Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Intracellular Galectin-3
Extracellular Galectin-3
Organ Fibrosis
Cardiac Fibrosis and Heart...
Atherosclerosis and Diabetes...
Galectin-3 Activation
Galectin-3 Inhibition
Conclusion
Abbreviations
Acknowledgements
References
Introduction
Intracellular Galectin-3
Extracellular Galectin-3
Organ Fibrosis
Cardiac Fibrosis and Heart...
Atherosclerosis and Diabetes...
Galectin-3 Activation
Galectin-3 Inhibition
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):593-609. doi:10.7150/thno.22196 This issue Cite
Review
Galectin-3 Activation and Inhibition in Heart Failure and Cardiovascular Disease: An Update
Navin Suthahar1, Wouter C. Meijers1, Herman H.W. Silljé1, Jennifer E. Ho2, Fu-Tong Liu3, Rudolf A. de Boer1 ![]()
1. University Medical Center Groningen, University of Groningen, Department of Cardiology, PO Box 30.001, 9700 RB Groningen, the Netherlands
2. Massachusetts General Hospital, Cardiovascular Research Center, Boston, MA, USA
3. Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan
Received 2017-8-2; Accepted 2017-9-24; Published 2018-1-1
Citation:
Suthahar N, Meijers WC, Silljé HHW, Ho JE, Liu FT, de Boer RA. Galectin-3 Activation and Inhibition in Heart Failure and Cardiovascular Disease: An Update. Theranostics 2018; 8(3):593-609. doi:10.7150/thno.22196. https://www.thno.org/v08p0593.htm
Other stylesAbstract

Galectin-3 is a versatile protein orchestrating several physiological and pathophysiological processes in the human body. In the last decade, considerable interest in galectin-3 has emerged because of its potential role as a biotarget. Galectin-3 is differentially expressed depending on the tissue type, however its expression can be induced under conditions of tissue injury or stress. Galectin-3 overexpression and secretion is associated with several diseases and is extensively studied in the context of fibrosis, heart failure, atherosclerosis and diabetes mellitus. Monomeric (extracellular) galectin-3 usually undergoes further “activation” which significantly broadens the spectrum of biological activity mainly by modifying its carbohydrate-binding properties. Self-interactions of this protein appear to play a crucial role in regulating the extracellular activities of this protein, however there is limited and controversial data on the mechanisms involved. We therefore summarize (recent) literature in this area and describe galectin-3 from a binding perspective providing novel insights into mechanisms by which galectin-3 is known to be “activated” and how such activation may be regulated in pathophysiological scenarios.
Keywords: Galectin-3, fibrosis, extracellular matrix, heart failure, renal disease, cardiovascular disease, interaction, cell-cell adhesion, carbohydrate binding domain
Introduction
Lectins are carbohydrate-binding proteins found in plants and animals that are specific for sugar moieties, and were initially defined as “sugar-binding proteins of non-immune origin that have the ability to agglutinate cells and precipitate glycoconjugates” [1]. Galectins belong to the family of animal-lectins and are a group of water soluble, non-glycosylated globular proteins that can interact with carbohydrates in a divalent cation-independent manner [2]. Two characteristic properties distinguish them from other animal-lectins: affinity for ß-galactoside derivatives and consensus amino-acid sequences [3,4]. Galectins are synthesized in the cytoplasm and function in both nuclear and cytoplasmic compartments. They are also secreted to the outer plasma membrane and the extracellular matrix (ECM), and are present in the circulation. Fifteen different galectins have been identified and characterized in humans, and are classified into three groups: proto-, chimera-, and tandem-repeat types based on their structure. The biological significance of galectins is paramount due to their vital role in several developmental and defence processes. The role of galectins as decipherers of glycocode has also been acknowledged for more than twenty years [5].
As the only chimeric galectin member of the vertebrate family, galectin-3 has a very interesting physico-chemical and biological profile: it exhibits sequence similarity to B-cell lymphoma-2 (Bcl-2) protein and is the only galectin containing a C-terminal anti-death NWGR motif [6]. Galectin-3 orchestrates several physiological processes and has also been identified as a “culprit-molecule” in the pathogenesis of various diseases, especially fibrosis, cardiovascular disease and cancer.
In this review, we summarize the function of galectin-3 in physiology and focus on its role in pathophysiological scenarios involving fibrosis, heart failure (HF), atherosclerosis and diabetes mellitus (DM). As galectin-3 is an emerging biotarget, we also describe its structure from a binding perspective paying special attention to the carbohydrate-recognition domain (CRD). Monomeric (extracellular) galectin-3 usually undergoes further “activation” which significantly broadens the spectrum of biological activity of this protein. However, precise knowledge in this direction is inadequate and the main aim of this article is to provide a deeper insight into mechanisms by which galectin-3 is known to be “activated”, and how such “activation” may be regulated in pathophysiological scenarios.
Galectin-3: One Protein With Different Names
Galectin-3 was previously known by several names including Mac-2 antigen [7], IgE-binding protein [8], L-29 [9,10] and CBP30 [11]. Galectin-3 was first identified by Ho and Springer as a macrophage sub-population specific marker (Mac-2 antigen) that was distributed in the cytosol, extracellular medium and also in membrane fractions of these cells [7]. Galectin-3 was also independently described as IgE-binding protein (eBP) [12], and analysis of eBP revealed two important constituent domains: CRD and amino domain carrying potential recognition sites for collagenase cleavage (N-terminal) [8]. Interactions of L-29 (galectin-3) with laminin and requirement of N-terminal in positive co-operative binding to laminin and other glycoconjugates were reported by Massa et al. [9], while in experiments involving baby hamster kidney cell extracts, the name carbohydrate binding protein-30 (CBP30) was frequently used (Figure 1) [11].
LGALS3: The Galectin-3 Gene And Its Regulation
LGALS3 is the single gene coding for galectin-3 in the human genome and is situated on chromosome 14, locus q21-22, and is composed of six exons and five introns spanning about 17 kilobases. Promoter methylation status of LGALS3, and elements such as CRE motifs, nuclear factor- κB (NF-κB) like sites and GC boxes located within the galectin-3 promoter regulate galectin-3 expression [13]. Galectin-3 also contains a special regulatory element called galig (galectin-3 internal gene) located within the second intron of LGALS3 gene [14]. The transcripts of galectin-3 and the internal promoter galig share common coding sequences but use alternative reading frames. Galig is a cell death gene coding for two proteins: a cytoplasmic protein, cytogaligin and a mitochondrial protein, mitogaligin. The interaction of mitogaligin with cardiolipin is believed to disrupt the mitochondrial membrane, and cell death induced by galig can be opposed by overexpression of myeloid cell leukaemia sequence 1 protein (MCL-1), which belongs to the anti-apoptotic Bcl-2 family [15]. Galectin-3 transcription can also be repressed by proteins such as Krüppel-like factor 3 (KLF-3), which belongs to the family of zinc finger transcription factors [16], or up-regulated by other transcription factors such as runt-related transcription factor 2 (RUNX2) [17].
Figure 1
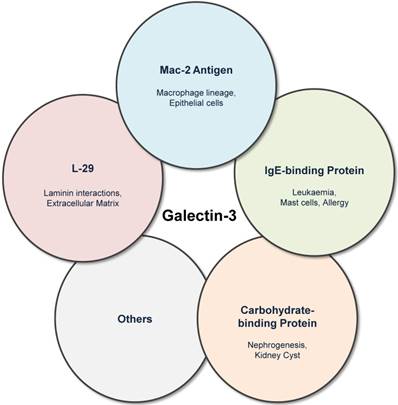
Galectin-3 was previously known by several names including Mac-2 antigen, IgE-binding protein, carbohydrate-binding protein and L-29. Although the historical nomenclature is obsolete, it highlights the various fields in which galectin-3 research has evolved.
Temporospatial Expression Of Galectin-3 Is Variable And Complex
In adults, galectin-3 is ubiquitously distributed in hematopoietic tissue, thymus, lymph nodes, skin, respiratory tract, digestive tract, reproductive tract and urinary tract [18,19], and baseline galectin-3 expression varies depending on tissue-type and tissue-maturity (Figure 2) [20]. Even within a tissue, different cell types express galectin-3 differentially: within the hematopoietic tissue, monocytes express galectin-3, and a higher amount is expressed in macrophages [18,21]. However, galectin-3 is virtually undetectable in human peripheral blood lymphocytes [6,22].
Although baseline galectin-3 expression is variable in different tissues, its expression is inducible. For instance, healthy cardiac tissue has a very low baseline galectin-3 expression, but during cardiac injury its expression is rapidly induced. Galectin-3 up-regulation plays a crucial role in the initial phases of tissue repair; however, sustained over-expression results in fibrosis of the heart [23,24]. Increased galectin-3 expression can also be induced in other tissues after injury, and this is significantly associated with organ fibrosis [25-27].
Figure 2
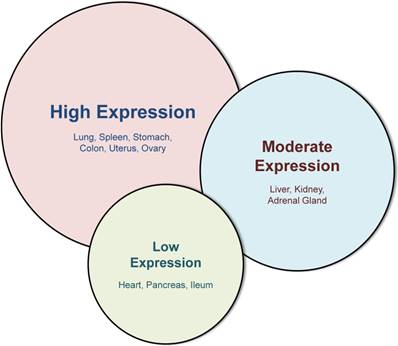
Western blot analysis of different tissues, adapted from Kim et al [20]. This figure illustrates the variability of galectin-3 in different murine tissues with the highest expression in lung, spleen, stomach, colon, uterus and ovary. While liver, kidney and adrenal gland display a moderate galectin-3 expression, baseline expression in the heart, pancreas and ileum is very low.
Secretion and Translocation of Galectin-3
Galectin-3 bypasses the classical “endoplasmic reticulum-Golgi apparatus” pathway [28] and is secreted via a non-classical mechanism. Although many factors are known to influence galectin-3 secretion, e.g., heat shock, calcium ionophores, acylation and phosphorylation [29-31], the exact mechanisms remain to be elucidated. Intracellularly, phosphorylation and importin-mediated mechanisms appear to be involved in nucleo-cytoplasmic shuttling of galectin-3 [32-35], while synexin-mediated mechanisms are indicated in galectin-3 translocation to the mitochondria [36].
Intracellular Galectin-3
Galectin-3 interacts with various ligands within the cell to elicit several biological processes. Potential intracellular binding partners include anti-apoptotic molecules such as Bcl-2 [6], and signalling molecules such as Gemin4 [37] and ß-catenin [38]. Intracellular binding usually occurs via protein-protein interactions utilizing either N-terminal or CRD without involving sugar moieties, i.e., no protein-sugar interactions. However, in in vitro experiments, certain protein-to-protein interactions (e.g., galectin-3-Bcl-2 interaction, galectin-3-ß-catenin interaction) can also be inhibited by lactose [6,38]; this could be explained by the involvement of CRD in protein-protein interactions or conformational changes induced by lactose.
Physiological Functions
Intracellular galectin-3 has several biological functions related to growth and development such as implantation of the embryo [39] and renal morphogenesis [40,41]. Increased galectin-3 expression is also found in the notochord, cartilage and bone during development [42], and appears to play a regulatory role in cellular fusion (e.g., osteoclast differentiation) [43], and cellular longevity (e.g., chondrocyte survival) [44,45]. However, most of this knowledge is obtained from murine experimental models.
Pathophysiological functions
Sustained galectin-3 expression, e.g., after tissue injury, could result in organ fibrosis. In vitro studies demonstrate that galectin-3-mediated fibrosis could be due to galectin-3 overexpression in several cell types: when murine and human hepatic stellate cells (HSCs) were activated by culturing on tissue culture plastic, a significant up-regulation of intracellular galectin-3 was observed. However, protein expression of α-smooth muscle actin (α-SMA, marker of HSC activation) in galectin-3-/- HSCs was insignificant compared to wild type (WT) HSCs [25]. This was also validated in an in vivo hepatic fibrosis model: liver sections from animals exposed to chronic chemical injury with CCl4 (8 weeks) displayed an intense signal for galectin-3, while controls expressed virtually no galectin-3. Furthermore, galectin-3 knockout (KO) mice treated with CCl4 also displayed a very low amount of collagen and α-SMA in hepatic tissue, while the WT mice demonstrated a significant increase in expression of these proteins [25]. Galectin-3 overexpression is also a characteristic feature of “profibrotic” M2 macrophages: naïve macrophages stimulated with interleukin-4 (IL-4) and IL-13 express higher levels of galectin-3, together with other markers of collagen turnover such as mannose receptors [46]. Although intracellular galectin-3 levels correlate with tissue repair [47,48] and subside over time, uncontrolled galectin-3 expression could result in sustained myofibroblast and macrophage activation leading to tissue fibrosis, possibly through intracellular and also extracellular signalling pathways.
Intracellular galectin-3 levels are also known to affect the inflammatory response through various mechanisms [49]. However, limited data exist regarding the function of intracellular galectin-3 in neutrophil apoptosis. A recent study performed in a galectin-3 KO mouse model indicates that there is reduced apoptosis of neutrophils and also reduced neutrophil clearance by macrophages [50], suggesting that galectin-3 might be an important player in resolving the “neutrophil-phase” of inflammation. It is speculated that when exported to the neutrophil surface, galectin-3 could act as an opsonin and initiate clearance by promoting macrophage efferocytosis [51]. Macrophage galectin-3 expression also appears to have a crucial role in phagocytosis of apoptotic bodies [52].
Recent studies also suggest that intracellular galectin-3 could have a greater role in the pathophysiology of DM type 1 by inducing β-cell apoptosis: β-cells from galectin-3 KO mice were resistant to inflammation-induced cell death by counteracting mitochondrial apoptotic pathways [53]. This is in contrast to previous research that demonstrated that intracellular galectin-3 supresses mitochondrial apoptotic pathways by preserving mitochondrial integrity [36].
In summary, the final outcome of the fibro-inflammatory response is determined by a dynamic balance between neutrophil apoptosis, macrophage and T-cell responses, fibroblast activation and myofibroblast persistence, and intracellular galectin-3 seems to be involved in many of these responses (Figure 3). However, our current understanding of galectin-3-mediated apoptotic mechanisms is limited and further studies are warranted to characterize the role of intracellular galectin-3 in apoptosis of different cell types, especially in immune-cells and collagen-producing cells.
Figure 3
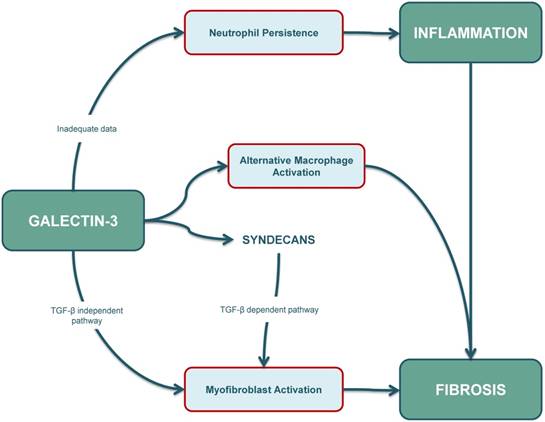
The role of galectin-3 in inflammation is ambiguous. Some studies suggest that apoptosis of neutrophils and their clearance by macrophages is reduced in galectin-3 KO mouse models. However, further research needs to be conducted as increased intracellular galectin-3 levels are usually associated with cellular longevity. The role of galectin-3 in fibrosis is well-established, and increased galectin-3 levels contribute to (myo)fibroblast activation through a TGF-β independent pathway and also through a TGF-β dependent pathway. Syndecans also play an important role, especially by affecting profibrotic signalling in cardiac fibroblasts, and possibly also by interacting with galectin-3. Furthermore, galectin-3 can also affect the fibrotic pathway by inducing alternative (M2) activation in macrophages. KO: knockout; TGF-β: transforming growth factor β
Extracellular Galectin-3
Galectin-3 can be secreted to the cell surface where it binds to glycan-rich molecules in cell-surface glycoproteins and glycolipids. When exported to the ECM, it interacts with various glycosylated matricellular binding partners such as laminin, tenascin and fibronectin [54-56]. Extracellular galectin-3-binding usually occurs through protein-carbohydrate interactions, orchestrated by the galectin-3 CRD; this results in lectin-saccharide bonds, typically inhibited by lactose [57].
Physiological functions
Extracellular galectin-3 plays an important role in embryogenesis, growth and development, and also in maintaining homeostasis [58,59]. For instance, CD98 heavy chain is a glycosylated transmembrane protein expressed in developing human trophoblasts. In in vitro conditions, galectin-3 associates with CD98 heavy-chain, possibly through lectin-glycan interactions, and facilitates placental (BeWo) cell fusion [58]; blocking galectin-3 CRD with lactose reduces cell fusion in these cells. Extracellular galectin-3 also appears to be an indispensable player in cartilage and bone-matrix remodelling in and around the period of endochondral ossification [60]. Furthermore, association of extracellular galectin-3 with glycoproteins such as von Willebrand factor (vWF) [61], factor VIII [62] and hensin is believed to regulate homeostatic mechanisms, i.e., galectin-3-vWF interaction modulates thrombus formation and galectin-3-hensin interaction is shown to promote hensin oligomerization, which is considered to be essential for renal adaptation during metabolic acidosis [63].
Interaction of galectin-3 with heavily glycosylated CD13 has been shown to aggregate monocytes and this process can be inhibited by lactose and anti-galectin-3 antibodies [64]; galectin-3 binding to other glycosylated partners such as CD98 has also been reported, and this mechanism is indicated in alternative activation of macrophages during wound healing [46]. Extracellular galectin-3 is also believed to facilitate monocyte migration by functioning as a chemotactic factor [65]. However, many of these extracellular functions were demonstrated by using recombinant protein added to cultured cells; whether these represent functions of endogenous galectin-3 remains to be established.
Extracellular galectin-3 also acts as an interpreter of glycocodes [5]. The sugar molecules from glycoproteins and glycolipids form the glycocalyx; unlike codes encrypted in amino-acids and nucleotides, these glycocodes (saccharides) are highly heterogeneous secondary gene products escaping direct control of genes. Due to the structural variability and complexity of carbohydrates, glycocodes carry a substantial amount of information and are able to influence a broad spectrum of biological activities by regulating various cellular processes [66]. They are also hypothesised to influence cell-survival by regulating entry of biological agents such as T-lymphocytes and viruses [67-69].
Pathophysiological functions
Galectin-3 is a “culprit” protein associated with several diseases. Herein, we focus on the role of extracellular galectin-3 in the pathogenesis of fibrosis, HF, atherosclerosis and DM type 2.
Organ Fibrosis
Genetic disruption of galectin-3 reduces or abolishes the development of fibrosis in various organs, e.g., heart, vessels, lung, liver and kidney [24-27,70,71]. Although profibrotic properties of galectin-3 can be studied using genetic KO models, it is not possible to conclude if it is intracellular or extracellular blockade (or both) that has resulted in reduction or loss of fibrosis. Other methods by which fibrotic effects of extracellular galectin-3 can be characterized is by adding recombinant galectin-3 to cells in in vitro experiments or through in vivo experiments involving pharmacological galectin-3 blockade; several existing galectin-3 inhibitors appear to reduce fibrosis by acting extracellularly and can therefore be utilized to understand galectin-3 function at the cellular membrane and in the extracellular matrix.
There are various mechanisms by which extracellular galectin-3 contributes to tissue fibrosis. Galectin-3 secreted by several cells, including monocytes and macrophages, can activate quiescent fibroblasts to myofibroblasts [26], which is the hallmark event in tissue fibrosis. Alternative macrophage activation (M2), implicated in tissue fibrosis also results in increased expression and secretion of galectin-3, and a positive feedback loop involving extracellular galectin-3 is thought to be responsible for sustained M2 activation of macrophages [46]. Galectin-3 could also promote fibrosis by modulating immuno-inflammatory responses and angiogenesis [72,73]. Furthermore, galectin-3 is hypothesized to form lectin-saccharide lattices on cell surfaces and transforming growth factor- ß (TGF-ß) receptor entrapment within the lattices could amplify profibrotic signalling [74].
Galectin-3 modulates TGF-ß function and also appears to be a crucial regulator of pulmonary fibrosis by activating macrophages and fibroblasts. Both genetic inhibition and treatment with galectin-3 inhibitor TD-139 reduced TGF-ß-induced and bleomycin-induced pulmonary fibrosis by reducing myofibroblast activation and collagen secretion [75]. In a hepatic fibrosis model, galectin-3 was required for TGF-ß-mediated myofibroblast activation and matrix production, and galectin-3 inhibition through in vivo siRNA knockdown prevented myofibroblast activation and hepatic injury [25]. In another model, disruption of galectin-3 gene resulted in reduced hepatic fibrosis although TGF-ß expression levels were not affected, suggesting that TGF-ß-mediated myofibroblast production and subsequent hepatic fibrosis required galectin-3, and galectin-3 could cause “TGF-ß-independent fibrosis” under certain circumstances (Figure 3) [25,76].
Galectin-3 secretion from macrophages appears to be a main driver of renal fibrosis in animal models of kidney injury [26,77]. In a unilateral ureteral obstruction (UUO) model, galectin-3 KO mice developed reduced interstitial fibrosis compared to sham-operated WT mice: staining of kidney tissue using picrosirius red displayed increased collagen-deposition in WT mice with UUO compared to galectin-3 KO counterparts, and immunohistological staining for α-SMA was markedly reduced in galectin-3-KO mice [26]. In chronic allograft injury models, there was also a marked reduction of renal interstitial fibrosis (reduced collagen I and α-SMA) in galectin-3 KO mice compared to WT mice: although the number of infiltrating leukocytes in the renal tissue were comparable between WT and KO mice, alternative macrophage (M2) activation and CD4+ T cells were significantly reduced in galectin-3 KO mice suggesting that galectin-3 may promote M2 macrophage activation and renal fibrosis post-transplantation [78]. The study conducted by Frenay and colleagues on REN2 rats added further evidence to the macrophage-galectin-3-fibrosis axis, and also highlighted the potential of pharmacological galectin-3 inhibition in ameliorating fibrosis: compared to untreated controls, inhibition of galectin-3 with N-acetyllactosamine (LacNAc) attenuated proteinuria, improved kidney function and reduced renal damage by significantly reducing macrophage infiltration, galectin-3 expression and α-SMA expression in this hypertensive nephropathy / HF model [79].
Cardiac Fibrosis and Heart Failure
Several studies performed in the last decade in healthy population as well as in HF patients demonstrate the close relationship between galectin-3, cardiac fibrosis and HF [80-84]. Galectin-3 has a class II recommendation in HF management according to ACCF/AHA 2013 guidelines [85]. Furthermore, it is an emerging target in the treatment of cardiac fibrosis and HF [86], and in the following sections we summarize the role of galectin-3 as a biotarget.
Animal Models of Cardiac Injury
In a pioneering study, Sharma et al. observed that some hypertensive rats that overexpressed murine renin gene (REN-2 rats) developed overt HF after about 4 months, while others did not decompensate. Analysis of decompensated hearts revealed that galectin-3 was the strongest upregulated gene and its expression was about 5x higher compared to compensated hearts. Causality was tested by infusion of galectin-3 in pericardial sacs of normal rats; this resulted in increased collagen I/III ratio and also led to cardiac remodelling and dysfunction [23]. Following studies by Liu et al. also yielded similar results: galectin-3 infusion into pericardial sac resulted in inflammation, ventricular remodelling and cardiac dysfunction in male adult rats [87]. Conclusive evidence for the role of galectin-3 in cardiac remodelling and fibrosis was accrued in the study conducted by Yu and colleagues [24]. Cardiac remodelling in mice was induced pharmacologically using angiotensin II infusion, or surgically through transverse aortic constriction (TAC); although galectin-3 KO mice developed left ventricular (LV) hypertrophy, they displayed no LV dysfunction and fibrosis. Pharmacological inhibition of galectin-3 with LacNAc also attenuated LV dysfunction and fibrosis in WT mice. On the other hand, untreated WT mice developed LV hypertrophy, fibrosis and adverse remodelling, highlighting that galectin-3 was not only a culprit protein but also a potential therapeutic target to counteract adverse cardiac remodelling.
Other models of HF are also useful to further understand galectin-3-mediated mechanisms of cardiac dysfunction. In a murine angiotensin II-induced hypertension model, genetic deletion of galectin-3 reduced cardiac inflammation (decreased macrophage infiltration) and fibrosis while WT mice exhibited severe myocardial fibrosis. However, myocyte cross-sectional area, an indicator of cardiac hypertrophy, was significantly increased in both WT and galectin-3 KO groups suggesting that angiotensin II induced cardiac hypertrophy in both groups, but reduced fibrosis only in galectin-3 KO groups. LV function was also well preserved in galectin-3 KO mice while WT mice exhibited a decline in LV systolic function (ejection fraction, EF reduced from 84% ± 1% to 61% ± 3% and systolic function reduced from 71% ± 0.3% to 57% ± 2%) [88]. Some reports suggest that galectin-3 could also be associated with cardiac injury and fibrosis in a non-hypertensive setting: when WT and galectin-3 KO mice were treated with aldosterone (1 mg/kg/day using osmotic minipump) for 3 weeks, galectin-3 KO mice displayed virtually no cardiac fibrosis. Similar effects were also elicited when aldosterone exposed WT mice were treated with a galectin-3 inhibitor, modified citrus pectin (MCP) [89].
Recently, in a canine model of HF with preserved ejection fraction (HFpEF) induced by aortic banding, myocardial galectin-3 was significantly upregulated after two weeks. Increase in galectin-3 expression positively correlated to the severity of diastolic dysfunction assessed with the echocardiographic diastolic parameter - early transmitral flow velocity to early diastolic tissue velocity (E/Em) ratio [90]. Although it is evident that galectin-3 plays a crucial role in cardiac fibrosis and in HF, some studies have demonstrated the significance of galectin-3 in maintaining the integrity of cardiac tissue after necrosis. In a mouse model of myocardial infarction (MI), galectin-3 KO displayed an increased trend towards mortality, chiefly due to ventricular rupture [48], emphasizing that galectin-3 is necessary for normal wound healing, especially during the initial phases of cardiac repair.
In vitro studies
Cardiac fibroblasts exposed to recombinant galectin-3 resulted in proliferation, differentiation and increased production of collagen; this was blocked by galectin-3 knockdown [91]. Recent studies suggest that transmembrane proteoglycans such as syndecans are also involved in cardiac fibrosis: syndecans have several heparan sulphate GAG chains that could potentially interact with galectin-3 and affect profibrotic syndecan signalling in cardiac fibroblasts (Figure 3) [92,93].
Although not directly involved in collagen production, M2 macrophages have an important role in collagen turnover affecting wound remodelling, and increased galectin-3 expression in activated macrophages has been observed in several in vitro studies [26,46,94-96]. Furthermore, galectin-3 also co-localized with activated macrophages in myocardial biopsies from failure-prone rats, suggesting that macrophage-derived galectin-3 could be an important player in cardiac remodelling [23].
Current studies indicate that galectin-3 can also be secreted by cardiomyocytes: mechanical stretching of cardiomyocytes in a cellular model of HFpEF resulted in increased galectin-3 expression in these cells, and also a significant increase in galectin-3 secretion [90]. In a different study investigating effects of protein kinase C (PKC) in cardiac hypertrophy, exposure of rat cardiomyocytes (HL-1 cells) to the PKC activator, phorbol dibutyrate (PDBu), resulted in hypertrophy and increased galectin-3 protein expression and collagen production [97]; pretreatment of HL-1 cells with galectin-3 inhibitor (β-lactose) blocked collagen production, indicating that galectin-3 expression and collagen secretion may be closely associated in cardiomyocytes.
In conclusion, although galectin-3 is a fibrogenic protein necessary for normal healing, sustained expression and secretion of galectin-3 within the cardiac tissue leads to adverse cardiac remodelling resulting in progressive fibrosis and HF. Genetic and pharmacological inhibition of galectin-3 reduces cardiac fibrosis in several animal models, and specific inhibitors that target galectin-3 mediated fibrosis appear to be quite promising in HF management.
Atherosclerosis and Diabetes Mellitus type 2
Atherosclerosis is a major cause of cardiovascular disease and galectin-3 levels are generally increased in atherosclerotic lesions. Foam cells, which are fat-laden macrophages, are abundantly present within atherosclerotic lesions and actively secrete galectin-3 [21]. This local increase in galectin-3 concentration could potentially be responsible for enhanced recruitment of monocytes and macrophages to the artery wall [65], exacerbating the pro-inflammatory state in atherosclerotic lesions [98]. However, galectin-3 might also contribute to pathogenesis through other mechanisms [99], including amplification of inflammatory pathways by intracellular mechanisms in macrophages [100]. Both genetic and pharmacological inhibition of galectin-3 resulted in reduced atherosclerotic lesions and slowed atherosclerotic plaque-progression in apolipoprotein E-KO mice [101,102].
The role of galectin-3 in DM type 2 is ambiguous: some studies claim that galectin-3 deficiency is associated with insulin resistance, and galectin-3 elicits a protective effect in DM type 2 by acting as a receptor for advanced glycation end products (AGEs) [103,104]. However, a very recent study demonstrated that knocking out galectin-3 gene in mice fed with a high-fat diet significantly reduced the development of insulin resistance. Furthermore, this study also provides preliminary evidence that extracellular galectin-3 binds the insulin receptor directly and attenuates downstream pathways, suggesting galectin-3 to be a novel targetable link in insulin resistance and DM type 2 [105].
Galectin-3 Activation
Monomeric galectin-3 undergoes further physicochemical modifications that increase its range of biological functionality, especially extracellular activity. The most important mechanisms leading to “bio-activation” of galectin-3 are self-associations (multimerization) and formation of galectin-3 lattices.
Self-associations of Galectin-3
Self-association increases the spectrum of biological activity of galectin-3, and can be divided into intra-molecular associations, within one galectin-3 molecule, and intermolecular associations, between different galectin-3 molecules. Galectin-3 self-association usually depends upon protein concentration and interaction with binding partners (ligands). Before delving into bio-activation of galectin-3 by such mechanisms, it is imperative to develop a general understanding of its structure and binding sites.
Galectin-3 Structure: A Binding Perspective
Galectin-3 molecule has a globular head with a diameter of about 3-4 nm attached to a slender 45-50 nm long tail that has great conformational flexibility [106]. The globular head harbours the carbohydrate recognition domain (CRD); the long tail contains the collagenase cleavable H-domain and culminates as the amino N-terminal (NT). Although some authors prefer to use “collagen-like” domain, galectin-3 does not have the Gly-X-Y characteristic of collagens.
From a chemical point of view, CRD is divided into five subsites (A-E): subsites C and D are responsible for binding ß-galactosides and the other subsites A, B and E are poorly characterized [107]. The CRD binds carbohydrate ligands, plays a role in C-type self-interactions [108] and is usually responsible for interactions occurring in the extracellular milieu [8,13]. The CRD is also known to interact with protein ligands such as β-catenin [38]. Galectin-3-functions are also modulated by the NT through various mechanisms including phosphorylation and self-interactions involving the NT region, e.g., NT-NT interactions and NT-CRD interactions [106,109]. Although the amino-terminal interacts with many ligands, it displays no carbohydrate-binding activity. The H-domain is the site of action of matrix metalloproteinases (MMPs) such as MMP 9 and MMP 2, and other proteases resulting in galectin-3 cleavage [110].
The classical ß-galactoside-binding region of galectin-3 CRD, called the canonical binding site or S-face, binds saccharides such as lactose. The other frequently described site is the non-canonical sugar binding site, also called the F-face [111]. The non-canonical site is reported to bind “sugars” with a larger carbohydrate foot-print such as MCPs and galactomannans (GMs) (Figure 4). These two different carbohydrate binding sites are not mutually “ligand” exclusive: this implies that binding of a ligand to the canonical site does not exclude the binding of a ligand to the non-canonical F site and vice versa [108,112]. However, data suggest that ligand-binding to the canonical site weakens the affinity of a ligand to the non-canonical site and conversely, binding of a ligand to the non-canonical site weakens ligand affinity to the canonical-site [111].
Figure 4
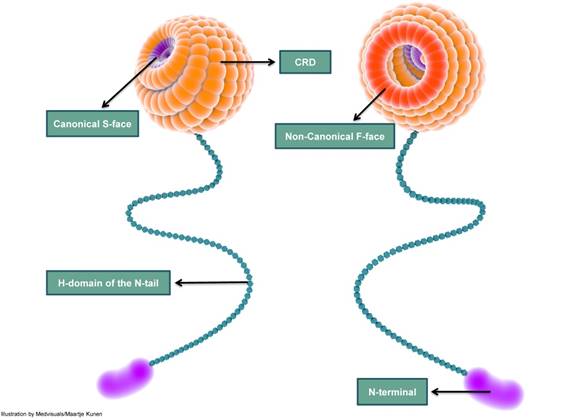
A simplified depiction of galectin-3 structure indicating the carbohydrate recognition domain (CRD), H-domain and the amino-terminal (N-terminal). The CRD is globular and consists of several carbohydrate binding-grooves. The most frequently described carbohydrate-binding sites are the canonical S-face and the non-canonical F-face. S-face binds β-galactosides such as lactose, while larger carbohydrates such as MCPs and GMs are reported to bind to the F-face. The CRD continues as a long and slender tail which ends in the N-terminal; the N-terminal does not exhibit carbohydrate-binding activity. CRD: carbohydrate recognition domain; N-terminal: amino terminal; MCP: modified citrus pectin; GM: galactomannan
Galectin-3 Self-interactions: Role in Activation
Galectin-3 self-interactions can be classified into intra-molecular interactions involving NT-CRD interactions and intermolecular interactions involving NT-NT and CRD-CRD interactions, and are usually modulated by various ligands [108,113]. Intra-molecular interactions lead to a relatively biologically inert galectin-3 molecule and intermolecular interactions result in bio-activation of galectin-3 by the formation of multimers.
NT-CRD interactions
Recent studies provide experimental evidence on intra-molecular interactions of galectin-3 resulting in a “closed conformer” and an “open conformer”. In the closed conformer, intra-molecular interactions occur between NT and F-face of the galectin-3 CRD resulting in stabilization of the molecule, rendering it relatively inert [114]: the NT effectively shields the CRD F-face in the closed conformation and could potentially block various interactions occurring in the non-canonical carbohydrate binding site. The canonical site is, however, open for interaction with classic ß-galactoside ligands such as lactose [115]. The liberation of NT from the CRD results in “opening-up” of (CRD) F-face to various binding partners. This “open conformer” also appears to be a prerequisite for intermolecular associations of galectin-3 resulting in dimerization and oligomerization (Figure 5).
High resolution nuclear magnetic resonance (NMR) studies showed that there are significant intra-molecular interactions between N-terminal domain and CRD. This was based on reduction in movement of NT and shielding of nuclei in intact hamster galectin-3 molecule [106]. A previous modelling study done by Barboni et al. proposed two different models for NT-CRD intra-molecular interactions [116]. Recently, Halimi et al. also demonstrated the interactions between NT and CRD of human galectin-3 using NMR [109]. Further evidence for NT-CRD association was obtained from the study conducted by Berbis et al: they worked on peptides derived from human galectin-3 N-terminal and their interactions with galectin-3 CRD, and elegantly demonstrated the role of N-terminal phosphorylation in NT-CRD interactions. Phosphorylation of N-terminal peptides elicited resonance shifts in that part of the lectin that was opposite to the canonical ß-galactoside binding site, providing preliminary proof that NT interacts with the non-canonical CRD binding site (NT-CRD F-face interaction). It was also demonstrated that the canonical ß-galactoside binding site was left open for interaction with sugars such as lactose [115].
NT-CRD interactions could have functional consequences for galectin-3 CRD interactions. Ochieng et al. observed that cleaved galectin-3 CRD terminal binds laminin more tightly [117], suggesting a regulatory role of the N-terminal in CRD-glycoconjugate interactions. However, it is still to be elucidated if it is the open N-tail (open-conformer) that is responsible for this regulatory role or if it is the NT-CRD F-face interaction (closed-conformer) that contributes to such an effect.
NT-NT interactions
Early evidence for NT-NT interactions was obtained from experiments utilizing monoclonal antibodies to target the amino region of the intact galectin-3 molecule: binding of antibodies to NT modulated the lectin activities of galectin-3. Certain antibodies such as MAb B2C10 inhibited galectin-3-IgE interaction, galectin-3-induced hemagglutination activity and also galectin-3-mediated superoxide production by human neutrophils, while other antibodies such as MAb A3A12 potentiated these activities. Facilitation or inhibition of dimerization (oligomerization) by the NT or direct self-association of galectin-3 via the amino terminal was held responsible for such effects [118].
FIGURE 5
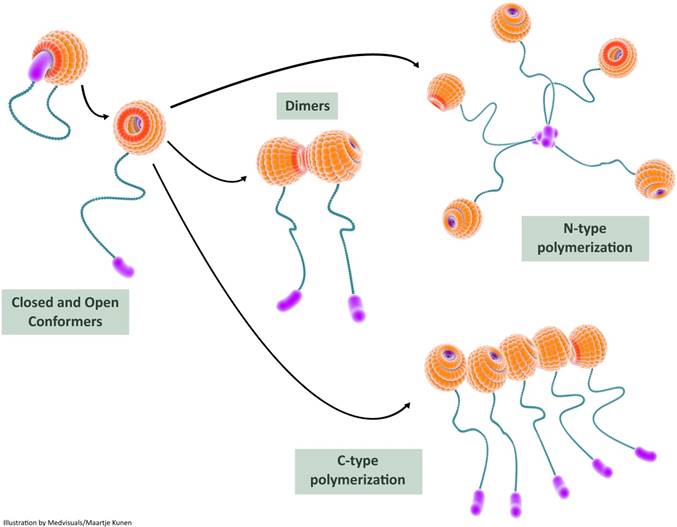
In this illustration, we depict the role of self-interactions in galectin-3 bioactivation. Intramolecular interactions between the carbohydrate recognition domain (CRD) and the N-terminal render the galectin-3 molecule relatively inert in the closed conformer state; the galectin-3 molecule can still bind S-face ligands such as lactose in this state. Release of the N-terminal from the F-face results in the open conformer which is biologically more active. The open conformer can bind to various ligands (both S-face ligands and F-face ligands) and can also undergo dimerization or oligomerization. Two types of intermolecular interactions, N-terminal interactions and CRD-CRD interactions, are usually observed during multimerization and this results in increased biological activity of galectin-3. CRD: carbohydrate recognition domain
Electron microscopy of NT fragments showed that their structure was fibrillar. Two types of N-terminal interactions were observed: side-side associations with “thickening” of fibrils suggesting oligomerization, and head-to-tail interactions resulting in considerable variation in fibril length [106]. Direct analysis of galectin-3 self-association using a solid-phase radioligand binding assay revealed that homophilic interactions of galectin-3 are mediated by the CRD in addition to the amino terminal. Moreover, these homophilic interactions could be potentiated by asialofetuin (ASF), which is a multivalent glycoprotein, and inhibited by lactose [119].
The existence of galectin-3 as a pentamer formed by N-terminal association has been described in the seminal paper by Ahmad et al. They also reported that galectin-3 shuttles rapidly between the monomeric and pentameric forms (equilibrium state) and precipitates as a pentamer with a series of divalent pentasaccharides with terminal LacNac residues [113]. A monomer-pentamer model thus emerged and several articles have adopted the N-terminal pentameric form as the classical (and only) form of oligomerization. Further experimental evidence for NT-NT oligomerization in the biological setting was accumulated by Fermino et al. Fluorescein isothiocyanate (FITC)-labelled full length galectin-3, unlike truncated galectin-3 (Gal-3C), exhibited a non-saturable binding to neutrophils and enhanced neutrophil activation, indicating that galectin-3 oligomerization induced by its interaction with lipopolysaccharide (LPS) was mediated by N-terminal during neutrophil activation [120].
Evidence for galectin-3 self-association utilizing NT-NT interactions induced by the CRD-ligand LNnT (lacto-N-neoTetraose) was accrued by Halimi and colleagues [109]. Pentamers and oligomers were observed using dynamic light scattering (DLS), and LNnT induced such effects only in full length protein, but not in recombinant CRD: LNnT, a neo-glycan, associates with galectin-3-CRD and probably releases the N-terminal domain from the CRD-F face, facilitating the formation of the “open” from of galectin-3. The free N-terminals in the “open conformer” could then interact with each other and form multimers through NT-NT interactions.
Although in many of the above-mentioned experiments, NT self-interactions required (CRD) ligands, a very recent NMR-based study evaluating the intrinsic propensity of (human) galectin-3 to self-associate reported that NT-NT interactions could also occur in a ligand-independent, concentration-dependent manner, through fuzzy interactions [121]. An earlier NMR study performed by Ippel et al. on (human) galectin-3 did not observe such NT-NT interactions, but concluded that intermolecular interactions occurred between F-faces of CRD, and NT facilitated such interactions [114].
CRD-CRD interactions
It has been generally accepted that NT is responsible for galectin-3 oligomerization, and ligand-binding to CRD could potentiate this effect. However, several studies including a recent study by Lepur et al. indicate that direct CRD-CRD interactions can also lead to galectin-3 oligomerization.
Yang et al. demonstrated, for the first time, self-association of galectin-3 utilizing CRD in the absence of saccharide ligands [122]. Further evidence for CRD-CRD interactions was accrued from the study conducted by Birdsall et al. in which intact hamster galectin-3 and also the CRD fragments were visualized in monomeric, dimeric and trimeric forms using electron microscopy [106]. A recent study by Lepur et al. using fluorescence anisotropy assay demonstrates the role of ASF (ligand / nucleating agent) in inducing galectin-3 multimerization through its CRD, and this has been named as C-type association. Efficient C-type oligomerization was not observed in solution previously and this would be the first study to describe such an effect. A monovalent or divalent LacNAc containing glycan was able to induce this C-type association, emphasizing that for efficient C-type self-association, a step of initiation or nucleation is necessary [108]. Precipitation also occurred with a divalent LacNAc containing ligand at very high concentrations (>50 µM) and this could be due to N-type association.
Thus, it appears that galectin-3 self-aggregation occurs either due to NT-NT interactions, [106,113] CRD-CRD interactions [108,109] or a mixture of both depending on galectin-3 concentration and availability of ligands and nucleating agents.
Galectin-3 Lattice: A Higher Level of Activation
Although galectin-3 can influence a variety of biological processes through self-interactions, a higher level of biological functionality is achieved through the formation of three dimensional frameworks consisting of galectin-3 in its different forms and several types of saccharide ligands [68]. This multi-dimensional organization, together with other components of the cell surface, is usually referred to as the cell surface “galectin-glycoprotein lattice” [123] (Figure 6). As self-interaction(s) of galectin-3 and also surface expression of glycosylated proteins and lipids keep changing continuously, this lattice is envisioned to be a highly dynamic structure similar to the cell membrane, and it has been described as an additional layer of membrane organization on extracellular surfaces of cell membranes [74]. The lattice formed by galectin-3 is also perceived to be stable, and the stability of these lattices at the surface of endothelial cells was visually demonstrated by Nieminen and colleagues while studying galectin-3 oligomerization; stability of galectin-3 lattice was further tested using fluorescence recovery after photobleaching (FRAP) technique [123].
Galectin-3 self-association and its interaction with various surface glycoproteins and glycolipids are important in the formation and maintenance of the lattice structure, and also in the regulation and distribution of glycoproteins at the cell-surface [74]. For cross-linking by galectin-3, glycoproteins and glycolipids should have appropriate glycosylation patterns. For instance, if the end terminal is sialic acid, the formation of a cross-linked lattice is disrupted [68]. Affinity of N-glycans for galectins in this lattice also increases with branching and poly-N-acetyllactosamine extensions. Uridine diphosphate-N-acetylglucosamine (UDP-GlcNac) is essential for these processes, and as glucose, glutamine and acetyl-CoA are necessary for the biosynthesis of UDP-GlcNAc, and N-glycan branching is dependent on UDP-GlcNac, such factors could potentially regulate glycoprotein retention in the lattice [124]. Receptors with five or more glycosylated sites are largely associated within the lattice [125].
Galectin lattice acts as a physical barrier and a biological sensor by regulating the entry of various pathogens by constantly surveying the extracellular milieu. It also associates with various receptor kinases (which are usually glycosylated) to bring about changes in intracellular biological processes [125]. Moreover, galectin lattice could potentially regulate metabolic homeostasis by changing the number and activity of cell surface receptors such as glucagon receptors and solute transporters [124]. It is also speculated to critically regulate T-cell receptor sensitivity, which can have a crucial role in the development of autoimmune diseases and cancer [67,69]. Galectin-3 lattice could also have a role in TGF-ß receptor entrapment, especially those receptors with N-linked glycosylation patterns [126] and this could possibly amplify TGF-ß mediated profibrotic signalling [125]. Thus galectin-3 through its self-associating, lattice-forming behaviour could influence innumerable biological processes, many of which still remain to be elucidated.
FIGURE 6
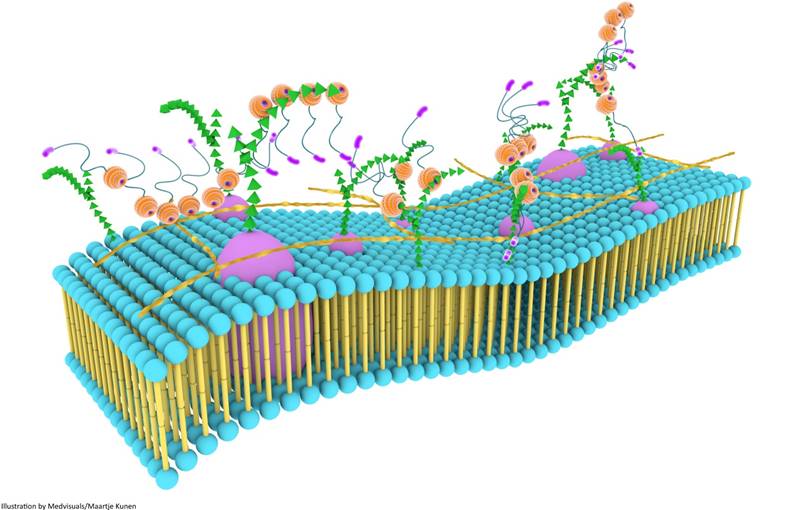
Galectin-3 lattices are focal, three-dimensional frameworks consisting of galectin-3 in its different forms and multimerization states, and is envisioned to be an additional layer of membrane organization. Galectin-3 interacts with various binding partners, usually carbohydrate molecules that project from glycoproteins and glycolipids, regulating several important biological processes. Galectin-3 lattices are a part of the larger “lectin-saccharide” lattices.
Galectin-3 Inhibition
As the carbohydrate recognition region of (extracellular) galectin-3 is responsible for several pathological effects orchestrated by this protein, pharmacological inhibition of galectin-3 has almost exclusively targeted the CRD for inhibiting (extracellular) activities of this protein. The CRD can be inhibited using carbohydrates that compete for the binding site or allosterically modulate it so as to render the CRD incapable of binding to ligands. Steric hindrance offered by high-molecular-weight compounds has also been exploited to confer additional galectin-3 inhibiting properties. Heparin-based inhibitors, truncated galectin-3 and peptide inhibitor G3-C12 have only been evaluated in cancer setting; however, we also discuss them briefly as they could evolve as promising candidates in the therapy of organ fibrosis and HF.
Carbohydrate-based compounds
Simple Sugars
Pharmacological inhibition of galectin-3 with LacNAc prevented LV dysfunction in failure-prone REN2 rats [24] and also had a protective effect against hypertensive nephropathy in REN2 rats [79]. Low-molecular-weight sugars such as lactose or LacNAc, however, cannot be used as “drugs” as they are rapidly absorbed and metabolized.
Galactomannans and Modified Citrus Pectin
GMs are galectin antagonists derived from plants. GM-CT-01, known by its trade name Davanat® is a GM (MW ~50kDa) with a half-life between 12 h and 18 h [128]. The safety profile of GM-CT-01 has been established, and half-life of Davanat® is much higher than that of LacNAc, making it more suitable for clinical use. Experimental evidence from Demotte et al. demonstrates that Davanat® is able to improve the activity of human tumour infiltrating lymphocytes, and disrupts galectin-glycoprotein lattices [127,128]; however, the exact mechanism of action remains unclear. In another study by Traber et al., two complex carbohydrate anti-galectin-3 drugs (GM-CT-01 and GR-MD-02) were used to treat nonalcoholic steatohepatitis (NASH) and fibrosis in a murine model. GM-CT-01 is a GM polysaccharide while GR-MD-02 is a galactoarabinorhamnogalacturonan polysaccharide polymer; GR-MD-02 performed better than GM-CT-01 in reducing hepatocellular damage, inflammation and fibrosis in the experimental NASH mouse model and both drugs reduced galectin-3 expression in macrophages [129].
Different types of MCPs (MW > 1000 kDa), e.g., GCS-100 and PectaSol-C®, are available commercially and several experiments performed in the past years successfully employ MCPs for targeting fibrosis. The galectin-3 inhibitory effects of MCPs have been investigated in various cellular and animal models: these include inhibition of galectin-3-mediated hemagglutination, reduction of cardiac inflammation, attenuation of organ fibrosis and reduction of atherosclerosis in apolipoprotein E-deficient mice [70,89,101,130-134]. Furthermore, MCPs have an acceptable human safety profile [134] and have been evaluated as cancer therapeutic agents [127].
Although not clearly established, it was assumed that inhibition of galectin-3 at the classical ß-galactoside binding site was responsible for anti-galectin-3 effects elicited by GMs and MCPs. However, a recent study by Miller et al. suggests that MCPs and polysaccharides with a larger carbohydrate foot print such as GMs bind to the non-canonical site in the CRD (F-face) instead of the canonical S-face [111]. This is also in line with the study conducted by Stegmayr et al. who demonstrated that the biological inhibitory effects of several different MCPs and GMs could not be due to inhibition of the canonical carbohydrate binding site [112]. If the established biological inhibitory effects of pectins and GMs can be conclusively attributed to the inhibition of the non-canonical binding site as suggested by Miller and colleagues, it would emerge as an attractive target for designing novel galectin-3 inhibitors.
Several studies claim that GMs and MCPs inhibit hemagglutination mediated by galectin-3 [70,132,133]; however, Stegmayr and colleagues demonstrated for the first time that Davanat® and MCPs including Pectasol-C® did not block galectin-3-induced hemagglutination [112]. Compounds inhibiting hemagglutination could block galectin-3 activation by disrupting the lattice-forming behaviour of this protein, and hence it is necessary to carry out further studies to precisely understand the mode of action of antifibrotic MCPs and GMs. It should also be noted that in many in vivo studies, the specificity of MCPs was not established and the possibility that their inhibitory activity was due to effects on targets other than galectin-3 also needs to be addressed.
Thiodigalactosides
Recently, thiodigalactoside derivatives targeting novel CRD sites other than the canonical binding site have emerged. TD-139 is a thiodigalactoside analogue that has been approved by the USFDA for the treatment of idiopathic pulmonary fibrosis as an inhaled powder, and is speculated to antagonize galectin-3 by binding to subsites B and E [107]. It is a small molecule (C28H30F2N6O8S) with a molecular weight of approximately 648 g/mol and can bind with high affinity to both galectin-3 and galectin-1. Although the mechanism of action is unclear, it is possible that this molecule allosterically modulates galectin-3 CRD. Some research groups also demonstrate that thiodigalactosides could be preferentially “tuned” to create more specific galectin-3 inhibitors [135].
Heparin-based inhibitors
They are a relatively new and attractive group of galectin-3 inhibitors that are sulphated or acetylated derivatives of heparin. In vitro results demonstrated that they were non-cytotoxic and galectin-3 selective (i.e., did not inhibit galectin-1, -4 and -8), and in in vivo experiments with nude mice these compounds significantly reduced galectin-3-mediated lung metastasis of human melanoma and colon cancer cells. Furthermore, these compounds exhibited no detectable anticoagulant activity, and appear to be promising therapeutic agents [136]. However, they have been tested only in in vivo models of metastasis and future studies need to be conducted to evaluate their potential as antifibrotic agents.
Neoglycoconjugates
Galectin-3 binds to branched-chain sugars with increased avidity and large lactose functionalized dendrimers provide an “excess of ligands” for galectin-3 binding. Michel and colleagues studied the effects of different types of lactose-functionalized dendrimers on cancer-cell aggregation and found that smaller dendrimers inhibited homotypic cellular aggregation, probably through competitive inhibition, while larger dendrimers with several lactose end groups enhanced aggregation by providing multiple galectin-3 binding sites [137]. Other chemically engineered glycoproteins (neoglycoproteins) have also been developed recently and have the potential to be used as novel therapeutic molecules against fibrosis by effectively targeting galectin-3. They not only serve as high affinity ligands but can also be modulated to achieve selectivity to galectin-3 over other galectins. Their use in the clinical setting is yet to be evaluated [138].
Peptide-based Compounds
NH2 terminally truncated galectin-3 (Gal-3C) has been evaluated in the therapy of galectin-3 related-tumours and it appears to be a promising agent with a low-toxicity profile [139-141]. However, there are no studies evaluating its potential in treating other galectin-3 related pathologies.
Recently, Sun and colleagues utilized galectin-3 binding peptide, G3-C12 to inhibit intracellular galectin-3 in cancer cells. As G3-C12 has a high selectivity to galectin-3 over other galectins, it functions as a selective galectin-3 targeting ligand. When this peptide is conjugated to a drug using a versatile drug carrier such as N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer, the resulting G3-C12-HPMA-drug conjugate could easily internalize into galectin-3 overexpressing cells [142]. This galectin-3 selective intracellular delivery concept could possibly also find utility in other galectin-3-related disease scenarios such as organ fibrosis and HF.
Conclusion
Activation of monomeric galectin-3 increases the spectrum of biological activity of this pleiotropic protein in various physiological and patho-physiological processes. It is however essential to understand that there could be more components to galectin-3 activation. It remains unclear how galectin-3 function might be modulated by other galectin members, signalling molecules such as syndecans and other biologically active molecules. Current understanding of various binding partners of galectin-3 is also incomplete. Further characterization and visualization of the galectin-3 lattice utilizing advanced (optical) techniques is necessary to understand the exact mechanisms by which this regulatory protein influences various (extra)cellular processes.
Although galectin-3 has been implicated in several debilitating disorders, only a few galectin-3 inhibitors have been developed that are of clinical relevance. There is an urgent need to develop galectin-3 inhibitors that have a high oral bioavailability and a low toxicity profile to combat progressive tissue fibrosis and galectin-3-related HF. While treating such disorders, it is necessary to pay attention to the “window of opportunity” as overexpression of galectin-3 could be protective in certain scenarios, especially during initial stages of wound healing after injury, and galectin-3 inhibition during this phase may result in loss of tissue integrity. Finally, galectin-3 is a pleiotropic protein with several physiological functions, and galectin-3 blockade could also inhibit such functions resulting in off-target side-effects. Thus, the timing of treatment, eligibility of patients and their genetics must be carefully considered before initiating therapies with galectin-3 inhibitors.
Abbreviations
ECM: extracellular matrix; Bcl-2: B-cell lymphoma-2; HF: heart failure; DM: diabetes mellitus; CRD: carbohydrate-recognition domain; NT: amino terminal; Mac-2: macrophage-2; eBP: IgE-binding protein; Ig: immunoglobulin; CBP: carbohydrate-binding protein; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; Galig: galectin-3 internal gene; MCL-1: myeloid cell leukaemia sequence 1 protein; KLF-3: Krüppel-like factor 3; RUNX-2: runt-related transcription factor-2; HSC: hepatic stellate cells; α-SMA: alpha-smooth muscle actin; CCl4: carbon tetrachloride; KO: knockout; IL: interleukin; CD: cluster of differentiation; vWF: von Willebrand factor; TGF-β: transforming growth factor beta; UUO: unilateral ureteral obstruction; TD: thiodigalactoside; LacNAc: N-acetyl-D-lactosamine; ACCF/AHA: American College of Cardiololgy/American Heart Association; TAC: transverse aortic constriction; LV: left ventricular; EF: ejection fraction; MCP: modified citrus pectin; HFpEF: heart failure with preserved ejection fraction; PKC: protein kinase C; PDBu: phorbol dibutyrate; AGE: advanced glycation end product; MMP: metalloprotease; GM: galactomannan; NMR: nuclear magnetic resonance; MAb: monoclonal antibody; ASF: asialofetuin; FITC: fluorescein isothiocyanate; LPS: lipopolysaccharide; LNnT: lacto-N-neoTetraose; acetyl-CoA: acetyl coenzyme A; DLS: dynamic light scattering; FRAP: fluorescence recovery after photobleaching; UDP-GlcNac: uridine diphosphate-N-acetylglucosamine; NASH: nonalcoholic steatohepatitis; Gal-3C: truncated galectin-3; HPMA: N-(2-hydroxypropyl)methacrylamide.
Acknowledgements
Dr. Suthahar is supported by a grant from the University Medical Center Groningen. This work was supported by the Netherlands Heart Foundation (CVON-DOSIS, grant 2014-40, to Dr. de Boer) and the Innovational Research Incentives Scheme program of the Netherlands Organization for Scientific Research (NWO VIDI, grant 917.13.350, to Dr. de Boer).
Conflict of Interest
Dr. de Boer is employed by the UMC Groningen, that received research funding and consultancy fees from AstraZeneca, Bristol-Myers Squibb, Trevena, Roche, Thermo Fisher and Novartis. Dr. de Boer received speaker honoraria from Novartis. Dr. de Boer is a scientific founder of, consultant to, and has stock options of G3 Pharmaceuticals, a company that aims to develop galectin-3 inhibitors. Dr. Ho is a receiver of research supplies (modified citrus pectin) from Econugenics.
References
1. Suseelan KN, Bhatia AR, Mitra R. Purification and characterization of two major lectins from Vigna mungo (blackgram). J Biosci. 1997;22:439-55
2. Drickamer K, Taylor ME. Biology of animal lectins. Annu Rev Cell Biol. 1993;9:237-64
3. Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T. et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell. 1994;76:597-8
4. Liu F-T, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5:29-41
5. Kasai K, Hirabayashi J. Galectins: a family of animal lectins that decipher glycocodes. J Biochem. 1996;119:1-8
6. Yang RY, Hsu DK, Liu FT. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc Natl Acad Sci U S A. 1996;93:6737-42
7. Ho MK, Springer TA. Mac-2, a novel 32,000 Mr mouse macrophage subpopulation-specific antigen defined by monoclonal antibodies. J Immunol. 1982;128:1221-8
8. Hsu DK, Zuberi RI, Liu FT. Biochemical and biophysical characterization of human recombinant IgE-binding protein, an S-type animal lectin. J Biol Chem. 1992;267:14167-74
9. Massa SM, Cooper DNW, Leffler H, Barondes SH. L-29, an endogenous lectin, binds to glycoconjugate ligands with positive cooperativity. Biochemistry. 1993;32:260-7
10. Leffler H, Masiarz FR, Barondes SH. Soluble lactose-binding vertebrate lectins: a growing family. Biochemistry. 1989;28:9222-9
11. Mehul B, Bawumia S, Martin SR, Hughes RC. Structure of baby hamster kidney carbohydrate-binding protein CBP30, an S-type animal lectin. J Biol Chem. 1994;269:18250-8
12. Cherayil BJ, Weiner SJ, Pillai S. The Mac-2 antigen is a galactose-specific lectin that binds IgE. J Exp Med. 1989;170:1959-72
13. Dumic J, Dabelic S, Flögel M. Galectin-3: an open-ended story. Biochim Biophys Acta. 2006;1760:616-35
14. Raimond J, Rouleux F, Monsigny M, Legrand A. The second intron of the human galectin-3 gene has a strong promoter activity down-regulated by p53. FEBS Lett. 1995;363:165-9
15. Duneau M, Boyer-Guittaut M, Gonzalez P, Charpentier S, Normand T, Dubois M. et al. Galig, a novel cell death gene that encodes a mitochondrial protein promoting cytochrome c release. Exp Cell Res. 2005;302:194-205
16. Knights AJ, Yik JJ, Mat Jusoh H, Norton LJ, Funnell APW, Pearson RCM. et al. Krüppel-like Factor 3 (KLF3/BKLF) Is Required for Widespread Repression of the Inflammatory Modulator Galectin-3 (Lgals3). J Biol Chem. 2016;291:16048-58
17. Stock M, Schäfer H, Stricker S, Gross G, Mundlos S, Otto F. Expression of galectin-3 in skeletal tissues is controlled by Runx2. J Biol Chem. 2003;278:17360-7
18. Sundblad V, Croci DO, Rabinovich GA. Regulated expression of galectin-3, a multifunctional glycan-binding protein, in haematopoietic and non-haematopoietic tissues. Histol Histopathol. 2011;26:247-65
19. Larsen L, Chen H-Y, Saegusa J, Liu F-T. Galectin-3 and the skin. J Dermatol Sci. 2011;64:85-91
20. Kim H, Lee J, Hyun JW, Park JW, Joo H, Shin T. Expression and immunohistochemical localization of galectin-3 in various mouse tissues. Cell Biol Int. 2007;31:655-62
21. Kim K, Mayer EP, Nachtigal M. Galectin-3 expression in macrophages is signaled by Ras/MAP kinase pathway and up-regulated by modified lipoproteins. Biochim Biophys Acta. 2003;1641:13-23
22. Hsu DK, Hammes SR, Kuwabara I, Greene WC, Liu FT. Human T lymphotropic virus-I infection of human T lymphocytes induces expression of the beta-galactoside-binding lectin, galectin-3. Am J Pathol. 1996;148:1661-70
23. Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JPM, Schroen B. et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. 2004;110:3121-8
24. Yu L, Ruifrok WPT, Meissner M, Bos EM, van Goor H, Sanjabi B. et al. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ Heart Fail. 2013;6:107-17
25. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP. et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci U S A. 2006;103:5060-5
26. Henderson NC, Mackinnon AC, Farnworth SL, Kipari T, Haslett C, Iredale JP. et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol. 2008;172:288-98
27. Kolatsi-Joannou M, Price KL, Winyard PJ, Long DA. Modified citrus pectin reduces galectin-3 expression and disease severity in experimental acute kidney injury. PLoS One. 2011;6:e18683
28. Menon RP, Hughes RC. Determinants in the N-terminal domains of galectin-3 for secretion by a novel pathway circumventing the endoplasmic reticulum-Golgi complex. Eur J Biochem. 1999;264:569-76
29. Sato S, Burdett I, Hughes RC. Secretion of the baby hamster kidney 30-kDa galactose-binding lectin from polarized and nonpolarized cells: a pathway independent of the endoplasmic reticulum-Golgi complex. Exp Cell Res. 1993;207:8-18
30. Mehul B, Hughes RC. Plasma membrane targetting, vesicular budding and release of galectin 3 from the cytoplasm of mammalian cells during secretion. J Cell Sci. 1997;110:1169-78
31. Menon S, Kang C-M, Beningo KA. Galectin-3 secretion and tyrosine phosphorylation is dependent on the calpain small subunit, Calpain 4. Biochem Biophys Res Commun. 2011;410:91-6
32. Hamann KK, Cowles EA, Wang JL, Anderson RL. Expression of carbohydrate binding protein 35 in human fibroblasts: variations in the levels of mRNA, protein, and isoelectric species as a function of replicative competence. Exp Cell Res. 1991;196:82-91
33. Funasaka T, Raz A, Nangia-Makker P. Nuclear transport of galectin-3 and its therapeutic implications. Semin Cancer Biol. 2014;27:30-8
34. Funasaka T, Raz A, Nangia-Makker P. Galectin-3 in angiogenesis and metastasis. Glycobiology. 2014;24:886-91
35. Takenaka Y, Fukumori T, Yoshii T, Oka N, Inohara H, Kim H-RC. et al. Nuclear export of phosphorylated galectin-3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol Cell Biol. 2004;24:4395-406
36. Yu F, Finley RL, Raz A, Kim H-RC. Galectin-3 translocates to the perinuclear membranes and inhibits cytochrome c release from the mitochondria. A role for synexin in galectin-3 translocation. J Biol Chem. 2002;277:15819-27
37. Park JW, Voss PG, Grabski S, Wang JL, Patterson RJ. Association of galectin-1 and galectin-3 with Gemin4 in complexes containing the SMN protein. Nucleic Acids Res. 2001;29:3595-602
38. Shimura T, Takenaka Y, Tsutsumi S, Hogan V, Kikuchi A, Raz A. Galectin-3, a novel binding partner of beta-catenin. Cancer Res. 2004;64:6363-7
39. Yang H, Lei C, Zhang W. Expression of galectin-3 in mouse endometrium and its effect during embryo implantation. Reprod Biomed Online. 2012;24:116-22
40. Koch A, Poirier F, Jacob R, Delacour D. Galectin-3, a novel centrosome-associated protein, required for epithelial morphogenesis. Mol Biol Cell. 2010;21:219-31
41. Bullock SL, Johnson TM, Bao Q, Hughes RC, Winyard PJ, Woolf a S. Galectin-3 modulates ureteric bud branching in organ culture of the developing mouse kidney. J Am Soc Nephrol. 2001;12:515-23
42. Fowlis D, Colnot C, Ripoche M ???A, Poirier F. Galectin-3 is expressed in the notochord, developing bones, and skin of the postimplantation mouse embryo. Dev Dyn. 1995;203:241-51
43. Li Y-J, Kukita A, Teramachi J, Nagata K, Wu Z, Akamine A. et al. A possible suppressive role of galectin-3 in upregulated osteoclastogenesis accompanying adjuvant-induced arthritis in rats. Lab Invest. 2009;89:26-37
44. Colnot C, Sidhu SS, Balmain N, Poirier F. Uncoupling of chondrocyte death and vascular invasion in mouse galectin 3 null mutant bones. Dev Biol. 2001;229:203-14
45. Boileau C, Poirier F, Pelletier J-P, Guévremont M, Duval N, Martel-Pelletier J. et al. Intracellular localisation of galectin-3 has a protective role in chondrocyte survival. Ann Rheum Dis. 2008;67:175-81
46. MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H. et al. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008;180:2650-8
47. Akimoto Y, Ikehara S, Yamaguchi T, Kim J, Kawakami H, Shimizu N. et al. Galectin expression in healing wounded skin treated with low-temperature plasma: Comparison with treatment by electronical coagulation. Arch Biochem Biophys. 2016;605:86-94
48. González GE, Cassaglia P, Noli Truant S, Fernández MM, Wilensky L, Volberg V. et al. Galectin-3 is essential for early wound healing and ventricular remodeling after myocardial infarction in mice. Int J Cardiol. 2014;176:1423-5
49. Liu F-T, Hsu DK. The role of galectin-3 in promotion of the inflammatory response. Drug News Perspect. 2007;20:455-60
50. Wright RD, Souza PR, Flak MB, Thedchanamoorthy P, Norling L V, Cooper D. Galectin-3-null mice display defective neutrophil clearance during acute inflammation. J Leukoc Biol. 2017;101:717-26
51. Karlsson A, Christenson K, Matlak M, Björstad A, Brown KL, Telemo E. et al. Galectin-3 functions as an opsonin and enhances the macrophage clearance of apoptotic neutrophils. Glycobiology. 2009;19:16-20
52. Sano H, Hsu DK, Apgar JR, Yu L, Sharma BB, Kuwabara I. et al. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest. 2003;112:389-97
53. Saksida T, Nikolic I, Vujicic M, Nilsson UJ, Leffler H, Lukic ML. et al. Galectin-3 deficiency protects pancreatic islet cells from cytokine-triggered apoptosis in vitro. J Cell Physiol. 2013;228:1568-76
54. Rosenberg I, Cherayil BJ, Isselbacher KJ, Pillai S. Mac-2-binding glycoproteins. Putative ligands for a cytosolic beta-galactoside lectin. J Biol Chem. 1991;266:18731-6
55. Sato S, Hughes RC. Binding specificity of a baby hamster kidney lectin for H type I and II chains, polylactosamine glycans, and appropriately glycosylated forms of laminin and fibronectin. J Biol Chem. 1992;267:6983-90
56. Ochieng J, Warfield P. Galectin-3 binding potentials of mouse tumor EHS and human placental laminins. Biochem Biophys Res Commun. 1995;217:402-6
57. Ochieng J, Leite-Browning ML, Warfield P. Regulation of cellular adhesion to extracellular matrix proteins by galectin-3. Biochem Biophys Res Commun. 1998;246:788-91
58. Dalton P, Christian HC, Redman CWG, Sargent IL, Boyd CAR. Membrane trafficking of CD98 and its ligand galectin 3 in BeWo cells-implication for placental cell fusion. FEBS J. 2007;274:2715-27
59. Van den Brûle FA, Fernandez PL, Buicu C, Liu FT, Jackers P, Lambotte R. et al. Differential expression of galectin-1 and galectin-3 during first trimester human embryogenesis. Dev Dyn. 1997;209:399-405
60. Ortega N, Behonick DJ, Colnot C, Cooper DNW, Werb Z. Galectin-3 is a downstream regulator of matrix metalloproteinase-9 function during endochondral bone formation. Mol Biol Cell. 2005;16:3028-39
61. Saint-Lu N, Oortwijn BD, Pegon JN, Odouard S, Christophe OD, de Groot PG. et al. Identification of galectin-1 and galectin-3 as novel partners for von Willebrand factor. Arterioscler Thromb Vasc Biol. 2012;32:894-901
62. O'Sullivan JM, Jenkins PV, Rawley O, Gegenbauer K, Chion A, Lavin M. et al. Galectin-1 and Galectin-3 Constitute Novel-Binding Partners for Factor VIII. Arterioscler Thromb Vasc Biol. 2016;36:855-63
63. Vijayakumar S, Peng H, Schwartz GJ. Galectin-3 mediates oligomerization of secreted hensin using its carbohydrate-recognition domain. Am J Physiol Renal Physiol. 2013;305:F90-9
64. Mina-Osorio P, Soto-Cruz I, Ortega E. A role for galectin-3 in CD13-mediated homotypic aggregation of monocytes. Biochem Biophys Res Commun. 2007;353:605-10
65. Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T. et al. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165:2156-64
66. Gabius H-J, Kayser K. Introduction to glycopathology: the concept, the tools and the perspectives. Diagn Pathol. 2014;9:4
67. Garner OB, Baum LG. Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem Soc Trans. 2008;36:1472-7
68. Brewer CF, Miceli MC, Baum LG. Clusters, bundles, arrays and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr Opin Struct Biol. 2002;12:616-23
69. Hsu DK, Chen H-Y, Liu F-T. Galectin-3 regulates T-cell functions. Immunol Rev. 2009;230:114-27
70. Sathisha U V, Jayaram S, Harish Nayaka MA, Dharmesh SM. Inhibition of galectin-3 mediated cellular interactions by pectic polysaccharides from dietary sources. Glycoconj J. 2007;24:497-507
71. Martínez-Martínez E, Calvier L, Fernández-Celis A, Rousseau E, Jurado-López R, Rossoni L V. et al. Galectin-3 blockade inhibits cardiac inflammation and fibrosis in experimental hyperaldosteronism and hypertension. Hypertension. 2015;66:767-75
72. Chen W-S, Cao Z, Leffler H, Nilsson UJ, Panjwani N. Galectin-3 Inhibition by a Small-Molecule Inhibitor Reduces Both Pathological Corneal Neovascularization and Fibrosis. Invest Ophthalmol Vis Sci. 2017;58:9-20
73. Nangia-Makker P, Honjo Y, Sarvis R, Akahani S, Hogan V, Pienta KJ. et al. Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am J Pathol. 2000;156:899-909
74. Nabi IR, Shankar J, Dennis JW. The galectin lattice at a glance. J Cell Sci. 2015;128:2213-9
75. Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T. et al. Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med. 2012;185:537-46
76. Martínez-Martínez E, López-Ándres N, Jurado-López R, Rousseau E, Bartolomé MV, Fernández-Celis A. et al. Galectin-3 Participates in Cardiovascular Remodeling Associated With Obesity. Hypertension. 2015;66:961-9
77. Desmedt V, Desmedt S, Delanghe JR, Speeckaert R, Speeckaert MM. Galectin-3 in Renal Pathology: More Than Just an Innocent Bystander. Am J Nephrol. 2016;43:305-17
78. Dang Z, MacKinnon A, Marson LP, Sethi T. Tubular Atrophy and Interstitial Fibrosis After Renal Transplantation Is Dependent on Galectin-3. Transplantation. 2012;93:477-84
79. Frenay A-RS, Yu L, van der Velde a R, Vreeswijk-Baudoin I, López-Andrés N, van Goor H. et al. Pharmacological inhibition of galectin-3 protects against hypertensive nephropathy. Am J Physiol Renal Physiol. 2015;308:F500-9
80. de Boer RA, van Veldhuisen DJ, Gansevoort RT, Muller Kobold AC, van Gilst WH, Hillege HL. et al. The fibrosis marker galectin-3 and outcome in the general population. J Intern Med. 2012;272:55-64
81. Ho JE, Liu C, Lyass A, Courchesne P, Pencina MJ, Vasan RS. et al. Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol. 2012;60:1249-56
82. van der Velde AR, Gullestad L, Ueland T, Aukrust P, Guo Y, Adourian A. et al. Prognostic value of changes in galectin-3 levels over time in patients with heart failure: data from CORONA and COACH. Circ Heart Fail. 2013;6:219-26
83. Lok DJ, Lok SI, Bruggink-André de la Porte PW, Badings E, Lipsic E, van Wijngaarden J. et al. Galectin-3 is an independent marker for ventricular remodeling and mortality in patients with chronic heart failure. Clin Res Cardiol. 2013;102:103-10
84. de Boer RA, Edelmann F, Cohen-Solal A, Mamas MA, Maisel A, Pieske B. Galectin-3 in heart failure with preserved ejection fraction. Eur J Heart Fail. 2013;15:1095-101
85. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH. et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;62:1495-539
86. de Boer RA, van der Velde AR, Mueller C, van Veldhuisen DJ, Anker SD, Peacock WF. et al. Galectin-3: a modifiable risk factor in heart failure. Cardiovasc Drugs Ther. 2014;28:237-46
87. Liu Y-H, D'Ambrosio M, Liao T, Peng H, Rhaleb N-E, Sharma U. et al. N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiol Heart Circ Physiol. 2009;296:H404-12
88. González GE, Rhaleb N-E, D'Ambrosio MA, Nakagawa P, Liao T-D, Peterson EL. et al. Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol. 2016;311:H1287-96
89. Calvier L, Martinez-Martinez E, Miana M, Cachofeiro V, Rousseau E, Sádaba JR. et al. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart Fail. 2015;3:59-67
90. Wu C-K, Su M-Y, Lee J-K, Chiang F-T, Hwang J-J, Lin J-L. et al. Galectin-3 level and the severity of cardiac diastolic dysfunction using cellular and animal models and clinical indices. Sci Rep. 2015;5:17007
91. He J, Li X, Luo H, Li T, Zhao L, Qi Q. et al. Galectin-3 mediates the pulmonary arterial hypertension-induced right ventricular remodeling through interacting with NADPH oxidase 4. J Am Soc Hypertens. 2017;11:275-289.e2
92. Talaga ML, Fan N, Fueri AL, Brown RK, Bandyopadhyay P, Dam TK. Multitasking Human Lectin Galectin-3 Interacts with Sulfated Glycosaminoglycans and Chondroitin Sulfate Proteoglycans. Biochemistry. 2016;55:4541-51
93. Lunde IG, Herum KM, Carlson CC, Christensen G. Syndecans in heart fibrosis. Cell Tissue Res. 2016;365:539-52
94. Jia W, Kidoya H, Yamakawa D, Naito H, Takakura N. Galectin-3 accelerates M2 macrophage infiltration and angiogenesis in tumors. Am J Pathol. 2013;182:1821-31
95. Novak R, Dabelic S, Dumic J. Galectin-1 and galectin-3 expression profiles in classically and alternatively activated human macrophages. Biochim Biophys Acta. 2012;1820:1383-90
96. Rőszer T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm. 2015;2015:816460
97. Song X, Qian X, Shen M, Jiang R, Wagner MB, Ding G. et al. Protein kinase C promotes cardiac fibrosis and heart failure by modulating galectin-3 expression. Biochim Biophys Acta. 2015;1853:513-21
98. Papaspyridonos M, McNeill E, de Bono JP, Smith A, Burnand KG, Channon KM. et al. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler Thromb Vasc Biol. 2008;28:433-40
99. Tian L, Chen K, Cao J, Han Z, Gao L, Wang Y. et al. Galectin-3-induced oxidized low-density lipoprotein promotes the phenotypic transformation of vascular smooth muscle cells. Mol Med Rep. 2015;12:4995-5002
100. Henderson NC, Sethi T. The regulation of inflammation by galectin-3. Immunol Rev. 2009;230:160-71
101. MacKinnon AC, Liu X, Hadoke PW, Miller MR, Newby DE, Sethi T. Inhibition of galectin-3 reduces atherosclerosis in apolipoprotein E-deficient mice. Glycobiology. 2013;23:654-63
102. Nachtigal M, Ghaffar A, Mayer EP. Galectin-3 gene inactivation reduces atherosclerotic lesions and adventitial inflammation in ApoE-deficient mice. Am J Pathol. 2008;172:247-55
103. Pang J, Rhodes DH, Pini M, Akasheh RT, Castellanos KJ, Cabay RJ. et al. Increased adiposity, dysregulated glucose metabolism and systemic inflammation in Galectin-3 KO mice. PLoS One. 2013;8:e57915
104. Pejnovic NN, Pantic JM, Jovanovic IP, Radosavljevic GD, Milovanovic MZ, Nikolic IG. et al. Galectin-3 deficiency accelerates high-fat diet-induced obesity and amplifies inflammation in adipose tissue and pancreatic islets. Diabetes. 2013;62:1932-44
105. Li P, Liu S, Lu M, Bandyopadhyay G, Oh D, Imamura T. et al. Hematopoietic-Derived Galectin-3 Causes Cellular and Systemic Insulin Resistance. Cell. 2016;167:973-984.e12
106. Birdsall B, Feeney J, Burdett ID, Bawumia S, Barboni EA, Hughes RC. NMR solution studies of hamster galectin-3 and electron microscopic visualization of surface-adsorbed complexes: evidence for interactions between the N- and C-terminal domains. Biochemistry. 2001;40:4859-66
107. Hsieh TJ, Lin H-Y, Tu Z, Lin T-C, Wu S, Tseng Y. et al. Dual thio-digalactoside-binding modes of human galectins as the structural basis for the design of potent and selective inhibitors. Sci Rep. 2016;6:29457
108. Lepur A, Salomonsson E, Nilsson UJ, Leffler H. Ligand induced galectin-3 protein self-association. J Biol Chem. 2012;287:21751-6
109. Halimi H, Rigato A, Byrne D, Ferracci G, Sebban-Kreuzer C, ElAntak L. et al. Glycan dependence of Galectin-3 self-association properties. PLoS One. 2014;9:e111836
110. Monaco S, Sparano V, Gioia M, Sbardella D, Di Pierro D, Marini S. et al. Enzymatic processing of collagen IV by MMP-2 (gelatinase A) affects neutrophil migration and it is modulated by extracatalytic domains. Protein Sci. 2006;15:2805-15
111. Miller MC, Ippel H, Suylen D, Klyosov AA, Traber PG, Hackeng T. et al. Binding of polysaccharides to human galectin-3 at a noncanonical site in its carbohydrate recognition domain. Glycobiology. 2016;26:88-99
112. Stegmayr J, Lepur A, Kahl-Knutson B, Aguilar-Moncayo M, Klyosov AA, Field RA. et al. Low or No Inhibitory Potency of the Canonical Galectin Carbohydrate-binding Site by Pectins and Galactomannans. J Biol Chem. 2016;291:13318-34
113. Ahmad N, Gabius H-J, André S, Kaltner H, Sabesan S, Roy R. et al. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem. 2004;279:10841-7
114. Ippel H, Miller MC, Vértesy S, Zheng Y, Cañada FJ, Suylen D. et al. Intra- and intermolecular interactions of human galectin-3: assessment by full-assignment-based NMR. Glycobiology. 2016;26:888-903
115. Berbís MÁ, André S, Cañada FJ, Pipkorn R, Ippel H, Mayo KH. et al. Peptides derived from human galectin-3 N-terminal tail interact with its carbohydrate recognition domain in a phosphorylation-dependent manner. Biochem Biophys Res Commun. 2014;443:126-31
116. Barboni EA, Bawumia S, Henrick K, Hughes RC. Molecular modeling and mutagenesis studies of the N-terminal domains of galectin-3: evidence for participation with the C-terminal carbohydrate recognition domain in oligosaccharide binding. Glycobiology. 2000;10:1201-8
117. Ochieng J, Green B, Evans S, James O, Warfield P. Modulation of the biological functions of galectin-3 by matrix metalloproteinases. Biochim Biophys Acta. 1998;1379:97-106
118. Liu FT, Hsu DK, Zuberi RI, Hill PN, Shenhav A, Kuwabara I. et al. Modulation of functional properties of galectin-3 by monoclonal antibodies binding to the non-lectin domains. Biochemistry. 1996;35:6073-9
119. Kuklinski S, Probstmeier R. Homophilic binding properties of galectin-3: involvement of the carbohydrate recognition domain. J Neurochem. 1998;70:814-23
120. Fermino ML, Polli CD, Toledo KA, Liu F-T, Hsu DK, Roque-Barreira MC. et al. LPS-induced galectin-3 oligomerization results in enhancement of neutrophil activation. PLoS One. 2011;6:e26004
121. Lin Y-H, Qiu D-C, Chang W-H, Yeh Y-Q, Jeng U-S, Liu F-T. et al. The intrinsically disordered N-terminal domain of galectin-3 dynamically mediates multisite self-association of the protein through fuzzy interactions. J Biol Chem; Sep 11. pii: jbc.M117.802793. doi: 10.1074/jbc.M117.802793. [Epub ahead of print].
122. Yang RY, Hill PN, Hsu DK, Liu FT. Role of the carboxyl-terminal lectin domain in self-association of galectin-3. Biochemistry. 1998;37:4086-92
123. Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of galectin-3 oligomerization on the surface of neutrophils and endothelial cells using fluorescence resonance energy transfer. J Biol Chem. 2007;282:1374-83
124. Dennis JW, Nabi IR, Demetriou M. Metabolism, cell surface organization, and disease. Cell. 2009;139:1229-41
125. Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M. et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123-34
126. Kim Y-W, Park J, Lee H-J, Lee S-Y, Kim S-J. TGF-β sensitivity is determined by N-linked glycosylation of the type II TGF-β receptor. Biochem J. 2012;445:403-11
127. Demotte N, Wieërs G, Van Der Smissen P, Moser M, Schmidt C, Thielemans K. et al. A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res. 2010;70:7476-88
128. Demotte N, Bigirimana R, Wieërs G, Stroobant V, Squifflet J-L, Carrasco J. et al. A short treatment with galactomannan GM-CT-01 corrects the functions of freshly isolated human tumor-infiltrating lymphocytes. Clin Cancer Res. 2014;20:1823-33
129. Traber PG, Zomer E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS One. 2013;8:e83481
130. Chauhan D, Li G, Podar K, Hideshima T, Neri P, He D. et al. A novel carbohydrate-based therapeutic GCS-100 overcomes bortezomib resistance and enhances dexamethasone-induced apoptosis in multiple myeloma cells. Cancer Res. 2005;65:8350-8
131. Cheng H, Zhang Z, Leng J, Liu D, Hao M, Gao X. et al. The inhibitory effects and mechanisms of rhamnogalacturonan I pectin from potato on HT-29 colon cancer cell proliferation and cell cycle progression. Int J Food Sci Nutr. 2013;64:36-43
132. Gao X, Zhi Y, Sun L, Peng X, Zhang T, Xue H. et al. The inhibitory effects of a rhamnogalacturonan I (RG-I) domain from ginseng pectin on galectin-3 and its structure-activity relationship. J Biol Chem. 2013;288:33953-65
133. Gao X, Zhi Y, Zhang T, Xue H, Wang X, Foday AD. et al. Analysis of the neutral polysaccharide fraction of MCP and its inhibitory activity on galectin-3. Glycoconj J. 2012;29:159-65
134. Streetly MJ, Maharaj L, Joel S, Schey SA, Gribben JG, Cotter FE. GCS-100, a novel galectin-3 antagonist, modulates MCL-1, NOXA, and cell cycle to induce myeloma cell death. Blood. 2010;115:3939-48
135. van Hattum H, Branderhorst HM, Moret EE, Nilsson UJ, Leffler H, Pieters RJ. Tuning the preference of thiodigalactoside- and lactosamine-based ligands to galectin-3 over galectin-1. J Med Chem. 2013;56:1350-4
136. Duckworth CA, Guimond SE, Sindrewicz P, Hughes AJ, French NS, Lian L-Y. et al. Chemically modified, non-anticoagulant heparin derivatives are potent galectin-3 binding inhibitors and inhibit circulating galectin-3-promoted metastasis. Oncotarget. 2015;6:23671-87
137. Michel AK, Nangia-Makker P, Raz A, Cloninger MJ. Lactose-functionalized dendrimers arbitrate the interaction of galectin-3/MUC1 mediated cancer cellular aggregation. Chembiochem. 2014;15:2106-12
138. Böcker S, Laaf D, Elling L. Galectin Binding to Neo-Glycoproteins: LacDiNAc Conjugated BSA as Ligand for Human Galectin-3. Biomolecules. 2015;5:1671-96
139. John CM, Leffler H, Kahl-Knutsson B, Svensson I, Jarvis GA. Truncated galectin-3 inhibits tumor growth and metastasis in orthotopic nude mouse model of human breast cancer. Clin Cancer Res. 2003;9:2374-83
140. Mirandola L, Yu Y, Chui K, Jenkins MR, Cobos E, John CM. et al. Galectin-3C inhibits tumor growth and increases the anticancer activity of bortezomib in a murine model of human multiple myeloma. PLoS One. 2011;6:e21811
141. Mirandola L, Nguyen DD, Rahman RL, Grizzi F, Yuefei Y, Figueroa JA. et al. Anti-galectin-3 therapy: a new chance for multiple myeloma and ovarian cancer? Int Rev Immunol. 2014;33:417-27
142. Sun W, Li L, Yang Q, Shan W, Zhang Z, Huang Y. G3-C12 Peptide Reverses Galectin-3 from Foe to Friend for Active Targeting Cancer Treatment. Mol Pharm. 2015;12:4124-36
Author contact
![]() Corresponding author: Professor R.A. de Boer MD PhD FESC FHFA; Department of Cardiology, University Medical Center Groningen, AB 43, PO Box 30.001, 9700 RB, Groningen, The Netherlands. E-mail r.a.de.boernl
Corresponding author: Professor R.A. de Boer MD PhD FESC FHFA; Department of Cardiology, University Medical Center Groningen, AB 43, PO Box 30.001, 9700 RB, Groningen, The Netherlands. E-mail r.a.de.boernl
Citation styles
APA
Suthahar, N., Meijers, W.C., Silljé, H.H.W., Ho, J.E., Liu, F.T., de Boer, R.A. (2018). Galectin-3 Activation and Inhibition in Heart Failure and Cardiovascular Disease: An Update. Theranostics, 8(3), 593-609. https://doi.org/10.7150/thno.22196.
ACS
Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; Ho, J.E.; Liu, F.T.; de Boer, R.A. Galectin-3 Activation and Inhibition in Heart Failure and Cardiovascular Disease: An Update. Theranostics 2018, 8 (3), 593-609. DOI: 10.7150/thno.22196.
NLM
Suthahar N, Meijers WC, Silljé HHW, Ho JE, Liu FT, de Boer RA. Galectin-3 Activation and Inhibition in Heart Failure and Cardiovascular Disease: An Update. Theranostics 2018; 8(3):593-609. doi:10.7150/thno.22196. https://www.thno.org/v08p0593.htm
CSE
Suthahar N, Meijers WC, Silljé HHW, Ho JE, Liu FT, de Boer RA. 2018. Galectin-3 Activation and Inhibition in Heart Failure and Cardiovascular Disease: An Update. Theranostics. 8(3):593-609.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.