Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(1):13-30. doi:10.7150/thno.21089 This issue Cite
Research Paper
Changes in the microarchitecture of the pancreatic cancer stroma are linked to neutrophil-dependent reprogramming of stellate cells and reflected by diffusion-weighted magnetic resonance imaging
Philipp Mayer1, Christine Dinkic2, Ralf Jesenofsky3, Miriam Klauss1, Peter Schirmacher4, Ulrike Dapunt5, Thilo Hackert6, Florian Uhle7, G. Maria Hänsch8, Matthias M. Gaida4 ![]()
1. Clinic for Diagnostic and Interventional Radiology, University Hospital Heidelberg, Heidelberg, Germany
2. Clinic for Gynecology and Obstetrics, University Hospital Heidelberg, Heidelberg, Germany
3. Clinic for Internal Medicine II, University Hospital Mannheim, Medical Faculty Heidelberg, Mannheim, Germany
4. Institute of Pathology, University Hospital Heidelberg, Heidelberg, Germany
5. Clinic for Orthopedics and Trauma Surgery, University Hospital Heidelberg, Heidelberg, Germany
6. Department of General, Visceral, and Transplantation Surgery, University Hospital Heidelberg, Heidelberg, Germany
7. Clinic for Anesthesiology, University Hospital Heidelberg, Heidelberg, Germany
8. Institute of Immunology, University Heidelberg, Heidelberg, Germany
Received 2017-5-19; Accepted 2017-9-13; Published 2018-1-1
Abstract

In pancreatic cancer (PDAC) intratumor infiltration of polymorphonuclear neutrophils (PMN) is associated with histologically apparent alterations of the tumor growth pattern. The aim of this study was to examine possible associations between PMN infiltration, tumor microarchitecture, and water diffusivity in diffusion-weighted magnetic resonance imaging (DW-MRI), and to further asses the underlying mechanisms.
Methods: DW-MRI was performed in 33 PDAC patients prior to surgery. In parallel, tissue specimen were examined histologically for growth pattern, azurocidin-positive PMN infiltrates, and the presence of alpha-smooth muscle actin (α-SMA) and metalloproteinase 9 (MMP9)-positive myofibroblastic cells. For confirmation of the histological findings, a tissue microarray of a second cohort of patients (n=109) was prepared and examined similarly. For in vitro studies, the pancreatic stellate cell line RLT was co-cultivated either with isolated PMN, PMN-lysates, or recombinant azurocidin and characterized by Western blot, flow cytometry, and proteome profiler arrays.
Results: Tumors with high PMN density showed restricted water diffusion in DW-MRI and histologic apparent alterations of the tumor microarchitecture (microglandular, micropapillary, or overall poorly differentiated growth pattern) as opposed to tumors with scattered PMN. Areas with altered growth pattern lacked α-SMA-positive myofibroblastic cells. Tissue microarrays confirmed a close association of high PMN density with alterations of the tumor microarchitecture and revealed a significant association of high PMN density with poor histologic grade of differentiation (G3). In vitro experiments provided evidence for direct effects of PMN on stellate cells, where a change to a spindle shaped cell morphology in response to PMN and to PMN-derived azurocidin was seen. Azurocidin incorporated into stellate cells, where it associated with F-actin. Down-regulation of α-SMA was seen within hours, as was activation of the p38-cofilin axis, up-regulation of MMP9, and acquisition of intracellular lipid droplets, which together indicate a phenotype switch of the stellate cells.
Conclusion: In PDAC, PMN infiltrates are associated with alterations of the tumor microarchitecture. As a causal relationship, we propose a reprogramming of stellate cells by PMN-derived azurocidin towards a phenotype, which affects the microarchitecture of the tumor.
Keywords: pancreatic cancer, tumor infiltrating neutrophils, desmoplastic stroma, stellate cells, azurocidin, diffusion-weighted magnetic resonance imaging
Introduction
Pancreatic cancer (PDAC) is a highly aggressive neoplasm with a 5-year survival rate of ~5%, and very limited therapeutic options [1]. A characteristic feature of PDAC is its extensive desmoplastic tumor stroma that may comprise up to 90% of the whole tumor mass [2], and which - as judged by histologic examination - is rather heterogeneous. The stromal components consist of a plethora of extracellular matrix proteins including collagens, decorin, or fibronectin that are produced locally by activated pancreatic stellate cells and cancer-associated fibroblasts [2]. Inflammatory cells, such as T cells, macrophages, and polymorphonuclear neutrophils (PMN) are interspersed in various densities and activation stages [3-7]. The abundance of the extracellular desmoplastic stroma is one of the key factors that determines tumor progression in PDAC [8].
In clinical diagnostics, diffusion weighted magnetic resonance imaging (DW-MRI), which measures the diffusion of water molecules within the tumor tissue, is often performed [9] and is useful to evaluate a focal pancreatic mass [10]. Because of increased cellular density and fibrosis, diffusion is usually more restricted in PDAC compared to healthy pancreatic tissue [10]. Restricted diffusion is expressed by lower values of the apparent diffusion coefficient (ADC) and/or the structural diffusion coefficient D, the difference being that ADC is derived from a monoexponential diffusion model and quantifies the combined effects of both, diffusion and capillary perfusion, while D is derived from the biexponential intravoxel incoherent motion (IVIM) model and constitutes perfusion-free diffusion [11]. Marked variations in ADC and D have been reported for PDAC [9, 12].
Previous studies, including our own, addressing the radio-pathological correlation of DW-MRI with histological parameters of PDAC, showed that the relative amount of tumor stroma or cancer cells, as well as the specific growth pattern of the cancer cells (glandular versus solid) influenced the DW-MRI parameters ADC and D, though the results are not yet conclusive [13-17].
In PDAC, restricted water diffusivity is associated with poor survival [18], as is intratumor inflammation [3, 7]. In particular, infiltrates of the most prominent innate immune cell type, polymorphonuclear neutrophil granulocytes, were associated with poor prognosis and poor histological grade of differentiation [7, 19]. These findings gave rise to the question how inflammatory infiltrates interrelate with water diffusivity. As reported previously by us and others, PMN are found in the tumor micromilieu of PDAC, predominantly in the desmoplastic stroma, and less frequently in the direct vicinity of the tumor cells themselves [4, 19]. As the source of cytokines, chemokines, proteases, and multiple growth factors, the infiltrated immune cells generate a “fibro-inflammatory micromilieu”, which is thought to contribute greatly to the development and progression of PDAC [3]. One explanation for the association of PMN infiltrates with poor prognosis and a poor histological grade of differentiation is that PMN-derived factors induce tumor-associated angiogenesis, loss of tumor cell adhesion, and promote tumor cell migration [4, 6, 20-23]. Histomorphologically, PDAC samples with dense PMN infiltrates display a distinct, usually microglandular or micropapillary, or overall poorly differentiated growth pattern, as opposed to the conventional glandular appearance of PDAC [19]. Possibly, PMN-derived proteases digest extracellular matrix proteins and cellular adhesion molecules, thus destroying and changing the conventional architecture of the tumor. PMN-mediated digestion of the cellular adhesion molecule E-cadherin, for example, induces an epithelial-to-mesenchymal transition of pancreatic cancer cells, thus facilitating their local and systemic spread [4, 6].
Considering the association of PDAC-associated PMN infiltrates with changes in growth pattern and prognosis on one side, and the possible links between water diffusivity and growth pattern as well as survival on the other, the aim of our study was to examine possible associations between PMN infiltrates and water diffusion, and to asses underlying mechanisms.
Materials and Methods
Patients
For a radio-pathological correlation, the database of the Clinic for Diagnostic and Interventional Radiology of the University Hospital of Heidelberg was searched retrospectively for patients who had undergone surgical resection of PDAC between 03/2013 and 08/2016 and who had undergone a DW-MRI scan of the pancreas the day before surgery. Thirty-three patients (12 females, 21 males; mean age 66.67 ± 9.83 years) with PDAC were included in this first cohort (MRI cohort). Exclusion criteria were neoadjuvant therapy or induction treatment. The study was approved by the ethics committee of Heidelberg University. Because of the known tumor heterogeneity, for the tissue-based analysis two whole tumor slides per patient were recovered. Tissue samples were stored in the tissue bank of the National Center for Tumor Diseases (NCT, Heidelberg, Germany) in accordance with the regulations of the tissue bank and the approval of the ethics committee of Heidelberg University. Patient characteristics, DW-MRI and histological parameters, tumor-node-metastasis (TNM) classification, residual tumor (R) classification and grading (G) are summarized in Table 1.
Mean DWI-parameters and histological parameters
| patient number | f (%) | D (×10-3 mm2/sec) | D* (×10-3 mm2/sec) | ADCall (×10-3 mm2/sec) | ADC800 (×10-3 mm2/sec) | azurocidin-positive cells/ HPF | CD15-positive cells/ HPF | ASMA-positive cells (Allred score) | tumor cell: stroma - ratio |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 20.61 | 0.97 | 18.51 | 1.48 | 1.28 | 23.2 | 21 | 7 | 45:55 |
| 2 | 9.49 | 0.99 | 12.55 | 1.17 | 1.09 | 88.7 | 63.9 | 6 | 45:55 |
| 3 | 8.54 | 1.04 | 47.94 | 1.18 | 1.16 | 30.3 | 28.8 | 6 | 35:65 |
| 4 | 12.62 | 1.04 | 8.45 | 1.40 | 1.39 | 8 | 9.8 | 7 | 35:65 |
| 5 | 9.82 | 1.04 | 7.33 | 1.25 | 1.29 | 41.7 | 41.1 | 8 | 30:70 |
| 6 | 9.55 | 1.06 | 58.74 | 1.32 | 1.15 | 49.9 | 53.3 | 5 | 15:85 |
| 7 | 6.72 | 1.07 | 26.33 | 1.20 | 1.04 | 78.2 | 72.2 | 7 | 50:50 |
| 8 | 10.69 | 1.11 | 4.35 | 1.32 | 1.23 | 1.6 | 1.8 | 4 | 30:70 |
| 9 | 10.06 | 1.14 | 6.22 | 1.31 | 1.17 | 9.1 | 8.8 | 6 | 70:30 |
| 10 | 7.50 | 1.15 | 7.33 | 1.29 | 1.23 | 26.6 | 24.2 | 5 | 40:60 |
| 11 | 8.26 | 1.16 | 27.45 | 1.33 | 1.21 | 49.7 | 54 | 4 | 35:65 |
| 12 | 18.55 | 1.20 | 18.88 | 1.61 | 1.48 | 8.6 | 9.3 | 8 | 40:60 |
| 13 | 5.89 | 1.21 | 6.96 | 1.35 | 1.27 | 1.5 | 1.4 | 6 | 25:75 |
| 14 | 11.90 | 1.22 | 13.29 | 1.45 | 1.36 | 14.4 | 8.8 | 7 | 30:70 |
| 15 | 8.53 | 1.24 | 9.94 | 1.40 | 1.35 | 3.8 | 4.7 | 7 | 20:80 |
| 16 | 8.55 | 1.24 | 148.51 | 1.45 | 1.40 | 9.7 | 5.4 | 6 | 50:50 |
| 17 | 12.12 | 1.25 | 15.53 | 1.48 | 1.35 | 44.7 | 41.1 | 5 | 30:70 |
| 18 | 9.04 | 1.26 | 148.51 | 1.44 | 1.29 | 3.7 | 3.4 | 7 | 30:70 |
| 19 | 12.07 | 1.27 | 15.90 | 1.49 | 1.39 | 1.4 | 1.9 | 7 | 30:70 |
| 20 | 14.01 | 1.30 | 9.57 | 1.61 | 1.45 | 4.5 | 4.8 | 6 | 30:70 |
| 21 | 28.07 | 1.33 | 12.18 | 1.92 | 1.53 | 1.1 | 1.4 | 8 | 50:50 |
| 22 | 5.00 | 1.34 | 31.55 | 1.45 | 1.34 | 1.1 | 1.8 | 7 | 30:70 |
| 23 | 9.52 | 1.36 | 103.07 | 1.55 | 1.45 | 9.5 | 10.3 | 6 | 25:75 |
| 24 | 16.11 | 1.40 | 127.28 | 1.75 | 1.61 | 5.2 | 6.3 | 5 | 35:65 |
| 25 | 10.42 | 1.40 | 93.38 | 1.69 | 1.62 | 1.4 | 1.2 | 6 | 30:70 |
| 26 | 7.61 | 1.42 | 6.22 | 1.53 | 1.49 | 5.6 | 3.8 | 5 | 25:75 |
| 27 | 9.21 | 1.42 | 6.22 | 1.59 | 1.47 | 0.9 | 0.4 | 8 | 40:60 |
| 28 | 7.04 | 1.43 | 18.88 | 1.55 | 1.45 | 1.9 | 1.6 | 7 | 20:80 |
| 29 | 10.57 | 1.47 | 9.57 | 1.67 | 1.48 | 1.1 | 1.4 | 7 | 30:70 |
| 30 | 3.67 | 1.50 | 75.87 | 1.59 | 1.61 | 1.8 | 0.6 | 5 | 80:20 |
| 31 | 7.23 | 1.52 | 45.70 | 1.66 | 1.47 | 1.1 | 0.9 | 7 | 20:80 |
| 32 | 9.87 | 1.53 | 55.76 | 1.74 | 1.68 | 1.4 | 1.7 | 6 | 35:65 |
| 33 | 7.51 | 1.79 | 80.72 | 1.93 | 1.81 | 4 | 6.6 | 6 | 30:70 |
| average | 10.50 | 1.27 | 38.75 | 1.49 | 1.38 | 16.2 | 15.1 | 6.3 | 35:65 |
| standard deviation | 4.73 | 0.19 | 42.74 | 0.19 | 0.18 | 23.0 | 20.5 | 1.1 | n.a. |
| median | 9.52 | 1.25 | 18.51 | 1.48 | 1.39 | 5.2 | 5.4 | 6 | 30:70 |
For high throughput tissue analysis, tissue microarrays with a core diameter of 1.5 mm (two cores per specimen, areas chosen randomly) were prepared of a second cohort of subsequent patients (n=109; non-MRI cohort; patients´ characteristics in Table S1A) who underwent pancreatic surgery due to PDAC in the time frame between 02/2004 and 09/2008 at the University Hospital of Heidelberg. Tissue specimens were formalin-fixed and paraffin-embedded, and stained with hematoxylin/eosin following standard protocols. Tissue samples were stored in the tissue bank of the National Center for Tumor Diseases (NCT, Heidelberg, Germany) in accordance with the regulations of the tissue bank and the approval of the ethics committee of Heidelberg University.
MR Imaging and Post-Processing
MR imaging was performed using a 1.5 T scanner (Aera, Siemens Medical Solutions, Erlangen, Germany) with a six-element body-phased array coil, a 24-channel spine array coil and a maximum gradient strength of 45 mT/m. The imaging protocol comprised breath-hold HASTE-IR T2-weighted imaging (TR/TE = 1000/80 ms) in coronal orientation, breath-hold T1-weighted in/opposed phase imaging in transversal orientation (TR/TE = 115/2.27 and 4.78 ms), and breath-hold (expiration) HASTE T2-weighted imaging in transversal orientation (TR/TE = 680/95 ms). As previously described [24], axial diffusion-weighted images were acquired using a single shot echo-planar imaging (SE2d-EPI) pulse sequence in expiratory breath-hold. The following imaging parameters were used: FOV = 350 × 248 mm², matrix size = 130 × 92, 14 slices, slice thickness/gap = 5/0.25 mm, TR = 2200 ms, TE = 58 ms, spectral fat saturation, bandwidth = 2262 Hz/pixel, k-space based parallel imaging technique (GRAPPA) acceleration factor of two, b-values = 0, 50, 100, 150, 200, 300, 400, 600 and 800 s/mm². To avoid motion artifacts, the acquisition was separated into blocks (b0, b50), (b0, b100), … , (b0, b800), and each block was acquired in a single breath-hold (TA = 22 s).
Post processing was performed using MITK Diffusion software (Medical Imaging Interaction Toolkit, Version 2014.10.02, DKFZ, Heidelberg, Germany; www.MITK.org) [25]. To overcome the limitation of the monoexponential diffusion model and the ADC, which is that diffusion measurements can be considerably confounded by blood perfusion due to the incoherent motion of blood, we made use of the biexponential model based on the intravoxel incoherent motion (IVIM) theory. The IVIM theory allows for the combined quantification of a diffusion coefficient D, a perfusion fraction f, and a pseudodiffusion coefficient D* [11], as follows:
S / S0 = (1 - f) x exp(-b x D) + f x exp(-b x (D + D*)) .
In this equation, S constitutes the signal with diffusion weighting and S0 constitutes the signal without diffusion weighting. The estimation of the parameters was based on the assumption that the diffusion measurement is influenced primarily by two effects, extravascular effects of passive diffusion (D) as well as a perfusion related effect attributable to the movement of molecules in the capillary network (pseudodiffusion coefficient, D*). A simultaneous nonlinear fit for all parameters D, D*, and the weighting coefficient f is potentially unstable. Therefore measurements at b-values greater than 170 s/mm2 were used in a first step to estimate f and D, as previously suggested [26]. In a second step, D* was calculated by using exhaustive search, which means that a search algorithm systematically tried all possible values for D* one after another in order to find the best fit.
In summary, D reflects diffusion without perfusion effects, D* is dependent on the vascular architecture as well as the velocity of the flowing blood, and f is linked to the blood volume in the analyzed tissue [27].
Similarly, ADC values were calculated using a monoexponential model, as follows:
S / S0 = exp (-b x ADC)
Again, S0 is the signal magnitude with no diffusion weighting and S is the signal magnitude with diffusion weighting. ADC values were calculated from b0 and b800 (ADC800) as well as from all b-values (ADCall).
Quantitate analysis of DW-MRI was performed independently by 2 radiologist with 10 years (MK, reader 1) and 5 years (PM, reader 2) of experience in abdominal imaging. Using the MITK Diffusion software a freehand volume of interest (VOI) was drawn directly on multiple slices. Before calculating the fits, the signal intensities within each volume of interest (VOI) were averaged. This VOI based approach was chosen since a pixel-by-pixel approach can be problematic in a biexponential diffusion model due to the signal to noise ratio of diffusion-weighted images [28] and can result in greater fitting errors [29]. A potential disadvantage of the VOI based approach is that it masks inherent heterogeneity of the tissue [28]. However, the aim of the current study was not to address tumor heterogeneity by a pixel-by-pixel radio-pathological correlation, but to correlate radiological parameters extracted from the whole tumor volume with whole tumor tissue slides.
The accurate anatomical outline of the tumor was determined with the help of conventional biliary-pancreatic MR images.
Immunohistochemistry
For the first cohort of patients (whole slides) and the second cohort (tissue microarrays), specimens were stained with standard hematoxylin/eosin and Masson-Goldner Trichrome. In addition, the following primary antibodies were used: anti-CD15 (DAKO, Hamburg, Germany, IS062, 1:50, retrieval condition: pH 6.0), anti- α-SMA (DAKO, M0851, 1:500, retrieval condition: pH 9.0), anti-azurocidin (Abcam, Cambridge, UK ab167336, 1:100, retrieval condition: pH 6.0), pan-cytokeratin antibody AE1/3 (M3515, Dako, 1:200, retrieval condition: pH 9.0), anti-MMP9 (Becton and Dickinson, Heidelberg, Germany, 1:1000, retrieval condition pH 9.0); anti-CD3 (Thermo Fischer Scientific, Darmstadt, Germany, SP7, 1:200, retrieval condition pH 6.0). For detection, the Histofine, Simple Stain universal polymer was used (Nichirei, Tokyo, Japan) followed by the color reaction with liquid permanent red (Zytomed, Berlin, Germany), or the DAKO-EnVISION Kit (DAKO) and counter stain with hematoxylin. The amount of α-SMA expression was determined using the Allred score [30], PMN (identified by expression of CD15) were counted, and a semiquantitative scoring system was used (0 - 20 PMN per array spot = score: 1; 21- 40 score: 2, 41 - 60 score: 3, 61 - 80 score: 4; 81 - 100 score: 5; > 101 score: 6). For the MRI cohort, 10 high power fields of whole slides were chosen randomly, and PMN were counted. PMN in blood vessels or in necrotic areas were excluded. Areas with accumulations of PMN (≥50 µm; ≥10 PMN per focus) with a micropapillary, microglandular, solid, or single cellular growth pattern were quantified as present or absent.
Immunofluorescence of PDAC tissue
Tissue microarrays of 32 patients were de-paraffinized and rehydrated using xylene and graded alcohols. After washing with Tris-buffered saline/Tween, the tissues were blocked with 5% normal goat serum (Dako) and the slides were incubated with mouse anti-azurocidin (Abcam, 1:100) for 1 h at room temperature. As secondary antibody the anti-mouse Cy3 (Dianova, Hamburg, Germany) was incubated for 30 min (1:400). Then, actin was visualized by Phalloidin-Alexa 488 (Life Technologies, Darmstadt, Germany) and the nuclear staining was performed with Hoechst33342. The samples were mounted with Moviol (Sigma-Aldrich) and viewed by laser scanning microscopy (Nikon, Düsseldorf, Germany).
Methods for in vitro experiments
Isolation of PMN
Peripheral blood from healthy human volunteers was obtained by peripheral venous puncture and collected in heparin-NH4-coated tubes (Sarstedt, Nürnbrecht, Germany). PMN were isolated by centrifugation on PolymorphPrep (Axis-Shield PoC AS, Oslo, Norway) yielding an 85-95% pure PMN population. The PMN were suspended in Hanks balanced salt solution and used within 1 h. For PMN lysates, 1x108 cells were resuspended in 1 mL phosphate buffer and then lysed by the nitrogen cavitation method for 20 min at 380 psi.
Cell lines: To generate the pancreatic stellate cell line RLT, the cells were isolated by the outgrowth method and were immortalized by transfection with SV40 large T antigen and human telomerase (hTERT) [31]. The cells were propagated in DMEM High glucose (Cell Concepts, Umkirch, Germany), containing 10% fetal bovine serum, 1% L-glutamine (Invitrogen, Karlsruhe, Germany) and 50 μg/mL gentamycin (Merck, Darmstadt, Germany) and were incubated at 37°C in a 5% CO2 humidified atmosphere. LX-2 cell line (a gift of Prof. Ralf Weiskirchen, University of Aachen, Germany, with permission of Prof. Scott Friedman, Mount Sinai Hospital, NY) was propagated in DMEM High Glucose (Cell Concepts, containing foetal calf serum (10%), L-glutamine (2%) and penicillin-streptomycin (1%). HL60 (ATCC, Rockville, USA) were propagated in RPMI, containing foetal calf serum (10%), L-glutamine (1%) and penicillin-streptomycin (1%).
Immunofluorescence of cells
30,000 RLT cells were seeded in chambered slides (Nunc, purchased from Sigma). After washing with phosphate-buffered saline, the cells were fixed using 2% paraformaldehyde for 15 min at room temperature. The cells were blocked with 5% goat serum for 30 min (Dako) then incubated with mouse anti-azurocidin (Abcam, 1:100) for 1 h at room temperature. As secondary antibody the anti-mouse Cy3 (Dianova) was incubated for 30 min (1:400). Then, the actin-cytoskeleton was visualized using Phalloidin-Alexa 488 (Life technologies) and the nuclear staining was performed with Hoechst33342. Slides were viewed by laser scanning microscopy, as described above.
Dyshesion assays with PMN
RLT cells or LX-2 (3 × 105 in 2 mL) were cultivated in six-well plates (Nunc, Roskilde, Denmark) for 24 h until they reached confluence. Then, isolated PMN (1 × 107), 100 µl PMN lysate (equivalent to 1x107 cells), 100 µl PMN lysate with 200 µg/mL heparin (>180 I.U./mg Ph. Eur.; Carl Roth, Karlsruhe, Germany), PMN lysate (equivalent to 1x107 cells) after release through a HiTrap Heparin column (used according the manufacturers protocol; GE Healthcare, Uppsala, Sweden), or recombinant human azurocidin (ACRO Biosystems, London, UK) at a concentration of 20 µg/mL were added to the cell cultures.
Quantification of lipid droplets
Lipid droplets were visualized by incorporating Nile Red. RLT cells (300,000) were plated either on plastic dishes (NUNC) or on Matrigel (Becton and Dickinson) and incubated with azurocidin (20 µg/mL) for 5 days and then incubated with Nile Red (1 µM; Bioquest) for 10 min at 37°C. Cells were harvested, fixed with 1 % PFA and viewed by laser scanning microscopy (see above). Droplets were quantified by flow cytometry using FACScalibur and CellQuest Pro software (Becton and Dickinson). 10,000 events were counted.
Protein isolation, SDS-PAGE, and Western blot
Proteins from 3 × 105 RLT cells were isolated with 150 µL RIPA-buffer with protease and phosphatase inhibitors according to the manufacturer's recommendation (Santa Cruz). Protein samples were heated for 10 min at 95°C and separated by SDS-PAGE (7%). After blotting to a PVDF (Merck/Millipore, Darmstadt, Germany) the following antibodies were used: mouse anti-azurocidin (Abcam, 1:1000), mouse anti-α-SMA (Sigma, 1:500), rabbit anti-p-cofilin (Cell Signaling, Danvars, MA, USA, 1:1000), mouse anti-MMP9 (1:500), rabbit anti-pp38 (Biovision, Milpita, CA, USA, 1:500), and rabbit anti-p-MAPKAP-2 (Santa Cruz; 1:300). After washing, membranes were incubated using a goat anti rabbit IgG POX or, goat anti mouse IgG POX, respectively (Becton and Dickinson) as secondary antibodies (room temperature for 30 min). To control for equal loading, β-actin was determined using anti-actin (Abcam). For detection, Amersham ECL plus Western Blotting Detection System (GE Healthcare, Munich, Germany) was used. Densitometrical analysis was performed using Quantity one software (Biorad, Hercules, CA, USA).
Proteome Profiler array
The Proteome Profiler Human Phospho-Kinase Array Kit and the Human Protease/Protease Inhibitor Array Kit were used according to the manufacturer's instructions (RnD Systems, Darmstadt, Germany), followed by densitometrical analysis with the Quantity one software (Biorad, Dreieich, Germany).
Statistical analysis
Statistical data analysis was performed using GraphPad Prism Version 7.03 (GraphPad Software, La Jolla, CA, USA) and MedCalc Version 17.6 (MedCalc Software, Ostend, Belgium). Results of the diffusion-weighted imaging (DWI)-analysis from both readers were averaged to create mean values for ADCall, ADC800, D, D*, and f. Correlations were performed using the Spearman's rank correlation test, unless specified differently. Inter-reader reliability was assessed by using the Intra-class Correlation Coefficient (ICC) with 95% confidence intervals (CI) and applying a 2-way ICC with random raters' assumption reproducibility. Correlations were calculated between the Allred score, numbers of azurocidin- and CD-15-positive cells per high power field (HPF) on the one side, and mean ADCall, ADC800, D, D*, and f values on the other side. Additionally, partial rank correlations were calculated between histopathological parameters and DWI parameters, adjusting for histopathological grading and tumor-stroma-ratio. A two-sided Student t test was used to test the differences in mean D, ADCall, and ADC800 values between groups with low and high numbers of azurocidin-positive or CD15-positive cells per HPF (≤ median number and > median number, respectively), and between G2- and G3-tumors. Further, differences in azurocidin-positive and CD15-positive cells per HPF between groups with low and high D values, ADCall values, or ADC800 values (≤ median D versus > median D, ≤ median ADCall versus > median ADCall, or ≤ median ADC800 versus > median ADC800), were tested with a two-sided Student t test. Associations between PMN density and growth pattern as well as PMN density and grading were calculated using Fisher's exact test, and the correlation between azurocidin-positive stellate cells and PMN by Kendall rank test. Progression-Free Survival (PFS) was estimated from the date of surgery to the date of progression (locoregional or metastatic, based on follow-up computed tomography scans) or last follow-up. Follow-up data regarding PFS were available from 62 patients (44 patients from the non-MRI cohort and 18 patients from the MRI cohort). PFS curves were estimated with the Kaplan-Meier method and compared by two-sided log-rank test. Additionally, the Cox proportional hazard model was used for multivariate analysis. The variables T (extent of primary tumor, T1-2 vs. T3), N (regional lymph node metastases, N0 vs. N+), M (distant metastases, M0 vs. M1), G (grading, G1-2 vs. G3), and CD15-score (score 1-2 vs. score 3-6) were entered in the Cox model in one single step. The variables D and ADCall/ADC800 were not entered in the Cox model since PFS data were only available for 18 patients from the MRI cohort.
P < 0.05 were considered to be statistically significant.
Results
Diffusion weighted magnetic resonance imaging (DW-MRI) of pancreatic cancer tissue and radio-pathological correlation
In the first set of experiments, DW-MRI with 9 b-values was performed in 33 patients with histologically confirmed PDAC. The values varied among the patients. Mean DWI-parameter values for both readers as well as histopathological parameters from the corresponding tissue specimens are presented in Table 1 and Table S1A. Agreement between reader 1 and reader 2 was excellent for f (ICC = 0.7962), D (ICC = 0.8420), ADCall (ICC = 0.8701), and ADC800 (ICC = 0.8317), and fair for D* (ICC = 0.5150).
There was no significant rank correlation between the mean perfusion fraction f and the mean pseudodiffusion coefficient D* on one side, and the number of PMNs per HPF on the other (0.036 ≤ ρ ≤ 0.128, p ≥ 0.4775). But, there were strong negative rank correlations between mean D, ADCall, and ADC800 values on one side, and the number of infiltrated PMNs on the other (-0.723 ≤ ρ ≤ -0.601, p < 0.0002) (Figures 1 and 2; Table 2). Partial rank correlation coefficients between mean D / ADCall / ADC800 values and PMN density, adjusting for histopathological grading and tumor cell-stroma-ratio, only minimally differed from unadjusted rank correlation coefficients (Table 2). This shows that rank correlation coefficients were not biased by an indirect effect of the relationship between PMN density and histopathological grading or tumor cell-stroma-ratio.
Representative data from two patients, one with high, one with low number of PMN: (A) MRI images of a representative patient with low diffusion constant D (patient number 3 from Table 1, left column) and a representative patient with high D (patient number 31 from Table 1, right column). The morphological images (axial native T1 VIBE fat saturated and T2 HASTE) show upstream dilatation of the main pancreatic ducts (green arrows) due to obstructing tumors in the pancreatic heads (red arrows). Stents in the distal common bile ducts which are not dilated (blue arrows). Diffusion-weighted MR images (b=600) caudad to the morphological images with the freehand VOIs (red). (B) Representative plots of signal decay and calculated DWI parameters for reader 1. Biexponential IVIM fits are in black, monoexponential fits (ADCall) are in red. Calculated D and ADCall values for reader 1 were 1.02 × 10-3 mm2/sec and 1.12 × 10-3 mm2/sec for patient number 3 (left) and 1.51 × 10-3 mm2/sec and 1.65 × 10-3 mm2/sec for patient number 31 (right). (C): Dependency of the structural diffusion coefficient D on the number of azurocidin-positive cells per HPF or on the number of CD15-positive cells per HPF. Abbreviations: D: structural diffusion coefficient; HPF: high power field; S: signal with diffusion weighting; S0: signal without diffusion weighting.
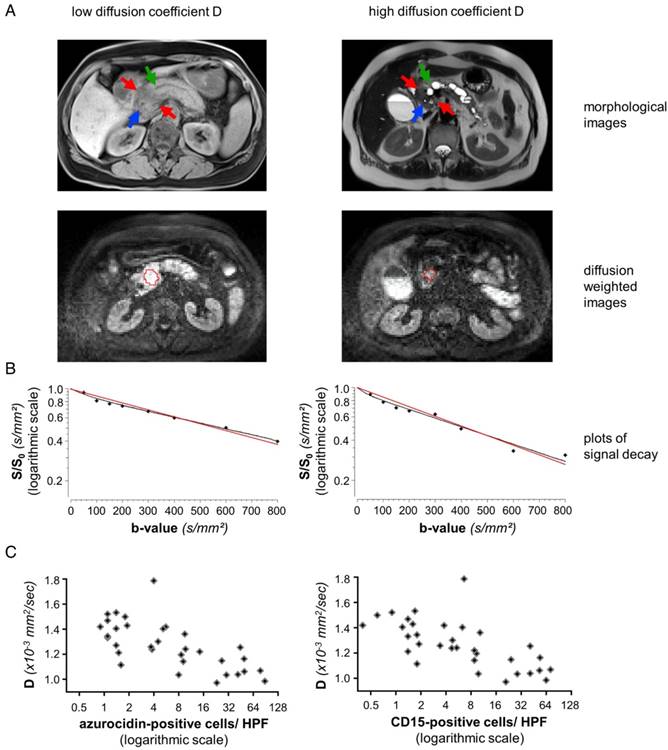
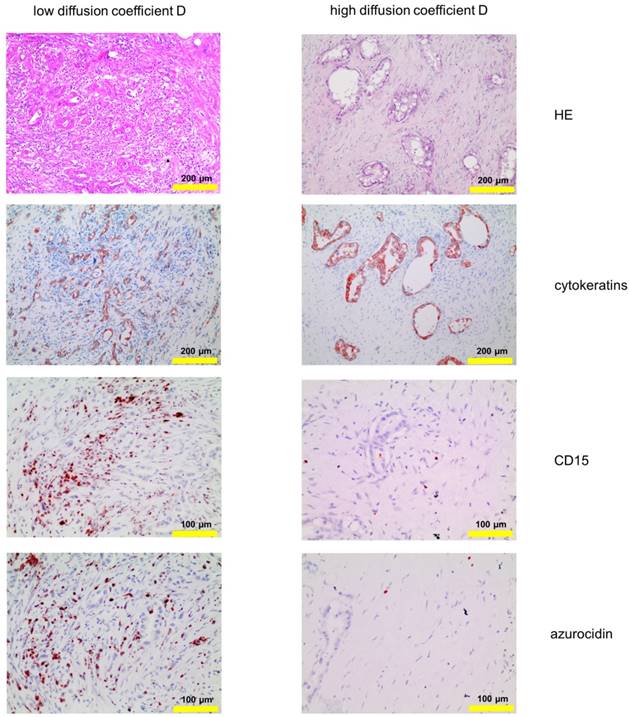
Corresponding tissue staining of the patients shown in Fig. 1: Shown is a conventional hematoxylin/eosin staining, staining for cytokeratin and for PMN, the latter identified by expression of CD15 and azurocidin (reddish-brown). Abbreviations: CD15: cluster of differentiation antigen 15; D: structural diffusion coefficient; HE: hematoxylin and eosin.
Rank correlation coefficients and partial rank correlation coefficients between average D and ADC values for both readers on the one side, and PMN density on the other.
| Rank correlation coefficients | Partial rank correlation coefficients (adjusted for histopathological grading and tumor cell-stroma-ratio) | |||
|---|---|---|---|---|
| CD15-positive cells | azurocidin-positive cells | CD15-positive cells | azurocidin-positive cells | |
| D | -0.723 | -0.703 | -0.713 | -0.692 |
| ADCall | -0.601 | -0.625 | -0.596 | -0.623 |
| ADC800 | -0.616 | -0.605 | -0.616 | -0.607 |
All coefficients shown in Table 2 were significantly different from zero (p < 0.05).
There were strong significant positive rank correlations between mean D and mean ADCall / ADC800 values (ρ = 0.821 and ρ = 0.821, p ≤ 0.0006). Rank correlation between D and the number of PMNs per HPF was not statistically different from rank correlation between ADCall / ADC800 and the number of PMNs per HPF (p ≥ 0.1148).
Average D values were significantly higher in tumors with PMN density values less than or equal to median PMN density (n=17) compared to tumors with PMN density values greater than median PMN density (n=16) (1.35 ± 0.12 × 10-3 mm2/sec vs. 1.18 ± 0.20 × 10-3 mm2/sec, p= 0.0071 when CD15 was used, and 1.38 ± 0.16 × 10-3 mm2/sec vs. 1.15 ± 0.13 × 10-3 mm2/sec, p= 4.4*10-5 when azurocidin positive cells were evaluated). Similarly, average ADCall and ADC800 values were significantly higher in tumors with PMN density values less than or equal to median PMN density compared to tumors with PMN density values greater than median PMN density (p ≤ 0.0447).
Vice versa, the mean numbers of PMNs per HPF were significantly higher in tumors with D values less than or equal to the median D value of 1.25 × 10-3 mm2/sec (n=17) compared to tumors with D > 1.25 × 10-3 mm2/sec (n=16) (26.45 ± 23.44 vs. 3.01 ± 2.74, p = 0.0008 for CD15 positive cells, and 28.81 ± 26.55 vs. 2.86 ± 10.41, p= 0.0010 for azurocidin positive cells).
Equivalently, tumors with ADC values less than or equal to median ADC values exhibited significantly more PMNs per HPF (n=17) than tumors with ADC values greater than median ADC values (n=16) (p ≤ 0.0104 for ADCall, p ≤ 0.0029 for ADC800).
Mean D, ADCall, and ADC800 values did not differ significantly between well or moderately differentiated (G2) tumors (n=18) and poorly differentiated (G3) tumors (n=15) (1.27 ± 0.21 × 10-3 mm2/sec vs. 1.27 ± 0.16 × 10-3 mm2/sec, p= 0.9331 for D, 1.49 ± 0.23 × 10-3 mm2/sec vs. 1.49 ± 0.16 × 10-3 mm2/sec, p= 0.9966 for ADCall, and 1.38 ± 0.19 × 10-3 mm2/sec vs. 1.39 ± 0.16 × 10-3 mm2/sec, p= 0.8675 for ADC800).
PMN in tissue of patients with PDAC
The number of PMN and their distribution within the tissue sample varied greatly within patients, and also within the same tissue specimen. Therefore, to look for distinct patterns, tissue micro arrays (diameter: 1.5 mm per spot) of 109 PDAC patients were analyzed. As before, PMN were identified by expression of CD15 and azurocidin, in addition to conventional hematoxylin/eosin staining. In all tumor samples, PMN were detected, with density varying from a single cell to 368 cells per array spot (examples in Figure S1; data summarized in Table S2). Tumors with high density (≥41 PMN per spot; score 3 - 6) were found in 50 patients. Here, PMN were clustered in areas with altered architecture of the stroma, especially apparent after visualizing the tumor cells by staining of cytokeratins. The morphological alterations included micropapillary, microglandular, solid, or single-cellular tumor growth, and poor surrounding stroma, or even areas devoid of stroma, with interspersed T cells (examples in Figure S2 and S3). The tumor cell / stroma ratio was not associated with the numbers of infiltrated PMN (Figure S4). In tumors with localized accumulations of PMN (≥50 µm; ≥10 PMN per focus) the cells formed small clusters, which again were associated with an altered growth pattern. The association between PMN clusters and altered growth pattern was highly significant according to Fisher's test (Table S3A). Density of PMN did not correlate with tumor size or with the presence of lymph node metastases; there was, however, an association between PMN density and grade of histological tumor differentiation. More patients with poorly differentiated tumors (G3) had high PMN (scores 3 to 6), compared to patients with well or moderately differentiated tumors (G1 or G2) (the association was statistically significant according to Fisher's test; data summarized in Table S3B).
Kaplan-Meier-Analysis revealed trends towards shorter Progression-Free Survival (PFS) in patients with high PMN density (CD15-score 3-6, median PFS 286 days) compared to patients with low PMN density (CD15-score 1-2, median PFS 394 days) although the difference was not statistically significant according to the log-rank test (p = 0.3248) (Figure S5). Adjusting for TNM classification and histopathological grading in the Cox regression model, the hazard ratio (HR) for PFS in patients with high PMN density was 1.69 (95% confidence interval (CI), 0.88 - 3.26, p= 0.1170) compared to patients with low PMN density. HRs, 95% CIs, and p-values for the variables CD15-score and histopathological grading from the Cox model are summarized in Table S1A, B.
Evaluation of alpha-smooth muscle actin positive cells in PDAC tissue
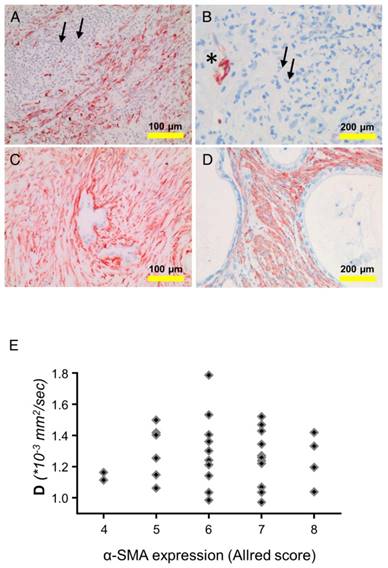
Alpha-smooth muscle actin (α-SMA) is expressed by smooth muscle cells, myofibroblastic cells, and also by stellate cells. The latter cell type is considered the main extracellular matrix producer in PDAC. Expression of α-SMA was examined in tissue samples of PDAC patients (n=142; both cohorts). The overall α-SMA expression as determined by an established quantification score (Allred score) was not associated with PMN density. The rank correlation between the overall α-SMA expression and the structural diffusion coefficient D (33 patients from cohort 1) was very weak (ρ=0.0087). However, as shown in the examples in Figure 3, in areas with low PMN-density (≤10 cells per spot) the number of α-SMA-positive cells was higher, compared to areas with high density (≥41 PMN per spot). Particularly, in the direct vicinity of PMN, the number of α-SMA-positive cells was low, suggesting highly localized interactions of PMN or PMN-derived mediators with tissue cells (examples in Figure 3).
Detection of α-smooth muscle actin (SMA) and PMN in tissue of different patients: (A, B) show examples for tissue with a prominent PMN infiltrate (small arrows). This area is devoid of α-SMA-positive cells (brown). α-SMA is seen only in a blood vessel (asterisk). (C,D) show areas with low PMN density and intense α-SMA staining (brown). (E) shows the dependency of the structural diffusion constant D on α-SMA expression (as determined using the Allred score), analyzing the tissue of the patients of cohort 1. Abbreviations: D: structural diffusion coefficient; α-SMA: alpha smooth muscle actin.
Effect of PMN on stellate cells and identification of azurocidin as effector molecule
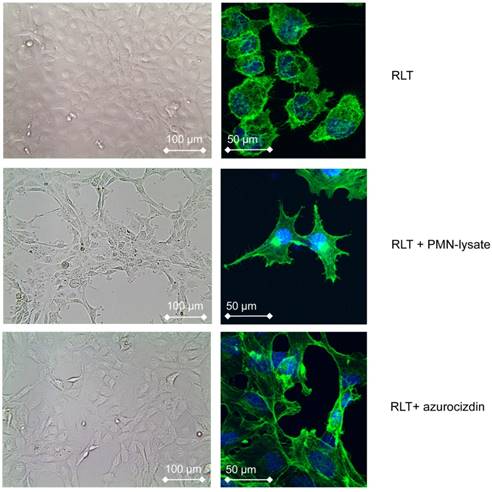
To gain information on possible interactions between PMN and stellate cells, PMN derived from healthy donors were co-cultivated with the pancreatic stellate cell line RLT, the latter grown as monolayer. Within 6 h, the morphology of stellate cells began to change to spindle shaped cells. After 24 h, all cells had acquired a spindle-cell phenotype (Figure 4). Pre-incubation of PMN with cytochalasin D, which is known to inhibit degranulation of neutrophils, abolished this effect, suggesting that a molecule released from PMN granula induced the morphological changes (data not shown). Indeed, lysates of PMN, obtained by nitrogen cavitation, which disrupts the cell but leaves the granules intact, induced a similar shape change, as did lysates of the promyeloid cell line HL-60. These data support the notion that the shape shift of stellate cells is caused by a preformed mediator or enzyme stored in PMN and in myeloid precursors as well. Addition to the culture medium of heparin (200 µg/mL) prevented the PMN-induced effect on stellate cells, as did removing heparin-binding proteins from PMN-lysates by adsorption to heparin-coated beads. These data pointed to azurocidin, also known as cationic antimicrobial protein CAP37 or heparin-binding protein (HBP), as effector molecule. Indeed, recombinant azurocidin also induced a similar shape change of stellate cells (Figure 4). Other PMN-derived proteases, such as neutrophil elastase, lactoferrin, or MMPs could be ruled out, because neither commercially available inhibitors to these factors prevented the shape change, nor were the respective isolated or recombinant proteins effective (data not shown). Experiments using the hepatic stellate cell line LX-2 yielded essentially similar results (data not shown).
Effect of PMN-lysates or azurocidin on the pancreatic stellate cell line RLT: In response to either PMN-lysates or azurocidin within 24 h a drastic shape change is seen (left: bright field images, right: single cells with blue nuclear staining, and green staining of actin (Phalloidin-FITC)). Abbreviations: PMN: polymorphonuclear neutrophil granulocytes.
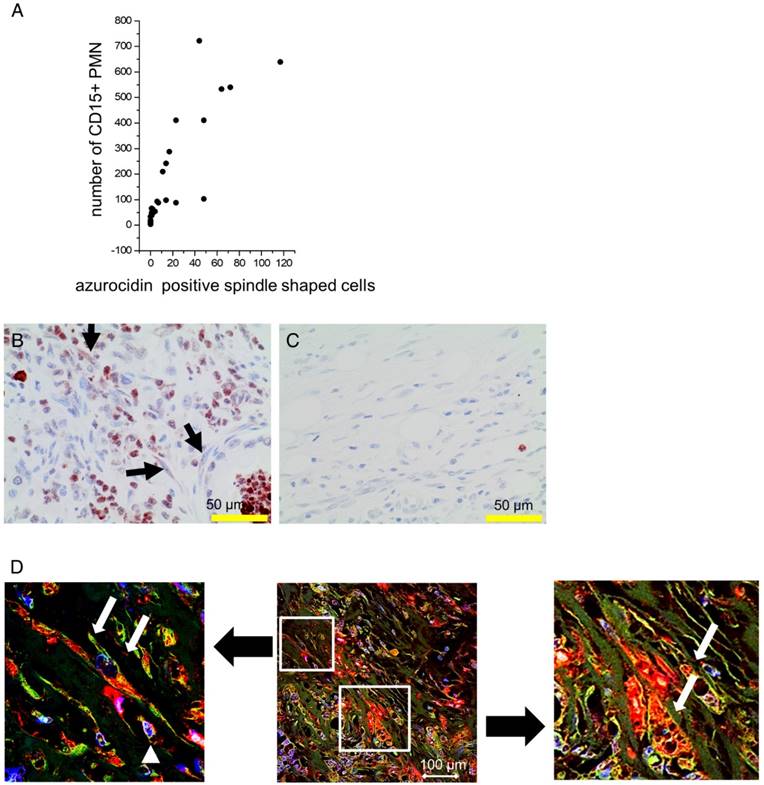
Cultivation of stellate cells with PMN lysates or recombinant azurocidin resulted in uptake of the protein, as seen by Western blotting of cell lysates and by laser scanning microscopy. Azurocidin was predominantly associated with F-actin, as visualized by co-localization of azurocidin with actin labeled with phalloidin-Alexa 488 (Figure 5). In tissue of PDAC there was a close correlation between the number of PMN and the number of azurocidin-positive spindle shaped cells (Pearson correlation coefficient 0.829; Kendall rank test; p <0.05); data derived from analyzing whole tissue sections of the patients of the MRI cohort) (Figure 6). By laser scanning microscopy, presence of azurocidin within spindle cells could be confirmed, and for some cells a co-localization with actin filaments was seen (Figure 6). The azurocidin-induced shape change of stellate cells and the association of azurocidin with F-actin suggested effects on signaling pathways that control the cytoskeleton. Indeed, azurocidin induced phosphorylation of p38α, of its downstream target MAPKAP-2, and de-phosphorylation of cofilin, the latter directly linking azurocidin effects to the cytoskeleton (Figure 7).
Uptake of azurocidin by stellate cells: Stellate cells were cultivated with PMN lysates for 24 h in the presence or absence of heparin. (A) Western blot of stellate lysates with an antibody to azurocidin, which appears with an apparent molecular weight of 37 kDa (marked by arrow). (B) Cells viewed by laser scanning microscopy. Nuclei are stained in blue; actin is visualized by phalloidin-Alexa 488 (green), and azurocidin in red (detected by labeled antibody). Merging the images shows azurocidin intracellularly (upper panel). Azurocidin was not detected when the PMN lysate was adsorbed to heparin (lower panel). (C) When single cells are zoomed in, an association of azurocidin with actin is seen, apparent as intense yellow staining. Abbreviations: PMN: polymorphonuclear neutrophil granulocytes.
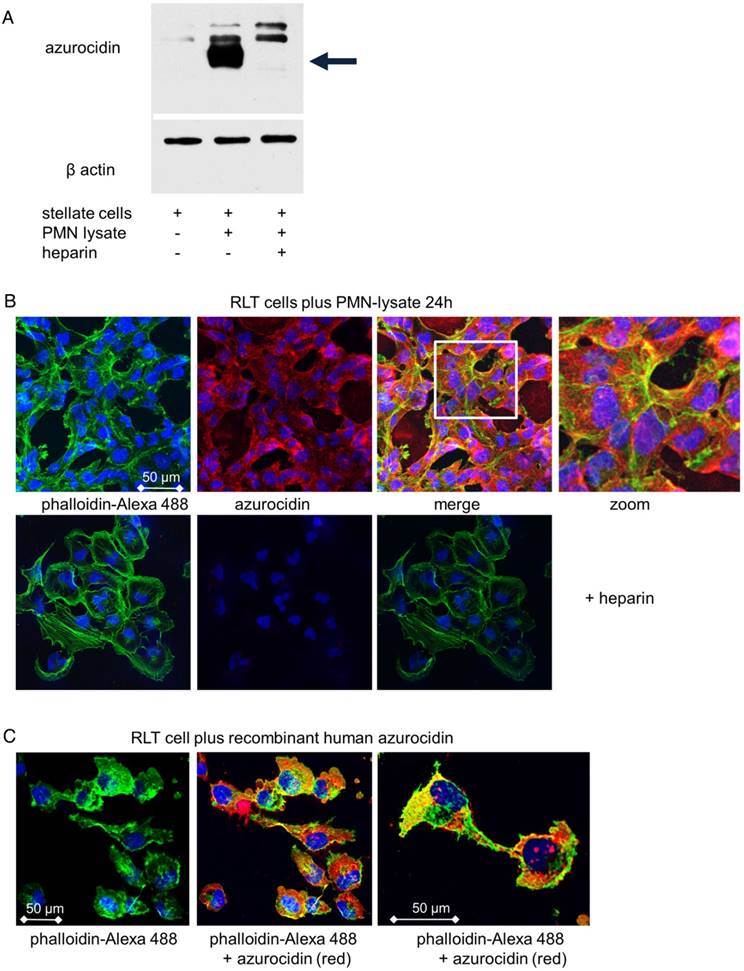
Association of numbers of CD15-positive PMN with azurocidin-positive spindle shaped cells: (A): In PDAC tissues of the MRI cohort, the number of PMN correlated with the number of spindle shaped cells (Pearson correlation coefficient: 0.85). (B,C) Representative examples of tissues with high (B) and low (C) numbers of PMN are shown. Arrows indicate azurocidin-positive spindle shaped cells. (D) By laser scanning microscopy of PDAC tissue, azurocidin is seen in spindle shaped cells (red, marked by white arrows in the zoomed images), in part also associated with actin (intense yellow). The smaller cells (marked by white arrowhead) are azurocidin-positive PMN. Abbreviations: CD15: cluster of differentiation antigen 15; PMN: polymorphonuclear neutrophil granulocytes.
Effects of PMN lysates or azurocidin on stellate cells in culture: (A) By Western blotting, phosphorylation of p38 and MAPKAP2 was seen (measured after 4 h). The effect of the PMN lysates was abolished by heparin. (B) Moreover, de-phosphorylation of cofilin was seen after 24 h. (C) Down-regulation of smooth-muscle actin and up-regulation of MMP9 was detected. The MMP9 up-regulation was confirmed by a proteome profiler assay. Abbreviations: α-SMA: alpha smooth muscle actin; MMP9: matrix metalloproteinase 9; p-MAPKAP2: phosphorylated mitogen activated protein kinase-activated protein kinase 2; PMN: polymorphonuclear neutrophil granulocytes.
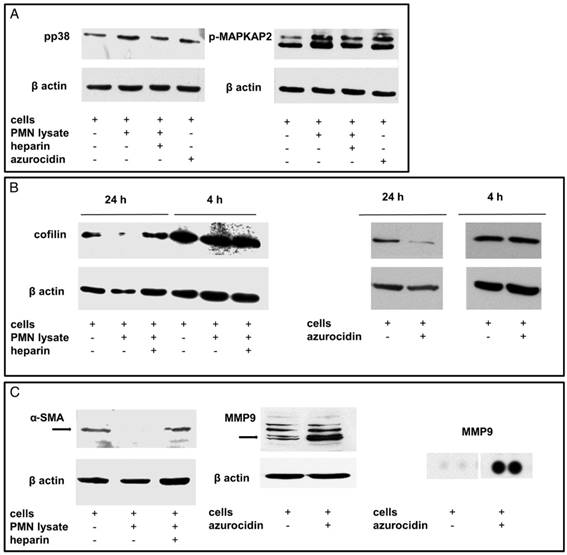
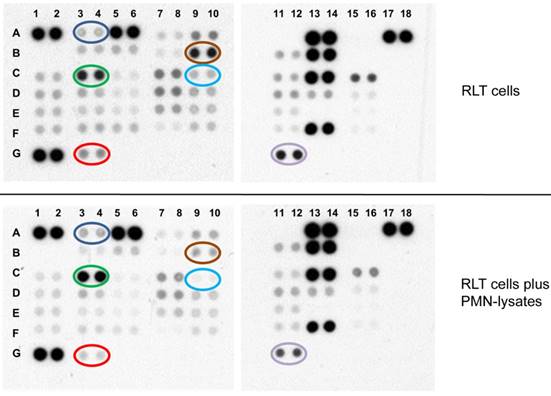
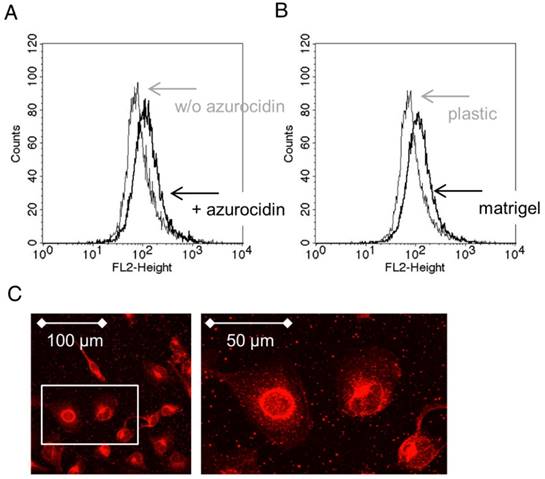
A protein profiler assay designed to detect phosphokinases confirmed phosphorylation of p38α (T180/Y182). Moreover, phosphorylation of CREB (S133) was detected, whereas the phosphorylated forms of PRAS40 (T246) and AKT1,2,3 (S473) were down-regulated, as was β-catenin (Figure 8). According to the literature, these phosphorylation patterns are associated with “silencing” of stellate cells [32-34]. To further assess “silencing”, the presence of cytoplasmic lipid droplets was assessed. Under our culture conditions, cultivated stellate cells contained lipid droplets as seen by incorporation of Nile red (Figure 9). When quantified by flow cytometry, an increase of Nile red incorporation was seen in cells cultivated with azurocidin. For comparison, cells were cultivated on Matrigel, which is known to enhance the number of lipid droplets [31] (Figure 9). Again in line with this “silencing” phenomenon, the abundance α-SMA was reduced in response to azurocidin (Figure 7C). The latter finding corresponds to our data derived from examining PDAC tissue, where stromal cells expressing low α-SMA were found in the vicinity of PMN clusters, and in association with altered stroma pattern or poor stroma (compare Figure 3).
A protein profiler assay is shown. The precoated membranes (rows 1-8; columns A-G) were incubated with stellate cell lysates (in duplicates) (upper panel) of stellates cell lysates treated with PMN (lower panel). Differentially expressed proteins are marked: (A3/4; blue circle) pp38, (B9/10; brown circle) Akt1/2/3; (C3/4; green circle) CREB; (C9/10; light blue circle) β-catenin; (G3/4; red) PRAS40; (G11/12; purple circle) HSP60. Abbreviations: PMN: polymorphonuclear neutrophil granulocytes.
Identification of lipid droplets in RLT cells: Lipid droplets were identified by staining with Nile red, and quantified by flow cytometry. (A) Cells after cultivation with azurocidin and (B) after culture on Matrigel (black lines; grey lines show cultivation of RLT cells on plastic). (C) Lipid droplets in RLT cells as seen by laser scanning microscopy. Abbreviations: w/o: without.
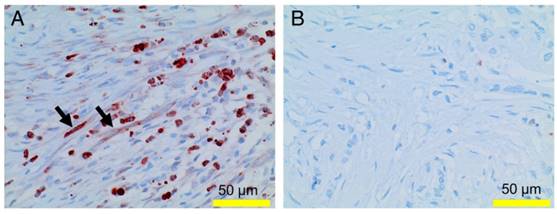
Taken together, our findings suggest a PMN-induced reprogramming of stellate cell function, which according to the literature [35-37], also results in reduced collagen production and in increased protease synthesis. Indeed, incubation of stellate cells with recombinant azurocidin resulted in an up-regulation of the tissue degrading protease MMP9 (5.9-fold; average of three independent experiments; and a marked increase in a proteome profiler assay) (Figure 7). Examining whole tissue sections of PDAC patients showed expression of MMP9 not only in tumor cells and infiltrating immune cells, but in myofibroblastic cells as well (Figure 10).
Expression of MMP9 in PDAC tissue: MMP9 (reddish-brown staining) is found in PMN and in spindle shaped cells (the latter indicated by arrows) in PDAC tissue with high PMN infiltrates (A), but not in tissue with low numbers of PMN (B).
Discussion
Diffusion-weighted magnetic resonance imaging (DW-MRI) permits relative statements on tissue quality because water diffusion depends on a variety of factors including vascularity of the tissue, three-dimensional structure, and, in the case of pancreatic cancer, also on the composition of the desmoplastic stroma and the interspersed cells [38, 39]. We now report a close association between water diffusion and PMN density. In tumors with high PMN-density, diffusion, as expressed by the structural diffusion coefficient D and ADC values, was significantly restricted compared to tumors with low PMN density. Because areas with high PMN density present with an altered growth pattern, including micropapillary, microglandular, solid, or single-cellular tumor growth, the restricted water diffusivity most likely reflects alterations of the microarchitecture. We cannot exclude completely that the infiltrated PMN due to enhanced cellularity contribute directly to the restricted water diffusion; however, we assume that the effects of an altered growth pattern prevail.
In order to compensate for intratumor heterogeneity, whole tumor volumes were analyzed for quantification of DW-MRI parameters and whole tissue slides were used for histopathological analyses in all patients of the MRI cohort. For the non-MRI cohort in which histopathological analyses were carried out on tissue microarrays, intratumor heterogeneity may be less relevant due to the high number of tissue specimens analyzed.
Various diffusion parameters obtained from mono- and biexponential diffusion models were correlated with histopathological parameters. To ensure that the DW-MRI parameters collected in the study are correct representations of the variables measured, inter-reader reliability was calculated between 2 radiologists.
Inter-reader reliability was excellent for the DWI-parameters f, D, ADCall, and ADC800, but inferior for the pseudodiffusion coefficient D* which is in line with the fact that the stability of the estimate of D* was reported to be inferior to that of f and D [24, 40].
The structural diffusion constant D corresponded to PMN density somewhat better compared to ADC800/ADCall values. An explanation for this might be that ADC values are confounded by contributions from moving blood while D constitutes perfusion-free diffusion [11].
There was no significant association of PMN density with tissue perfusion measured by means of IVIM perfusion parameters f and D* (f reflects microvessel density in PDAC [24]).
The structural diffusion coefficient D and ADC values did not differ significantly between well and moderately (G2) and poorly (G3) differentiated PDAC, which is in agreement with the results of Rosenkrantz et al. [41]. Poorly differentiated (G3) tumors, however, showed denser PMN infiltrates (scores 3 and higher). This association was only seen when all patients (n=140) were evaluated histologically, but not when only the patients of the MRI cohort (n=33 patients) were considered. Presumably, the heterogeneity among the patients and the inherent limitations of scoring requires analysis of a large number of patients to avoid underpowered statistics. This might also explain why we saw a correlation of PMN density and histological grading on one hand, and PMN density and structural diffusion coefficient on the other, but not between grading and structural diffusion coefficient, because for the latter analysis only 33 patients were available.
There was a trend towards shorter progression-free survival (PFS) in patients with high PMN density compared to patients with low PMN density. Similarly, in multivariate survival analysis using Cox's regression model, high PMN density was non-significantly associated with higher risk of early recurrence.
Given the fact that high PMN density is associated with restricted diffusion as well as with the histologically-apparent alteration of the tumor stroma, the question of a causal relationship arose. As described in more detail in the introduction, infiltrated, and hence activated, PMN have various means to alter the tumor micromilieu and to affect the tumor cells, e.g., by inducing an epithelial-to-mesenchymal transition [4, 6] (reviewed in [23]). The occurrence of α-SMA negative cells in PMN-dense areas, however, suggested a further mechanism. α-SMA is expressed in stellate cells and indicates an “active” status; down-regulation of α-SMA is interpreted as a “silencing” or “inactivation” of the cells [36, 37]. The term “silencing” is somewhat misleading because the cells actually terminate some functions, but acquire others, for which reason we prefer the term “reprogramming”. Based on morphology only and due to the lack of specific markers, stellate cells cannot be differentiated from cancer associated fibroblasts, characterized as myofibroblastic cells, which is a heterogeneous cell population that displays high plasticity. Although these cells acquire α-SMA and release lipid droplets [36], there is also evidence for α-SMA negative myofibroblasts [42]. Our in vitro data using an established stellate cell line, however, point to a direct interaction of stellate cells with PMN. PMN-derived azurocidin had profound effects on stellate cells, apparent as a drastic shape change, down-regulation of α-SMA, β-catenin, and of transcription factors linked to the PI3K/AKT pathway, as well as re-establishing lipid droplets, all indicators of “quiescence” as it is described in hepatic stellate cells [32-34]. We are well aware of the limitations of in vitro experiments. However, our in vitro findings are in line with our observation that in biopsies with high PMN density and altered growth pattern, low numbers of α-SMA-positive cells were found.
PMN store an abundance of proteases, bactericidal and cytotoxic proteins, and receptors in their granules, among those is azurocidin (also known as CAP37 or heparin-binding protein). It is a member of the serine protease family, such as neutrophil elastase or proteinase-3, but lacks proteolytic activity, and it is mainly studied for its role in host defense against microbial infection [43, 44]. Interestingly, azurocidin is taken up by tissue cells [45], and a single study with fibroblasts described effects on cell morphology, possibly due to interactions with actin [46]. Also, under our experimental conditions, azurocidin was incorporated into pancreatic stellate cells and was found in association with F-actin, especially in cells with altered, spindle-shaped morphology. Furthermore, also in the tissue specimen, an association of azurocidin with spindle-shaped stroma cells was seen.
How stellate cells take up azurocidin and the ensuing signaling pathway is still under investigation. First data suggest an active process, involving phosphorylation of p38 and of its downstream target MAPKAP-2. Moreover, in response to azurocidin, dephosphorylation of cofilin was seen, which indicates cofilin activation. Activated cofilin is crucial for the intracellular actin reorganization, and thus it is directly linked to changes in cell morphology. Moreover, cofilin is also linked to down-regulation of α-SMA [47], fitting into the concept that PMN via azurocidin induces the so-called “quiescent” or “inactivated” phenotype. That these cells are indeed not “quiescent” but rather “reprogrammed” was shown in the experiment where stellate cells in response to azurocidin up-regulated production of the tissue-degrading protease MMP9. In the tissue specimen of PDAC, MMP9 expression was not only seen in tumor cells, but also in spindle-shaped stroma cells as well.
Based on our findings and on data in the literature, we propose the following concept: In pancreatic cancer, infiltrated PMN alter the functional status of stellate cells towards matrix-degradation, the latter is apparent as altered architecture of the tumor with depletion of stroma and distinct changes in the tumor cells' growth patterns. Alteration of stroma then affects the water diffusivity, as measured by DW-MRI. In conclusion, reprogramming of stellate cells by infiltrated PMN could be a key event in alteration of the desmoplastic stroma, and hence the tumor microenvironment.
Abbreviations
α-SMA: alpha smooth muscle actin; ADC: apparent diffusion coefficient; ADC800: apparent diffusion coefficient calculated from b-values 0 and 800; ADCall: apparent diffusion coefficient calculated from all b-values; CD: cluster of differentiation antigen; CI: confidence interval; CREB: cyclic adenosine monophosphate response element-binding protein; D: diffusion coefficient; D*: pseudodiffusion coefficent; DKFZ: Deutsches Krebsforschungszentrum/ German Cancer Research Center; DMEM: Dulbecco's modified Eagle's medium; DW-MRI: diffusion-weighted magnetic resonance imaging; f: perfusion fraction; FOV: field of view; G: histological grading; GRAPPA: generalized autocalibrating partially parallel acquisitions; HASTE: half-Fourier acquisition single-shot turbo spin-echo; HBP: heparin binding protein; HE: hematoxylin and eosin; HPF: high power field; HR: hazard ratio; hTERT: human telomerase reverse transcriptase; ICC: intra-class correlation coefficient; IR: inversion recovery; IVIM: intravoxel incoherent motion; MITK: Medical Imaging Interaction Toolkit; MMP9: matrix metalloproteinase 9; MRI: magnetic resonance imaging; NCT: National Center for Tumor Diseases; PDAC: pancreatic ductal adenocarcinoma; PFA: paraformaldehyde; PFS: progression-free survival; p-MAPKAP-2: phosphorylated mitogen-activated protein kinase-activated protein kinase 2; PMN: polymorphonuclear neutrophil granulocytes; PRAS40: proline-rich AKT substrate of 40 kilodalton; PVDF: polyvinylidene fluoride; R: residual tumor; RIPA: radioimmunoprecipitation assay; RPMI medium: Roswell Park Memorial Institute medium; S: signal with diffusion weighting; S0: signal without diffusion weighting; SE2d-EPI: 2D spin echo- echo planar imaging; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; TA: acquisition time; TE: echo time; TMA: tissue microarray; TNM: tumor node metastasis; TR: repetition time; VOI: volume of interest.
Acknowledgements
The authors thank Birgit Prior, Sabine Stegmaier, Diana Lutz, Karin Rebholz, and Nicole Marnet for excellent technical support. MMG was supported by the German Research Foundation (Ga 1818/2-1). CD was supported by a fellowship of the Medical Faculty Heidelberg.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, Detlefsen S. et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 2012;61:172-8
3. Alam MS, Gaida MM, Bergmann F, Lasitschka F, Giese T, Giese NA. et al. Selective inhibition of the p38 alternative activation pathway in infiltrating T cells inhibits pancreatic cancer progression. Nat Med. 2015;21:1337-43
4. Gaida MM, Steffen TG, Gunther F, Tschaharganeh DF, Felix K, Bergmann F. et al. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur J Immunol. 2012;42:3369-80
5. Gaida MM, Welsch T, Herpel E, Tschaharganeh DF, Fischer L, Schirmacher P. et al. MHC class II expression in pancreatic tumors: a link to intratumoral inflammation. Virchows Arch. 2012;460:47-60
6. Grosse-Steffen T, Giese T, Giese N, Longerich T, Schirmacher P, Hansch GM. et al. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: the role of neutrophils and neutrophil-derived elastase. Clin Dev Immunol. 2012;2012:720768
7. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y. et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108:914-23
8. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719-34
9. Barral M, Taouli B, Guiu B, Koh DM, Luciani A, Manfredi R. et al. Diffusion-weighted MR imaging of the pancreas: current status and recommendations. Radiology. 2015;274:45-63
10. Fattahi R, Balci NC, Perman WH, Hsueh EC, Alkaade S, Havlioglu N. et al. Pancreatic diffusion-weighted imaging (DWI): comparison between mass-forming focal pancreatitis (FP), pancreatic cancer (PC), and normal pancreas. J Magn Reson Imaging. 2009;29:350-6
11. Le Bihan D, Breton E, Lallemand D, Aubin ML, Vignaud J, Laval-Jeantet M. Separation of diffusion and perfusion in intravoxel incoherent motion MR imaging. Radiology. 1988;168:497-505
12. Lee SS, Byun JH, Park BJ, Park SH, Kim N, Park B. et al. Quantitative analysis of diffusion-weighted magnetic resonance imaging of the pancreas: usefulness in characterizing solid pancreatic masses. J Magn Reson Imaging. 2008;28:928-36
13. Waghray M, Yalamanchili M, di Magliano MP, Simeone DM. Deciphering the role of stroma in pancreatic cancer. Curr Opin Gastroenterol. 2013;29:537-43
14. Muraoka N, Uematsu H, Kimura H, Imamura Y, Fujiwara Y, Murakami M. et al. Apparent diffusion coefficient in pancreatic cancer: characterization and histopathological correlations. J Magn Reson Imaging. 2008;27:1302-8
15. Legrand L, Duchatelle V, Molinie V, Boulay-Coletta I, Sibileau E, Zins M. Pancreatic adenocarcinoma: MRI conspicuity and pathologic correlations. Abdom Imaging. 2015;40:85-94
16. Klauss M, Lemke A, Grunberg K, Simon D, Re TJ, Wente MN. et al. Intravoxel incoherent motion MRI for the differentiation between mass forming chronic pancreatitis and pancreatic carcinoma. Invest Radiol. 2011;46:57-63
17. Heid I, Steiger K, Trajkovic-Arsic M, Settles M, Esswein MR, Erkan M. et al. Co-clinical Assessment of Tumor Cellularity in Pancreatic Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:1461-70
18. Kurosawa J, Tawada K, Mikata R, Ishihara T, Tsuyuguchi T, Saito M. et al. Prognostic relevance of apparent diffusion coefficient obtained by diffusion-weighted MRI in pancreatic cancer. J Magn Reson Imaging. 2015;42:1532-7
19. Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P. et al. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod Pathol. 2011;24:1612-9
20. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104:20262-7
21. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest. 2010;120:1151-64
22. Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z. et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. 2011;54:948-55
23. Felix K, Gaida MM. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression. Int J Biol Sci. 2016;12:302-13
24. Klauss M, Mayer P, Bergmann F, Maier-Hein K, Hase J, Hackert T. et al. Correlation of Histological Vessel Characteristics and Diffusion-Weighted Imaging Intravoxel Incoherent Motion-Derived Parameters in Pancreatic Ductal Adenocarcinomas and Pancreatic Neuroendocrine Tumors. Invest Radiol. 2015;50:792-7
25. Fritzsche KH, Neher PF, Reicht I, van Bruggen T, Goch C, Reisert M. et al. MITK diffusion imaging. Methods Inf Med. 2012;51:441-8
26. Patel J, Sigmund EE, Rusinek H, Oei M, Babb JS, Taouli B. Diagnosis of cirrhosis with intravoxel incoherent motion diffusion MRI and dynamic contrast-enhanced MRI alone and in combination: preliminary experience. J Magn Reson Imaging. 2010;31:589-600
27. Koh DM, Collins DJ, Orton MR. Intravoxel incoherent motion in body diffusion-weighted MRI: reality and challenges. AJR Am J Roentgenol. 2011;196:1351-61
28. Riches SF, Hawtin K, Charles-Edwards EM, de Souza NM. Diffusion-weighted imaging of the prostate and rectal wall: comparison of biexponential and monoexponential modelled diffusion and associated perfusion coefficients. NMR Biomed. 2009;22:318-25
29. Fujima N, Yoshida D, Sakashita T, Homma A, Tsukahara A, Tha KK. et al. Intravoxel incoherent motion diffusion-weighted imaging in head and neck squamous cell carcinoma: assessment of perfusion-related parameters compared to dynamic contrast-enhanced MRI. Magn Reson Imaging. 2014;32:1206-13
30. Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155-68
31. Jesnowski R, Furst D, Ringel J, Chen Y, Schrodel A, Kleeff J. et al. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: deactivation is induced by matrigel and N-acetylcysteine. Lab Invest. 2005;85:1276-91
32. Houglum K, Lee KS, Chojkier M. Proliferation of hepatic stellate cells is inhibited by phosphorylation of CREB on serine 133. J Clin Invest. 1997;99:1322-8
33. Ge WS, Wang YJ, Wu JX, Fan JG, Chen YW, Zhu L. beta-catenin is overexpressed in hepatic fibrosis and blockage of Wnt/beta-catenin signaling inhibits hepatic stellate cell activation. Mol Med Rep. 2014;9:2145-51
34. Reyes-Gordillo K, Shah R, Popratiloff A, Fu S, Hindle A, Brody F. et al. Thymosin-beta4 (Tbeta4) blunts PDGF-dependent phosphorylation and binding of AKT to actin in hepatic stellate cells. Am J Pathol. 2011;178:2100-8
35. Haber PS, Keogh GW, Apte MV, Moran CS, Stewart NL, Crawford DH. et al. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am J Pathol. 1999;155:1087-95
36. Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA. et al. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534-41
37. Mews P, Phillips P, Fahmy R, Korsten M, Pirola R, Wilson J. et al. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut. 2002;50:535-41
38. Klauss M, Gaida MM, Lemke A, Grunberg K, Simon D, Wente MN. et al. Fibrosis and pancreatic lesions: counterintuitive behavior of the diffusion imaging-derived structural diffusion coefficient d. Invest Radiol. 2013;48:129-33
39. Wegner CS, Gaustad JV, Andersen LM, Simonsen TG, Rofstad EK. Diffusion-weighted and dynamic contrast-enhanced MRI of pancreatic adenocarcinoma xenografts: associations with tumor differentiation and collagen content. J Transl Med. 2016;14:161
40. Lemke A, Stieltjes B, Schad LR, Laun FB. Toward an optimal distribution of b values for intravoxel incoherent motion imaging. Magn Reson Imaging. 2011;29:766-76
41. Rosenkrantz AB, Matza BW, Sabach A, Hajdu CH, Hindman N. Pancreatic cancer: lack of association between apparent diffusion coefficient values and adverse pathological features. Clin Radiol. 2013;68:e191-7
42. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. The Journal of experimental medicine. 2017;214:579-96
43. Iversen LF, Kastrup JS, Bjorn SE, Rasmussen PB, Wiberg FC, Flodgaard HJ. et al. Structure of HBP, a multifunctional protein with a serine proteinase fold. Nat Struct Biol. 1997;4:265-8
44. Linder A, Soehnlein O, Åkesson P. Roles of Heparin-Binding Protein in Bacterial Infections. Journal of Innate Immunity. 2010;2:431-8
45. Olofsson AM, Vestberg M, Herwald H, Rygaard J, David G, Arfors KE. et al. Heparin-binding protein targeted to mitochondrial compartments protects endothelial cells from apoptosis. J Clin Invest. 1999;104:885-94
46. Ostergaard E, Flodgaard H. A neutrophil-derived proteolytic inactive elastase homologue (hHBP) mediates reversible contraction of fibroblasts and endothelial cell monolayers and stimulates monocyte survival and thrombospondin secretion. J Leukoc Biol. 1992;51:316-23
47. Pho M, Lee W, Watt DR, Laschinger C, Simmons CA, McCulloch CA. Cofilin is a marker of myofibroblast differentiation in cells from porcine aortic cardiac valves. Am J Physiol Heart Circ Physiol. 2008;294:H1767-78
Author contact
![]() Corresponding author: Matthias M. Gaida, M.D., Institute of Pathology, University Hospital Heidelberg, Im Neuenheimer Feld 224, 69120 Heidelberg, Germany. Phone:+49-6221-56-4160; Fax: +49-6221-56-5251; Email: Matthias.Gaidauni-heidelberg.de
Corresponding author: Matthias M. Gaida, M.D., Institute of Pathology, University Hospital Heidelberg, Im Neuenheimer Feld 224, 69120 Heidelberg, Germany. Phone:+49-6221-56-4160; Fax: +49-6221-56-5251; Email: Matthias.Gaidauni-heidelberg.de