissue-like organoid in mice. (A) Fluorescence imaging of transplant tissues. Red, PNAd (one of the HEV-specific adhesion molecules) and green, B220. (B) Immunohistochemical staining of spleen and transplant tissues. Red, PECAM-1 (adhesion molecule expressed on endothelial cells of blood vessel) and bluish purple, B220. (C) Schematic of the procedure to study formation of antigen-specific IgG1 antibody under various conditions. (D) Specific IgG1 antibody was detected by ELISA in sera from SCID mice after various treatment as indicated. Reprinted with permission from ref. [131]. Copyright 2004 Nature Publishing Group.
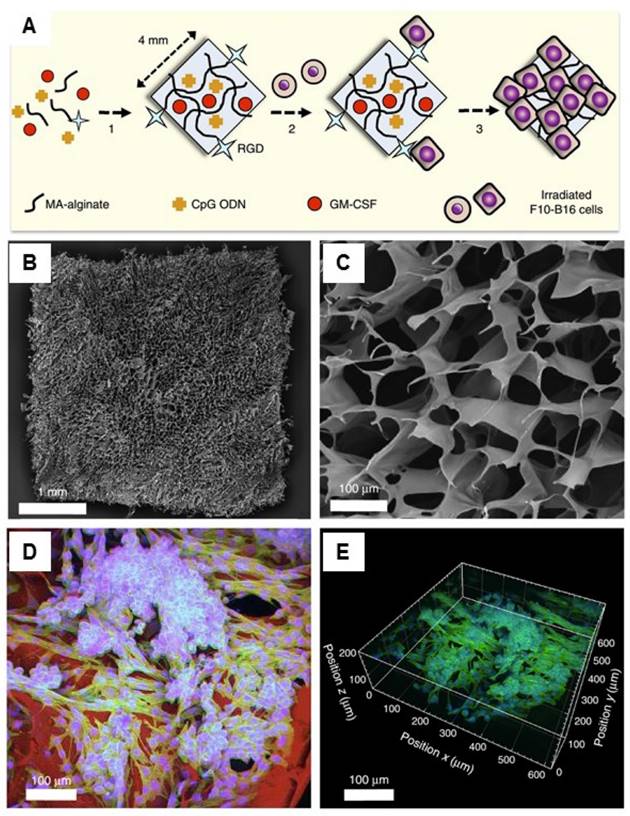
Another therapeutic strategy is the implantation of cell-free systems saturated with immunomodulators to reprogram the specific lymphocytes.[33, 127, 133-138] For example, Mooney and coworkers designed a macroporous PLG matrix to present granulocyte macrophage colony-stimulating factor (GM-CSF), danger signals, and tumor antigens for the recruitment and activation of DCs, leading to significant anti-tumor immunity.[134] Additionally, the same group also incorporated the cytokine GM-CSF, autologous tumor lysate, and Toll-like receptor 9 (TLR9) agonist CpG oligodeoxynucleotides (CpG ODN) into a porous PLG scaffold for the local recruitment and activation of DCs by subcutaneous implantation.[136] Vaccination using this method induced both local and systemic CTL responses, leading to regression of established local and metastatic tumors. The enhanced efficacy of this vaccine could be a result of the promoted local activity of DCs induced by danger signals and antigens at the vaccination site. The same group recently reported cryogel-based whole-cell cancer vaccines for cancer immunotherapy.[133] The authors similarly encapsulated GM-CSF and CpG ODN into sponge-like macroporous cryogels, which were formulated using alginate containing covalently conjugated Arg-Gly-Asp (RGD) peptides (Figure 6). These cryogels were then subcutaneously injected into mice, leading to significant anti-tumor T cell responses in a melanoma model. In addition, inorganic mesoporous silica rods (MSRs) with a high-aspect ratio were found to assemble in vivo after subcutaneous injection, forming macroporous structures.[127] This macroporous structure could create a 3D cellular microenvironment which could recruit the host immune cells. The authors found that the MSR scaffolds could greatly increase the number of recruited cells compared to polymer scaffolds. High level of CD8+ T cells were found in the spleens of treated mice, indicating that the MSR-vaccine induced antigen-specific cellular responses.
Injectable cryogel-based whole-cell cancer vaccines. (A) Schematic of an alginate-derived active vaccine formulation. (B-C) SEM imaging (B) and SEM cross-sectional image (C) of the alginate cryogel. (D-E) 2-D confocal micrograph (D) and 3-D reconstructed confocal fluorescence micrograph (E) of the alginate cryogel. Irradiated B16-F10 cells on a typical RGD-containing cryogel was showed in the picture. Reprinted with permission from ref. [133]. Copyright 2015 Nature Publishing Group.
Thymus
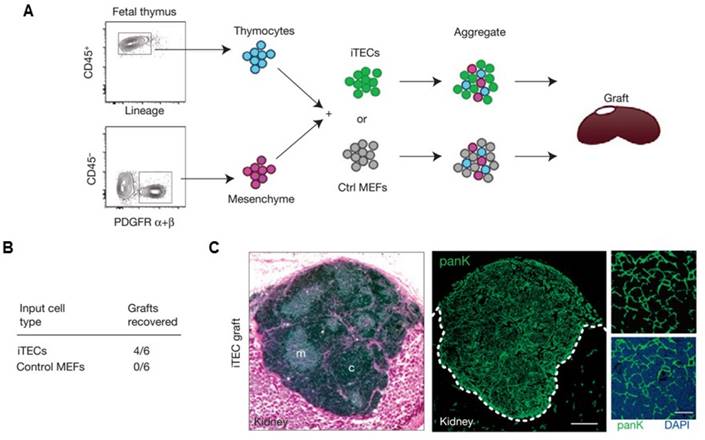
Other artificial lymphoid organs, such as an artificial thymus, are also highly attractive targets because they also play important roles in immune system. For example, thymus evolution with age results in a decreased output of naïve T lymphocytes, which contributes to weaker immunosurveillance in the elderly for various diseases, including cancer[139]. Although in vivo models have yet been reported, artificial thymic organoids have been developed in vitro.[140-142] In a recent study, Blackburn and co-workers[142] described a system for producing thymic epithelial cells (TECs), a key cell type of the thymic stroma, by reprogramming mouse embryonic fibroblasts (MEF) that expressed Foxn1 in vitro. It has been found that with Foxn1 expression, MEFs acquired a normal TECs phenotype, characterized by surface markers (EpCAM) and genes for factors that are important for their functional activity (Dll4, CCL25). Co-cultivation of the Foxn1 transformed MEF and T cell progenitors led to the generation of a transient population of CD4+ CD8+ thymocytes, as well as terminally differentiated CD4+ and CD8+ T cells compared to the TECs isolated from embryonic mouse thymus. This work provides opportunity of applying thymus transplantation to boost immune function, which is an essential step towards bioengineering of an artificial thymus (Figure 7).[142]
Artificial thymus generated from FOXN1-reprogrammed fibroblasts. (A) Schematic of the procedure to show in vivo grafting assay. (B) Summary of recovered grafts after various treatment as indicated. (C) H&E staining (left), and pan-cytokeratin (right) staining of the iTEC-derived kidney graft, indicating that FOXN1-induced TECs (iTECs) established a complete, fully organized and functional thymus. Reprinted with permission from ref. [142] Copyright 2014 Nature Publishing Group.
Spleen and mucosal tissue
The construction of an engineered spleen has been successfully realized in a mouse model. [143] In this spleen transplantation technology, spleen units from neonatal rats were loaded onto a scaffold made of polyglycolic acid coated with collagen and then transplanted into the omentum.[143] The transplanted spleen resulted in the formation of a normal splenic structure. In addition, immunological function of the implanted spleen was also demonstrated. Similarly, mucosal immune tissue has also been demonstrated through neointestine synthesis.[144] In this technique, neonatal rat small bowel was seeded onto biodegradable polymer tubes made by polyglycolic acid fibers coated with collagen. These constructs were transplanted into syngeneic adult recipients. After 20 weeks, intraepithelial and lamina propria immunocyte were detected in transplanted intestines, suggesting their capacity to form a mucosal immune system.
Conclusion and Outlook
As an interdisciplinary interface of cancer biology, immunology, bioengineering and materials science, the development of artificial immune cells and artificial lymphoid organs holds great promise in enhancing immunotherapy. It can overcome limitations of natural immune cells and lymphoid organs, creating desired therapy options for patients.
For the bioengineering of aAPCs (Table 1), ex vivo use of these systems for T cell expansion and activation have showed advantages and effectiveness compared to the use of natural APCs, which have been often used in clinical applications for T cell transfer therapy. Such biomimetic particles can specifically encapsulate and release soluble small molecules and present biological proteins to trigger cell signaling of T cells. Further development of ex vivo used aAPCs optimized for clinical applications should be with improved flexibility and efficient antigen presentation. Although still limited by current knowledge, optimization directions include adjustment of surface ligands, cytokines, material size and shape, ligand mobility and orientation, and anisotropy. For example, the design of 3D-SLB particles could be made by coating natural cell membranes with ligands and signals, as lipid bilayers to PLGA particles would be a promising strategy to mimic the natural DCs. Furthermore, the incorporation of magnetic particles can be readily applied to separate particles and cells after expansion, improving the clinical efficiency for the T cells expanded ex vivo.
Summary of representative aAPCs used for immunotherapy.
| Types of aAPC | Advantage | Potential limitation | Immunomodulators used | Ref. | |
|---|---|---|---|---|---|
| Lipid-based | Liposomes | Easy preparation Fluid membrane | Unstable Low T cell activation | MHC | [58] |
| Microdomain liposomes | Fluid membrane Ligand pre-clustering | Unstable | MHC, αCD3, αCD28, αLFA-1, | [59-61] | |
| SLB particles | Stable Fluid membrane | Not extensively applied | pMHC | [71] | |
| Polymeric | PLGA particles | Easy preparation Cytokine release Shape change Biodegradable | Rigid surface | αCD3, αCD28, MHC, IL-2 | [77-80, 82, 83] |
| Sepharose or polystyrene beads | Easy preparation Widely used | Non-biodegradable Rigid surface | MHC, αCD3, CD80, αCD28, CD54, CD83, α4-1BB, 4-1BBL | [84-93] | |
| Filamentous polymers | Flexibility and multivalency Biocompatible | Not extensively applied | αCD3 | [103] | |
| Inorganic | Magnetic particles | Easy preparation, Easy enrichment and separation Controlled by magnetic field Widely used | Non-biodegradable Rigid surface | αCD3, αCD28, MHC, CD80 | [86, 104-108, 111-113, 115, 154, 155] |
| Carbon nanotubes | High surface area Ligand pre-clustering | Non-biodegradable Rigid surface Large clusters | αCD3, MHC, αCD28 | [116-119] | |
The development of safe and biocompatible systems should be taken into consideration when used for the in vivo aAPC immunotherapy. Biodegradable polymer particles have often demonstrated excellent biocompatibility and are useful for the release of cytokines or other immunomodulatory factors. However, how to enhance their interaction with T cells in vivo should also be further studied. Additionally, developing nanosized particles[145, 146] that can accumulate in the lymph nodes and organs may boost the efficacy of in vivo aAPC therapy. Tumor heterogeneity is another major challenge when training T cells for ACT.[12, 147, 148] Treating a heterogeneous tumor with single antigen-specific T cells can lead to limited antitumor effects. Particularly, attention has shifted to neoantigens, which are an individual's tumor-specific mutations antigens that are new to the immune system and are not found in normal tissues.[12, 149] Further development of aAPCs that can elicit broad-spectrum and neoantigen-specific T cells for personalized cancer immunotherapies is one of the emerging trend in this field.[150] Additionally, during the translation of these therapies to a clinical setting, the application of good manufacturing practices (GMP) of such biomaterials or biomimetic materials may present significant challenges. The concerns of compliance within GMP standardization and associated costs need to be addressed for further translation.
Moreover, as the natural organs are complex, further thorough understanding of lymphoid organogenesis and molecular signaling is highly important. One strategy considers implantation of biomaterial-based cell-free scaffolds containing immunotherapy agents as a “node” for immunization, while the other strategy is more complex and looks to re-program or introduce cells into lymphoid organ structures, for long-term function as a lymphoid organ after implantation. However, it should be noted that while many approaches relying on gene-modified mouse cell lines are useful for basic research, they could not be applied to patients. These problems can be avoided by replacing the cells with patient-derived induced pluripotent stem cells (iPSC) as a source of stromal cells for generation of artificial lymphoid organs.[151, 152] Furthermore, the development of high-performance lymphoid organs is feasible by combining these two strategies together.
Acknowledgements
This work was supported by grants from the Alfred P. Sloan Foundation (Sloan Research Fellowship) and the pilot grant from the UNC Cancer Center.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Parham P. The immune system. Garland Science. 2014
2. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121:1-14
3. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-8
4. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annual Review of Immunology. 2007;25:267-96
5. Speiser DE, Ho P-C, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16:599-611
6. Zhang P, Su DM, Liang M, Fu J. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol Immunol. 2008;45:1470-6
7. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463-9
8. Klevorn LE, Teague RM. Adapting Cancer Immunotherapy Models for the Real World. Trends Immunol. 2016;37:354-63
9. Littman DR. Releasing the Brakes on Cancer Immunotherapy. Cell. 2015;162:1186-90
10. Couzin-Frankel J. Cancer immunotherapy. Science. 2013;342:1432-3
11. O'Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. 2015;36:71-80
12. Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS. et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536:91-5
13. Hori S, Takahashi T, Sakaguchi S. Control of autoimmunity by naturally arising regulatory CD4+ T cells. Adv Immunol. 2003;81:331-71
14. Karim M, Kingsley CI, Bushell AR, Sawitzki BS, Wood KJ. Alloantigen-induced CD25+ CD4+ regulatory T cells can develop in vivo from CD25- CD4+ precursors in a thymus-independent process. J Immunol. 2004;172:923-8
15. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8:337-50
16. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566-81
17. Bender A, Sapp M, Schuler G, Steinman RM, Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J Immunol Methods. 1996;196:121-35
18. Tan JK, Watanabe T. Artificial engineering of secondary lymphoid organs. Adv Immunol. 2010;105:131-57
19. Nosenko MA, Drutskaya MS, Moisenovich MM, Nedospasov SA. Bioengineering of Artificial Lymphoid Organs. Acta Naturae. 2016;8:10-23
20. Kim JV, Latouche JB, Riviere I, Sadelain M. The ABCs of artificial antigen presentation. Nat Biotechnol. 2004;22:403-10
21. Xu C, Hu S, Chen X. Artificial cells: from basic science to applications. Mater Today. 2016
22. Ye Y, Wang J, Hu Q, Hochu GM, Xin H, Wang C. et al. Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano. 2016;10:8956-63
23. Wang C, Ye Y, Hochu GM, Sadeghifar H, Gu Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016;16:2334-40
24. Jeanbart L, Swartz MA. Engineering opportunities in cancer immunotherapy. Proc Natl Acad Sci USA. 2015;112:14467-72
25. Koshy ST, Mooney DJ. Biomaterials for enhancing anti-cancer immunity. Current opinion in biotechnology. 2016;40:1-8
26. Gu L, Mooney DJ. Biomaterials and emerging anticancer therapeutics: engineering the microenvironment. Nat Rev Cancer. 2016;16:56-66
27. Liang C, Xu L, Song G, Liu Z. Emerging nanomedicine approaches fighting tumor metastasis: animal models, metastasis-targeted drug delivery, phototherapy, and immunotherapy. Chem Soc Rev. 2016;45:6250-69
28. Cheung AS, Mooney DJ. Engineered Materials for Cancer Immunotherapy. Nano today. 2015;10:511-31
29. Goldberg MS. Immunoengineering: how nanotechnology can enhance cancer immunotherapy. Cell. 2015;161:201-4
30. Hotaling NA, Tang L, Irvine DJ, Babensee JE. Biomaterial Strategies for Immunomodulation. Annu Rev Biomed Eng. 2015;17:317-49
31. Lu Y, Aimetti AA, Langer R, Gu Z. Bioresponsive materials. Nat Rev Mater. 2016;1:16075
32. Scott EA, Karabin NB, Augsornworawat P. Overcoming Immune Dysregulation with Immunoengineered Nanobiomaterials. Annu Rev Biomed Eng. 2016:19
33. Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2016
34. Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat Biomed Eng. 2017;1:0011
35. Sunshine JC, Green JJ. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine. 2013;8:1173-89
36. Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014;32:456-65
37. Perica K, Kosmides AK, Schneck JP. Linking form to function: Biophysical aspects of artificial antigen presenting cell design. Biochim Biophys Acta. 2015;1853:781-90
38. Meyer RA, Sunshine JC, Green JJ. Biomimetic particles as therapeutics. Trends Biotechnol. 2015;33:514-24
39. Irvine DJ, Stachowiak AN, Hori Y. Lymphoid tissue engineering: Invoking lymphoid tissue neogenesis in immunotherapy and models of immunity. Sem Immunol. 2008;20:137-46
40. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245-52
41. Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296-306
42. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74-80
43. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557-69
44. Oelke M, Krueger C, Giuntoli RL, Schneck JP. Artificial antigen-presenting cells: artificial solutions for real diseases. Trends Mol Med. 2005;11:412-20
45. Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC. et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15:981-8
46. Tostanoski LH, Gosselin EA, Jewell CM. Engineering Tolerance Using Biomaterials to Target and Control Antigen Presenting Cells. Discovery Medicine. 2016;21:403-10
47. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D. et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453-64
48. Liu ZY, Li Z. Molecular Imaging in Tracking Tumor-Specific Cytotoxic T Lymphocytes (CTLs). Theranostics. 2014;4:990-1001
49. Turtle CJ, Riddell SR. Artificial antigen-presenting cells for use in adoptive immunotherapy. Cancer journal. 2010;16:374-81
50. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol. 2014;14:435-46
51. Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3:984-93
52. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E. et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573-84
53. Tian Y, Zajac AJ. IL-21 and T Cell Differentiation: Consider the Context. Trends Immunol. 2016;37:557-68
54. Einsele H. Killer aAPCs as tools for immunosuppression. Blood. 2008;111:3305 -
55. Schutz C, Fleck M, Mackensen A, Zoso A, Halbritter D, Schneck JP. et al. Killer artificial antigen-presenting cells: a novel strategy to delete specific T cells. Blood. 2008;111:3546-52
56. Schütz C, Fleck M, Schneck JP, Oelke M. Killer artificial antigen presenting cells (KaAPC) for efficient in vitro depletion of human antigen-specific T cells. J Vis Exp. 2014
57. McLaughlin S, Wang JY, Gambhir A, Murray D. PIP2 AND proteins: Interactions, organization, and information flow. Annual Review of Biophysics and Biomolecular Structure. 2002;31:151-75
58. Prakken B, Wauben M, Genini D, Samodal R, Barnett J, Mendivil A. et al. Artificial antigen-presenting cells as a tool to exploit the immune 'synapse'. Nat Med. 2000;6:1406-10
59. Ding Q, Chen J, Wei XH, Sun WQ, Mai JH, Yang YZ. et al. RAFTsomes Containing Epitope-MHC-II Complexes Mediated CD4+T Cell Activation and Antigen-Specific Immune Responses. Pharm Res-Dordr. 2013;30:60-9
60. Giannoni F, Barnett J, Bi K, Samodal R, Lanza P, Marchese P. et al. Clustering of T cell ligands on artificial APC membranes influences T cell activation and protein kinase C theta translocation to the T cell plasma membrane. Journal of immunology. 2005;174:3204-11
61. Zappasodi R, Di Nicola M, Carlo-Stella C, Mortarini R, Molla A, Vegetti C. et al. The effect of artificial antigen-presenting cells with preclustered anti-CD28/-CD3/-LFA-1 monoclonal antibodies on the induction of ex vivo expansion of functional human antitumor T cells. Haematologica. 2008;93:1523-34
62. Zhang PF, Chen YX, Zeng Y, Shen CG, Li R, Guo ZD. et al. Virus-mimetic nanovesicles as a versatile antigen-delivery system. Proc Natl Acad Sci USA. 2015;112:E6129-E38
63. Alarcon B, Swamy M, van Santen HM, Schamel WW. T-cell antigen-receptor stoichiometry: pre-clustering for sensitivity. EMBO Rep. 2006;7:490-5
64. Castellana ET, Cremer PS. Solid supported lipid bilayers: From biophysical studies to sensor design. Surf Sci Rep. 2006;61:429-44
65. Fang RH, Hu CMJ, Luk BT, Gao WW, Copp JA, Tai YY. et al. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett. 2014;14:2181-8
66. Dehaini D, Fang RH, Zhang L. Biomimetic strategies for targeted nanoparticle delivery. Bioeng Transl Med. 2016
67. Fang RH, Zhang L. Nanoparticle-Based Modulation of the Immune System. Annu Rev Chem Biomol Eng. 2016;7:305-26
68. Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamphiwatana S, Dehaini D. et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118-21
69. Tan SW, Li X, Guo YJ, Zhang ZP. Lipid-enveloped hybrid nanoparticles for drug delivery. Nanoscale. 2013;5:860-72
70. Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA. et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers (vol 10, pg 389, 2011). Nat Mater. 2011;10:476 -
71. Goldstein SAN, Mescher MF. Cell-Sized, Supported Artificial Membranes (Pseudocytes) - Response of Precursor Cytotoxic Lymphocytes-T to Class-I Mhc Proteins. J Immunol. 1986;137:3383-92
72. Luk BT, Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. Journal of controlled release: official journal of the Controlled Release Society. 2015;220:600-7
73. Chen W, Zhang Q, Luk BT, Fang RH, Liu Y, Gao W. et al. Coating nanofiber scaffolds with beta cell membrane to promote cell proliferation and function. Nanoscale. 2016;8:10364-70
74. Wei X, Gao J, Fang RH, Luk BT, Kroll AV, Dehaini D. et al. Nanoparticles camouflaged in platelet membrane coating as an antibody decoy for the treatment of immune thrombocytopenia. Biomaterials. 2016;111:116-23
75. Tan S, Wu T, Zhang D, Zhang Z. Cell or cell membrane-based drug delivery systems. Theranostics. 2015;5:863
76. Mescher MF, Savelieva E. Stimulation of tumor-specific immunity using tumor cell plasma membrane antigen. Methods. 1997;12:155-64
77. Han H, Peng JR, Chen PC, Gong L, Qiao SS, Wang WZ. et al. A novel system of artificial antigen-presenting cells efficiently stimulates Flu peptide-specific cytotoxic T cells in vitro. Biochem Biophys Res Commun. 2011;411:530-5
78. Steenblock ER, Fadel T, Labowsky M, Pober JS, Fahmy TM. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. The Journal of biological chemistry. 2011;286:34883-92
79. Steenblock ER, Fahmy TM. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Mol Ther. 2008;16:765-72
80. Sunshine JC, Perica K, Schneck JP, Green JJ. Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells. Biomaterials. 2014;35:269-77
81. Kosmides AK, Meyer RA, Hickey JW, Aje K, Cheung KN, Green JJ. et al. Biomimetic biodegradable artificial antigen presenting cells synergize with PD-1 blockade to treat melanoma. Biomaterials. 2017;118:16-26
82. Fahmy TM, Samstein RM, Harness CC, Mark Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26:5727-36
83. Steenblock ER, Wrzesinski SH, Flavell RA, Fahmy TM. Antigen presentation on artificial acellular substrates: modular systems for flexible, adaptable immunotherapy (vol 9, pg 451, 2009). Expert Opin Biol Th. 2009;9:1115 -
84. Mescher M. Surface contact requirements for activation of cytotoxic T lymphocytes. J Immunol. 1992;149:2402-5
85. Deeths MJ, Mescher MF. B7-1-dependent co-stimulation results in qualitatively and quantitatively different responses by CD4+ and CD8+ T cells. European journal of immunology. 1997;27:598-608
86. Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat Med. 2003;9:619-24
87. Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D. et al. Potent costimulation of human CD8 T cells by anti-4-1BB and anti-CD28 on synthetic artificial antigen presenting cells. Cancer immunology, immunotherapy: CII. 2008;57:175-83
88. Deeths MJ, Mescher MF. ICAM-1 and B7-1 provide similar but distinct costimulation for CD8(+) T cells, while CD4(+) T cells are poorly costimulated by ICAM-1. European journal of immunology. 1999;29:45-53
89. Lu X, Jiang X, Liu R, Zhao H, Liang Z. Adoptive transfer of pTRP2-specific CTLs expanding by bead-based artificial antigen-presenting cells mediates anti-melanoma response. Cancer Lett. 2008;271:129-39
90. Oelke M, Schneck JP. Overview of a HLA-Ig based "Lego-like system" for T cell monitoring, modulation and expansion. Immunol Res. 2010;47:248-56
91. Jiang X, Lu X, Liu R, Zhang F, Zhao H. HLA Tetramer Based Artificial Antigen-Presenting Cells Efficiently Stimulate CTLs Specific for Malignant Glioma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:7329-34
92. Shen C, Cheng K, Miao S, Wang W, He Y, Meng F. et al. Latex bead-based artificial antigen-presenting cells induce tumor-specific CTL responses in the native T-cell repertoires and inhibit tumor growth. Immunology letters. 2013;150:1-11
93. Shen C, Zhang J, Xia L, Meng F, Xie W. Induction of tumor antigen-specific cytotoxic T cell responses in naive mice by latex microspheres-based artificial antigen-presenting cell constructs. Cell Immunol. 2007;247:28-35
94. Walter S, Herrgen L, Schoor O, Jung G, Wernet D, Bühring H-J. et al. Cutting edge: predetermined avidity of human CD8 T cells expanded on calibrated MHC/anti-CD28-coated microspheres. J Immunol. 2003;171:4974-8
95. Schilbach K, Kerst G, Walter S, Eyrich M, Wernet D, Handgretinger R. et al. Cytotoxic minor histocompatibility antigen HA-1-specific CD8+ effector memory T cells: artificial APCs pave the way for clinical application by potent primary in vitro induction. Blood. 2005;106:144-9
96. Deeths MJ, Mescher MF. B7-1-dependent co-stimulation results in qualitatively and quantitatively different responses by CD4(+) and CD8(+) T cells. European journal of immunology. 1997;27:598-608
97. Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK. et al. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. Journal of immunology. 1999;162:3256-62
98. Crispe IN, Bevan MJ, Staerz UD. Selective activation of Lyt 2+ precursor T cells by ligation of the antigen receptor. Nature. 1985;317:627-9
99. Curtsinger JM, Lins DC, Mescher MF. CD8+ memory T cells (CD44high, Ly-6C+) are more sensitive than naive cells (CD44low, Ly-6C-) to TCR/CD8 signaling in response to antigen. J Immunol. 1998;160:3236-43
100. Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485-92
101. Champion JA, Katare YK, Mitragotri S. Making polymeric micro- and nanoparticles of complex shapes. Proc Natl Acad Sci USA. 2007;104:11901-4
102. Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP. et al. Biodegradable Nanoellipsoidal Artificial Antigen Presenting Cells for Antigen Specific T-Cell Activation. Small. 2015;11:1519-25
103. Mandal S, Eksteen-Akeroyd ZH, Jacobs MJ, Hammink R, Koepf M, Lambeck AJA. et al. Therapeutic nanoworms: towards novel synthetic dendritic cells for immunotherapy. Chem Sci. 2013;4:4168-74
104. Ugel S, Zoso A, De Santo C, Li Y, Marigo I, Zanovello P. et al. In vivo administration of artificial antigen-presenting cells activates low-avidity T cells for treatment of cancer. Cancer Res. 2009;69:9376-84
105. Maus MV, Riley JL, Kwok WW, Nepom GT, June CH. HLA tetramer-based artificial antigen-presenting cells for stimulation of CD4+ T cells. Clin Immunol. 2003;106:16-22
106. Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB. et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921-30
107. Perica K, Tu A, Richter A, Bieler JG, Edidin M, Schneck JP. Magnetic Field-Induced T Cell Receptor Clustering by Nanoparticles Enhances T Cell Activation and Stimulates Antitumor Activity. ACS Nano. 2014;8:2252-60
108. Perica K, Bieler JG, Schutz C, Varela JC, Douglass J, Skora A. et al. Enrichment and Expansion with Nanoscale Artificial Antigen Presenting Cells for Adoptive Immunotherapy. ACS Nano. 2015;9:6861-71
109. Laport GG, Levine BL, Stadtmauer EA, Schuster SJ, Luger SM, Grupp S. et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood. 2003;102:2004-13
110. Rapoport AP, Levine BL, Badros A, Meisenberg B, Ruehle K, Nandi A. et al. Molecular remission of CML after autotransplantation followed by adoptive transfer of costimulated autologous T cells. Bone Marrow Transplant. 2004;33:53-60
111. Rapoport AP, Stadtmauer EA, Aqui N, Vogl D, Chew A, Fang HB. et al. Rapid immune recovery and graft-versus-host disease-like engraftment syndrome following adoptive transfer of Costimulated autologous T cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:4499-507
112. Hardy NM, Mossoba ME, Steinberg SM, Fellowes V, Yan XY, Hakim FT. et al. Phase I trial of adoptive cell transfer with mixed-profile type-I/type-II allogeneic T cells for metastatic breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:6878-87
113. Porter DL, Levine BL, Bunin N, Stadtmauer EA, Luger SM, Goldstein S. et al. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood. 2006;107:1325-31
114. Walther A, Müller AH. Janus particles. Soft Matter. 2008;4:663-8
115. Lee K, Yi Y, Yu Y. Remote Control of T Cell Activation Using Magnetic Janus Particles. Angew Chem Int Ed Engl. 2016;55:7384-7
116. Fadel TR, Steenblock ER, Stern E, Li N, Wang XM, Haller GL. et al. Enhanced cellular activation with single walled carbon nanotube bundles presenting antibody stimuli. Nano Lett. 2008;8:2070-6
117. Fadel TR, Look M, Staffier PA, Haller GL, Pfefferle LD, Fahmy TM. Clustering of stimuli on single-walled carbon nanotube bundles enhances cellular activation. Langmuir. 2009;26:5645-54
118. Fadel TR, Sharp FA, Vudattu N, Ragheb R, Garyu J, Kim D. et al. A carbon nanotube- polymer composite for T-cell therapy. Nat Nanotechnol. 2014;9:723 -
119. Fadel TR, Li N, Shah S, Look M, Pfefferle LD, Haller GL. et al. Adsorption of Multimeric T Cell Antigens on Carbon Nanotubes: Effect on Protein Structure and Antigen-Specific T Cell Stimulation. Small. 2013;9:666-72
120. Giese C, Demmler CD, Ammer R, Hartmann S, Lubitz A, Miller L. et al. A human lymph node in vitro-challenges and progress. Artif Organs. 2006;30:803-8
121. Roh KH, Roy K. Engineering approaches for regeneration of T lymphopoiesis. Biomater Res. 2016;20:20
122. Cupedo T, Vondenhoff MF, Heeregrave EJ, De Weerd AE, Jansen W, Jackson DG. et al. Presumptive lymph node organizers are differentially represented in developing mesenteric and peripheral nodes. Journal of immunology. 2004;173:2968-75
123. Altman GH, Diaz F, Jakuba C, Calabro T, Horan RL, Chen J. et al. Silk-based biomaterials. Biomaterials. 2003;24:401-16
124. Shapiro L, Cohen S. Novel alginate sponges for cell culture and transplantation. Biomaterials. 1997;18:583-90
125. Perez RA, Kim M, Kim T-H, Kim J-H, Lee JH, Park J-H. et al. Utilizing core-shell fibrous collagen-alginate hydrogel cell delivery system for bone tissue engineering. Tissue Eng Part A. 2013;20:103-14
126. Lu T, Li Y, Chen T. Techniques for fabrication and construction of three-dimensional scaffolds for tissue engineering. Int J Nanomedicine. 2013;8:337-50
127. Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G. et al. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat Biotechnol. 2015;33:64-72
128. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V. et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355-60
129. Purwada A, Jaiswal MK, Ahn H, Nojima T, Kitamura D, Gaharwar AK. et al. Ex vivo engineered immune organoids for controlled germinal center reactions. Biomaterials. 2015;63:24-34
130. Hori Y, Winans AM, Huang CC, Horrigan EM, Irvine DJ. Injectable dendritic cell-carrying alginate gels for immunization and immunotherapy. Biomaterials. 2008;29:3671-82
131. Suematsu S, Watanabe T. Generation of a synthetic lymphoid tissue-like organoid in mice. Nat Biotechnol. 2004;22:1539-45
132. Okamoto N, Chihara R, Shimizu C, Nishimoto S, Watanabe T. Artificial lymph nodes induce potent secondary immune responses in naive and immunodeficient mice. J Clin Invest. 2007;117:997-1007
133. Bencherif SA, Warren Sands R, Ali OA, Li WA, Lewin SA, Braschler TM. et al. Injectable cryogel-based whole-cell cancer vaccines. Nat Commun. 2015;6:7556
134. Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nat Mater. 2009;8:151-8
135. Ali OA, Tayalia P, Shvartsman D, Lewin S, Mooney DJ. Inflammatory cytokines presented from polymer matrices differentially generate and activate DCs in situ. Adv Funct Mater. 2013;23:4621-8
136. Ali OA, Emerich D, Dranoff G, Mooney DJ. In situ regulation of DC subsets and T cells mediates tumor regression in mice. Sci Transl Med. 2009;1:8ra19-8ra
137. Tang L, Zheng Y, Mabardi L, Irvine DJ. T lymphocyte engineering with cytokine nanogels for enhanced cancer immunotherapy. J Immunother Cancer. 2015;3:1
138. Kobayashi Y, Watanabe T. Gel-Trapped Lymphorganogenic Chemokines Trigger Artificial Tertiary Lymphoid Organs and Mount Adaptive Immune Responses In Vivo. Front Immunol. 2016;7:316
139. Lynch HE, Goldberg GL, Chidgey A, Van den Brink MRM, Boyd R, Sempowski GD. Thymic involution and immune reconstitution. Trends in Immunol. 2009;30:366-73
140. Clark RA, Yamanaka K, Bai M, Dowgiert R, Kupper TS. Human skin cells support thymus-independent T cell development. J Clin Invest. 2005;115:3239-49
141. Poznansky MC, Evans RH, Foxall RB, Olszak IT, Piascik AH, Hartman KE. et al. Efficient generation of human T cells from a tissue-engineered thymic organoid. Nat Biotechnol. 2000;18:729
142. Bredenkamp N, Ulyanchenko S, O'Neill KE, Manley NR, Vaidya HJ, Blackburn CC. An organized and functional thymus generated from FOXN1-reprogrammed fibroblasts. Nat Cell Biol. 2014;16:902-8
143. Grikscheit TC, Sala FG, Ogilvie J, Bower KA, Ochoa ER, Alsberg E. et al. Tissue-engineered spleen protects against overwhelming pneumococcal sepsis in a rodent model. J Surg Res. 2008;149:214-8
144. Perez A, Grikscheit TC, Blumberg RS, Ashley SW, Vacanti JP, Whang EE. Tissue-engineered small intestine: ontogeny of the Immune System12. Transplantation. 2002;74:619-23
145. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
146. Jiang W, von Roemeling CA, Chen Y, Qie Y, Liu X, Chen J. et al. Designing nanomedicine for immuno-oncology. Nat Biomed Eng. 2017;1:0029
147. Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC. et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther. 2014;22:623-33
148. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283-96
149. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74
150. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17:209-22
151. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917-20
152. Bredenkamp N, Jin X, Liu D, O'Neill KE, Manley NR, Blackburn CC. Construction of a functional thymic microenvironment from pluripotent stem cells for the induction of central tolerance. Regen Med. 2015;10:317-29
153. Liechtenstein T, Dufait I, Bricogne C, Lanna A, Pen J, Breckpot K. et al. PD-L1/PD-1 Co-Stimulation, a Brake for T cell Activation and a T cell Differentiation Signal. J Clin Cell Immunol. 2012 S12
154. Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB. et al. Effects of CD28 costimulation on long-term proliferation of CD4(+) T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921-30
155. Lum LG, LeFever AV, Treisman JS, Garlie NK, Hanson JP. Immune modulation in cancer patients after adoptive transfer of anti-CD3/anti-CD28-costimulated T cells - Phase I clinical trial. J Immunother. 2001;24:408-19
Author contact
![]() Corresponding author: E-mail: zguunc.edu
Corresponding author: E-mail: zguunc.edu