Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Photoactuators
Photosensors
Innovation
Limitations
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(14):3539-3558. doi:10.7150/thno.20593 This issue Cite
Review
Genetically Encoded Photoactuators and Photosensors for Characterization and Manipulation of Pluripotent Stem Cells
Jordan E. Pomeroy1,2, Hung X. Nguyen1, Brenton D. Hoffman1, Nenad Bursac1 ![]()
1. Department of Biomedical Engineering, Duke University, 101 Science Drive, Room 1427, Fitzpatrick CIEMAS, Durham, North Carolina 27708, USA.
2. Division of Cardiology, Department of Medicine, Duke University Health System, Durham, North Carolina, USA.
Received 2017-4-15; Accepted 2017-7-14; Published 2017-8-18
Abstract

Our knowledge of pluripotent stem cell biology has advanced considerably in the past four decades, but it has yet to deliver on the great promise of regenerative medicine. The slow progress can be mainly attributed to our incomplete understanding of the complex biologic processes regulating the dynamic developmental pathways from pluripotency to fully-differentiated states of functional somatic cells. Much of the difficulty arises from our lack of specific tools to query, or manipulate, the molecular scale circuitry on both single-cell and organismal levels. Fortunately, the last two decades of progress in the field of optogenetics have produced a variety of genetically encoded, light-mediated tools that enable visualization and control of the spatiotemporal regulation of cellular function. The merging of optogenetics and pluripotent stem cell biology could thus be an important step toward realization of the clinical potential of pluripotent stem cells. In this review, we have surveyed available genetically encoded photoactuators and photosensors, a rapidly expanding toolbox, with particular attention to those with utility for studying pluripotent stem cells.
Keywords: optogenetics, photoactuator, photosensor, pluripotent, stem cell, CRISPR
Introduction
We are now in the fourth decade since Evans and Kaufman first derived pluripotent stem cells from the inner cell mass of mouse blastocysts [1]. It was not until the second decade after this seminal discovery that Thomson et al. [2] successfully established the first human embryonic stem cell (hESC) lines ushering in a wave of optimism for the use of human pluripotent stem cells (hPSCs) in regenerative medicine. A decade ago, Yamanaka and colleagues [3] demonstrated potential for an unlimited supply of pluripotent stem cells by deriving the first induced pluripotent stem cell (iPSC) lines from fully differentiated, mouse somatic cells. Reprogramming human somatic cells to hiPSCs followed shortly thereafter [4, 5]. Since then, the relatively slow trajectory toward expected realization of pluripotent stem cells in clinics has been grounded on our limited understanding of the developmental blueprint from naïve pluripotency to diverse, differentiated, and functionally stable cell states [6-13].
Excitingly, the last two decades have also witnessed unparalleled development of molecular scale, genetically-encoded photoactuators and photosensors [14, 15] in a field broadly named optogenetics. The ability to investigate and manipulate molecular interactions through discrete delivery and detection of light has created new avenues for exploration. The fact that these tools are genetically encoded facilitates design of functional cassettes that can be targeted to specific cell populations or subcellular locations in living cells. While neurobiology has claimed the majority of scientific advances using optogenetic tools [16, 17], light-mediated interrogation of complex biologic processes holds promise for any branch of life science, and may eventually lead to novel therapeutics for treatment of human disease. Utilizing optogenetic tools to address knowledge gaps in pluripotent stem cell biology will hopefully propel the next phase of research on an accelerated course.
By definition, photoactuators are made to function as light-stimulated effectors of cellular processes. Ion channel activity, cell-signaling pathways, gene and protein expression and cellular distribution have all been manipulated using photoactuated systems. Analogously, photosensors function as light-stimulated, real-time monitors of cellular activity. Cell-signaling pathways, ion flux, voltage shifts, tension dynamics, and epigenome regulation have all been investigated by fluorescence based photosensors. As the optogenetic toolbox evolves to create spectral diversity in photoactuators and photosensors, the ability to multiplex light excitation and emission signals when using these tools will lead to routine, all-optical interrogation of cell circuitry [18-20]. In this review, we have surveyed the current state of available genetically encoded photoactuators and photosensors with commentary on their current and future utility for characterization and manipulation of pluripotent stem cells.
Photoactuators
Photoactuators are light activated molecules, proteins in the practice of optogenetics, which effectuate cellular processes upon light stimulation. Genetically encoded photoactuators are intriguing given the ability to use tailored genetic cassettes to target the expressed photoactuator to specific cell populations or subcellular compartments. Additionally, the inherent reversibility of the system permits interrogation of cellular processes, in discrete temporal segments, in living cells. This contrasts with prior methodologies which rely on more imprecise chemical inhibition or stimulation strategies to manipulate cell signaling that, at best, function on a timescale of seconds, cannot be easily localized to single cells or subcellular compartments, have non-specific side-effects and potentially lead to non-physiologic function. Genetically encoded photoactuators coupled with precision optics delivering millisecond timescale, single-cell targeted pulses of light have thus dramatically increased our understanding of the molecular scale events that control vital cellular processes.
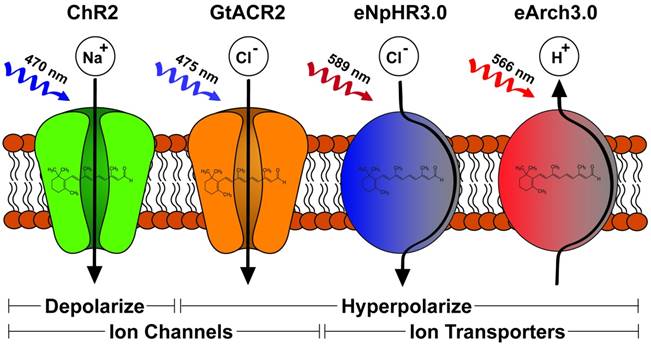
The field of optogenetics was first coined with the use of a native photoactuator, the light-gated cation channel Channelrhodopsin 2 (ChR2; Fig. 1) isolated from the algae Chlamydomonas reinhardtii, successfully xeno-expressed in amphibian and mammalian cells [21]. The proof-of-concept utility of ChR2 was successfully demonstrated through precise, temporal control of murine [22] and embryonic chick [23] neuronal activity via light stimulation. Since these initial experiments less than two decades ago, the photoactuator toolbox has grown to encompass a multitude of light modulated ion channels, cell signaling pathways, protein expression and genome engineering methods (Table 1). Harnessing this expanding toolbox for the field of pluripotent stem cell biology would allow researchers to characterize and manipulate the processes of pluripotency, self-renewal, proliferation, differentiation as well as macro-scale phenomena of embryogenesis and morphogenesis. The following sections detail many of the available genetically encoded photoactuators of interest for stem cell biologists.
Diverse light-gating characteristics and ion selectivity of microbial opsins. The schematic depicts four opsins and their functions. ChR2 is a passive cation channel excited by blue light leading to membrane depolarization. GtACR2 is a passive anion channel also excited by blue light leading to membrane hyperpolarization. Both eNpHR3.0 and eArchT3.0 are active ion transporters excited by red light and lead to membrane hyperpolarization. eNPHR3.0 accomplishes this by inward transport of chloride ions while eArchT3.0 performs outward proton transport.
Examples of Genetically Encoded Photoactuators
| Ion Flow (channel/transporter) | Ion | Membrane Transport | Activation λ (nm) | Function | Ref |
|---|---|---|---|---|---|
| ChR1 | proton (in) | passive | Blue (500) | depolarization | [24] |
| ChR2 | cation | passive | Blue (470) | depolarization | [21] |
| ChIEF | cation | passive | Blue (470) | depolarization | [29] |
| VChR1 | cation | passive | Red (540-570) | depolarization | [32] |
| Chrimson | cation | passive | Red (590) | depolarization | [31] |
| eNpHR 3.0 | anion | active | Red (589) | hyperpolarization | [37] |
| eArchT 3.0 | proton (out) | active | Red (566) | hyperpolarization | [38] |
| GtACR1/2 | anion | passive | 515,475 | hyperpolarization | [42] |
| BLINK1 | cation | passive | Blue (455) | hyperpolarization | [43] |
| Intracellular Signaling | Pathway | Photoprotein | Act λ (nm) | Photoactuation Method | |
| Raf1 | MAPK/ERK | CRY2-CIB1 | Blue (488) | translocation | [68] |
| Raf1 | MAPK/ERK | CRY2olig | Blue (488) | oligomerization | [69] |
| Ras | MAPK/ERK | PHYB-PIF6 | (650)/(750) | translocation | [70] |
| RAF1/MEK1/MEK2 | MAPK/ERK | Dronpa | (500)/(400) | photoswitch/photocage | [60] |
| P85β (PI3K) | PI3K/AKT | PhyB-PIF6 | (650)/(750) | photoswitch | [75] |
| P85β (PI3K) | PI3K/AKT | CRY2-CIBN | Blue (442) | translocation | [76] |
| 5-ptase(OCRL) | PI3K/AKT | CRY2-CIBN | Blue (488) | translocation | [77] |
| Rac1, Cdc42, RhoA | Rho GTPase | PhyB-PIF | Red (650)/(750) | photoswitch | [61] |
| Rac1 | Rho GTPase | LOVpep-ePDZ | Blue (440-473) | translocation | [57] |
| Opto-FGFR1 | RTKs | CRY2olig | Blue (488) | oligomerization | [84] |
| Opto-RTKs | RTKs | LOVdimer | Blue (450) | translocation | [85] |
| Opto-α1AR/β2AR | GPCR | Bovine Rhodopsin | Green (500) | direct activation | [53] |
| RGS4Δ | GPCR | CRY2-CIBN | Blue (445) | translocation | [87] |
| Caspase-7 | Apoptosis | LOV | Blue (438) | photocaging | [90] |
| Bax/Caspase-3 | Apoptosis | CRY2-CIB | Blue (488) | translocation | [91] |
| KillerRed | Apoptosis | KR | Red (585) | ROS production | [52] |
| Transcription/Editing | Function | Photoprotein | Act λ (nm) | Photoactuation Method | |
| splitGAL4 | Transcription | PhyB-PIF | Red (650)/(750) | photoswitch | [62] |
| VP16 | Transcription | LOV | Blue (450-465) | photocaging/dimerize | [59] |
| splitGAL4/VP16 | Transcription | CRY2-CIBN | Blue (461) | translocation | [97, 98] |
| TALEN | Transcription | LOV/GI | Blue (450) | translocation | [99] |
| Cas9/VP64 | Transcription | CRY2-CIBN | Blue (450) | translocation | [100] |
| Histone Effectors | Silencing | CRY2-CIB1 | Blue (450-475) | translocation | [101] |
| splitCas9/CRISPR | Editing | Magnets | Blue (470) | translocation | [102] |
| Other Photoactuation Targets | Function | Photoprotein | Act λ (nm) | Photoactuation Method | |
| Intein/Extein | Splice Activation | PhyB-PIF3 | (660)/(750) | translocation | [103] |
| Degron/Proteasome | Degradation | LOV2 | Blue (465) | photocaging | [104] |
| Trapping | Sequestering | CRY2-CIB1 | Blue (457-488) | translocation | [106] |
| Clathrin | Endocytosis | CRY2olig | Blue (488) | translocation | [107] |
| Actin/Diaphonous | Actin Dynamics | LOV | Blue (405) | photocaging | [109] |
Light-gated ion channels and transporters
Depolarizing opsins
Microbial opsins can be utilized to activate or inhibit mammalian excitability via depolarizing or hyperpolarizing current, respectively. Channelrhodopsins (ChR) represent the major optogenetic class for membrane depolarization, with the first two channels discovered in C. reinhardtii: the light-gated proton channel ChR1 [24] and the light-gated cation channel ChR2 [21]. Since the first studies demonstrating the use of ChR2 to activate neurons using blue light [22, 23], various modifications have been performed on this channel to improve its performances for specific experimental requirements. Photocurrent amplitude was shown to increase two-fold via the single mutation H134R [25], while light sensitivity could be enhanced via C128A/T/S [26] or D156A [27] mutations. In addition, the variant ChR2 E123T/A (or ChETA) exhibited faster deactivation kinetics [28], making it suitable for use in high-frequency stimulation. Furthermore, chimeragenesis of ChR1 and ChR2 yielded the ChIEF variant [29] displaying stable photocurrent under prolonged illumination. Similarly, high steady-state/peak photocurrent ratio was observed in the ChR2 L132C variant (CatCh), which also showed higher permeability to Ca2+ [30]. By using de novo sequencing of 127 algal transcriptomes, Klapoetke et al. further discovered 61 new ChR homologs [31]. Of note, this study identified one homolog from Stigeoclonium helveticum (“Chronos”) displaying fastest reported kinetics to date and another homolog from Chlamydomonas noctigama (“Chrimson”) exhibiting 45-nm more red-shift than previously engineered red-shifted variants VChR1 [32], C1V1 [33], and ReaChR [34], which could facilitate deep tissue stimulation in vivo.
Hyperpolarizing opsins
Inhibition of cell excitability via membrane hyperpolarization has traditionally been achieved using either light-gated inward chloride pumps (halorhodopsin, HR; Fig. 1) [35] or light-gated outward proton pumps (bacteriorhodopsin, BR). The most popular HR ortholog, NpHR from Natronobacterium pharaonis, showed significant intracellular aggregation in its wild-type form upon expression in mammalian cells [36]. The addition of an endoplasmic reticulum export motif and trafficking signal from Kir2.1 channel in the eNpHR3.0 (Fig. 1) variant yielded drastically improved membrane expression and enhanced photocurrent amplitude for efficient neuronal silencing [37]. This approach was also shown to significantly improve membrane trafficking of different BR orthologs, including eBR, eArch3.0, eArchT3.0 (Fig. 1), and eMac3.0 [37, 38]. Compared to the proton pumps, eNpHR3.0 displayed slower activation kinetics, higher stability under prolonged illumination, and more red-shifted excitation spectrum making it more suitable for sustained inhibition and combination with most depolarizing ChR variants which are activated by blue light [38]. Nevertheless, because both BR and HR employ active ion transport, they are only able to pump one ion per photocycle. This inefficient mode of transport requires continuous and high-intensity illumination, limiting effective inhibition to a small region [39]. With the goal of improving inhibition efficiency, efforts have been made to convert the cation-conducting ChR variants into light-gated chloride channels, via either altering the channel pore [40] or modifying residues involved in its photocycle [41]. More recently, Govorunova et al. have identified two naturally occurring ChR homologs from Guillardia theta, GtACR1 and GtACR2, which strictly conduct anions, allowing membrane hyperpolarization with rapid kinetics and significantly higher efficiency than previous hyperpolarizing channels [42]. The exciting development of a non-opsin based, light-gated potassium channel was also recently described by Cosentino et al. [43]. The authors engineered a fusion construct of a blue-light activity domain of the plant protein LOV2-Jα with the small viral potassium channel, Kcv, creating a reversible, light-activated, hyperpolarizing potassium channel named BLINK1. Its relatively slow activation and inactivation kinetics (minutes) are highly suitable for experiments requiring prolonged hyperpolarization for behavioral loss-of-function studies. Overall, careful consideration of the diverse physiologic properties of both depolarizing and hyperpolarizing, light-activated ion channels and transporters [38] will be necessary for informed experimentation in various types of stem cells and their progeny.
Microbial opsins in pluripotent stem cells
The ability to non-invasively and selectively activate or inhibit cells and tissues at high temporal and spatial resolutions make microbial opsins suitable candidates for assessing functional integration between stem cell-derived tissue grafts and host tissue in situ or ex vivo. In acute brain slice preparations, optogenetic manipulation combined with electrophysiological recordings were utilized to reveal bi-directional functional integration between PSC-derived neuronal grafts and host neurons as well as extensive synaptic connectivity within the graft [44-46]. These ex vivo results were further corroborated in an in vivo murine Parkinson's disease model [47], whereby transplantation of hESC-derived mesencephalic dopaminergic neurons expressing eNpHR3.0 into lesioned striatum rescued Parkinsonian motor deficits 4-6 months after transplantation, while delivering 543-nm light via fiber optic cannula silenced graft activity and re-introduced motor deficits in awake animals. In addition to assessing functional integration of transplanted cells in CNS networks, microbial opsins have also been used to examine connectivity between PSC-derived neurons and skeletal or cardiac [48] myocytes in in vitro co-cultures, suggesting formation of functional neuro-muscular junctions. Importantly, in an in vivo murine model with sciatic nerve ligation, engrafted mESC-derived motor neurons expressing ChR2 successfully reinnervated the denervated muscle fibers and, upon illumination with 470-nm light pulses, triggered muscle contraction activity similar to uninjured animals [49]. The authors further suggested that combination of stem cells and optogenetics could provide potential therapies for loss of muscle function following injuries or neurodegenerative diseases. Similarly, for cardiac cell therapies, in vivo optogenetic activation and inhibition of implanted hPSC-derived cardiomyocytes in a feedback system could enable electrical synchronization of engrafted cells and host myocardium in the absence of gap junctional coupling to minimize risk of arrhythmia development [50].
Photoactuation for intracellular processes
Light mediated control of intracellular processes is an unprecedented development in the field of cell biology [51]. Successfully developing cell signaling photoactuators (examples shown in Table 1) first requires identification of the appropriate light activated protein chaperone for fusion to an activity domain of a signaling pathway or cellular function of interest. Several light activated protein motifs have been genetically modified to fit experimental need, each with unique protein component (1 v. 2 binding partners), chromophore requirement, and activation/inactivation wavelengths and time scales (Table 2) [15]. Currently available methods for photoactuation mostly involve photocaging that sterically inhibits an activity domain, or translocation that results in association or dissociation of a photoactuator upon light stimulation. Other photoactuators act through the ability to produce reactive oxygen species (ROS) [52] or directly affect second messenger molecules such as G-proteins [53].
Common Photoactuator Systems
| Photoactuating Protein | Chromophore | Genetic Cassettes | Act/Inact λ (nm) | Act/ Inact Time | Photoactuation Method | Refs |
|---|---|---|---|---|---|---|
| PhyB-PIF6 | PCB | 2 | 650/750 | millisec/millisec | translocation | [61] |
| CRY2-CIB1 | Flavin | 2 | 450/Dark | seconds/minutes | translocation | [63] |
| CRY2 olig | Flavin | 1 | 450/Dark | seconds/minutes | translocation | [107] |
| LOV | Flavin | 1 | 450/Dark | seconds/seconds | photocaging | [54-56] |
| LOVdimer | Flavin | 1-2 | 450/Dark | seconds/seconds | translocation | [57-59] |
| Dronpa | None | 1 | 400/500 | seconds/seconds | photocaging | [60] |
| UVR8 | None | 1 | Dark/280 | hours/seconds | translocation | [64, 65] |
Photocaging
One of the first genetically encoded photoactuator systems developed is the light, oxygen and voltage (LOV) domain. LOV domains are blue-light (400nm - 473nm) stimulated proteins, isolated from multiple plant, bacterial and fungal species, that have most typically been utilized for photocaging. In the unexposed state, LOV domains sterically inhibit fused activity domains which then become “uncaged” with illumination and are free to interact with desired targets [54-56]. Additional utility of this system has come in the form of heterodimerization to recruit desired signaling domains [57] and homodimerization to actuate DNA binding leading to transcription [58, 59]. Advantages of the LOV photocaging system are the use of a single genetic cassette, presence of an endogenous chromophore, Flavin, and the short activation timescale on the order of seconds with fast inactivation in dark conditions. The photoactuator protein Dronpa has also been used for photocaging [60] to create a photoswitch capable of turning kinase activity on and off with light stimulation of 500nm and 400nm, respectively. Potential drawbacks of photocaging modalities mostly lie in the difficulty of bioengineering steric hindrance to a diverse range of signaling molecules.
Translocation
A second, more widely applied, technique is translocation. In this system, two light activated proteins either associate or dissociate when illuminated with the appropriate wavelength of light. Several plant-derived photoactuator systems have been co-opted to achieve this goal. The phytochrome B (PhyB) system, derived from Arabidopsis thaliana, associates with phytochrome interacting factor (PIF) proteins on red light (650nm) stimulation and disassociates on infrared light (750nm) stimulation [61, 62]. The power of this system lies in the ability to rapidly (seconds) photoswitch between associated and dissociated states via light stimulation. The drawback of this system is the need for exogenous production of the chromophore phycocyanobilin (PCB) in non-photosynthetic organisms which necessitates introduction of a three-component system in mammalian cells. Another A. thaliana derived system is cryptochrome 2 (CRY2) which both homo-oligomerizes with itself and heterodimerizes to its cryptochrome-interacting basic helix-loop-helix (CIB1) binding partner when stimulated by blue light (405nm-488nm) [63]. The light mediated association occurs within seconds and dissociation occurs after five minutes of dark conditions. A significant feature of CRY2's ability to homo-oligomerize is the potential to eliminate the need for a two-component genetic system since aggregated, homo-oligomerized fusion proteins may independently activate target signaling pathways. In addition, CRY2 utilizes the chromophore, Flavin, found endogenously in mammalian cells. While PhyB-PIF and CRY2-CIB1 actively associate on light stimulation, both UV-resistance locus 8 (UVR8) [64, 65] and Dronpa [66] systems perform translocation functions through dissociation on UV (280nm-315nm) and cyan/green (490nm) spectrum illumination, respectively. UVR8 and Dronpa utilize tryptophan residues as chromophores which allows their application in multiple systems since a separate chromophore is not necessary for activation. Dronpa also re-associates with violet-light stimulation (390nm) allowing for rapid photoswitching in both translocation and photocaging methods.
Photoactuated cell signaling pathways
Pluripotent stem cell applications
The cell signaling pathways involved in the maintenance of pluripotency, self-renewal, proliferation and differentiation have been extensively studied and expertly reviewed [67]. Nevertheless, our understanding of the complexity and dynamism responsible for maintaining the pluripotent state remains underdeveloped. Most of our knowledge arises from the shift to chemically defined cell-culture media from mouse embryonic fibroblast (MEF)-conditioned media, MEF feeder cells and serum media additives. Chief among the pathways regulating pluripotency are stimulation of MAPK/ERK via FGF2 and PI3K/AKT through insulin/IGF. The complex cross-talk between these pathways is clear, but expanded comprehension has been limited by inability to investigate the incident pathways at the single-cell and subcellular level. Thus, the potential application of genetically encoded photoactuators to modulate pluripotent cell signaling pathways is high. Below, we highlight several of existing photoactuators targeted to specific pathways, use of which would be likely to increase our understanding of dynamic pluripotent cell-signaling networks.
MAPK/ERK
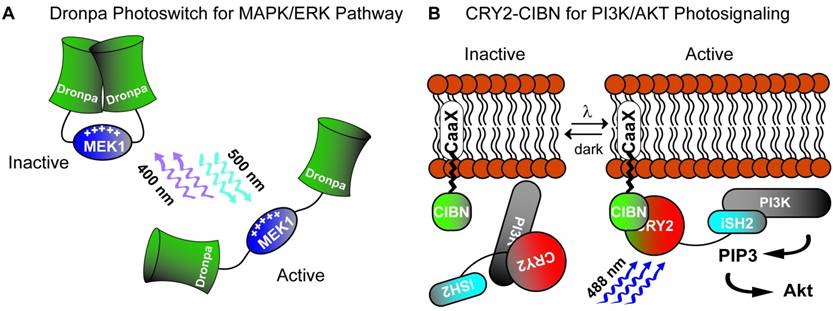
Mitogen-activated protein kinase (MAPK) pathway activation is involved in diverse cell-signaling functions including cell proliferation, differentiation, survival and apoptosis. Several groups have utilized photoactuated domains to successfully recruit components of MAPK modules and activate downstream signaling pathways [60, 68-70]. Given that mammalian cells produce the endogenous chromophore Flavin, the use of the CRY2-CIB1 translocation system has great practicality in human pluripotent stem cells. For example, CIB1 can be anchored to the plasma membrane to recruit the CRY2-Raf1 fusion gene upon blue light stimulation [68]. This results in the subcellular compartmentalization of effect by recruiting a MAPK effector, Raf1, to the cytosolic membrane surface and improving its association with endogenous effectors in the Ras/Raf/MEK/ERK pathway. The final result of this photosignaling system was the induction of neurite outgrowth in PC12 cells [68]. Another example is the use of CRY2 homo-oligomerization to create a single genetic photoactuator cassette containing the CRY2-Raf1 fusion product. Blue light stimulation in this system promotes cytosolic aggregations sufficient to activate downstream Raf/MEK/ERK [69]. Although of likely diminished utility in mammalian systems owing to the need for the exogenous chromophore PCB, Toettcher et al [70] demonstrated use of PhyB-PIF6 translocation by anchoring PhyB to the plasma membrane with recruitment of a PIF6-SOS fusion product resulting in Ras activation on red light stimulation. The most promising part of this assay was the tandem, live-cell assay for downstream ERK activation demonstrating single-cell dose response relationship, a key to effective interrogation of pluripotency signaling networks. A more recent study reported the creation of photoswitch kinases (psRaf1, psMEK1 and psMEK2) through novel engineering of the Dronpa system from a tetrameric to a dimeric photo-dissociable (pdDronpa) system [60]. Two pdDronpa proteins are fused on opposite ends of the target kinase which is inactive with pdDronpa dimerization at baseline (Fig. 2A). On illumination with cyan-light (500nm), the pdDronpa domains dissociate, lose fluorescence and open the fused kinase activity domain to effector binding. Illumination with violet-light (400nM) restores dimerization and fluorescence to pdDronpa, and inactivates the fused kinase motif completing the photoswitch cycle.
Two strategies for photoactivation of cell signaling pathways. A) A Dronpa dimer photoswitch which uncages the MEK1 activity domain on cyan light (500 nm) and photocages MEK1 on violet light (400 nM). B) Utilization of the CRY2-CIBN blue light (488 nm) stimulated photoactuator pair to recruit iSH2 fused CRY2 to membrane anchored CIBN to activate the PI3K pathway.
PI3K/AKT
The phosphoinositide 3-kinase (PI3K)-Akt pathway transmits cell-surface stimuli through a diverse set of receptors to regulate several biologic functions such as cell growth, survival, migration and cell-cycle progression [71, 72]. Pluripotent stem cell culture media often utilizes insulin and/or IGF supplementation for maintenance of pluripotency and self-renewal. Activation of insulin/IGF receptors stimulates the PI3K/AKT pathway which has been hypothesized to stabilize ERK dependent pathways downstream of MAPK [67]. Alternatively, suppression of PI3K/AKT supports early differentiation as shown in a study demonstrating Activin-a stimulation leading to definitive endodermal specification [73] when PI3K is suppressed. This complex, dynamic cross-talk between two major cell-signaling pathways requires more defined resolution which has not been afforded by current knockdown, mutational or chemical studies [74]. To this end, many groups have employed genetically encoded photoactuators to probe the PI3K/AKT pathway in non-pluripotent stem cell systems. Most have used membrane PI3K recruitment strategies to facilitate production of PIP3 such as PhyB-PIF6 red-light induced translocation [75] of the iSH2 domain of the p85β regulatory subunit of PI3K or blue-light induced translocation through CRY2-CIBN [76]. Alternatively, deactivation of the PI3K pathway has been achieved by dephosphorylating PIP3 [77]. In this study, the inositol 5-phosphatase domain of OCRL (5-ptase(OCRL)) was fused to CRY2 and translocated to membrane bound CIBN on blue-light stimulation. More recently, Xu et al [78] created a fusion CRY2-iSH2 protein, iSH2 recruiting PI3K p110α catalytic subunits, which translocated to a membrane anchored n-terminal CIB1 fusion protein (CaaX-CIBN) on blue light stimulation (Fig. 2B). Effective PI3K pathway photoactivation was confirmed using a downstream Akt reporter. Importantly, the authors demonstrated similar Akt activation in response to both light and insulin exposure signifying physiologic activation of the pathway via light. They further demonstrated localization of a downstream PI3K/AKT effect through a CRY2-Akt2 fusion protein which led to localized exocytosis when translocated to membrane anchored CaaX-CIBN on blue light stimulation. Such utility, on a single-cell and subcellular level, is exactly the type of tool necessary to decipher cell-signaling networks involved in pluripotent cells.
Rho GTPase
Rho GTPases are small signaling G-proteins existing as a subfamily of the Ras superfamily. Primarily active in regulating actin dynamics, signaling through Rho GTPases has downstream effects on cell migration, cell polarity and organelle development [79]. For example, hematopoietic stem cells are migratory by nature and dependent on Rho GTPase activity for their cell motility [80] which makes this signaling pathway an attractive target to study in this system. Genetically encoded photoactuator systems to regulate the Rho GTPases Rac1, Cdc42 and RhoA were first engineered by Levskaya et al [61]. The authors fused PIF motifs to specific guanine nucleotide exchange factors (PIF-GEFs) which, upon red-light stimulation, translocated to membrane bound PhyB leading to formation of lamellipodia and filopodia in NIH3T3 cells. Published nearly simultaneous with the above study, Yazawa et al [55] reported similar success in Rac1 membrane recruitment and activation using another A. thaliana derived blue-light stimulated heterodimerizing pair of proteins, FKF1 and GIGANTEA. The large size of these photactuated proteins, however, have limited their use. Nevertheless, these proof-of-concept studies led to further photoactuated translocation methods using a LOVpep and ePDZ binding pair where Rac1 was fused to a LOV2-Jα sequence (LOV2-Jα-Rac1) and translocated to cell membrane bound ePDZ domains on blue-light stimulation [57]. The authors described this as a powerful system given small size of the LOVpep, endogenous Flavin chromophore and fast photoswitching kinetics. The LOV domain was also used in a photocaging technique where a LOV-Jα-Rac1 fusion protein led to Rac1 steric inhibition in the dark, but activation of Rac1 on blue-light stimulation, leading to increased motility of mouse embryonic fibroblasts [54].
Receptor Tyrosine Kinase / GPCR Signaling
The above cell signaling pathways are largely activated by transmembrane proteins who function to transduce external cellular signals to internal signaling pathways. Receptor Tyrosine Kinases (RTK) are one class of transmembrane protein that phosphorylate initial signal transduction proteins after ligand binding. Ligand-gated activation of RTKs stimulates downstream pathways such as Ras/Raf/MEK/ERK, PI3K/AKT and Phospholipase C. Another class of receptors, G-Protein Coupled Receptors (GPCRs), activate intracellular associated guanine nucleotide binding proteins (G proteins) in response to ligand binding resulting in diverse downstream signaling effects. The important roles of RTK [81] and GPCR [82, 83] activation in pluripotent stem cells are well documented, which makes these molecules an attractive target for the application of genetically encoded photoactuators. The RTK receptor FGFR1 became a photoactuator (Opto-FGFR1) by utilizing a CRY2 homo-oligomerization translocation strategy. To create Opto-FGFR1, a membrane targeting sequence and the c-terminal catalytic domain of FGFR1 were fused to opposite ends of CRY2 [84]. Subsequent blue-light stimulation of Opto-FGFR1 modified cell polarity and induced directed cell migration. A LOV homodimerization strategy was also employed with FGFR1, EGFR and RET receptors (Opto-RTKs) leading to blue-light stimulation of downstream ERK, AKT, and PLCγ pathways [85]. Here, the c-terminal activity domains of RTKs were fused to LOV peptides to achieve signal transduction by promoting stability of RTK activity domain on blue-light stimulation. Given that FGF2 supplementation is crucial for self-renewal and maintenance of pluripotency in several culture systems, the use of Opto-FGFRs could help elucidate the dynamics of this signal transduction pathway in pluripotent stem cells.
Like Opto-RTKs, work to establish a photoactivated set of GPCRs is a burgeoning field [86]. Specifically, Opto-α1AR and Opto-β2AR adrenergic receptors [53] termed Opto-XRs were formed by engineering a bovine G(t)-protein coupled rhodopsin stimulated by green light (504nm) by replacing its intracellular loops with those of the Gq-coupled α1a-adrenergic receptor (α1AR) or Gs-coupled β2-adrenergic receptor (β2AR). Green-light stimulation of Opto-α1AR resulted in Gq activation and increase in intracellular calcium. For the Opto-β2AR system, co-transfection of the cAMP-gated calcium channel CNGA2-C460W/E583M was necessary to translate the increase in cAMP after Gs activation to an increased cytosolic calcium signal. Inhibition of GPCR signaling has also been accomplished through a CRY2-CIBN translocation system. Translocation of a CRY2 fused, truncated RGS4 subunit (CRY2-RGS4Δ) deactivated Gαi/βγ subunits through GTP hydrolysis when recruited to a cell-membrane bound CIBN-CaaX on blue light (445nm) stimulation [87]. The authors of this study concluded that a gradient of active G-protein subunits generates directed cell migration and that Gi signaling is crucial for cell directional sensing and adaptation.
Apoptosis
While apoptosis is not classically associated with maintenance of pluripotent stem cells, its role in development and morphogenesis through programmed cell death is clear [88]. Recent studies have also demonstrated that incomplete activation of apoptotic caspases may produce mitogenic signals stimulating stem/progenitor cells in tissue regeneration in a process deemed “compensatory proliferation” [89]. Accordingly, photoactivation of selected caspases may provide a unique tool for the interrogation of apoptosis during development, tissue morphogenesis and regeneration. Recently, the LOV2 domain was fused to the apoptosis-executing domain of caspase-7 [90]. Blue light stimulation initiated steric disinhibition of caspase-7 leading to cell death within one hour of constant illumination. Interestingly, illumination times less than 5 minutes or oscillating one-minute exposures did not induce cell death. Optogenetic control of Bax induced caspase-3 cleavage has also been demonstrated through CRY2-CIB translocation of a CRY2-Bax fusion protein to a CIB-Tom20 fusion protein anchored in the outer mitochondrial membrane [91]. Subsequent blue-light stimulation induced apoptosis in HeLa cells. Another interesting method is the use of the fluorescent protein, KillerRed (KR), which produces ROS with green light (545nm) illumination [52]. Introduction of KR into Xenopus embryos allowed for induction of ROS-mediated apoptosis in target tissues during development, a feat which has wide utility in studying embryogenesis and morphogenesis through targeted cell ablation.
Photoactivated DNA transcription and editing
Genome engineering is a rapidly expanding field that has seen recognition in both scientific and public circles. Methods for design of zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regulatory interspaced short palindromic repeat (CRISPR)/Cas-based RNA guided DNA endonucleases [92] and their applications in fundamental biology [93] and, potentially, translational medicine [94] have been extensively reviewed. The use of gene editing in iPSCs was the subject of a recent review by Hockemeyer and Jaenisch [95] who concluded that much work remains to optimize the system, not withstanding the inherent ethical considerations. With their tremendous potential, it was inevitable that genome engineering technologies and optogenetics would be combined. Below we detail some of the earliest efforts in this pioneering field.
DNA transcription
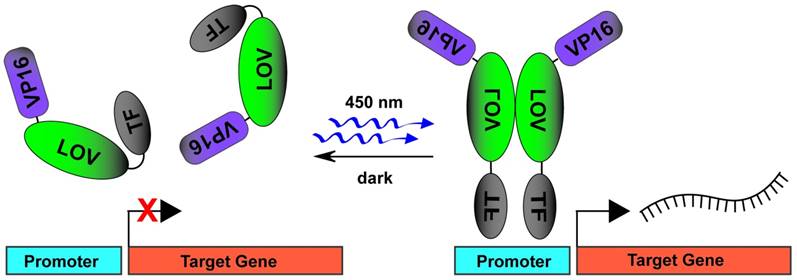
A plethora of technologies for light mediated recruitment of transcription factors to promote or inhibit DNA transcription exist in practice [96]. One of the earliest techniques utilized the PhyB-PIF system whereby GAL4 DNA binding domain was fused to PhyB (PhyB-GBD) and GAL4 activation domains were fused to PIF3 (PIF3-GAD) [62]. Upon red light stimulation, expression of marker genes modulated by GAL4 DNA binding sites increased with the ability to photoswitch to repression with infrared light illumination. The need for an exogenous chromophore (PCB) in this system, prompted the further development of multiple light mediated DNA transcription systems utilizing CRY2-CIB1 [97, 98] and LOV2 [55, 59, 99] domains (Fig. 3A). The first use of photactivated zinc finger proteins (ZFPs) to promote DNA transcription used a ZFP fused to a GI light binding partner protein (ZFP-GI) which then heterodimerized on blue light stimulation with the VSP16 transcriptional activation domain fused to a LOV domain (LOV-VSP16) [99]. This genetically encoded photoactuator system was called light-inducible transcription using engineered zinc finger proteins (LITEZ). CRY2-CIB1 system was further combined with the CRISPR/Cas9 [100] to allow light-induced gene activation by fusing full-length CRY2 to the VP64 transactivation domain (CRY2FL-VP64). Blue light stimulation then resulted in translocation of a dual fused CIBN to dCas9 protein (CIBN-dCas9-CIBN), which together with a sequence specific gRNA, induced transcription of target genes. The ability to rapidly target this system to multiple DNA areas through sequence specific gRNAs combined with precision control of light activation makes this an extremely attractive approach for use in pluripotent stem cells.
DNA transcription induced by blue light stimulated TF uncaging and dimerization of LOV domains. In dark conditions, the fusion protein of VP16-LOV-TF exists in a monomeric state. The TF activator domain is photocaged by an inactive LOV domain. On blue light stimulation, the TF activator domain is uncaged and the LOV domains dimerize to allow for VP16 mediated recruitment of transcription machinery.
Genome editing
Given the utility of TALEN and CRISPR/Cas9 technologies, many groups are evolving photoactivated, epigenetic and DNA modification systems. The first report of effective light-mediated histone modification came from Konermann et al [101]. In this study the authors modified LITEZ to use the CRY2-CIB1 instead of LOV system in order to recruit histone effector domains to site-specific epigentic loci (open heterochromatin) thus inducing transcriptional repression of Grm2 in neurons. Following this work, Nihongaki et al [102] introduced a split Cas9 system that was fused to a novel light induced translocation (dimerization) system called Magnets which associates on blue-light illumination. The Magnets split-Cas9 system effectively induced indel mutations through non-homologous end joining and site specific genome modification through homology directed repair mechanisms. Interestingly, the authors originally used the CRY2-CIB1 system, but were unable to demonstrate light-induced Cas9 activity. They hypothesized that steric hindrance may have precluded interaction of the two split Cas9 halves or that homo-oligomerization of CRY2 may have decreased heterodimerization with CIB1 and effective reunion of the split Cas9. Regardless, the above techniques clearly demonstrate the broad potential of genetically encoded photactuators for modulation of the cellular transcriptome of pluripotent cells and their differentiated progeny.
Despite the strong promise of light-activated DNA transcription and editing technologies, low efficiency of these systems will require further improvements. While use of CRISPR/Cas9 technology and VP64 transactivation domains [100] may pave the way for more efficient photoactivatable transcription, efficient transgene delivery in primary cells is still challenging. The incomplete penetrance of genome editing due to inherent limits of DNA recombination, and limited light penetration in 3D cell cultures and native tissues are additional obstacles to achieving high efficiency in these systems.
Other photoactuation targets
Individual proteins not involved in classic cell signaling pathways are additional targets for genetically encoded photoactuators. Strategies include recruitment of intein/extein protein self-cleavage domains [103], degron cued proteasomal degradation [104, 105], creation of protein traps through light-stimulated “trap” protein aggregation [106], and promotion of clathrin-mediated endocytosis [107]. Intracellular protein trafficking [65], organelle transport [108], actin dynamics [109] and cell polarity [110] have also been successfully manipulated using genetically encoded photoactuators. While none of the above mechanisms have been reported directly for the modulation of pluripotent stem cell self-renewal and differentiation, it is likely that these vital processes will be the targets of future studies.
Photosensors
Photosensors are fluorescent molecules or proteins that, in response to specific cellular processes, exhibit changes in optical characteristics which can be monitored in real time. Genetically encoded photosensors, analogous to photoactuators, allow use of tailored genetic cassettes to target specific cell populations or subcellular compartments. Previously developed sensing probes, such as calcium sensitive dyes, have been highly effective, but do not allow precise cell targeting. Moreover, chemical dyes are often toxic to cells thus compelling terminal evaluation. Genetically encoded photosensors, conversely, have improved our ability to sense physiologic, live-cell processes in multiple cell types spanning the spectrum from pluripotency to fully differentiated states. The following text details the available classes of genetically encoded photosensors and many of their current uses (Table 3).
Selected Genetically Encoded Photosensors
| Signaling Sensors | Pathway | FRET Pair | Ex/Em λ (nm) | Function | Ref |
|---|---|---|---|---|---|
| Rac | Rho GTPase | GFP/Alexa546 | 480/568 | visualize Rac | [113] |
| RhoA | Rho GTPas | CFP/YFP | 436/535 | visualize RhoA | [115] |
| Ras | MAPK/ERK | CFP/YFP | 433/527 | visualize Ras/Raf | [114] |
| ERK | MAPK/ERK | EGFP/mRFP1 | 910(2p)/635 | visualize ERK | [116] |
| AKT | PI3K/AKT | CFP/YFP | 440/535 | visualize AKT | [117] |
| GECIs | Sensor | Reporter | Ex/Em λ (nm) | Feature | |
| Cameleon | CaM-M13 | EYFP/ECFP | 440/480(535) | First GECI | [118] |
| TN-XXL | Troponin C | CFP/cpCitrine | 505/475(527) | Ratiometric | [121] |
| GCaMP6 | CaM-M13 | cpEGFP | 485/515 | High ΔF/F | [127] |
| jRGECO1a | CaM-M13 | mApple | 570/600 | Red-shifted | [129] |
| GEVIs | Sensor | Reporter | Ex/Em λ (nm) | Feature | |
| VSFP2.3 | Ci-VSD | CFP/YFP | 440/470(530) | Ratiometric | [138] |
| ArcLight | Ci-VSD | pHluorin | 485/515 | High ΔF/F in vitro | [139] |
| ASAP1 | Gg-VSD | cpGFP | 485/515 | Fast kinetics | [140] |
| Ace2N-mNeon | Ace2N | Ace/mNeonGreen | 500/520 | High ΔF/F in vivo | [147] |
| FlicR1 | Ci-VSD | cpmApple | 570/597 | Red-shifted | [142] |
| Tension Sensors | Tension Element | FRET Pair | Ex/Em λ (nm) | Function | |
| Vinculin | flagelliform | mTFP1/Venus | 462/528 | focal adhesions | [172] |
| Talin | villin | YPet/mCherry | 508/610-710 | focal adhesions | [173] |
| Actinin/Spectrin/Filamin | α-helical | Cerulean/Venus | 433/527 | linker validation | [174] |
| Actinin | spectrin | Cerulean/Venus | 433/527 | stress sensor | [175] |
| E-Cadherin | flagelliform | mTFP/Venus | 458/528 | cadherin force | [177] |
| Integrin | flagelliform | mTFP/Venus | 458/528 | integrin force | [179] |
| Epigenome Sensors | Mechanism | FRET Pair | Ex/Em λ (nm) | Function | |
| HP1α | heterochromatin binding | CFP/PA-GFP//YFP | 405/488//535 | transcription | [186] |
| Histone | methyltransferase | CFP/YFP | 433/527 | methylation | [187] |
Basics of Förster Resonance Energy Transfer (FRET)
Most genetically-encoded photosensors are based on a phenomenon termed Förster Resonance Energy transfer (FRET), which involves non-radiative transfer of energy between an excited donor fluorophore and an adjacent acceptor fluorophore. Specifically, FRET results in the quenching of donor fluorescence and the sensitized emission of the acceptor fluorophore in response to donor excitation light. The amount of FRET that occurs is determined by the optical properties, separation distance, relative orientation, and mobility of the fluorophores. It is commonly assumed that the fluorophores diffuse randomly and have no preferred orientation [111, 112]. In this limit, FRET Efficiency (E) can be well-described with the simple equation:
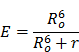
where Ro, the Förster distance, is defined as the fluorophore separation distance where 50% FRET efficiency is achieved and r is the separation distance of the two fluorophores. Virtually all genetically encoded FRET-based photosensors function on the principle that a biologically interesting event, such as the presence of calcium, induces a conformation change in the sensing construct that alters the separation distance of the two fluorophores, resulting in a readily measurable change in FRET.
Cell signaling photosensors
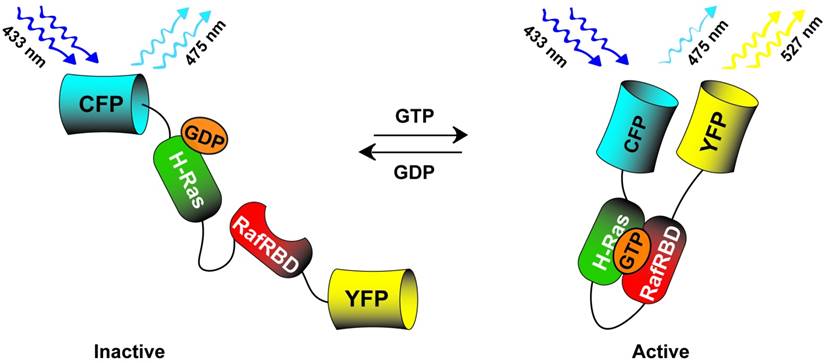
Many of the cell signaling pathways involved in pluripotency, as discussed in the Photoactuator section, were targets for the early genetically encoded photosensors. Kraynov et al [113] created a two piece FRET system which visualized activation of the GTPase Rac at the leading edge of motile cells. In this system, Rac fused to GFP (Rac-GFP) was transiently expressed as a FRET donor while an activated Rac-GTP p21 binding domain (PBD), covalently labeled with Alexa546 as a FRET acceptor, was microinjected into the cell cytoplasm. This study was closely followed by Mochizuki et al [114] who probed the dynamics of epidermal growth factor mediated Ras activation through a novel, single-cassette genetically encoded photosensor. This system consisted of H-Ras fused to the Ras-binding domain of Raf (Raf RBD), both of which were flanked by the FRET pair of CFP/YFP (YFP-HRas-RafRBD-CFP). When activated Ras binds GTP, its higher affinity for Raf RBD brings the normally diametrically opposed CFP/YFP pair into a FRET participating position (schematic in Fig. 4). Similarly, Pertz et al [115] published a study detailing the dynamics of RhoA activity in migrating cells. The authors fused the Rho binding domain (RBD) of rhotekin, which has binding affinity for active GTP-RhoA, to a cyan and yellow FRET pair followed by a full length RhoA (RBD-CFP-YFP-RhoA). Active GTP-RhoA binds to the neighboring, fused RBD bringing the CFP/YFP fluorophores in closer FRET proximity. Using this tool, the authors demonstrated that RhoA is active in a distinct band at the edge of cell protrusion along with additional subcellular compartments. Similar, single-cassette fusion protein FRET systems were also utilized with downstream effectors of MAPK and PI3K signaling such as ERK [116] and AKT [117], to detail activation kinetics and subcellular compartmentalization upon growth factor stimulation. The above genetically encoded cell signaling photosensors, used in concert with their photoactuating counterparts, hold immense potential for molecular scale investigation of signal pathway dynamics by combining the ability to directly activate and then monitor specific activity states of a signaling network.
CFP-HRas-RafBD-YFP single cassette Ras photosensor. The sensor detects active GTP bound Ras by associating with the fused RafRBD thus increasing the FRET in the CFP-YFP FRET pair. When phosphatases deactivate the pathway, GDP bound Ras does not associate with the fused RafRBD leading to decreased FRET signal.
Genetically encoded calcium indicators
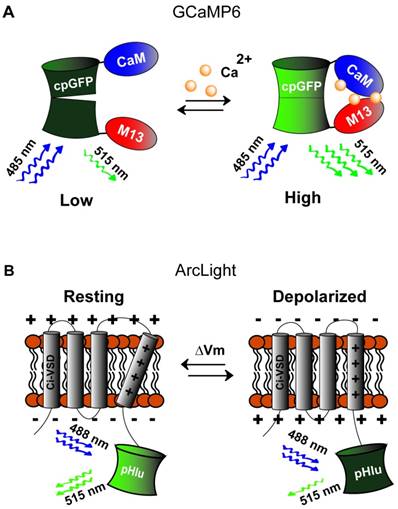
The first engineered genetically-encoded calcium indicator (GECI) was Cameleon [118], which contains M13 and calmodulin (CaM) domains fused between EYFP and ECFP. Binding of calcium induces the formation of the CaM-M13 complex, bringing the two FPs closer together and enhancing the FRET efficiency between them. This design was preserved in subsequent improved variants, most notably in the YC-Nano series [119]. Alternatively, the M13 and CaM domains could be replaced by the calcium-binding protein Troponin C, which was demonstrated in the TN-XL [120], TN-XXL [121], and the more recent Twitch indicators [122]. Moreover, improvement in signal response amplitude was achieved using single-FP GECIs, including the Camgaroo [123], Case [124], Pericam [125], and GCaMP [126] (Fig. 5A) families. These constructs consist of M13 and CaM domains fused to EYFP or circularly-permuted GFP. The most popular GECIs currently are those from the GCaMP6 series, with GCaMP6f exhibiting fast decay kinetics while GCaMP6s and GCaMP6m displaying larger response amplitudes [127]. Also of particular interest is the development of red-shifted GECIs, which could facilitate deep-tissue in vivo studies and multi-color experiments involving GFP-based indicators or optogenetic actuators [18, 128, 129]. Recent examples of red GECIs include jRGECO1a based on mApple and jRGCaMP1a,b based on mRuby, showing comparable sensitivity to GCaMP6 and better tissue penetration [129].
Genetically encoded calcium and voltage indicators. A) Green (515 nm) fluorescence intensity of calcium sensor, GCaMP (activated by blue light, 488 nm), is increased when calcium levels increase causing association of CaM and M13 thus restoring tertiary structure of the GFP fluorophore. B) Voltage sensor, ArcLight, is composed of the C-terminal voltage sensing domain of the Kv2.1 channel (CI-VSD) fused to super ecliptic pHluorin reporter activated by blue (485 nm) light. Depolarization causes shifting of the voltage sensing domain to decrease green (515 nm) fluorescence.
Genetically encoded voltage indicators
The earliest examples of genetically-encoded voltage indicators (GEVI) involved fusion of GFP to the C-terminus of a voltage-gated ion channel. This was first demonstrated in the development of FlaSh using a non-conducting mutant of the Shaker Kv channel [130]. In order to improve upon the slow kinetics of FlaSh, another GEVI construct (SPARC) was created by inserting GFP at the DII-DIII loop of rat Nav1.4 channel [131]. As voltage-gated ion channels consist of voltage-sensitive (S1-S4 segments) and pore-forming (S5-S6 segments) domains, the inclusion of S1-S4 transmembrane segments alone was proposed to be sufficient to confer voltage-sensing ability in GEVI. This was demonstrated in VSFP1, the first generation of the voltage-sensitive fluorescent protein (VSFP) family, which consists of a FRET pair of cyan and yellow reporters fused to the C terminus of the voltage-sensing domain (VSD) of Kv2.1 channel [132]. Despite promising proof-of-concept results in Xenopus oocytes, early GEVI constructs yielded very poor membrane targeting and significant intracellular accumulation in mammalian cells [133].
The discovery of voltage-sensing phosphatase from Ciona intestinalis (Ci-VSP) provided the first evidence of VSD outside of voltage-gated ion channel families [134]. This led to development of second-generation VSFP2 constructs, showing drastically improved membrane trafficking in mammalian cells [135, 136]. Further enhancements in VSFP2 performance were achieved by flanking Ci-VSD by the FRET donor and acceptor FPs in the VSFP Butterfly family [137]. While the use of FRET pair of FPs enables ratiometric assessment of fluorescence signals for improved readout and reduced motion artifacts, many GEVI families have been developed using single FP reporter, including third-generation VSFP3 [138], ArcLight [139], ASAP [140], ElectricPk [141], and FlicR1 [142]. In particular, ArcLight constructs comprise the super-ecliptic pHluorin reporter attached to the C-terminus of Ci-VSD (schematic in Fig. 5B). Similarly, ElectricPk and FlicR1 were constructed via C-terminal fusion of this voltage-sensing domain to circularly-permuted fluorescent protein (cpFP). Derived from a red fluorescent protein, FlicR1 exhibits red-shifted spectrum compared to GFP-based indicators, making it a more suitable candidate for deep tissue imaging or all-optical experiments involving blue light-activated microbial opsins [142]. Interestingly, in ASAP, cpFP is inserted into the S3-S4 loop of the VSD from Gallus gallus voltage-sensing phosphatase (Gg-VSP), making it the only GEVI family bearing FP located on the extracellular side [140].
Furthermore, another class of GEVI does not involve VSD but rather relies on voltage-sensitive proton transfer of the retinal chromophore in microbial opsins. These constructs, including the original Arch [143] and improved versions QuasAr1 and QuasAr2 [144], have fast photoactivation kinetics and robust resistance to photobleaching. However, fluorescence signals from opsin-based GEVIs are weak due to the low quantum yields of retinal chromophore, precluding potential in vivo applications [143, 145]. This limitation was addressed by the fusion of FP to the opsin-based GEVI to form a FRET pair [146, 147]. These fused opsin constructs showed significantly improved fluorescence dynamic range and fast activation kinetics, with the most recent Ace2N-mNeon indicator capable of reporting single action potential (APs) in live mammals [147].
Photoindicators of calcium and voltage for pluripotent cell biology
The ability of GEVI and GECI to enable visual assessment of action potential characteristics and calcium transients make them promising candidates for high-throughput, high-resolution, and long-term functional phenotyping of pluripotent stem cell-derived cardiomyocytes (PSC-CMs) and neurons. Transduction of human PSC-CMs with ArcLight A242 lentivirus allowed rapid and simultaneous AP recordings in differentiating cardiomyocyte populations, displaying distinct cardiac subtypes (ventricular, atrial, and nodal) [148]. Furthermore, ArcLight optical recordings also confirmed pro-arrhythmic APD prolongation and development of early afterdepolarizations (EAD) due to application of pharmaceutical agents or in hiPSC-CMs derived from a patient with the long QT syndrome type 2 (LQT2) [149]. Expression of GCaMP5G, on the other hand, was used to evaluate impaired calcium-handling properties following application of ouabain, or in hiPSC-CMs bearing the catecholaminergic polymorphic ventricular tachycardia type 2 (CPVT-2) phenotype [149]. Using co-expression of ArcLight and the red-shifted R-GECO1, Song et al. demonstrated simultaneous voltage and intracellular calcium imaging to reveal both prolonged APD and irregular calcium transients in iPSC-CMs derived from patients with Timothy Syndrome [150]. Furthermore, use of CM subtype-specific promoters to express VSFP-CR allowed distinct drug tests in ventricular, atrial, and nodal cells derived from healthy and LQT1 individuals [151]. Recently, combined use of virally expressed photoactuators (ChR2) and voltage and calcium-sensitive dyes allowed the development of an automated all-optical electrophysiology for drug testing in hPSC-CMs [152]. The non-invasive and high-throughput nature of these approaches could also facilitate development of methods to improve the efficiency of stem cell differentiation (or direct cardiac reprogramming from fibroblasts [153]) towards cardiac lineage and screening of factors to enhance maturation and homogeneity of derived populations for potential cardiac cell therapies. Of note, GCaMP3 and GCaMP7 expression in human and Macaca PSC-CMs has also been utilized to confirm electrical coupling between transplanted hESC-CM grafts and host cardiomyocytes [154-156].
Tension photosensors
The mechanical nature of the cellular microenvironment is now recognized as a key determinant of many developmental, physiological, and pathophysiological processes [157-159] involving stem cells, as well as an important variable in tissue engineering and regenerative medicine [160-162]. In these scenarios, cells adhere to a deformable extracellular matrix (ECM) that is both a source of applied forces and a means of mechanical support [163]. Cells detect, interpret, and respond to these mechanical signals through a poorly understood process called mechanotransduction [164, 165]. Mechanosensitive signaling affects stem cell differentiation [166], and efforts to control the physical nature of tissue engineering scaffolds are often aimed at manipulating the behavior of stem cells and their progeny.
Mechanotransduction in general represents the conversion of mechanical to biochemical signals [164], which is often mediated through the deformation of key proteins and the exposure of their cryptic binding sites or kinase substrate domains [167, 168]. To probe when and where these types of events occur, a variety of approaches for studying mechanotransduction at the molecular scale have been developed [169]. Of particular relevance are the FRET-based sensors designed to report the mechanical loads experienced by specific proteins in living cells [170, 171]. These are designed in direct analogy to photosensors described above, where the separation of fluorophores is induced by a biological interesting event, in this case the application of mechanical load.
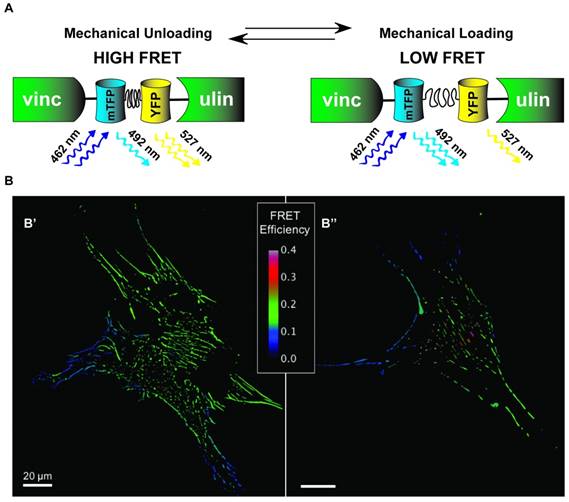
Although there are several existing designs for genetically-encoded FRET-based tension sensors, most use extensible domains placed between two fluorophores [172-174]. While the actual situation is more complex, the general principle can be understood from Hooke's Law, F=kx. As force (F) is applied to the sensor, the deformable domain will extend, increasing the separation distance of the fluorophores (x), and reducing the amount of FRET. If the mechanical properties of the extensible domain are known, represented by the spring constant k in this simplified example, then the magnitude of the force, also referred to as the tension, can be inferred. Extensible domains used to date include alpha-helical domains [174], spectrin-repeat domains [175], domains based on a component (flagelliform) of spider silk [172], and the villin head piece [173]. Notably, the flagelliform- and villin-based tension sensors enable particularly quantitative measurements as single-molecule biophysical techniques were used to calibrate their force sensitivity. Furthermore, there are several versions of these sensors that allow measurement of forces spanning a range from 1 - 10 pN [173, 176]. For reference, a single myosin motor generates in the range of 2-7 pN per stroke. To date, tension sensitive biosensors based on extensible domains have been developed for actinin [174], spectrin [174], filamin [174], vinculin [172] (schematic in Fig. 6), E-Cadherin [177], VE-Cadherin [178], PE-CAM, actin, talin [173], integrin [179], and alpha-catenin [180]. This diverse assortment of proteins enables characterization of the tension experiences by critical mechanical linker proteins in a variety of sub-cellular structures and should enable a wide variety of studies into the molecular mechanisms used by stem cells to detect and respond to mechanical cues.
Distribution of vinculin tension in Mesenchymal Stem Cells. A) Schematic of VinTS where mTFP1 and YFP FRET pairs are linked by flagelliform tension sensing element and inserted inside vinculin. As vinculin experiences increasing tension causing separation of mTFP1-flagelliform-YFP, the force environment is inferred by changes in FRET signal intensity. B) Human mesenchymal stem cells (hMSCs) transduced to stably express a vinculin FRET-based tension sensor (VinTS); B' and B'' represent two individual MSCs. FRET efficiency (shown) is inversely related to load across vinculin, a tension-sensitive protein that plays a critical role in mediating cell-matrix and cell-cell adhesion.
Epigenomic photosensors
Pluripotent cells exist in an epigenetically disinhibited state [181, 182]. The process of reprogramming a differentiated cell to an iPSC is likely driven through the removal of epigenetic imprinting [183, 184] with many iPSCs demonstrating epigenetic remnants of their former differentiated self [185]. Developing genetically encoded photosensors to study proteins involved in maintenance of the epigenome is thus an exciting field of study in pluripotent stem cell biology. One of the first forays into production of epigenomic photosensors was a study by Demarco et al [186] which evaluated the heterochromatin dynamics of mouse pituitary cells by using a YFP fused heterochromatin-1 alpha (YFP-HP1α) as a FRET acceptor for the donor CFP fused transcription factor CCAAT/enhancer binding protein alpha (CFP-C/EBPα) and demonstrated nuclear co-localization. They also show the use of a photoactivated GFP (PA-GFP) which switches from a dim to brightly fluorescent state on brief, intense 400nm light stimulation. By targeting the light to discrete areas of the cell nucleus, the authors tracked the diffusion/mobility of PA-GFP-HP1α to the remaining nuclear area over time. Interestingly, they also detailed a novel technique called photo-quenching FRET (PQ-FRET) in which PA-GFP is used as a FRET acceptor to quench CFP donor fluorescence. This allowed for simultaneous monitoring of acceptor protein mobility along with measures of acceptor protein exchange (rate of quenching) and donor-acceptor distance (amount of donor quenching). In another study, Lin et al [187] devised a fusion protein containing a histone peptide linked to a methyllysine binding domain sandwiched between a CFP/YFP FRET pair. This method allowed for active surveillance of histone methylation as the newly methylated photosensor increased FRET fluorescence. Overall, the production of genetically encoded epigenomic photosensors is an emerging field, but one that has immense potential for applications in the areas of stem cell differentiation, maturation, and reprogramming.
Innovation
Through the rapidly expanding field of genetically encoded photoactuators and photosensors, researchers now have available tools to fine tune our understanding of cellular and molecular biology. On their own, each technology described in this review recounts extraordinary insight and ingenuity on the part of the investigators. Additional innovative power will come from the ability to combine various photoactuators and photosensors in the same cell system to multiplex the control and monitoring of biological processes. Specific to pluripotent stem cell research, optogenetic systems have vast potential to help improve quality control of new cell lines, manipulate and monitor processes of cell differentiation, maturation, and function, and once in vivo imaging techniques improve, assess outcomes of stem cell-based therapies.
One major roadblock to putting multiplexed systems in place is the need for spectral divergence between multiplexed probes to ensure specificity of activation and emission readouts. Work to achieve this goal is rapidly progressing. As described above and reviewed by Emiliani et al. [17] the multitude of red-shifted photosensors now available for combination with blue-light photactuators [18, 19] are facilitating such advancements. Itoh et al. [188] have recently described a novel “multistepped” approach using a light-induced heat shock protein expression system, named infrared laser-evoked gene operator (IR-LEGO), that uses infrared light to induce expression of blue-light activated ChR2. While the photoactuator and photosensor are not active simultaneously, the authors have designed a system for cell lineage tracing combined with the power to interrogate destination neural circuitry of labelled cells. Uses for optogenetic functional lineage tracing techniques should have wide use beyond neurobiology.
A powerful prediction from Welch et al [189], in 2011, was that the evolution of molecular biosensors (photosensors) will facilitate computational multiplexing technology capable of deciphering comprehensive molecular pathways. Such computational analysis could occur via integration of data from independent, yet overlapping experiments using FRET based sensors to demonstrate proximal relationships between up- and downstream members in cell signaling pathways. These experiments, as freely noted by the authors, come with significant challenges including the need for thorough vetting of reproducibility and consideration of non-physiologic responses from transgene expression of critical pathway proteins. Regardless, computational multiplexing has the potential to dramatically increase our understanding of complex cell signaling pathways of high relevance for pluripotent stem cell research.
Another innovation that has immediate implications for pluripotent stem cell biology is the direct control of differentiation by optogenetic technologies. Sokolik et al [190] have shown proof-of-concept studies inducing neural differentiation through a genetically encoded photoactuator expression system. Here the authors used a LOV domain dimerization strategy (GAVPO) [58] to photoactuate expression of Brn2 initiating neural differentiation by competing with Oct4 for Sox2 binding and subsequent loss of Nanog expression. Successful differentiation was only initiated with appropriate intensity and duration of illumination highlighting the complex positive feedback loops present in pluripotent stem cell-fate decision making. Given that the dynamics of pluripotent cell signaling pathways are well-studied, the application of genetically encoded photoactuator systems to query the initial, and successive, stimuli required to define differentiation pathways is ripe for exploration. Moreover, as the ability to deliver patterned light stimuli develops [191], a logical leap toward inducing spatiotemporal differentiation cues in 2- and 3-dimensions is not far from imagination. Recently, Guglielmi et al [192] published an excellent review discussing potential to optogenetically regulate tissue morphogenesis as a foundation for the field of “synthetic morphology” [193]. Specifically, creating and utilizing a library of intracellular light-sensitive sensor and effector modules would allow us to better grasp the fundamental principles of organ development and engineer superior tissue constructs for basic research and regenerative medicine applications.
Limitations
The use of genetically encoded photoactuators and photosensors can greatly aid fundamental research in stem cell biology; however, successful implementation must come with recognition of challenges intrinsic to these techniques. First and foremost, transgenes are non-endogenous constructs that cannot be considered biologically inert, and should only be utilized in well-controlled systems after ensuring no interference with native cell differentiation or function. One such example of direct functional interference is the competition for calcium binding when using calmodulin based photosensors such as GCaMP. At high expression levels, these constructs could disrupt intracellular calcium homeostasis leading to complex physiologic effects including cardiac hypertrophy, as observed in mice expressing GCaMP2 [194, 195]. Furthermore, while optogenetic approaches have multiple advantages over chemical actuators or sensors, they are still limited with respect to transgene stability and toxicity, resulting signal-to-noise ratio at non-toxic expression levels, relatively slow signal kinetics, potential interference from autofluorescence, and non-specific effects caused by prolonged, strong illumination or heating. One strategy to mitigate transgene dosing and toxicity would be the use of genome editing approaches to target the optogenetic construct into the endogenous locus of query such that expression is mediated by physiologic activation, or to a safe harbor site to prevent random transgene integration. Additionally, more sophisticated multi-probe applications may necessitate expert knowledge in optical instruments, spectral deconvolution, and signal analysis. Overall, the future uses of optogenetics in stem cell research will require careful consideration of past and prospective limitations for each individual probe and its application.
Conclusion
Genetically encoded photoactuators and photosensors have the ability to characterize and manipulate the processes of reprogramming, pluripotency, self-renewal, differentiation, maturation, migration, and morphogenesis. Photoactuators comprise a diverse set of light-activated systems from light-gated ion channels and transporters to cell-signaling, DNA transcription, and genome engineering. Photosensors come in an equally diverse set of offerings from genetically encoded calcium and voltage indicators to cell signaling proteins, epigenome modifiers, and tension sensors. While we were unable to showcase all the currently available genetically encoded photoactuators and photosensors in this rapidly expanding field (and apologize to those whose outstanding work we have neglected), we have attempted to review the main optogenetic techniques with direct utility for pluripotent stem cell research. Our inquiry has demonstrated that future avenues for innovation in the field of optogenetics have potential to transform pluripotent cell research. Particularly, the co-expression of genetically encoded photoactuators and photosensors in a cell-specific, spatiotemporally defined fashion would enable all-optical interrogation of fate and function of pluripotent stem cells and their progeny. Combined with powerful imaging and computational analysis tools, optogenetic multiplexing of cell signaling pathways will allow for improved deconstruction of the relevant events transmitting stimuli to cellular processes. Moreover, light-mediated activation and monitoring of target cells and tissues in vivo, accomplished through minimally invasive techniques, promises to elucidate organism level effects originating from molecular scale perturbations. We are hopeful that this intersection of ideas will propel future discoveries leading to improved human therapies.
Abbreviations
hESC: human embryonic stem cell; hPSC: human pluripotent stem cell; iPSC: induced pluripotent stem cell; ChR: channelrhodopsin; HR: halorhodopsin; BR: bacteriorhodopsin; PCB: phycocyanobilin; MEF: mouse embryonic fibroblast; MAPK: mitogen activated protein kinase; ROS: reactive oxygen species; LOV: light oxygen and voltage; PhyB: phytochrome B; PIF: phytochrome interacting factor; CRY2: crytptochrome 2; CIB1: cryptochrome-interacting basic helix-loop-helix; UVR8: UV resistance locus 8; MEF: mouse embryonic fibroblast; ERK: extracellular signal related kinase; MEK: map kinase; PI3K: phosphoinositide 3 kinase; AKT: serine threonine kinase; FGF: fibroblast growth factor; IGF: insulin-like growth factor; RTK: receptor tyrosine kinase; GPCR: G-protein coupled receptor; PLC: phospholipase C; ZFN: zinc finger nuclease; TALEN: transcription activator-like effector nucleases; CRISPR: clustered regulatory interspaced short palindromic repeats; FRET: Förster resonance energy transfer; GFP: green fluorescent protein; CFP: cyan fluorescent protein; YFP: yellow fluorescent protein; GTP: guanosine triphosphate; GDP: guanosine diphosphate; GECI: genetically encoded calcium indicator; GEVI: genetically encoded voltage indicator; VSFP: voltage sensing fluorescent protein; VSD: voltage sensing domain; ECM: extracellular matrix.
Acknowledgements
The authors would like to thank Christopher Gilchrist for the image contribution to Figure 6B. This work is supported by NIH grants HL126524, HL132389 to NB, T32 HL007101-41 to JEP and a National Science Foundation CAREER Award (NSF-1454257) and NIH grant EB022166 to BDH.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154-6
2. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS. et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145-7
3. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76
4. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861-72
5. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917-20
6. Weinberger L, Ayyash M, Novershtern N, Hanna JH. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat Rev Mol Cell Biol. 2016;17:155-69
7. Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016;17:183-93
8. Mummery CL, Zhang J, Ng ES, Elliott DA, Elefanty AG, Kamp TJ. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ Res. 2012;111:344-58
9. Abdelalim EM, Emara MM. Advances and challenges in the differentiation of pluripotent stem cells into pancreatic beta cells. World J Stem Cells. 2015;7:174-81
10. Lim WF, Inoue-Yokoo T, Tan KS, Lai MI, Sugiyama D. Hematopoietic cell differentiation from embryonic and induced pluripotent stem cells. Stem Cell Res Ther. 2013;4:71
11. Meng X, Leslie P, Zhang Y, Dong J. Stem cells in a three-dimensional scaffold environment. Springerplus. 2014;3:80
12. Cohen DM, Chen CS. Mechanical control of stem cell differentiation. StemBook. Cambridge (MA). 2008
13. Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol. 2016;17:194-200
14. Deisseroth K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat Neurosci. 2015;18:1213-25
15. Tischer D, Weiner OD. Illuminating cell signalling with optogenetic tools. Nat Rev Mol Cell Biol. 2014;15:551-8
16. Smedemark-Margulies N, Trapani JG. Tools, methods, and applications for optophysiology in neuroscience. Front Mol Neurosci. 2013;6:18
17. Emiliani V, Cohen AE, Deisseroth K, Hausser M. All-Optical Interrogation of Neural Circuits. J Neurosci. 2015;35:13917-26
18. Akerboom J, Carreras Calderon N, Tian L, Wabnig S, Prigge M, Tolo J. et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci. 2013;6:2
19. Inoue M, Takeuchi A, Horigane S, Ohkura M, Gengyo-Ando K, Fujii H. et al. Rational design of a high-affinity, fast, red calcium indicator R-CaMP2. Nat Methods. 2015;12:64-70
20. Mirzapour Delavar H, Karamzadeh A, Pahlavanneshan S. Shining Light on the Sprout of Life: Optogenetics Applications in Stem Cell Research and Therapy. J Membr Biol. 2016;249:215-20
21. Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P. et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940-5
22. Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263-8
23. Li X, Gutierrez DV, Hanson MG, Han J, Mark MD, Chiel H. et al. Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc Natl Acad Sci U S A. 2005;102:17816-21
24. Nagel G, Ollig D, Fuhrmann M, Kateriya S, Musti AM, Bamberg E. et al. Channelrhodopsin-1: a light-gated proton channel in green algae. Science. 2002;296:2395-8
25. Gradinaru V, Thompson KR, Zhang F, Mogri M, Kay K, Schneider MB. et al. Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci. 2007;27:14231-8
26. Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. Bi-stable neural state switches. Nat Neurosci. 2009;12:229-34
27. Bamann C, Gueta R, Kleinlogel S, Nagel G, Bamberg E. Structural guidance of the photocycle of channelrhodopsin-2 by an interhelical hydrogen bond. Biochemistry. 2010;49:267-78
28. Gunaydin LA, Yizhar O, Berndt A, Sohal VS, Deisseroth K, Hegemann P. Ultrafast optogenetic control. Nat Neurosci. 2010;13:387-92
29. Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96:1803-14
30. Kleinlogel S, Feldbauer K, Dempski RE, Fotis H, Wood PG, Bamann C. et al. Ultra light-sensitive and fast neuronal activation with the Ca(2)+-permeable channelrhodopsin CatCh. Nat Neurosci. 2011;14:513-8
31. Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK. et al. Independent optical excitation of distinct neural populations. Nat Methods. 2014;11:338-46
32. Zhang F, Prigge M, Beyriere F, Tsunoda SP, Mattis J, Yizhar O. et al. Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat Neurosci. 2008;11:631-3
33. Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ. et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171-8
34. Lin JY, Knutsen PM, Muller A, Kleinfeld D, Tsien RY. ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat Neurosci. 2013;16:1499-508
35. Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N. et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633-9
36. Gradinaru V, Thompson KR, Deisseroth K. eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain cell biology. 2008;36:129-39
37. Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I. et al. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154-65
38. Mattis J, Tye KM, Ferenczi EA, Ramakrishnan C, O'Shea DJ, Prakash R. et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods. 2011;9:159-72
39. Berndt A, Deisseroth K. OPTOGENETICS. Expanding the optogenetics toolkit. Science. 2015;349:590-1
40. Berndt A, Lee SY, Ramakrishnan C, Deisseroth K. Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science. 2014;344:420-4
41. Wietek J, Wiegert JS, Adeishvili N, Schneider F, Watanabe H, Tsunoda SP. et al. Conversion of channelrhodopsin into a light-gated chloride channel. Science. 2014;344:409-12
42. Govorunova EG, Sineshchekov OA, Janz R, Liu X, Spudich JL. NEUROSCIENCE. Natural light-gated anion channels: A family of microbial rhodopsins for advanced optogenetics. Science. 2015;349:647-50
43. Cosentino C, Alberio L, Gazzarrini S, Aquila M, Romano E, Cermenati S. et al. Optogenetics. Engineering of a light-gated potassium channel. Science. 2015;348:707-10
44. Weick JP, Johnson MA, Skroch SP, Williams JC, Deisseroth K, Zhang SC. Functional control of transplantable human ESC-derived neurons via optogenetic targeting. Stem Cells. 2010;28:2008-16
45. Tonnesen J, Parish CL, Sorensen AT, Andersson A, Lundberg C, Deisseroth K. et al. Functional integration of grafted neural stem cell-derived dopaminergic neurons monitored by optogenetics in an in vitro Parkinson model. PLoS One. 2011;6:e17560
46. Pina-Crespo JC, Talantova M, Cho EG, Soussou W, Dolatabadi N, Ryan SD. et al. High-frequency hippocampal oscillations activated by optogenetic stimulation of transplanted human ESC-derived neurons. J Neurosci. 2012;32:15837-42
47. Steinbeck JA, Choi SJ, Mrejeru A, Ganat Y, Deisseroth K, Sulzer D. et al. Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson's disease model. Nat Biotechnol. 2015;33:204-9
48. Steinbeck JA, Jaiswal MK, Calder EL, Kishinevsky S, Weishaupt A, Toyka KV. et al. Functional Connectivity under Optogenetic Control Allows Modeling of Human Neuromuscular Disease. Cell Stem Cell. 2016;18:134-43
49. Bryson JB, Machado CB, Crossley M, Stevenson D, Bros-Facer V, Burrone J. et al. Optical control of muscle function by transplantation of stem cell-derived motor neurons in mice. Science. 2014;344:94-7
50. Zhuge Y, Patlolla B, Ramakrishnan C, Beygui RE, Zarins CK, Deisseroth K. et al. Human pluripotent stem cell tools for cardiac optogenetics. Conf Proc IEEE Eng Med Biol Soc. 2014;2014:6171-4
51. Zhang K, Cui B. Optogenetic control of intracellular signaling pathways. Trends Biotechnol. 2015;33:92-100
52. Jewhurst K, Levin M, McLaughlin KA. Optogenetic Control of Apoptosis in Targeted Tissues of Xenopus laevis Embryos. J Cell Death. 2014;7:25-31
53. Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458:1025-9
54. Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B. et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461:104-8
55. Yazawa M, Sadaghiani AM, Hsueh B, Dolmetsch RE. Induction of protein-protein interactions in live cells using light. Nat Biotechnol. 2009;27:941-5
56. Strickland D, Yao X, Gawlak G, Rosen MK, Gardner KH, Sosnick TR. Rationally improving LOV domain-based photoswitches. Nat Methods. 2010;7:623-6
57. Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C. et al. TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nat Methods. 2012;9:379-84
58. Wang X, Chen X, Yang Y. Spatiotemporal control of gene expression by a light-switchable transgene system. Nat Methods. 2012;9:266-9
59. Motta-Mena LB, Reade A, Mallory MJ, Glantz S, Weiner OD, Lynch KW. et al. An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat Chem Biol. 2014;10:196-202
60. Zhou XX, Fan LZ, Li P, Shen K, Lin MZ. Optical control of cell signaling by single-chain photoswitchable kinases. Science. 2017;355:836-42
61. Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997-1001
62. Shimizu-Sato S, Huq E, Tepperman JM, Quail PH. A light-switchable gene promoter system. Nat Biotechnol. 2002;20:1041-4
63. Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7:973-5
64. Crefcoeur RP, Yin R, Ulm R, Halazonetis TD. Ultraviolet-B-mediated induction of protein-protein interactions in mammalian cells. Nat Commun. 2013;4:1779
65. Chen D, Gibson ES, Kennedy MJ. A light-triggered protein secretion system. J Cell Biol. 2013;201:631-40
66. Zhou XX, Chung HK, Lam AJ, Lin MZ. Optical control of protein activity by fluorescent protein domains. Science. 2012;338:810-4
67. Dalton S. Signaling networks in human pluripotent stem cells. Curr Opin Cell Biol. 2013;25:241-6
68. Zhang K, Duan L, Ong Q, Lin Z, Varman PM, Sung K. et al. Light-mediated kinetic control reveals the temporal effect of the Raf/MEK/ERK pathway in PC12 cell neurite outgrowth. PLoS One. 2014;9:e92917
69. Bugaj LJ, Choksi AT, Mesuda CK, Kane RS, Schaffer DV. Optogenetic protein clustering and signaling activation in mammalian cells. Nat Methods. 2013;10:249-52
70. Toettcher JE, Weiner OD, Lim WA. Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell. 2013;155:1422-34
71. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-7
72. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329-41
73. McLean AB, D'Amour KA, Jones KL, Krishnamoorthy M, Kulik MJ, Reynolds DM. et al. Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells. 2007;25:29-38
74. Idevall-Hagren O, De Camilli P. Detection and manipulation of phosphoinositides. Biochim Biophys Acta. 2015;1851:736-45
75. Toettcher JE, Gong D, Lim WA, Weiner OD. Light-based feedback for controlling intracellular signaling dynamics. Nat Methods. 2011;8:837-9
76. Kakumoto T, Nakata T. Optogenetic control of PIP3: PIP3 is sufficient to induce the actin-based active part of growth cones and is regulated via endocytosis. PLoS One. 2013;8:e70861
77. Idevall-Hagren O, Dickson EJ, Hille B, Toomre DK, De Camilli P. Optogenetic control of phosphoinositide metabolism. Proc Natl Acad Sci U S A. 2012;109:E2316-23
78. Xu Y, Nan D, Fan J, Bogan JS, Toomre D. Optogenetic activation reveals distinct roles of PIP3 and Akt in adipocyte insulin action. J Cell Sci. 2016;129:2085-95
79. Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol. 2016;17:496-510
80. Nayak RC, Chang KH, Vaitinadin NS, Cancelas JA. Rho GTPases control specific cytoskeleton-dependent functions of hematopoietic stem cells. Immunol Rev. 2013;256:255-68
81. Anneren C. Tyrosine kinase signalling in embryonic stem cells. Clin Sci (Lond). 2008;115:43-55
82. Layden BT, Newman M, Chen F, Fisher A, Lowe WL Jr. G protein coupled receptors in embryonic stem cells: a role for Gs-alpha signaling. PLoS One. 2010;5:e9105
83. Doze VA, Perez DM. GPCRs in stem cell function. Prog Mol Biol Transl Sci. 2013;115:175-216
84. Kim N, Kim JM, Lee M, Kim CY, Chang KY, Heo WD. Spatiotemporal control of fibroblast growth factor receptor signals by blue light. Chem Biol. 2014;21:903-12
85. Grusch M, Schelch K, Riedler R, Reichhart E, Differ C, Berger W. et al. Spatio-temporally precise activation of engineered receptor tyrosine kinases by light. EMBO J. 2014;33:1713-26
86. Kleinlogel S. Optogenetic user's guide to Opto-GPCRs. Front Biosci (Landmark Ed). 2016;21:794-805
87. O'Neill PR, Gautam N. Subcellular optogenetic inhibition of G proteins generates signaling gradients and cell migration. Mol Biol Cell. 2014;25:2305-14
88. Suzanne M, Steller H. Shaping organisms with apoptosis. Cell Death Differ. 2013;20:669-75
89. Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal. 2010;3:re8
90. Mills E, Chen X, Pham E, Wong S, Truong K. Engineering a photoactivated caspase-7 for rapid induction of apoptosis. ACS Synth Biol. 2012;1:75-82
91. Hughes RM, Freeman DJ, Lamb KN, Pollet RM, Smith WJ, Lawrence DS. Optogenetic apoptosis: light-triggered cell death. Angew Chem Int Ed Engl. 2015;54:12064-8
92. Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397-405
93. Wang H, La Russa M, Qi LS. CRISPR/Cas9 in Genome Editing and Beyond. Annu Rev Biochem. 2016;85:227-64
94. Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21:121-31
95. Hockemeyer D, Jaenisch R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell. 2016;18:573-86
96. Muller K, Naumann S, Weber W, Zurbriggen MD. Optogenetics for gene expression in mammalian cells. Biol Chem. 2015;396:145-52
97. Hughes RM, Bolger S, Tapadia H, Tucker CL. Light-mediated control of DNA transcription in yeast. Methods. 2012;58:385-91
98. Liu H, Gomez G, Lin S, Lin S, Lin C. Optogenetic control of transcription in zebrafish. PLoS One. 2012;7:e50738
99. Polstein LR, Gersbach CA. Light-inducible spatiotemporal control of gene activation by customizable zinc finger transcription factors. J Am Chem Soc. 2012;134:16480-3
100. Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015;11:198-200
101. Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L. et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472-6
102. Nihongaki Y, Kawano F, Nakajima T, Sato M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol. 2015;33:755-60
103. Tyszkiewicz AB, Muir TW. Activation of protein splicing with light in yeast. Nat Methods. 2008;5:303-5
104. Renicke C, Schuster D, Usherenko S, Essen LO, Taxis C. A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem Biol. 2013;20:619-26
105. Bonger KM, Rakhit R, Payumo AY, Chen JK, Wandless TJ. General method for regulating protein stability with light. ACS Chem Biol. 2014;9:111-5
106. Lee S, Park H, Kyung T, Kim NY, Kim S, Kim J. et al. Reversible protein inactivation by optogenetic trapping in cells. Nat Methods. 2014;11:633-6
107. Taslimi A, Vrana JD, Chen D, Borinskaya S, Mayer BJ, Kennedy MJ. et al. An optimized optogenetic clustering tool for probing protein interaction and function. Nat Commun. 2014;5:4925
108. Harterink M, van Bergeijk P, Allier C, de Haan B, van den Heuvel S, Hoogenraad CC. et al. Light-controlled intracellular transport in Caenorhabditis elegans. Curr Biol. 2016;26:R153-4
109. Rao MV, Chu PH, Hahn KM, Zaidel-Bar R. An optogenetic tool for the activation of endogenous diaphanous-related formins induces thickening of stress fibers without an increase in contractility. Cytoskeleton (Hoboken). 2013;70:394-407
110. Buckley CE, Moore RE, Reade A, Goldberg AR, Weiner OD, Clarke JD. Reversible Optogenetic Control of Subcellular Protein Localization in a Live Vertebrate Embryo. Dev Cell. 2016;36:117-26
111. Evers TH, van Dongen EM, Faesen AC, Meijer EW, Merkx M. Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry. 2006;45:13183-92
112. Lakowicz JR. Principles of fluorescence spectroscopy. 3rd ed. New York: Springer. 2006
113. Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac activation dynamics visualized in living cells. Science. 2000;290:333-7
114. Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A. et al. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 2001;411:1065-8
115. Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069-72
116. Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ. et al. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci U S A. 2008;105:19264-9
117. Miura H, Matsuda M, Aoki K. Development of a FRET biosensor with high specificity for Akt. Cell Struct Funct. 2014;39:9-20
118. Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M. et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882-7
119. Horikawa K, Yamada Y, Matsuda T, Kobayashi K, Hashimoto M, Matsu-ura T. et al. Spontaneous network activity visualized by ultrasensitive Ca(2+) indicators, yellow Cameleon-Nano. Nat Methods. 2010;7:729-32
120. Mank M, Reiff DF, Heim N, Friedrich MW, Borst A, Griesbeck O. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophys J. 2006;90:1790-6
121. Mank M, Santos AF, Direnberger S, Mrsic-Flogel TD, Hofer SB, Stein V. et al. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat Methods. 2008;5:805-11
122. Thestrup T, Litzlbauer J, Bartholomaus I, Mues M, Russo L, Dana H. et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat Methods. 2014;11:175-82
123. Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci U S A. 1999;96:11241-6
124. Souslova EA, Belousov VV, Lock JG, Stromblad S, Kasparov S, Bolshakov AP. et al. Single fluorescent protein-based Ca2+ sensors with increased dynamic range. BMC biotechnology. 2007;7:37
125. Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc Natl Acad Sci U S A. 2001;98:3197-202
126. Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137-41
127. Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295-300
128. Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M. et al. An expanded palette of genetically encoded Ca(2)(+) indicators. Science. 2011;333:1888-91
129. Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP. et al. Sensitive red protein calcium indicators for imaging neural activity. eLife. 2016:5
130. Siegel MS, Isacoff EY. A genetically encoded optical probe of membrane voltage. Neuron. 1997;19:735-41
131. Ataka K, Pieribone VA. A genetically targetable fluorescent probe of channel gating with rapid kinetics. Biophys J. 2002;82:509-16
132. Sakai R, Repunte-Canonigo V, Raj CD, Knopfel T. Design and characterization of a DNA-encoded, voltage-sensitive fluorescent protein. Eur J Neurosci. 2001;13:2314-8
133. Baker BJ, Lee H, Pieribone VA, Cohen LB, Isacoff EY, Knopfel T. et al. Three fluorescent protein voltage sensors exhibit low plasma membrane expression in mammalian cells. Journal of neuroscience methods. 2007;161:32-8
134. Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239-43
135. Lundby A, Akemann W, Knopfel T. Biophysical characterization of the fluorescent protein voltage probe VSFP2.3 based on the voltage-sensing domain of Ci-VSP. Eur Biophys J. 2010;39:1625-35
136. Dimitrov D, He Y, Mutoh H, Baker BJ, Cohen L, Akemann W. et al. Engineering and characterization of an enhanced fluorescent protein voltage sensor. PloS one. 2007;2:e440
137. Akemann W, Mutoh H, Perron A, Park YK, Iwamoto Y, Knopfel T. Imaging neural circuit dynamics with a voltage-sensitive fluorescent protein. Journal of neurophysiology. 2012;108:2323-37
138. Lundby A, Mutoh H, Dimitrov D, Akemann W, Knopfel T. Engineering of a genetically encodable fluorescent voltage sensor exploiting fast Ci-VSP voltage-sensing movements. PloS one. 2008;3:e2514
139. Jin L, Han Z, Platisa J, Wooltorton JR, Cohen LB, Pieribone VA. Single action potentials and subthreshold electrical events imaged in neurons with a fluorescent protein voltage probe. Neuron. 2012;75:779-85
140. St-Pierre F, Marshall JD, Yang Y, Gong Y, Schnitzer MJ, Lin MZ. High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nat Neurosci. 2014;17:884-9
141. Barnett L, Platisa J, Popovic M, Pieribone VA, Hughes T. A fluorescent, genetically-encoded voltage probe capable of resolving action potentials. PloS one. 2012;7:e43454
142. Abdelfattah AS, Farhi SL, Zhao Y, Brinks D, Zou P, Ruangkittisakul A. et al. A Bright and Fast Red Fluorescent Protein Voltage Indicator That Reports Neuronal Activity in Organotypic Brain Slices. J Neurosci. 2016;36:2458-72
143. Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D, Cohen AE. Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nat Methods. 2011;9:90-5
144. Hochbaum DR, Zhao Y, Farhi SL, Klapoetke N, Werley CA, Kapoor V. et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat Methods. 2014;11:825-33
145. Lin MZ, Schnitzer MJ. Genetically encoded indicators of neuronal activity. Nat Neurosci. 2016;19:1142-53
146. Gong Y, Wagner MJ, Zhong Li J, Schnitzer MJ. Imaging neural spiking in brain tissue using FRET-opsin protein voltage sensors. Nat Commun. 2014;5:3674
147. Gong Y, Huang C, Li JZ, Grewe BF, Zhang Y, Eismann S. et al. High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science. 2015;350:1361-6
148. Leyton-Mange JS, Mills RW, Macri VS, Jang MY, Butte FN, Ellinor PT. et al. Rapid cellular phenotyping of human pluripotent stem cell-derived cardiomyocytes using a genetically encoded fluorescent voltage sensor. Stem Cell Reports. 2014;2:163-70
149. Shinnawi R, Huber I, Maizels L, Shaheen N, Gepstein A, Arbel G. et al. Monitoring Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes with Genetically Encoded Calcium and Voltage Fluorescent Reporters. Stem Cell Reports. 2015;5:582-96
150. Song L, Awari DW, Han EY, Uche-Anya E, Park SH, Yabe YA. et al. Dual optical recordings for action potentials and calcium handling in induced pluripotent stem cell models of cardiac arrhythmias using genetically encoded fluorescent indicators. Stem Cells Transl Med. 2015;4:468-75
151. Chen Z, Xian W, Bellin M, Dorn T, Tian Q, Goedel A. et al. Subtype-specific promoter-driven action potential imaging for precise disease modelling and drug testing in hiPSC-derived cardiomyocytes. Eur Heart J. 2016
152. Klimas A, Ambrosi CM, Yu J, Williams JC, Bien H, Entcheva E. OptoDyCE as an automated system for high-throughput all-optical dynamic cardiac electrophysiology. Nat Commun. 2016;7:11542
153. Addis RC, Ifkovits JL, Pinto F, Kellam LD, Esteso P, Rentschler S. et al. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J Mol Cell Cardiol. 2013;60:97-106
154. Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J. et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012;489:322-5
155. Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ. et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273-7
156. Shiba Y, Gomibuchi T, Seto T, Wada Y, Ichimura H, Tanaka Y. et al. Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature. 2016;538:388-91
157. Wozniak MA, Chen CS. Mechanotransduction in development: a growing role for contractility. Nat Rev Mol Cell Biol. 2009;10:34-43
158. Polacheck WJ, Li R, Uzel SG, Kamm RD. Microfluidic platforms for mechanobiology. Lab on a chip. 2013;13:2252-67
159. Mammoto T, Mammoto A, Ingber DE. Mechanobiology and developmental control. Annu Rev Cell Dev Biol. 2013;29:27-61
160. Nemir S, West JL. Synthetic materials in the study of cell response to substrate rigidity. Ann Biomed Eng. 2010;38:2-20
161. Sun Y, Fu J. Mechanobiology: a new frontier for human pluripotent stem cells. Integr Biol (Camb). 2013;5:450-7
162. Takahashi K, Kakimoto Y, Toda K, Naruse K. Mechanobiology in cardiac physiology and diseases. J Cell Mol Med. 2013;17:225-32
163. Schoen I, Pruitt BL, Vogel V. The Yin-Yang of Rigidity Sensing: How Forces and Mechanical Properties Regulate the Cellular Response to Materials. Annu Rev Mater Res. 2013;43:589-618
164. Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316-23
165. Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21-33
166. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677-89
167. Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S. et al. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 2006;127:1015-26
168. del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323:638-41
169. LaCroix AS, Rothenberg KE, Hoffman BD. Molecular-Scale Tools for Studying Mechanotransduction. Annu Rev Biomed Eng. 2015;17:287-316
170. Hoffman BD. The detection and role of molecular tension in focal adhesion dynamics. Prog Mol Biol Transl Sci. 2014;126:3-24
171. LaCroix AS, Rothenberg KE, Berginski ME, Urs AN, Hoffman BD. Construction, imaging, and analysis of FRET-based tension sensors in living cells. Methods in cell biology. 2015;125:161-86
172. Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT. et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263-6
173. Austen K, Ringer P, Mehlich A, Chrostek-Grashoff A, Kluger C, Klingner C. et al. Extracellular rigidity sensing by talin isoform-specific mechanical linkages. Nature cell biology. 2015;17:1597-606
174. Meng F, Suchyna TM, Sachs F. A fluorescence energy transfer-based mechanical stress sensor for specific proteins in situ. The FEBS journal. 2008;275:3072-87
175. Meng F, Sachs F. Visualizing dynamic cytoplasmic forces with a compliance-matched FRET sensor. J Cell Sci. 2011;124:261-9
176. Brenner MD, Zhou R, Conway DE, Lanzano L, Gratton E, Schwartz MA. et al. Spider Silk Peptide Is a Compact, Linear Nanospring Ideal for Intracellular Tension Sensing. Nano Lett. 2016;16:2096-102
177. Borghi N, Sorokina M, Shcherbakova OG, Weis WI, Pruitt BL, Nelson WJ. et al. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc Natl Acad Sci U S A. 2012;109:12568-73
178. Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol. 2013;23:1024-30
179. Nordenfelt P, Elliott HL, Springer TA. Coordinated integrin activation by actin-dependent force during T-cell migration. Nat Commun. 2016;7:13119
180. Acharya BR, Wu SK, Lieu ZZ, Parton RG, Grill SW, Bershadsky AD. et al. Mammalian Diaphanous 1 Mediates a Pathway for E-cadherin to Stabilize Epithelial Barriers through Junctional Contractility. Cell reports. 2017;18:2854-67
181. Medvedev SP, Pokushalov EA, Zakian SM. Epigenetics of pluripotent cells. Acta Naturae. 2012;4:28-46
182. Boland MJ, Nazor KL, Loring JF. Epigenetic regulation of pluripotency and differentiation. Circ Res. 2014;115:311-24
183. Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development. 2009;136:509-23
184. Papp B, Plath K. Epigenetics of reprogramming to induced pluripotency. Cell. 2013;152:1324-43
185. Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P. et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285-90
186. Demarco IA, Periasamy A, Booker CF, Day RN. Monitoring dynamic protein interactions with photoquenching FRET. Nat Methods. 2006;3:519-24
187. Lin CW, Jao CY, Ting AY. Genetically encoded fluorescent reporters of histone methylation in living cells. J Am Chem Soc. 2004;126:5982-3
188. Itoh M, Yamamoto T, Nakajima Y, Hatta K. Multistepped optogenetics connects neurons and behavior. Curr Biol. 2014;24:R1155-6
189. Welch CM, Elliott H, Danuser G, Hahn KM. Imaging the coordination of multiple signalling activities in living cells. Nat Rev Mol Cell Biol. 2011;12:749-56
190. Sokolik C, Liu Y, Bauer D, McPherson J, Broeker M, Heimberg G. et al. Transcription factor competition allows embryonic stem cells to distinguish authentic signals from noise. Cell Syst. 2015;1:117-29
191. Chiou PY, Ohta AT, Wu MC. Massively parallel manipulation of single cells and microparticles using optical images. Nature. 2005;436:370-2
192. Guglielmi G, Falk HJ, De Renzis S. Optogenetic Control of Protein Function: From Intracellular Processes to Tissue Morphogenesis. Trends Cell Biol. 2016;26:864-74
193. Davies JA. Synthetic morphology: prospects for engineered, self-constructing anatomies. J Anat. 2008;212:707-19
194. Tallini YN, Ohkura M, Choi BR, Ji G, Imoto K, Doran R. et al. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci U S A. 2006;103:4753-8
195. Chang Liao ML, de Boer TP, Mutoh H, Raad N, Richter C, Wagner E. et al. Sensing Cardiac Electrical Activity with a Cardiac Myocyte-Targeted Optogenetic Voltage Indicator. Circ Res. 2015;117:401-12
Author contact
![]() Corresponding author: nenad.bursacedu; phone: 919-660-5510
Corresponding author: nenad.bursacedu; phone: 919-660-5510