Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Cellular reprogramming: recent...
Targeted genome-editing...
Repurposing bacterial adaptive...
CRISPR editing and iPSC...
Cellular reprogramming together...
CRISPR epigenome editing
Concluding remarks
Abbreviations
Acknowledgements
References
Footnotes
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(18):4445-4469. doi:10.7150/thno.18456 This issue Cite
Review
CRISPR Genome Engineering for Human Pluripotent Stem Cell Research
Somali Chaterji1 ![]() , Eun Hyun Ahn2, 4, Deok-Ho Kim3, 4, 5
, Eun Hyun Ahn2, 4, Deok-Ho Kim3, 4, 5 ![]()
1. Department of Computer Science, Purdue University, West Lafayette, IN 47907, USA;
2. Department of Pathology, University of Washington, Seattle, WA 98195, USA;
3. Department of Bioengineering, University of Washington, Seattle, WA 98195, USA;
4. Institute for Stem Cell and Regenerative Medicine, University of Washington, Seattle, WA 98109, USA;
5. Center for Cardiovascular Biology, University of Washington, Seattle, WA 98109, USA.
Received 2016-11-22; Accepted 2017-8-24; Published 2017-10-7
Abstract

The emergence of targeted and efficient genome editing technologies, such as repurposed bacterial programmable nucleases (e.g., CRISPR-Cas systems), has abetted the development of cell engineering approaches. Lessons learned from the development of RNA-interference (RNA-i) therapies can spur the translation of genome editing, such as those enabling the translation of human pluripotent stem cell engineering. In this review, we discuss the opportunities and the challenges of repurposing bacterial nucleases for genome editing, while appreciating their roles, primarily at the epigenomic granularity. First, we discuss the evolution of high-precision, genome editing technologies, highlighting CRISPR-Cas9. They exist in the form of programmable nucleases, engineered with sequence-specific localizing domains, and with the ability to revolutionize human stem cell technologies through precision targeting with greater on-target activities. Next, we highlight the major challenges that need to be met prior to bench-to-bedside translation, often learning from the path-to-clinic of complementary technologies, such as RNA-i. Finally, we suggest potential bioinformatics developments and CRISPR delivery vehicles that can be deployed to circumvent some of the challenges confronting genome editing technologies en route to the clinic.
Introduction
Cells navigate the environment differentially in response to evolving ambient stimuli [1]. With this in mind, we as engineers, seek to predictably reprogram this ability of cells. This is accomplished by precisely constructing or finetuning cellular gene circuits [2], and of late, the cellular non-coding genome with the accrued knowledge of cis- (e.g., genomic enhancers [3]) and trans-regulators (e.g., microRNA [4, 5] and transcription factors (TFs) [6]), to rewire them to meet our end goals. The desire to induce stemness, or pluripotency, in this regard, has long been a dream for researchers. Toward this end, TFs have comprised the oft-trodden route for seeking such cellular transformations, specifically, from differentiated cellular states to progenitor or stem cell types. While the use of TFs has resulted in several success stories in the recent past, their limited precision in binding to specific DNA regulatory sequences, and the resultant unintended consequences of promiscuous binding to multiple such regulatory sites has been a stumbling block.
In terms of successes in inducing stemness, the initial creation of induced pluripotent stem cells (iPSCs), wherein a mature cell can be transformed into a pluripotent cell using a potpourri of carefully selected TFs, sparked off several use cases of such reprogrammed cells for diverse downstream applications. These range from cell-based therapies to disease modeling−from monogenic ones to complex, polygenic diseases, such as Alzheimer's and cardiovascular diseases [7, 8]. Further, the ability to transdifferentiate cells pushed the boundaries of cellular reprogramming, by forcing cells to switch lineages, without explicit dedifferentiation [9]. It is now known that the trans-differentiation events, triggered by transient exposure to pluripotency-associated factors, occur via a latent iPSC-like stage [10]. Hereby, cells navigate two so-called valleys or steady-state “creodes” in the Waddington epigenetic landscape and the process itself is inherently inefficient. Such a landscape is represented by a series of branching valleys and ridges that depict stable cellular states and the barriers that exist between those states, respectively [11]. It is coined after the proponent of epigenetics, Conrad Hal Waddington, who in 1942, described the molecular mechanisms by which the genotype modulates the cellular phenotype, recognizing for the first time that the epigenetic landscape has a causal mechanism of action on cell behavior.
In this review, we will use the word “reprogramming” specifically in reference to the formation of pluripotent stem cells (PSCs) from differentiated cell states, especially focusing on the iPSC technology. The virtual immortality of iPSC lines, coupled with their ability to preserve the pathophysiologic mechanistic features of the person they were derived from, makes them an attractive source of cells for disease modeling and personalized cell therapy.
Moving on to CRISPR synthetic endonucleases
Biologists have long been able to edit genomes with a menagerie of molecular tools. The ability to modify the genome precisely is essential to dissect the mechanistic basis of diseases. Genome editing, which first surfaced in the late 1980s [12], with further refinements in mammalian cells in the 1990s [13], is synonymously used with the terms genome engineering or gene editing technologies. The early experiments demonstrated that an exogenously provided template could result in the integration of the new strand of DNA into the genome. These early experiments used classic homologous recombination and had lower off-targeting rates. However, the low efficiency of these classic methods has prodded researchers to design more efficient approaches.
Initial use of TFs as reprogramming factors primed the field to look toward improving the precision and efficiency of the technology, with TFs giving way to zinc finger nucleases (ZFNs) and transcription activator-like effector (TALE) nucleases, or TALENs. This in turn paved the way for the repurposing of the adaptive prokaryotic immune system, consisting of clustered regularly interspaced short palindromic repeats (CRISPRs), which house short invader-derived sequence strings and the CRISPR-associated (Cas) protein-coding genes [14, 15]. This trajectory was motivated by the need to overcome the complicated design, cost, and size issues with the earlier versions of genome editing, such as meganucleases, ZFNs, and TALENs. For example, zinc fingers, which promised genome editing accurately and efficiently, cost US $5,000 or higher to order and were not widely adopted because of the difficulty to engineer them. Because CRISPR relies on an enzyme called Cas9 that uses a guide RNA (sgRNA) for targeting and then editing the nucleotide, scientists often need to order only the RNA fragment. The other components can be bought off the shelf with a total cost of as little as $30. This is what essentially paved the way forward for CRISPR as a disruptor technology and democratized the field of genome editing. This is akin to the way next-generation benchtop sequencers got us closer to the thousand-dollar genome. The Cas9 nuclease in Type II CRISPR-Cas systems [16] can cleave target DNA (or RNA) at specific sites, via double-stranded breaks (DSBs), which can then be repaired using DNA repair mechanisms, such as non-homologous end joining (NHEJ) [17] or homology-directed repair (HDR), the latter being the traditional approach of yore [12].
Adapting from the prokaryotic immune system
Interestingly, CRISPR/Cas9 systems are foreign to the eukaryotic genome and were adapted from prokaryotic immune systems [18]. The CRISPR array consists of multiple copies of a short repeat sequence, typically 25 to 40 nucleotides, separated by similar-sized variable sequences that are derived from viral or plasmid invaders. CRISPR loci are present in nearly all archaeal genomes that have been sequenced thus far and roughly half the bacterial genomes and serve as genomic memory from invading pathogens that the organism chooses to retain. These loci are transcribed and then processed to form mature target-specific CRISPR RNA (crRNA) effector complexes, which in collaboration with the scaffold-like, trans-activating RNA (tracrRNA) bind to the Cas9 endonuclease, enabling the correct conformation of the Cas9 endonuclease for cleavage to occur downstream. Thus, Cas genes are strictly found in CRISPR-containing prokaryotic genomes, and mostly, in operons in close proximity to the CRISPR loci. In their native format, CRISPRs and Cas genes function toward protecting the prokaryotic genomes from the continual onslaught of invaders. In particular, exposure of CRISPR-Cas possessing microbes to invaders results in the addition of new invader-derived sequences at the leader-proximal end of CRISPR loci in the microbial genomes. The ultimate products of the CRISPR loci are small RNAs, around 42 nucleotides in length. In the type II system, in particular, the Cas9 protein recognizes the crRNA, which then Watson-Crick base-pairs with the sequence adjacent to a protospacer adjacent motif (PAM), and also to the 80-nucleotide tracrRNA [19]. Biosynthetically, a single 102-nucleotide sgRNA, constructed as a crRNA and tracrRNA chimera was shown to enhance cleavage, in contrast to the original two-component RNA system [20, 21]. On the surface then, the CRISPR-Cas system is reminiscent of the RNA-interference (RNA-i) pathway, which has been used primarily to repress gene function, without the complete ablation of the gene. However, RNA-i piggybacks on microRNA (miRNA) and other endogenous small RNA processing pathways, including small interfering RNAs (siRNAs) and PIWI-interacting RNAs (piRNAs).
Precursor technologies prior to genome editing
RNA-i is a naturally occurring post-transcriptional gene regulatory process that is used in diverse organisms to modulate gene expression. It hinges on the use of endogenous small non-coding RNA pathways. In the native process, miRNAs are expressed from the genome as long, double-stranded primary miRNAs. These then undergo nuclear processing by the proteins Drosha and Pasha/DGCR8 (Microprocessor complex subunit), to form a stem looped pre-miRNA, which is then exported to the cytoplasm. Next, further processing by another enzyme, Dicer, generates the mature miRNA. In contrast, small interfering RNA (siRNAs) are typically exogenous in mammalian cells, often introduced by infecting viral particles. Double-stranded viral RNA is cleaved by the same pre-miRNA-processing enzyme, Dicer, to form short siRNA fragments. Thus, both siRNAs and miRNAs share similar machinery downstream of their initial processing steps. Using these interfering RNA molecules as its mediator, RNA-i carved out a niche for itself in the early 2000s. However, the technology was replete with major obstacles, with a major one being the lack of targeting precision. Furthermore, the target, for the most part, is the protein-coding genome.
The imprecision in miRNA targeting stems from the rather relaxed requirement for sequence complementarity, for example, between the miRNA's 5' end and the corresponding mRNA's 3' untranslated region (UTR). This scenario in living systems would solicit the advantage of being able to use the same regulatory RNA strings for diverse targets in order to regulate gene expression. In contrast, in a therapeutic setting, such non-canonical, or so-called seedless interactions, defined as regulatory RNA-mRNA interactions not requiring miRNA seed-based complementarity result in harder-to-predict interactions, and consequently, imprecise targeting protocols. These would then call for sophisticated predictive algorithms to rectify, as used in our recent work [4].
Motivated by the trajectory of maturation of some of these related technologies, this review first summarizes the initial successes of cellular reprogramming, wherein cells can be efficiently reprogrammed from various initial cell types (Figure 1). Next, we highlight the differences between RNA-i and genome editing technologies, focusing on CRISPR-Cas9, in relation to fostering novel cell engineering approaches. We also discuss the lessons learned from the development trajectory of RNA-i technology and how this information can whet the transition of the CRISPR-Cas9 technology to the clinic. We present some of the technical roadblocks presented in a field where CRISPR has democratized genome editing technologies. Finally, we discuss how the precision and delivery of genome editing technologies, specifically that of CRISPR-Cas9, can be improved to enable bench-to-bedside translation.
Genetically engineered stem cells and their downstream applications. Top panel: Adult stem cells, induced pluripotent stem cells (iPSCs), and embryonic stem cells (ESCs), alongside genome editing, can be used in downstream applications, e.g., tissue repair, drug discovery and safety profiling, and disease modeling. However, there are some barriers to translation and these include immunogenicity, tumorigenicity, cost-effective scalability in clinical-grade production, epigenetic variability, and clonal or subtype phenotypic diversity. Bottom panel: iPSCs need to be optimized in terms of their genetic and epigenetic features through an appropriate balance of reprogramming factors and continuous passaging [94], such that the ideal iPSC phenotype is observed. This is essentially a fully reprogrammed phenotype that abrogates epigenetic and functional differences between iPSCs generated from different somatic cell types.
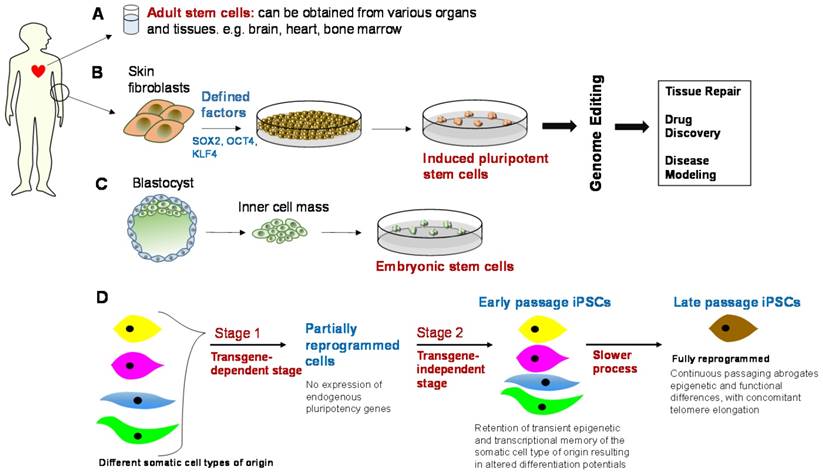
Cellular reprogramming: recent progress and challenges
Pluripotency, an evanescent attribute of embryonic stem cells (ESCs), is first encountered in the inner cell mass (ICM) of pre-implantation blastocysts and is gradually superseded by the overt differentiation of the cells into diverse somatic lineages. PSCs can give rise to all somatic lineages, that is, cells that could have arisen from all three embryonic layers, including ectoderm (e.g., neurons), mesoderm (e.g., blood or muscle), or endoderm (e.g., pancreas), and possibly, even primordial germ cells, but not an extra-embryonic trophoblast lineage. Experimental chimeras are widely recognized as the gold standard for assessing pluripotency. In vivo, pluripotency is a transient state, however, ex vivo, pluripotent cells can be derived from early embryos and can be maintained indefinitely via an optimized microenvironment of exogenous cues. Thus, pluripotency is not an irreversible feature intrinsically resident in cells, but is a transient feature in evolving cells, at different stages of pre- and post-implantation. Overall, the goal in the creation of PSCs ex vivo is to reduce the genomic or epigenomic “distance” between embryonic stem cells (ESCs), conceivably, the PSC gold standard, and synthetically derived PSCs. In addition, a reliable, essentially inexhaustible supply of quality-controlled PSCs, both for disease modeling and regenerative medicine, is required.
The different facets of pluripotency
PSCs, which include ESCs, somatic cell nuclear transfer (SCNT)-ESCs, and iPSCs, have the combined property of self-renewal and differentiation into multiple lineages; although, this clinically appealing self-renewal property needs to be preserved via the right stimuli. This is primarily achieved by promoting the expression of ESC-specific genes and suppressing differentiation-related genes [22]. ESCs are obtained from the inner cell mass (ICM) of discarded in vitro fertilization (IVF) embryos1 in their blastocyst stage [23]. In fact, by culturing an ICM of blastocysts, mouse ES cells were first generated in 1981, proliferating infinitely, while maintaining pluripotency [24, 25]. The unexpected finding that somatic cells can revert all the way back to the embryonic state using a carefully selected menagerie of TFs led to the chemical manipulation of signaling pathways to reprogram cells [26] and even to trans-differentiation events, such as transdifferentiating pancreatic exocrine cells to β-cells [27] and other transformations [9, 28].
Another induction mechanism, the SCNT induction of pluripotency, was first demonstrated in sheep with the birth of Megan and Morag in 1995, followed with the birth of Dolly, the sheep [29]. In this mechanism, the careful removal of the nucleus of an egg cell results in an enucleated oocyte. This is followed by replacement with a somatic cell's nucleus, at which point the egg's cytoplasmic, meiosis factors enable the formation of a fertilized egg nucleus [30, 31]. The altered somatic cell is then allowed to develop to the blastocyst stage and SCNT-ESCs are obtained from its ICM. Interestingly, while the SCNT process has demonstrated that epigenetic, rather than genetic, alterations underlie most differentiation processes during cellular development, cell fusion experiments (where somatic cells have been fused with pluripotent cells) have demonstrated that the pluripotent state is dominant over the somatic state in the context of cell hybrids [32]. Together, these observations led to the evolution of the iPSC technology, circumventing the limited supply of human oocytes, necessary for both ESCs and SCNT-ESCs. iPSCs are generated using the retroviral mediated insertion of the TF cocktail: Oct-4 (octamer binding protein 4) also known as POU5F1 (POU domain, class 5, transcription factor 1), SOX2, KLF4 (Krüppel-like factor 4), and MYC (collectively, OSKM) [33-35]. This cocktail was determined to be sufficient to establish a de novo pluripotency program, producing embryoid2 bodies in vitro and teratomas in vivo, and formation of diverse tissues in chimeric embryos in mouse blastocysts, and with more refinement of the protocols, complete mice upon injection into tetraploid mice blastocysts [36]. The selected TFs in the generation of these iPSCs are essentially transcriptional regulators, which were found to be active in ESCs. OSKM, or its variants, have been found to be sufficient to convert mature cells into iPSCs, affording a magnifying glass into the mechanisms driving this remarkable cellular fate change and a powerful means to model cell development in a dish. This is important because iPSCs represented a game changer in the ability to model the development processes that a defective stem cell (e.g., clinical-grade iPSC line acquired from a patient) would undergo, potentially revealing all the mechanistic transformations that can occur in the development of the pathological manifestations of the specific genotype. This could then supplement, and potentially replace in part, experiments with mice and other model organisms, typically carried out in exclusion of human cell-based experiments. This is because human stem-cell based models, recapitulating specific diseases, were simply not available. In this context, while mouse experiments have been the de facto standard for drug testing and mechanistic assays, interestingly, even for ESCs derived from mouse versus human, there are distinct differences in signaling processes manifested by the two cell types. Thinking at a coarser granularity, the number of times a mouse heart beats per minute is 600, while the human heart is roughly one-tenth of that number! It has been shown that the mouse epiblast stem cells (EpiSCs), derived from the pluripotent epiblast tissue of early post-implantation mouse embryos and possibly the in vitro counterparts of anterior primitive-streak cells, are temporally distinct from mouse ESCs and may serve as the missing link between mouse and human embryos [37].
Reversibility of “stemness” and alternate sources of PSCs
The hypothesis that the “stemness” property of a living cell is reversible was first validated via a series of seminal experiments performed by Sir John Gurdon in 1962 [38]. Other classic studies then followed, including those conducted on cells from Drosophila melanogaster, in which the “transdetermination” phenomenon was observed. Specifically, it was shown that cells from the fruit fly's genital structures could give rise to leg or head structures, and eventually, to wings [39]. In 2006 and 2007, Takahashi and Yamanaka made landmark contributions to the field by creating mouse and human iPSCs, respectively, with the introduction of several reprogramming factors, specifically the OSKM cocktail [33, 34]. Since the publication of this groundbreaking work, other TF cocktails, consisting of factors such as OCT4, Nanog, SOX2, and LIN28 (ONSL), and of OSK, have been reported [40]. By altering the composition and stoichiometry of the iPSC-generating cocktail, among other input factors, the efficiency of the iPSC-generation process and the quality of the iPSCs can be controlled effectively. Attempts have also been made to computationally finetune this process by using a recently developed network-biology platform, CellNet, to assess the gene-regulatory networks produced by different TFs [41]. The revolutionary discoveries that initialized the generation of these iPSCs won John Gurdon and Shinya Yamanaka the Nobel Prize for Physiology or Medicine in 2012. With this discovery, and others [42], came the ability to surmount the ethical controversies encircling the use of ESCs, notwithstanding that ESCs are derived from the ICM of blastocysts. Although, technical challenges exist in the use of ESCs. For example, ESCs from an allogeneic source can result in HLA (human leukocyte antigen)-based immune rejection, limiting their utility for cell-replacement therapies. The ability to generate patient-specific iPSCs alleviates this immunogenicity problem, effectively removing a significant barrier in the translation of cell-based therapies to the clinic. Another, less easy issue to address with ESCs, is their tumorigenicity [43], which can be thought to be intricately tied to the very hallmarks of pluripotency.
iPSC quality control
Since the initial generation of iPSCs in 2006, many research groups have created iPSC lines, extending to different cell types and emanating from different patient pools, including from other species, and identifying new TF combinations that increase the efficiency of iPSC production (see [35] for a recent review). Further, the most common source of human iPSCs is dermal fibroblasts [44], stemming from their ease of access and programmability efficiency, with peripheral blood [45], cord blood [46], and Epstein-Barr virus-immortalized B-cell lines also emerging as practically attractive sources, with the latter affording the opportunity to acquire iPSC donors from biobanks [47]. The most technically rewarding feature of iPSCs is that they can recapitulate the pathophysiologic background of the patient from which they are derived, for example, ordinary skin cells can be derived from a patient and then converted to iPSCs for further processing. This results in the potential to create personalized disease models to recapitulate distinct human disease phenotypes, and then, to be able to perform gene corrections (e.g., genome editing), and to personalize therapeutic screenings. This feature is also shared by SCNT-ESCs, albeit, constrained by the limited supply of embryos for research use. Notwithstanding, the SCNT technique is now back as the alternate PSC-generating technology on the block, given its more recent accomplishments in primate and human SCNT systems, stymied in the past by the arrested growth of SCNT-derived embryos [31]. This resurgence of SCNT-ESCs was also fueled by findings related to the presence of hotspots of aberrant epigenetic programming, such as regions around telomeres or centromeres or aberrant imprinting at specific gene clusters (e.g., Dlk1-Dio3 cluster [40]). Such aberrations may result in greater molecular differences between iPSCs and ESCs than desirable for clinical applications. Furthermore, not all PSCs are alike [37], they could be naïve or they could be primed, for example [48], and in their naïve state they are closer to ESCs (for a recent review, see [49]), and therefore, possibly more beneficial from a translation standpoint. Thought-provokingly though, there could be a dark side to this naïve pluripotency. Rat embryonic stem cells (ES cells), expanded in cytokine leukemia inhibitory factor (LIF)-containing feeders, tend to acquire genetic abnormalities. This could potentially arise from the increased activity of endogenous retroviral elements or jumping genes [50], or, the reduced activity of repressive methylating marks, which is what confers the naïve pluripotency in the first place [51]. This, in fact, points to the possibility that with the appropriate use of predictive technologies, coupled with laboratory validation, iPSCs could be made more similar to SCNT-ESCs. Thus, SCNT-ESCs can be viewed as a complementary technology, rather than as a competing technology, wherein differences in the epigenomic profiles of SCNT-ESCs and iPSCs could be minimized using the right combination of TFs, supplementing or supplanting parts of the original OSKM cocktail, as the need may be with other TFs or miRNAs or small chemical compounds. In addition, while initially SCNT-ESCs were considered a panacea for patients with mitochondrial disorders, recent studies have shown that mitochondrial-mismatched stem cells when reintroduced into the cells from which the donor nuclei were obtained can cause mitochondria-related antigenicity [52]. Notwithstanding these technical caveats, this increased availability of disease-reminiscent cells from actual patients has a transformative potential in disease modeling, informing both drug discovery and cell-therapy advances, and there are pros and cons of both technologies that can be harnessed to improve the state of the art. Further, the combination of high-throughput karyotyping assays alongside algorithmic tuning can hone the potential of these derived pluripotent cells. In this context, unlike the more elusive differentiation recipes for ESCs and the still-emerging advances in SCNT-ESCs, the iPSC technology has already attained significant maturity, is elegant in its simplicity, and may also afford greater reproducibility in differentiating into various target phenotypes. Thus, iPSCs have been used to develop “disease-in-a-dish” models for benchmarking various proposed therapies, in a patient-specific and disease-specific manner and can benefit from an alliance with the slew of maturing genome editing technologies, such as variants of the CRISPR-Cas components [53, 54]. Furthermore, newer versions of the endonucleases, such as CasX and CasY, both of which are smaller than the conventional type will, in effect, possibly make the recent CRISPR-related patent litigations over CRISPR-Cas technology moot. Moving forward, we first discuss the translational challenges of the iPSC technology and the evolution of the CRISPR-based genome editing technologies, to understand the burgeoning potential of such an alliance, some of which is just beginning to be unearthed with the march toward precision medicine, and with it, the ensuing flourish of novel computing infrastructures and technologies, such as those engineered by our group [55, 56].
A relatively recent success story from such an alliance is the NHEJ-mediated correction of iPSCs derived from dominant dystrophic epidermolysis bullosa (DDEB) [57], a rare, dominant negative, blistering skin disorder, with no current cure. Given that CRISPR genome editing technologies are at the brink of new and improved clinical trials, especially in the realm of cell-based technologies [8], we will focus this review on featuring the advances in CRISPR-based genome editing technologies.
Targeted genome-editing technologies and their modus operandi
Designer nucleases have energized advances in genomic medicine by enabling the targeted manipulation of specific genomic sequences. Their basic strategy is essentially the same. It involves directing a DSB in the desired genomic locus in an RNA-guided fashion, followed by post-scission repair mechanisms. As described earlier, this could be via the error-prone, albeit predominant mechanism, NHEJ, or, the less frequent, HDR pathway. The CRISPR technology, the latest of several customizable genome-editing approaches, is a more flexible gene editing platform, with the most commonly used form being the SpCas9 nuclease, acquired from Streptococcus pyogenes. Cas9 molecules from other species, Cas9-like CRISPR nucleases and engineered versions of Cas9 with novel functions have also been established and can convey particular advantages in diverse settings, as described in a recent review [54].
The origins of the CRISPR-related research can be traced back to 1987 when Nakata and colleagues discovered a set of interspaced short repeat sequences, in proximity to the Escherichia coli iap (Inhibitor of Apoptosis) gene, which is responsible for the isozyme conversion of alkaline phosphatase [58]. As a programmable form of bacterial immunity molecular machinery, with the first experimental evidence of the existence of these type II CRISPR-Cas systems in 2007 [15], repurposing for use in mammalian cells was pioneered around the same time, in 2013, by two groups [21, 59]. This class of nucleases differs from the three other major classes of nucleases: meganucleases, ZFNs, and TALE nucleases (or TALENs), primarily in that the CRISPR-Cas system does not require extensive protein engineering and can be tailored simply by altering the guide RNAs (sgRNAs). ZFNs and TALENs are chimeric enzymes, consisting of a DNA-binding sequence fused to a sequence-agnostic FokI DNA-cleaving nuclease domain. This FokI enzyme, naturally found in Flavobacterium okeanokoites, is a restriction endonuclease that must dimerize for DNA cleavage to occur [60]. ZFNs have proven difficult for non-specialists to synthesize from scratch because of the challenge in assembling zinc finger domains that can bind to a string of nucleotides. Attempts by the Zinc Finger Consortium (http://zincfingers.org/) have enabled efforts at improving the technology while bounding the costs associated with the technology. So, while re-targeting of these ZFNs and meganucleases requires elaborate protein engineering and TALENs require complex molecular cloning, the use of in vitro transcribed nuclear localization signals (sgRNAs) that can be made to target any 20-bp nucleotide sequence, makes the CRISPR-based systems essentially cloning-free [61]. Further, unlike the FokI enzyme that operates as a dimer in ZFNs and TALENs, the Cas9 endonuclease acts as a monomer to induce the DSB in the target nucleotide. Also, the Cas9 gene is small compared to a pair of TALEN genes, roughly 4.1 kbp versus 6 kbp, making delivery simpler. Finally, due to the small size of the CRISPR guide RNA, it is possible to multiplex gene targeting, simultaneously affect multiple genetic loci [21, 62], and potentially dissect the mechanisms of a swath of complex, polygenic diseases in their native contexts, such as via saturation mutagenesis [63].
Repurposing bacterial adaptive immunity for genome editing in mammalian cells
In more advanced living organisms, such as humans, pathogens are detected by antibodies and cells, such as B- and T-lymphocytes. In lower living organisms, such as in bacteria, other creative processes fight the constant onslaught of predators. Predation by mobile genetic elements, such as phages, plasmids, and transposons, on bacteria, is ubiquitous. This has promoted the deployment of creative defense systems, including CRISPR-Cas arrays, in bacteria and other prokaryotes to fight predation. Now, the type II CRISPR-Cas systems have been adapted to enable efficient genome editing in a wide range of cultured cells and organisms, with its most widely used form consisting of the Cas9 enzyme and a single guide RNA (sgRNA, which is ~20 nucleotides in length) that mimics the natural hybrid of the crRNA and the tracrRNA. Target recognition by the Cas9-sgRNA complex requires Watson-Crick base pairing with the sgRNA's 5' end as well as a short PAM sequence, located immediate downstream of the target DNA sequence and varying in sequence features among CRISPR-Cas orthologs found in different bacterial species. Further, the Cas9 nuclease contains two conserved HNH and RuvC endonuclease domains, which when inactivated (via point mutations), results in the dCas9 enzyme variant, with removal of one of the domains creating a Cas9 nickase. The dCas9 nuclease-deficient variant retains the full DNA binding activity, albeit, losing the DNA cleaving activity.
Stages in the CRISPR-attack mechanism in bacteria and how to engineer the sgRNA
The CRISPR-Cas system, as a second-line-of-defense, confers adaptive immunity to the bacteria, presenting a heritable and chronologically-captured account of past invasions, without sacrificing fitness.
The CRISPR-attack mechanism can be summarized in three execution steps, where the first stage can be likened to an “information-processing subsystem” and the second and third stages can be grouped into an “executive subsystem.”
Stage 1 - CRISPR adaptation
This stage involves the genetic memory and recognition of alien DNA by dedicated Cas proteins, followed by processing and integration into the CRISPR locus (Figure 5). This stage can be subdivided into two steps: the selection of a protospacer−short piece of DNA typically around 30 bp in length homologous to viral or plasmid DNA−followed by the generation of spacer material and the integration into the CRISPR array with the synthesis of new flanking repeat sequences. The short (3 or 4 bp) PAMs located immediately downstream of the protospacer appear to determine the protospacer selection, followed by integration into a pre-existing CRISPR array. The alien DNA is processed into small spacer elements by assistive Cas proteins and encoded into the CRISPR-Cas system. The elements are subsequently inserted into the CRISPR locus toward its leader sequence, which may, in some cases, necessitate the destruction of some obsolete spacers in the array in order to bound the size of the CRISPR array. Therefore, a chronological record of the integration of the spacer sequences reflects the hierarchy of previous encounters with mobile genetic elements (bacterial pathogens). The CRISPR array evolves by deleting redundant or obsolete spacers. Each of these new spacer sequences matches some section of the infecting phage genome, referred to as a protospacer. Although the location of the protospacer sequence in the pathogen's genome is random, it is always just a few base pairs from the short PAM that is recognized by the CRISPR system. This latter step is a way for discriminating “non-self” from “self” genetic material, thus making the system self-protecting.
Primary steps of CRISPR-Cas-based immunity. The mechanism of CRISPR-mediated interference can be summarized in three execution steps consisting of an information-processing subsystem (CRISPR adaptation) and a two-part executive subsystem (CRISPR expression and CRISPR-based interference). In the first step, adaptation, new spacers are inserted into the CRISPR locus and can either be naïve or primed acquisition, the latter resulting in acquisition of spacers from the same mobile genetic element. In the latter two steps, transcription of the CRISPR locus and processing of CRISPR RNA occurs, followed by the detection and degradation of the pathogen or mobile genetic elements by CRISPR RNA.
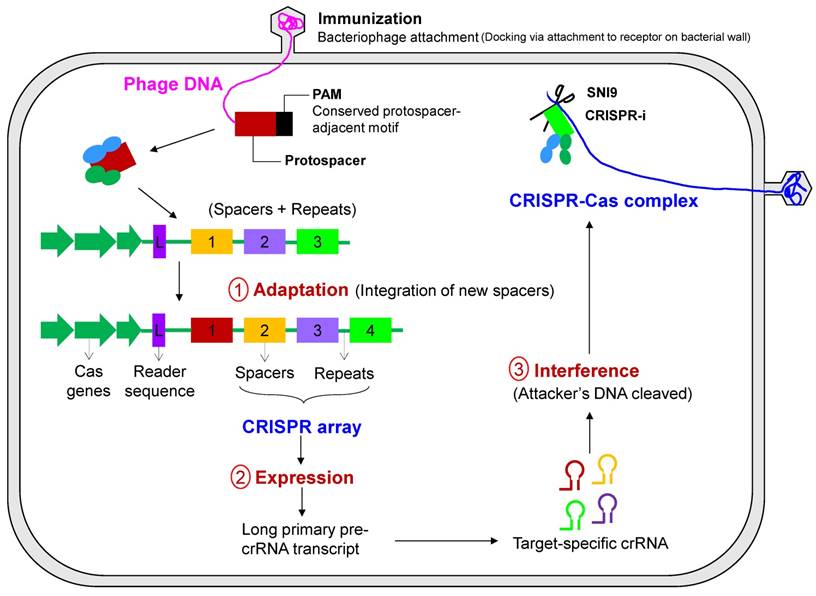
Stage 2 - CRISPR expression and processing
CRISPR expression is the transcription of the precursor CRISPR-RNA, pre-crRNA (longer transcripts) that are sequentially processed to small crRNAs that actually do the work. This step is catalyzed by endoribonucleases encoded by the Cas genes that may either operate as a subunit of a larger complex, as in the Cascade complex in E. coli [64], or may operate as a stand-alone enzyme as in Cas6 in the archaeon P. furiosus [65]. These crRNAs act as guide RNAs for different interference modules that target and cleave genetic material after annealing to the complementary protospacer sequence of the invading (pathogenic) element.
Stage 3 - CRISPR interference
To correctly position the CRISPR attack, CRISPR interference involves the potential degradation of the target nucleic acid (DNA or RNA) by the CRISPR nuclease that recognizes the pathogen's DNA (target DNA sequence) and the corresponding PAM. The cleavage by the ribonucleoprotein complex, consisting of the crRNA guide RNA and a set of Cas proteins, occurs at or in the vicinity of the PAM sequence. Interestingly, while bacteria have devised mechanisms to ward off infection, viruses can sometimes deceive the CRISPR-Cas system of the host by randomly mutating key bases in the CRISPR-RNA interaction or PAM recognition step. As an example, the integration of a virus protospacer into the host DNA can actually spur the pathogen's invasion, such as in the case of the cholera virus, tricking the CRISPR-Cas system into actually enabling the viral infection [66].
When deciphering the above CRISPR-Cas mechanisms, researchers noticed that the guide RNA in the system, which recognizes the viral nucleotide, can be engineered to recognize any target nucleotide, not just viral nucleotides, guiding the nuclease to snip the specific target, at which point the mutant gene can be replaced with a healthy copy. This is the basis of the use of CRISPR-Cas in eukaryotic systems, all of which can be done in cultured cells and fertilized eggs, allowing for the generation of transgenic animals with genes knocked out.
Barriers to clinical translation of the repurposed guide RNA
While the CRISPR-Cas system presents promising approaches to the evolution of genomic medicine, there is increasing concern that changes introduced by genome editing can be heritable, making off-target effects more alarming.
CRISPR off-targeting
Off-target effects of these systems in the bacterial and archaeal worlds, from where these were derived, may in fact be a beneficial mechanism. Specifically, off-targeting can help these prokaryotic organisms recognize and cleave hypervariable DNA from predators, optimizing immune surveillance using some optimization function, e.g., genetic algorithms. However, when repurposed in eukaryotic systems, these same defensive mechanisms undermine specificity [19, 67] and reduce cellular fitness [68]. Attempts are being made to improve the specificity of CRISPR-Cas systems through both biochemical methods [21] and computational algorithms [69]. In particular, Cas9 targeting is modulated by the 5' variable region of the sgRNA, which hybridizes to the complementary protospacer motif [70]. While scoring models have been developed based off experimental binding data [71], genome-wide unique sgRNA sites have also been identified [72]. While in the S. pyogenes Cas9 variant, the PAM sequence is an “NGG”, tools for coming up with optimal protospacer designs are also available, based off the criteria of on-target editing efficiency and off-targeting at undesired genomic locations [71, 72]. Thus, the presence of off-target effects and possible genome editing-derived oncogenicity indicates that the technology is still in a fledgling state, as indicated by the observation that the CRISPR/Cas9 technology used to modify the hemoglobin B locus in human zygotes was at a highly inefficient frequency [73]. In this context, it may be mentioned that the number of off-target events that can be tolerated is application-dependent. When the off-target effects are introduced by a Cas9 endonuclease versus when using dCas9, the latter may be less deleterious. This is because dCas9 typically affects the transcriptional properties of the genome without introducing permanent (heritable) changes. Finally, sgRNAs themselves may vary in specificity, from being highly specific to being the poster child of a “promiscuous” sgRNA. Thus, modified sgRNAs may be required, as illustrated by the sgRNA for VEGF-A, whose off-target effects were studied using Digenome-seq [74], which can detect indels with a frequency as low as 0.1% and lower. Indels can also be identified using Guide-seq (genome-wide DSB detection), where barcoded DNA pieces are inserted, followed by high-throughput sequencing [75]. Also, translocation events can be determined by Guide-seq and high-throughput, genome-wide, translocation sequencing (HTGTS) [76]. This, notwithstanding the fact that the mining of glorious volumes of these NGS datasets, which are both varied and on the rise, is expensive. Complicating this scenario further is the fact that there could be cell-type specific DSB hotspots and even unique DSB hotspots in the same cell type from different individuals. For the former, cell-type specific empirical validation is called for, and, for the latter, recombination initiation, high-resolution, individual-specific maps are useful.
Finally, the premise for genome editing of human somatic cells is that corrective changes to a sufficient number of defective cells could offer a once-and-done therapy for the patients. However, while increasing the dose of the nuclease may increase the probability of the mutated gene being corrected, it comes at the cost of simultaneously increasing the risk of cuts being made elsewhere in the genome, especially when contemplating in vivo applications. Thus, it is safer to start with the application of genome-editing technologies on somatic cells, rather than in human germline cells, as is recently becoming a more visible application, albeit, with limited success [77].
CRISPR editing and iPSC technology: An alliance to foster technology translation
The CRISPR technology has been advancing rapidly, and since 2013, there have been multiple reports of successful repurposing of the CRISPR technology for human gene editing [20, 21, 61, 67, 78]. Given the ubiquitous use of murine models for researching human disease phenotypes and the known genomic and physiologic differences between mice and men, iPSCs, which are normal primary cell lines, bring forth a radically new way of understanding human disease mechanisms. In particular, human iPSCs offer an unprecedented means to perform both disease modeling and personalized cell replacement therapy. Such applications have received a further boost, fostered by the alliance of iPSC technology with genome editing and with the refinement of the genome editing protocol, initially reported to be roughly 1-2% in human iPSCs3 [21]. What started off in human cells with the introduction of the Cas9 expression vector and the crRNA and tracrRNA ensemble was further simplified by combining the two RNAs into one chimeric RNA [61].
Notably, while immortalized human tumor cell lines have been edited with very high efficiency [79], the success rates in human iPSCs have been much lower [59, 80], which may presumably be the resilience to DNA damage in tumor cell lines. Thus, efforts have been made to maximize the efficiency of genome editing in iPSC cell lines [81]. These design considerations start from the very choice of the iPSC cell lines. For example, low passage number iPSCs would have low karyotypic abnormalities. However, these cell lines may also retain greater degrees of similarity to the differentiated cell type from which the iPSC may have been derived. There may be a sweet spot here that can be algorithmically determined, for example. Next, the plasmid donor vectors need to be carefully selected and it has been found that polymorphic differences between the plasmid vectors and genomic loci will decrease targeting efficiencies [82]. Also, somewhat intuitively, insertion vectors are preferred over deletion vectors [83]. Finally, once the genome of the iPSC cell line has been edited, validation of the editing efficiency is needed and some guidelines from past efforts at characterization can be employed [84], such as pluripotency tests, karyotype analysis, gene expression profiling and epigenetic analysis.
iPSC Technology and its Challenges
In terms of PSCs, iPSCs have come under fire from researchers, calling for a rigorous evaluation of the safety profile of stem cells before banking on iPSCs, quite literally [85], for use in downstream applications. Even from the standpoint of iPSC creation, reliance on viral vectors as the most efficient means of delivering reprogramming factors risks insertional mutagenesis, which could also affect downstream differentiation [86]. In addition, random integration of these foreign viral elements into the iPSC genome can create distinct iPSC lines, which is not desirable. While alternate approaches are being researched [87, 88], including the first reports of reprogramming using non-integrating vectors [89, 90], the creation process will need to be streamlined, patient recruitment protocols need to be established [91], and possibly more convenient sources of cells need to be identified (e.g., cryo-preserved blood samples [92]), prior to industrializing the process for clinical translation.
Furthermore, recent reports have debated as to whether SCNT-ESCs may afford a better source of derived-ESCs [30]. This is important because SCNT-derived ESCs have been thought to be very similar to conventional ESCs, as determined by tetraploid complementation assay (TCA), the most stringent test of pluripotency, compounded by the recent exome-sequencing findings that have indicated significantly lower mutational load in SCNT ESCs relative to iPSCs of syngeneic background [93]. Further, a bottleneck here has been the ability to reduce the variability between created iPSC lines. Such differences are mostly caused by differences in the genetic backgrounds of the cell types, and more so, on the reprogramming protocol that the cell lines have undergone because the effects of varying genetic backgrounds can be abrogated via prolonged culture [94]. Here, the ability to genome-edit otherwise isogenic cell lines to ensure that control and diseased cell lines have the same genetic background is beneficial. This presence of isogenic cell lines is crucial for low-effect loci discovered through the genome-wide association studies (GWAS) because, in such cases, as is often encountered for complex, polygenic diseases, the difference between the normal and diseased phenotypes may be subtler, and that too, attributable to multiple genomic loci. Thus, such isogenic pairs of disease-specific and control iPSC cell lines, such as in creative clinical trials-in-a-dish formats [21], would enable the cost-effective translation of iPSC-based technologies, even in the context of harder-to-model, complex diseases, with multiple low-effect disease loci.
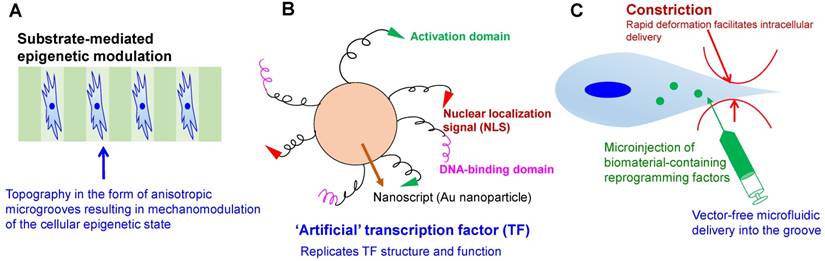
Another requirement for the clinical adoption of iPSCs is the use of non-integrating approaches to generate iPSCs, which in fact have been demonstrated to have different reprogramming efficiencies, success rates, and genomic integrities [95]. Further, yet another, and harder-to-surmount challenge, is the contested similarity of iPSCs to ESCs in their gene expression profiles [96], epigenetic lineage memories [97], and proteomic profiles [98]. This propels the controversy surrounding the use of iPSCs as ESC surrogates in the first place. In this context, spatio-temporal control of reprogramming factors released using various biomaterial-based strategies may afford a novel and more versatile channel of delivering reprogramming factors or small molecules for generating iPSCs [99] (Figure 2). In addition, mechanistic explorations [21], abetted by bioinformatics tools [41], can also help maintain the genomic stability of an otherwise physiologically unstable process of cellular reprogramming. Finally, in the past iPSCs were considered mostly for monogenic diseases. However, given the surge of genome editing-based technologies, genome editing can enable the use of iPSCs toward the cure of polygenic, complex diseases, which result from the presence of multiple mutations in the genome, as opposed to a single mutation [100]. In this context, it may be appreciated that the ability to source iPSCs from the diseased cell mass in a patient will also preserve tumor heterogeneity, which has been increasingly studied boosted by the surge in single-cell omics technologies [101]. This is important because such clinical extracts afford a realistic window into diverse populations of diseased cellular ensembles.
Engineering strategies toward enhanced efficiency and safety in cellular reprogramming. (A) Microtopography-induced mesenchymal-to-epithelial transition in adult fibroblasts, as seen in [196]. (B) Artificial transcription factor-based transcriptional activation and consequent reprogramming; e.g., Nanoscript [99], which regulates the multi-domain structure and gene-regulatory function of natural TFs. (C) High-throughput microfluidic technology for rapid mechanical disruption of the cell, enabling the intracellular localization of reprogramming factors. Alternatively, microinjection or microfluidic electroporation can be used to localize the effects of pulsed electric fields.
Major applications of CRISPR editing in cell engineering approaches
CRISPR-based refinement of iPSC creation and refinement protocols
One of the primary challenges of the iPSC technology is the epigenomic variation of the derived iPSC cells [102] and this is one of the areas where the dCas9 mutant, in conjunction with epigenetic modifiers [103], can facilitate quality control of the generated iPSCs. Further, gene activation using the same dCas9, albeit, in activator mode, can be used to control differentiation regimens of the iPSCs [104]. While, in theory, iPSCs can result in a slew of differentiated cell types, such as neurons, hepatocytes, or cardiomyocytes [105, 106], a lot of these processes are still inefficient, produce heterogeneous cell populations, and need optimization for high efficiency, reproducibility, and scaling up.
CRISPR-based genome screening
Classic genetic screens, whether forward or backward screens, ascribe functionality to the different genes. While forward genetics identifies genes responsible for a specific trait or phenotype, reverse genetics analyzes the phenotype of an organism following the disruption of a known gene. It is hard to simultaneously gauge the effects of the 22,000 or so genes in the human genome and this is where the advantages of high-throughput screening using CRISPR comes to the fore. While RNA-i has played a prominent role in high-throughput screens in the past, incomplete knockdown, ability to target only coding regions [107], and off-targeting have dampened the results. In comparison, CRISPR can create frameshift mutations in the coding regions, using NHEJ [108], or even mutate non-coding regions [109], genome-wide [110]. This is important, given the surge of findings showing the importance of the non-coding regions as disease drivers; see the recent review on the influence of non-coding variants in cancer for a summary of some of these revelations [111]. Importantly, in the case of editing non-coding genomic elements, functional knockouts with a single sgRNA is not practical, with exceptions [112]. Instead, two sgRNA have been used to precipitate simultaneous breaks flanking the target region, resulting in a well-defined genomic deletion [113] and scalable tools for designing these paired sgRNAs are also on the horizon [114]. Thus, CRISPR screens have been used for identifying non-coding cis-regulatory elements, such as in [103]. Furthermore, CRISPR-mediated screens can be both loss-of-function or gain-of-function screens, depending on the type of domain that is fused to the denatured Cas9 enzyme [115]. Finally, the ability of CRISPR-based screens to interrogate gene regulatory networks [116] is a useful tool for validating bioinformatics-based mapping of such networks.
Chimeras and organoids
While genome editing has the power to study complex diseases and even remove the scourge of organ shortages by bringing Margaret Atwood's pigoons4 from the novel “Oryx and Crake” to life, it is important to move with caution to avoid enthusiasm in genome editing from biasing the necessary scientific exploitations and explorations needed to sound out the technology. For one, it may be better to term these so-called human-pig chimeras, “genome-edited pigs”, rather than “pigoons” to avoid the cynicism associated with the creation of animal chimeras, in general. Consider this, raising genome-edited pigs for acquiring organ transplants, rather than having to wait for donated organs from fatally injured young humans, such as those killed in road accidents. Every day, about 22 people in the United States die waiting for an organ transplant (https://www.organdonor.gov/statistics-stories/statistics.html). In the arena of inter-specific chimeras, a team, led by Izpisua Belmonte of Salk Institute for Biological Studies, began by combining genome editing with stem cell biology, two revolutionary platform technologies [117], and using the pig as an “animal incubator”. A similar experiment was first carried out in 2010, when Japanese scientists produced a mouse from rat PSCs, so inter-specific chimeras, contributing to xenogeneic development [118]. Such experiments, motivated by the fact that it is challenging to regenerate entire organs (for organ replacement needs) in vitro, have not steered clear of controversy. Notwithstanding, these experiments are promising because interactions between cells and tissues is critical for organogenesis and creating the complex microenvironment in vitro for actual transplantation purposes is daunting. This, is different from the need to generate synthetic microenvironments for mechanistic revelations, where simplified environments may actually be desirable when teasing out the individual mechanistic factors, rather than being on the lookout for holistic revelations about the entire niche. Importantly, the 2010 experiments used Pdx1-/- mice for the studies to knock out the pancreatic and duodenal homeobox1 TF essential for pancreatic development and β cell maturation, and thus, created an open pancreatic developmental niche for accepting the donor cells. Of note here is that homozygous deletion of Pdx1 in mice results in death due to pancreatic insufficiency. These rat-mice pairings were followed by demonstrations of organ-specific pairings in other interspecies chimera, in close succession [119, 120], essentially indicating that developing an organ of a certain species inside the body of a different species is possible if the correct microenvironment is established. Belmonte's group took these experiments a step forward by using CRISPR editing to turn off the mouse gene that makes the pancreas [117]. Then, rat PSCs containing the intact pancreas gene were inserted into the surrogate mice, resulting in the mice “incubating” rat (xenogeneic) pancreases.
DSB repair mechanisms and endonuclease variants
Using genome editing, one can create either indel mutations at the break site using NHEJ or introduce “knockin” alterations using HDR. NHEJ is error-prone and introduces SNPs or small insertions or deletions (indels). These indels cause frameshift mutations, resulting in functional gene knock-outs [121]. In comparison, HDR inserts a desired sequence, combining a target locus with an exogenously supplied genomic fragment at one or multiple locations of the genome [21]. Further, while NHEJ is active throughout the entire cell cycle, and therefore easier to exploit, HDR faces competition from NHEJ, being active primarily during the S/G2 phase. This competition is worrisome because in the context of gene editing to treat sickle cell anemia, for example, the repair of the DSB by NHEJ, as opposed to HDR, could result in an allele similar to that seen in β-thalassemia, requiring suppression of NHEJ, or enhancement of HDR-like mechanisms [122].
Cas9 variants
While the wild-type Cas9 introduces a DSB via its two nuclease domains, namely, RuvC and HNH domains, Cas9 nickases with a point mutation in one of the two domains only cleave a single DNA strand. Alternately, these nickases can be paired such that a pair of offset sgRNAs, complementary to opposite DNA strands are nicked [123], and then, these nicked sites are repaired by the high-fidelity base excision repair mechanism (BER); BER reactions in cells are extremely fast, with an individual BER event often occurring in a matter of minutes [59]. Notably, most nicks result in very low indel rates resulting in effective targeted gene disruption [124, 125]. Now, if instead of mutating one of the nuclease domains, both domains are mutated, the result is a dCas9 without any nuclease activity. The dCas9 is created via two point mutations in both its RuvC-like (D10A) and HNH nuclease (H840A) domains, and this mutant Cas9 is devoid of endonuclease activity. However, this dCas9-sgRNA complex can terminate transcription elongation, as confirmed by native elongating transcript sequencing (NET-seq) experiments. In this case, the binding of the sgRNA to the promoter region can sterically prevent the association between integral cis-acting DNA motifs and their cognate trans-acting TFs, switching off transcription initiation. In this mechanism, commonly termed CRISPR-i, and in many ways analogous to RNA-i, repression efficiency can be modulated by the number of mismatches in the sgRNA base-pairing region [110, 126]. Furthermore, using a complementary mechanism (CRISPR-a), effector domains to activate the transcription of target genes can be employed [127].
Evolution of genome editing: CRISPR lessons from RNA-i and CRISPR-i
RNA-i is a conserved endogenous pathway that affords the sequence-specific silencing of the defective gene by knockdown of the target mRNA. However, as described before, its utility has been hampered by incomplete gene knockdown, extensive off-target effects, requirement of host-cell factors, and experiment-to-experiment variability, making downstream phenotype prediction challenging. On the other hand, the CRISPR-Cas system, combines the permanent mutagenicity of conventional mutagens with the relatively simple RNA-i programmability. Further, whereas RNA-i is not useful for diseases in which the complete ablation of gene expression is essential, the CRISPR technology can completely knock out genes, without leaving behind “scar” sequences. Alternately, it is also able to function in an RNA-i-reminiscent manner when deploying a catalytically dead, or, nuclease-deficient, hence non-cleaving, Cas9 mutant, dCas9, from catalytically deadCas9 [128], resulting in CRISPR-interference or CRISPR-i. Thus, in addition to Cas9-based total loss-of-function, CRISPR-i and CRISPR-a can facilitate partial loss or gain of function [127].
The safe and effective delivery of these RNA-i molecules is another issue [129]. With lessons from the delivery issues encountered in the development of RNA-based therapeutics [130], scientists can use the CRISPR technology to directly manipulate any gene in diverse cell types and organisms with enhanced precision and completeness. In this context, it may be noted that, in the case of CRISPR, the delivery of these nucleases may either be carried out on their own or with so-called donor DNA. The latter process resulting in new genetic information being added through the surrogate HDR substrata. Further, alternate forms of programmable enzymes can also be complexed to the DNA-binding domain, when using dCas9 variants, resulting in site-specific recombinases [131] and transposases [132]. If desired, CRISPR-technology variants can also cleave RNA, instead of DNA, thus resulting in non-heritable changes [65], adding to the versatility of the technology.
Mechanistic differences between RNA-i and CRISPR-Cas9
RNA-i and CRISPR-Cas9 technologies share similarities, with both methods utilizing small RNAs with high desirable levels of on-target specificity [14, 59]. However, their molecular mechanisms are intriguingly different, as outlined in Table 1. RNA-i operates by honing in on the endogenous miRNA-processing pathway, using near-perfect complementarity with the target mRNA [133]. In fact, depending on the degree of sequence identity between the miRNA and its target, the nature of the regulatory effect can be different. For example, limited complementarity can result in mRNA deadenylation or decay, while extensive complementarity can result in slicing, meaning complete cleavage. Further, seedless miRNA interactions account for upwards of 90% interactions, as gleaned from cross-linked immunoprecipitation followed by high-throughput sequencing (CLIP-seq) data. Driven by this, we have developed machine learning (ML) algorithms, specifically kernel support vector machine (SVM) models, to predict these accurately and in a computationally efficient manner [4]. Similar technologies are on the rise to predict the off-target bindings of CRISPR and other adaptive nucleases [69].
Comparison between RNA-i and CRISPR action
| RNAi | CRISPR action | |
|---|---|---|
| Target molecule; On-target nucleotide size | mRNA; 18-20 nucleotides | DNA, mRNA; 18-20 nucleotides |
| Source of system | Human endogenous miRNA-processing pathway | System for resistance against viral infections in bacteria |
| Outcome | Silencing of genes at mRNA level; Reversible knockdown (But, not applicable for phase III therapies) | Inactivation of genes at DNA level; Blockage of RNA polymerase; Reversible knockdown |
| Loss of function mechanism | Post-transcriptional RNA degradation. Target mRNA is sequestered or degraded via endonucleolytic cleavage or deadenylation | Regulates gene expression mainly on the transcriptional level. Repression of transcription: steric blockage of RNA polymerase; action of optional repressive chromatin modifying transcriptional repressors |
| Guiding sequence | siRNA or shRNA | sgRNA |
| Number of required components; Transgenes involved | One; siRNA or shRNA | Two or three; dCas9, sgRNA, optional transcription repressor (e.g., KRAB of Kox1, CS of HP1α, WPRW motif of Hes1) |
| Required sequence info | Transcriptome | TSS |
| Off-target effects | Extensive | Limited |
| Affected off-target space | Transcriptome | Window around TSS |
| Ability to target small RNAs | No | Yes |
| Used in pooled genome-wide screens | Yes | Yes |
| Requirements for targeting | RNA sequence complementarity | RNA sequence complementarity, PAM immediately 3' to the target sequence |
| Transcript variants | mRNA of the transcriptome with partial sequence complementarity | Only variants resulting from cleavage at a narrow window around the TSS of genes |
| References | [133, 138, 197-199] | [14, 15, 61, 158, 200] |
Abbreviations used are: miRNA, micro RNA; dCas9, dead CRISPR associated protein 9; KRAB, Krüppel-associated box; CS, chromoshadow; siRNA, short interfering RNA; sgRNA, single chimeric guide RNA; PAM, protospacer adjacent motif
Molecular machinery of CRISPR-Cas systems
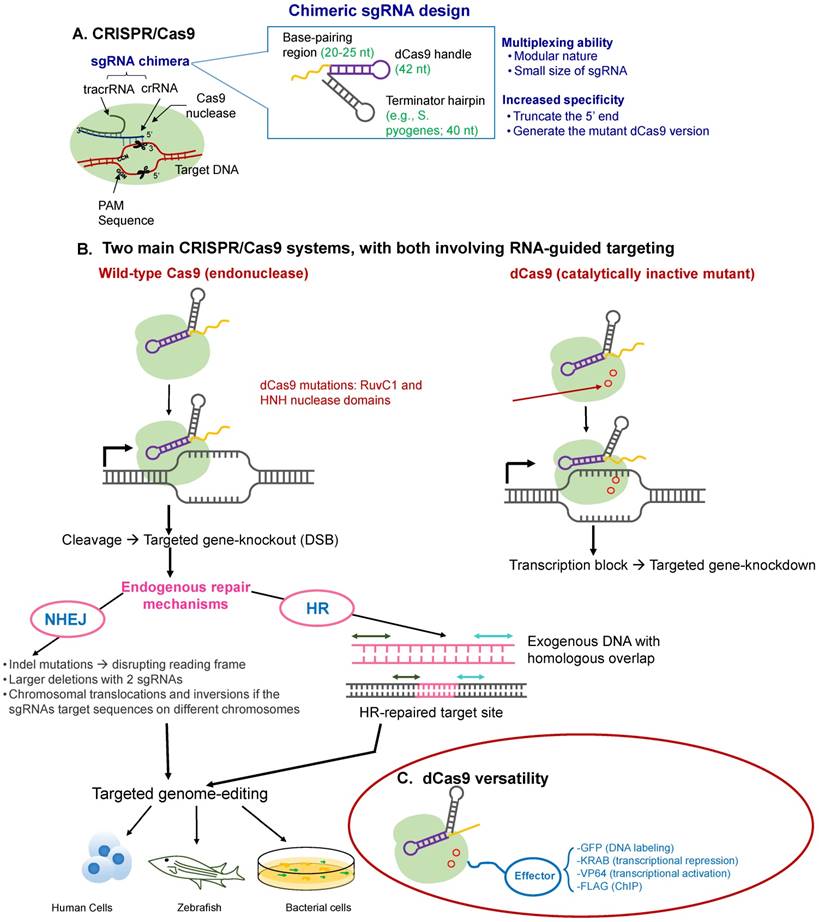
Genome editing operates by using site-specific endonucleases to drive desired genetic alterations, whether single-nucleotide polymorphisms (SNPs), or, whole gene addition or removal, again with a high degree of precision. Among the prevalent genome-editing technologies, the type II CRISPR system from Streptococcus pyogenes (SpCas9), targets a 20-nucleotide DNA sequence, immediately followed by a 5'-NGG-3' PAM, generating a blunt-ended DSB. This system was touted as the simplest to program and use. However, recent studies have found that SpCas9 may be less specific in action in comparison to a Cas9 ortholog from another species, S. aureus (SaCas9), as assessed by BLESS (direct in situ breaks labeling, enrichment on streptavidin and next-generation sequencing) [100]. The molecular machinery of the CRISPR-Cas9 bacterial adaptive immunity can be repurposed to alter (in the dCas9 variant form) or abrogate the transcription of any gene, be it in eukaryotic or in prokaryotic systems. The use of genome editing in prokaryotes affords an unprecedented advance in engineering synthetic biological systems.
At its simplest, the CRISPR-Cas9 system consists of the chimeric single guide RNA (sgRNA), consisting of the combination of CRISPR-associated RNA (crRNA) and partially complementary trans-activating RNA (tracrRNA). The crRNA has a variable guide sequence that directs the Cas9 endonuclease action in a sequence-directed manner. Cas9 is directed to DNA sequences complementary to the protospacer, and then Cas9 creates a double-stranded break (DSB) at the genomic locus to be modified, which triggers cellular DNA repair by one of two methods. In the first, this DSB is repaired by the imprecise NHEJ pathway, the predominant DSB repair pathway in mammalian cells, creating frameshift mutations, disrupting the reading frame of a coding sequence or the binding sites of trans-acting factors (e.g., TFs) on DNA sequences that act as cis-regulatory elements (e.g., enhancers or promoters). Such mutations either intentionally knockout a gene, facilitating reverse genetics and assignment of gene function, or correct a disrupted reading frame. This mode is typically useful when a loss-of-function event is desired. In the second, more precise HDR pathway [134], there is a simple deletion of the targeted sequence, and then, this deleted sequence is filled up by any desired exogenous sequence (Figure 3). This repair template contains homologous sequences to the regions flanking the DSB, resulting in scarless DNA insertion, including the addition of whole genes. The latter process, HDR, is however the less preferred route selected by the cellular machinery. Therefore, in order to coax the cell toward selecting the HDR pathway over NHEJ, various innovative approaches have been developed, such as the use of small molecule activators of the HDR pathway [135]. Alternately, the higher chance of the cell using its NHEJ machinery has also been put to advantage by the simultaneous use of two Cas nucleases, excising the intervening sequence, as has been recently used for the removal of a premature STOP codon in Duchene muscular dystrophy [136].
Overview of the CRISPR-Cas9 mechanism of action. (A) CRISPR-Cas proteins, derived from the prokaryotic adaptive immune system, can target foreign DNA for cleavage using the CRISPR RNA (crRNA). Cas9 is obtained from Type II CRISPR-Cas systems and creates breaks in an approximately 20-nt strand of DNA that is complementary to crRNA, whose maturity is dependent on the trans-activating RNA, tracrRNA. TracrRNA is the RNA that shares partial complementarity with crRNA and binds to the Cas9 endonuclease. (B) Chimeric design of single-guide RNA (sgRNA) by fusing crRNA and tracrRNA, with multiplexing capability, and possible design considerations that can increase its on-target specificity. The sgRNA targets the Cas9 endonuclease to genomic sites complementary to its 5' end. Further, the target DNA sequence needs to be followed in sequence by a protospacer adjacent sequence (PAM), typically the NGG sequence. The five nucleotides that are upstream of the PAM sequence constitute the seed region for target recognition. (C) Two main CRISPR-Cas systems: (i) the wild-type Cas9 resulting in targeted gene knockout and (ii) the catalytically-inactive (non-cleaving) mutant dCas9 gene, with two silencing mutations of the RuvC1 and HNH nuclease domains (D10A and H841A), resulting in targeted gene knockdown (can be thought of as CRIPSR-i); both of which can be used for targeted genome editing in various species, including human cells. (D) The dCas9 mutant can be tagged to various effector molecules, resulting in DNA labeling, transcriptional activation or repression, or chromatin immunoprecipitation (ChIP).
Learning from non-canonical interactions
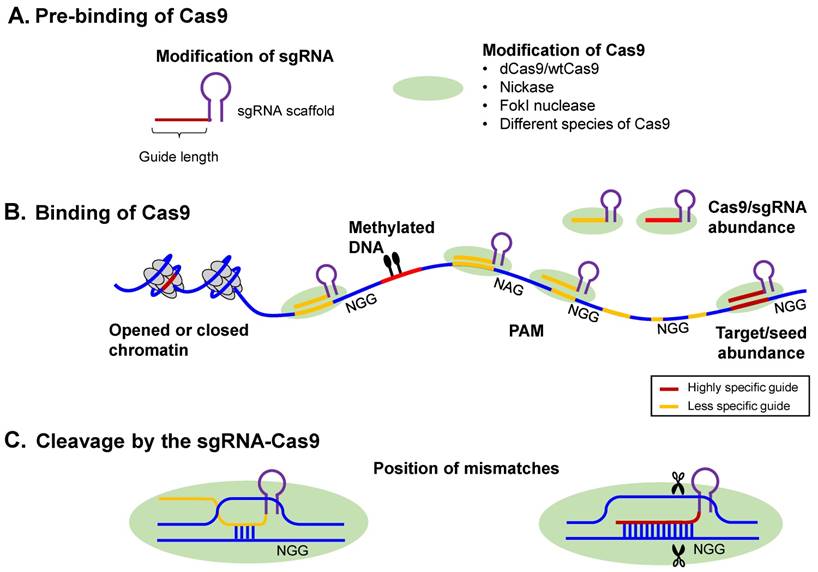
Stemming from the similarities of these two technologies, the promises and roadblocks in the evolution of RNA-i have informed the development of the CRISPR technology [14, 137] [Table 1]. As an example, near-perfect complementarity was assumed to be required for RNA-i processes to work. However, now it is known that non-canonical RNA-i interactions may be the primary driver of off-target interactions [138]. Recently, great strides have been made in solving this problem by using predictive algorithms to learn from high-throughput sequencing data (e.g., CLIP-seq), while taking the widespread, non-canonical regulatory RNA-mRNA interactions into account [4]. While in the RNA-i world, this occurrence of non-canonical interactions was discovered with the maturation of the technology, bioinformatics algorithms have been at the forefront of this realization from the outset of CRISPR technology [69, 74]. Specifically, it has been demonstrated that the CRISPR-Cas9 system can allow for multiple mismatches between the sgRNA and cognate nucleotide sequence, modulated by the quantity, position, and base identity of the mismatch, resulting in off-target effects [21, 59, 139]. Furthermore, crRNAs have been demonstrated to vary widely in their efficiency, with variable indel rates of 5 to 65% [21]. Thus, efforts are being made to refine the precision of the technology and to design safeguards to reduce off-target lesions [74], increasing the specificity of the CRISPR-Cas9 systems. Such unwanted mutations are especially disruptive for applications where high precision levels are desired, such as in creating isogenic cell lines for testing causal sequence variants [140] or in clinical applications. Figure 4 summarizes some of the factors that need to be taken into account while designing a more specific CRISPR-Cas9 system [141].
Factors affecting CRISPR-Cas9 specificity, adapted from [141]. The Cas9-sgRNA targeting specificity can be broadly classified into: (i) The intrinsic targeting specificity that is encoded in the Cas9 endonuclease and (ii) The relative abundance of the Cas9-sgRNA complex relative to the target concentration, with the Cas9-induced cleavage becoming less specific at higher Cas9-sgRNA concentrations, that is, with mismatches in the target sites being better tolerated. This is akin to the RNA-i non-canonical interference mechanisms. Further, CpG methylation and chromatin accessibility, the latter evidenced by DHS peaks, were found to affect off-target binding, such that lower CpG methylation and higher chromatin accessibility promoted off-target binding. Consequently, such off-target effects were significantly enriched at the regulatory elements of active genes, for example.
Consider patients with genetic mutations that make them vulnerable to a cardiomyopathic phenotype. This phenotype is manifested by weakened heart muscles and a proclivity to heart failure. As an example, there could be a mutation in the phospholamban (PLN) gene, which is an important regulator of calcium cycling and critical to cardiac health [142]. Skin cells from such a patient can be isolated and then converted to iPSCs via cellular reprogramming. Next, these iPSCs can be differentiated into cardiomyocytes (iCMs, meaning induced cardiomyocytes), which then carry the genetic history of the patient with the specific cardiomyopathy phenotype. Then, targeted genome editing nucleases can convert the faulty iCMs into healed iCMs, which can subsequently be transplanted back into the patient. This is exactly what was done in a recent study [143] where the cells from a patient with a hereditary dilated cardiomyopathy associated with a PLN R14del mutation were edited to restore a wild-type phenotype. In this way, targeted genome editing offers the ability to isolate the patient's diseased cells and exogenously correct their phenotype. Corrected autologous cells could then potentially be re-introduced into the patient's body to ameliorate or even cure the condition.
This process sounds attractive and the progress, in recent years, bodes well [144], especially in adoptive cell therapies, where T-cells are harvested from the patient, modified ex vivo, expanded, and then reinfused into the patient [145]. Realistically, however, reprogramming the patient's cells, differentiating them, correcting them, and reintroducing the corrected cells into the patient's body is a tall order in which all the parts of the pipeline need to be juxtaposed in a fail-safe, perfectly elegant manner. In today's clinic, introducing cells-as-drugs [74, 146], in order to coax the faulty cells to rewire in their native niche, post transplantation, is gaining traction, especially in the realm of cancer immunotherapy [147]. In this capacity, the living cells, or even artificially-synthesized cells [148], or cell-derived systems [149], would act as information processing modules, programmed to carry out the dictated tasks. Examples would be living cells that could achieve programmed cell death in the host [150] or those that are armed with combinatorial sensing circuits for multi-input autonomous decision making [151]. Regardless, the marriage of genome editing and iPSCs, if successful, holds the promise to alter the face of medicine. As a proof-of-principle experiment, non-genome-edited iPSCs have, for the first time, been used in a patient for treating age-related macular degeneration [152], which alongside the use of genome-edited iPSCs in cell-based models [153], promises an exciting path forward for genome-edited iPSCs.
Cellular reprogramming together with genome editing: Magnum opus for cell-based therapeutics?
As applications of genome editing extend into sensitive areas, such as stem cell therapeutics, it is critical to thoroughly examine whether this approach causes unwanted genetic changes through the rigorous assessment of the genome-editing efficiency in terms of both on-target events (e.g., scission or epigenome editing) and off-target lesions, potentially resulting in cytotoxicity. One of the earliest examples of CRISPR applications in stem cell research was in the functional repair of cystic fibrosis transmembrane conductor receptor (CFTR) in intestinal stem cell organoids of cystic fibrosis patients [153], affording a proof-of-concept for genome editing by HDR in patients with a single-gene hereditary defect [153], followed by many other studies involving blood and neuromuscular disorders, as summarized in a recent review [154]. However, before translation to the clinic, the following are some of the most pressing issues to be resolved.
Minimizing mutational load due to unanticipated off-target effects
While initial attempts have been made to minimize mutational load [155, 156], the effects of unanticipated off-target lesions introduced by genome editing is unclear. Most studies investigated off-target effects of this approach in cultured human transformed or immortalized cells, such as in 293T and K562 cells [21, 157]. In cancer cell lines, Cas9-gRNAs caused higher-than-expected levels of off-target mutagenesis [21], raising concerns regarding the application of genome targeting for therapeutic purposes. More recent genotyping studies examined the off-target effects of genome-editing methods (CRISPR-Cas9, TALENs, and ZFNs) in the entire genome of human iPSCs [158] or PSCs [157] by using whole genome sequencing (WGS). Although these recent WGS studies suggested that the genome-editing approach exhibited low levels of sequence changes in iPSCs and other PSCs, these modified clones are not 100% isogenic compared to their parental cell lines. This is because they seem to have acquired other genetic variations during clonal expansion. Furthermore, due to the limitations of current next-generation sequencing (NGS)-based WGS methodology, the entire genome-wide analysis of off-target mutations induced by genome editing remains unresolved. Conventional NGS methods are not able to detect low-frequency off-target mutations due to their high background error frequency (0.1%) [159]. Sequencing artifacts make it difficult to discern nuclease editing-induced alterations. In addition, bioinformatics filtration can eliminate some genuine mutations. Unlike conventional sequencing technologies that sequence only a single strand of DNA, Duplex Sequencing sequences both strands of DNA and scores mutations only if they are present in both strands of the same DNA molecule as complementary substitutions. This approach significantly reduces the background error frequency (5x10˗8 to 10˗8) [159] and thereby accurately identifies the low-frequency off-target mutations. An unbiased and genome-wide method that accurately detects even ultra-low frequency off-target mutations would be required to define the changes induced by genome editing. In addition, it is not easy to interpret many sub-chromosomal changes, copy number variations, or point mutations that are not clearly associated with genetic abnormalities of known diseases. High-throughput functional genomic analyses would be necessary to examine the effects of new genetic lesions induced by genome editing on the growth, differentiation, tumorigenicity, or functionality of stem cells. On the bright side, however, given the low frequency of off-targeting at any given locus, a prudent study design with multiple wild-type clones compared to multiple targeted clones of interest, would mitigate genetic heterogeneity concerns. This is because it is unlikely that multiple targeted clones will have the same off-target lesion. Further, software programs aimed at the rational design of sgRNAs in CRISPR-Cas9 systems (e.g., [69]) can help determine the levels of on-target and off-target cleavages. It is worth noting that even with the same genomic sequence, different steric contexts [160], or varied genomic contexts, such as different epigenetic modifications at the genomic loci, can alter the effects of the nuclease (Figure 4), and such factors can be built into the software's input feature space. In fact, the catalytically deactivated Cas9 (dCas9 from dead-Cas9) can be fused with various effector domains, e.g., epigenetic modifiers [32] to specifically alter the on-target activity of the nuclease, as in [161] (Figure 3D). This kind of fusion extends the scope of CRISPR way beyond loss-of-function experiments [115]. For example, when fused with epigenetic modifiers, dCas9 can act as a transcriptional repressor [115] or as a transcriptional activator [67, 125], transcriptional activation facilitating CRISPR's use in gain-of-function experiments. Also, dCas9 can facilitate programmable chromatin [162] and RNA [163] pulldown. Finally, in a recent bid toward improving the methods to directly visualize genomics loci in the 3D nucleus, the nuclease-deficient dCas9 variant was used as a probe to label sequence-specific genomic loci fluorescently without globally denaturing DNA [164]. One such method, CASFISH [165], which is Cas9-mediated fluorescence in situ hybridization, is a rapid cost-effective method. It does not require heat and formamide treatment to globally denature DNA as generic DNA FISH technologies do.
Nuclease delivery challenges
The successful delivery of the guide RNAs and nuclease (Cas9) is essential for efficient genome editing. In this context, genome editing can be thought of as an easier task, given that unlike RNA-i, genome-editing therapeutics do not necessitate sustained transgene expression, increasing the portfolio of delivery agents. However, the selection of the delivery agent also depends on the desired method of DNA repair after the DSB. While for NHEJ, only the endonuclease needs to be delivered, for HDR, the donor DNA needs to be co-delivered for a sustained period of time, with adenoviral vectors demonstrating high accuracy as HDR donors, albeit, with low efficiency [166]. Furthermore, unlike RNA-i, which piggybacks on endogenous RNA-based pathways, delivery of CRISPR-Cas9-based therapeutics requires the delivery of the Cas9 gene or protein, which can be quite bulky. Traditionally, small-molecule synthetic drugs are below 500 kDa in weight while bulkier antibodies that can be successfully delivered do not require intracellular delivery. A solution here is to use gene therapy vectors to express both the endonuclease domain and the sgRNA domain. Here, while constitutive transgene expression is an advantage, increasing the potential for on-target cleavage, extended persistence of these nuclease components may result in higher off-target mutations, presenting somewhat of a double-edged sword.
Conventional delivery agents include both viral and non-viral delivery approaches. On the viral side, the usual vehicles can be used, including lentiviruses (e.g., integrase-defective lentiviruses [167]), adenoviruses, and adeno-associated viruses (AAV) [168]. While AAV-based viral vectors and electroporation have been widely used in preclinical animal models in vivo, as listed here [169], with promising results in hemophilia [170], muscular dystrophies [171], and other hereditary diseases, the use of AAV-based systems can bear the risk of residual nuclease expression and integration of the viral vectors into the host genome, although not observed in preclinical studies. With this mind, non-viral modalities, including, cell-penetrating peptides, lipid nanoparticles, semiconductor quantum dots [172], or cationic lipids [173] can be used, with electroporation and rapid mechanical deformation being more recent forms of delivery. However, electroporation, which involves pulsed electric fields, can permanently disrupt cell membrane integrity, especially in cells of the blood and immune system [174]. So, rapid mechanical deformation with microfluidic devices appears to be safer and more precise, at the single-cell level, delivering a higher throughput and the ability to transfect hard-to-transfect cells, such as human ESCs or iPSCs, relative to immortalized tumor cell lines [175].
Improved precision of sgRNAs
A critical need in biology is to identify the sets of genes underlying specific biological processes. The feasibility of large-scale, loss-of-function screens in mammalian cells is very useful from this standpoint, wherein the DNA-level inactivation of genes and the ability to edit non-coding parts of the genome would be informative. Such DNA-level, genome-wide screens are beyond the scope of RNA-i, even when combined with NGS technologies. An end result like this necessitates the efficient cleavage of both copies of targeted loci by single copies of high-scoring, highly effective sgRNA. This is in contrast to the high concentrations of sgRNA required for transfection-based experiments [176]. When extended to clinical applications, off-target lesions, especially in cancer cell lines [32], and oncogene activation are some of the concerns of genome editing that need to be addressed before promoting the more mainstream use of this technology in medicine. In genome editing, the sgRNAs need to be designed to be precise and powerful such that the desired gene locus can be effectively repaired, as in the case of the gene Tafazzin (TAZ), which was shown to be a necessary and sufficient mutation for Barth syndrome-related cardiomyopathy [177].
Cell-based models to chimeric animal models
The iPSC technology by itself can recapitulate morphological and functional phenotypes of various diseases. As an example, from the standpoint of tissue regeneration, the optimal differentiation of PSCs to the target cell type of interest is critical. To this end, there are established differentiation protocols, such as those that were demonstrated to result in clinical-scale production (>1 billion cells/batch) of cardiomyocytes (CMs) from human ESCs, e.g., in [178]. Here, the differentiated CMs were shown to display sound structural and functional properties in an infarcted primate heart, demonstrating the promise of remuscularizing a human heart. However, when such technologies, are deployed in combination with genome editing and extracellular matrix (ECM)-mimicking technologies [7], the mechanistic insights gleaned from such experiments will shine further. This is because genome editing will result in appropriate isogenic controls for the test cases.
The extraordinary tour de force of genome editing in cellular engineering can be unleashed by deploying robust study designs in concert with combinatorial modeling algorithms. At the most basic mechanistic level, this could involve using genome editing to both insert a disease-causing mutation in wild-type cell lines (e.g., using HDR) and to correct disease-causing mutations in patient-specific iPSC cell lines (e.g., using NHEJ), testing both the sufficiency and necessity of the mutation, respectively. Further, by introducing disease mutations in iPSC lines with different genetic backgrounds, the extent of the lethality of the mutation can also be tested. Finally, the ability of genome editing to transcend the boundaries of cell-based models can be probed by incorporating iPSCs into chimeric animal models [165], offering an ability to interrogate the effect of the mutation in a whole-animal model. In addition, model systems, such as stem cell organoids and microfluidic systems, with ECM-mimicking scaffolds armed with a combinatorial library of input variables, offer a way to precisely and predictively control the cellular microenvironment, approximating whole-body responses.
CRISPR epigenome editing
How are different cell types so unique, collaborating in life processes by virtue of their exquisite specializations, in spite of their identical genomes? These cellular specializations may be attributed to the “epigenome”, with cell type-specific gene-expression levels being modulated by it. With the human genome project demonstrating the feasibility and initial success of large-scale sequencing projects toward solving the overflowing trove of puzzles deciphering human health and diseases, the scientific community started mapping cellular epigenomes and annotating them using sophisticated machine learning techniques [136]. The epigenome, which includes DNA methylation, post-translational histone modifications, chromatin remodeling, and non-coding RNAs, can regulate chromatin accessibility (to chromatin modifiers), and thus, the expression of genes. In fact, the cellular epigenome is a reversible and heritable “layer” of regulation. With the ENCODE (Encyclopedia of DNA Elements) project being a trailblazer in the epigenetics community [179], pioneering many of the technologies to identify regulatory elements in the human genome, modENCODE further added to the repository of experiments. modENCODE houses datasets of the epigenomes of model organisms [180], which are mostly easier to experiment with, and hence, useful for validating and finetuning initial computational predictions. Abetted by this increasing awareness of the importance of the epigenome, the International Human Epigenome Consortium (IHEC) (http://www.ihec-epigenomes.org/) was started in 2010. With a vision to map epigenomes, and more specifically, to generate 1,000 reference epigenomes, using both primary tissues and cell lines (ENCODE uses cell lines), IHEC now has nine organizations under its umbrella, including ENCODE (USA) and BLUEPRINT (European Union), the latter having chosen to focus on the blood system [181], given the faster-route-to-market of blood products in general.
Alongside this surge of epigenomic datasets, and perhaps, motivated by the axiom: “Correlation does not imply Causation” (whereby it is seen that while current studies can infer functionality for epigenomic marks via correlation, it is a challenge to determine which marks contribute to which functions), there has been a need for the targeted manipulation of epigenomic features. To this end, uses of small-molecule inhibitors (e.g., DNA methyltransferase or histone deacetylase inhibitors [182]) and nucleases have been underway. In terms of small-molecule inhibitors, desirable properties include rapidly reversible and dose-dependent effects; the ability to conduct phenotypic screening of small-molecule libraries [183], resulting in flexibility of design; and their relative ease of handling and with more established delivery protocols. However, the in vivo targets of these chemical inhibitors are often multitudinous, and this, naturally, can lead to multifaceted side effects, in vivo. Thus, comes the excitement encircling the ability of CRISPR-Cas, and other designer nuclease systems, to edit the epigenome, such as, in the form of the targeted perturbation of histone modifications [161]. Further, simultaneous interrogation is often important given the structure of the human genome. The eukaryotic genome is highly compact and has a functionally responsive three-dimensional (3D) structure, as is becoming more and more evident with the sophistication of chromosome-conformation capture techniques, as typified by chromosome conformation capture (3C) and derivative (4C, 5C, Hi-C) methods. 3C methods, with their variants [184], afford us an unprecedented insight into looping three-dimensional epigenomic landscapes, such as the loops that initiate enhancer-promoter contact, and to topologically interacting domains. In this developing view of the topologically folded genome, the local chromatin architecture is segmented into distinct modules called physical domains or topologically associated domains (TADs) using CTCF-binding sites, which function as insulators between TADs, with the regions demarcated by TADs often times containing coordinately regulated genes. Now, these TADs contain hundreds, or even thousands, of candidate marks and interrogating them simultaneously is the next frontier to be conquered.
In addition, epigenome editing can be thought of as being at the forefront of synthetic biology, enabling an engineering framework toward using chromatin logic, and having seen an initial entrance to the clinical scene, mostly in the form of small-molecule, chromatin modifiers [185]. Epigenome editing basically involves altering the chromatin state, and ensuing gene expression, without bringing about changes in the genomic sequence, which can essentially unravel the regulatory sophistication of the chromatin. So, while for genome editing, there are gene-silencing or activating factors, fused to the targeting module; in epigenome editing, the targeting module is fused to chromatin-modifying modules, such as, DNA methyltranferases, or demethylases, and histone acetyltransferases (HATs, e.g., p300), or deacetylases (HDACs), to name a few. Such editing, in conjunction with powerful ML algorithms to map out the very presence of these genomic regulatory elements, such as in [136], will go a long way in teasing out the mechanistic basis of diseases. Further, is it possible to consider epigenome editing as a tool to efface the dark side of induced pluripotency−the latter, in the form of aberrant epigenomic programming? Can some of the epigenetic variations, stemming from the induction of pluripotency, be undermined by CRISPR endonucleases, such as, some of the new tools on the horizon, unearthed by the marvels of metagenomics and technological discovery engines [186]? This would, of course, again, be dependent on the on-target specificity of the genome editing processes and on the ability to tease out the epigenetic signatures in such reprogrammed cells, forestalling undesirable cancer-ridden trajectories.
Finally, it is worth mentioning here that 93% of GWAS hits, both disease- and phenotype-associated hits, are found in the non-coding genome [187]. Thus, naturally occurring mutations that confer resistance to different diseases have been found in these non-coding regions. Many such non-coding mutations involve loss-of-function alleles, which can be catalyzed using NHEJ-based frameshift mutations, when genome editing. Given the high efficiency of NHEJ-based gene editing, this strategy has been in the works for the treatment of HIV [188]. However, as in the case of more traditional antiviral therapies, the nature of these lentiviral infections, whereby the virus, such as the HIV virus, invading T cells and integrating into the T-cell genome, for example, makes it a challenge to eliminate the latent viral genome from the host cells [189]. In principle, T-cells could be programmed to generate the nuclease, when invaded by the HIV genome, but genome editing the T-cells has been a challenge. For one, while the NHEJ-based mutations ideally result in inactivating the HIV genome, in some cases, the indel may not inactivate the virus, and in fact, prime the virus for enhanced survival and that is enough to make the treatment go awry. However, the ability of the Cas9 nuclease to excise latent viruses from the host cell's genome is a big fillip for the use of these technologies for HIV-related pathologies because in HIV large reservoirs of latent provirus often persist after the end of the antiviral regimen and these could reactivate the infection once the treatment ceases [81]. Further, and more relevant to our article here, is the combined use of iPSCs and CRISPR toward being able to confer resistance to HIV. It is known that individuals homozygous for the C-C chemokine receptor type 5 (CCR5) gene with 32-basepair deletions (labeled CCR5-delta32, a deletion mutation of the gene) are immune to HIV-1 infection and only 1% of the total population has two copies of this gene with relatively high frequencies in Europe. Further, around 20% of the population carry only one copy of the mutation, and although, they can still contract HIV, its progress is greatly impeded. Thus, CCR5 disruption in iPSCs is a feasible route for developing HIV resistance [190]. In the past, however, incomplete protection from HIV-1 using shRNA-mediated knockdown and the concomitant potential for mutagenesis from the integrated viral vectors required for constitutive shRNA expression [191], concerns about the fitness of the transduced cells [188], and the off-target damage [192] have sullied the initial enthusiasm and these need to be resolved prior to further translation. In this regard, the potentially permanent mutation of the gene by CRISPR-Cas9 editing and the lower chance of off-targeting, especially in relation to zinc finger nucleases, may be somewhat of a panacea.
Concluding remarks
The envisioned ability of two rapidly evolving technologies−creation and maintenance of pluripotency (e.g., iPSC technology) and genome editing (e.g., CRISPR-Cas9 technology)−to alter the face of disease on earth is breathtaking, albeit, with some unresolved scientific quandaries. Technologies for reading the genome (NGS technologies), writing the genome (synthesizing millions of basepairs), and high-precision editing of the genome, and of the epigenome, have all developed at a frenetic pace over the last decade. Indeed, these fast-evolving technologies complement each other to enhance current strides in today's genomic medicine era. For example, the fast pace of development of genome editing technologies−a dream in the world of medicine since the recognition of genes as units of heredity−holds the promise of eradicating congenital diseases, modeling the effects of non-coding genomic variants, and slowing the onslaught of long-standing epidemics, especially multigenic diseases and viral epidemics. With it, comes the ability to target and manipulate genomes (and epigenomes) that were largely refractory to editing in the years predating genome engineering. Combined with therapeutic progenitor cells, they can address a wide swath of pathologies and answer fundamental scientific questions. However, recent efforts at genome editing of the human embryo [77, 193] raise both technical and ethical challenges. On the technical front, it is important to investigate the ramifications of off-targeting, mosaicism (somatic and possible germline), allelic complexity, the possibility of germline perturbation via mitochondrial replacement [194], and the biology of DNA-repair mechanisms [195], among other aspects. Looking askance, with a more ethical slant, one may wonder what the more far-flung effects of editing the human germline might be. Thus, the time is ripe to accelerate the path-to-the-clinic course of genome-edited and engineered cell-based technologies in a rigorous, albeit cautious, manner. Clinical translation of genome editing, alongside cellular reprogramming can cause a paradigm shift in gene therapy, permanently eliminating disease symptoms with engineered endonucleases. Thus, one can be cautiously optimistic that via extensive design and high-throughput experimentation, comprehensive bioinformatics filtration, and the use of creative, high-efficiency biomimetic platforms, maturation of the synergistic technologies described in this review will be intensely rewarding.
Abbreviations
PSC: pluripotent stem cell; iPSC: induced pluripotent stem cell; ESC: embryonic stem cell; TF: transcription factor; ZFN: zinc finger nuclease; TALEN: transcription activator-like effector nuclease; CRISPR: clustered regularly interspaced short palindromic repeats; Cas: CRISPR-associated; crRNA: CRISPR RNA; tracrRNA: trans-activating RNA; PAM: protospacer adjacent motif; NHEJ: non-homologous end joining; HDR: homology-directed repair; siRNA: small interfering RNA; piRNA: PIWI-interacting RNA; RNA-i: RNA interference; sgRNA: single guide RNA; DSB: double-stranded break; GWAS: genome-wide association studies; BER: base excision repair mechanism; dCas9: (catalytically) dead Cas9; ML: machine learning; CFTR: cystic fibrosis transmembrane conductor receptor; WGS: whole genome sequencing; CASFISH: Cas9-mediated fluorescence in situ hybridization; AAV: adeno-associated viruses; ECM: extracellular matrix; ENCODE: Encyclopedia of DNA Elements; IHEC: International Human Epigenome Consortium; 3C: chromosome conformation capture; TAD: topologically associated domain; NGS: next-generation sequencing.
Acknowledgements
This work was supported by the following National Institutes of Health grants: R01HL135143 and R01NS094388 (to D.-H. K.), and R01AI123037 (to S.C.).
Competing Interests
D-H.K. is a co-founder and scientific board member at NanoSurface Biomedical Inc.
References
1. Scadden David T. Nice Neighborhood: Emerging Concepts of the Stem Cell Niche. Cell. 2014;157:41-50
2. Weinberg BH, Pham NH, Caraballo LD, Lozanoski T, Engel A, Bhatia S. et al. Large-scale design of robust genetic circuits with multiple inputs and outputs for mammalian cells. Nature Biotechnol. 2017;35:453-62
3. Kim SG, Ampornpunt NT, Fang C-H, Harwani M, Grama AY, Chaterji S. Opening up the blackbox: An interpretable deep neural network-based classifier for cell-type specific enhancer predictions. BMC Syst Biol. 2016
4. Ghoshal A, Grama A, Bagchi S, Chaterji S. An Ensemble SVM Model for the Accurate Prediction of Non-Canonical MicroRNA Targets. ACM-BCB Best Paper Award: ACM. 2015:403-12
5. Ghoshal A, Shankar R, Bagchi S, Grama A, Chaterji S. MicroRNA target prediction using thermodynamic and sequence curves. BMC Genomics. 2015;16:999
6. Wang D, Yan K-K, Sisu C, Cheng C, Rozowsky J, Meyerson W. et al. Loregic: a method to characterize the cooperative logic of regulatory factors. PLoS Comput Biol. 2015;11:e1004132
7. Chaterji S, Kim P, Choe S, Tsui J, Lam C, Ho D. et al. Synergistic Effects of Matrix Nanotopography and Stiffness on Vascular Smooth Muscle Cell Function. Tissue Eng Pt A. 2014
8. Esensten JH, Bluestone JA, Lim WA. Engineering therapeutic T cells: from synthetic biology to clinical trials. Annu Rev Pathol. 2017;12:305-30
9. Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375-86
10. Bar-Nur O, Verheul C, Sommer AG, Brumbaugh J, Schwarz BA, Lipchina I. et al. Lineage conversion induced by pluripotency factors involves transient passage through an iPSC stage. Nat Biotechnol. 2015;33:761-8
11. Bhattacharya S, Zhang Q, Andersen ME. A deterministic map of Waddington's epigenetic landscape for cell fate specification. BMC Syst Biol. 2011;5:85
12. Capecchi MR. Altering the Genome by Homologous Recombination. Science. 1989;244:1288-92
13. Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A. 1994;91:6064-8
14. Barrangou R, Birmingham A, Wiemann S, Beijersbergen RL, Hornung V, Smith Anja vB. Advances in CRISPR-Cas9 genome engineering: lessons learned from RNA interference. Nucleic Acids Res. 2015
15. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S. et al. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science. 2007;315:1709-12
16. Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ. et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Micro. 2015;13:722-36
17. Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169-75
18. Makarova KS, Wolf YI, Van der Oost J, Koonin EV. Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol Direct. 2009;4:29
19. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816-21
20. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE. et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823-6
21. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N. et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819-23
22. Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049-61
23. Lerou PH, Yabuuchi A, Huo H, Takeuchi A, Shea J, Cimini T. et al. Human embryonic stem cell derivation from poor-quality embryos. Nat Biotechnol. 2008;26:212-4
24. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154-6
25. Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A. 1981;78:7634-8
26. Hou P, Li Y, Zhang X, Liu C, Guan J, Li H. et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651-4
27. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to [beta]-cells. Nature. 2008;455:627
28. Sekiya S, Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature. 2011;475:390
29. Campbell KH, McWhir J, Ritchie WA, Wilmut I. Sheep cloned by nuclear transfer from a cultured cell line. Nature. 1996;380:64
30. Sancho-Martinez I. Will SCNT-ESCs be better than iPSCs for personalized regenerative medicine. Cell Stem Cell. 2013;13:141-2
31. Tachibana M, Amato P, Sparman M, Gutierrez Nuria M, Tippner-Hedges R, Ma H. et al. Human Embryonic Stem Cells Derived by Somatic Cell Nuclear Transfer. Cell. 2013;153:1228-38
32. Apostolou E, Hochedlinger K. Chromatin dynamics during cellular reprogramming. Nature. 2013;502:462-71
33. Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663-76
34. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K. et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861-72
35. Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Bio. 2016;17:183-93
36. Zhao X-y, Li W, Lv Z, Liu L, Tong M, Hai T. et al. iPS cells produce viable mice through tetraploid complementation. Nature. 2009;461:86
37. Wu J, Okamura D, Li M, Suzuki K, Luo C, Ma L. et al. An alternative pluripotent state confers interspecies chimaeric competency. Nature. 2015;521:316
38. Gurdon JB. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. Development. 1962;10:622-40
39. Gehring W. Clonal analysis of determination dynamics in cultures of imaginal disks in Drosophila melanogaster. Dev Biol. 1967;16:438-56
40. Carey BW, Markoulaki S, Hanna JH, Faddah DA, Buganim Y, Kim J. et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell. 2011;9:588-98
41. Cahan P, Li H, Morris SA, da Rocha EL, Daley GQ, Collins JJ. CellNet: Network Biology Applied to Stem Cell Engineering. Cell. 2014;158:903-15
42. Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704-12
43. Hentze H, Graichen R, Colman A. Cell therapy and the safety of embryonic stem cell-derived grafts. Trends Biotechnol. 2007;25:24-32
44. Lowry W, Richter L, Yachechko R, Pyle A, Tchieu J, Sridharan R. et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proceedings of the National Academy of Sciences. 2008;105:2883-8
45. Loh Y-H, Agarwal S, Park I-H, Urbach A, Huo H, Heffner GC. et al. Generation of induced pluripotent stem cells from human blood. Blood. 2009;113:5476-9
46. Haase A, Olmer R, Schwanke K, Wunderlich S, Merkert S, Hess C. et al. Generation of induced pluripotent stem cells from human cord blood. Cell stem cell. 2009;5:434-41
47. Choi SM, Liu H, Chaudhari P, Kim Y, Cheng L, Feng J. et al. Reprogramming of EBV-immortalized B-lymphocyte cell lines into induced pluripotent stem cells. Blood. 2011;118:1801-5
48. Nichols J, Smith A. Naive and Primed Pluripotent States. Cell Stem Cell. 2009;4:487-92
49. Weinberger L, Ayyash M, Novershtern N, Hanna JH. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nature Reviews Molecular Cell Biology. 2016
50. Wang J, Xie G, Singh M, Ghanbarian AT, Raskó T, Szvetnik A. et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature. 2014;516:405
51. Buehr M, Meek S, Blair K, Yang J, Ure J, Silva J. et al. Capture of Authentic Embryonic Stem Cells from Rat Blastocysts. Cell. 2008;135:1287-98
52. Deuse T, Wang D, Stubbendorff M, Itagaki R, Grabosch A, Greaves Laura C. et al. SCNT-Derived ESCs with Mismatched Mitochondria Trigger an Immune Response in Allogeneic Hosts. Cell Stem Cell. 2015;16:33-8
53. Burstein D, Harrington LB, Strutt SC, Probst AJ, Anantharaman K, Thomas BC. et al. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017;542:237-41
54. Fellmann C, Gowen BG, Lin P-C, Doudna JA, Corn JE. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nature Reviews Drug Discovery. 2017;16:89-100
55. Mahadik K, Chaterji S, Zhou B, Kulkarni M, Bagchi S. Orion: Scaling Genomic Sequence Matching with Fine-Grained Parallelization. Supercomputing 2014 (The International Conference for High Peformance Computing, Networking, Storage and Analysis): IEEE. 2014 1-11
56. Mahadik K, Wright C, Zhang J, Kulkarni M, Bagchi S, Chaterji S. SARVAVID: A Domain Specific Language for Developing Scalable Computational Genomics Applications. Proceedings of the 2016 International Conference on Supercomputing: ACM. 2016:34
57. Shinkuma S, Guo Z, Christiano AM. Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proceedings of the National Academy of Sciences. 2016;113:5676-81
58. Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of bacteriology. 1987;169:5429-33
59. Dianov GL, Hübscher U. Mammalian Base Excision Repair: the Forgotten Archangel. Nucleic Acids Research. 2013;41:3483-90
60. Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10570-5
61. Cho SW, Kim S, Kim JM, Kim J-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature Biotechnol. 2013;31:230-2
62. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. cell. 2013;153:910-8
63. Findlay GM, Boyle EA, Hause RJ, Klein J, Shendure J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014;513:120
64. Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP. et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960-4
65. Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L. et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945-56
66. Seed KD, Lazinski DW, Calderwood SB, Camilli A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. 2013;494:489-91
67. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnol. 2013;31:822-6
68. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnol. 2013;31:827-32
69. Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401-W7
70. Mojica F, Diez-Villasenor C, Garcia-Martinez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733-40
71. Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I. et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnol. 2014;32:1262-7
72. Hodgkins A, Farne A, Perera S, Grego T, Parry-Smith DJ, Skarnes WC. et al. WGE: a CRISPR database for genome engineering. Bioinformatics. 2015;31:3078-80
73. Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E. et al. Highly efficient Cas9-mediated transcriptional programming. Nat Meth. 2015;12:326-8
74. Kim D, Bae S, Park J, Kim E, Kim S, Yu HR. et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Meth. 2015;12:237-43
75. Kleinstiver BP, Prew MS, Tsai SQ, Topkar V, Nguyen NT, Zheng Z. et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481
76. Frock RL, Hu J, Meyers RM, Ho Y-J, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnol. 2015;33:179-86
77. Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z. et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & Cell. 2015;6:363-72
78. Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. elife. 2013;2:e00471
79. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature Biotechnol. 2014;32:279-84
80. Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A, Mali P. et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41:9049-61
81. Byrne SM, Mali P, Church GM. Genome Editing in Human Stem Cells. Method Enzymol. 2014;546:119-38
82. Deyle DR, Li LB, Ren G, Russell DW. The effects of polymorphisms on human gene targeting. Nucleic Acids Res. 2014;42:3119-24
83. Russell DW, Hirata RK. Human gene targeting favors insertions over deletions. Hum Gene Ther. 2008;19:907-14
84. Martí M, Mulero L, Pardo C, Morera C, Carrió M, Laricchia-Robbio L. et al. Characterization of pluripotent stem cells. Nat Protoc. 2013;8:223
85. Turner M, Leslie S, Martin Nicholas G, Peschanski M, Rao M, Taylor Craig J. et al. Toward the Development of a Global Induced Pluripotent Stem Cell Library. Cell Stem Cell. 2013;13:382-4
86. Sommer CA, Sommer AG, Longmire TA, Christodoulou C, Thomas DD, Gostissa M. et al. Excision of reprogramming transgenes improves the differentiation potential of iPS cells generated with a single excisable vector. Stem Cells. 2010;28:64-74
87. Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771
88. Cho H-J, Lee C-S, Kwon Y-W, Paek JS, Lee S-H, Hur J. et al. Induction of pluripotent stem cells from adult somatic cells by protein-based reprogramming without genetic manipulation. Blood. 2010;116:386-95
89. Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949-53
90. Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945-9
91. Aalto-Setälä K, Conklin BR, Lo B. Obtaining consent for future research with induced pluripotent cells: opportunities and challenges. PLoS Biol. 2009;7:e1000042
92. Giorgetti A, Montserrat N, Aasen T, Gonzalez F, Rodríguez-Pizà I, Vassena R. et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell. 2009;5:353
93. Li Z, Lu H, Yang W, Yong J, Zhang Z-n, Zhang K. et al. Mouse SCNT ESCs Have Lower Somatic Mutation Load Than Syngeneic iPSCs. Stem Cell Reports. 2014;2:399-405
94. Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY. et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nature Biotechnol. 2010;28:848-55
95. Schlaeger TM, Daheron L, Brickler TR, Entwisle S, Chan K, Cianci A. et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015;33:58-63
96. Chin MH, Mason MJ, Xie W, Volinia S, Singer M, Peterson C. et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111-23
97. Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS. et al. The genetic landscape of a cell. Science. 2010;327:425-31
98. Phanstiel DH, Brumbaugh J, Wenger CD, Tian S, Probasco MD, Bailey DJ. et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat Methods. 2011;8:821-7
99. Patel S, Jung D, Yin PT, Carlton P, Yamamoto M, Bando T. et al. NanoScript: a nanoparticle-based artificial transcription factor for effective gene regulation. ACS Nano. 2014;8:8959-67
100. Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16:197-212
101. Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V. et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nature Biotechnol. 2010;28:839-47
102. Nishizawa M, Chonabayashi K, Nomura M, Tanaka A, Nakamura M, Inagaki A. et al. Epigenetic variation between human induced pluripotent stem cell lines is an indicator of differentiation capacity. Cell Stem Cell. 2016;19:341-54
103. Thakore PI, D'Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM. et al. Highly Specific Epigenome Editing by CRISPR/Cas9 Repressors for Silencing of Distal Regulatory Elements. Nat Meth. 2015;12:1143-9
104. Chakraborty S, Ji H, Kabadi AM, Gersbach CA, Christoforou N, Leong KW. A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Reports. 2014;3:940-7
105. Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP. et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30-e41
106. Macadangdang J, Guan X, Smith AST, Lucero R, Czerniecki S, Childers MK. et al. Nanopatterned Human iPSC-based Model of a Dystrophin-Null Cardiomyopathic Phenotype. Cell Mol Bioeng. 2015;8:320-32
107. Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K. et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nature Biotechnol. 2016;34:192-8
108. Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G. et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515-26
109. Canver MC, Bauer DE, Orkin SH. Functional interrogation of non-coding DNA through CRISPR genome editing. Methods. 2017
110. Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM. et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769-73
111. Khurana E, Fu Y, Chakravarty D, Demichelis F, Rubin MA, Gerstein M. Role of non-coding sequence variants in cancer. Nature Rev Genet. 2016;17:93-108
112. Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O. et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192
113. Aparicio-Prat E, Arnan C, Sala I, Bosch N, Guigó R, Johnson R. DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genomics. 2015;16:846
114. Pulido-Quetglas C, Aparicio-Prat E, Arnan C, Polidori T, Hermoso T, Palumbo E. et al. Scalable Design of Paired CRISPR Guide RNAs for Genomic Deletion. PLoS Comput Biol. 2017;13:e1005341
115. Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442-51
116. Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ. et al. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell. 2015;162:675-86
117. Wu J, Platero-Luengo A, Sakurai M, Sugawara A, Gil MA, Yamauchi T. et al. Interspecies Chimerism with Mammalian Pluripotent Stem Cells. Cell. 2017;168:473-86.e15
118. Kobayashi T, Yamaguchi T, Hamanaka S, Kato-Itoh M, Yamazaki Y, Ibata M. et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell. 2010;142:787-99
119. Isotani A, Hatayama H, Kaseda K, Ikawa M, Okabe M. Formation of a thymus from rat ES cells in xenogeneic nude mouse↔ rat ES chimeras. Genes to Cells. 2011;16:397-405
120. Usui J-i, Kobayashi T, Yamaguchi T, Knisely A, Nishinakamura R, Nakauchi H. Generation of kidney from pluripotent stem cells via blastocyst complementation. Am J Pathol. 2012;180:2417-26
121. Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Rivera RMC. et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403-7
122. Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538-42
123. Ran FA, Hsu Patrick D, Lin C-Y, Gootenberg Jonathan S, Konermann S, Trevino AE. et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell. 2013;154:1380-9
124. Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour JV, Rawlings DJ. et al. Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods. 2011;8:671-6
125. Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S. et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotechnol. 2013;31:833-8
126. Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013;8:2180-96
127. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH. et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647-61
128. Chou CH, Lin FM, Chou MT, Hsu SD, Chang TH, Weng SL. A computational approach for identifying microRNA-target interactions using high-throughput clip and par-clip sequencing. BMC Genomics. 2013:14
129. Meade BR, Gogoi K, Hamil AS, Palm-Apergi C, Berg Avd, Hagopian JC. et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat Biotechnol. 2014;32:1256-61
130. Corrigan-Curay J, O'Reilly M, Kohn DB, Cannon PM, Bao G, Bushman FD. et al. Genome Editing Technologies: Defining a Path to Clinic. Mol Ther. 2015;23:796-806
131. Abi-Ghanem J, Chusainow J, Karimova M, Spiegel C, Hofmann-Sieber H, Hauber J. et al. Engineering of a target site-specific recombinase by a combined evolution-and structure-guided approach. Nucleic Acids Res. 2012:gks1308
132. Yant SR, Huang Y, Akache B, Kay MA. Site-directed transposon integration in human cells. Nucleic Acids Res. 2007;35:e50
133. Siomi H, Siomi MC. On the road to reading the RNA-interference code. Nature. 2009;457:396-404
134. Urnov FD, Miller JC, Lee Y-L, Beausejour CM, Rock JM, Augustus S. et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646-51
135. Pinder J, Salsman J, Dellaire G. Nuclear domain 'knock-in' screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015
136. Kim S, Harwani M, Grama A, Chaterji S. EP-DNN: A Deep Neural Network-Based Global Enhancer Prediction Algorithm. Sci Rep. 2016;6:1-13
137. Cox DBT, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21:121-31
138. Gumienny R, Zavolan M. Accurate transcriptome-wide prediction of microRNA targets and small interfering RNA off-targets with MIRZA-G. Nucleic Acids Res. 2015;43:1380-91
139. Hsu PD, Zhang F. Dissecting neural function using targeted genome engineering technologies. ACS Chem Neurosci. 2012;3:603-10
140. Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R. et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318-31
141. Buganim Y, Markoulaki S, van Wietmarschen N, Hoke H, Wu T, Ganz K. et al. The developmental potential of iPSCs is greatly influenced by reprogramming factor selection. Cell Stem Cell. 2014;15:295-309
142. Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U. et al. Dilated Cardiomyopathy and Heart Failure Caused by a Mutation in Phospholamban. Science. 2003;299:1410-3
143. Karakikes I, Stillitano F, Nonnenmacher M, Tzimas C, Sanoudou D, Termglinchan V. et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nature Commun. 2015:6
144. Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17:170-82
145. June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: A race to the finish line. Sci Transl Med. 2015;7:280ps7-ps7
146. Ankrum JA, Miranda OR, Ng KS, Sarkar D, Xu C, Karp JM. Engineering cells with intracellular agent-loaded microparticles to control cell phenotype. Nat Protoc. 2014;9:233-45
147. Chakravarti D, Wong WW. Synthetic biology in cell-based cancer immunotherapy. Trends Biotechnol. 2015;33:449-61
148. Karzbrun E, Tayar AM, Noireaux V, Bar-Ziv RH. Programmable on-chip DNA compartments as artificial cells. Science. 2014;345:829-32
149. Fuhrmann G, Herrmann IK, Stevens MM. Cell-derived vesicles for drug therapy and diagnostics: Opportunities and challenges. Nano Today. 2015;10:397-409
150. Caliando BJ, Voigt CA. Targeted DNA degradation using a CRISPR device stably carried in the host genome. Nat Commun. 2015:6
151. Roybal Kole T, Rupp Levi J, Morsut L, Walker Whitney J, McNally Krista A, Park Jason S. et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016
152. Nakano-Okuno M, Borah BR, Nakano I. Ethics of iPSC-based clinical research for age-related macular degeneration: patient-centered risk-benefit analysis. Stem Cell Rev Rep. 2014;10:743-52
153. Schwank G, Koo B-K, Sasselli V, Dekkers Johanna F, Heo I, Demircan T. et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell. 2013;13:653-8
154. Shui B, Hernandez Matias L, Guo Y, Peng Y. The Rise of CRISPR/Cas for Genome Editing in Stem Cells. Stem Cells Int. 2016;2016:8140168
155. Guilinger JP, Pattanayak V, Reyon D, Tsai SQ, Sander JD, Joung JK. et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA cleavage specificity. Nat Methods. 2014;11:429
156. Suzuki K, Yu C, Qu J, Li M, Yao X, Yuan T. et al. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell. 2014;15:31-6
157. Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H. et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27-30
158. Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C. et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15:12
159. Kennedy SR, Schmitt MW, Fox EJ, Kohrn BF, Salk JJ, Ahn EH. et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat Protoc. 2014;9:2586-606
160. Sternberg SH, LaFrance B, Kaplan M, Doudna JA. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature. 2015;527:110-3
161. Hilton IB, D'Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE. et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nature Biotechnol. 2015;33:510-7
162. Fujita T, Fujii H. Isolation of Specific Genomic Regions and Identification of Associated Molecules by Engineered DNA-Binding Molecule-Mediated Chromatin Immunoprecipitation (enChIP) Using CRISPR. In: (ed.) Chellappan SP. Chromatin Protocols. New York, NY: Springer New York. 2015:43-52
163. O'Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, Doudna JA. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014;516:263-6
164. Chen B, Hu J, Almeida R, Liu H, Balakrishnan S, Covill-Cooke C. et al. Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci. Nucleic Acids Res. 2016;44:e75-e
165. Deng W, Shi X, Tjian R, Lionnet T, Singer RH. CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells. Proc Natl Acad Sci USA. 2015;112:11870-5
166. Holkers M, Maggio I, Henriques SFD, Janssen JM, Cathomen T, Goncalves MAFV. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat Meth. 2014;11:1051-7
167. Hoban MD, Cost GJ, Mendel MC, Romero Z, Kaufman ML, Joglekar AV. et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597-604
168. Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE. et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384-9
169. Cornu TI, Mussolino C, Cathomen T. Refining strategies to translate genome editing to the clinic. Nat Med. 2017;23:415-23
170. Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS. et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217-21
171. Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E. et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400-3
172. Onoshima D, Yukawa H, Baba Y. Multifunctional quantum dots-based cancer diagnostics and stem cell therapeutics for regenerative medicine. Adv Drug Deliv Rev. 2015;95:2-14
173. Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH. et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnol. 2015;33:73-80
174. Hornung V, Latz E. Intracellular DNA recognition. Nat Rev Immunol. 2010;10:123-30
175. Chandradoss Stanley D, Schirle Nicole T, Szczepaniak M, MacRae Ian J, Joo C. A Dynamic Search Process Underlies MicroRNA Targeting. Cell. 2015;162:96-107
176. Graham DB, Root DE. Resources for the design of CRISPR gene editing experiments. Genome Biol. 2015;16:260
177. Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A. et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616-23
178. Chong JJ, Yang X, Don CW, Minami E, Liu Y-W, Weyers JJ. et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273-7
179. Consortium EP. The ENCODE (ENCyclopedia of DNA elements) project. Science. 2004;306:636-40
180. Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML. et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787-97
181. Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A. et al. BLUEPRINT to decode the epigenetic signature written in blood. Nature Biotechnol. 2012;30:224-6
182. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673
183. Xu Y, Shi Y, Ding S. A chemical approach to stem-cell biology and regenerative medicine. Nature. 2008;453:338-44
184. Neems DS, Garza-Gongora AG, Smith ED, Kosak ST. Topologically associated domains enriched for lineage-specific genes reveal expression-dependent nuclear topologies during myogenesis. Proc Natl Acad Sci USA. 2016;113:E1691-E700
185. Fierz B, Muir TW. Chromatin as an expansive canvas for chemical biology. Nat Chem Biol. 2012;8:417-27
186. Wilke A, Bischof J, Gerlach W, Glass E, Harrison T, Keegan KP. et al. The MG-RAST metagenomics database and portal in 2015. Nucleic Acids Res. 2015;44:D590-D4
187. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190-5
188. Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G. et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901-10
189. Liao H-K, Gu Y, Diaz A, Marlett J, Takahashi Y, Li M. et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat Commun. 2015;6:6413
190. Kang H, Minder P, Park MA, Mesquitta W-T, Torbett BE, Slukvin II. CCR5 disruption in induced pluripotent stem cells using CRISPR/Cas9 provides selective resistance of immune cells to CCR5-tropic HIV-1 virus. Mol Ther Nucleic Acids. 2015;4:e268
191. Liang M, Kamata M, Chen KN, Pariente N, An DS, Chen IS. Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J Gene Med. 2010;12:255-65
192. Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765-70
193. Kang X, He W, Huang Y, Yu Q, Chen Y, Gao X. et al. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. J Assist Reprod Gen. 2016;33:581-8
194. Adashi EY, Cohen IG. Going Germline: Mitochondrial Replacement as a Guide to Genome Editing. Cell. 2016;164:832-5
195. Stirling PC, Hieter P. Canonical DNA Repair Pathways Influence R-Loop-Driven Genome Instability. J Mol Biol. 2016
196. Downing TL, Soto J, Morez C, Houssin T, Fritz A, Yuan F. et al. Biophysical regulation of epigenetic state and cell reprogramming. Nat Mater. 2013;12:1154-62
197. Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57-67
198. Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447-59
199. Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B. et al. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Meth. 2012;9:363-6
200. Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186-91
Footnotes
IVF embryos are human embryos carrying specific mutations or chromosomal aberrations identified by pre-implantation genetic diagnosis (PGD) or pre-implantation genetic screening (PGS).
Cellular ensembles derived from pluripotent cells, formed by growing PSCs in suspension, in the absence of self-renewal-promoting growth factors. Once aggregates are formed, these cells start differentiating, in some ways replaying early embryonic development.
At low transfection efficiencies, stemming from the large size of the Cas9 construct, positive selection of targeted clones in iPSCs is a labor-intensive process.
Margaret Atwood, often called the prophet of dystopia, wrote a speculative fictional trilogy about scientific advancement spiraling out of control.
Author contact
![]() Corresponding authors: Deok-Ho Kim (deokhoedu) or Somali Chaterji (schaterjiorg)
Corresponding authors: Deok-Ho Kim (deokhoedu) or Somali Chaterji (schaterjiorg)