Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviatons
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):830-845. doi:10.7150/thno.22012 This issue Cite
Research Paper
Drug Repurposing Screening Identifies Tioconazole as an ATG4 Inhibitor that Suppresses Autophagy and Sensitizes Cancer Cells to Chemotherapy
Pei-Feng Liu1*, Kun-Lin Tsai2*, Chien-Jen Hsu1*, Wei-Lun Tsai3,4, Jin-Shiung Cheng3, Hsueh-Wei Chang5,6, Chung-Wai Shiau7, Yih-Gang Goan8, Ho-Hsing Tseng1, Chih-Hsuan Wu1, John C. Reed9#, Lee-Wei Yang2,10 ![]() , Chih-Wen Shu1
, Chih-Wen Shu1 ![]()
1. Department of Medical Education and Research, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan;
2. Institute of Bioinformatics and Structural Biology, National Tsing-Hua University, Hsinchu, Taiwan;
3. Division of Gastroenterology and Hepatology, Department of Internal Medicine, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan;
4. School of Medicine, National Yang-Ming University, Taipei, Taiwan;
5. Department of Biomedical Science and Environmental Biology, Kaohsiung Medical University, Kaohsiung, Taiwan;
6. Institute of Medical Science and Technology, National Sun Yat-Sen University, Kaohsiung, Taiwan;
7. Institute of Biopharmaceutical Sciences, National Yang-Ming University, Taipei, Taiwan.
8. Division of Thoracic Surgery, Department of Surgery, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan;
9. Sanford-Burnham Medical Research Institute, Program on Apoptosis and Cell Death Research, and Conrad Prebys Center for Chemical Genomics, La Jolla, CA, USA.
10. Physics Division, National Center for Theoretical Sciences, Hsinchu 30013, Taiwan.
* Co-first authors
# Present address: Roche. Basel, Switzerland
Received 2017-7-20; Accepted 2017-10-31; Published 2018-1-1
Abstract

Background: Tumor cells require proficient autophagy to meet high metabolic demands and resist chemotherapy, which suggests that reducing autophagic flux might be an attractive route for cancer therapy. However, this theory in clinical cancer research remains controversial due to the limited number of drugs that specifically inhibit autophagy-related (ATG) proteins.
Methods: We screened FDA-approved drugs using a novel platform that integrates computational docking and simulations as well as biochemical and cellular reporter assays to identify potential drugs that inhibit autophagy-required cysteine proteases of the ATG4 family. The effects of ATG4 inhibitors on autophagy and tumor suppression were examined using cell culture and a tumor xenograft mouse model.
Results: Tioconazole was found to inhibit activities of ATG4A and ATG4B with an IC50 of 1.3 µM and 1.8 µM, respectively. Further studies based on docking and molecular dynamics (MD) simulations supported that tioconazole can stably occupy the active site of ATG4 in its open form and transiently interact with the allosteric regulation site in LC3, which explained the experimentally observed obstruction of substrate binding and reduced autophagic flux in cells in the presence of tioconazole. Moreover, tioconazole diminished tumor cell viability and sensitized cancer cells to autophagy-inducing conditions, including starvation and treatment with chemotherapeutic agents.
Conclusion: Tioconazole inhibited ATG4 and autophagy to enhance chemotherapeutic drug-induced cytotoxicity in cancer cell culture and tumor xenografts. These results suggest that the antifungal drug tioconazole might be repositioned as an anticancer drug or chemosensitizer.
Keywords: Drug repurposing screen, ATG4 and autophagy inhibitory drug, cancer therapy, docking, molecular dynamics simulations.
Introduction
Macroautophagy (herein referred to as autophagy) is an evolutionarily conserved process by which cells utilize double-membraned autophagosomes to recruit dysfunctional components for their bulk degradation in lysosomes and ultimate recycling [1]. Autophagy plays an important role in cellular homeostasis; therefore, the dysregulation of autophagy contributes to many diseases, including cancer [2-4]. Autophagy can function as a tumor promoter to induce tumor growth, progression and resistance to microenvironmental stresses, such as starvation, hypoxia and epithelial-to-mesenchymal transition [2]. Moreover, abundant evidence has emerged to show that autophagy inhibition synergizes with chemotherapy to trigger tumor cell death [5-7]. Chloroquine (CQ) and its derivative hydroxychloroquine (HCQ), which are clinically used antimalarial drugs, also inhibit autophagy by preventing the acidification of the lysosomal compartment [8]. CQ and HCQ have recently been administered independently or in combination with anticancer drugs in clinical trials for certain types of cancers [7]. Moreover, some data show that HCQ may aid cancer treatments [9]. Nevertheless, the chemosensitizing effects of CQ/HCQ may be autophagy independent and attributable to the destabilization of lysosomes [10] and tumor vasculature [11]. Thus, drugs that target autophagy-related (ATG) proteins could provide an opportunity to test the role of autophagy in clinical studies.
At least thirty-eight ATG genes that are involved in the autophagy machinery have been identified [12]. Specifically, ATG4 is a cysteine protease required for the initiation of ATG8 conjugation to phosphatidylethanolamine (PE) and the deconjugation of PE-ATG8 (ATG8-II) from membranes of autophagosomes or non-autophagosomes to facilitate autophagy [13, 14]. The human genome contains four ATG4 genes (ATG4A, ATG4B, ATG4C and ATG4D) [15] and seven ATG8 genes (two isoforms of LC3A, LC3B/C, GABARAP, GABARAPL1 and GABARAPL2) [16]. Although ATG4B is the most proteolytically active ATG4 protein and exhibits the broadest specificity for substrates among the four ATG4 members, the remaining ATG4 members exhibit proteolytic activity on certain substrates of the GABARAP subfamily [17, 18]. ATG4A cleaves all GABARAP subfamily members (GABARAP, GABARAPL1 and GABARAPL2), whereas ATG4D requires caspase-3 to activate and hydrolyze GABARAPL1 and GABARAPL2 [19]. Overall, currently available information suggests that ATG4 family members have both overlapping and unique functions.
In this study, we employed a platform that integrates computational docking and molecular dynamics (MD) simulations to screen FDA-approved drugs for ATG4 inhibitors. Subsequently, biochemical and cellular ATG4B reporter assays were used to confirm tioconazole as an ATG4 inhibitor. Tioconazole is predicted to occupy the active site of ATG4A/B, and it diminishes autophagic flux in cancer cells. Furthermore, tioconazole suppressed tumor growth and enhanced chemotherapy-induced apoptosis in cancer cells and tumor xenografts. Our results show that tioconazole, a clinical antifungal drug, can inhibit ATG4 to diminish autophagic flux and ultimately sensitize cancer cells to chemotherapeutic drugs.
Materials and Methods
Reagents and Cell Culture
We obtained a list of 1312 FDA-approved drugs from MedChem Express (MCE). To validate hits, drugs purchased from Enzo Life Science were used to screen drugs that inhibit ATG4 with biochemical, yeast and mammalian cell-based reporter assays. Human colorectal cancer HCT116 cells, glioblastoma H4 cells and breast cancer MDA-MB-231 from the ATCC were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, 12100-046) supplemented with 10% HyClone fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 mg/mL). The cells were seeded into two- or three-dimensional culture dishes and treated with Doxorubicin (Dox, Millipore, 324380) or Camptothecin (CPT, Millipore, C9911) with or without tioconazole (Sigma-Aldrich, FL-32099). The treated cells were harvested to determine cell viability, apoptosis and immunoblotting. The detailed information previously described is provided in Supplementary Material. For gene knockdown, cells were transfected in the presence of 5 nM scrambled siRNA (Sigma, SIC002) or siRNA against ATG4 (Ambion, 35623 and 121998 for ATG4A, 20218, s23245 and s23246 for ATG4B, 34931 and 121984 for ATG4C, and 34865 and 149022 for ATG4D) using RNAiMAX (Life Technologies, 13778-150). To generate the shRNA stable cell line, shRNAs against ATG4B (TRCN0000073801), ATG5 (TRCN0000151963) and ATG7 (TRCN0000007584) obtained from The RNAi Consortium (TRC, Taiwan) were infected into HCT116 cells for stable selection. Plasmids for GFP-LC3 (21073) and Lamp1-RFP (1817) were purchased from Addgene and used to evaluate the fusion between autophagosomes and lysosomes via confocal microscopy as described below.
Docking and Explicit Solvent MD Simulations Used in Drug Screening and Inhibitory Mechanism Studies
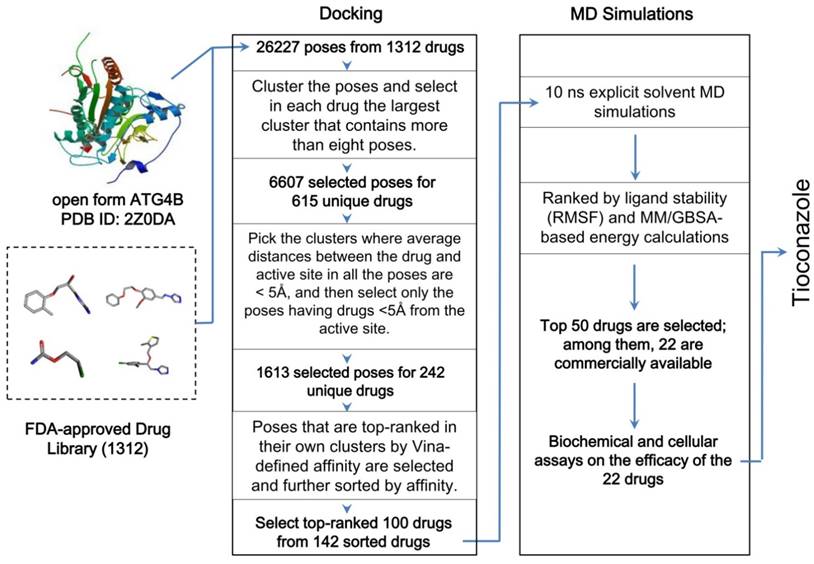
In silico drug screening for 1312 FDA-approved drugs were conducted in two stages. At first, docking software Vina [20] was used to evaluate potentially effective drug candidates based on three factors: (1) how many slightly different docking poses co-occupy the same binding site; (2) how far away these poses are from the active site of the open form of ATG4B, or ATG4B(O), and; (3) how favorable the Vina-defined energies of the individual poses are. Poses belonging to a large cluster (containing > 8 poses in a binding pocket), having a short distance (< 5 Å) from the active site, and bearing a low binding energy with ATG4B were selected and ranked by their docking affinity with the ATG4B(O). Each of the top 100 candidates was then subject to MD simulations for the binding stability check. The drugs that left the binding pocket within 10 ns were deprioritized and those that stayed in the pockets were rank-ordered based on binding energies calculated from MM/GBSA [21] as well as root mean square fluctuations (RMSF) of the distances between drugs and the active site. The trajectories of explicit solvent simulations at body-temperature were analyzed by Generalized-Born (GB) model augmented with the hydrophobic solvent accessible surface area (SA) to obtain the binding energy of the drugs. The 50 top-ranked drugs that have the lowest binding energy (per GBSA) and the highest stability (per RMSF) were selected and 22 of them could be readily purchased from the market for subsequent biochemical and cellular reporter assays (see below). More details on docking and simulations can be found in Supplementary Material.
To understand the molecular mechanism of drug-mediated inhibition of LC3 proteolysis, we conducted further docking experiments for the best drug derived from our screening protocols by AutoDock4 [22]. Open and closed ATG4B as well as the substrate LC3 served as the targets for the small molecule docking. The stability of important docking poses was then examined by standard explicit-solvent MD simulations at 37 oC, 1 atm, for tens or hundreds of nanoseconds.
Structure Preparation
The open (PDB ID: 2Z0D) and closed (PDB ID: 2CY7) conformations of ATG4B feature two major distinctions: (A) In the closed form, the N-terminal tail of ATG4B folds in and covers the active site; in the open form, the tip of the LC3 C-terminus is situated at the ATG4B active site and the N-terminus of ATG4B is held open by another crystallographically adjacent LC3 molecule [45]. (B) In the closed form, the substrate-binding residue Trp142 forms close contacts with Pro260 in the regulatory loop (G257-A263), which makes the active site inaccessible to the substrate LC3. In the open form, Pro260 detaches from Trp142 and allows the LC3 C-terminus to dock. As a result, the open and closed conformations also suggest an “active” and “inactive” ATG4B, respectively. The preparation of 3D structures of FDA-approved drugs ready for docking and MD simulations as well as additional details are available in the Supplementary Material.
ATG4 Reporter Assays
Biochemical ATG4 reporter assays were conducted as previously reported [18, 23]: recombinant ATG4B or ATG4A was mixed with substrate, 100 nM LC3B-PLA2 or GATE16-PLA2, in the presence or absence of tioconazole in the assay buffer. The fluorescence intensity was kinetically monitored for 1 h to determine the ATG4 activity at room temperature with excitation and emission wavelengths of 485 nm and 530 nm, respectively. 40 μL of the ATG4B reporter yeast cells previously described [24] were seeded in a 384-well white plate and grown on minimal synthetic dropout (SD) medium that contained 1% galactose, 0.2% raffinose, BU salts and tryptophan for 24 h. 10 μL of Beta-Glo, a luminescent substrate for β-galactosidase, was added into each well to measure the β-galactosidase activity to reflect the ATG4B proteolysis activity. For the ATG4-cleavable reporter assay in mammalian cells, the N-terminus (NLuc, residues 2-416) and C-terminus (CLuc, residues 398-550) of the luciferase gene were constructed in the expression vector pcDNA3.0. The full length of LC3 was inserted to split NLuc and CLuc with two linkers (GGGGS)2 as shown in Figure 2F. The wild-type reporter vector was transfected into HEK293T cells in a 96-well white plate (Grinner) overnight and treated with tioconazole for 6 h. The luciferase activity was measured via One-Glo (Promega) according to the instruction manual. The non-cleavable mutant G120A was used to normalize the luminescent signal and determine the ATG4 activity.
Autophagic Flux and Immunoblotting
To monitor autophagy activity, the cells were treated with tioconazole (Sigma-Aldrich) in the presence or absence of 20 μM CQ (Sigma-Aldrich, C6628). The cells were briefly rinsed in PBS (Biological Industries, 02-023-1) and lysed with RIPA buffer (1% NP40 (MDBio, 101-9016-45-9), 50 mM Tris HCl, pH 7.5, 150 mM NaCl, 0.25% sodium deoxycholate (Sigma-Aldrich, D6750), 1% sodium dodecyl sulfate (SDS; Calbiochem, 428015), and protease inhibitor cocktail (Roche, 11873580001)). The cell lysates were used for immunoblotting with antibodies against the primary antibodies against ATG4B (A2981), LC3 (L7543), and ACTB (β-actin, A5441) (all purchased from Sigma-Aldrich). The differential accumulations of LC3 in cells with and without CQ were used to quantitate the autophagic flux [25]. For cleavage of GABARAPL2-PLA2 and caspase-3 activation, reaction mixtures or cell lysates were immunoblotted with antibodies against Myc (Roche, 11667149001), caspase-3 (Cell Signaling, 9661), poly (ADP-ribose) polymerase (PARP) (Cell Signaling, 9532) and GAPDH (Cell Signaling, 5174). The proteins were probed with an HRP-labeled secondary antibody (Santa Cruz, sc-2004 or sc-2005) and detected with an ECL reagent. The membrane was scanned and analyzed for the protein expression level with the ChemiDoc XRS Imaging System (Bio-Rad).
Tumor Xenograft
Human colorectal cancer HCT116 cells (2 × 106) were mixed with Matrigel (1:1) and subcutaneously implanted into six-week-old immunodeficient mice (nu/nu, female). Tioconazole (60 mg/kg) and Dox (1 mg/kg) were administered via intraperitoneal injection into the xenografted mice every other day starting at day 3 post-implantation. The tumor size in each mouse was measured every 3 to 4 days with vernier calipers, and the tumor volumes were calculated using the formula (larger diameter) × (smaller diameter)2 × π/6. Tumors were further cut from euthanized mice and embedded in paraffin. The tumors were sectioned into 3 µm for antigen retrieval with EDTA buffer, pH 9.0, using a pressure cooker, followed by immunohistochemistry staining using an anti-LC3 monoclonal antibody (Nano Tools, 5F10, 1:50) and active caspase-3 (Asp175) (Cell signaling, 9661, 1:100), respectively. The protein levels in the tumor sections were determined using the UltraVision™ Quanto Detection System HRP DAB (Thermo Scientific) and observed under microscopy. All animal experiments were approved by the Institutional Animal Care and Use Committee at Kaohsiung Veterans General Hospital.
Bimolecular Fluorescence Complementation Assay
N- and C-terminal Venus expression plasmids, kindly provided by Gordon Mills (51), were used to construct the ATG4B and LC3 chimera genes, respectively. The plasmids (1 μg/well) were transfected with lipofectamine 2000 transfection reagent (Life Technologies, 11668-027) into HEK293T cells in a 6-well plate for 24 h. The cells were imaged via fluorescence microscopy or harvested to quantify the fluorescence intensity via a flow cytometer (Becton Dickinson).
Fluorescence Microscopy
Human glioma H4 cells that harbored GFP-LC3 were seeded onto 0.2% gelatin-coated glass dishes for 48 h and treated with tioconazole. The treated cells were fixed to observe the GFP-LC3 puncta via fluorescence microscopy. For protein colocalization, the H4 stable cells previously described were transfected with 1 μg plasmids Cherry tagged ATG4B or Lamp1-RFP (Addgene, 1817) for 36 h using Lipofectamine 2000 (Invitrogen). The cells transfected with ATG4B-Cherry or Lamp1-RFP were treated with tioconazole, fixed with 3.7% paraformaldehyde at room temperature for 15 min, and subsequently washed with PBS three times prior to observation using a confocal microscope.
Transmission Electron Microscopy (TEM)
HCT116 cells treated with tioconazole (40 μM), CQ (20 μM) for 8 h were rinsed with PBS once and fixed in 2% glutaraldehyde at room temperature for 30 min. The cells were harvested to postfix with 1% osmium tetroxide and stained with 2% uranyl acetate (Agar Scientific) at room temperature for 1 h. The cell pellets were embedded to obtain 80 nm sections with a diamond knife. The sections were stained with 2% uranyl acetate and 0.3% lead citrate (Agar Scientific) and imaged with a JEM1400 PLUS transmission electron microscope (JEOL).
Cell Viability Assay
Cells were seeded into a 384-well plate overnight and treated with various drugs for 24 h. CellTiter-Glo (Promega, Madison, WI, USA) was subsequently added to the treated cells, and the luminescence was read using a Fluoroskan Ascent FL reader (Thermo Fisher Scientific). The following equation was used to evaluate drug interaction (additive, synergistic, or antagonistic): α = SFA × SFB/SFA+B, where SFA and SFB are surviving fractions of drugs A and B, respectively, and SFA+B is the surviving fraction following combined treatment. α = 1, > 1 and < 1 represents additive, synergistic and antagonistic effect, respectively. On the other hand, the cell viability was monitored with an impedance-based instrument system (iCELLigence, ACEA Biosciences) for live cells. Briefly, HCT116 cells (4×104 cells/well) were seeded into electronic plates (E-Plates L8, ACEA Biosciences) with 400 μL of DMEM that contained 10% FBS and incubated for 30 h. The cells were pretreated with tioconazole (40 μM) for 1 h and subsequently treated with Dox (1 μM). The cellular impedance was periodically measured every 15 min until 80 h.
Real-Time PCR
The cells transfected with siRNA were used to extract the total RNA with TRIzol Reagent (Invitrogen, 15596-018). A total of 1 μg RNA was reverse-transcribed with SuperScript II RNase H-Reverse Transcriptase (Invitrogen, 18064-014) for cDNA synthesis. The amount of ATG4A, ATG4B, ATG4C and ATG4D mRNA relative to GAPDH was analyzed by real-time PCR performed in a StepOnePlusTM system (Applied Biosystems) with the SYBR Green Master Mix (Applied Biosystems, 4385612). The primers for the genes are as follows: ATG4A forward 5'-TGCTGGTTGGGGATGTATGC-3' and reverse 5'- GCGTTGGT ATTCTTTGGGTTGT -3', ATG4B forward 5'-GATAGCGCAAATGGGAGTTGG -3' and reverse 5'-CCACGTATCGAAGACAGCAAG-3', ATG4C forward 5'- TAGAGGATCACGTAATTGCAGGA-3' and reverse 5'- GTTGTCAAAGCTGAGCCTTCTAT-3', and ATG4D forward 5'- GGAACAACGTCAAGTACGGTT-3' and reverse 5'-CTCGCCCTCGAAACGGTAG using GAPDH as normalization control.
Flow Cytometry for Mitochondrial Membrane Potential and Apoptosis
For the MMP analysis, the cells were treated with JC-1 (5,5,6,6-tetrachloro-1,1,3,3-tetraethyl- benzimidazolylcarbocyanine iodide, Invitrogen) at 37 °C for 20 min prior to harvesting. The JC-1 aggregates (red) and monomer (green) were used to determine the loss of MMP in cells following treatment. To determine the apoptotic cells, the cells were treated with Dox and tioconazole for 24 h and detached with accutase (eBioscience). The cells were stained with 5 μL of annexin V (AV)-fluorescein isothiocyanate (FITC) and 1 μL of propidium iodide (PI, 100 μg/mL) for 15 min according to the instruction manual (Invitrogen). The stained cells were analyzed for apoptosis and necrosis using FACScan (Becton Dickinson) and FlowJo (Tree Star) software.
Spheroid Cell Culture and Live/ Dead Assay
The cells (4000 cells/well) were seeded into an ultra-low attachment, 96-well plate (Costar®, USA) and grown overnight to form spheroid cells. The cells were treated with tioconazole (40 μM) in the presence or absence of Dox (1 μM) for 48 h. The spheroid cells were stained with Calcein AM (1 μM) and Ethidium homodimer-1 (EthD-1, 2 μM) (LIVE/DEAD® Viability/Cytotoxicity Kit, ThermoFisher Scientific) for 30 minutes. The live (green) and dead (red) spheroid cells were imaged via fluorescence microscopy and quantitated using a Fluoroskan Ascent FL reader (Thermo Fisher Scientific) with excitation at 485 nm and emissions at 530 nm and 645 nm for calcein AM and EthD-1, respectively.
Statistical Analysis
All data are expressed as the mean ± SEM from at least 3 individual experiments. The statistical analyses were performed using a nonparametric 2-tailed Student's t-test or an ANOVA with Tukey's post hoc test. P values less than 0.05 were considered significant (*P<0.05, **P<0.01, ***P<0.001).
Results
In Silico Drug-repurposing Screening to Identify Tioconazole as an ATG4 Inhibitor
Taking the open form of ATG4B structure as the drug target, we used the docking software Vina [20] and AMBER16 package with ff14SB force field to screen a library of 1312 FDA-approved drugs in silico. Vina provided at most 20 docking poses for each drug, which resulted in a total of 26,227 poses. To consider the important entropic effect [26], we clustered the poses in similar loci and binding orientations and prioritized drugs whose largest cluster contained more than 8 poses. Subsequently, we selected the poses that were sufficiently close to the active site (1613 poses of 242 unique drugs) and exhibited favorable Vina-defined binding energy to obtain 142 poses with the most favorable binding mode for each of the 142 unique drugs. The top-ranked 100 drugs with the best binding affinity were subjected to further body-temperature, explicit-solvent MD simulations for stability and binding free energy evaluations based on MM/GBSA [27] (see Methods and Supplementary Material), and the most favorable and purchasable 22 drugs were obtained for the subsequent biochemical and cellular assays to confirm their inhibitory efficacy (Figure 1).
Workflow of the in silico drug screening for 1312 FDA-approved drugs. The drugs were docked into the open form of ATG4B, and the results were sorted by clustered poses, distances between drugs and the active site, and the Vina-defined binding affinity. The top-ranked 100 drugs were further screened for their binding stability using 10 ns explicit-solvent MD simulations at body temperature and subsequent MM/GBSA energy calculations. Additional details are provided in the Methods and Supplementary Material.
LC3-PLA2 and the LC3 cleavable transcription factor were used as ATG4 reporter substrates for biochemical and yeast cell-based assays, respectively, as previously reported [18, 24]. The yeast cell-based assay was initially improved with luminescent substrate and optimized for the robustness of the screening (Figure S1). Four of the 22 drugs screened from the computational platform inhibited the activity of ATG4B by >70% (red points <30% in Figure 2A) in the biochemical reporter assays: Tolfenamic acid, Mefenamic acid, Tioconazole, and Entacapone. The rankings for these four drugs predicted by using only docking results were 30, 36, 82, and 62, which were notably improved to 14, 11, 48, and 1 after further examination by MD simulations and MM/GBSA calculations for binding stability. Tioconazole is the only one of these four compounds to also exhibit strong inhibition in the yeast cell-based assay (Figure 2A). To avoid interference due to antifungal effects in the yeast-based assay, a low dose of tioconazole was subjected to further validation using caspase-1 yeast cell-based assay and caspase-3 biochemical assay as counter screens (Figure 2B) (caspases belong to another class of cysteine proteases), and the results of these assays further suggested that tioconazole selectively inhibited ATG4B. A highly similar analog of tioconazole, miconazole, that originally ranked 83 based on MD simulations (MM/GBSA + ligand RMSF) failed to exhibit as good inhibitory effect on ATG4B as tioconazole (Figure 2C). Entacapone, the highest-ranked drug based on computation, was as effective as tioconazole in the biochemical reporter assay but not cellular assay (Figure 2A), possibly due to its limited membrane permeability.
Screening and Evaluation of Drugs for ATG4 Inhibition. (A) The ability of the 22 hits obtained from the in silico drug screening to inhibit ATG4 was evaluated using LC3-PLA2 biochemical assays (red dots) and ATG4 reporter yeast cells (green dots). EGTA (5 mM, red dot line) and NEM (10 mM, green dot line) were used as positive controls for biochemical and cellular ATG4 reporter assays, respectively. (B) Tioconazole (Tc, 2.5 μM) was confirmed with ATG4 reporter yeast cells and LC3-PLA2 biochemical assay and counter assayed with caspase-1 reporter yeast cells for selectivity (left panel). Various concentrations of Tc were mixed with 0.5 nM caspase-3 and 100 μM Ac-DEVD-AFC to determine the effects of Tc on the caspase-3 activity (right panel). (C) The activities of 2-fold serially titrated Tc and its analog miconazole (Mc) on ATG4B were compared with an LC3-PLA2 assay. (D) ATG4B (0.1 nM) or (E) ATG4A (5 nM) were mixed with 2-fold serially diluted Tc and 100 nM GABARAPL2-PLA2 to determine the IC50 of Tc for ATG4 members. Quantitative results are shown in each panel (n=4). (D) ATG4B (1 nM) or (E) ATG4A (50 nM) were mixed with 10-fold serially diluted Tc and 500 nM GABARAPL2-PLA2 for 1 h. The cleavage of GABARAPL2-PLA2 was validated with immunoblotting using an anti-Myc antibody. (F) The expression vector that encoded ATG4-cleavable luciferase was constructed with LC3 and a luciferase chimera gene, as shown in the schematic diagram (upper panel). HEK293T cells were transfected with scramble siRNA (siCtrl) or siRNA against ATG4B (siATG4B) for 48 h, followed by transfection with the ATG4 reporter vector for 24 h. The luciferase activity was measured with a luminescent reader (n=6), and the knockdown efficiency of ATG4B was determined using immunoblotting. (G) HEK293T cells were transfected overnight with the ATG4 reporter vector as described in (I) and treated with Tc for 8 h to measure the luciferase activity (lower panel). The cleavage of LC3 and luciferase chimera protein by ATG4 in cells treated with Tc or untreated cells was verified via immunoblotting (upper panel). (H) N-terminal Venus fused with ATG4B and C-terminal Venus fused with LC3 were co-transfected into HEK 293T cells. After overnight culture, the cells were fixed to observe Venus complementary under fluorescence microscopy (right panel). Bar: 100 μm. (I) The transfected cells were treated with Tc for 8 h and fixed for flow cytometry to quantify Venus fluorescence using GFP as a counter assay. The results are expressed as the mean ± SEM from at least 3 individual experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
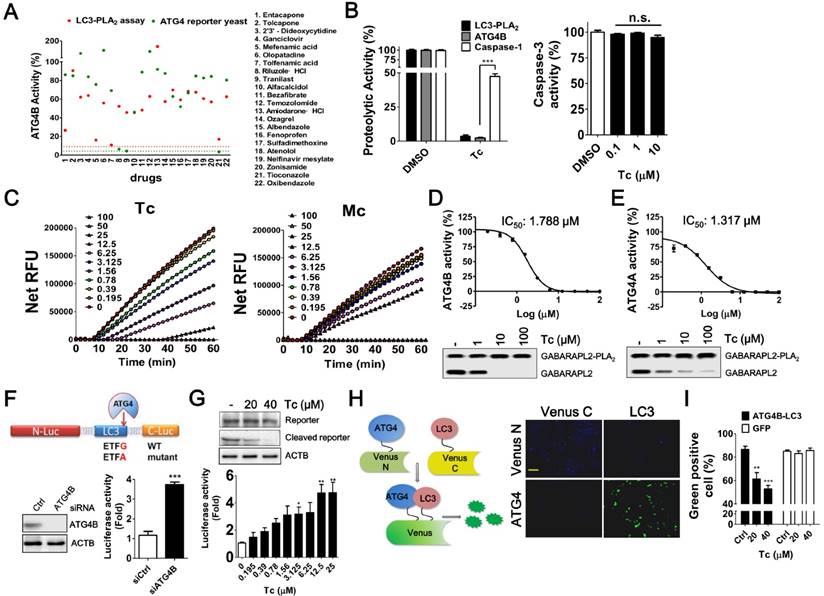
The ability of tioconazole to inhibit ATG4B and ATG4A proteolytic activity were assessed with a biochemical assay using GABARAL2-PLA2 as a common substrate reporter for both ATG4B and ATG4A (Figure 2D-E). The half-maximal inhibitory concentrations (IC50) of tioconazole for ATG4B and ATG4A were 1.8±0.16 μM and 1.3±0.18 μM, respectively, which indicates that tioconazole is ~30-fold more potent than currently reported ATG4 inhibitors [28]. Moreover, immunoblotting results confirmed that tioconazole blocked the cleavage of GABARAL2-PLA2 by ATGA and ATG4B (Figure 2D-E).
Next, the ability of tioconazole to inhibit ATG4 proteolysis in mammalian cells was examined using ATG4 cleavable luciferase assay (Figure 2F). Briefly, LC3 was constructed to split N-terminal and C-terminal luciferase in a mammalian expression vector, as indicated (Figure 2F). Silencing ATG4B and treatment with tioconazole increased the luciferase activity and attenuated the cleavage of chimera luciferase in the reporter cells (Figure 2G), which suggests that tioconazole might inhibit ATG4 in mammalian cells.
To further examine the effects of tioconazole on the binding of ATG4 to LC3, the Venus N-terminal-ATG4B and Venus C-terminal-LC3 chimera expression vectors were transfected into HEK293T cells (Figure 2H), and the interaction between ATG4B and LC3 in the presence or absence of tioconazole was assessed using flow cytometry (Figure 2I). The bimolecular fluorescence complementation (BiFC) results indicated that tioconazole reduced the fluorescence emitted from the formation of the ATG4B and LC3 complex and did not affect GFP fluorescence (Figure 2I), which suggests that tioconazole interfered with the binding between ATG4 and LC3 in living cells.
Docking and MD Simulations Further Support Direct Blockage of the ATG4B Active Site as the Primary Inhibitory Effect of Tioconazole
To investigate the molecular origin of the tioconazole-mediated inhibition of ATG4 proteolysis, we conducted further docking experiments for both the open (active, Figure 3A, PDB code 2Z0D) and closed (inactive, Figure 3B, PDB code 2CY7) forms of ATG4B using AutoDock4 [22]; the results are shown in Figure 3. Tioconazole, which consists of 3-, 5- and 6-membered rings, namely, dichlorophenyl, chlorothiophenyl and imidazole rings (PubChem CID: 5482), has been shown to preferably dock at the active site of the open form of ATG4B (ATG4B (O), Figure 3C). Similar results were obtained for the modeled open form of ATG4A (Figure S2), which confirms the previously demonstrated specificity. We also found that tioconazole was able to interact with Asp278, a key catalytic residue, even in the closed form of ATG4B (Figure 3D), when most of the active site is shielded by the enzyme's N-terminus. This finding is consistent with the previously reported ability of several lead compounds to interact with part of the active site, even in the closed state of ATG4B [28]. However, most interactions were predicted to occur in a canyon flanked by the N-terminus and the main body of ATG4B toward the root of the N-terminus (Figure 3D). The top-ranked docking poses (Tables S1) are summarized in Supplementary Material. Interestingly, we also identified highly scored docking poses in a cluster that occupies the binding site for ATG4B's N-terminus in LC3 (Figure 3E). LC3's seizure of ATG4B's N-terminus helps maintain the enzyme in an open form and consequently active state. Hence, moderately populated tioconazole in the N-term binding site of LC3 could possibly impair the stabilization of an active enzyme by the substrate and consequently reduce its proteolytic activity.
Docking and MD Simulations for Tioconazole Binding to ATG4. (A) The structures of the open/active (PDB code: 2Z0D, ATG4B (O)) and (B) closed/inactive (PDB code: 2CY7, ATG4B (C)) forms of ATG4B are shown in panels A and B (in white), respectively. Together with the open form, LC3 with a cleaved C-terminus docked into the active site (red protein) and LC3 from one of the adjacent crystallographic unit cell (green protein) are shown. The active site of ATG4B consists of the residues Cys74 (the catalytic cysteine), Asp278 and His280, shown in licorice. The adjacent residues, Trp142, Pro260 and Asn261 (in licorice), form a substrate-binding cavity, with the latter two residues located at a regulatory loop that spatially flanks the cavity. Note the differences in the N-terminus positions in the open and closed forms of ATG4B. Panels C, D and E show the 100 docking poses obtained from AutoDock for the active (C) and inactive (D) forms of ATG4B as well as LC3 (E). The residues of LC3 that interact with the N-terminal tail of ATG4B, observed via crystallography and later confirmed by NMR[45], are shown in thick licorice. Tioconazole is shown as a transparent green ball-and-stick structure, and residues in proteins are color-coded in blue (infrequent) → white → red (frequent) by the number of times they are in contact with ligands for the examined 100 docking poses (within 4 Å). The spatial “regions” that indicate the locations of clustered poses identified by AutoDock (see the rightmost column in Table S1) are numbered in red. The docking results for clusters 1, 2, 3 and 6, rank-ordered, are shown in panel (F). Tioconazole is shown as a ball-and-stick structure, and the active site residues Cys74, Asn278, and His280 as well as the adjacent residues Trp142, Pro260 and Asn261 are presented in thick licorice. Surrounding residues with atoms in close contact (< 4 Å) with tioconazole are shown in thin licorice. Transparent clouds are colored in blue → white → red to show atoms with an increased frequency of contacts with tioconazole, summing all 100 docking poses. Also see Figure S2 and the supplemental movies.
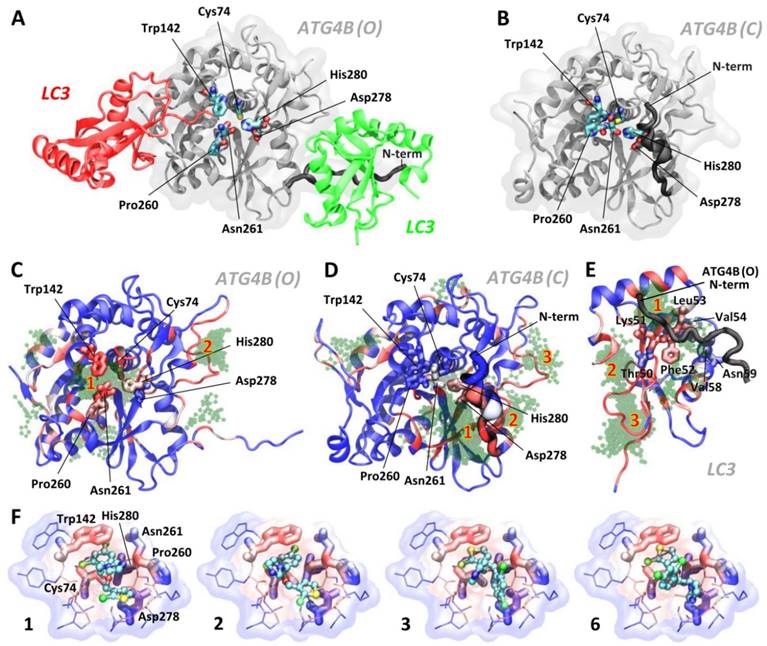
The stability of several highly ranked docking poses (in terms of the binding free energy and the size of the cluster) in ATG4B(O) was subsequently assessed using MD simulations. The highest scored poses (ranks 1, 2, 3 and 6) suggested that tioconazole blocked the entry of the C-terminus of LC3 into the catalytic pocket and most frequently contacted the catalytic Cys74 and Trp142 residues via its dichlorophenyl or chlorothiophenyl rings (Figure 3F). MD simulations demonstrated that the ligands of ranks 1 and 2 remained with the enzyme at the active site throughout the entire 100 ns simulations (Movies S1 and S2), whereas the ligands of ranks 3 and 6 left the enzyme at 23.3 ns and 10.5 ns, respectively (Movies S3 and S4). Interestingly, the imidazole ring of tioconazole in rank 1 swung to the opposite side of the active site at 2.8 ns and subsequently mimicked the rank 2 pose (Movies S1 and S2). Conversely, ligands in the rank 1 pose in the “closed” ATG4B and those in LC3's N-terminal binding site left the binding site after 8.5 ns and 13.0 ns, respectively, suggesting that binding was only transient (Movies S5 and S6). Based on these findings, we conclude that tioconazole primarily inhibits proteolytic function by directly blocking the active site in the open form of ATG4B.
Tioconazole Accumulates Autophagosomes and Diminishes Autophagic Flux in Cancer Cells
Inhibiting ATG4 may increase or decrease LC3 association with the autophagosome because ATG4 plays a dual role: it first mediates conjugation of LC3 to lipids and subsequently deconjugates LC3-II by acting as a hydrolase to remove the lipids [29, 30]. We found that the siRNA-mediated silencing of ATG4 family members or treatment with tioconazole resulted in the accumulation of GFP-LC3-II puncta (Figure 4A-B). Similarly, transmission electron microscopy results showed that the number of autophagosomes was increased in the tioconazole-treated cells, whereas very few autophagosomes were observed in untreated cells (Figure 4E-F).
Effects of Tioconazole on Autophagic Activity in Cancer Cells. (A) Human glioma H4 cells that stably express GFP-LC3 were transfected with 5 nM non-targeting siRNA (Ctrl) or siRNA against ATG4 family members (ATG4) for 48 h, and the knockdown efficiency of ATG4 was verified with real time PCR. (B) The knocked-down cells were fixed for observation via fluorescence microscopy, and the number of GFP-LC3 puncta is quantified in the right panel. Bar: 20 μm. (C) The cells that stably express GFP-LC3 were treated with tioconazole for 8 h and fixed to observe the GFP-LC3 puncta, which were (D) quantified via fluorescence microscopy. The autophagy inhibitor CQ was used as a positive control. Bar: 20 μm. (E) HCT16 cells treated with tioconazole (Tc, 40 μM) or CQ (20 μM) for 6 h were fixed and imaged with TEM. Representative autophagic vacuoles (AVs) are shown. Arrowhead: autophagosome. Bar: 200 nm. (F) The numbers of AVs for each optical section (9 μm2) are quantified (n=8). (G) HCT116 cells expressing GFP-LC3 and RFP-Lamp1 were treated with Tc for 6 h and fixed to observe colocalization of GFP-LC3 and RFP-Lamp1 with confocal microscopy. GFP-LC3 that had colocalized with or was surrounded by RFP-Lamp1 was identified as fusion between autophagosomes and lysosomes. The colocalization coefficients of images were quantified by the Ziess LSM 710 Software and are shown in the right panel. Bar: 20 μm. (H) HCT16 cells were transfected with siRNA (5 nM) for 66 h and then treated with CQ (20 μM) for 2 h. Cells were harvested for immunoblotting using antibodies against LC3, ATG4B or ACTB, and autophagy flux was quantified in untreated cells and cells treated with CQ based on changes in LC3-II. (I) H4, HCT116 or MDA-MB-231 cells were treated with Tc for 6 h in the presence (+) or absence (-) of CQ (20 μM) and harvested for immunoblotting. (J) The autophagic flux was quantified as described in (I). Representative data are shown, and the quantified results are expressed as the mean ± SEM from at least 3 individual experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
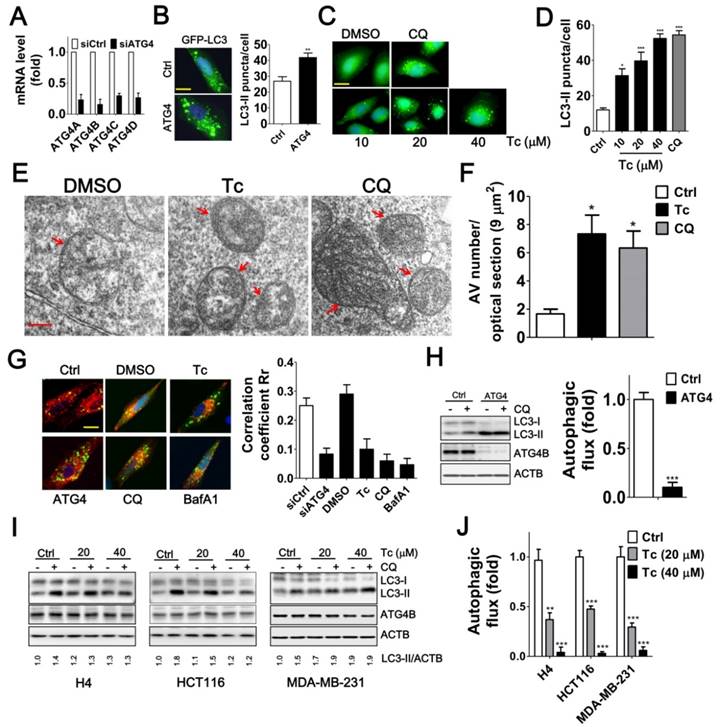
Moreover, ATG4 deconjugates LC3-II from the autophagosome to facilitate autophagosome maturation for fusion with lysosomes in cells [14, 31]. Likewise, silencing ATG4 and treatment with tioconazole decreased the co-localization of LC3 and LAMP1, which are typical markers of autophagosomes and lysosomes, respectively (Figure 4G and S3). In addition, since both autophagy inducers and a block in downstream steps increase the levels of LC3-II and autophagosomes, the differential amount of LC3-II between cells with and without autophagy inhibitor CQ was used to precisely measure LC3-II turnover [32], which is defined as autophagic flux. Immunoblotting results showed that silencing ATG4 or tioconazole failed to increase LC3-II concentration in cells treated with CQ in which its degradation is inhibited, suggesting that silencing ATG4 or tioconazole treatment inhibited LC3-II degradation instead of stimulating its formation (Figure 4H-J). These results indicate that tioconazole may inhibit ATG4 to result in the accumulation of LC3-II and impair the fusion of autophagosomes and lysosomes, which, in turn, diminishes autophagic activity.
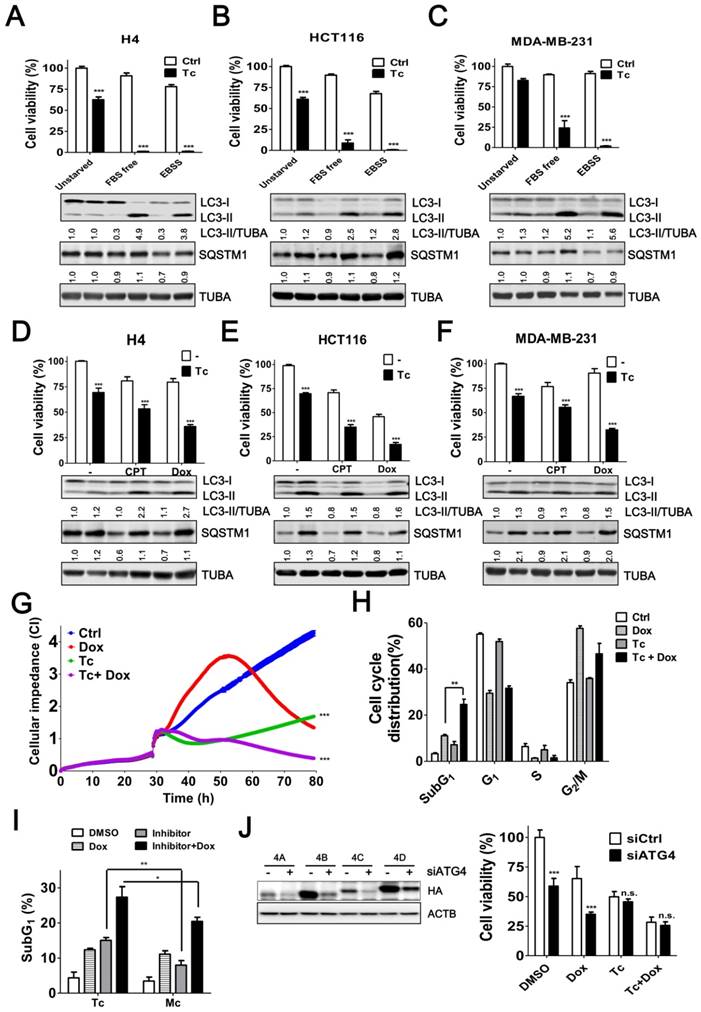
Tioconazole Sensitizes Cancer Cells to Starvation and Chemotherapeutic Drugs
Induced autophagy plays an important role in the resistance to starvation and chemotherapy in cancer cells [6, 33, 34]. To assess the effects of tioconazole on the autophagy inhibition and cytotoxicity in cancer cells during treatment of starvation or chemotherapeutic drugs, the levels of autophagy marker LC3-II and adaptor sequestosome-1 (SQSTM1, also known as p62) were examined by immunoblotting (Figure 5A-F) and cell viability was estimated based on the cellular ATP levels (Figure 5A-F) or cellular impedance (Figure 5G). Tioconazole blocked LC3-II turnover and SQSTM1 degradation, which was triggered by starvation and chemotherapeutic drugs in cancer cells (Figure 5A-F). Tioconazole significantly reduced the cell viability and synergized the cytotoxic effects of starvation in cancer cells, including serum free medium (α values are 38.5±7.2, 6.3±2.7 and 3.1±1.1 for H4, HCT116 and MDA-MB-231 cells, respectively) and starvation buffer Earle's balanced salt solution (EBSS) (α values are 35.6±7.2, 29.4±7.1 and 31.1±3.7 for H4, HCT116 and MDA-MB-231cells, respectively) (Figure 5A-C). The synergistic effects of tioconazole and Dox are shown in Figure 5D-F (α values are 1.6±0.1, 2.0±0.2 and 1.87±0.2 for H4, HCT116 and MDA-MB-231cells, respectively), whereas additive effects between tioconazole and CPT were observed in cancer cells (α values are 1.1±0.1 and 0.9±0.2 for H4 and MDA-MB-231cells, respectively). Combined treatment with tioconazole and Dox significantly increased cell death compared with cells treated with either Dox or tioconazole alone, as indicated by the subG1 proportion (Figure 5H). As observed for tioconazole and its analog miconazole in vitro (Figure 2C), miconazole less potently enhanced cytotoxicity of Dox to result in tumor cell death (Figure 5I). Moreover, silencing ATG4 reduced tumor viability and increased Dox-induced cell death, whereas it did not further enhance the effects of tioconazole (Figure 5J).
Tioconazole Sensitizes Cancer Cells to Starvation and Chemotherapeutic Drugs. (A) H4, (B) HCT116 or (C) MDA-MB-231 cells were starved in FBS-free media or EBSS in the presence or absence of tioconazole (40 μM) for 24 h. The cytotoxicity of these treatments was assessed using CellTiter-Glo. (D) H4, (E) HCT116 and (F) MDA-MB-231 cells were treated with the anticancer drug CPT (1.5 μM) or Dox (1 μM) for 24 h in the presence or absence of tioconazole, and cell viability was measured with CellTiter-Glo. The immunoblotting results of LC3-II and SQSTM1 for the cells as aforementioned are shown in each panel. (G) HCT116 cells were cultured in electronic plates and treated with Dox (1 μM) in the presence or absence of Tc (40 μM) to monitor cell viability in live cells with an impedance-based system. (H) HCT116 cells treated with Tc or (I) its analog Mc in the presence or absence of Dox (1 μM) for 24 h were harvested for FACS-based cell cycle distribution analysis or to quantify the subG1 population with FlowJo. (J) HCT116 cells were transfected with 5 nM scramble siRNA (siCtrl) or siRNA against ATG4 (siATG4) for 56 h. The transfected cells were treated with Dox or Tc for 24 h, and the cell viability was measured with CellTiter-Glo. The knockdown efficiency of siRNA against ATG4 was confirmed by immunoblotting in cells transfected with HA-tagged ATG4 members. The results are expressed as the mean ± SEM from 3 individual experiments. n.s., p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
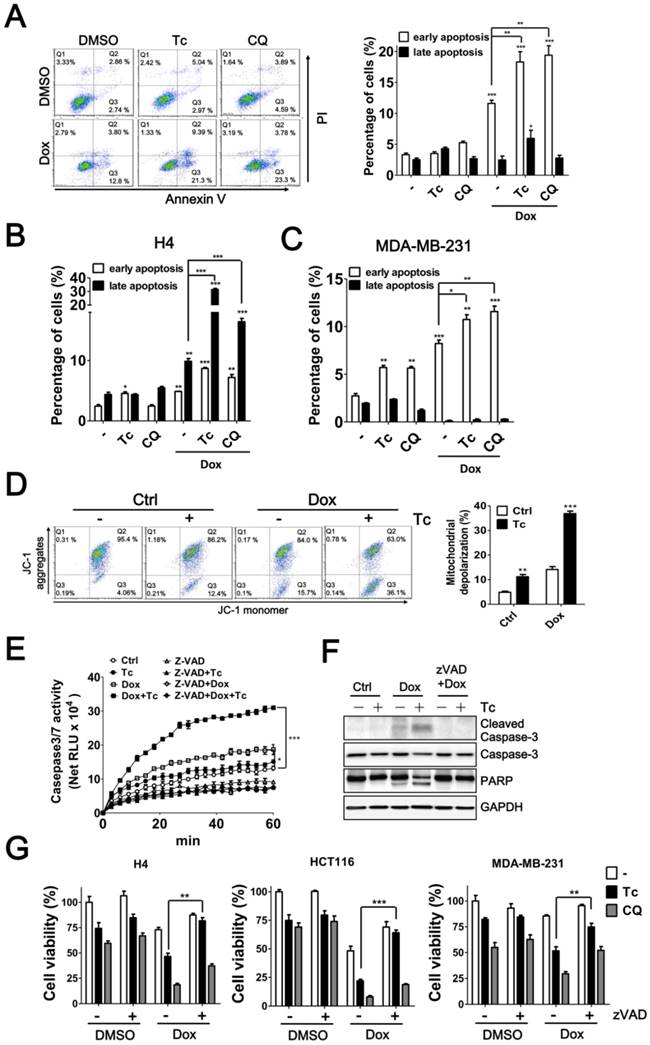
Autophagy serves as a defense mechanism in cancer cells in response to chemotherapy-induced apoptosis. Thus, we further examined the effect of tioconazole on apoptosis in cancer cells treated with Dox. Similar to the autophagy inhibitor CQ, tioconazole increased the percentage of early apoptotic cells (AV+PI-) in the Dox-treated HCT116, MDA-MB-231 and H4 cells (Figure 6A-C and S4), likely due to reduction of mitochondrial membrane potential (MMP) in these cells (Figure 6D and S4C). In addition, caspase-3/7 activity and cleavage of caspase-3 and its substrate PARP were significantly elevated in cells exposed to Dox alone or co-treated with tioconazole (Figure 6E-F). Pretreatment with z-VAD-fmk blocked caspase-3 activation and rescued the apoptotic effects in tumor cell cultures treated with tioconazole and Dox (Figure 6G-I). Overall, these results indicated that tioconazole sensitizes tumor cell lines to chemotherapeutic drug-induced apoptosis.
Effects of Tioconazole on Chemotherapy-Induced Apoptosis in Cancer Cells. (A) HCT116, (B) H4 and (C) MDA-MB-231 cells were treated with Dox (1 μM) for 24 h in the presence or absence of tioconazole (Tc, 40 μM), and apoptotic cells were stained with PI/AV. The apoptotic cells were analyzed and quantified with Prism 5.0. (D) The treated cells as in (A) were stained with JC-1 to determine the mitochondrial membrane potential. The representative data and quantitative results are shown in the left and right panels, respectively. (E) Untreated HCT116 cells or HCT116 cells treated with zVAD-FMK (50 μM) as described in (A) were harvested to assess caspase-3 activation with Caspase-Glo 3/7 luminescent assay. (F) HCT116 cells treated as in (C) were harvested for immunoblotting using antibodies against caspase-3 or PARP. (G) H4, HCT116 and MDA-MB-231 cells were pretreated with zVAD-FMK (50 μM), Tc (40 μM) or CQ (20 μM) prior to treatment with Dox (1 μM) for 24 h, and the cell viability was measured. The results are expressed as the mean ± SEM from at least 3 individual experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
Tioconazole Enhances Chemotherapy Efficacy in Spheroid Cell Culture and Xenografted Tumors
To precisely assess the effects of tioconazole in tumors, HCT116 cells were cultured as spheroids and then treated with tioconazole alone or in combination with Dox (Figure 7A-C). Tioconazole significantly decreased the size of tumor spheroids and enhanced the cytotoxicity of Dox, as assessed based on the ATP levels and number of dead cells (Figure 7). Moreover, tioconazole enhanced Dox-induced cell death both in cells treated with scramble shRNA and shRNA against ATG4B (Figure 7D-E). Single ATG gene knockdown is likely not sufficient to block autophagy, as previously reported [35], which may explain why tioconazole appears to be more potent than ATG4B shRNA.
Effects of Tioconazole on Chemosensitivity in Tumor Spheroid Culture and Xenograft Mouse Model. (A) HCT116 cells were cultured in an ultra-low attachment dish for 24 h to form spheres. The cells were treated with Dox (1 μM) in the presence or absence of Tc (40 μM) for 48 h and stained with Hoechst 33342 (1 μg/mL) to image the spheres. Scale bar: 400 μm. (B) The relative sphere volume was quantified using DMSO-treated cells as the normalized control. (C) HCT116 sphere-forming cells treated as in (A) were lysed to measure ATP levels and assess cell viability. (D) HCT116 cells harboring shRNA against ATG4B, ATG5 or ATG7 formed spheres and were treated with Tc (40 μM) or CQ (20 μM) in the presence or absence of Dox (1 μM) for 48 h. The viable and dead spheres were imaged with a LIVE/DEAD staining kit. The knockdown efficiency of ATG genes was verified by immunoblotting (left panel) (E) The red fluorescence of the spheres in (D) was quantitated with a reader to assess the dead cell population (n = 6). The quantified results are expressed as the mean ± SEM from 3 individual experiments. (F) Mice injected with 2 × 106 human colorectal cancer HCT116 cells were treated with Tc (60 mg/kg) in the presence (+) or absence (-) of Dox (1 mg/kg) every other day. The tumor volume (circle) in each mouse was measured every 3 to 4 days (5 per group). The p values were determined with an ANOVA. (G) The representative pictures of the xenograft tumors and tumor weight at day 21 after injection are shown in the left and right panels, respectively. Scale bar: 1 cm. (H) The xenografted tumor was harvested and embedded for immunohistochemistry using an antibody against LC3. Arrow: cells with LC3 puncta. Scale bar: 40 μm. (I) The number of LC3 puncta in each cell was counted for at least 150 cells and quantified. (J) The level of cleaved caspase-3 in sections prepared as in (H) was determined with immunohistochemistry as shown in the left panel. Arrow: apoptotic cell. Scale bar: 20 μm. n.s. (K) The cleaved caspase-3-positive cells with condensed nuclei were counted as apoptotic cells. The quantified results were obtained from at least 1500 cells and are shown in the right panel. p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
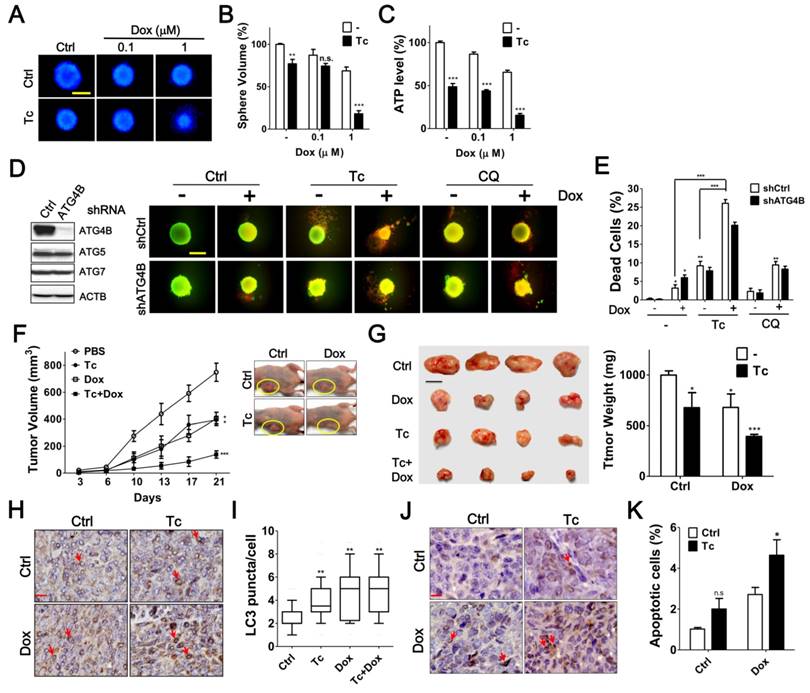
To assess the antitumor effects of tioconazole in vivo, HCT116 cells were xenografted into nude mice, and the animals were treated with tioconazole alone or in combination with Dox (Figure 7F). Tioconazole reduced the xenograft tumor volumes and sensitized xenograft tumors to Dox (Figure 7G). Moreover, the number of LC3 puncta and levels of cleaved caspase-3 were significantly increased in tumor tissues of the tioconazole-treated mice (Figure 7H-K). Overall, these findings are consistent with the cell culture model and support the notion that tioconazole may inhibit ATG4 and autophagy to enhance chemotherapy efficacy.
Discussion
Autophagy modulation has been suggested as a potential cancer therapy. However, this theory remains controversial in clinical settings due to the limited number of drugs available to modulate autophagy. ATG4 is a key component of autophagy signaling, and its levels are elevated in cancer cells to promote tumorigenesis and malignancy [36, 37], which suggests that ATG4 is a suitable drug target to interrogate the role of autophagy in cancer treatment. In this study, we employed a novel platform that integrates docking/simulations/binding free energy calculations and biochemical/cellular assays to screen a library of FDA-approved drugs and reported the following findings. First, a drug-repositioning screen indicated that tioconazole might obstruct the active site to inhibit ATG4 proteolytic activity. Second, tioconazole accumulated autophagosomes and diminished autophagic flux. Third, tioconazole decreased tumor viability and sensitized cancer cells to starvation and chemotherapeutic drugs. These findings support tioconazole as a potential drug that inhibits ATG4 and autophagy. In addition, tioconazole is an FDA-approved drug, which suggests that tioconazole may have a new indication in cancer therapy.
ATG4 not only cleaves the C-terminus of LC3 precursor for lipidation, but also deconjugates LC3-II from the membrane for recycling LC3 [25], implying the effects of ATG4 on LC3-II puncta in cells could be varied. Indeed, LC3-II and GABARAP-II puncta are decreased in atg4b-/- mouse embryonic fibroblast (MEF) cells [38]. In contrast, knockdown of ATG4B elevates GFP-LC3 puncta in several cancer cells, including colorectal cancer HCT116 cells [36], breast cancer MCF-7 cells [29], prostate cancer PPC1 and neuroglioma H4 cells (unpublished results). Moreover, the ectopic expression of dominant negative ATG4 mutants reduces fusion between autophagosomes and late compartments in human differentiating erythrocytes [31]. Our present data also showed that both knockdown of ATG4 and treatment with the ATG4 inhibitor tioconazole increased GFP-LC3 puncta. Tioconazole further increased the number of accumulated autophagosomes and diminished the fusion step of autophagosomes and lysosomes to attenuate autophagic flux. These results suggest that the effects of ATG4 on LC3 lipidation and delipidation for autophagosome formation or fusion with lysosome might be varied in different cell types.
On the other hand, the impairment of lysosomes may increase the number of GFP-LC3 puncta and autophagosomes. Thus, we measured the lysosomal cathepsin B activity in ATG4-silenced or tioconazole-treated cells (Figure S5). Silencing ATG4 or treatment with tioconazole did not inhibit cathepsin B activity in H4 and HCT116 cells, but slight inhibition was observed in MDA-MB-231 cells. These results imply that tioconazole increases the number of LC3 puncta and accumulates autophagosomes in cancer cells primarily by inhibiting ATG4 instead of destabilizing lysosomal function.
Autophagy promotes tumor malignance via several mechanisms, such as the maintenance of cancer stemness, survival of senescent cancer cells, resistance to the hypoxic microenvironment and therapy-induced death [2], supporting our notion that tioconazole may suppress tumor malignancy mainly through autophagy inhibition. Moreover, tioconazole not only inhibits lanosterol 14a-demethylase to block fungal ergosterol but also inhibits mammalian cytochrome P450 3A4 [39]. Our results showed that tioconazole inhibited HCT116 cell viability by ~50%, which is ~9% more than the inhibition observed in ATG4-silenced cells (Figure 5J).
Tioconazole also significantly enhanced the cytotoxicity of Dox both in cells treated with scramble shRNA and cells treated with shRNA against ATG4B (Figure 6D), suggesting that the slight inhibition of cytochrome P450 or other molecules may also be involved in the tumor-suppressive effects of tioconazole, which requires additional studies to examine this possibility.
The clinical association of autophagy in cancer has been reported previously. The LC3 protein level is positively correlated with BECN1 expression and is associated with poor survival in breast cancer patients following chemotherapy [40]. LC3 puncta are increased in metastatic breast cancer and melanoma and are highly associated with poor outcomes in breast cancer [41]. Gene silencing ATG5 or treatment with CQ significantly suppresses pancreatic cancer cell growth in vitro and in vivo [42]. These findings indicate that autophagy is required for tumor growth, metastasis and resistance to chemotherapy. Small molecule inhibitors against essential proteins for autophagy machinery, including Vps34 and ULK1, also suppress autophagy and tumor cells [33, 43]. However, the safety and efficacy of these inhibitors in clinics are not clear yet. Thus, to date, there is no approved drug target ATG protein for cancer treatment. Herein, we employed a novel drug screening platform to screen FDA-approved drugs and validated tioconazole as a potential ATG4 inhibitor (IC50 for ATG4A and ATG4B are 1.3±0.18 μM and 1.8±0.16 μM, respectively). Tioconazole suppressed tumor growth and enhanced chemotherapy-induced apoptosis in cancer cells and tumor xenografts in mice. The pharmacokinetics of orally administered tioconazole have been tested in dogs and mice [44], which would accelerate the novel use of tioconazole for cancer therapy in clinical settings.
Abbreviatons
ATG: autophagy-related; ATG4B: autophagy related 4B, cysteine peptidase; CQ: chloroquine; LC3: microtubule-associated protein 1 light chain 3; Tc: tioconazole; Mc: miconazole; Dox: doxorubicin; SQSTM1: sequestosome 1; MD simulation: molecular dynamics simulation; GABARAPL2: GABA(A) receptor-associated protein-like 2; ACTB: β-actin; TUBA: α-tubulin.
Acknowledgements
We thank Dr. Tamotsu Yoshimori and Dr. Walther Mothes for sharing the plasmids pEGFP-LC3 and Lamp1-RFP, respectively. We also thank the National Center for High-performance Computing of National Applied Research Laboratories of Taiwan for providing the vast computational resources required for this work.
Grant Support
This work was supported by the Ministry of Science and Technology (105 and 102-2311-B-075B-001 to C.W. Shu) (103-2627-M-007-001 to L.W. Yang), the Kaohsiung Veterans General Hospital and the Yen Tjing Ling Medical Foundation (VGHKS102-007 to C.W. Shu, VGHKS103-G01-1 to C.J. Hsu, and CI-103-11, 2014 to C.W. Shu).
Supplementary Material
Supplementary figures and tables.
Supplemental Movie S1-S5 are available online. http://dyn.life.nthu.edu.tw/Tioconazole/movie
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr. 2007;27:19-40
2. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F. et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856-80
3. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69-79
4. Renna M, Jimenez-Sanchez M, Sarkar S, Rubinsztein DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem. 2010;285:11061-7
5. Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500-10
6. Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK. et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4:773-80
7. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA. et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255-63
8. Yang ZJ, Chee CE, Huang S, Sinicrope F. Autophagy modulation for cancer therapy. Cancer Biol Ther. 2011;11:169-76
9. Solitro AR, MacKeigan JP. Leaving the lysosome behind: novel developments in autophagy inhibition. Future Med Chem. 2016;8:73-86
10. Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200-12
11. Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K. et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. 2014;26:190-206
12. Klionsky DJ. Citing recent declines in the discovery of new ATG genes, some scientists now suggest that the end of autophagy research may be within sight. Autophagy. 2014;10:715-6
13. Nakatogawa H, Ishii J, Asai E, Ohsumi Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy. 2012;8:177-86
14. Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S. et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy. 2012;8:883-92
15. Marino G, Uria JA, Puente XS, Quesada V, Bordallo J, Lopez-Otin C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem. 2003;278:3671-8
16. Schaaf MB, Keulers TG, Vooijs MA, Rouschop KM. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 2016;30:3961-78
17. Li M, Hou Y, Wang J, Chen X, Shao ZM, Yin XM. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286:7327-38
18. Shu CW, Drag M, Bekes M, Zhai D, Salvesen GS, Reed JC. Synthetic substrates for measuring activity of autophagy proteases: autophagins (Atg4). Autophagy. 2010;6:936-47
19. Betin VM, Lane JD. Atg4D at the interface between autophagy and apoptosis. Autophagy. 2009;5:1057-9
20. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455-61
21. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10:449-61
22. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785-91
23. Shu CW, Madiraju C, Zhai D, Welsh K, Diaz P, Sergienko E. et al. High-throughput fluorescence assay for small-molecule inhibitors of autophagins/Atg4. J Biomol Screen. 2011;16:174-82
24. Hayashi H, Cuddy M, Shu VC, Yip KW, Madiraju C, Diaz P. et al. Versatile assays for high throughput screening for activators or inhibitors of intracellular proteases and their cellular regulators. PLoS One. 2009;4:e7655
25. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313-26
26. Chang CE, Chen W, Gilson MK. Ligand configurational entropy and protein binding. Proc Natl Acad Sci U S A. 2007;104:1534-9
27. Tsui V, Case DA. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 2000;56:275-91
28. Akin D, Wang SK, Habibzadegah-Tari P, Law B, Ostrov D, Li M. et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10:2021-35
29. Bortnik S, Choutka C, Horlings HM, Leung S, Baker JH, Lebovitz C. et al. Identification of breast cancer cell subtypes sensitive to ATG4B inhibition. Oncotarget. 2016;7:66970-88
30. Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T. et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651-9
31. Betin VM, Singleton BK, Parsons SF, Anstee DJ, Lane JD. Autophagy facilitates organelle clearance during differentiation of human erythroblasts: evidence for a role for ATG4 paralogs during autophagosome maturation. Autophagy. 2013;9:881-93
32. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
33. Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ. et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59:285-97
34. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9-15
35. Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T. et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654-8
36. Liu PF, Leung CM, Chang YH, Cheng JS, Chen JJ, Weng CJ. et al. ATG4B promotes colorectal cancer growth independent of autophagic flux. Autophagy. 2014;10:1454-65
37. Rothe K, Lin H, Lin KB, Leung A, Wang HM, Malekesmaeili M. et al. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood. 2014;123:3622-34
38. Marino G, Fernandez AF, Cabrera S, Lundberg YW, Cabanillas R, Rodriguez F. et al. Autophagy is essential for mouse sense of balance. J Clin Invest. 2010;120:2331-44
39. Zhang W, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM. Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab Dispos. 2002;30:314-8
40. Chen S, Jiang YZ, Huang L, Zhou RJ, Yu KD, Liu Y. et al. The residual tumor autophagy marker LC3B serves as a prognostic marker in local advanced breast cancer after neoadjuvant chemotherapy. Clin Cancer Res. 2013;19:6853-62
41. Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18:370-9
42. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717-29
43. Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F. et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013-9
44. Jevons S, Gymer GE, Brammer KW, Cox DA, Leeming MR. Antifungal activity of tioconazole (UK-20,349), a new imidazole derivative. Antimicrob Agents Chemother. 1979;15:597-602
45. Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y. et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009;28:1341-50
Author contact
![]() Corresponding authors: Chih-Wen Shu, Department of Medical Education and Research, Kaohsiung Veterans General Hospital, 386, Ta-Chung First Road, Kaohsiung, Taiwan, R.O.C. 813 Tel: 886-7-3422121 ext 1503; Fax: 886-7-3468056 E-mail: cwshugov.tw or Lee-Wei Yang, Institute of Bioinformatics and Structural Biology, National Tsing-Hua University, 101, Sec 2, Kuang-Fu Rd., Hsinchu, Taiwan, R.O.C.300 Tel: 886-3-5742467; Fax: 886-3-5715934 E-mail: lwyangnthu.edu.tw
Corresponding authors: Chih-Wen Shu, Department of Medical Education and Research, Kaohsiung Veterans General Hospital, 386, Ta-Chung First Road, Kaohsiung, Taiwan, R.O.C. 813 Tel: 886-7-3422121 ext 1503; Fax: 886-7-3468056 E-mail: cwshugov.tw or Lee-Wei Yang, Institute of Bioinformatics and Structural Biology, National Tsing-Hua University, 101, Sec 2, Kuang-Fu Rd., Hsinchu, Taiwan, R.O.C.300 Tel: 886-3-5742467; Fax: 886-3-5715934 E-mail: lwyangnthu.edu.tw