Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2017; 7(17):4192-4203. doi:10.7150/thno.21400 This issue Cite
Research Paper
Pro-Inflammatory CXCR3 Impairs Mitochondrial Function in Experimental Non-Alcoholic Steatohepatitis
Jinghua Du1, 2*, Xiang Zhang1*, Juqiang Han1, 3, Kwan Man4, Yanquan Zhang1, Eagle SH Chu1, Yuemin Nan2 ![]() , Jun Yu1
, Jun Yu1 ![]()
1. Institute of Digestive Disease and The Department of Medicine and Therapeutics, State Key Laboratory of Digestive Disease, Li Ka Shing Institute of Health Sciences, CUHK-Shenzhen Research Institute, The Chinese University of Hong Kong, Hong Kong
2. Department of Traditional and Western Medical Hepatology, Third Hospital of Hebei Medical University, Shijiazhuang, China
3. Institute of liver disease, Beijing Military General Hospital, Beijing, China
4. Department of Surgery, LKS Faculty of Medicine, The University of Hong Kong, Hong Kong
* These authors contributed equally to this work.
Received 2017-6-9; Accepted 2017-8-14; Published 2017-9-26
Abstract

Mitochondrial dysfunction plays a crucial role in the development of non-alcoholic steatohepatitis (NASH). However, the regulator of mitochondrial dysfunction in the pathogenesis of NASH is still largely unclear. CXCR3 is an essential pro-inflammatory factor in chronic liver diseases. We explored the significance of CXCR3 in regulating mitochondrial function during NASH development in animal models and cultured hepatocytes.
Methods: The effects of CXCR3 on mitochondrial function were evaluated by genetic knockout or pharmacological inhibition in mouse models and in vitro. The ultrastructural changes of mitochondria were assessed by transmission electron microscopy (TEM). Hepatic levels of mitochondrial reactive oxygen species (ROS), DNA damage, membrane potential and ATP were examined.
Results: CXCR3 ablation by genetic knockout or pharmacological inhibition in mice protected against NASH development by influencing mitochondrial function. Similarly, depletion of CXCR3 reduced steatohepatitis injury in cultured hepatocytes. TEM analysis revealed that liver mitochondrial integrity was much improved in CXCR3 knockout (CXCR3-/-) compared to wildtype (WT) mice. In agreement with this, impaired mitochondrial function was pronounced in WT mice compared to CXCR3-/- mice, evidenced by increased protein expression of dynamic-related protein-1 (DRP1) and fission-1 (FIS1) and decreased protein expression of mitofusin-1 (MFN1). Mitochondrial dysfunction was induced in AML-12 hepatocytes by methionine and choline deficient medium and in HepG2 cells by palmitic acid. The impaired mitochondrial function in both cell lines was evidenced by reduced membrane potential and ATP content, and by increased mitochondrial ROS accumulation and DNA damage. However, CXCR3 knockdown by siCXCR3 significantly diminished the mitochondrial dysfunction in both AML-12 and HepG2 hepatocytes. In addition, inhibition of CXCR3 by CXCR3 specific antagonists SCH546738 and AMG487 restored mitochondrial function and inhibited mitochondrial-dependent apoptosis in the liver of WT mice fed with methionine and choline deficient diet.
Conclusion: CXCR3 induces mitochondrial dysfunction, which contributes to the pathogenesis of steatohepatitis. Pharmacologic blockade of CXCR3 prevents mitochondrial dysfunction and restores the severity of steatohepatitis, indicating a potential clinical impact for controlling the disease.
Keywords: CXCR3, non-alcoholic fatty liver diseases, mitochondria, apoptosis.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease worldwide. Non-alcoholic steatohepatitis (NASH) is a more severe form of NAFLD that is characterized by lobular inflammation, hepatocellular ballooning, and fibrosis with an inherent risk of progression to end-stage hepatocellular carcinoma (HCC) (1). Although the mechanisms of NASH progression have not been fully understood, accumulating evidence revealed a pivotal role of mitochondrial dysfunction in the development of steatohepatitis (2, 3). Mitochondria are the primary cellular organelles for the oxidation and metabolism of fatty acid and glucose. Mitochondrial dysfunction not only impairs fat homeostasis in the liver but also leads to the accumulation of reactive oxygen species (ROS) that trigger lipid peroxidation, cytokine overproduction and cell death. Abnormal morphologic changes and increased production of ROS in liver mitochondria have been observed in patients and animal models with NASH (4, 5). However, the molecular mechanisms underlying the regulation of mitochondria in the pathogenesis of NASH are still largely unclear.
Chemokines are ubiquitous chemotactic molecules that play major roles in acute and chronic inflammatory conditions (6). CXC chemokines are the largest families, members of which are expressed in acutely inflamed and fibrotic livers (7). CXCR3 is a G protein-coupled receptor of cytokine CXCL10, CXCL11 and CXCL9, and is essential in chronic liver inflammation (8, 9). CXCR3 is highly expressed in T lymphocytes, monocytes/macrophages and resident cells (Kupffer cells, hepatocytes and hepatic stellate cells) (10, 11). It could promote lipid accumulation, inflammatory cell infiltration and endoplasmic reticulum (ER) stress in the development of steatohepatitis (12). However, whether CXCR3 could act as a key regulator of mitochondrial dysfunction in NASH development is still unknown. In this study, we explored the effect and molecular mechanisms of CXCR3 on mitochondrial dysfunction during the progression of NASH using CXCR3 knockout (CXCR3-/-) mouse model and hepatocytes cell line models. Here we demonstrate for the first time that CXCR3 impairs mitochondrial integrity and function in the development of nutritional steatohepatitis through the induction of mitochondrial ROS, mitochondrial DNA damage and mitochondrial-related apoptosis. We show that the elimination of CXCR3 by genetic depletion or drug antagonists could restore mitochondrial integrity and therefore its function in NASH.
Methods
Animals and treatments
CXCR3-/- mice were established as previously described (12). Male CXCR3-/- mice and age-matched wild-type (WT) C57BL/6J mice (8-9 weeks old) were fed ad libitum with methionine and choline deficient (MCD) diet (ICN Biomedical, Costa Mesa, CA) for 4 weeks or high fat-high carbohydrate-high cholesterol (HFHC) diet (Specialty feeds, Glen forest, WA, Australia) for 12 weeks to induce steatohepatitis (12). N=5-8/group. Normal chow and corresponding control diet supplemented with choline bitartate (2 g/kg) and DL-methionine (3 g/kg) were fed to control group.
In a separate experiment, C57BL/6 WT mice were given AMG487 (5 mg/kg daily, R&D systems, Minneapolis, MN) or SCH546738 (10 mg/kg daily, Medchemexpress LLC, Princeton, NJ) for 10 days after 2 week MCD diet as previous described (13).
All animals were maintained in an environmentally controlled facility with diurnal light cycle and free access to water. All animal studies were performed in accordance with guidelines approved by the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong.
Transmission electron microscopy
Mitochondrial ultrastructure in mouse liver was evaluated using transmission electron microscopy (TEM). Liver was dissected and fixed in 2.5% glutaraldehyde for 2 h at 4°C, post-fixed in 1% osmium tetroxide for 1 h at 4 °C, dehydrated, and embedded in epoxy resin. The tissue was sliced using an ultramicrotome (RMC/MTX; Elexience). Ultrathin sections (60-80 nm) were mounted on copper grids, contrasted with 8% uranyl acetate and lead citrate, and observed with a Philips CM100 transmission electron microscope equipped with a TENGRA 2.3K X 2.3K TEM camera. Analysis was performed with the Soft Imaging System.
Isolation of mitochondria from liver tissue
Liver mitochondria and cytosol fractions were separated according to the instructions included with the Qproteome Mitochondria Isolation Kit (catalog No. 37612, QIAGEN, Hong Kong). Briefly, the liver cells were suspended in ice-cold Lysis Buffer and incubated for 10 min at 4 °C on a shaker. The lysate was then centrifuged at 1000 x g for 10 min and the supernatant was collected as cytosolic proteins. The cell pellet was suspended in ice-cold Disruption Buffer and disrupted by a blunt-ended needle and a syringe. The lysate was then centrifuged at 1000 x g for 10 min. After that, the supernatant was collected and centrifuged again at 6000 x g for 10 min. The pellet contains mitochondria.
Western blot analysis
Liver tissues or cells were lysed in RIPA buffer. Lysates were centrifuged (14,000 x g for 10 min) and the supernatant was collected. The proteins were size-separated by 10% SDS-PAGE, transferred to PVDF membranes, and blocked with 5% non-fat dry milk before being incubated with primary antibodies: mitofusin-1 (MFN1) (Proteintech, Chicago, IL), dynamic-related protein-1 (DRP1) (Proteintech), fission-1 (FIS1) (Santa Cruz Biotechnology, Santa Cruz, CA), apoptosis signal-regulating kinase 1 (ASK1) (Cell Signaling Technology, Danvers, MA), phosphorated-c-Jun N-terminal Kinase (p-JNK) (Cell Signaling Technology), phosphorated-c-Jun (p-c-Jun) (Cell Signaling Technology), apoptosis protease activating factor-1 (Apaf-1) (Proteintech), cytochrome c (Cyt c) (Cell Signaling Technology), caspase 3 (Cell Signaling Technology), cleaved caspase 3 (Cell Signaling Technology), PARP (Cell Signaling Technology), cleaved PARP (Cell Signaling Technology), apoptosis-inducing factor (AIF) (Cell Signaling Technology), and endonuclease G (EndoG) (Cell Signaling Technology). Secondary antibodies were anti-mouse IgG and anti-rabbit IgG (1:5000 dilutions, Cell Signaling Technology). The membrane was visualized using the ECL-Plus chemiluminescence reagent (GE Healthcare, Buckinghamshire, UK).
RNA isolation and real-time reverse transcription (RT)-PCR
Total RNA was isolated from cells with TRIzol (Thermo Fisher Scientific, Waltham, MA), and 1 μg total RNA was reverse transcribed to cDNA with First-Strand cDNA Synthesis Kit (Roche Applied Science, Mannheim, Germany), according to the manufacturers' instructions. Real-time PCR was performed in a 15 μL reaction by using Maxima SYBR Green/ROX qPCR Master Mix (Roche), mouse CXCR3 primer (F: ATCAGCGCTTCAATGCCAC; R: TGGCTTTCTCGACCACAGTT), human CXCR3 primer (F: CTGTGGCCGAGAAAGCAG; R: AGGCGCAAGAGCAGCATC), human tumor necrosis factor-α (TNF-α) primer (F:AACCTCCTCTCTGCCATCAA; R: GGAAGACCCCTCCCAGATAG), human interleukin (IL)-6 primer (F: TTGGTGTTGTGCAAGGGTCT; R: GAGAAGGCAACTGGACCGAA), mouse GAPDH primer: (F:AACGACCCCTTCATTGAC; R:TCCACGACATACTCAGCA), human GAPDH primer (F: AGGCAACTAGGATGGTGTGG; R:TTGATTTTGGAGGGATCTCG) and a Light Cycler 480 real-time PCR system (Roche). A three-step cycling protocol (initial denaturation at 95 °C for 10 min, 45 cycles of 15 s denaturation at 95 °C, 30 s annealing at 60 °C, and 30 s extension at 72 °C) was used to amplify the genes. Relative difference in fold change between an experimental and calibrator sample were calculated by using the comparative cycle threshold (2-ΔΔCt) method. GAPDH was used as internal control to calculate the relative expression.
Cell culture
Mouse immortalized hepatocytes AML-12 were cultured in DMEM/Ham's F12 supplemented with 10% fetal bovine serum (FBS), a mixture of insulin-transferrin-selenium (ITS), 0.1 mM dexamethasone, penicillin and streptomycin. Human hepatocytes HepG2 were cultured in DMEM supplemented with 10% FBS, penicillin and streptomycin. The cells were maintained under a humidified atmosphere of 95% air and 5% CO2 at 37 °C. They were routinely subcultured using trypsin- ethylenediaminetetraacetic acid every 5-7 days. AML-12 cells were exposed to control medium or DMEM/F12 medium deficient in methionine and choline (MCD medium) for 24 h. HepG2 cells were treated with 200 μM palmitic acid or bovine serum albumin (BSA) control for 24 h.
Knockdown of CXCR3 by siRNA transfection
Double-stranded siRNA oligonucleotides for CXCR3 (GenePharma, Shanghai, China) were transfected using Lipofectamine 2000 (Thermo Fisher Scientific) as recommended by the manufacturer (Thermo Fisher Scientific). The sense and antisense sequences of the siRNA are as follows: Mouse siCXCR3-1: 5'- GGAUUUCAGCCUGAACUUUTT-3' (sense), 5'-AAAGUUCAGGCUGAAAUCCTT-3' (antisense); Mouse siCXCR3-2: 5'- GCUAGAUGCCUCGGACUUUTT-3' (sense), 5'-AAAGUCCGAGGCAUCUAGCTT-3' (antisense); Human siCXCR3-1: 5'- CCUUCCUGCUGGCUUGUAUTT-3'(sense),5'-AUACAAGCCAGCAGGAAGGTT-3' (antisense); Human siCXCR3-2: 5'- GGUCAUGGCCUACUGCUAUTT -3' (sense), 5'- AUAGCAGUAGGCCAUGACCTT-3' (antisense). AML-12 and HepG2 cells were plated with 70-90% confluent in 6-well plate at the time of transfection. 5 μL Lipofectamine 2000 (Thermo Fisher Scientific) was diluted in 250 μL Opti-MEM Medium and siRNA plasmid was diluted into 250 μL Opti-MEM medium. The diluted DNA plasmid was then added to the diluted Lipofectamine 2000 and incubated for 20 min. After that, the plasmid-lipid complex was dispensed into plates. Fresh medium was added 6 h after transfection. Cells were treated with MCD medium or palmitic acid 48 h after transfection and processed for immunofluorescence or protein extraction 72 h after transfection.
Confocal microscopy
AML-12 and HepG2 hepatocytes were plated on coverslips and stained with MitoTracker red (Thermo Fisher Scientific). After washing twice with PBS, the cells were fixed with 4% paraformaldehyde in PBS for 15 min and then washed three times with PBS. Cell images were captured with an LSM 510 Zeiss confocal microscope (ZEISS, Göttingen, Germany) equipped with a 63x oil immersion objective and data were analyzed using LSM Meta software.
Mitochondrial membrane potential
Mitochondrial membrane potential (ΔΨm) was assessed by tetramethylrhodamine ethyl ester (TMRM) (Thermo Fisher Scientific), a specific fluorescent cationic dye that is readily sequestered by active mitochondria, depending on their transmembrane potential. AML-12 and HepG2 were treated with MCD medium and palmitic acid, respectively, for 24 h. Cells were then incubated with 10 nM TMRM for 30 min at 37 °C. The fluorescence levels were analyzed by flow cytometry.
Flow cytometry
Cells were incubated with TMRM fluorescent dye for 20 min and washed with PBS. For the quantification of TMRM, the stained cells were collected by trypsinization, and the fluorescence intensities were measured by Accuri C6 cytometer (BD Biosciences, Franklin Lakes, NJ). The live cells were gated using forward scatter (FSC) and side scatter (SSC). The debris and dead cells with low FSC and high SSC were excluded. The presented results are the average of at least three independent experiments.
ATP determination
ATP levels of cells were determined by ATP Colorimetric/Fluorometric Assay kit (ab83355, Abcam, Hong Kong) for mitochondrial dysfunction. Measurements were performed with the colorimetric assay, following the manufacturer's instructions, on 106 cells. ATP contents were measured in triplicate and expressed as % control.
Mitochondrial DNA damage analysis
Mitochondrial DNA (mtDNA) was extracted using an mtDNA isolation Kit (BioVision, Milpitas, CA). Mitochondrial DNA was digested with DNAse I and alkaline phosphatase (Cayman, Ann Arbor, MI). The amount of 8-hydroxydeoxyguanosine (8-OHdG) in the DNA digestion was measured by EIA kit (Cayman). Standard samples of 8-OHdG were analyzed to ensure good separation.
Statistical analysis
Quantitative analyses using cultured cells are represented as histograms. Values were obtained from three independent experiments, and expressed as mean ± standard deviation (SD). Statistical analysis was carried out using one-way ANOVA or Student's t test, with P < 0.05 considered as significant. All statistical data were calculated with the SPSS 17.0 software.
Results
CXCR3 damages mitochondrial integrity in nutritional steatohepatitis in mice
WT mice fed with MCD diet for 4 weeks or HFHC diet for 12 weeks developed steatohepatitis, whilst CXCR3-/- mice fed with MCD or HFHC diet showed significantly ameliorated hepatic steatosis and inflammation (Figure 1A) (12). To examine the effect of CXCR3 on mitochondria in the development of steatohepatitis, we analyzed hepatic mitochondrial morphology in HFHC-fed WT and CXCR3-/- mice. Using TEM to analyze ultrastructural changes in the liver, we found that the mitochondria were swollen, round-shaped, and the mitochondrial cristae were disrupted in severe steatohepatitis in HFHC-fed WT mice; whereas mitochondria were less swollen with well-organized cristae in CXCR3-/- mice with mild steatohepatitis (Figure 1B). In keeping with the ameliorated steatohepatitis, HFHC-fed CXCR3-/- mice diet showed improved mitochondrial structure of less swollen and relatively more organized mitochondrial cristae compared to WT mice fed the same diet. Decreased lipid vesicle droplets were also observed in HFHC-fed CXCR3-/- mice compared to HFHC-fed WT mice (Figure 1B). These results support a role of CXCR3 in damaging mitochondrial integrity in the pathogenesis of nutritional steatohepatitis.
Deficiency of CXCR3 protects against dietary-induced steatohepatitis and mitochondrial injury. (A) Representative H&E staining of liver sections from WT and CXCR3-/- mice fed with control or MCD diet for 4 weeks, or HFHC diet for 12 weeks; scale bar = 40 μm; (B) Representative transmission electron micrographs of liver sections from WT and CXCR3-/- mice fed with control or HFHC diet. The mitochondria from liver sections of HFHC fed WT mice were swollen and cristae appeared disrupted (red arrows). In CXCR3-/- mice fed HFHC diet, a mixed population composed of disrupted and organized cristae was present. Magnifications: upper panel, 3900×; lower panel, 8900×; (C) Western blot analysis of mitochondrial fusion protein mitofusin-1 (MFN1), mitochondrial fission proteins dynamic-related protein-1 (DRP1) and fission-1 (FIS1) in liver tissues from WT and CXCR3-/- mice fed with control or MCD diet and normal chow or HFHC diet. Values are mean ± SD (n=5/group). MCD, methionine-and-choline-deficient; HFHC, high-fat high-carbohydrate high-cholesterol.
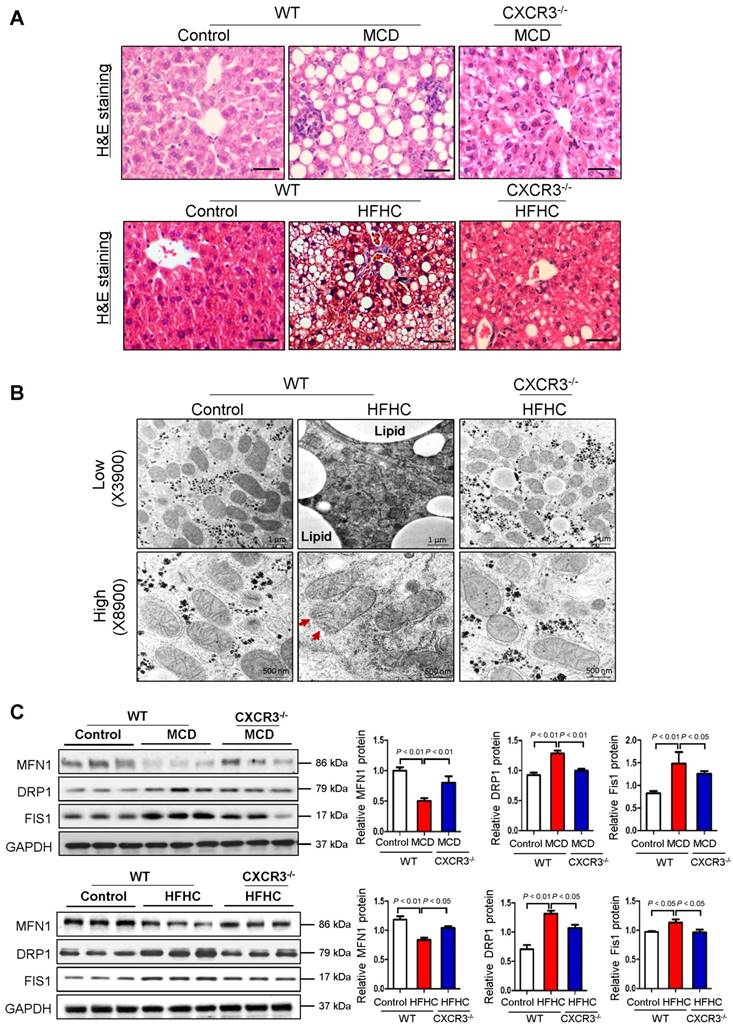
Considering the observed effect of CXCR3 on reducing mitochondrial integrity in steatohepatitis, we tested whether CXCR3 would alter the expression of genes that control mitochondrial integrity. Mitochondrial metabolic function is regulated by a family of mitochondrial GTPases, including MFN1, DRP1 and FIS1 (14). We found decreased protein expression of MFN1, increased protein expression of DRP1 and FIS1 in MCD- or HFHC- fed WT mice with steatohepatitis compared to WT mice fed with control diet with normal liver histology (Figure 1C). However, hepatic expression of MFN1 was induced, while DRP1 and FIS1 were reduced in MCD-fed CXCR3-/- compared to MCD-fed WT mice (Figure 1C). The results were confirmed in HFHC-fed CXCR3-/- mice compared to WT mice fed with the same diet (Figure 1C). Collectively, these results demonstrate that CXCR3 contributes to mitochondrial disintegration in the evolution of steatohepatitis by regulating key mitochondrial morphology regulators.
CXCR3 knockdown prevents mitochondrial dysfunction in hepatocytes
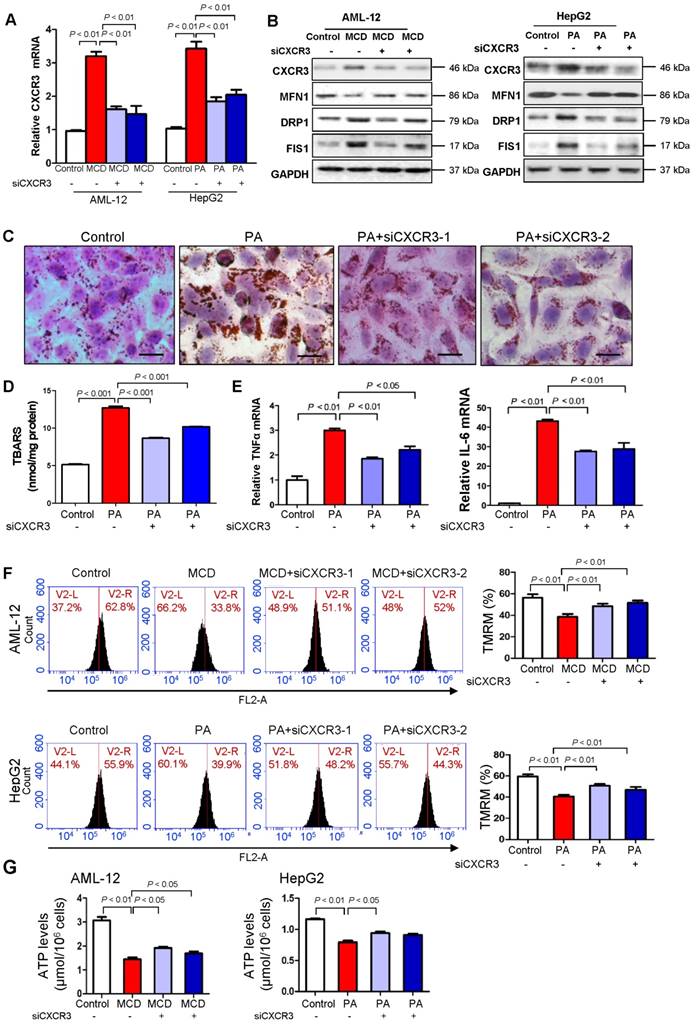
To elucidate the molecular mechanisms underlying the harmful effect of CXCR3 on mitochondrial integrity, we investigated the effect of CXCR3 on mitochondrial function in hepatocytes. CXCR3 was knocked down by small interfering RNA in both MCD medium-treated mouse AML-12 hepatocytes and palmitic acid-treated human HepG2 hepatocytes. The knockdown efficiencies of si-CXCR3-1 and si-CXCR3-2 were confirmed by quantitative RT-PCR (Figure 2A) and Western blot (Figure 2B). When AML-12 cells were exposed to MCD medium and HepG2 cells were treated with palmitic acid, mitochondrial integrity was reduced as shown by decreased expression of MFN1 and increased expression of DRP1 and FIS1 (Figure 2B), concomitant with higher CXCR3 mRNA and protein levels (Figure 2A-B) and increased lipid accumulation (Figure 2C), lipid peroxidation (Figure 2D) and production of inflammatory cytokines TNF-α and IL-6 (Figure 2E). However, CXCR3 knockdown by si-CXCR3-1 and si-CXCR3-2 could abolish the reduction of MFN1 and induction of DRP1 and FIS1 in MCD medium-treated AML-12 and palmitic acid-treated HepG2 hepatocytes (Figure 2B), paralleled with ameliorated lipid peroxide levels (Figure 2D). These data further confirm the effect of CXCR3 in reducing mitochondrial integrity in hepatocytes. We next analyzed mitochondrial function by measuring mitochondrial membrane permeability in hepatocytes using TMRM staining, a potentiometric dye taken up only by functional mitochondria (15). Using flow cytometric analysis, we observed a marked reduction of TMRM fluorescence intensity in both MCD medium-treated AML-12 hepatocytes and palmitic acid-treated HepG2 hepatocytes compared with control cells (Figure 2F). However, CXCR3 knockdown by siCXCR3-1 and siCXCR3-2 significantly restored the levels of TMRM in AML-12 exposed to MCD medium and HepG2 exposed to palmitic acid (P < 0.01, Figure 2F), inferring an increase in active mitochondria with intact membrane potentials by CXCR3 knockdown. In addition, CXCR3 knockdown increased ATP content in MCD medium-treated AML-12 hepatocytes and palmitic acid-treated HepG2 hepatocytes transfected with si-CXCR3 compared with hepatocytes transfected with control siRNA (Figure 2G). These results suggest that CXCR3 depletion prevents mitochondrial dysfunction in hepatocytes.
Knockdown of CXCR3 prevents mitochondrial dysfunction in hepatocytes with steatohepatitis changes. (A) AML-12 hepatocytes were exposed to MCD medium and HepG2 hepatocytes were treated with palmitic acid. CXCR3 knockdown was performed by transfection with two siRNAs. The transfection efficiency was determined by qRT-PCR. Data are presented as representative results of three independent experiments; (B) Hepatic CXCR3, mitochondrial fusion marker MFN1 and fission marker DRP1 and FIS1 were analyzed by western blot after siCXCR3 transfection in AML-12 and HepG2 hepatocytes; (C) Representative Oil red O staining pictures (scale bar = 20 μm) (D) cellular lipid hydroperoxide levels, (E) TNFα mRNA and IL-6 mRNA levels in palmitic acid-treated HepG2 cells with CXCR3 knockdown by si-CXCR3; (F) Mitochondrial membrane potential was stained with TMRM for 30 min and analyzed using flow cytometry in MCD medium-cultured AML-12 cells and palmitic acid-treated HepG2 cells with CXCR3 knockdown; (G) The ATP content of AML-12 and HepG2 treated with MCD medium or palmitic acid with the transfection of siCXCR3 was analyzed by ATP assay kit. MCD, methionine-and-choline-deficient; PA, palmitic acid.
CXCR3 knockdown prevents mitochondrial ROS accumulation in hepatocytes
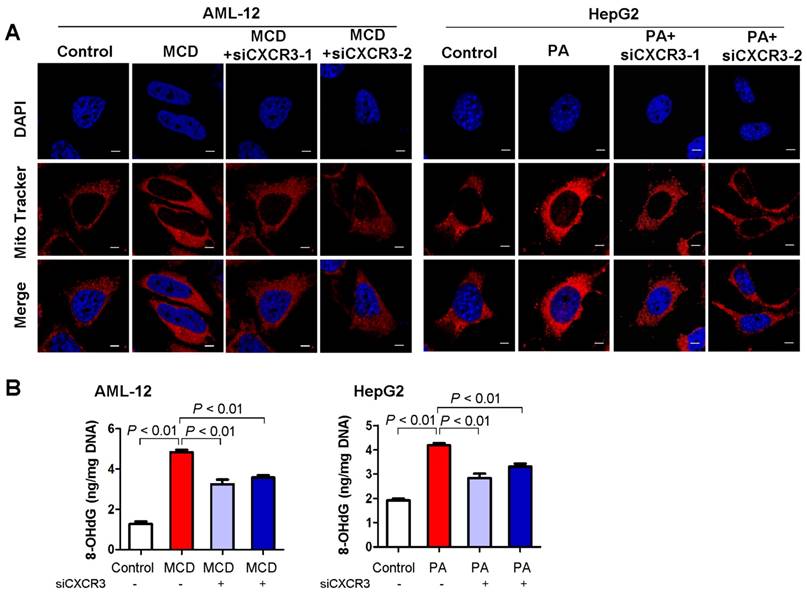
As mitochondria are the major producer of ROS in cells, we examined mitochondrial ROS production in hepatocytes with MCD medium- or palmitic acid- induced steatohepatitic changes. Confocal microscopy analysis of fluorescent stained live AML-12 and HepG2 hepatocytes showed increased mitochondrial ROS in MCD medium-exposed AML-12 culture and palmitic acid-treated HepG2 cells compared to control cells (Figure 3A). Conversely, CXCR3 knockdown abolished the induction of mitochondrial ROS in MCD medium-exposed AML-12 and palmitic acid-treated HepG2 cells (Figure 3A). These results suggest that CXCR3 increases ROS production by the electron transport chain of dysfunctional mitochondria in steatohepatitis. CXCR3 knockdown could prevent mitochondrial ROS accumulation.
CXCR3 knockdown prevents mitochondrial ROS accumulation and mitochondrial DNA damage in in vitro steatohepatitis. (A) After transfection with siCXCR3 and treatment with MCD medium or palmitic acid for 24 h, AML-12 and HepG2 were stained with MitoTracker red for 30 min and analyzed using confocal microscopy. Data are shown as representative results of three independent experiments; scale bar = 5 μm; (B) The oxidative mitochondrial DNA damage assay was performed to measure the levels of oxidative DNA damage marker 8-OHdG in MCD medium cultured AML-12 and palmitic acid treated HepG2 hepatocytes transfected with si-CXCR3 or si-control. Data shown are representative of three independent experiments. MCD, methionine-and-choline-deficient; PA, palmitic acid.
CXCR3 knockdown reduces mitochondrial DNA damage in hepatocytes
Mitochondrial ROS production could cause mtDNA damage (16). We thus examined the effect of CXCR3 on mitochondrial DNA damage by oxidative mtDNA damage assay to measure the levels of 8-OHdG, a marker for oxidative DNA damage (17). AML-12 hepatocytes exposed to MCD medium and HepG2 hepatocytes treated with palmitic acid showed increased 8-OHdG levels, indicating damaged mtDNA. Conversely, knockdown of CXCR3 by siRNA significantly reduced mtDNA damage in the two hepatocyte cell lines (Figure 3B).
CXCR3 deficiency prevents mitochondria-mediated apoptosis in steatohepatitis in mice
Mitochondria are pivotal in controlling cell death through the regulation of mitochondrial intrinsic apoptosis pathway. To identify the molecular mechanism for the mitochondrial dysfunction induced by CXCR3 in steatohepatitis, we examined hepatic apoptosis in liver tissues from WT and CXCR3-/- mice. As shown in Figure 4A, the hepatic protein expressions of ASK1, p-JNK, p-c-Jun, cleaved caspase 3, and cleaved PARP were upregulated in WT mice fed with MCD diet, while these inductions were abolished by CXCR3 knockout. Similarly, the protein expression of p-JNK, cleaved caspase 3, and cleaved PARP was suppressed in HFHC-fed CXCR3-/- mice compared to HFHC-fed WT mice (Figure 4B). These results suggest that CXCR3 deficiency prevents caspase-dependent apoptosis in dietary steatohepatitis.
Ablation of CXCR3 inhibits caspase-dependent and caspase-independent apoptosis signaling pathways and inhibition of CXCR3 by its antagonists prevents mitochondrial damage in steatohepatitis. (A) Hepatic apoptosis signal-regulating kinase 1 (ASK1), p-JNK, p-c-Jun, cleaved caspase 3, caspase 3, cleaved PARP, PARP, apoptosome-containing adaptor apoptosis protease activating factor-1 (Apaf-1) protein levels in the livers from WT and CXCR3-/- mice fed with control or MCD diet; (B) Hepatic p-JNK, JNK, cleaved caspase 3, caspase 3, cleaved PARP, and PARP protein levels in the livers from WT and CXCR3-/- mice fed with control or HFHC diet; (C) Hepatic mitochondrial and cytosolic Cyt c, AIF and EndoG protein levels from WT and CXCR3-/- mice fed with MCD diet. HSP60 was used as a mitochondrial internal control and GAPDH as a cytosolic internal control. (D) Hepatic CXCR3, mitochondrial fusion marker MFN1 and fission marker DRP1 and FIS1 were analyzed by western blot; (E) Apoptosis related markers, ASK1, p-JNK, cleaved caspase 3 and cleaved PARP were measured by western blot in MCD-fed WT mice treated with vehicle or CXCR3 antagonists SCH546738 and AMG487.
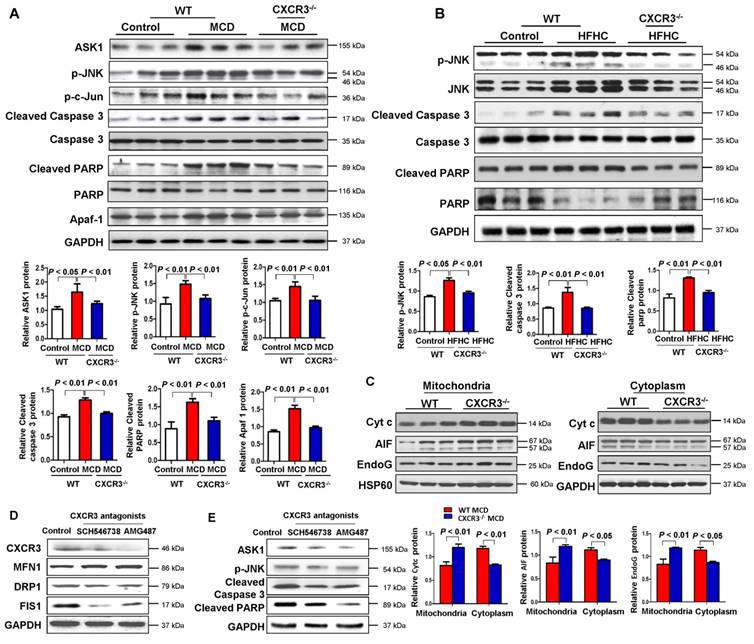
Apart from the ASK1/JNK pathway, caspase-dependent apoptosis could also be caused by the formation of apoptosome-containing adaptor Apaf-1, which could bind to Cyt c after its release from mitochondria to cytoplasm (18). We found that hepatic accumulation of Apaf-1 was significantly reduced in MCD-fed CXCR3-/- compared to MCD-fed WT mice (Figure 4A). To determine whether CXCR3 could inhibit the release of Cyt c from mitochondria, we isolated mitochondria from liver tissues of MCD-fed WT and CXCR3-/- mice. Our result showed that Cyt c expression increased in the mitochondria and decreased in the cytoplasm in CXCR3-/- mice compared to WT mice (Figure 4C), suggesting the decreased release of Cyt c from mitochondria to cytosol by depletion of CXCR3.
In addition, mitochondria have been reported to regulate caspase-independent death effectors AIF and EndoG. To further dissect the role of CXCR3 in modulating mitochondrial function, we analyzed the effect of CXCR3 on caspase-independent apoptosis. The hepatic expression levels of AIF and EndoG in MCD-fed CXCR3-/- mice were significantly increased in mitochondrial fragment and decreased in cytosol protein compared to MCD-fed WT mice (Figure 4C), suggesting that CXCR3 deficiency also prevent caspase-independent apoptosis.
CXCR3 antagonists protect against mitochondrial dysfunction by regulating mitochondrial apoptosis in steatohepatitis mice
We further investigated whether treatment with CXCR3 antagonists could reverse mitochondrial dysfunction in established steatohepatitis in mice. Two CXCR3 antagonists, AMG487 and SCH546738, were administrated to C57BL/6 mice for 10 days after 2 weeks of MCD diet. Our results showed that both AMG487 and SCH546738 significantly up-regulated the mitochondrial fusion protein MFN1 and down-regulated the fission protein DRP1 and FIS1 concomitant with decreased CXCR3 levels in MCD-fed WT mice (Figure 4D), indicating improved mitochondrial integrity by CXCR3 antagonists. Moreover, the effects of CXCR3 antagonists on mitochondrial apoptosis were also investigated by evaluating the protein expression of the key mitochondrial apoptosis-related factors. The results showed that both AMG487 and SCH546738 suppressed protein expression of ASK1, p-JNK, cleaved caspase 3 and cleaved PARP in MCD-fed mice (Figure 4E).
Discussion
In the present study, we demonstrated that CXCR3 deficiency significantly ameliorated mitochondrial integrity observed under TEM in experimental NASH mice models for the first time. Mitochondria exist as a dynamic network that undergoes constant fission and fusion to maintain biogenesis balance (19-21). Mitochondrial fusion is mediated by fusion protein MFN1, a dynamic-related GTPase that is responsible for the fusion of outer mitochondrial membranes (22). Mitochondrial fission is controlled by another large GTPase DRP1, which could interact with mitochondrial receptor protein FIS1. Increased mitochondrial fission and decreased mitochondrial fusion indicate mitochondrial fragmentation (22). In this connection, we revealed that hepatic MFN1 fusion protein level was significantly increased and DRP1, FIS1 fission protein levels were decreased in the CXCR3-/- mice compared with WT mice in two dietary models of steatohepatitis, indicating the induction of mitochondrial fragmentation by CXCR3 in the disease development. This was consistent with mitochondrial swelling and the disarray of mitochondrial cristae by CXCR3 observed under TEM (Figure 1B). Therefore, our findings suggest that CXCR3 is involved in the mitochondrial fragmentation and dysfunction in steatohepatitis.
To confirm the impaired mitochondrial function by CXCR3 in steatohepatitis, we evaluated the effects of CXCR3 on mitochondrial function in hepatocytes with steatohepatitis changes. In vitro steatohepatitis models were established in AML-12 cells cultured in MCD medium and HepG2 cells treated with saturated fatty acids. We observed impaired mitochondrial integration, mitochondrial membrane potential, ATP production, and enhanced mitochondrial ROS production and mtDNA damage in hepatocytes with steatohepatitis changes (23-25). However, CXCR3 knockdown by siCXCR3 significantly diminished mitochondrial fragmentation, mitochondrial ROS accumulation, mtDNA damage and restored mitochondrial membrane potential and ATP content. Determination of the levels of 8-OHdG, a marker for oxidative DNA damage (17), further confirmed the significantly reduced 8-OHdG level by CXCR3 knockdown. Moreover, restored ATP by CXCR3 knockdown inhibited the release of Cyt c from mitochondria to the cytosol in steatohepatitis. Sustained hepatic oxidative stress and mtDNA damage have been recently reported in human NASH (2). ROS-induced damage of mtDNA severely affects mitochondrial function and induces hepatic steatosis and lesion (26). Collectively, our findings indicate that CXCR3 promotes mitochondrial dysfunction by inducing mitochondrial ROS and mtDNA damage, which contribute to NASH development (Figure 5). CXCR3 deficiency may prevent steatohepatitis by increasing mitochondrial membrane potential and ATP content, and by decreasing mitochondrial ROS and ROS induced-mtDNA damage.
Schematic diagram for the mechanisms of CXCR3 in the induction of mitochondrial dysfunction in experimental steatohepatitis. CXCR3-induced mitochondrial dysfunction promoted NASH development by inducing DNA damage, mitochondrial apoptosis and lipid accumulation. CXCR3 impaired mitochondrial function in mice as evidenced by increased DRP1 and FIS1 and decreased MFN1. CXCR3-impaired mitochondrial function was confirmed in AML-12 and HepG2 cells demonstrated by reduced membrane potential, ATP content, increased mitochondrial ROS accumulation and DNA damage. CXCR3 also induced mitochondria-mediated apoptosis through ASK1/JNK pathway and AIF/EndoG pathway, which contributed to the evolution of steatohepatitis.
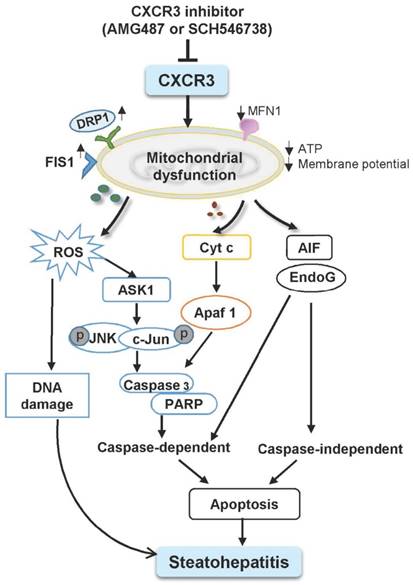
The mechanisms underlying mitochondrial dysfunction by CXCR3 in steatohepatitis were further investigated. The balance between fission and fusion is critical for mitochondrial homeostasis and is linked with apoptosis and mitochondrial fragmentation, an early phenomenon during apoptotic cell death (27). Mitochondrial dynamic alteration causes enhanced JNK phosphorylation through activation of ASK1 (28). Dietary induced steatohepatitis increased the activation of ASK1, JNK, and the JNK-associated transcription factor c-Jun. However, CXCR3 deficiency suppressed the activation of ASK1, p-JNK and p-c-Jun in both MCD-induced and HFHC-induced steatohepatitis. ASK1 is a member of the mitogen-activated protein kinase (MAPK) family, and is an upstream activator of JNK signaling cascades (29). Stimulation of the JNK pathway has been hypothesized as a concurrent pro-apoptotic mechanism in NASH and lipotoxicity (30). These observations suggest that the activation of ASK1/JNK signaling pathway was involved in CXCR3 associated steatohepatitis.
Mitochondria have been shown to be the target of many pro-apoptotic proteins (31) and play a crucial role in apoptosis through the regulation of apoptotic cascade. The present study showed that JNK activation increased the leakage of Cyt c from mitochondria to the cytosol and activated caspase 3 “cell execution pathway”. Deficiency of CXCR3 reduced caspase-dependent apoptosis in both dietary steatohepatitis mouse models. Moreover, mitochondrial dysfunction induced the release of AIF and EndoG from mitochondria to cytosol. AIF and EndoG are mitochondrial proteins that mediate caspase-independent apoptosis in a number of models after translocation into the cytosol or into the nucleus (32, 33). When AIF and EndoG were released from the mitochondria in response to apoptotic stimuli, they became active executioners of the cells by causing condensation of chromatin in the nuclei and large-scale fragmentation of DNA (34, 35). These processes were significantly blunted by CXCR3 deficiency, suggesting the effect of CXCR3 deletion in inhibiting caspase-independent apoptosis in steatohepatitis. These findings collectively reveal that CXCR3 deficiency could prevent non-alcoholic steatohepatitis through both caspase-dependent and caspase-independent apoptosis signaling pathways (Figure 5).
Based on the above findings by CXCR3 depletion, we conducted intervention experiments using two CXCR3 antagonists on steatohepatitis. Both AMG487 and SCH546738 ameliorated mitochondrial dysfunction in steatohepatitis induced by MCD and HFHC, as indicated by induced MFN1 and reduced DRP1 and FIS1 levels. This protection was associated with decreased mitochondrial-related apoptosis. Therefore, CXCR3 antagonists may prevent steatohepatitis by ameliorating mitochondrial dysfunction and have a potential therapeutic impact for NASH treatment.
In conclusion, we have demonstrated that CXCR3 deficiency could prevent mitochondrial dysfunction during the pathogenesis of steatohepatitis, as shown by decreased mitochondrial fragmentation, mitochondrial ROS accumulation, mtDNA damage and increased membrane potential and ATP content. CXCR3 induced mitochondrial dysfunction promoted caspase dependent and independent apoptosis cascades, thereby inducing steatohepatitis. Our findings provide new insights into the role of CXCR3 in mitochondrial dysfunction in steatohepatitis, and suggest that pharmacologic blockade of CXCR3 could serve as a potential therapeutic approach for NASH treatment through the restoration of mitochondrial function.
Abbreviations
NASH, non-alcoholic steatohepatitis; CXCR3-/-, CXCR3 knockout; WT, wild-type; DRP1, dynamic-related protein-1; FIS1, fission-1; MFN1, mitofusin-1; ROS, reactive oxygen species; NAFLD, non-alcoholic fatty liver disease; HCC, hepatocellular carcinoma; ER, endoplasmic reticulum; MCD, methionine and choline deficient diet; HFHC, high-fat high-carbohydrate high-cholesterol; TEM, transmission electron microscopy; ASK1, apoptosis signal-regulating kinase 1; p-JNK, phosphorated-c-Jun N-terminal Kinase; p-c-Jun, phosphorated-c-Jun; Apaf-1, apoptosis protease activating factor-1; Cyt c, cytochrome c; AIF, apoptosis-inducing factor; EndoG, endonuclease G; TNF-α, tumor necrosis factor-α; IL, interleukin; FBS, fetal bovine serum; ITS, insulin-transferrin-selenium; BSA, bovine serum albumin; TMRM, tetramethylrhodamin ethyl ester; FSC, forward scatter; SSC, side scatter; mtDNA, Mitochondrial DNA; 8-OHdG, 8-hydroxydeoxyguanosine; SD, standard deviation.
Acknowledgements
The authors thank Dr Olabisi O. Coker for assistance with English language editing.
Authors' contributions
JD and XZ were involved in study design, experiments conduct, and data analysis; JH, YZ and ESHC performed the experiments; KM provided material support; YN commented on the study; JY designed, supervised the study and wrote the paper. All authors reviewed the manuscript.
Financial Support
The project was supported by the RGC-GRF Hong Kong (766613, 14106415, 14111216, 14163817); National Basic Research Program of China (973 Program, 2013CB531401); RGC-Theme-based Research Scheme Hong Kong (T12-403-11); Shenzhen Virtual University Park Support Scheme to CUHK Shenzhen Research Institute; CUHK Direct Grant.
All experiments protocols were approved by the Chinese University of Hong Kong.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56:1384-1391
2. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C. et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739-746
3. Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58:1497-1507
4. Krahenbuhl S. Alterations in mitochondrial function and morphology in chronic liver disease: pathogenesis and potential for therapeutic intervention. Pharmacol Ther. 1993;60:1-38
5. Galloway CA, Lee H, Brookes PS, Yoon Y. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2014;307:G632-641
6. Xu L, Kitade H, Ni Y, Ota T. Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease. Biomolecules. 2015;5:1563-1579
7. Wasmuth HE, Lammert F, Zaldivar MM, Weiskirchen R, Hellerbrand C, Scholten D, Berres ML. et al. Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology. 2009;137:309-319 319 e301-303
8. Larrubia JR, Benito-Martinez S, Calvino M, Sanz-de-Villalobos E, Parra-Cid T. Role of chemokines and their receptors in viral persistence and liver damage during chronic hepatitis C virus infection. World J Gastroenterol. 2008;14:7149-7159
9. Casrouge A, Decalf J, Ahloulay M, Lababidi C, Mansour H, Vallet-Pichard A, Mallet V. et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest. 2011;121:308-317
10. Zeremski M, Petrovic LM, Chiriboga L, Brown QB, Yee HT, Kinkhabwala M, Jacobson IM. et al. Intrahepatic levels of CXCR3-associated chemokines correlate with liver inflammation and fibrosis in chronic hepatitis C. Hepatology. 2008;48:1440-1450
11. Zeremski M, Dimova R, Brown Q, Jacobson IM, Markatou M, Talal AH. Peripheral CXCR3-associated chemokines as biomarkers of fibrosis in chronic hepatitis C virus infection. J Infect Dis. 2009;200:1774-1780
12. Zhang X, Han J, Man K, Li X, Du J, Chu ES, Go MY. et al. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophage and autophagy. J Hepatol. 2016;64:160-170
13. Walser TC, Rifat S, Ma X, Kundu N, Ward C, Goloubeva O, Johnson MG. et al. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res. 2006;66:7701-7707
14. Romanello V, Sandri M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front Physiol. 2015;6:422
15. Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J. et al. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32:2570-2584
16. Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct. 2012;2012:646354
17. Kasai H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2'-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat Res. 1997;387:147-163
18. Baliga B, Kumar S. Apaf-1/cytochrome c apoptosome: an essential initiator of caspase activation or just a sideshow? Cell Death Differ. 2003;10:16-18
19. Yoon Y, Galloway CA, Jhun BS, Yu T. Mitochondrial dynamics in diabetes. Antioxid Redox Signal. 2011;14:439-457
20. Figge MT, Reichert AS, Meyer-Hermann M, Osiewacz HD. Deceleration of fusion-fission cycles improves mitochondrial quality control during aging. PLoS Comput Biol. 2012;8:e1002576
21. Patel PK, Shirihai O, Huang KC. Optimal dynamics for quality control in spatially distributed mitochondrial networks. PLoS Comput Biol. 2013;9:e1003108
22. Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6-13
23. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA. et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183-1192
24. Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, Parker WD Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31:430-434
25. Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659-1664
26. Demeilliers C, Maisonneuve C, Grodet A, Mansouri A, Nguyen R, Tinel M, Letteron P. et al. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology. 2002;123:1278-1290
27. Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH. et al. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47:547-557
28. Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A. et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A. 2012;109:5523-5528
29. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239-252
30. Leamy AK, Egnatchik RA, Young JD. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog Lipid Res. 2013;52:165-174
31. Lauricella M, Emanuele S, D'Anneo A, Calvaruso G, Vassallo B, Carlisi D, Portanova P. et al. JNK and AP-1 mediate apoptosis induced by bortezomib in HepG2 cells via FasL/caspase-8 and mitochondria-dependent pathways. Apoptosis. 2006;11:607-625
32. Bras M, Yuste VJ, Roue G, Barbier S, Sancho P, Virely C, Rubio M. et al. Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Mol Cell Biol. 2007;27:7073-7088
33. Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, Newmeyer DD. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell. 2009;20:4871-4884
34. Furre IE, Moller MT, Shahzidi S, Nesland JM, Peng Q. Involvement of both caspase-dependent and -independent pathways in apoptotic induction by hexaminolevulinate-mediated photodynamic therapy in human lymphoma cells. Apoptosis. 2006;11:2031-2042
35. Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587-1592
Author contact
![]() Corresponding authors: Professor Yuemin Nan, Department of Traditional and Western Medical Hepatology, Third Hospital of Hebei Medical University, Shijiazhuang, China. Phone: (86) 311 66781227, E-mail: nanyuemincom or Professor Jun Yu, Department of Medicine and Therapeutics, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, NT, Hong Kong. Phone: (852) 37636099, Fax: (852) 21445330, Email:junyuedu.hk
Corresponding authors: Professor Yuemin Nan, Department of Traditional and Western Medical Hepatology, Third Hospital of Hebei Medical University, Shijiazhuang, China. Phone: (86) 311 66781227, E-mail: nanyuemincom or Professor Jun Yu, Department of Medicine and Therapeutics, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, NT, Hong Kong. Phone: (852) 37636099, Fax: (852) 21445330, Email:junyuedu.hk