Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(18):4632-4642. doi:10.7150/thno.18630 This issue Cite
Research Paper
MMPP Attenuates Non-Small Cell Lung Cancer Growth by Inhibiting the STAT3 DNA-Binding Activity via Direct Binding to the STAT3 DNA-Binding Domain
Dong Ju Son1*, Jie Zheng1*, Yu Yeon Jung2*, Chul Ju Hwang1, Hee Pom Lee1, Ju Rang Woo3, Song Yi Baek3, Young Wan Ham4, Min Woong Kang5, Minho Shong5, Gi Ryang Kweon5, Min Jong Song6, Jae Kyung Jung1, Sang-Bae Han1, Bo Yeon Kim7, Do Young Yoon8, Bu Young Choi9 ![]() , Jin Tae Hong1
, Jin Tae Hong1 ![]()
1. College of Pharmacy & Medical Research Center, Chungbuk National University, Cheongju, Chungbuk 28160, Republic of Korea;
2. Department of Dental Hygiene, Gwangyang Health Sciences University, Gwangyang, Jeonnam 57764, Korea;
3. New Drug Development Center, Osong Medical Innovation Foundation, Cheongju, Chungbuk 28160, Republic of Korea;
4. Department of Chemistry, Utah Valley University, 800 W University Pkwy, Orem, UT 84058, USA;
5. Chungnam National University Hospital, Chungnam National University School of Medicine, Daejeon, Chungnam 34134, Republic of Korea;
6. Department of Obstetrics and Gynecology, Daejeon St. Mary's Hospital, College of Medicine, The Catholic University of Korea, Daejeon 34943, Republic of Korea;
7. World Class Institute, Korea Research Institute of Bioscience and Biotechnology, Cheongju, Chungbuk 28116, Republic of Korea;
8. Department of Bioscience and Biotechnology, Konkuk University, Seoul 05029, Republic of Korea;
9. Department of Pharmaceutical Science and Engineering, Seowon University, Cheongju, Chungbuk, 28674, Republic of Korea.
* These authors contributed equally to this work
Received 2016-12-5; Accepted 2017-2-13; Published 2017-10-16
Abstract

Rationale: Signal transducer and activator of transcription-3 (STAT3) plays a pivotal role in cancer biology. Many small-molecule inhibitors that target STAT3 have been developed as potential anticancer drugs. While designing small-molecule inhibitors that target the SH2 domain of STAT3 remains the leading focus for drug discovery, there has been a growing interest in targeting the DNA-binding domain (DBD) of the protein.
Methods: We demonstrated the potential antitumor activity of a novel, small-molecule (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol (MMPP) that directly binds to the DBD of STAT3, in patient-derived non-small cell lung cancer (NSCLC) xenograft model as well as in NCI-H460 cell xenograft model in nude mice.
Results: MMPP effectively inhibited the phosphorylation of STAT3 and its DNA binding activity in vitro and in vivo. It induced G1-phase cell cycle arrest and apoptosis through the regulation of cell cycle- and apoptosis-regulating genes by directly binding to the hydroxyl residue of threonine 456 in the DBD of STAT3. Furthermore, MMPP showed a similar or better antitumor activity than that of docetaxel or cisplatin.
Conclusion: MMPP is suggested to be a potential candidate for further development as an anticancer drug that targets the DBD of STAT3.
Keywords: (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol, STAT3, DNA-binding domain, anticancer, NSCLC.
Introduction
Signal transducer and activator of transcription-3 (STAT3) is a key mediator of intracellular signaling stimulated by various cytokines, hormones, growth factors, and oncoproteins [1]. After activation, STAT3 translocates to the nucleus where it binds to consensus STAT3-binding sequences located in the promoters of target genes, inducing transcriptional activation of various growth promoting genes [1]. Compelling evidence suggests that constitutive activation of STAT3 occurs frequently in many human cancers and is critical for cancer progression and aggressiveness [2]. In addition, activated STAT3 contributes in oncogenic transformation via transcriptional regulation of its critical downstream signaling elements that are important for cell cycle, proliferation, survival, migration, invasion, angiogenesis, and immune system evasion [3-5]. Therefore, STAT3 represents an attractive target for designing anticancer therapy [6].
The most common anticancer drug discovery approaches for turning off the STAT3 signaling largely focus on kinases upstream of STAT3; for example, pg130 complex recruits Janus kinases (JAKs), thereby disrupting STAT3 signaling and activity [6]. Although targeting the kinases responsible for STAT3 activation can successfully reduce STAT3 activity, off-target toxicities are still a concern related to this therapeutic approach. The off-target unwanted effects can be avoided, while retaining effective anticancer activity, by directly targeting STAT3 protein. Recently, a number of small molecules have been developed that inhibit the STAT3 function by interrupting the formation of STAT3 dimers through direct binding to the SH2 domain [6, 7]. Some of these compounds showed remarkable preclinical anticancer activity and a few have entered the clinical trials. However, none of them has been approved for clinical use yet due to the high concentrations required for STAT3 inhibition, which increases the likelihood for off-target toxicities. Moreover, protein-protein interactions make it more difficult and complex. While STAT3 SH2 domain remains the main focus for designing small-molecule inhibitors, there has been a growing interest in developing specific inhibitors that can target the DNA-binding domain (DBD) of STAT3 [8-11].
In our previous studies, we have shown that (E)-2,4-bis(p-hydroxyphenyl)-2-butenal (BHPB), a bioactive compound derived from tyrosine-fructose Maillard reaction product [12], effectively inhibited the activation of STAT3 and its downstream signaling pathway, and attenuated tumor growth and arthritis [13, 14]. However, this hit compound had some limitations in terms of its chemical stability and drug-like properties, mainly due to the presence of α,β-unsaturated aldehyde moiety in its chemical structure. The presence of α,β-unsaturated carbonyl groups in a drug candidate makes it a strong electrophile for Michael addition with a variety of nucleophiles present in living systems (e.g. cysteine, nucleic acids), resulting in the formation of a non-selective covalent bond. This could lead to severe side effects, thereby limiting the therapeutic potential of the compound.
In this study, we performed an extensive structure and activity-guided hit optimization and mechanistic study to overcome these problems. We designed and synthesized 16 BHPB analogs, and identified a novel and improved lead compound, (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol (MMPP). The antitumor effects of MMPP and the underlying mechanism were evaluated in vitro and in vivo.
Materials and Methods
The materials and methods are available in the Methods section of the Supplementary Material.
Results
Identification of active analogs of BHPB targeting STAT3
In an effort to improve drug-likeness and therapeutic efficacy, we designed and synthesized a library of 16 BHPB analogs by modifying the conjugated α,β-unsaturated aldehyde moiety or protecting phenolic alcoholic moieties to various ethers or both (Supplementary Fig. S1). First, molecular dynamics simulation analysis was performed to identify a hit compound that had potential for STAT3-binding. Of all 16 BHPB analogs, compound 13 (MMPP; Fig 1a) showed the strongest binding affinity to STAT3; it had the lowest binding free energy (-8.2 kcal/mol) compared with that of BHPB (-6.4 kcal/mol) (Supplementary Table S1). In addition, MMPP potently suppressed STAT3-dependent luciferase reporter expression in and viability of NCI-H460 NSCLC cells at the lowest IC50 (1.95 and 12.3 μg/mL, respectively). These inhibitory activities of MMPP were more potent than those of the original compound BHPB (Supplementary Table S1), suggesting that MMPP exhibits a higher inhibition of STAT3 activity and tumor growth. These findings led us to further investigate the anticancer activity, possible mechanism, and drug-like properties of MMPP.
Chemical structure of MMPP, Pull-down assay, and surface plasmon resonance assay. (a) Chemical structure of MMPP. (b) Pull-down assay showing an interaction between MMPP and STAT3 or STAT1. MMPP was conjugated with epoxy-activated Sepharose 6B. (c) Surface plasmon resonance binding kinetic traces for the binding between MMPP immobilized on the chip surface and STAT3 recombinant protein. MMPP: (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol; STAT: signal transducer and activator of transcription
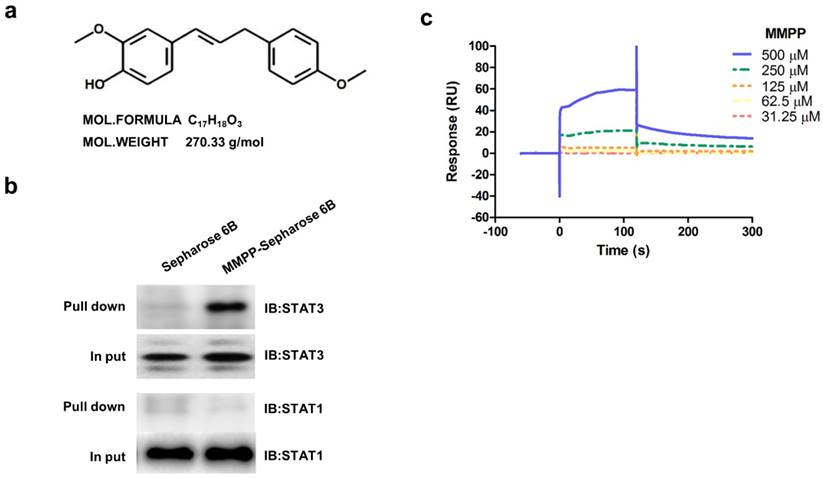
MMPP directly binds to the DBD of STAT3
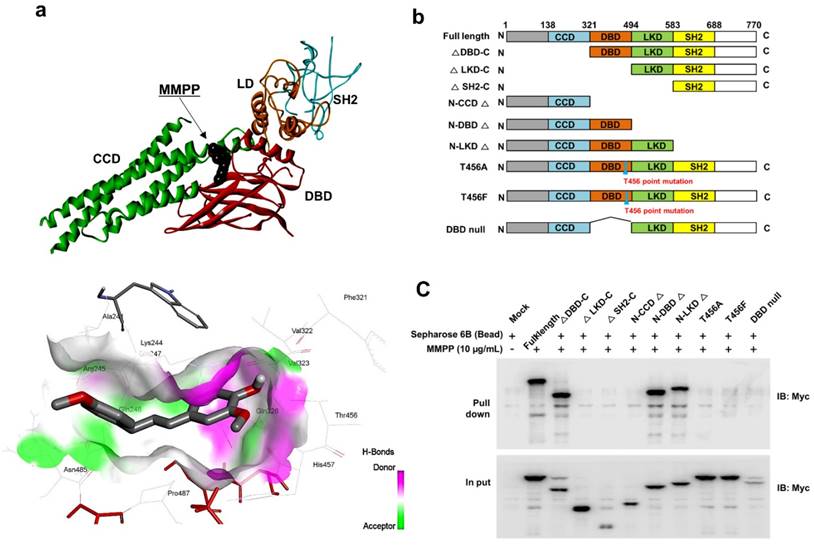
As MMPP was designed to target STAT3 and it exhibited inhibitory effects on STAT3 activation, we investigated if it could physically interact with STAT3. The pull-down assay using MMPP-conjugated epoxy-activated Sepharose 6B beads and A549 cell lysates showed that MMPP-conjugated beads successfully pulled down STAT3, whereas the vehicle control beads did not (Fig. 1b). STAT1 was not affected. Moreover, a surface plasmon resonance (SPR) analysis showed that the binding of MMPP to STAT3 protein increased with an increase in concentration of MMPP, suggesting that MMPP binds to STAT3 protein with a high affinity in a concentration-dependent manner (Fig. 1c). To determine the potential binding mode of MMPP to STAT3 and the precise conformation of MMPP at the binding site in STAT3, virtual docking analysis was performed between MMPP and the crystal structure of STAT3. Fig. 2a shows a surface rendering of the DBD of STAT3 with MMPP. It shows that MMPP directly binds to the hydroxyl residues in the DBD of STAT3 (inside a flight pocket comprised of Ala241, Lys244, Arg245, Gln247, Gln248, Phe321, Val322, Cal323, Gln326, Thr456, His457, Asn485, and Pro487), although it weakly interacts with STAT1 (Supplementary Fig. S2). To further elucidate the interaction between MMPP and the DBD of STAT3, GST pull-down assays were performed. Briefly, a series of GST-STAT3-fusion protein-expressing vectors with different domains as well as T456 mutant (Fig. 2b) were constructed and used to investigate whether DBD is the specific binding site for MMPP. The result showed that STAT3 fragments lacking DBD were not pulled down by MMPP-conjugated beads, whereas those with deletion of other domains, including N-terminal domain, CCD, LD, SH2 domain, or TAD domain were successfully pulled down by MMPP-conjugated beads (Fig. 2c). These findings suggested that MMPP could directly bind to STAT3 and its binding site was likely located in the DBD of STAT3, as anticipated. Furthermore, MMPP did not bind to STAT3 when a threonine residue (T456, a core amino acid of the DBD) in the DBD was mutated (T456 of the DBD was replaced with an unrelated amino acid A or F), suggesting that T456 residue in DBD is critical for direct binding between MMPP and STAT3 (Fig. 2c).
MMPP directly binds to the DBD of STAT3. (a) Docking model of MMPP with STAT3. (b) Schematic domain structures of STAT3 recombinant proteins. (c) Pull-down assay with deletion of different binding sites of STAT3 was performed to determine whether MMPP binds to DBD of STAT3. DBD: DNA binding domain; MMPP: (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol; STAT: signal transducer and activator of transcription
MMPP selectively inhibits STAT3 DNA binding activity, but not its dimerization
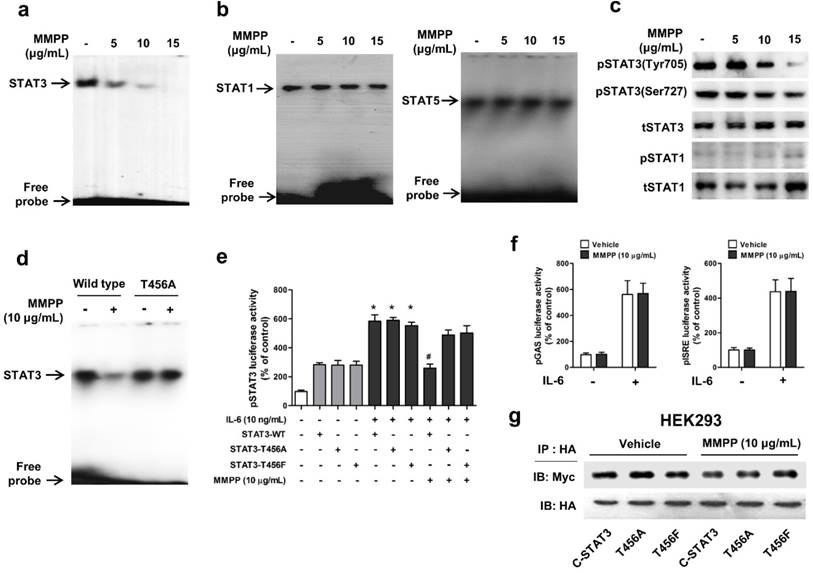
To verify the above findings and to determine the importance of T456 residue in DBD in MMPP-mediated suppression of STAT3 activity, we performed a STAT3-dependent luciferase reporter gene assay and an electrophoretic mobility shift assay (EMSA) in NSCLC cells transiently transfected with T456 mutant plasmids (T456A or T456F). First, we evaluated the effect of MMPP on STAT3 DNA binding activity by EMSA. MMPP inhibited the STAT3 DNA-binding activity in NCI-H460 and A549 cells (Fig. 3a, Supplementary Fig. S3a). In contrast, the STAT1 and STAT5 DNA-binding activity remained unaffected by treatment with MMPP (Fig. 3b), indicating STAT3 selectivity of MMPP over STAT1 and STAT5. Moreover, phosphorylation of STAT3 (Tyr705 and Ser727), but not that of STAT1, as assessed via western blot, was effectively inhibited by MMPP treatment in a concentration-dependent manner (Fig 3c, Supplementary Fig. S3b). MMPP treatment significantly decreased the STAT3 DNA binding activity in control cells (wild-type STAT3 vector transfected); however, T456 mutation abolished the inhibitory effect of MMPP on STAT3 DNA binding activity (Fig. 3d). MMPP-mediated suppression of STAT3 luciferase activity was markedly reduced by T456 mutation (Fig. 3e). Moreover, the luciferase activities of STAT1 (pGAS) and STAT5 (pISRE) were not affected by MMPP treatment (Fig. 3f).We developed an assay that directly measures STAT3-STAT3 dimerization in intact cells. For this, HA-tagged STAT3 and Myc-tagged STAT3 were cloned into a vector and HEK293 cells that stably co-expressed HA-STAT3 and Myc-STAT3 were generated. As shown in Fig. 3g, Myc-STAT3 co-immunoprecipitated with HA-STAT3 in HEK293 cells that co-express Myc-STAT3 and HA-STAT3 in vehicle treated cells. The T456 mutation or treatment with MMPP did not affect the co-expression of HA-STAT3 and Myc-STAT3, indicating that MMPP did not inhibit STAT3-STAT3 dimerization, at concentration as high as 10 μg/mL. Based on these findings, we suggest that MMPP binds directly and selectively to DBD of STAT3 by interacting with hydroxyl residues, especially of T456, thereby resulting in the inhibition of STAT3 DNA binding activity but not STAT3 dimerization.
MMPP inhibits STAT3 DNA-binding activity in NCI-H460 cells. (a-b) NCI-H460 NSCLC cells were treated with MMPP (0-15 μg/mL) for 6 h and then lysed with buffers A and C. Nuclear extracts were incubated with ³²p-end-labeled oligonucleotides containing STAT3 (a) or STAT1 (b) sequence. EMSA was performed to assess the effect of MMPP on DNA binding activity. (c) Total cell lysates were used to determine the expression of Tyr705 phosphorylated-STAT3, Ser727 phosphorylated-STAT3, total-STAT3, phosphorylated-STAT1, and total-STAT1 in NCI-H460 cells. Western blot analysis was carried out to assess the effect of MMPP on STAT3 nuclear translocation. (d) Cells were transfected with wild type STAT3 plasmid or STAT3 mutant T456A plasmid. EMSA was performed to assess the effect of STAT3 mutant T456A on the inhibitory effect of MMPP on the DNA binding activity of STAT3. (e) Luciferase assay was carried out to determine the effects of STAT3 mutants T456A or T456F on the inhibitory effect of MMPP on IL-6-induced STAT3 luciferase activity (n= 6, data shown as mean ± SEM, *p < 0.05, paired t-test). (f) Luciferase assay was conducted to determine the effects of MMPP on IL-6-induced STAT1 (pGAS) and STAT5 (pISRE) luciferase activity. (n= 6, data shown as mean ± SEM, *p < 0.05, paired t-test). (g) Co-immunoprecipitation assay was performed to assess the effect of MMPP on the STAT3-STAT3 dimerization. EMSA: electrophoretic mobility shift assay; IL: interleukin; MMPP: (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol; STAT: signal transducer and activator of transcription.
MMPP inhibits cancer cell survival and induces apoptosis
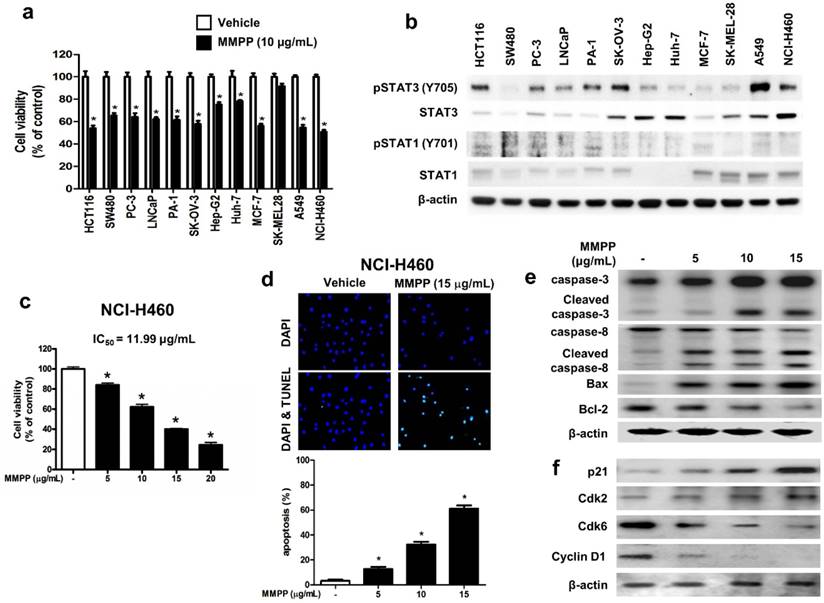
To assess the effects of MMPP on tumor cell survival, various cancer cells with different tissue origin were treated with the compound. Results showed that MMPP significantly inhibited the viability of cancer cells including that of colon (HCT116, SW480), prostate (PC3, LNCaP), ovary (PA-1, SK-OV-3), liver (Hep-G2, Huh-7), breast (MCF-7), skin (SK-MEL-28), and lung cells (A549, NCI-H460) (Fig. 4a). In addition, MMPP effectively suppressed STAT3-dependent luciferase reporter gene expression in these cancer cells (Supplementary Fig. S4). The NSCLC cell lines, A549 and NCI-H460, showed constitutively activated STAT3 as well as total STAT3, whereas STAT1 and STAT5 were expressed at low levels (Fig. 4b). Therefore, the effect of MMPP on cell viability and apoptosis in these NSCLC cells as well as in a non-cancerous lung epithelial cell line (LL-24) was also evaluated. MMPP decreased the viability of both NSCLC cells in a concentration-dependent manner, but exhibited no toxicity in LL-24 normal lung epithelial cells at the tested concentrations (Fig. 4c, Supplementary Fig. S5a). To determine whether apoptosis plays a part in MMPP-induced reduction in cell viability, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed. In both NCI-H460 and A549 cells, remarkable induction of apoptosis was observed following treatment with MMPP (Fig. 4d, Supplementary Fig. S5b). Moreover, MMPP elevated the expression of apoptotic proteins, such as Bax, cleaved caspase-3, and cleaved caspase-8, in a concentration-dependent manner, while it suppressed the expression of the antiapoptotic protein Bcl-2 in both NCI-H460 (Fig. 4e) and A549 cells (Supplementary Fig. S6a). Results also showed that MMPP effectively regulated the expression of G1-phase cell cycle regulatory proteins including p21, CDK6, and cyclin D1 in both NCI-H460 (Fig. 4f) and A549 cells (Supplementary Fig. S6b).
MMPP inhibits cell growth and induces apoptosis in various cancer cell lines. (a) HCT116 and SW480 colon cancer cells, PC-3 and LNCaP prostate cancer cells, PA-1 and SKOV-3 ovarian cancer cells, Hep G2 and Huh-7 liver cancer cells, MCF-7 breast cancer cells, SK-MEL-28 human melanoma cells, A549 and NCI-H460 NSCLC cells were treated with MMPP (10 μg/mL) for 24 h. The relative cell survival rate was determined by MTT assay. (n= 10, data shown as mean ± SEM, *p < 0.05, paired t-test). (b) Cells were lysed and analyzed by western blotting with antibodies against pSTAT3 (Y705), total STAT3, pSTAT1 (Y701), and total STAT1, PCNA, using β-actin as a loading control. (c) NCI-H460 cells were treated with MMPP (0-20 μg/mL) for 24 h and the relative cell survival rate was determined by MTT assay (n= 10, data shown as mean ± SEM, *p < 0.05, paired t-test). (d) NCI-H460 cells were cultured in an eight chambered glass culture slide, serum starved for 12 h, and then treated with MMPP (0-15 μg/mL) for 24 h. Cell apoptosis was determined by TUNEL-assay. The apoptotic index was determined as the TUNEL-positive cell number divided by the total cell number (n= 3, data shown as mean ± SEM, *p < 0.05, paired t-test). (e-f) Cells were treated with MMPP (0-15 μg/mL) for 24 h, lysed and analyzed by western blotting with antibodies against multiple apoptosis regulatory proteins (e) and cell cycle regulatory proteins (f), using β-actin as a loading control. MMPP: (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol; PCNA: proliferating cell nuclear antigen; STAT: signal transducer and activator of transcription, TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling
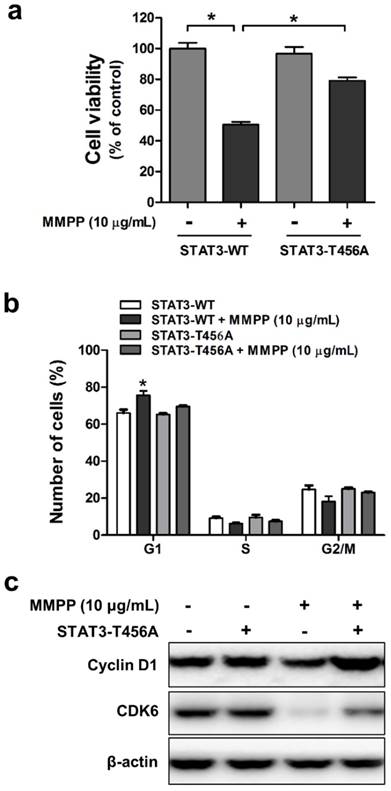
The DBD is critical for MMPP-mediated inhibition of cancer cell viability and cell cycle arrest
Since we confirmed that MMPP suppresses STAT3 activity through direct binding to DBD, we next evaluated whether DBD is critical for inhibitory effect of MMPP on cancer cell viability and cell cycle. For this study, mutant STAT3 plasmid (T456A) was transiently transfected into NCI-H460 and A549 cells, which were then used for MTT assay and fluorescence-activated cell sorting (FACS) flow cytometry analysis. MMPP inhibited the viability of and cell cycle progression in wild-type STAT3 plasmid-transfected cells. However, its inhibitory effects were attenuated by the T456 residue mutation in the DBD of STAT3 in both NCI-H460 (Fig. 5a and b) and A549 cells (Supplementary Fig. S7a and b). Furthermore, MMPP effectively decreased the expression of G1-phase cell cycle regulatory proteins such as CDK6 and cyclin D1, but its inhibitory effect was attenuated by the mutation in the DBD of STAT3 (Fig. 5c, Supplementary Fig. S7c). Moreover, treatment with Stattic, an inhibitor of STAT3 activation, or STAT3 siRNA augmented the inhibitory effect of MMPP on lung cancer cell growth and expression of CDK6 and cyclin D1 in both NCI-H460 and A549 cells (Supplementary Fig. S8).
Effects of mutant STAT3 (T456A) on MMPP-induced cell growth inhibition, CDK6 expression, and cyclin D1/G1 cell cycle arrest, and combinatorial effect of MMPP and STAT3 inhibition on the expression of G1 cell cycle regulatory proteins in NCI-H460 cells. (a) NCI-H460 NSCLC cells were transfected with mutant STAT3 (T456A) plasmid for 24 h and then treated with MMPP (10 μg/mL) for 24 h. Cell viability was determined by MTT assay (n = 10, data shown as mean ± SEM, *p < 0.05, paired t-test). (b-c) Cells were transfected with wild type STAT3 plasmid or STAT3 mutant T456A plasmid. (b) FACS analysis (PI staining) was conducted to assess the effect of STAT3 mutant T456A on the inhibitory effect of MMPP on the cell cycle arrest. (c) Cells were lysed and analyzed by western blotting with antibodies against cyclin D1 and CDK6, using β-actin as a loading control. MMPP: (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol; STAT: signal transducer and activator of transcription
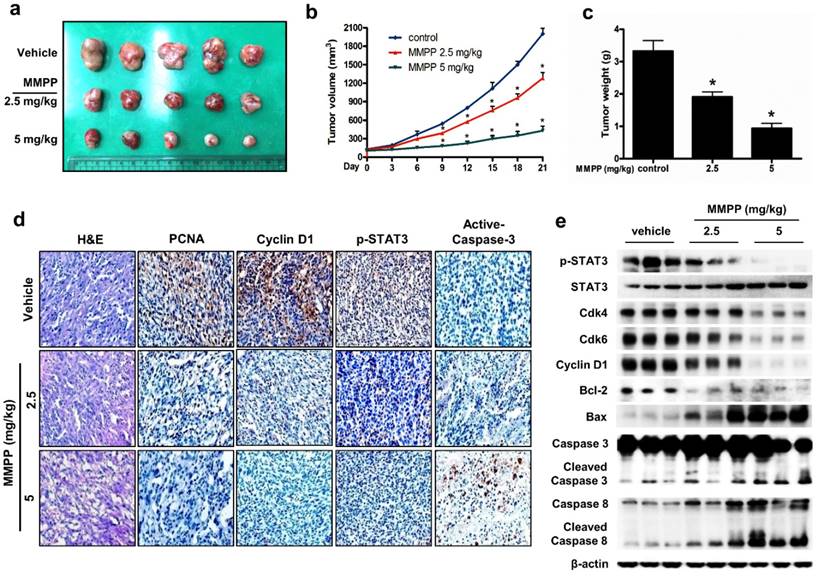
MMPP inhibits lung tumor growth in vivo
Finally, we tested the antitumor efficacy of MMPP in vivo using two different xenograft mouse models of NSCLC: NCI-H460 xenograft model and patient-derived xenograft (PDX) model. In the NCI-H460 xenograft model, treatment with MMPP (2.5-5 mg/kg, i.p. twice a week for 3 weeks) significantly and dose-dependently reduced tumor volume and weight compared with that reported for the vehicle controls (Fig. 6a-c). Similarly, MMPP treatment via oral dosing (5 mg/kg, three times a week for 3 weeks) significantly inhibited tumor growth in the NCI-H460 xenograft model (Supplementary Fig. S9a, b). The immunohistochemical staining of xenograft tumors showed that the expression of p-STAT3, proliferating cell nuclear antigen (PCNA), and cyclin D1 markedly decreased, whereas that of active-caspase-3 increased in MMPP-treated group compared with that in the vehicle-treated control group (Figs. 6d, Supplementary Fig. S9c). In addition, western blot analysis of tumor tissues revealed that MMPP treatment decreased the expression of Cdk4, Cdk6, cyclin D1, and Bcl-2, whereas that of apoptotic proteins such as Bax, cleaved caspase-3, and cleaved caspase-8 increased compared with the results of the vehicle-treated control group (Fig. 6e). MMPP treatment effectively inhibited the DNA-binding activity of STAT3 and nuclear translocation of p-STAT3; it also increased soluble nuclear STAT3 in nuclear fraction of the tumor tissue (Fig. 6e, Supplementary Fig. S9d and e). Administration of MMPP (5 mg/kg, i.p. twice a week for 3 weeks) also effectively suppressed tumor growth and STAT3 activity in xenograft models of A549 NSCLC, HCT116 colon cancer, and PA-1 ovarian cancer cells (Supplementary Fig. S10a-c). The antitumor activity of MMPP was superior to that of cisplatin (5 mg/kg) but similar to that of docetaxel (5 mg/kg) (Supplementary Fig. S10d).
Antitumor activity of MMPP in NCI-H460 xenograft model. (a-c) Growth inhibition of subcutaneously transplanted NCI-H460 xenografts in BALB/c mice treated with MMPP (2.5 mg/kg and 5 mg/kg twice a week) for 3 weeks. The mice were administered 0.01% DMSO or MMPP (2.5 mg/kg and 5 mg/kg) by i.p. injection. Tumor burden was measured once per week using a caliper, and volume was calculated (n = 10, data shown as mean ± SEM, *p < 0.05, paired t-test). (d) Expression levels of PCNA, active caspase-3, p-STAT3, and cyclin D1 in xenograft tissues after different treatments as determined by immunohistochemistry. (e) Tumor tissues were lysed and analyzed by western blotting with antibodies against p-STAT3, total STAT3, and multiple cell cycle and apoptosis regulatory proteins, using β-actin as a loading control. MMPP: (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol; PCNA: proliferating cell nuclear antigen; STAT: signal transducer and activator of transcription
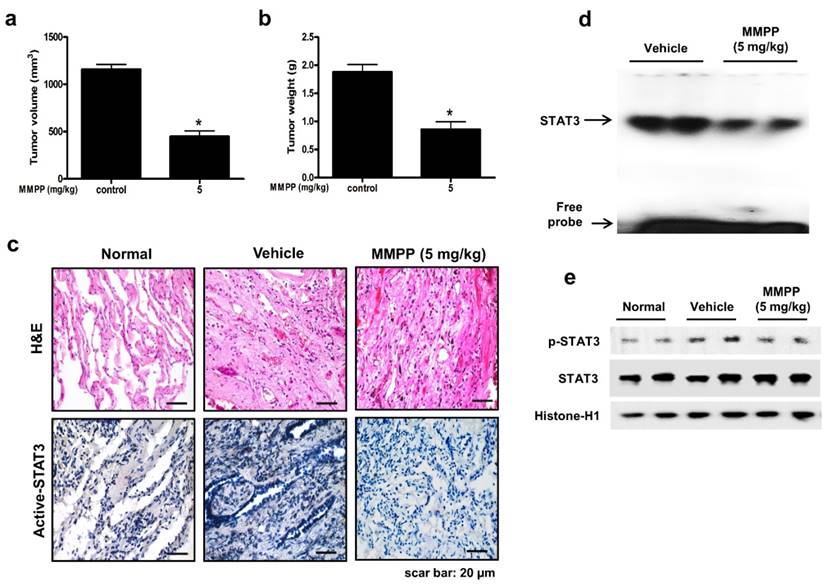
To evaluate the antitumor efficacy of MMPP using a cancer model that more closely mimics the conditions of a tumor inside a human body and is useful for predicting clinical outcomes, a PDX model was established by direct transplantation of fresh human tumor specimen (1-2 mm3) from 6 NSCLC patients into the dorsal subcutaneous space of immunodeficient mice (n = 6 for each tumor specimen). The PDX models are maintained by passaging cells directly from mouse to mouse to reach 1000-1500 mm3 tumor volume. After the tumor tissue had been passaged three times, PDX tumor tissues were divided into small pieces, directly re-transplanted into mice to establish PDX models for evaluating the antitumor efficacy of MMPP. In the NSCLC PDX model established in this study, the final volume and weight of tumors in the MMPP-treated mice (5 mg/kg, oral, three times a week for 1 month) were significantly lesser than those in the vehicle-treated control mice (Fig. 7a, b). Immunohistochemical analysis of tumor sections also revealed that MMPP suppressed tumor growth and expression of p-STAT3 in PDX tumor tissues (Fig. 7c), and the DNA-binding activity (Fig. 7d) and nuclear translocation of p-STAT3 (Fig. 7e). Importantly, the MMPP-treated animals did not show any signs of body weight differences or liver toxicity (Supplementary Fig. S11). These findings demonstrated a potent antitumor effect of MMPP in two different murine models of NSCLC.
Antitumor activity of MMPP in NSCLC patient-derived xenograft model. Xenograft mice bearing patient-derived tumors were administered 0.01% DMSO or MMPP (5 mg/kg) for 1 month. Tumor volume (a) and tumor weight (b) were measured after necropsy (n= 5, data shown as mean ± SEM, *p < 0.05, paired t-test). (c) p-STAT3 expression in xenografts as determined by immunohistochemistry. (d) DNA-binding activity of STAT3 as determined by EMSA. Results are representative of three experiments. (e) Expression of STAT3 and p-STAT3 as determined by western blot analysis. Results are representative of three experiment. EMSA: electrophoretic mobility shift assay; MMPP: (E)-2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1-yl)phenol; STAT: signal transducer and activator of transcription
MMPP is predicted to have drug-like properties
To investigate whether MMPP has favorable drug-like properties to be considered for further clinical development, in silico toxicology and absorption, distribution, metabolism, and excretion (ADME) studies were carried out. The in silico analysis predicted that MMPP was markedly less toxic than BHPB was, which was predicted to have mutagenic and hepatotoxic potential, indicating the remarkably improved toxicological profile of MMPP (Supplementary Table S2). MMPP showed highly improved drug-likeness profiles compared to those of BHPB.
Discussion
NSCLC has a high mortality rate. Although several tyrosine kinase inhibitors are currently used for its treatment, the rate of relapse and therapy failure has increased [15]. Similar to other organ-specific cancers, NSCLC is also characterized by elevated levels of STAT3 and inappropriate activation of STAT3-mediated signaling. Aberrant STAT3 signaling is a key factor responsible for maintaining various cancer hallmarks, such as abnormal cell proliferation, evasion from apoptosis, enhanced angiogenesis, and escape from host antitumor response. In fact, constitutively activated STAT3 has been identified in most human tumors including NSCLC [2]. Various studies have demonstrated that inhibitors of the STAT3 signaling pathway could be useful for the treatment of lung cancer. Small-molecule STAT3 inhibitors such as A18, A26, A69, and BP-1-102 were shown to suppress tumor growth of NSCLC and STAT3 activation in vivo [9, 16]. Moreover, cyclosporine A promoted gefitinib-induced apoptosis through inhibition of STAT3 in NSCLC in vivo and in vitro [17]. Similarly, garcinol suppressed the growth of hepatocellular carcinoma through inhibition of STAT3 dimerization in vivo and in vitro [18]. HO-3867, a safe STAT3 inhibitor, selectively killed ovarian cancer cells [19]. However, the drug-like properties of these compounds remain unknown. Thus, drug development using STAT3 as a target remains a viable strategy for eradicating NSCLCs. Considering STAT3 as a validated target for developing novel anticancer therapies, we synthesized a number of BHPB analogs as potential small-molecule inhibitors of STAT3 in the present study. Of all the analogs, MMPP showed the highest STAT3-binding and -inhibitory activities, indicating its potential as a drug. Therefore, in silico modeling was used to understand the binding pattern of MMPP with STAT3 and to evaluate its potential in suppressing NSCLC growth in mouse xenograft models.
The present study revealed that MMPP suppressed tumor growth in a dose-dependent manner (2.5 or 5 mg/kg body weight) through the inhibition of STAT3 activity in NCI-H460 xenograft and PDX models in mice. MMPP also inhibited NSCLC cell growth through cell cycle arrest and apoptosis induction via inhibition of STAT3 activity in A549 and NCI-H460 NSCLC cells in vitro. It has been reported that BHPB inhibits the growth of both A549 and NCI-H460 NSCLC cells with IC50 values of 31.0 μg/mL and 31.4 μg/mL, respectively [20]. MMPP inhibited the growth of these cells with IC50 values of 12.80 μg/mL and 11.99 μg/mL, respectively. Moreover, BHPB showed mutagenicity in vivo and chromosome damage in vitro, but MMPP did not, indicating its better efficacy and safety. BHPB was hepatotoxic, whereas MMPP treatment (5 mg/kg body weight) did not show any signs of toxicity in normal cells or hepatotoxic effects. Moreover, there were no significant differences in the rule of five, CMC-like rule, WDI-like rule, Caco2 cell permeability, and plasma protein binding, MMPP showed better aqueous solubility, human intestinal absorption, and skin permeability. Similar to i.p. administration, orally administered MMPP (5 mg/kg body weight) suppressed tumor growth in NCI-H460 xenograft model. Thus, MMPP is suggested have good oral bioavailability. Since it did not cause any toxicity to the host, the antitumor effect of MMPP was better than that of cisplatin, and similar to that of docetaxel (all at 5 mg/kg body weight). Owing to these excellent properties and antitumor activities, MMPP could be potentially developed as an anticancer drug.
Numerous studies have reported that compounds designed to target cell cycle arrest have an anticancer effect in various cancer cell lines including NSCLC cells [21, 22]. A mechanistic investigation showed that MMPP-induced G0/G1 arrest is mainly regulated by the down-regulation of cyclin D1 and CDK6. Cyclin D1 is a rate-limiting kinase for cell cycle progression from the G1 to the S phase, during which DNA replication occurs [23]. It has been reported that STAT3 plays a key role in the G1/S phase transition induced by the cytokine receptor subunit gp130 [24]. A novel STAT3 inhibitor, fluacrypyrim, was reported to induce cell cycle arrest and apoptosis by blocking STAT3 activation [25]. The present study also showed that MMPP induced apoptosis and G1 cell cycle arrest via inhibition of the STAT3 signaling pathway. In fact, treatment with MMPP in combination with STAT3 inhibition by Stattic or siRNA intensified the inhibitory effect of MMPP on cancer cell growth and expression of CDK6 and cyclin D1 (Supplementary Fig. 3). Transfection of cells with the STAT3 mutant T456A partially inhibited these effects. These data further demonstrate that STAT3 plays a pivotal role in MMPP-induced cell cycle arrest and apoptosis.
The supershift assay showed that MMPP more selectively inhibits STAT3 DNA-binding activity compared to that reported for STAT1 or STAT5. BIAcore SPR analysis, pull-down assay, and immunoprecipitation assay also showed that MMPP more selectively binds to STAT3 than with STAT1, indicating its specificity for STAT3. Competition analysis suggested that MMPP binds to the DBD of STAT3. Molecular docking simulations revealed that MMPP directly binds to the hydroxyl residue in the DBD of STAT3 (inside a flight pocket comprised of Ala241, Lys244, Arg245, Gln247, Gln248, Phe321, Val322, Cal323, Gln326, Thr456, His457, Asn485, and Pro487). As shown by luciferase assay and EMSA, mutant STAT3 (T456A or T456F) partially abolished the inhibitory effect of MMPP on STAT3 activation; the T456A mutation also partially abolished MMPP-induced cell growth inhibition. These data suggest that the DBD is crucial to MMPP-STAT3 interaction and imply that MMPP may exert anticancer effects through inhibition of STAT3 by directly binding to the DBD. Several small-molecule inhibitors of STAT3 such as Stattic, STA-21, and S3I-201 have been reported [26-28]. Targeting of the SH2 domain of STAT3 is undesirable, because only un-phosphorylated STAT3 can bind to DNA, resulting in incomplete inhibition of STAT3 [29]. Moreover, several SH2 inhibitors such as phosphotyrosyl peptides, tripeptides, and peptidomimetics showed poor cell permeability and stability [30]. However targeting the N-terminal domain of STAT3 is complicated because its structure is not completely elucidated, and the genes regulated by the N-terminus of STAT3 have not been fully identified [31]. Therefore, inhibitors of the DBD hold promise for cancer treatment. MMPP could suppress the growth and DNA-binding activity of STAT3 in other cancer cell lines including colon and ovarian cancer cells. However, it is not clear how the inhibitory effect of MMPP on the DNA binding activity of STAT3 resulted in the inhibition of STAT3 phosphorylation. However, it is noteworthy that MMPP exerted inhibitory effect on the ERK pathway in ovarian cancer cells (data not shown). We also found that the MMPP analog BHPB affected p38 MAP kinase. Thus, the ERK pathway, which is upstream of STAT3, could also be influenced by STAT3 activity, thereby inhibiting the phosphorylation of STAT3. Taken together, these findings indicated that MMPP has potential as a STAT3 inhibitor for cancer treatment. Therefore, MMPP, as a specific inhibitor of the DBD of STAT3, could overcome the disadvantages, such as off-target effects, of previous cell cycle inhibitors. For development of MMPP as a clinical use drug, we were granted a National (Republic of Korea) patent, and have also filed an application for international patent (PCT/KR/2016/003696).
In conclusion, MMPP could inhibit the growth of NSCLC cells in vitro and in vivo via G1-phase cell cycle arrest by directly interacting with the DBD of STAT3. Therefore, MMPP holds promise for further clinical development as an anti-NSCLC agent.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was financially supported by the National Research Foundation of Korea [NRF] grant funded by the Korea government (MSIP) (No. MRC, 2017R1A5A2015541), the Ministry of Trade, Industry & Energy (MOTIE, 1415139249) through the fostering project of Osong Academy-Industry Convergence (BAIO), and by the Functional Districts of the Science Belt support program, Ministry of Science, ICT and Future Planning.
Author Contributions
D.J.S., J.Z. and Y.Y.J. carried out most of the experiments, performed data analysis, and were the primary writers of the manuscript. C.J.H. performed GST pull-down assays. H.P.L. performed in silico ADME analysis. J.R.W. and S.Y.B. performed Biacore SPR analysis. Y.W.H. designed and synthesized test compounds. M.W.K., M.H.S., G.R.K., and M.J.S. provided specimens of NSCLC patients and contributed in establishment of PDX model. J.K.J., S.B.H., B.Y.K., D.Y.Y., and B.Y.C. provided advice throughout the project. J.T.H. supervised the entire project and had a major role in experimental design, data interpretation, and writing the manuscript. All authors reviewed the manuscript. D.J.S., J.Z. and Y.Y.J contributed equally to this work. B.Y.C. and J.T.H. also contributed equally to this paper and are co-corresponding authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Darnell JE Jr. STATs and gene regulation. Science. 1997;277:1630-5
2. Song L, Turkson J, Karras JG, Jove R, Haura EB. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene. 2003;22:4150-65
3. Lee H, Pal SK, Reckamp K, Figlin RA, Yu H. STAT3: a target to enhance antitumor immune response. Curr Top Microbiol Immunol. 2011;344:41-59
4. Chen Z, Han ZC. STAT3: a critical transcription activator in angiogenesis. Med Res Rev. 2008;28:185-200
5. Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548-56
6. Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem Biol. 2016;11:308-18
7. Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H. et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene. 2016;35:802
8. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP. et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochimica et biophysica acta. 2014;1845:136-54
9. Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H. et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene. 2016;35:783-92
10. Leung CH, Chan DS, Ma VP, Ma DL. DNA-binding small molecules as inhibitors of transcription factors. Medicinal research reviews. 2013;33:823-46
11. Li H, Ban F, Dalal K, Leblanc E, Frewin K, Ma D. et al. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. Journal of medicinal chemistry. 2014;57:6458-67
12. Hwang IG, Kim HY, Lee SH, Woo KS, Ban JO, Hong JT. et al. Isolation and identification of an antiproliferative substance from fructose-tyrosine Maillard reaction products. Food chemistry. 2012;130:547-51
13. Cho SH, Park MH, Lee HP, Back MK, Sung HC, Chang HW. et al. (E)-2,4-Bis(p-hydroxyphenyl)-2-butenal enhanced TRAIL-induced apoptosis in ovarian cancer cells through downregulation of NF-kappaB/STAT3 pathway. Arch Pharm Res. 2014;37:652-61
14. Ban JO, Kim DH, Lee HP, Hwang CJ, Shim JH, Kim DJ. et al. Anti-arthritis effects of (E)-2,4-bis(p-hydroxyphenyl)-2-butenal are mediated by inhibition of the STAT3 pathway. British journal of pharmacology. 2014;171:2900-12
15. Abdelhamed S, Ogura K, Yokoyama S, Saiki I, Hayakawa Y. AKT-STAT3 Pathway as a Downstream Target of EGFR Signaling to Regulate PD-L1 Expression on NSCLC cells. J Cancer. 2016;7:1579-86
16. Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT. et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9623-8
17. Shou J, You L, Yao J, Xie J, Jing J, Jing Z. et al. Cyclosporine A sensitizes human non-small cell lung cancer cells to gefitinib through inhibition of STAT3. Cancer letters. 2016;379:124-33
18. Sethi G, Chatterjee S, Rajendran P, Li F, Shanmugam MK, Wong KF. et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Molecular cancer. 2014;13:66
19. Rath KS, Naidu SK, Lata P, Bid HK, Rivera BK, McCann GA. et al. HO-3867, a safe STAT3 inhibitor, is selectively cytotoxic to ovarian cancer. Cancer research. 2014;74:2316-27
20. Kollipara PS, Jeong HS, Han SB, Hong JT. (E)-2,4-bis(p-hydroxyphenyl)-2-butenal has an antiproliferative effect on NSCLC cells induced by p38 MAPK-mediated suppression of NF-kappaB and up-regulation of TNFRSF10B (DR5). British journal of pharmacology. 2013;168:1471-84
21. Johnson AJ, Smith LL, Zhu J, Heerema NA, Jefferson S, Mone A. et al. A novel celecoxib derivative, OSU03012, induces cytotoxicity in primary CLL cells and transformed B-cell lymphoma cell line via a caspase- and Bcl-2-independent mechanism. Blood. 2005;105:2504-9
22. Gao X, Liu Y, Deeb D, Arbab AS, Guo AM, Dulchavsky SA. et al. Synthetic oleanane triterpenoid, CDDO-Me, induces apoptosis in ovarian cancer cells by inhibiting prosurvival AKT/NF-kappaB/mTOR signaling. Anticancer Res. 2011;31:3673-81
23. Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends in pharmacological sciences. 2003;24:139-45
24. Fukada T, Ohtani T, Yoshida Y, Shirogane T, Nishida K, Nakajima K. et al. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. The EMBO journal. 1998;17:6670-7
25. Yu ZY, Huang R, Xiao H, Sun WF, Shan YJ, Wang B. et al. Fluacrypyrim, a novel STAT3 activation inhibitor, induces cell cycle arrest and apoptosis in cancer cells harboring constitutively-active STAT3. International journal of cancer. 2010;127:1259-70
26. Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4700-5
27. McMurray JS. A new small-molecule Stat3 inhibitor. Chemistry & biology. 2006;13:1123-4
28. Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR. et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7391-6
29. Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, Tarasov SG. et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. The Journal of biological chemistry. 2012;287:14192-200
30. Bhasin D, Cisek K, Pandharkar T, Regan N, Li C, Pandit B. et al. Design, synthesis, and studies of small molecule STAT3 inhibitors. Bioorganic & medicinal chemistry letters. 2008;18:391-5
31. Hu T, Yeh JE, Pinello L, Jacob J, Chakravarthy S, Yuan GC. et al. Impact of the N-Terminal Domain of STAT3 in STAT3-Dependent Transcriptional Activity. Molecular and cellular biology. 2015;35:3284-300
Author contact
![]() Corresponding authors: Jin Tae Hong, PhD, Professor, College of Pharmacy and Medical Research Center, Chungbuk National University, Osongsaengmyeong 1-ro 194-31, Osong-eup, Heungduk-gu, Cheongju, Chungbuk, 28160, Republic of Korea. Telephone: +82-43-261-2813. E-mail: jinthongac.kr or Bu Young Choi, PhD, Professor, Department of Pharmaceutical Science and Engineering, Seowon University, Musimseoro 377-3, Seowon-gu, Cheongju, Chungbuk, 28674, Republic of Korea. Telephone: +82-43-299-8411. E-mail: bychoiac.kr
Corresponding authors: Jin Tae Hong, PhD, Professor, College of Pharmacy and Medical Research Center, Chungbuk National University, Osongsaengmyeong 1-ro 194-31, Osong-eup, Heungduk-gu, Cheongju, Chungbuk, 28160, Republic of Korea. Telephone: +82-43-261-2813. E-mail: jinthongac.kr or Bu Young Choi, PhD, Professor, Department of Pharmaceutical Science and Engineering, Seowon University, Musimseoro 377-3, Seowon-gu, Cheongju, Chungbuk, 28674, Republic of Korea. Telephone: +82-43-299-8411. E-mail: bychoiac.kr