Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):749-766. doi:10.7150/thno.20183 This issue Cite
Research Paper
Atherosclerosis is exacerbated by chitinase-3-like-1 in amyloid precursor protein transgenic mice
Yu Yeon Jung1,2*, Ki Cheon Kim1*, Mi Hee Park1*, Youngsik Seo3, Heonyong Park3, Min Hee Park4,5, Jun Chang4, Dae Youn Hwang6, Sang Bae Han1, Sanghyeon Kim7, Dong Ju Son1,5* ![]() , Jin Tae Hong1
, Jin Tae Hong1 ![]()
1. College of Pharmacy & Medical Research Center, Chungbuk National University, Cheongju, Chungbuk 28160, Korea;
2. Department of Dental Hygiene, Gwangyang Health Sciences University, Gwangyang, Jeonnam 57764, Korea;
3. Department of Molecular Biology and Institute of Nanosensor Biotechnology, Dankook University, Yongin, Gyeonggi 16890, Korea;
4. Division of Life & Pharmaceutical Sciences, and the Center for Cell Signaling & Drug Discovery Research, Ewha Womans University, Ewhayeodae-gil, Seodaemun-gu, Seoul 03760, Korea;
5. Life Science Research Center, NovaKmed Co. Ltd., 1646 Yuseong-daero, Yuseong-gu, Daejeon 34054, Korea;
6. College of Biomaterials Science, Pusan National University, Miryang, Kyungnam 50463, Korea;
7. Stanley Brain Research Laboratory, Stanley Medical Research Institute, 9800 Medical Center Drive, Rockville, MD 20850, USA.
* These authors contributed equally to this work.
Received 2017-3-21; Accepted 2017-11-9; Published 2018-1-1
Abstract

Although the important role of amyloid precursor protein (APP) in vascular diseases associated with Alzheimer's disease (AD) has been demonstrated, the underlying molecular mechanisms and physiological consequences are unclear. We aimed to evaluate vascular inflammation and atherosclerosis in Swedish mutant of human APP transgenic (APPsw-Tg) and ApoE-/-/APPsw-Tg mice. We also aimed to explore the mechanisms underlying any changes observed in these mice compared with non-Tg controls. Methods: The transgenic and non-Tg mouse strains were subjected to partial ligation of the left carotid artery to induce atherosclerotic changes, which were measured using histological approaches, immunohistochemistry, quantitative polymerase chain reaction, and gene expression microarrays. Results: Our results showed increased vascular inflammation, arterial wall thickness, and atherosclerosis in APPsw-Tg and ApoE-/-/APPsw-Tg mice. We further found that the expression of chitinase-3-like-1 (Chi3l1) is increased in the APPsw-Tg mouse artery and Chi3l1 mediates endothelial cell (EC) inflammation and vascular smooth muscle cell (VSMC) activation, which in turn exacerbates atherosclerosis. In addition, using two publicly available microarray datasets from the dorsolateral prefrontal cortex of people with AD and unaffected controls as well as inflamed human umbilical vein endothelial cells, we found that Chi3l1 and associated inflammatory gene were significantly associated with AD, evaluated by co-expression network analysis and functional annotation. Knockdown of Chi3l1 in the arterial endothelium in vivo suppressed the development of atherosclerosis. We also show that microRNA 342-3p (miR-342-3p) inhibits EC inflammation and VSMC activation through directly targeting Chi3l1, and that APPsw increased Chi3l1 expression by reducing miR-342-3p expression in the arterial endothelium, promoting atherosclerosis. Conclusion: Our findings suggest that targeting Chi3l1 might provide new diagnostic and therapeutic strategies for vascular diseases in patients with AD.
Keywords: Alzheimer's diseases, amyloid precursor protein, atherosclerosis, chitinase-3-like-1, endothelial cells, microRNA 342-3p.
Introduction
Alzheimer's disease (AD), a progressive neurodegenerative disorder, remains a major cause of morbidity and mortality worldwide [1]. AD is characterized by extracellular amyloid plaques (senile plaques) composed of aggregated amyloid-β (Aβ) peptides and the intracellular formation of neurofibrillary tangles, consisting of aggregated, hyperphosphorylated tau protein, accompanying neuron loss, and eventual dementia [2]. Numerous studies have shown that vascular disease represents a major potential risk factor associated with the development and progression of AD; in particular, atherosclerosis has long been implicated in the pathogenesis of dementia [1, 3, 4]. Additionally, increasing evidence suggests that AD and atherosclerosis may not only share genetic and environmental risk factors but might also have common molecular mechanisms [5].
The amyloid precursor protein (APP) is an integral membrane protein expressed abundantly in the brain; the consecutive proteolysis of APP by β- and γ-secretases generates Aβ peptides [6, 7]. Several previous studies have demonstrated that APP is also present in arterial vascular cells and that it may play a role in the pathogenesis of vascular inflammation and atherosclerosis [8-14]. Considering that APP is overexpressed in atherosclerosis-prone regions of the human and mouse aorta [15, 16], it is plausible to assume that APP levels are associated with arterial plaque formation and thus, apart from epidemiological and pathophysiological factors, might serve as a biochemical link between AD and atherosclerosis [17]. Previously, the development of non-dietary induced early atherosclerotic lesions has been observed in double Swedish mutated human APP (APPsw) transgenic (Tg) mice [11]. Furthermore, APP overexpression induced endothelial dysfunction and accelerated aortic atherosclerotic development in apolipoprotein E knockout (ApoE-/-) mice [18, 19]. Conversely, genetic deletion of APP attenuated atherogenesis in ApoE-/- mice [20]. In addition, a recent study has shown that an increased level of APP mediates monocyte adhesion to the endothelium in ApoE-/- mice and in patients with AD, suggesting that endothelial APP function is involved in the vascular dysfunction associated with atherosclerosis and AD [19].
However, the underlying molecular mechanisms and physiological consequences of APP action in AD-associated vascular dysfunction and atherogenesis remain unclear. A previous study suggested that APP acts in this context through inducing the expression of inflammatory mediators such as monocyte chemoattractant protein-1, interleukin 6 (IL-6), vascular cell adhesion molecule 1 (VCAM1), and intercellular adhesion molecule 1 (ICAM1) in the aorta of APP23/ApoE-/- mice [18]. Another study demonstrated the decreased expression of endothelial nitric oxide synthase (eNOS) and superoxide dismutase and increased production of superoxide anions in the aorta of APP-Tg mice, and showed that these were recovered by treatment with the peroxisome proliferator-activated receptors δ ligand, resulting in decreased endothelial dysfunction [10]. Further studies are required to fully elucidate the exact molecular mechanisms underlying the pathophysiological role of APP in the development of atherosclerosis.
We have recently utilized a proteomics approach to identify novel markers for AD, demonstrating that the expression level of a chitinase-like-3 protein (CL3P) family member is markedly increased in the brain tissue of Tg mice overexpressing APPsw under the control of the neuron-specific enolase (NSE) promoter [21]. Although a number of CL3Ps have been identified in mammals, their roles have only recently begun to be elucidated. Previous studies have identified significantly increased expression levels of CL3P in chronic inflammation [22] and in the brain of AD patients [23]. In addition, the results from recent studies on human subjects have suggested that chitinase-3-like protein 1 (Chi3l1), also termed YKL-40, might serve as a potential biomarker in the blood plasma and cerebrospinal fluid for AD diagnosis [24-29]. Furthermore, recent studies have reported that the expression level of Chi3l1 is increased in atherosclerotic aorta [30, 31] and have demonstrated that increased plasma Chi3l1 is associated with endothelial dysfunction [32, 33] and thromboembolic stroke [34] and have suggested Chi3l1 as a diagnostic marker for cardiovascular diseases [35-37], whereas knockdown of Chi3l1 in ApoE-/- mice suppresses atherosclerotic plaque progression [30] and abdominal aortic aneurysm (AAA) development [38].
The expression of Chi3l1 has recently been shown to be regulated by a vascular function-regulatory microRNA (miRNA), miR-24, during AAA development [38]. Notably, miRNAs have been demonstrated to be involved in key physiological and pathophysiological aspects of vascular function and atherogenesis [39, 40]. However, the role of Chi3l1 with respect to APP and the correlation between Chi3l1 and miRs in atherosclerosis have not yet been studied. Accordingly, we investigated the role of Chi3l1 in APPsw-mediated vascular dysfunction and atherosclerosis progression using APPsw-Tg mice as well as ApoE-/-/APPsw-Tg mice. We also explored the involvement of microRNA regulatory mechanisms in Chi3l1-mediated vascular cell dysfunction.
Results
Increased vascular inflammation and arterial wall thickening in APPsw-Tg mice
To investigate the association between APPsw and vascular disease development and progression, we first determined whether APPsw regulates the mRNA expression of VCAM1 and ICAM1, vascular inflammatory marker genes, in the arterial endothelium under the pathophysiological conditions of vascular inflammation as well as atherogenesis. For this assessment, we utilized the partial carotid ligation model, a mouse model of vascular inflammation and atherosclerosis, and an intimal RNA isolation method [41, 42]. APPsw-Tg and non-Tg littermate control mice underwent partial carotid ligation surgery on left carotid artery (LCA), whereas the contralateral right carotid artery (RCA) remained non-manipulated to serve as a control, then endothelial-enriched RNA was collected from the LCA and RCA of APPsw-Tg and non-Tg mice at 48 h post-ligation, as presented in Figure S1A. The mRNA expression of VCAM1 and ICAM1 in the endothelium was found to be increased in partially-ligated LCA as compared to non-manipulated RCA in non-Tg mice (Figure 1A) as previously shown [42]. In comparison, APPsw-Tg mice also demonstrated increased expression of both VCAM1 and ICAM1 in LCA; however, the increment rates were much greater than those of non-Tg mice (Figure 1A). Notably, we observed that the basal expression levels of VCAM and ICAM1 in APPsw-Tg mice RCA were higher than those in the non-Tg mice RCA (Figure 1A).
Transgenic expression of APPsw increases vascular inflammation and arterial wall thickening in vivo. (A) The expression of VCAM1 and ICAM1 mRNA was determined by qPCR using endothelial-enriched RNA obtained from the left carotid artery (LCA) and right carotid artery (RCA) following partial carotid ligation in APPsw-Tg or non-Tg mice at 2 days post-ligation (n = 5 each, data shown as mean ± SEM, *p < 0.05 as determined by paired t-test). (B-J) For the assessment of arterial wall thickening, APPsw-Tg and non-Tg littermate control mice (n = 6 each) were partially ligated and fed a high-fat diet for 4 weeks. (B) Aortic trees including the carotid arteries were dissected and examined by bright-field imaging, and the (E) lesion area, (F) lesion size and intima-media thickness were quantified (n = 6 each, data shown as mean ± SEM, *p < 0.05 as determined by Student's t-test). Scale bar, 1 mm. Frozen sections prepared from the lesion area of LCA, denoted by red arrows in B, were stained with (C) H&E, (D) PCNA, (H) VCAM1, (I) ICAM1 and (J) MOMA2. Images shown are representative microscopy images (n = 6 each). Nuclei (blue) and protein expression (brown) are shown. Scale bar, 100 μm.
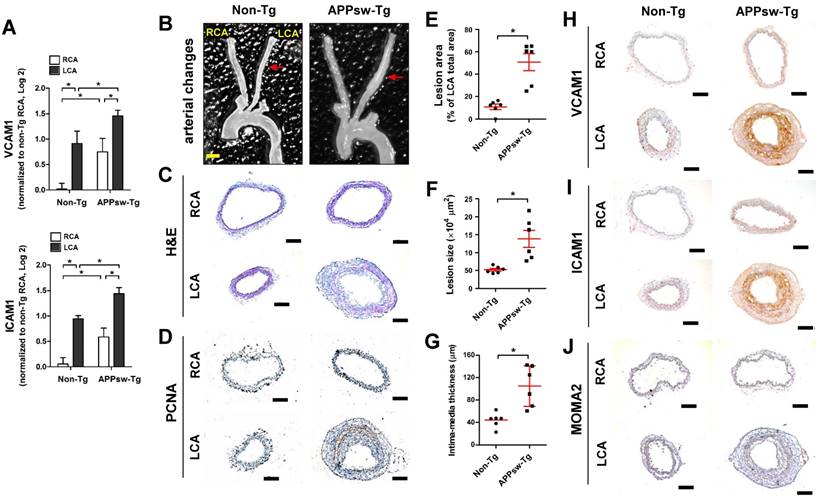
Next, we examined the effect of APPsw on wall-thickening and vascular inflammation using the same partial carotid ligation model in conjunction with a high-fat diet (HFD) for 4 weeks (Figure S1B). We observed minimal arterial changes in the LCA of non-Tg mice: decreased overall arterial lumen size was apparent compared with the RCA, but without significant intima-media thickening (Figures 1B, C).
In contrast, partial ligation of the LCA induced marked structural changes and robust arterial wall thickness in APPsw-Tg mice LCAs, whereas the unligated RCAs remained lesion free (Figures 1B, C). In addition, increased cell proliferation, as assessed by PCNA immunostaining, in the arterial wall was observed in APPsw-Tg mice LCA compared to non-Tg mice (Figure 1D). More specifically, the lesion area (Figure 1E) and lesion size (Figure 1F) in APPsw-Tg mice LCAs were significantly increased by 4.6-fold and 2.7-fold compared with non-Tg mice, respectively. Furthermore, the vessel intima-media thickness was significantly increased in the APPsw-Tg mice LCAs compared to that in the non-Tg control mice (Figure 1G). We further confirmed that the basal protein expression levels of VCAM1 and ICAM1 in the RCAs of APPsw mice were much higher than those of non-Tg mice, consistent with the mRNA expression levels shown in Figure 1A, and that the expression level changes in APPsw-Tg mice LCAs compared with RCA were much greater than those in non-Tg mice (Figures 1H, I). Furthermore, increased arterial wall thickening in the LCA of the APPsw-Tg mice was correlated with increased leukocyte infiltration, as assessed by MOMA2 immunostaining, whereas this association was not observed in non-Tg control mice (Figure 1J). Taken together, our findings demonstrated that overexpression of APPsw promotes a pro-atherogenic phenotype in the artery.
Identification of genes regulated by APPsw in the mouse arterial endothelium
To identify which genes were regulated by APPsw in the arterial endothelium in vivo, we performed an in vivo DNA microarray study using endothelial-enriched RNAs obtained from APPsw-Tg mice carotid arteries 48 h post partial ligation and RNAs from non-Tg mice for comparison as presented in Figure S1A. The microarray data showed 389 (290 upregulated and 99 downregulated) genes were altered by more than 2-fold in the non-Tg mice LCA compared with RCA endothelium by 48 h after partial ligation (Figure 2A, Table S1), whereas 522 (433 upregulated and 89 downregulated) genes were changed in the APPsw-Tg mice LCA (Figure 2B, Table S2). Interestingly, we found that 219 (210 upregulated and 9 downregulated) gene were changed (≥2-fold) in the non-manipulated RCA endothelium of APPsw-Tg mice compared with the non-Tg mice RCA (Figure 2C, Table S3). In addition, 288 genes were upregulated and 14 genes were downregulated by more than 2-fold in the partially-ligated LCA endothelium of APPsw-Tg mice compared with the non-Tg mice LCA (Figure 2D, Table S4). These findings suggest that APPsw modulates the expression of endothelial genes under both intact and atheroprone conditions.
Global gene expression profiles in the carotid artery endothelium of APPsw-Tg mice in vivo. Total RNAs were obtained from the intima of mouse LCA and RCA at 2 days after ligation. Affymetrix GeneChip® Mouse Gene 2.0 ST arrays containing 770,317 mouse gene probes were used. (A-D) Scatter plots show normalized intensities of each probe under 4 comparison conditions: (A) non-Tg LCA vs. non-Tg RCA, (B) APPsw-Tg LCA vs. APPsw-Tg RCA, (C) APPsw-Tg RCA vs. non-Tg RCA, and (D) APPsw-Tg LCA vs. non-Tg LCA after ligation. Genes are shown that were up-regulated (red) or down-regulated (blue) (≥ 2-fold difference) at a false discovery rate of ≤ 10% in LCA compared with RCA. Microarray results for selected (E) upregulated and (F) downregulated genes in both APPsw-Tg and non-Tg mice are shown as the fold-increase or fold-decrease of gene expression in LCAs compared with that in RCA on a log2 scale (mean ± SEM). (G-H) The expression of (G) Chi3l3 and (H) Chi3l1 mRNA was determined by qPCR using endothelial-enriched RNA obtained from the LCA and RCA following partial carotid ligation in APPsw-Tg or non-Tg mice at 2 days post-ligation (n = 5 each). Scale of Y-axis of panel G is one order of magnitude larger than that of panel H. (I) Frozen sections of carotid arteries (non-manipulated RCA) from APPsw-Tg and non-Tg mice were stained with an antibody against Chi3l1. Images shown are representative microscopy images (n = 4 each). The rectangle indicates the magnified area shown in the lower panel. Nuclei (blue) and protein expression (brown) are shown. Scale bar, 100 μm.
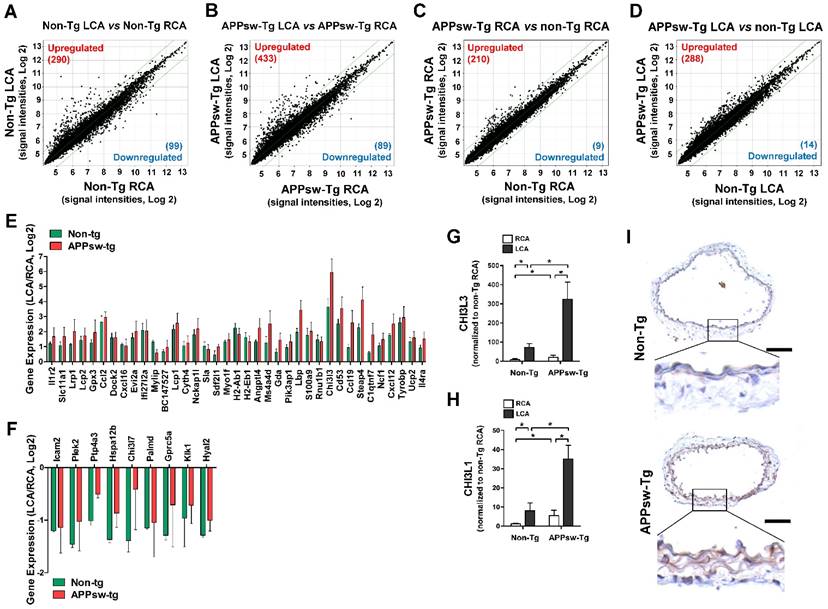
To explore the functional importance of the APPsw-regulated genes in more depth, we subjected the list of changed (≥2-fold) genes to functional annotation analysis. On the basis of array Gene Ontology, DAVID and Kyoto Encyclopedia of Genes and Genomics (KEGG) analysis, we found that genes involved in multiple pathways including immune system processes, inflammatory response, cell adhesion, response to stress, transport, signal transduction, and apoptotic process were affected by APPsw overexpression (Table S5). Biological processes enriched in genes whose expression differed between non-Tg mice LCA and non-Tg mice RCA (Table S6), APPsw-Tg mice LCA and APPsw-Tg mice RCA (Table S7), APPsw-Tg mice RCA and non-Tg mice LCA (Table S8), or APPsw-Tg mice LCA and non-Tg mice LCA (Table S9) were associated with APPsw expression in the mouse model of vascular inflammation and atherosclerosis. Among the upregulated (Figure 2E) or downregulated (Figure 2F) genes in both APPsw-Tg and non-Tg mice, we found that chitinase-3-like-3 (Chi3l3) was the most robustly changed (34-fold upregulated) gene in the partially-ligated LCA compared with the non-manipulated RCA in both APPsw-Tg and non-Tg mice. This microarray result was further confirmed by quantitative real-time PCR (qPCR) verification (Figure 2G).
Chi3I3 is known as a mouse-specific chitinase-3-like protein [43] and has considerable sequence homology with Chi3l1 [44], also known as YKL-40, which has homologs present in both humans and mice. Therefore, we further determined whether expression changes were observed in Chi3l1 as well. Our qPCR results showed that the expression of Chi3l1 was significantly upregulated in partially-ligated LCAs compared with non-manipulated RCAs in both APPsw-Tg and non-Tg mice, but its increment level in APPsw-Tg mice LCA was much greater than that in non-Tg mice LCA (Figure 2H). In addition, similar to that observed for Chi3l3 expression, we found that the basal mRNA and protein expression levels of Chi3l1 in non-manipulated APPsw-Tg mouse RCA were much higher than those in the non-Tg mouse RCA (Figure 2H, I). These results indicated that Chi3l3 and Chi3l1 represent genes that are robustly upregulated in the artery by APPsw expression under pro-atherogenic conditions in vivo. However, because Chi3l3 is a mouse-specific gene, we decided to focus on the potential pathophysiological importance of Chi3l1 on vascular inflammation and atherogenesis related to APPsw.
Co-expression network analysis of Chi3l1 expression in AD patient brain and inflamed human endothelial cells
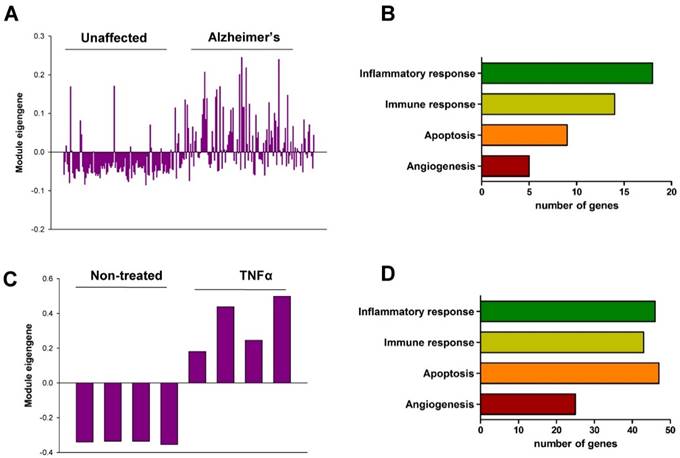
In order to validate the role of Chi3l1 in the pathophysiology of AD as well as vascular disease in humans, we performed co-expression network analysis and then compared two co-expression modules including Chi3l1. First, we generated co-expression modules using the microarray data from the dorsolateral prefrontal cortex (PFC) of people with AD (n = 129) and unaffected controls (n = 101). Of the 45 co-expression modules, an AZ_M32 module that included the Chi3l1 gene was significantly associated with AD (Table S10). The eigengene values in this module were significantly up-regulated in the PFC of AD patients as compared to unaffected controls (Figure 3A), and inflammatory response, immune response, apoptosis, and angiogenesis were significantly enriched in this module (Figure 3B, Table S11). Next, we generated co-expression modules from microarray data from tumor necrosis factor alpha (TNF-α)-treated human vascular endothelial cells (HUVECs, n = 4) and mock-treated HUVECs (n = 4). Of the 61 co-expression modules, an EC_M47 module that included the Chi3l1 gene was significantly associated with TNFα treatment in the HUVECs (Table S12). The eigengene values in this module were significantly up-regulated in the TNFα-treated HUVECs as compared to mock-treated cells (Figure 3C), and inflammatory response, immune response, angiogenesis, and apoptosis were significantly enriched in this module (Figure 3D, Table S13). This result suggests that up-regulation of Chi3l1 may be significantly associated with AD as well as atherosclerosis through inflammatory response, immune response, angiogenesis, and apoptosis.
Co-expression module associated with Chi3l1. (A) The eigengene values across samples in the AZ_M32. (B) Major biological processes (Gene Ontology) significantly enriched in the genes in the co-expression module AZ_M32. (C) The eigengene values across samples in the EC_M47. (D) Major biological processes (Gene Ontology) significantly enriched in the genes in the co-expression module EC_M47.
Chi3l1 induces endothelial inflammation and enhances PDGF-induced VSMC migration and proliferation
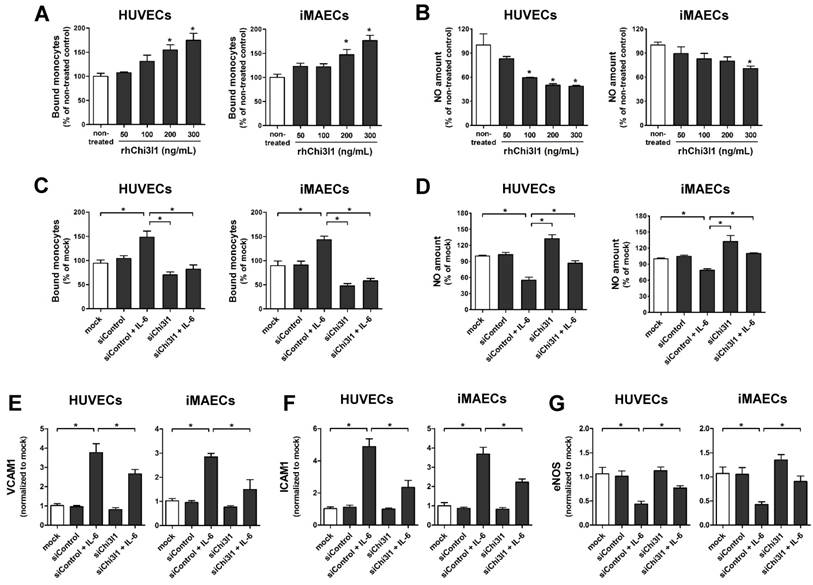
Next, we explored the role of Chi3l1 on vascular cell function in primary cultured arterial endothelial cells (ECs) and vascular smooth muscle cell (VSMCs) in vitro. First, to test whether Chi3l1 regulates endothelial inflammation, human umbilical vein ECs (HUVECs) and immortalized mouse arterial ECs (iMAECs) were treated with recombinant human Chi3l1 protein (rhChi3l1) and then monocyte-EC adhesion and NO generation assays were performed. Treatment of rhChi3l1 increased monocyte-EC adhesion in a dose-dependent manner in both HUVECs and iMAECs (Figure 4A), whereas it decreased NO levels (Figure 4B). We further tested whether these proinflammatory effects were able to be reversed by the Chi3l1 knockdown. For this, HUVECs and iMAECs were stimulated with recombinant human interleukin-6 (IL-6) to induce inflammation. In ECs, IL-6 treatment significantly increased the expression of Chi3l1, whereas this was significantly downregulated by Chi3l1 siRNA (Figure S2). We found that knockdown of Chi3l1 also inhibited IL-6-induced monocyte adhesion (Figure 4C) and reversed the reduction of NO affected by IL-6 in ECs (Figure 4D). In addition, knockdown of Chi3l1 reduced IL-6-induced expression of cell adhesion proteins such as VCAM1 (Figure 4E) and ICAM1 (Figure 4F) and recovered eNOS (Figure 4G) expression in ECs. These results suggest that Chi3l1 enhances IL-6-mediated endothelial inflammatory responses including VCAM1 and ICAM1 as well as eNOS expression and monocyte adhesion.
Chi3l1 induces endothelial inflammation in vitro. (A-B) HUVECs and iMAECs were treated with rhChi3l1 protein for 24 h, then (A) THP1 monocyte adhesion to ECs and (B) NO level were determined (n = 5). (C-D) HUVECs and iMAECs were transfected with Chi3l1 siRNA (siChi3l1, 150 nM) or control siRNA (siControl) for 24 h. Cells were then treated with IL-6 (10 ng/mL; 24 h), and (C) monocyte-EC adherence and (D) NO level were determined (n = 5). (E-G) HUVECs and iMAECs were transfected with siChi3l1 or siControl. After 24 h, cells were treated with IL-6 for 24 h, and the mRNA expression of (E) VCAM1, (F) ICAM1, and (G) eNOS was determined by qPCR (n = 5). All data are shown as mean ± SEM, *p < 0.05 as determined by paired t-test.
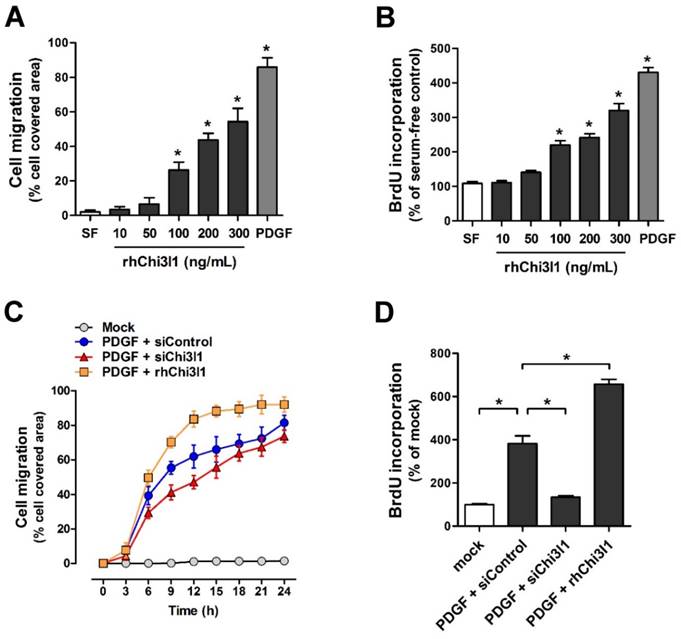
To examine cell migration and proliferation in primary cultured mouse aortic VSMCs from C57BL/6 mice aorta in response to rhChi3l1 treatment, VSMCs were treated with several doses of rhChi3l1 for 24 h and then subjected to wound healing and BrdU incorporation assays. We observed that high-doses of rhChi3l1 (≥ 100 ng/mL) significantly induced migration (Figure 5A) and proliferation (Figure 5B) whereas the cells did not respond to low-dose (≤ 50 ng/mL) rhChi3l1 treatment (Figures 5A, B). Notably, we found that co-treatment with a low-dose of rhChi3l1 (50 ng/mL), which did not induce significant cell responses alone, significantly enhanced PDGF-subunit B homodimer (PDGF-BB)-induced cell migration and proliferation, whereas knockdown of Chi3l1 led to significantly reduced PDGF-BB-stimulated VSMC responses (Figures 5C, D). These results suggested that Chi3l1 potentiates VSMC responses to PDGF-BB with respect to cell migration and proliferation.
Chi3l1 enhances vascular smooth muscle cell migration and proliferation. VSMCs were isolated from thoracic aortas of C57BL/6 mice, then primary cultured. (A-B) VSMCs were treated with rhChi3l1 (10-300 ng/mL) or PDGF-BB (25 ng/mL) in serum-free media for 24 h, then (A) the cell migration (assessed by area of migrated cells, n = 3) and (B) cell proliferation (assessed by BrdU incorporation, n = 6) was determined. (C-D) VSMCs were transfected with siChi3l1 (150 nM) or treated with rhChi3l1 (50 ng/mL) in serum-free medium for 24 h, then treated with PDGF-BB (25 ng/mL; 24 h), and (C) cell migration (n = 3) and (D) cell proliferation (n = 5) were determined. All data are shown as mean ± SEM, *p < 0.05 as determined by paired t-test.
miR-342-3p targets Chi3l1 and regulates EC inflammation as well as VSMC migration and proliferation
To investigate whether microRNA (miRNAs) are involved in Chi3l1-mediated enhancement of vascular inflammation and atherosclerosis in APPsw-Tg mice, we carried out an in silico analysis using the miRNA target prediction tools from miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/). The Gene-microRNA targets analysis resulted in a list of 5934 and 527 miRNAs that are predicted to bind to their target sequences in the 3' UTR region of APP and Chi3l1, respectively (Figure S3A). We compared these putative APP- or Chi3l1-targeting miRNAs to a list of miRNAs from publicly available microarray data of partially ligated mouse carotid arterial endothelium [45]. Among 537 miRNAs that were differentially expressed in partially ligated mouse carotid arterial endothelium, we identified 14 miRNAs that were predicted to target both of APP and Chi3l1 (Figure S3B). Of those 14 candidate miRNAs, we validated the expression of 11 miRNAs—3 (miR-103, miR-107, and miR-706) were excluded due to lack of specific primer availability—in endothelial RNA obtained from APPsw-Tg and non-Tg mouse carotid arteries at 48 h post partial ligation of LCA using the Qiagen miScript miRNA-specific primer assay. The qPCR results showed that expression of miR-24-1-5p, miR-26a, miR-26b, miR-107 and miR-125b-5p were found to be increased in partially-ligated LCA endothelium as compared to non-manipulated RCA in APPsw-Tg mice and expression levels of these miRNAs were higher than those of non-Tg mice LCA (Figure S4A), whereas miR-125a-3p, miR-296-3p, miR-541-5p and miR-342-3p were decreased in APPsw-Tg mice compared with non-Tg mice (Figure 6A, Figure S4B), but expression patterns of miR-242-5p and miR-26-3p were not different between APPsw-Tg mice and non-Tg mice (Figure S4C). As only miR-342-3p showed significant differences among changed miRNAs in APPsw-Tg mice LCA (Figure 6A), we focused our attention on miR-342-3p as a novel regulator of Chi3l1. Interestingly, very recent studies have suggested that miR-342-3p acts as a novel biomarker for AD and is downregulated in the blood serum/plasma of patients with AD [46, 47]. However, the functional role of miR-342-3p in vascular disease and AD, and in particular vascular inflammation and atherosclerosis through the regulation of Chi3l1 expression, has not previously been reported.
MicroRNA-342-3p targets Chi3l1 and inhibits endothelial inflammation and VSMC migration and proliferation. (A) The expression of miR-342-3p was determined by qPCR using endothelial-enriched RNA obtained from the LCA and RCA following partial carotid ligation in APPsw-Tg or non-Tg mice at 2 days post-ligation (n = 5 each, data are shown as mean ± SEM, *p < 0.05 as determined by Student's t-test). (B) iMAECs were treated with IL-6 (10 ng/mL; 24 h), then miR-342-3p expression was determined (n = 5, data shown as mean ± SEM, *p < 0.05 as determined by Student's t-test). Panel C shows the seed sequence of miR-342-3p and complementary 3'-UTR sequences of Chi3l1 wild-type and mutant. (D) iMAECs transfected with dual-luciferase reporter plasmids containing wild-type or mutant Chi3l1-3'-UTR were treated with miR-342-3p mimic or control mimic (Control-miR). Firefly luciferase activity (normalized to control Renilla luciferase) indicating Chi3l1 expression was determined using Luc-Pair miR Luciferase Assay Kit (n = 4 each, data shown as mean ± SEM, *p < 0.05 as determined by Student's t-test). (E-F) The expression of Chi3l1 (E) mRNA (n = 5, data are shown as mean ± SEM, *p < 0.05 as determined by Student's t-test) and (F) protein in IL-6-stimulated iMAECs in the presence of miR-342-3p mimic (50 nM) or control-miR were determined. (G-H) HUVECs and iMAECs were transfected with miR-342-3p (50 nM) for 24 h, then (G) THP1 monocyte adhesion to ECs and (H) NO level was determined (n = 5). (I) VSMCs were transfected with miR-342-3p (50 nM) or control-miR in serum-free medium for 24 h, then treated with PDGF-BB (25 ng/mL; 24 h), and cell migration (n = 3) and cell proliferation (n = 5) were determined.
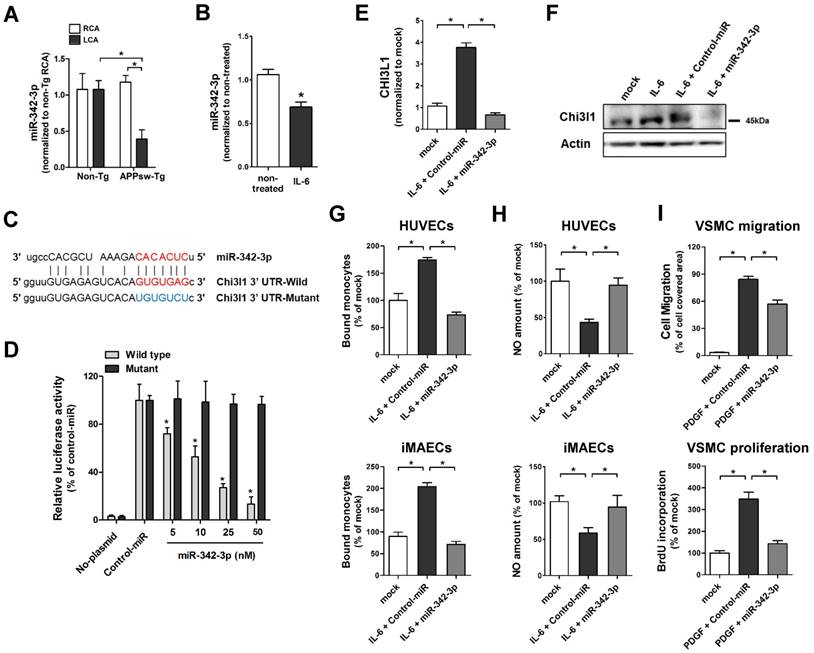
To investigate the biological role of the potential miR-342-3p regulation of Chi3l1 in ECs, we first tested whether the expression of miR-342-3p was influenced in inflamed ECs. We found that miR-342-3p expression was significantly decreased in IL-6-teated iMAECs (Figure 6B), which is consistent with our observations in vivo as shown above. Next, we examined whether miR-342-3p directly targets Chi3l1 as predicted by the in silico analysis. For this study, we employed a dual-luciferase reporter assay in iMAECs transfected with either wild-type or mutated Chi3l1 3'-UTR firefly luciferase constructs (Figure 6C). Overexpression of miR-342-3p by transfection of miR-342-3p mimic inhibited luciferase activity in a dose-dependent manner, whereas mutant or control-miR mimics had no effect (Figure 6D). This result suggested that miR-342-3p binds to the Chi3l1 3'-UTR and inhibits its expression. Next, we tested whether Chi3l1 expression was regulated in a miR-342-3p-dependent manner. Transfection of the miR-342-3p mimic effectively decreased IL-6-induced Chi3l1 expression in iMACEs (Figures 6E, F). Together, these findings demonstrated that the downregulation of miR-342-3p in inflamed ECs is directly responsible for the associated increase of Chi3l1 expression.
To investigate the biological relevance of downregulated miR-342-p in ECs and VSMCs, we examined the effects of miR-342-3p on monocyte adhesion and NO generation in ECs and on cell migration and proliferation in VSMCs. Overexpression of miR-342-3p by transfection with a miR-342-3p mimic significantly decreased IL-6-induced monocyte-EC adhesion both in iMAECs and HUVECs (Figure 6G). In contrast, miR-342-3p treatment reversed the reduction of NO caused by IL-6 (Figure 6H) in ECs. Furthermore, transfection of VSMCs with a miR-342-3p mimic effectively attenuated PDGF-BB-induced cell migration and proliferation (Figure 6I). Taken together, these results suggested that Chi3l1 functions downstream of miR-342-3p and that overexpression of miR-342-3p may have the potential to overcome EC inflammation and VSMC migration and proliferation in APPsw-Tg mice.
Knockdown of Chi3l1 decreases atherosclerotic lesions in ApoE-/-/APPsw-Tg mice
To investigate whether Chi3l1 plays a key role in the development of atherosclerosis in APPsw-Tg mice, we tested the effect of Chi3l1 knockdown on atherosclerosis development utilizing a Chi3l1 short hairpin RNA (shRNA)-expressing adenoviral vector (Ad-shRNA-Chi3l1) (Figure S5A). First, to test the transduction and knockdown efficiencies of Ad-Chi3l1-shRNA in the ECs in vitro, red fluorescent protein (RFP)-expressing Ad-shRNA-Chi3l1 was transduced to iMAECs. We found that Ad-Chi3l1-shRNA could be effectively transduced to the cultured iMAECs as demonstrated by fluorescence microscopy of RFP expression (Figure S5B). As qPCR and Western blot results showed that Ad-Chi3l1-shRNA effectively silenced Chi3l1 expression in the iMAECs (Figure S5C). We further tested knockdown efficiency of Ad-shRNA-Chi3l1 in the mouse carotid endothelium in vivo, and found that administration of Ad-shRNA-Chi3l1 via tail-vein 2 days prior to LCA partial ligation surgery effectively knocked-down Chil1 expression in the mouse carotid endothelium as compared with the control Ad vector (Figure S5D); this dose was used for all subsequent studies.
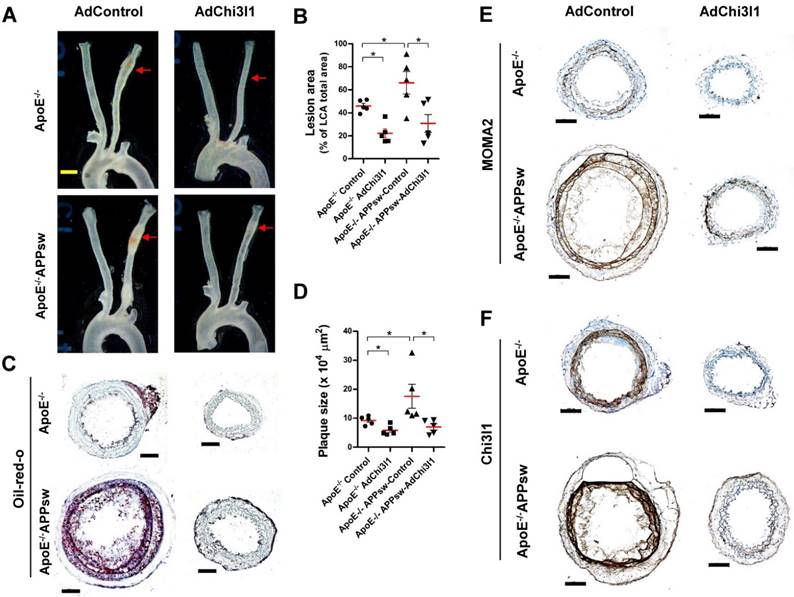
Finally, we tested the effect of Ad-shRNA-Chi3l1 in the partial carotid ligation atherosclerosis model using ApoE-/-/APPsw-Tg as well as ApoE-/- mice. In the control Ad vector-treated ApoE-/-/APPsw-Tg mice, the LCA developed robust atherosclerotic plaques at 10 days post-ligation when fed HFD compared with littermate control ApoE-/- mice (Figures 7A-D). Notably, treatment with Ad-shRNA-Chi3l1 significantly reduced atherosclerosis lesion development compared with the Ad-vector control group in both ApoE-/-/APPsw-Tg and ApoE-/- mice (Figure 7A-D). Increased atherosclerosis in the LCA model was correlated with increased leukocyte infiltration, as assessed by MOMA2 immunostaining (Figure 7E). Furthermore, we confirmed that the expression level of Chi3l1 in the LCA was significantly increased in both ApoE-/-/APPsw-Tg and ApoE-/- mice and subsequently markedly decreased following administration of Ad-Chi3l1-shRNA (Figure 7F). Taken together, these findings confirmed that Chi3l1 plays a key role in the development of atherosclerosis in APPsw-Tg mice.
Ad-shRNA-Chi3l1 reduces atherosclerosis in ApoE-/-/APPsw-Tg and ApoE-/- mice. For the assessment of atherosclerosis, ApoE-/-/APPsw-Tg and littermate ApoE-/-/non-Tg control mice were injected with Ad-shRNA-Chi3l1 (108 p.f.u. per animal, via the tail vein) or empty control vector (AdControl) 2 days prior to partial carotid ligation and fed a high-fat diet for 10 days. (A) Aortic trees including the carotid arteries were dissected and examined by bright-field imaging, and the atherosclerotic lesion area was quantified in B (n = 5 each, data shown as mean ± SEM, *p < 0.05 as determined by Student's t-test). Scale bar, 1 mm. (C) Frozen sections prepared from the lesion area of LCA, denoted by red arrows in A, were stained with oil-red-O and plaque size was quantified in D (n = 5 each, data shown as mean ± SEM, *p < 0.05 as determined by Student's t-test). Scale bar, 100 μm. (E-F) LCA frozen sections were used for immunostaining with antibodies specific to (E) MOMA2 or (F) Chi3l1.
Discussion
The association of APP with vascular inflammation and atherosclerosis has been suggested previously [11, 15-20]; however, compelling evidence for its functional role and underlying mechanism has not yet become available, largely because of a lack of adequate animal models and methods to test the functions of APP in vivo. In the present study, we employed a partial carotid ligation mouse model that rapidly induces gene expression changes, endothelial inflammation, and atherosclerosis [41, 42, 48]. We also used a powerful method to extract endothelium-enriched RNA samples from the mouse carotid intima [41], which allowed us to carry out genome-wide studies to identify APP regulating genes in APPsw-Tg mice. We further generated ApoE-/-/APPsw-Tg mice to provide direct evidence for the role of APPsw in atherosclerosis development in vivo. In particular, these analyses demonstrated the elevated expression of Chi3l1 in inflamed endothelium both in vivo and in vitro. We further found that the Chi3l1 gene is significantly associated with AD as well as EC inflammation through co-expression network analysis using microarray data from both AD patient brain tissues and inflamed ECs. Furthermore, through in silico analyses, we found that the Chi3l1 gene is related to multiple biological processes including immune/inflammatory response in the human brain with AD and inflamed ECs. It has long been recognized that immune/inflammatory response is a key event in the development of both AD and atherosclerosis [49]. In addition, pathological deposition of toxic Aβ activates immune/inflammatory response during the disease progression of both AD and atherosclerosis [19, 50, 51]. These observations suggest that Chi3l1 may have a significant association with APP-mediated vascular inflammation as well as atherosclerosis. However, the involvement of Chi3l1 in AD-associated vascular inflammation and atherogenesis remains unclear. We thus focused our efforts on investigating the relationship between Chi3l1 and the pathophysiological role of APPsw in the development of atherosclerosis.
Chi3l1, a chitinase-like protein member of family 18 chitinases that is expressed in innate immune cells, lacks the characteristic enzymatic activity of this family; rather, is has been found to be involved in endothelial dysfunction and tissue remodeling [52]. chi3l1 expression is increased in a variety of inflammatory and chronic diseases including obesity, diabetes, nephropathy disease, rheumatoid arthritis, inflammatory bowel disease, cancer, coronary arterial diseases, and acute ischemic stroke [52-58]. Furthermore, Chi3l1 was found to be secreted from differentiated macrophages in early-stage atherosclerosis lesions [59, 60] as well as from VSMCs during hyper cholesterol-induced coronary atherosclerotic plaque development in swine [31]. Notably, knockdown of the elevated Chi3l1 expression associated with the atherosclerotic aorta and endothelial dysfunction suppresses the progression of these characteristics in a mouse model [30-32, 34, 38, 61]. These data suggest that in addition to its potential role as a circulating biomarker for AD diagnosis [24-29], Chi3l1 might play an important role in vascular inflammation and the development of atherosclerosis in patients with AD. Therefore, we investigated whether the observed increased vascular inflammation, arterial wall thickening, and atherosclerosis in APPsw-Tg mice were mediated by Chi3l1 by performing supplementation and knockdown analyses. Briefly, rhChi3l1 treatment exhibited pro-atherogenic effects in vitro, as measured by monocyte-EC adhesion, inflammatory gene expression, and reduced NO generation in ECs along with enhanced VSMC proliferation and migration, whereas Chi3l1 knockdown yielded the opposite effects along with inhibiting atherosclerotic plaque development and vascular inflammation in vivo. Taken together, our findings suggest that elevated expression of Chi3l1 is significant for the development of atherosclerosis in the APPsw-Tg mouse artery.
miRNAs are small non-coding RNA molecules that modulate the stability and/or the translational efficiency of their target messenger RNAs, generally by binding to the respective 3'-UTRs [62]. miRNAs have been identified as key physiological and pathophysiological regulators of complex biological processes linked to multiple cardiovascular diseases including atherosclerosis [63]. Previously, several miRNAs have been identified to be associated with the expression of APP and its function in AD development and disease progression. For example, miR-20a, miR-17-5p, miR-106b, miR-101, miR-16, and miR-193b were identified as negative regulators of APP and furthermore are considered as potential diagnostic and prognostic markers for AD [64-67]. In addition, a few miRNAs such as miR-24 and miR-449a are known to target Chi3l1 and regulate its biological functions [38, 68, 69]. However, the association between miRNAs, APPsw, and Chi3l1 in vascular inflammation and atherosclerosis has not been addressed. Therefore, in this study, we searched for miRNAs that are regulated in the inflamed carotid endothelium in APPsw-Tg mice and explored their involvement in Chi3l1-mediated vascular inflammation and atherogenesis. Among 14 miRNAs identified by in silico target prediction analysis targeting miRNAs for both Chi3l1 and APP from among the list of genes previously identified as being differentially expressed in partially ligated carotid endothelium [45], we found that miR-342-3p, previously reported to be downregulated in the blood of AD patients [46, 47], was significantly downregulated in APPsw-Tg mice compared to non-Tg mice as well. It was further reported that miR-342-3p, which is also downregulated in a murine heart failure model [70], likely plays a role in cell viability, migration, and proliferation in human cervical cancer, the development of lymphoma, MYC transcriptional activity in human lung cancer, and in the adipogenesis of mesenchymal stem cells [71-77]. However, the functional role of miR-342-3p in vascular inflammation and atherosclerosis through regulating Chi3l1 expression has not previously been reported.
Here, we found that the overexpression of miR-342-3p mediated by a specific miRNA mimic generally reproduces the effects seen following the Chi3l1 knockdown, consistent with our demonstration that miR-342-3p binds to the Chi3l1 gene 3' UTR and represses its expression in ECs under inflammatory conditions. Taken together, these results suggested that decreased miR-342-3p expression in APPsw-Tg mice endothelium leads to a rise in Chi3l1 expression, resulting in the exacerbation of vascular inflammation and the development of atherosclerosis in APPsw-Tg mice.
In conclusion, we demonstrated that Chi3l1 plays a key role as a pro-atherogenic factor in vascular inflammation and atherosclerosis development in APPsw-Tg mice through enhancing EC inflammation and VSMC migration and proliferation. We further demonstrated that miR-342-3p acts as an anti-atherogenic miRNA through directly targeting Chi3l1, and that APPsw mediates increased expression of Chi3l1 by reducing miR-342-3p expression in the arterial endothelium, promoting atherogenesis in APPsw-Tg mice. Our findings suggest that the convergent targeting of Chi3l1 and miR-342-3p might provide new diagnostic and therapeutic strategies for vascular diseases in patients with AD.
Materials and Methods
Animals and ethics statement
Human APPsw (K595N/M596L)-expressing mice under the control of the NSE promoter in the C57BL/6 background strain and non-Tg littermates were kindly provided by the Korea National Institute of Food And Drug Safety Evaluation (Cheongju, Chungbuk, Korea) [78]. C57BL/6J and ApoE-/-mice (B6.129P2-ApoEtm1Unc/J) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). We bred APPsw-Tg mice with ApoE-/- mice (B6.129P2-Apoetm1Unc/J, Jackson Laboratory, Bar Harbor, ME, USA) to generate ApoE-/-/APPsw-Tg mice to generate ApoE-/-/APPsw-Tg mice. The transgenic status of each animal was confirmed by PCR from tail-tip DNA using specific primers for the human APP (5'-CTG AGT CTG CAG TCC TCG A-3' and 5'-CTC TTC TCA CTG CAT GTC TC-3' forward and reverse, respectively). The ApoE knockout was detected using a set of 3 primers (primer 1: 5'-GCC TAG CCG AGG GAG AGC CG-3'; primer 2: 5'-TGT GAC TTG GGA GCT CTG CAG C-3'; and primer 3: 5'-GCC GCC CCG ACT GCA TCT-3'). These primers amplify a 155 bp wild-type band (primers 1 and 2) and a 245 bp targeted (null) band (primers 1 and 3) (Figure S7). All animal studies followed the guidelines of the Animal Care and Use Committee of Chungbuk National University (Cheongju, Chungbuk, Korea). All protocols involving mice in this study were reviewed and approved by the Chungbuk National University Institutional Animal Care and Use Committee (IACUC) and complied with the Korean National Institute of Health Guide for the Care and Use of Laboratory Animals (CBNUA-792-15-01).
Animal studies with partial carotid ligation surgery
Mice were anesthetized initially with 3.5% isoflurane; this dose was then reduced to 1.5-2% during the entire procedure while the mice were subjected to partial carotid ligation of the LCA as previously described [41]. Briefly, the LCA bifurcation was exposed by blunt dissection and three of four caudal LCA branches (left external carotid, internal carotid, and occipital arteries) were ligated with 6-0 silk sutures, leaving the superior thyroid artery intact. The contralateral RCA was left intact as an internal control. The surgical incision was then closed with 6-0 monofilament sutures and the analgesic buprenorphine (0.1 mg/kg) was administrated subcutaneously. The mice were monitored until recovery in a chamber on a heating pad.
Microarray procedures
At 48 h after partial ligation, endothelial-enriched total carotid intimal RNAs were separately obtained from the LCA and RCA of APPsw-Tg and non-Tg mice as described previously [41]. Intimal RNA samples from the carotid arteries of 4 mice were pooled to obtain approximately 50 ng of total RNA as a single array sample, yielding 3 array samples from the 12 mice per group. All RNA samples used for the DNA microarray study passed an initial quality control test using a BioAnalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) and each sample was linearly amplified, according to our prior protocol [42]. Each sample was then used for microarray studies using GeneChip® Mouse Gene 2.0 ST arrays (Affymetrix, Santa Clara, CA, USA) at BioCore Co. (Seoul, Korea). After hybridization in a GeneChip® Hybridization Oven 645 (Affymetrix), washing and staining steps were performed in a GeneChip® Fluidics Station 450 (Affymetrix) and then the GeneChip® arrays were scanned on a GeneChip® 3000 7G scanners (Affymetrix) to determine the probe fluorescence intensity. The raw probe intensities were then normalized using the quantile normalization algorithm [79].
Microarray data analysis and bioinformatics
The normalized microarray data were statistically analyzed using GeneSpring GX 13.0 software (Agilent Technologies). The differentially expressed genes between LCA and RCA that showed ≥2.0-fold changes at P-values of <0.05 by 1-way ANOVA were identified. The lists of differentially expressed genes were interrogated for statistically significant overrepresented cellular functions and disorders using the Cluster 3.0, TreeView, DAVID, KEGG pathway, and GeneSpring GX 13.0 software programs.
Co-expression network analysis and functional annotation
We constructed co-expression networks using two publicly available microarray datasets (GSE44770 and GSE9647). The first microarray datasets (GSE44770) [80] from the PFC of people with Alzheimer´s disease (n = 129) and unaffected controls (n = 101) were downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). The normalized microarray data were used to generate gene co-expression networks using WGCNA [81]. The second data sets (GSE2639) were from TNF-α treated human vascular endothelial cells (HUVEC, n = 4) and mock-treated HUVEC (n = 4) for 5 h. The normalized expression values were used as input for Weighted Correlation Network Analysis (WGCNA) [81] to generate a network. DAVID (http://david.abcc.ncifcrf.gov/home.jsp) was used to identify the biological processes that were significantly enriched in the genes included in the co-expression modules [82]. P-values <0.05 were considered significant.
Arterial wall thickening and atherosclerotic plaque formation assessment
For studies of arterial wall thickening, APPsw-Tg and non-Tg littermate control mice were maintained on Paigen HFD [83] (Science Diets, Topeka, KS, USA) containing 1.25% cholesterol, 15% fat, and 0.5% cholic acid for 4 weeks following the partial carotid ligation surgery. Animals were euthanized by CO2 gas inhalation and perfused with saline containing heparin, then the aorta and carotid arteries were carefully excised and dissected free of surrounding fat tissue. Aortas and carotid arteries were photographed using a digital microscope device (ToupCam TPH02000A, Hangzhou, China) with a scale bar for reference. LCA and RCA tissues were embedded in Tissue-Tek® optimal cutting temperature compound (Sakura Finetek, Torrance, CA, USA), and stored at -80oC until used. Frozen tissue sections (7 μm) were prepared as previously described [45]. To visualize aortic wall thickness, sections were stained with H&E, then micrographs were taken using a light microscope (Axio Imager A2, Carl Zeiss, Oberkochen, Germany) at 10× magnification and images were analyzed using NIH ImageJ software as described previously [45]. Lesion areas (% opaque area of the total LCA area), lesion size (cross-sectional areas; intima-media area), and wall thickness (intima-media thickness) were quantified using three individual sections from each LCA segment as described previously [84].
For studies of atherosclerotic plaque formation, aorta and carotid arteries from partial carotid ligated ApoE-/-/APPsw-Tg and ApoE-/- mice fed an HFD for 10 days were carefully excised and the surrounding fat tissue was removed by dissection. The RCA and LCA were photographed using a digital microscope device (ToupCam TPH02000A) and then frozen tissue sections were prepared as described above. To visualize atherosclerotic plaques, oil-red-O staining of the tissue sections was carried out as previously described [85]; tissues were imaged using a light microscope (Axio Imager A2, Carl Zeiss, Oberkochen, Germany) at 10× magnification, and micrographs were obtained. Images were analyzed using NIH ImageJ software; lesion area (% opaque area covered by a plaque of the total LCA area) and plaque size were quantified using three individual sections from each LCA segment as previously described [45].
Immunohistochemistry
Frozen sections were fixed in a 1:1 mixture of methanol/acetone for 10 min at room temperature (RT) and then blocked (1 h at RT) using 10% (v/v) donkey serum in phosphate-buffered saline (PBS). Immunohistochemical staining was carried out using the following antibodies: Chi3l1 (1:100; Abcam, Cambridge, UK), VCAM (1:50; BD Pharmingen, Franklin Lakes, NJ, USA), ICAM1 (1:100; Santa Cruz Biotechnology, Dallas, TX, USA), PCNA (1:150; Cell Signaling, Danvers, MA, USA), and MOMA2 (1:150, Abcam) overnight at 4ºC. The sections were subject to incubation with biotinylated secondary antibodies (1:500, Santa Cruz Biotechnology) for 2 h at RT. After washing in PBS, the immunocomplex was visualized using DAB solution (Vector Laboratories, Burlingame, CA, USA) containing 0.08% (v/v) hydrogen peroxide in distilled water, following manufacturer protocol. Sections were dehydrated in a series of graded alcohols, cleared in xylene, and coverslipped using Cytoseal XYL mounting media (Richard-Allan Scientific, Kalamazoo, MI, USA). Micrographs were taken with a light microscope (Zeiss Axio Imager A2) at 10× or 40× magnification.
Adenoviral vector generation for Chi3l1 knockdown and Ad-shRNA-Chi3l1 injection in vivo
An adenoviral vector encoding mouse Chi3l1 shRNA (Ad-shRNA-Chi3l1) was used to knock down Chi3l1 expression. Ad-shRNA-Chi3l1 was constructed (shRNA sense sequence: 5'- GGT TTG ACA GAT ACA GCA ATG-3') by Sirion Biotech (Martinsried, Germany). The U6-shRNA-SV40-pA region of the pO6A5 shuttle vector (pO6A5-U6-mPGK-TagRFP) was transferred via recombination in a BAC vector containing the genome of the replication Ad5-based vector deleted for the E1/E3 genes. Adenoviral particles were produced by construction of the shRNA expression shuttle vector into the HEK-293 cells. Cloning success for the resultant vector was verified by restriction analysis and DNA sequencing. To investigate the role of Chi3l1 on atherosclerosis in vivo, ApoE-/-/APPsw-Tg and ApoE-/-/non-Tg littermate control mice were injected with either Ad-shRNA-Chi3l1 or the empty vector control (Ad-RFP) (108 p.f.u. per animal, via the tail vein) 5 days prior to partial carotid ligation and 10 days HFD exposure.
qPCR
Total RNA was collected from mouse carotid endothelium or EC cultures in 700 μL of QIAzol® and was purified using the miRNeasy Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol. For the measurement of mRNA expression, total RNA was reverse transcribed into complementary DNA (cDNA) using a High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA, USA). The resulting cDNA was subjected to qPCR using the QuantiFast® SYBR® Green PCR master mix (Qiagen) with custom-designed specific primers and incorporating 18S as a house-keeping control on a StepOnePlus™ Real-Time PCR System (Applied Biosystems). Primer sequences used for mRNA expression are listed in Table S14. For the detection of miRNA, cDNA was prepared in a reverse transcription reaction using a miScript II RT Kit (Qiagen). Expression of mature miRNA was determined using a miRNA-specific miScript Primer Assay and a miScript SYBR® Green PCR Kit (Qiagen), following the manufacturer's instructions. Relative fold changes in the expression of the target genes were calculated using RNU6B as an internal control. The fold changes between groups were determined for all targets using the 2ΔΔCt method.
Cell culture and treatment with siRNA and the miRNA mimic
iMAECs were generated from C57BL/6 mice as previously described [86]. HUVECs were purchased from Lonza (Walkersville, MD, USA) and were cultured and maintained as described previously [87]. Primary mouse arterial VSMCs were isolated from the thoracic aorta of C57BL/6 mice and maintained as described previously [88]. Chi3l1 siRNA (OriGene, Rockville, MD, USA) or a miR-342-3p mimic (mirVana™, Thermo Fisher Scientific, Waltham, MA, USA) were transfected into cells using the Lipofectamine® RNAiMAX transfection reagent (Thermo Fisher Scientific) in normal culture medium, following the manufacturer's protocol.
Monocyte-EC binding assay
Following the treatment of iMAECs or HUVECs with rhChi3l1 for 24 h, a monocyte-EC binding assay was performed using fluorescence-labeled THP-1 cells as described previously [89]. To test the effect of Chi3l1 knockdown or miR-342-3p overexpression on monocyte-EC adhesion, iMAECs or HUVECs were transfected with Chi3l1 siRNA or the miR-342-3p mimic for 24 h, then cells were treated with IL-6 (10 ng/mL) for 24 h and a monocyte-EC binding assay was performed.
NO measurement
Cellular NO in iMAECs and HUVECs was quantified using the fluorescence spectra of intracellular 4,5-diaminofluorescein diacetate (DAF-2 DA, Merck Millipore, Darmstadt, Germany) as described previously [90]. Cells were pre-incubated with HEPES buffer (5 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 5 mM glucose, pH7.4) containing 1 μM Ca2+ ionophore, A23187 (Sigma) for 20 min. Subsequently, cells were incubated at 37ºC and 5% CO2 with 10 μM DAF-2 DA for 15 min and were harvested and lysed by sonication. Supernatants were then obtained by centrifugal fractionation, diluted, and scanned using a spectrofluorophotometer (RF 5301PC, Shimadzu, Kyoto, Japan) at excitation and emission of 495 and 515 nm, respectively. NO was calculated using DAF-2 DA fluorescence intensity [90].
Smooth muscle cell proliferation and migration
To test the effect of rhChi3l1 on VSMC proliferation, VSMCs were placed in serum-free medium overnight and stimulated with rhChi3l1 for 24 h; then, cell proliferation was determined using a BrdU cell proliferation assay kit (BioVision, Milpitas, CA, USA) following the manufacturer's protocol. To test the effect of Chi3l1 knockdown or miR-342-3p overexpression on VSMC proliferation, cells were transfected with Chi3l1 siRNA or the miR-342-3p mimic for 24 h and then treated with 25 ng/mL PDGF-BB for 24 h, following which cell proliferation was determined. To assess migration, VSMCs were seeded in a μ-Dish with Culture-Insert for cell migration assays (ibidi, Martinsried, Germany) and were maintained in serum-free media overnight prior to the migration assay. VSMCs were stimulated with rhChi3l1 or PDGF-BB, then the migrated cells were monitored for 24 h by taking a picture every 3 h. The cell-covered area was quantified using NIH ImageJ software.
Preparation of whole-cell lysates and immunoblotting
Following treatment, cells were washed three times with ice-cold PBS and lysed with radioimmunoprecipitation assay buffer. The protein content of each sample was determined using a DC assay kit (Bio-Rad, Hercules, CA, USA). Aliquots of cell lysates were resolved by size by 10% SDS-PAGE and subsequently transferred to a PVDF membrane (EMD Millipore, Billerica, MA, USA). The membrane was incubated with a Chi3l1 antibody (Abcam; 1:1,000) at 4ºC, followed by incubation with the secondary antibody (1:5,000) for 1 h at RT. Protein expression was detected using a chemiluminescence method. Full-sized scans of all western blots are provided in Figure S8.
Dual-luciferase activity assays
Measurement of firefly luciferase activity was obtained at RT using the Luc-Pair miR Luciferase Assay Kit (GeneCopoeia, Rockville, MD, USA) and a luminometer (1420 Luminescence Counter, PerkinElmer, Waltham, MA, USA). Dual-luciferase reporter constructs containing the 3′-UTR of Chi3l1 containing a miR-342-3p-binding site (5′-GUG AGA GUC ACA GUG UGA G-3′) (GeneCopoeia) or a mutated miR-342-3p-binding site (5′-GUG AGA GUC ACA UGU GUC U-3′) (custom cloned at Cosmogenetech Co., Seoul, Korea) were transfected into iMAECs using a Nucleofection Kit (Lonza). iMAECs were transfected first with wild-type or mutated target gene 3′-UTR using the HUVEC Nucleofector Kit (Lonza) and were allowed to recover for 24 h. The second transfection was performed using increasing concentrations of miR-342-3p mimic and control-miR with the Lipofectamine® RNAiMAX transfection reagent (Thermo Fisher Scientific). Firefly and Renilla luciferase activities were measured using a Luciferase assay system (Promega, Madison, WI, USA) as per manufacturer recommendation.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA). Pairwise comparisons were performed using 1-way Student's t-tests. Data are presented as mean ± SEM of the indicated number of experiments. Differences between groups were considered significant at P-values below 0.05.
Abbreviations
APP: amyloid precursor protein; AD: Alzheimer's disease; APPsw: Swedish mutant of human amyloid precursor protein; Tg: transgenic; ApoE: apolipoprotein E; Chi3l1: chitinase-3-like-1; EC: endothelial cells; VSMC: vascular smooth muscle cell; miR: microRNA; Aβ: amyloid-β; ApoE-/-: apolipoprotein E knockout; VCAM1: vascular cell adhesion molecule 1; ICAM1: intercellular adhesion molecule 1; eNOS: endothelial nitric oxide synthase; CL3P: chitinase-like-3 protein; NSE: neuron-specific enolase; AAA: abdominal aortic aneurysm; RCA: right carotid artery; LCA: left carotid artery; HFD: high-fat diet; PCNA: proliferating cell nuclear antigen; KEGG: Kyoto Encyclopedia of Genes and Genomics; qPCR: quantitative real-time PCR; PFC: dorsolateral prefrontal cortex; TNF-α: tumor necrosis factor alpha; HUVECs: human vascular endothelial cells; iMAECs: immortalized mouse arterial ECs; rhChi3l1: recombinant human Chi3l1 protein; NO: nitric oxide; IL-6: interleukin-6; eNOS: endothelial nitric oxide synthase; PDGF: platelet-derived growth factor; Ad-shRNA: short hairpin RNA-expressing adenoviral vector; RFP: red fluorescent protein; cDNA: complementary DNA.
Acknowledgements
This work was financially supported by the National Research Foundation of Korea [NRF] grant funded by the Korea government (MSIP) (No. MRC, 2017R1A5A2015541), the Ministry of Trade, Industry & Energy (MOTIE, 1415139249) through the fostering project of Osong Academy-Industry Convergence (BAIO), the Functional Districts of the Science Belt support program, Ministry of Science, ICT and Future Planning, and by the INNOPOLIS Foundation funded with a grant in aid from the Korean government through NovaKmed Co., Ltd. (A2016-02-DD-022).
Author contributions
Dong Ju Son., Yu Yeon Jung, Ki Cheon Kim, and Mi Hee Park conducted most of the experiments, performed data analysis, generated most of the experimental mice and were the primary writers of the manuscript. Y.S.S. performed in vitro monocyte-EC adhesion assay and Western blotting assay for iMAECs and HUVECs samples. Sanghyeon Kim and Min Hee Park performed the bioinformatic analysis. Dae Youn Hwang provided APPsw-Tg mice. Sang Bae Han, Jun Chang, and Heonyong Park provided advice throughout the project. Jin Tae Hong supervised the entire project and had a major role in experimental design, data interpretation, and writing the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gupta A, Iadecola C. Impaired Abeta clearance: a potential link between atherosclerosis and Alzheimer's disease. Front Aging Neurosci. 2015;7:115
2. Cummings JL, Cole G. Alzheimer disease. Jama. 2002;287:2335-8
3. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844-66
4. Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F. et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer's disease in the Rotterdam Study. Lancet. 1997;349:151-4
5. Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer's disease: Inflammation, cholesterol, and misfolded proteins. Lancet. 2004;363:1139-46
6. Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P. et al. β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735-41
7. Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gomez MG, Langois CM. et al. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer's disease kindred: a cross-sectional study. Lancet Neurol. 2012;11:1057-65
8. Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS. et al. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:7606-10
9. Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin-1. Brain research Molecular brain research. 1992;16:128-34
10. d'Uscio LV, Das P, Santhanam AVR, He T, Younkin SG, Katusic ZS. Activation of PPARδ prevents endothelial dysfunction induced by overexpression of amyloid-β precursor protein. Cardiovascular research. 2012;96:504-12
11. Li L, Cao D, Garber DW, Kim H, Fukuchi K. Association of aortic atherosclerosis with cerebral beta-amyloidosis and learning deficits in a mouse model of Alzheimer's disease. The American journal of pathology. 2003;163:2155-64
12. Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nature medicine. 2006;12:1005-15
13. Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E. et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nature medicine. 2003;9:907-13
14. Puglielli L, Friedlich AL, Setchell KD, Nagano S, Opazo C, Cherny RA. et al. Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. The Journal of clinical investigation. 2005;115:2556-63
15. De Meyer GR, De Cleen DM, Cooper S, Knaapen MW, Jans DM, Martinet W. et al. Platelet phagocytosis and processing of beta-amyloid precursor protein as a mechanism of macrophage activation in atherosclerosis. Circulation research. 2002;90:1197-204
16. Austin SA, Sens MA, Combs CK. Amyloid precursor protein mediates a tyrosine kinase-dependent activation response in endothelial cells. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:14451-62
17. Jans DM, Martinet W, Van De Parre TJL, Herman AG, Bult H, Kockx MM. et al. Processing of amyloid precursor protein as a biochemical link between atherosclerosis and Alzheimer's disease. Cardiovascular and Hematological Disorders - Drug Targets. 2006;6:21-34
18. Tibolla G, Norata GD, Meda C, Arnaboldi L, Uboldi P, Piazza F. et al. Increased atherosclerosis and vascular inflammation in APP transgenic mice with apolipoprotein E deficiency. Atherosclerosis. 2010;210:78-87
19. Austin SA, Combs CK. Amyloid precursor protein mediates monocyte adhesion in AD tissue and apoE(-)/(-) mice. Neurobiology of aging. 2010;31:1854-66
20. Van De Parre TJ, Guns PJ, Fransen P, Martinet W, Bult H, Herman AG. et al. Attenuated atherogenesis in apolipoprotein E-deficient mice lacking amyloid precursor protein. Atherosclerosis. 2011;216:54-8
21. Hwang DY, Cho JS, Lee SH, Chae KR, Lim HJ, Min SH. et al. Aberrant expressions of pathogenic phenotype in Alzheimer's diseased transgenic mice carrying NSE-controlled APPsw. Exp Neurol. 2004;186:20-32
22. Gratchev A, Schmuttermaier C, Mamidi S, Gooi L, Goerdt S, Kzhyshkowska J. Expression of Osteoarthritis Marker YKL-39 is Stimulated by Transforming Growth Factor Beta (TGF-beta) and IL-4 in Differentiating Macrophages. Biomarker Insights. 2008;3:39-44
23. Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27
24. Choi J, Lee HW, Suk K. Plasma level of chitinase 3-like 1 protein increases in patients with early Alzheimer's disease. J Neurol. 2011;258:2181-5
25. Wildsmith KR, Schauer SP, Smith AM, Arnott D, Zhu Y, Haznedar J. et al. Identification of longitudinally dynamic biomarkers in Alzheimer's disease cerebrospinal fluid by targeted proteomics. Mol Neurodegener. 2014;9:22
26. Rosen C, Andersson CH, Andreasson U, Molinuevo JL, Bjerke M, Rami L. et al. Increased Levels of Chitotriosidase and YKL-40 in Cerebrospinal Fluid from Patients with Alzheimer's Disease. Dement Geriatr Cogn Dis Extra. 2014;4:297-304
27. Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG. et al. Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol. 2015;72:1029-42
28. Kester MI, Teunissen CE, Sutphen C, Herries EM, Ladenson JH, Xiong C. et al. Cerebrospinal fluid VILIP-1 and YKL-40, candidate biomarkers to diagnose, predict and monitor Alzheimer's disease in a memory clinic cohort. Alzheimers Res Ther. 2015;7:59
29. Alcolea D, Martinez-Lage P, Sanchez-Juan P, Olazaran J, Antunez C, Izagirre A. et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology. 2015;85:626-33
30. Gong Z, Xing S, Zheng F, Xing Q. Increased expression of chitinase 3-like 1 in aorta of patients with atherosclerosis and suppression of atherosclerosis in apolipoprotein E-knockout mice by chitinase 3-like 1 gene silencing. Mediators of inflammation. 2014;2014:905463
31. Rocchiccioli S, Cecchettini A, Ucciferri N, Terreni M, Viglione F, Trivella MG. et al. Site-Specific Secretome Map Evidences VSMC-Related Markers of Coronary Atherosclerosis Grade and Extent in the Hypercholesterolemic Swine. Dis Markers. 2015;2015:465242
32. Jafari B, Elias JA, Mohsenin V. Increased plasma YKL-40/chitinase-3-like-protein-1 is associated with endothelial dysfunction in obstructive sleep apnea. PloS one. 2014;9:e98629
33. Erfan G, Guzel S, Alpsoy S, Rifaioglu EN, Kaya S, Kucukyalcin V. et al. Serum YKL-40: a potential biomarker for psoriasis or endothelial dysfunction in psoriasis? Mol Cell Biochem. 2015;400:207-12
34. Ridker PM, Chasman DI, Rose L, Loscalzo J, Elias JA. Plasma levels of the proinflammatory chitin-binding glycoprotein YKL-40, variation in the chitinase 3-like 1 gene (CHI3L1), and incident cardiovascular events. Journal of the American Heart Association. 2014;3:e000897
35. Kjaergaard AD, Johansen JS, Bojesen SE, Nordestgaard BG. Role of inflammatory marker YKL-40 in the diagnosis, prognosis and cause of cardiovascular and liver diseases. Crit Rev Clin Lab Sci. 2016:1-13
36. Wu S, Hsu LA, Cheng ST, Teng MS, Yeh CH, Sun YC. et al. Circulating YKL-40 level, but not CHI3L1 gene variants, is associated with atherosclerosis-related quantitative traits and the risk of peripheral artery disease. International journal of molecular sciences. 2014;15:22421-37
37. Sui X, Gao C. Association of serum YKL-40 levels with the presence and severity of coronary artery disease in patients with obstructive sleep apnea syndrome. Clin Invest Med. 2013;36:E306-11
38. Maegdefessel L, Spin JM, Raaz U, Eken SM, Toh R, Azuma J. et al. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nature communications. 2014;5:5214
39. Romaine SP, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. 2015;101:921-8
40. Calway T, Kim GH. Harnessing the Therapeutic Potential of MicroRNAs for Cardiovascular Disease. J Cardiovasc Pharmacol Ther. 2015;20:131-43
41. Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A. et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. American journal of physiology Heart and circulatory physiology. 2009;297:H1535-43
42. Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ. et al. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood. 2010;116:e66-73
43. Maddens B, Ghesquiere B, Vanholder R, Demon D, Vanmassenhove J, Gevaert K. et al. Chitinase-like proteins are candidate biomarkers for sepsis-induced acute kidney injury. Mol Cell Proteomics. 2012;11:M111 013094
44. Jin HM, Copeland NG, Gilbert DJ, Jenkins NA, Kirkpatrick RB, Rosenberg M. Genetic characterization of the murine Ym1 gene and identification of a cluster of highly homologous genes. Genomics. 1998;54:316-22
45. Son DJ, Kumar S, Takabe W, Kim CW, Ni CW, Alberts-Grill N. et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nature communications. 2013;4:3000
46. Tan L, Yu JT, Tan MS, Liu QY, Wang HF, Zhang W. et al. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer's disease. J Alzheimers Dis. 2014;40:1017-27
47. Lugli G, Cohen AM, Bennett DA, Shah RC, Fields CJ, Hernandez AG. et al. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PloS one. 2015;10:e0139233
48. Alberts-Grill N, Rezvan A, Son DJ, Qiu H, Kim CW, Kemp ML. et al. Dynamic immune cell accumulation during flow-induced atherogenesis in mouse carotid artery: an expanded flow cytometry method. Arterioscler Thromb Vasc Biol. 2012;32:623-32
49. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826-37
50. Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J Neuroinflammation. 2005;2:9
51. Wilcock DM, Zhao Q, Morgan D, Gordon MN, Everhart A, Wilson JG. et al. Diverse inflammatory responses in transgenic mouse models of Alzheimer's disease and the effect of immunotherapy on these responses. ASN Neuro. 2011;3:249-58
52. Di Rosa M, Malaguarnera L. Chitinase 3 Like-1: An Emerging Molecule Involved in Diabetes and Diabetic Complications. Pathobiology. 2016;83:228-42
53. Hartl D, Lee CG, Da Silva CA, Chupp GL, Elias JA. Novel biomarkers in asthma: chemokines and chitinase-like proteins. Curr Opin Allergy Clin Immunol. 2009;9:60-6
54. Volck B, Price PA, Johansen JS, Sorensen O, Benfield TL, Nielsen HJ. et al. YKL-40, a mammalian member of the chitinase family, is a matrix protein of specific granules in human neutrophils. Proc Assoc Am Physicians. 1998;110:351-60
55. Vos K, Steenbakkers P, Miltenburg AM, Bos E, van Den Heuvel MW, van Hogezand RA. et al. Raised human cartilage glycoprotein-39 plasma levels in patients with rheumatoid arthritis and other inflammatory conditions. Ann Rheum Dis. 2000;59:544-8
56. Mizoguchi E. Chitinase 3-like-1 exacerbates intestinal inflammation by enhancing bacterial adhesion and invasion in colonic epithelial cells. Gastroenterology. 2006;130:398-411
57. Chupp GL, Lee CG, Jarjour N, Shim YM, Holm CT, He S. et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. The New England journal of medicine. 2007;357:2016-27
58. Kawada M, Seno H, Kanda K, Nakanishi Y, Akitake R, Komekado H. et al. Chitinase 3-like 1 promotes macrophage recruitment and angiogenesis in colorectal cancer. Oncogene. 2012;31:3111-23
59. Boot RG, van Achterberg TA, van Aken BE, Renkema GH, Jacobs MJ, Aerts JM. et al. Strong induction of members of the chitinase family of proteins in atherosclerosis: chitotriosidase and human cartilage gp-39 expressed in lesion macrophages. Arteriosclerosis, thrombosis, and vascular biology. 1999;19:687-94
60. Rehli M, Niller HH, Ammon C, Langmann S, Schwarzfischer L, Andreesen R. et al. Transcriptional regulation of CHI3L1, a marker gene for late stages of macrophage differentiation. The Journal of biological chemistry. 2003;278:44058-67
61. Bakirci EM, Unver E, Degirmenci H, Kivanc T, Gunay M, Hamur H. et al. Serum YKL-40/chitinase 3-like protein 1 level is an independent predictor of atherosclerosis development in patients with obstructive sleep apnea syndrome. Turk Kardiyol Dern Ars. 2015;43:333-9
62. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13:271-82
63. Hata A. Functions of microRNAs in cardiovascular biology and disease. Annual review of physiology. 2013;75:69-93
64. Vilardo E, Barbato C, Ciotti M, Cogoni C, Ruberti F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. The Journal of biological chemistry. 2010;285:18344-51
65. Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y. et al. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer's-associated pathogenesis in SAMP8 mice. Neurobiology of aging. 2012;33:522-34
66. Hebert SS, Horre K, Nicolai L, Bergmans B, Papadopoulou AS, Delacourte A. et al. MicroRNA regulation of Alzheimer's Amyloid precursor protein expression. Neurobiol Dis. 2009;33:422-8
67. Liu CG, Song J, Zhang YQ, Wang PC. MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer's disease. Molecular medicine reports. 2014;10:2395-400
68. Jin T, Lu Y, He QX, Wang H, Li BF, Zhu LY. et al. The Role of MicroRNA, miR-24, and Its Target CHI3L1 in Osteomyelitis Caused by Staphylococcus aureus. J Cell Biochem. 2015;116:2804-13
69. Sarma NJ, Tiriveedhi V, Subramanian V, Shenoy S, Crippin JS, Chapman WC. et al. Hepatitis C virus mediated changes in miRNA-449a modulates inflammatory biomarker YKL40 through components of the NOTCH signaling pathway. PloS one. 2012;7:e50826
70. Kaneko M, Satomi T, Fujiwara S, Uchiyama H, Kusumoto K, Nishimoto T. AT1 receptor blocker azilsartan medoxomil normalizes plasma miR-146a and miR-342-3p in a murine heart failure model. Biomarkers. 2016:1-8
71. Czimmerer Z, Varga T, Kiss M, Vazquez CO, Doan-Xuan QM, Ruckerl D. et al. The IL-4/STAT6 signaling axis establishes a conserved microRNA signature in human and mouse macrophages regulating cell survival via miR-342-3p. Genome Med. 2016;8:63
72. Tai MC, Kajino T, Nakatochi M, Arima C, Shimada Y, Suzuki M. et al. miR-342-3p regulates MYC transcriptional activity via direct repression of E2F1 in human lung cancer. Carcinogenesis. 2015;36:1464-73
73. Wang L, Xu L, Xu M, Liu G, Xing J, Sun C. et al. Obesity-Associated MiR-342-3p Promotes Adipogenesis of Mesenchymal Stem Cells by Suppressing CtBP2 and Releasing C/EBPalpha from CtBP2 Binding. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2015;35:2285-98
74. Xie X, Liu H, Wang M, Ding F, Xiao H, Hu F. et al. miR-342-3p targets RAP2B to suppress proliferation and invasion of non-small cell lung cancer cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:5031-8
75. Zhao L, Zhang Y. miR-342-3p affects hepatocellular carcinoma cell proliferation via regulating NF-kappaB pathway. Biochemical and biophysical research communications. 2015;457:370-7
76. Li XR, Chu HJ, Lv T, Wang L, Kong SF, Dai SZ. miR-342-3p suppresses proliferation, migration and invasion by targeting FOXM1 in human cervical cancer. FEBS letters. 2014;588:3298-307
77. Huang H, Fan L, Zhan R, Wu S, Niu W. Expression of microRNA-10a, microRNA-342-3p and their predicted target gene TIAM1 in extranodal NK/T-cell lymphoma, nasal type. Oncology letters. 2016;11:345-51
78. Hwang DY, Cho JS, Lee SH, Chae KR, Lim HJ, Min SH. et al. Aberrant expressions of pathogenic phenotype in Alzheimer's diseased transgenic mice carrying NSE-controlled APPsw. Experimental Neurology. 2004;186:20-32
79. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185-93
80. Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA. et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707-20
81. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559
82. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3
83. Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231-40
84. Son DJ, Kim SY, Han SS, Kim CW, Kumar S, Park BS. et al. Piperlongumine inhibits atherosclerotic plaque formation and vascular smooth muscle cell proliferation by suppressing PDGF receptor signaling. Biochemical and biophysical research communications. 2012;427:349-54
85. Nam D, Ni C-W, Rezvan A, Suo J, Budzyn K, Llanos A. et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. American Journal of Physiology - Heart and Circulatory Physiology. 2009;297:H1535-H43
86. Ni CW, Kumar S, Ankeny CJ, Jo H. Development of immortalized mouse aortic endothelial cell lines. Vasc Cell. 2014;6:7
87. Ni CW, Qiu H, Jo H. MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. American journal of physiology Heart and circulatory physiology. 2011;300:H1762-9
88. Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T. A simple method of isolating mouse aortic endothelial cells. Journal of atherosclerosis and thrombosis. 2005;12:138-42
89. Sorescu GP, Sykes M, Weiss D, Platt MO, Saha A, Hwang J. et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress stimulates an inflammatory response. The Journal of biological chemistry. 2003;278:31128-35
90. Kim J, Park J, Choi S, Chi SG, Mowbray AL, Jo H. et al. X-linked inhibitor of apoptosis protein is an important regulator of vascular endothelial growth factor-dependent bovine aortic endothelial cell survival. Circulation research. 2008;102:896-904
Author contact
![]() Corresponding authors: Jin Tae Hong, Ph.D., Professor/Chief of Director, College of Pharmacy & Medical Research Center, Chungbuk National University, 194-31 Osongsaengmyeong 1-ro, Osong-Biocampus Building Room 301, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Korea, Telephone: 82-43-261-2813, FAX: 82-43-268-2732, E-mail: jinthongac.kr or Dong Ju Son, Ph.D., Professor/Chief Technology Officer, Life Science Research Center, NovaKmed Co. Ltd., 1646 Yuseong-daero, Innobiz Park Suite 403, Yuseong-gu, Daejeon 34054, Korea, Telephone: 82-70-4852-2396, E-mail: sondj1ac.kr/sondj1co.kr
Corresponding authors: Jin Tae Hong, Ph.D., Professor/Chief of Director, College of Pharmacy & Medical Research Center, Chungbuk National University, 194-31 Osongsaengmyeong 1-ro, Osong-Biocampus Building Room 301, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Korea, Telephone: 82-43-261-2813, FAX: 82-43-268-2732, E-mail: jinthongac.kr or Dong Ju Son, Ph.D., Professor/Chief Technology Officer, Life Science Research Center, NovaKmed Co. Ltd., 1646 Yuseong-daero, Innobiz Park Suite 403, Yuseong-gu, Daejeon 34054, Korea, Telephone: 82-70-4852-2396, E-mail: sondj1ac.kr/sondj1co.kr