Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):846-859. doi:10.7150/thno.21793 This issue Cite
Research Paper
Gilz-Activin A as a Novel Signaling Axis Orchestrating Mesenchymal Stem Cell and Th17 Cell Interplay
Patricia Luz-Crawford1,4, Gabriel Espinosa-Carrasco1#, Natacha Ipseiz7#, Rafael Contreras1,4, Gautier Tejedor1, Daniel A. Medina5, Ana-Maria Vega-Letter6, Devi Ngo3, Eric F. Morand3, Jérôme Pène1, Javier Hernandez1*, Christian Jorgensen1,2*, Farida Djouad1 ![]()
1. IRMB, INSERM, Université Montpellier, Montpellier, France
2. Service d'Immuno-Rhumatologie, Hôpital Lapeyronie, Montpellier, France
3. School of Clinical Sciences, Monash University, Melbourne, Australia
4. Facultad de Medicina, Universidad de los Andes, Santiago, Chile
5. Department of Chemical and Bioprocess Engineering, Pontificia Universidad Católica de Chile, Santiago, Chile
6. Clinica Alemana Universidad del Desarrollo, Santiago, Chile
7. Cardiff University School of Medicine, United Kingdom.
# Equally contributing authors
* Equally contributing authors
Received 2017-7-6; Accepted 2017-11-9; Published 2018-1-1
Abstract

Mesenchymal stem cells (MSC) are highly immunosuppressive cells able to reduce chronic inflammation through the active release of mediators. Recently, we showed that glucocorticoid-induced leucine zipper (Gilz) expression by MSC is involved in their therapeutic effect by promoting the generation of regulatory T cells. However, the mechanisms underlying this pivotal role of Gilz remain elusive.
Methods and Results In this study, we have uncovered evidence that Gilz modulates the phenotype and function of Th1 and Th17 cells likely by upregulating the level of Activin A and NO2 secreted by MSC. Adoptive transfer experiments sustained this Gilz-dependent suppressive effect of MSC on Th1 and Th17 cell functions. In immunoregulatory MSC, obtained by priming with IFN-γ and TNF-α, Gilz was translocated to the nucleus and bound to the promoters of inos and Activin βA to induce their expression. The increased expression of Activin A directly impacted on Th17 cells fate by repressing their differentiation program through the activation of Smad3/2 and enhancing IL-10 production.
Conclusion Our results reveal how Gilz controls inos and Activin βA gene expression to ultimately assign immunoregulatory status to MSC able to repress the pathogenic Th17 cell differentiation program and uncover Activin A as a novel mediator of MSC in this process.
Keywords: Mesenchymal Stem Cells, Gilz, Activin A, Th17 cells, Immunosuppression.
Introduction
Th17 and Th1 cells are associated with the development of autoimmune diseases, including multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease, and psoriasis [1-7]. Presently, the development of new approaches for the treatment of autoimmune disorders aims to target different aspects of the generation and function of Th17 and Th1 cells and promising advances rely on T cell plasticity [8]. Indeed, a strong relationship between the regulatory T cell (Treg) and Th17 lineages has been reported, in part based on the observation that, in the presence of IL-6, endogenous TGF-β can induce the differentiation of Treg cells into Th17 cells [9, 10]. The converse possibility to generate T cells with regulatory function from Th17 cells has also been described [11-13], even if less evidenced. Moreover, although less described, a plasticity of Th1 cells has also been proposed. Indeed, a high dose allergen tolerance was shown to induce clonal expansion of specific IL-10-secreting T cells, which may switch in vivo from existing Th1-like allergen-specific cells [14].
An increasing number of results have highlighted the general capacity of polarized T cell subsets to adapt their phenotype and function in response to environmental tissue changes in the course of inflammatory reactions. In the attempts to force inflammatory Th17 cells to adopt a regulatory phenotype, mesenchymal stem cells (MSC) have been extensively explored due to their potent T cell suppressive properties, described both in vitro and in vivo a decade ago [15-17]. Among the possible mediators identified, inducible nitric oxide synthase (iNOS) [18] and prostaglandin E2 (PGE2) play important roles in murine MSC (mMSC) [19, 20]. MSC immunoregulatory functions are not constitutive, but require a priming step. Several cytokines, including IFN-γ, TNF-α, IL-1α, and IL-1β, trigger the expression of iNOS in mMSC [18]. iNOS production by mMSC exerts regulatory effects through the generation of toxic reactive nitrogen species, as well as through the nitration of other molecules proximate to its source of production [18]. MSC suppressive properties are in part mediated through the action of NO and result in the inhibition of CD4+ and CD8+ T cell proliferation as demonstrated both in vitro and in vivo [19, 21-23]. Using activated mouse CD4+ T cells under Th1 or Th17 skewing conditions in vitro, we previously reported that MSC inhibit the proliferation of Th1 and Th17 cells as well as their cytokine production. [24]. The generation of a Th1 cell population co-producing IFN-γ and IL-10 was recently proposed to be one of the mechanisms by which MSC exert their immunosuppressive properties [25]. More recently, we showed that MSC negative regulation of Th17 responses restores the balance between Th17 and Treg cells in a murine model of autoimmune disease [26]. Although the mechanisms by which MSC exert their immunosuppressive effects have been the subject of many studies, the molecular regulation of the mediators responsible for MSC immunosuppressive effects, as well as the signaling pathways they activate in Th1 and Th17 cells remain largely unknown.
Glucocorticoid-induced leucine zipper (Gilz or TSC22D3) is a protein that binds to and inhibits major pro-inflammatory transcription factors including nuclear factor κB (NF-κB) and activator protein 1 (AP-1) [27]. Moreover, GILZ regulates signaling kinases Ras and Raf-1 leading to the inhibition of MAP kinases [28, 29]. A body of evidence indicates that Gilz is involved in the suppression of both innate and adaptive immune responses [27]. Overexpression of Gilz drives the development of regulatory dendritic cells (DC), and thereby prevents the production of pro-inflammatory cytokines by DC activated by CD40 ligand [30]. Tolerance-inducing regulatory DC, which express Gilz, enhance the differentiation of IL-10-producing CD25highFoxp3+CTLA4+ Treg cells from naïve CD4+ T lymphocytes [31]. More recently, in an experimental model of arthritis, we have shown a pivotal role of Gilz in the therapeutic effects of MSC, through the generation of Treg cells bearing the CD4+RORγT+IL-17+IL-10+ signature [26]. In line with this study, the generation of Treg cells induced by MSC through the up-regulation of developmental endothelial locus-1 (Del-1) expression was also recently shown to be Gilz-dependent [32]. Although Gilz expression is required for MSC immunoregulatory activity, whether its expression directs the production of MSC immunosuppressive effect mediators remains to be demonstrated.
Activin A, a member of the TGF-β superfamily, exerts immunomodulatory effects on DC and T cells. DC both secrete and respond to Activin A. Autocrine actions of Activin A potently reduce DC capacity to produce pro-inflammatory cytokines as well as their T-cell stimulatory potential [33]. Moreover, Activin A exerts immunomodulatory functions through the induction of CD4+Foxp3- IL-10+ and CD4+Foxp3- IL-10- Treg cells that inhibit Th1-driven responses [34]. Activin A receptor type IIA (ActRIIA) expression in CD4+ cells is dependent on TGF-β1 and IL-6 and is specific of and restricted to Th17 cells [35]. Although these studies suggest that ActRIIA acts as a marker for Th17 cells and that Activin A might regulate the Th17/Treg balance, how pathogenic Th17 fate is shaped by extrinsic signals delivered by primed MSC focusing on their capacity to release Activin A has not been addressed.
In the present study, we investigated how Gilz, expressed by MSC, may affect the generation of Th1 and Th17 cells and addressed the role of Activin A in this aspect. Going further, we investigated the intracellular regulation of MSC immunosuppressive mediators directed by Gilz, identifying its binding sites on the promoters of inos and Activin βA. Finally, we explored signaling pathways and transcriptional events that skew Th17 differentiation and function. Our findings indicate that the expression of Gilz by MSC is essential for the release of the suppressive molecules NO2 and Activin A. These effects underpin key aspects of the suppressive effects of MSC on T cells.
Results
Gilz deficiency impairs MSC immunomodulatory function in vitro
To assess the role of Gilz in MSC-mediated immunosuppressive function, we isolated MSC from bone marrow of Gilz-deficient mice and their wild-type Gilz+/+ littermates, referred to as Gilz-/- MSC and WT MSC respectively, as previously described [23, 26, 36-38]. First, we investigated the phenotypic profile and multilineage differentiation potential of Gilz-/- MSC in vitro. Both WT MSC and Gilz-/- MSC were positive for Sca1, CD29, CD44 and CD105 and negative for CD11b and CD45 (Fig. S1A). The multipotent nature of the cells was confirmed by their capacity to differentiate into the three main lineages relying on the expression of lineage-specific markers quantified by RT-qPCR. WT and Gilz-/- MSC could be differentiated into chondrocytes, as shown by induction of collagen II (Col2b) and aggrecan (AGN), and into adipocytes, as revealed by increased expression of peroxysome proliferator-activated receptor (PPAR)-γ and fatty acid binding protein 4 (FABP4) (Fig. S1B). WT and Gilz-/- MSC also differentiated into osteoblasts, expressing osteocalcin (OC) and alkaline phosphatase (ALP), although the expression of these genes was significantly diminished in Gilz-/- MSC (Fig. S1 B).
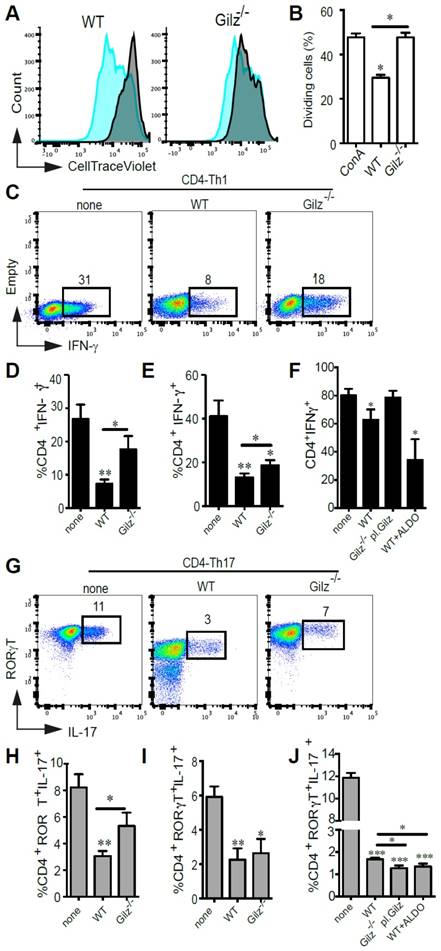
Next, we assessed in vitro the immunosuppressive properties of WT and Gilz-/- MSC on ConA-stimulated splenocytes by determining the percentage of proliferating cells stained with CellTrace Violet (CTV). After 3 days of co-culture, WT MSC significantly inhibited T cell proliferation, but in contrast Gilz-/- MSC did not inhibit T cell proliferation (Fig. 1A-B). Since MSC have been described to negatively regulate both Th1 and Th17 responses, we analysed the effects of WT and Gilz-/- MSC on the polarization of naïve CD4+ T cells toward Th1 and Th17 lineage. The specific combinations of cytokines and neutralizing antibodies used for each lineage (see Materials and Methods) induced IFN-γ-producing cells (Th1) (Fig. 1C-F) and IL-17-producing cells (Th17), respectively, the latter cells also being positive for the Th17 lineage-specific transcription factor ROR-γT (Fig. 1G-J). The addition of WT MSC at day 0 of T cell differentiation resulted in a significant decrease in the frequency of both Th1 and Th17 cells (Fig. 1C-D, G-H). In comparison, the capacity of Gilz-/- MSC to regulate CD4+ T cell differentiation into Th1 or Th17 cells was significantly impaired (Fig. 1C-D, G-H). The role of Gilz on MSC regulatory effect was further evaluated by rescue experiments using Gilz-/- MSC transfected with plasmid pCDNA3.1-GILZ (Gilz-/- pl. Gilz) (Fig. S1C). While Gilz-/- pl. Gilz MSC were significantly more suppressive than WT MSC on CD4+ T cells induced to differentiate into Th17 (Fig. 1J), they did not affect the generation of Th1 cells as compared to the Th1 differentiating cells cultured alone (Fig. 1F). These results reinforce the key role of Gilz on the capacity of MSC to repress CD4+ T cell differentiation toward Th17 lineage. We next assessed the effect of WT and Gilz-/- MSC on mature Th1 or Th17 cell function. WT and Gilz-/- MSC both significantly inhibited Th1 and Th17 cell cytokine expression after 3 days of co-culture (Fig. 1E, I). However, Gilz-/- MSC were significantly less effective in reducing Th1 signature cytokine production than WT MSC (Fig. 1E, I), while suppression of Th17 signature cytokine production by WT and Gilz-/- MSC was similar. Additionally, we tested the effect of another corticosteroid, aldosterone, on the capacity of MSC to repress the differentiation of CD4+ T cells into Th1 or Th17 cells. First, we showed a dose-dependent increase of Gilz expression in human MSC treated with Aldosterone from a dose of 0.1 µM as compared to the control untreated MSC (Fig. S2A). Moreover, the pre-treatment of MSC with aldosterone, TNFα and IFNɣ significantly enhanced the suppressive effect of human MSC as compared to untreated MSC and MSC primed with either aldosterone or TNFα and IFNɣ on PHA-activated PBMC proliferation (Fig. S2B-C). In parallel, using mouse WT MSC, we observed that a 24 h treatment with 1 µM of aldosterone significantly increased their immunoregulatory effect on CD4+ T cell induction to differentiate into Th1 (Fig. 1F) or Th17 cells (Fig. 1J). These results indicate that MSC immunosuppressive effects on Th1 and Th17 differentiation, and mature Th1 cell function, are dependent on MSC expression of Gilz, but that MSC immunosuppressive effects on mature Th17 cells are independent of Gilz.
In vitro evaluation of the immunomodulatory properties of WT MSC and Gilz-/- MSC. (A-B) CTV-labelled splenocytes were stimulated with ConA and the proliferation of cells was measured by CTV dilution. Results are expressed as the percentage of ConA-induced proliferation, which was assigned a value of 100%, or as the percentage of CTV+ dividing cells. If not indicated otherwise, P values refer to ConA-activated samples. (C-J) Cell differentiation was assessed using T cells induced to differentiate into Th1 or Th17 cells in the absence (none) or presence of WT, Gilz-/- or Gilz-/-pl.Gilz MSC added at day 0 (CD4-Th1 or CD4-Th17, respectively) (C, D, F and G, H, J) or at day 4 of the differentiation process (mature Th1 or Th17, respectively) (E and I), at a MSC:T ratio of 1:10. For T cells characterization, intracellular detection of IFN-γ (Th1) or ROR-γT and IL-17 (Th17) was performed by flow cytometry (C and G, representative dot plots). (F and J) Evaluation of T cell differentiation toward the Th1 or Th17 lineages in the presence of MSC pre-treated for 24 h with 1 µM of aldosterone. (B, D, E, F, H, I and J) Mean values of n≥3 independent experiments. P values refer to the condition without MSC (none). * P < 0.05, ** P < 0.01. All error bars indicate SEM.
Gilz deficiency impairs MSC immunomodulatory function in vivo
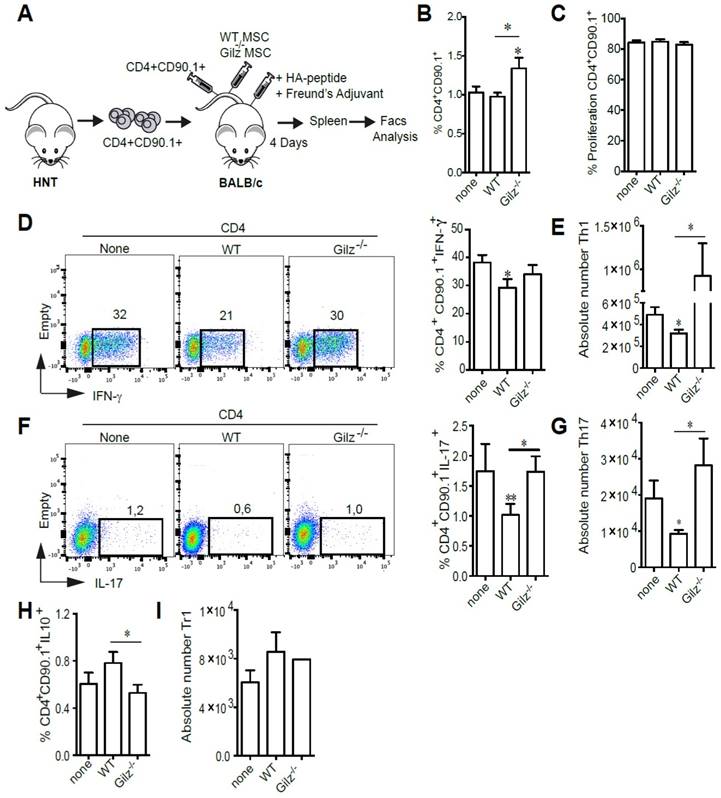
We next assessed the immunosuppressive properties of WT and Gilz-/- MSC in vivo using an adoptive T cell transfer model. To determine whether Gilz expressed by MSC affects proliferation and differentiation of Th1 and Th17 subsets in secondary lymphoid organs, we adoptively transferred CTV-labeled HNT TCR transgenic Thy1.1+ CD4+ T cells specific for the influenza virus hemagglutinin (HA) into BALB/c mice. Mice were then immunized with HA peptide prior to MSC treatment (Fig. 2A). At day 4 after transfer, while we did not observe any difference in the percentage of CD4+CD90.1+ cells in the spleen of MSC-treated mice as compared to the control mice, a significant increase was observed in mice treated with Gilz-/- MSC (Fig. 2B). However, the proliferation rate of CD4+CD90.1+ cells was the same in the 3 different groups (Fig. 2C). Additionally, HNT Th1 and Th17 cells were detectable in spleens of immunized mice, with Th1 cells predominating (Fig. 2D, F). Compared to non-treated mice, mice treated with WT MSC displayed a significant decrease in the frequency (Fig. 2D, F) and absolute numbers (Fig. 2E,G) of Th1 and Th17 cells. In contrast, treatment with Gilz-/- MSC had no effect (Fig. 2D-G). Moreover, we observed an increase in the frequency of donor CD90.1+ CD4+ T cells producing IL-10 in the spleens of mice treated with WT MSC, but not Gilz-/- MSC (Fig. 2H-I). These results extend to in vivo conditions the requirement for Gilz for MSC to inhibit differentiation of naïve T cells into Th1 and Th17 cells.
Gilz deficiency impaired the capacity of MSC to inhibit the generation of Th1 and Th17 in vivo. (A) Representative scheme of the in vivo experimental setting. BALB/c mice were adoptively transferred with naïve HNT Thy1.1+ CD4+ T cells and then immunized with HA peptide in complete Freund's adjuvant. The same day, mice were either treated with the indicated MSC or left untreated. At day 4 after transfer, mice were sacrificed and donor T cells were identified in the spleen by virtue of the expression of the congenic Thy1.1 marker. (B) Percentage of CD4+CD90.1+ cells in the spleen of the mice. (C) Proliferation rate of CD4+CD90.1+ cells isolated from the spleen of mice. (D-E) Percentage (D) and absolute number (E) of Th1 cells within the donor HNT Th1.1+ CD4+ T cell population from non-treated mice or mice treated with either WT or Gilz-/- MSC. (F-G) Percentage (F) and absolute number (G) of donor Th17 cells in the spleen of BALB/c mice treated or not with either WT or Gilz-/- MSC. (H-I) Percentage of CD4+CD90.1+IL10+ (H) and absolute number of Tr1 cells (I) within the donor HNT Th1.1+ CD4+ T cell population from non-treated mice or mice treated with either WT or Gilz-/- MSC in the spleen. Dot plots are representative of 1 out of ≥3 independent experiments. Mean values of n≥3 independent experiments. * P < 0.05, ** P < 0.01.
The secretion of MSC immunosuppressive mediators is Gilz-dependent
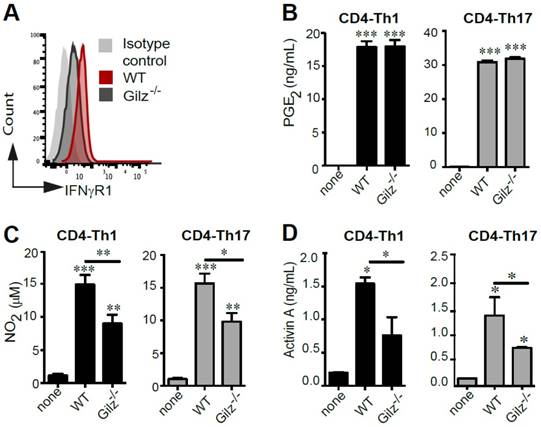
Since the critical role of IFN-γ in priming MSC-mediated suppression of immune cells is well established, we assessed the expression of the IFNγ receptor (IFNγR1) by WT and Gilz-/- MSC. FACS analysis indicated that IFNγR1 expression was higher in WT MSC (Fig. 3A). This low expression level of IFNγR1 in Gilz-/- MSC as compared to WT MSC could be related to their reduced immunosuppressive properties since IFNγR1 deficiency in MSC was described by their lack of immunosuppressive and therapeutic effects in an experimental model of GVHD [39]. Next, we analysed MSC production of PGE2 and NO2, which have been described to be associated with MSC immunomodulatory effects. WT and Gilz-/- MSC cultured for 3 days in the presence of differentiating CD4-Th1 or CD4-Th17 cells secreted similar levels of PGE2 (Fig. 3B). In contrast, after co-culture with differentiating CD4-Th1 or CD4-Th17 cells, Gilz-/- MSC secretion of NO2 was significantly lower than WT MSC (Fig. 3C). Finally, MSC secretion of Activin A was quantified. We observed abundant production of Activin A by WT MSC cultured in the presence of differentiating Th1 or Th17 cells, but significantly less Activin A was produced by Gilz-/- MSC under the same conditions (Fig. 3D). These results indicate that Gilz-dependent suppressive effect of MSC on T cell subsets is associated with Activin A and NO2 release.
Quantification of the mediators involved in MSC immunosuppressive properties. (A) Expression profile of IFNγR1 on WT and Gilz-/- MSC by FACS analysis. (B) PGE2 assessment by ELISA in the 3-day culture supernatants of T cells induced to differentiate toward Th1 (grey bar) or Th17 (black bar) lineages, cultured alone or with MSC (WT or Gilz-/-). (C) Quantification of NO2 production using a modified Griess reaction in the 3-day culture supernatants of T cells induced to differentiate toward Th1 (grey bar) or Th17 (black bar) lineages cultured alone or with MSC (WT or Gilz-/-). (D) Activin A quantification by ELISA in the 3-day culture supernatants of T cells induced to differentiate toward Th1 (grey bar) or Th17 (black bar) lineages cultured alone or with MSC (WT or Gilz-/-). If not indicated, P values refer to the condition when the different T cell subsets were cultured without MSC (none). Mean values of n≥3 independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001.
Identification of GILZ binding site motifs in Activin and inos promoter regions
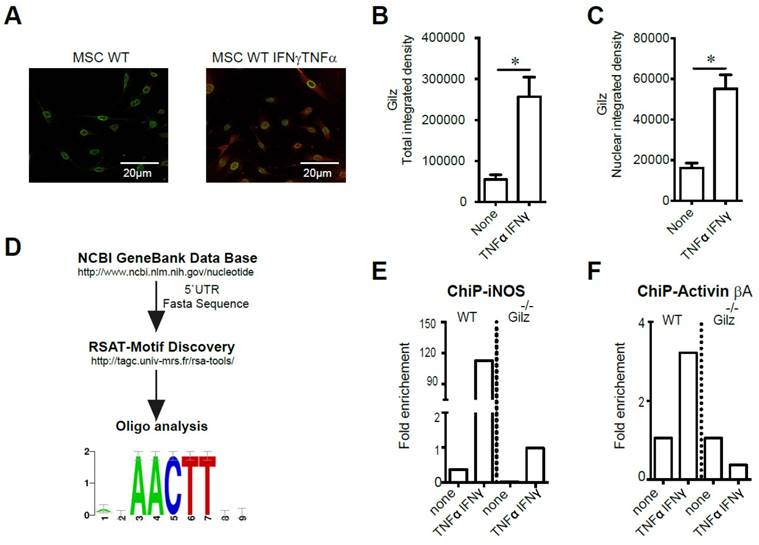
We next investigated potential mechanisms for Gilz modulation of MSC Activin A and NO2. First, we studied Gilz expression profile in MSC both in basal condition (none) and in response to activation with TNF-α and IFN-γ. We thus performed an immunofluorescence analysis of Gilz expression in MSC to assess the effect of MSC stimulation on Gilz expression level and cellular distribution. We found, in response to MSC activation, a significant increase of Gilz expression (Fig. 4A-B). Of note, the analysis of Gilz nuclear content revealed a significant increase of Gilz nuclear localization upon MSC stimulation as compared to non-activated cells (Fig. 4C). Next, we studied Gilz-specific transcriptional regulation of Activin βA and inos. To identify a putative DNA binding site, Mus musculus 5'UTR sequences for reported Gilz gene targets were retrieved from NCBI Nucleotide database [40]. The fasta sequences were used to look for over-represented motif enrichment between sequences using the Regulatory Sequence Analysis Tools, RSAT [41], as described in Materials and Methods. This analysis revealed the consensus sequence AACTT as the strongest binding site candidate (Fig. 4D). Accordingly, we designed primers covering each AACTT sequence in the regions close to the promoters of Activin βA and inos genes, and studied the recruitment of Gilz on these genes using chromatin immunoprecipitation (ChIP). In WT MSC, while no binding of Gilz at the inos promoter could be observed in unstimulated cells, activation with IFN-γ and TNF-α induced a massive recruitment of Gilz to the inos promoter at +22nt (Fig. 4E). As expected, in Gilz-/- MSC, no significant recruitment of Gilz to the inos promoter was observed (Fig. 4E). The stimulation of WT MSC by IFN-γ and TNF-α also induced substantial recruitment of Gilz at the Activin βA promoter at -129 nt, compared to unstimulated cells in which no binding of Gilz on Activin βA promoter was observed (Fig. 4F). Altogether these results suggest that under MSC activation, cytosolic Gilz is translocated to the nucleus where it binds to the inos and Activin βA promoters to regulate the expression of these two key mediators of MSC immunosuppressive effects.
Study of Gilz DNA binding motifs in primed MSC. (A-C) Immunodetection of Gilz localization in MSC by confocal microscopy. The cells were visualized and photographed using a confocal microscope (Leica TCS SP5; Leica, Heidelberg, Germany). All the analyses were performed using the ImageJ program. For quantification, the fluorescence intensity reflecting Gilz expression profile (either total or nuclear) was quantified in different fields of each image for each condition. (D) The fasta sequences of the promoters of iNOS and Activin were used to look for overrepresented motif enrichment through the Motif Discovery oligo-analysis from Regulatory Sequence Analysis Tools (RSAT) that could be a possible target for Gilz binding, selecting oligomer lengths up to 5 bases and count on single strand parameters. (E-F) WT and Gilz-/- MSC were treated at 80% confluence either with 20 ng/mL IFN-γ and 10 ng/mL TNF-α. Then, the cells were harvested to perform chromatin immunoprecipitation (ChIP) experiments and the binding of Gilz on inos or Activin βa promoter regions was analyzed. Mean values of n≥3 independent experiments. * P < 0.05. All error bars indicate SEM.
Gilz-dependent secretion of Activin A by MSC impairs Th17 cell differentiation and function
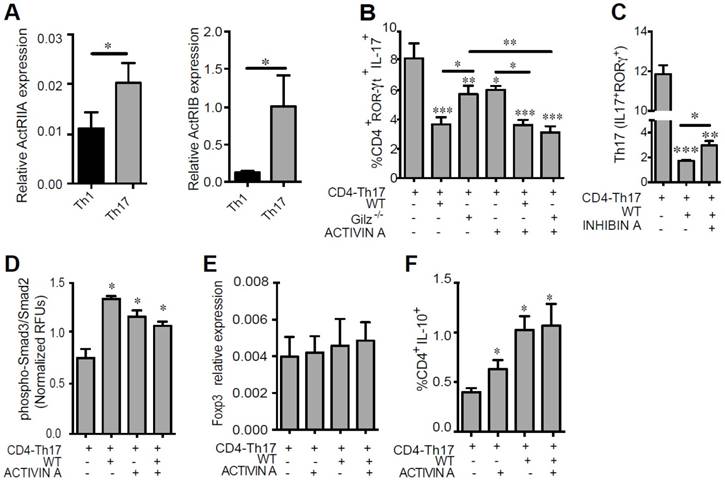
To determine whether Activin A produced by MSC is critical for their immunomodulatory effects on Th1 or Th17 polarization, we determined the expression of Activin receptors (ActRIIA and ActRIB) on these T cell subsets. Both Th1 and Th17 cells expressed ActRIIA and ActRIB, but receptor expression was significantly higher in Th17 cells (Fig. 5A). We thus further investigated the role of Activin A on the suppressive effect of MSC on Th17 cells. CD4+ T cells under Th17 polarizing conditions were co-cultured with WT or Gilz-/- MSC for 3 days, in the presence or absence of Activin A. Compared to WT MSC, Gilz-/- MSC inhibition of Th17 differentiation, as estimated by IL-17 and ROR-ɣT expression, was significantly impaired (Fig. 5B). The addition of Activin A restored the ability of Gilz-/- MSC to inhibit Th17 differentiation (Fig. 5B) but had no effect on WT MSC (Fig. 5B). Of note, the addition of Activin A to CD4+ T cells significantly reduced the frequency of Th17 cells, but these effects were less than the effects of co-culture with WT MSC (Fig. 5B). To further study the key role of Activin A on MSC immunoregulatory role, we assessed the effect of Inhibin A, a natural Activin antagonist. The addition of Inhibin A in the co-culture of MSC with differentiating Th17 cells partially but significantly reversed the inhibitory effect of MSC on Th17 cell differentiation (Fig. 5C). These results suggest that Gilz-dependent secretion of Activin A by MSC provides a mechanism through which MSC inhibit Th17 cell differentiation. Like other members of the TGF-β family, Activin A signals via phosphorylation of Smad2 and/or Smad3 proteins. Thus, we addressed the effects of MSC on the phosphorylation of these proteins in differentiating CD4-Th17 cells. Freshly isolated CD4+ T cells were cultured under Th17 skewing conditions with or without WT MSC and/or Activin A. Both WT MSC and Activin A enhanced Smad3/Smad2 phosphorylation in T cells (Fig. 5D). However, no additive effect of Activin A and MSC was observed (Fig. 5D). We previously showed increased expression of TGF-β1 in MSC when cultured with T cells undergoing Th17 differentiation [24]. Since Activin A acts as an enhancer of the TGF-β1-induced generation of iTreg, and that Treg and Th17 cells differentiate in a mutually exclusive fashion [42], we addressed whether IL-10-producing cells were generated under Th17-skewing conditions in presence of MSC and/or Activin A. In parallel with the suppression of Th17 differentiation by culture with MSC or Activin A shown above, a significant increase in the percentage of T cells producing IL-10 was observed in response to MSC or Activin A (Fig. 5F). No effect on Foxp3 expression level was observed in T cells cultured under the same conditions (Fig. 5E). Collectively, these data suggest that Activin A production by MSC represses the Th17 differentiation program accompanied by Smad3/2 activation and the generation of IL-10 producing T cells.
Role of Activin A on Th17 cell function. (A) The expression levels of the Activin A receptors ActRIIA and ActRIB were determined on Th1 and Th17 cells by RT-qPCR. (B-C) Cell differentiation was assessed in the absence or presence of WT MSC or Gilz-/- MSC added at day 0 (CD4-Th17) at a MSC:T ratio of 1:10. When indicated, 100 ng/mL of Activin A (B) or 30 ng/mL of Inhibin A (C) was added to the T cells, alone or in combination with the MSC. The effect of Activin A or Inhibin A on the generation of Th17 cells was assessed by measuring the percentage of the IL-17 producing T-cells positive for ROR-γT under Th17 skewing conditions (CD4-Th17). CD4+ T cells were cultured without or with MSC, alone or in combination with Activin A. Unless otherwise indicated, P values refer to values obtained for CD4-Th17 when cultured alone. (D) Smad3/Smad2 phosphorylation in CD4+ T cells cultured for 3 h under Th17 skewing conditions in absence or presence of WT MSC. When indicated, 100 ng/mL of Activin A was added to the T cells induced to differentiate, alone or in combination with the WT MSC. (E) Relative expression level of Foxp3 expressed by CD4+ T cells under Th17-skewing conditions cultured alone of in presence of MSC. (F) Percentage of T cells producing IL-10 after culture of CD4+ T cells under Th17-skewing conditions in presence of MSC or Activin A. Unless otherwise indicated, P values refer to values obtained for either CD4-Th1 or CD4-Th17 when cultured alone. * P < 0.05, ** P < 0.01, *** P < 0.001. Mean values of n≥3 independent experiments.
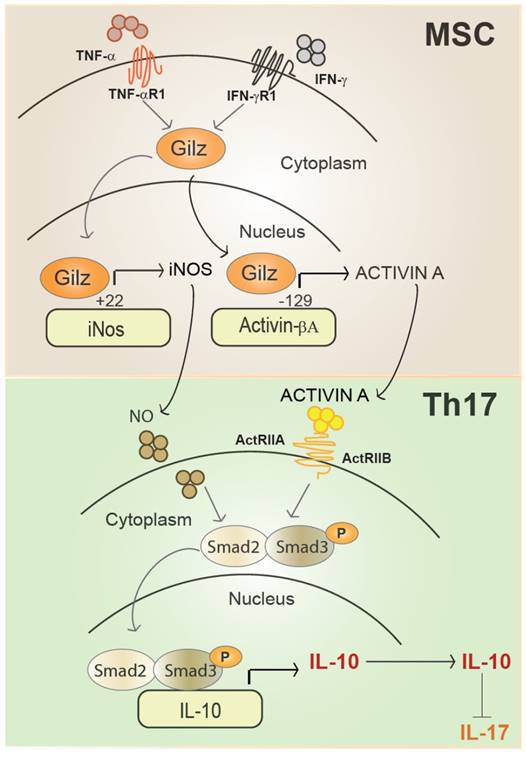
Representative scheme of Gilz nuclear translocation upon MSC activation. In the nucleus, Gilz binds to the binding sites in the promoter of the gene encoding inos and Activin and upregulates their expression levels. Activated MSC secrete high amounts of Activin A that binds to its receptors ActRIIA and ActRIB at the surface of T cells. Both Activin A and NO produced by primed MSC enhance Smad3/Smad2 phosphorylation in Th17 cells promoting IL-10 expression and secretion to the detriment of IL-17.
Discussion
In the present study, we investigated the mechanisms by which Gilz sustains the capacity of MSC to regulate Th1 and Th17 responses in vitro and in vivo. We show that this effect is mediated by an increased secretion of Activin A and NO2 by MSC. The priming of MSC with IFN-γ and TNF-α towards an immunosuppressive phenotype induces Gilz translocation from the cytosol to the nucleus, where it binds to Activin A and inos promoters. These data demonstrate a direct regulation of the transcription of these two genes associated with the immunosuppressive effect of Gilz in MSC. Moreover, our results show that Gilz-dependent release of Activin A by MSC induces the phosphorylation of Smad3/Smad2 in T cells, which promotes a switch from Th17 cells towards an IL-10 expressing regulatory T cell phenotype. We further demonstrated in vitro that the immunosuppressive effect of MSC on activated T cells undergoing Th1 or Th17 differentiation, and on mature Th1 cells, is dependent on Gilz expression. In an adoptive transfer study, we showed that MSC expression of Gilz was required for the inhibition of the transferred Th1 and Th17 cells in immunized mice as well as for the increase in CD4+ T cells producing IL-10 in response to MSC. These results are in line with our previous study showing that MSC prevention of CIA progression by repressing both Th1 and Th17 responses in secondary lymphoid organs, enabling the generation of Treg cells producing IL-10, required the expression of Gilz by MSC [26].
Th17 differentiation, induced by low concentrations of TGF-β, enables T cells to express low levels of T-bet and thereby increases the tendency of Th17 cells to give rise to Th1 cells [43]. In contrast, high concentrations of TGF-β inhibit Th17 cell differentiation [44]. Therefore, depending on the concentration, TGF-β might exhibit different functions by either promoting Th17 or Treg cell differentiation [44, 45]. It was also reported that Activin A and TGF-β signalling pathways converge with regard to Smad2 and 3 during generation of Treg cells [42]. We recently showed that CD4+ T cells under Th17-skewing conditions induce significant increases in TGF-β1 expression in MSC [24]. In the present study we demonstrate that under the same conditions Activin A production by MSC was significantly enhanced. We hypothesized that TGF-β1 and Activin A produced by MSC co-cultured with T cells under Th17 skewing conditions inhibits Th17 differentiation, in favour of Treg. In line with this hypothesis, we showed in the present study that Activin A alone significantly repressed the Th17 molecular program of T cells. In parallel, we demonstrated that addition of Activin A to CD4+ T cells skewed their differentiation into Th17 cells to a population of cells producing IL-10. In accordance with a study showing that Activin A induces the generation of Treg cells without changing the frequency of Foxp3-expressing CD25+CD4+ T cells [34], we did not observe any change in Foxp3 expression level in Activin A-induced CD4+ IL-10-producing cells. This suggests that the immunomodulatory effect of Activin A on Th17 cell responses relies on the induction of IL-10. Together, these results designate Activin A as an additional mediator of MSC immunosuppressive properties. Accordingly, Activin A treatment significantly decreased the frequency of Th17 cells, but to a lesser extent than the inhibition observed in response to co-culture with WT MSC, suggesting that factors produced by MSC additional to Activin A mediate their suppressive functions. However, Gilz-/- MSC lacked the effects of WT MSC on Th17 cell generation, confirming a requirement for Gilz in Th17 suppression mediated by MSC. Finally, Activin A treatment restored the immunomodulatory properties of Gilz-/- MSC, suggesting that the regulation of the Th17/Treg balance by MSC might be in part mediated by a Gilz-dependent enhancement of Activin A production. Our findings introduce Activin A as a novel member in the family of these regulatory factors produced by MSC.
To our knowledge, this study is the first one to report a Gilz-dependent regulation of Activin A and NO2 production by MSC. Thus, we provide evidence that the suppressive effects of MSC on T cells undergoing Th1 or Th17 differentiation and on mature Th1 cells are mandatory Gilz-dependent. Indeed, we dissected and highlighted a mechanism explaining the role of Gilz in MSC immunosuppressive effects on T cells. We demonstrated that, in inflammatory conditions, factors from the microenvironment such as TNF-α and IFN-γ, induce in MSC the translocation of Gilz protein from the cytosolic to the nuclear compartment where it directly modulates the expression of Activin A and NO2 by binding specific motifs in the promoters of Activin βA and inos, respectively. In primed MSC, Gilz might act as a molecular switch of Activin A synthesis that after its secretion by MSC is able to bind to its receptors ActRIIA and ActRIB present at the surface of Th17 cells. By signalling via the Smad2 and/or Smad3 protein pathway, Activin A ultimately induces the transcription of IL-10 in Th17 cells and inhibition of IL-17, switching the differentiation of the pro-inflammatory Th17 lineage toward a Treg cell phenotype and function. In conclusion, this study, by expanding the knowledge on the molecular mechanisms that mediate the immunosuppressive effects of MSC on T cells, suggests a new paradigm of immunomodulation by MSC. The inhibition of Th17 cell development by Gilz-expressing MSC offers a promising therapeutic strategy for the treatment of various autoimmune diseases that are mediated by Th17 cells.
Materials and Methods
Ethics statement
All animal studies were conducted according to the European guidelines for animal welfare (2010/63/EU). Protocols were approved by the Languedoc Roussillon Institutional Animal Care and Use Committee (CEEA-LR-12163).
Mice
DBA/1 and BALB/c mice were purchased from Charles River Laboratories. Gilz-deficient mice have been previously described [46]. HNT TCR (class II restricted and recognizes its epitope PR/8 HA 126-138 in the context of I-Ad) transgenic mice [47] were backcrossed with BALB/c mice for more than 10 generations. In addition, they were crossed with BALB/c Thy1.1+/+ congenic mice for two generations to achieve homozygosity for Thy1.1. Mice were maintained under specific pathogen free conditions at the animal facility of the Institute for Neurosciences of Montpellier.
Isolation and characterization of murine mesenchymal stem cells
For MSC isolation, bones from littermate C57BL/6 wild-type (WT MSC) and Gilz-deficient mice (Gilz-/- MSC) were used. Bone marrow were collected by flushing femurs and tibias and the cell suspension was plated at a concentration of 1x106 cells/cm2 in a modified minimum essential Eagle's medium (MEM) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Thermo Fisher Scientific), 2 mM glutamine, 100 U/mL penicillin, 100 mg/mL streptomycin (Lonza, Levallois-Perret, France) and 2 ng/mL human basic fibroblast growth factor (bFGF) (R&D Systems, Lille, France). MSC were then characterized by the expression of the typical mesodermal antigens CD29, Sca1 and CD44 and the absence of the hematopoietic markers CD45 and CD11b (all monoclonal antibodies from BD Pharmingen) by flow cytometry (FACS). Functionally, the differentiation potential of WT MSC and Gilz-/- MSC differentiation potential into adipocytes, chondrocytes and osteoblasts was assessed by RT-qPCR using specific primers for each lineage. Indeed, to assess the chondrogenic potential, we used the primers for type II collagen (Col IIB) and aggrecan (AGN) detection, to assess the adipogenic potential, we used the primers for fatty acid binding protein 4 (FABP4) and peroxisome proliferator-activated receptor- (PPAR)γ detection, and to assess the osteogenic potential, we used the primers for osteocalcin (OC) and Alcaline Phosphatase (ALP).
Rescue experiments
The plasmid pCDNA3.1-GILZ (kindly provided by Carlo Riccardi) was transfected in Gilz-/- MSC (Gilz-/-pl.Gilz MSC) using Lipofectamine according to the recommendations of the manufacturer (Invitrogen).
T cell isolation
CD4+ T cells were purified from freshly isolated splenocytes from female DBA/1 mice or from the lymph nodes (LN) and spleen of HNT TCR Thy1.1 transgenic mice using the Dynal® CD4 negative isolation kit according to the manufacturer's instructions (Invitrogen).
In vitro T cell differentiation and co-culture with MSC
Purified CD4+ T cells were then activated with anti-mouse CD3/CD28 Dynabeads (Invitrogen) and cultured in complete Iscove's Modified Dulbecco's Media (IMDM) containing 10% of heat-inactivated FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 20 mM HEPES and 50 µM of beta-mercaptoethanol (Invitrogen). Th1 differentiation was induced by adding 20 ng/mL IL-12 and 2.5 µg/mL anti-IL-4 monoclonal antibody, and Th17 differentiation was induced with 2.5 ng/mL TGF-β1, 50 ng/mL IL-6 and 2.5 µg/mL of both anti-IFN-γ, and anti-IL-4 capture antibodies (R&D Systems). Where indicated, 100 ng/mL of recombinant Activin A or 30 ng/mL of recombinant Inhibin A (R&D Systems) was added. For the pre-treatment of MSC with aldosterone (Sigma Aldrich) at 0.01, 0.1 and 1 µM, the drug was added to the MSC media for 24 h and then removed prior to co-culture of MSC with activated T cells. MSC were co-cultured with activated CD4+ T cells during differentiation towards Th1 or Th17 cells (CD4-Th1 or CD4-Th17), or with mature Th1 or Th17 cells (at day 4 of the differentiation processes). WT MSC or Gilz-/- MSC were added at day 0 or day 4, respectively, at a 1:10 MSC:T cells ratio. After 3 days of co-culture, the differentiation of T-cells was tested by intracellular staining using FACS analysis respectively.
Proliferation assay
Fresh splenocytes or purified CD4+ T cells were labeled with CellTrace Violet (CTV) (Life-Technology, Saint Aubin, France) prior to being cocultured with or without WT MSC or Gilz-/- MSC at a 1:10 MSC:T cell ratio in presence of 5 µg/mL of concanavalin (ConA) (Sigma-Aldrich). After 72 h, the proliferation of T cells was quantified by flow cytometry.
RT-qPCR analysis
Total RNA from WT MSC or Gilz-/- MSC were extracted using the RNeasy mini kit (Qiagen S.A.). RNA (500 ng) was reverse transcribed using the Multiscribe reverse transcriptase (Applied Biosystems). Real-time RT-qPCR was performed using the SYBR Green I Master kit and a LightCycler® 480 Detection system, following the manufacturer's recommendations (Roche Applied Science). Specific primers for Gilz, Col2b, AGN, FABP4, PPAR-γ, OC and ALP were designed using the Primer3 software. Data were normalized to the housekeeping gene ribosomal protein S9 (RPS9). Values are expressed as relative mRNA level of specific gene expression as obtained using the 2-ΔCt formulae.
Activin A, Prostaglandin E2 and NO2 quantification
The enzyme-linked immunosorbent assay (ELISA) for Activin A (R&D Systems) and the Enzyme Immunoassay kit for Prostaglandin E2 (PGE2) (Ann Arbor, Souffelweyersheim) were used. NO2 production was quantified using a modified Griess reagent (Sigma-Aldrich).
Gilz DNA binding motif identification
Briefly, the Mus musculus partial 5'UTR sequence for iNOS, Nos2, inducible nitric oxide synthase and Activin beta A subunit were retrieved from NCBI Nucleotide database (Nucleotide GeneBank codes U58677.1, L23806.1, AF427516.1 & D83214.2) [40]. The fasta sequences were used to look for overrepresented motif enrichment, through the Motif Discovery oligo-analysis provide by the Regulatory Sequence Analysis Tools, RSAT [41], selecting oligomer lengths up to 5 bases and count on single strand parameters. The motif with best expected relative frequency between the sequences was chosen.
Chromatin immunoprecipitation (ChIP)
MSC were treated at 80% confluence either with 20 ng/mL IFN-γ and 10 ng/mL TNF-α or with supernatant collected from Th1 or Th17 cells for 30 min. The cells were harvested and ChIP analysis was performed with a ChIP-IT® High Sensitivity kit (Active Motif) according to the manufacturer´s instructions. The following antibodies were used: mouse anti-Gilz (ab55015, Abcam) and normal mouse IgG (sc-2025, Santa Cruz, Heidelberg, Germany). Primers for inhibinβA were forward: 5'-CTGGAAAAACGAGTCATCTGCTG-3', reverse: 5'-GTCAGAGCTGTCTGAATTCCTCT-3'; and for inos were forward: 5'-CTGGTTTGAAACTTCTCAGCCAC-3', reverse: 5'-CAACGTTCTCCGTTCTCTTGCAG-3'.
Phospho-Smad3 and 2 immunoassays
Phospho-Smad3 and Smad2 were quantified with an ELISA-based assay using fluorogenic substrates kit (R&D Systems). T-cells were fixed with paraformaldehyde 8% and permeabilized following the manufacturer's instructions. Protein phosphorylation was finally measured using a double immunoenzymatic labeling procedure allowing the total protein and the phosphorylated protein to be quantified. Normalized results were determined by dividing the phospho-Smad3/Smad2 fluorescence measured at 600 nm by the total Smad3/Smad2 fluorescence at 450 nm for each sample.
Adoptive transfer experiments
Isolated T cells were labeled with 2 μM of Cell Trace Violet (CTV) (CellTrace CTV Cell Proliferation Kit, Invitrogen) in PBS for 10 min at 37°C. Then, 3,5x106 labeled HNT TCR Thy1.1+ CD4+ T cells were injected i.v. in PBS into female BALB/c mice (8-10 weeks old). In order to immunize the recipient mice, 50 μg of influenza PR/8 HA 126-138 peptide was injected intradermaly in complete freund adjuvant. 2 h after immunization, groups of mice were either left untreated or injected i.v. with 1x106 WT or Gilz-/- derived MSCs.
Flow cytometry analysis
After 3 days of culture or co-culture with MSC, T cells were stimulated for 4 h with 50 ng/mL phorbolmyristate acetate (PMA) (Sigma-Aldrich), 1 µg/mL ionomycin (Sigma-Aldrich), and 10 µg/mL brefeldin A (Sigma-Aldrich). For surface antigen staining, cells were first incubated with antibodies against CD4-APC-H7 and CD25-APC (BD Pharmingen) for 20 min at 4°C in the dark. They were then fixed overnight at 4°C with the FoxP3 staining buffer set (eBioscience) in order to perform intracellular staining. Then, cells were incubated for 30 min with anti-IFN-γ-PE, IL-17-Percp5.5, IL-10-PE (BD Pharmingen), ROR-γT-APC and FoxP3-FITC antibodies (eBioscience). For in vivo experiments, four days after injection of cells and immunization, LN and spleen from the recipient mice were collected and processed separately to obtain a single cell suspension by mechanical disruption on 40 μm filters (Corning) in PBS containing 2% FCS 0.02% sodium azide at 4°C. After counting, LN cells and splenocytes were cultured for 4 h in the prescence of PMA, Ionomycin and Brefeldin A (Sigma Aldrich) at 37º, 5% CO2. Then, cells were stained using specific surface antibodies against CD4 and CD90.1 (BD Pharmingen). For intracellular detection, the specific Fixation and Permeabilization Kit (eBioscience) was used following the manufacturer's instructions. Specific antibodies were used against Foxp3, IFN-γ, IL-17 and IL-10 in order to identify different subpopulations of T-CD4 cells. Acquisition was finally performed with the FACS Canto II and analyzed using the BD FACSdiva software (BD Pharmingen).
Statistical analysis
Results are expressed as the mean ± (SEM). Generated P values and post-analyses were performed first with Kruskal-Wallis test to analyze differences between all the groups and then using the Mann-Whitney test to compare two groups. P-values < 0.05 (*), P < 0.01 (**) or P < 0.001 (***) were considered statistically significant. Analysis and graphical representation were performed using Graph-Pad PrismTM software (Graphpad).
Abbreviations
GILZ: glucocorticoid-induced leucine zipper; IFN: Interferon; TNF: tumor necrosis factor; iNOS: inducible nitric oxide synthase; TREG: regulatory T cell; IL: inerleukin; TGF: transforming growth factor; MSC: mesenchymal stem cells; PGE2: prostaglandin 2; NF-κB: nuclear factor κB; DC: dendritic cell; AP-1: activator protein 1; MAP kinase: mitogen-activated protein kinase; CTLA4: cytotoxic T-lymphocyte-associated protein 4; RORγ: retinoid-related orphan receptor-γ; DEL-1: developmental endothelila locus-1; ACTRIIA: activin A receptor type IIA; NO2: nitric dioxide; FOXP3: forkhead box protein P3; WT: wild-type; COL2: collagen II; PPAR-γ: peroxisome proliferator-activated receptor-γ; FABP4: fatty acid binding protein; OC: osteocalcin; CONA: concanavalin A; CTV: CellTrace Violet; PHA: phytohemagglutinin; ALDO: aldosterone; ALP: alkaline phosphatase; FBS: fetal bovine serum; CHIP: chromatin immunoprecipitation; HA: hemagglutin.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by Inserm, the University of Montpellier I and grants from the Medical Research Foundation (projet FRM 2011 "Comité Languedoc-Roussillon-Rouergue"), the French National Research Agency as part of the "Investments for the Future", ECellFrance consortium, program n° ANR-11-INBS-0005, the Société Française de Rhumatologie (SFR) and funding from the European Community's seventh framework program for the collaborative project: "REGENER-AR: Bringing Regenerative Medicine into the market: Allogeneic eASCs Phase IB/IIA clinical trial for treating Rheumatoid Arthritis" (contract no. 279174 EC). EFM and DN were supported by a grant from the National Health and Medical Research Council of Australia. D.A.M was supported by Fondecyt postdoctorado grant n° 3160525 and PLC by CONICYT through the program becas Chile folio n° 74140021.
Author contributions
P.L.-C. and F.D. designed the all project and the experiments with the input of E.F.M, J.P, J.H. and C.J.
P.L.-C., G.E.-C., N.I., R.C., G.T., D.A.M., A.-M.V., D.N., F.D. performed the experiments and analysed the results.
G.E.-C. and J.H. designed the adoptive transfer experiment.
P.L.-C., J.P. and F.D. wrote the manuscript with the input of E.F.M, J.H. and C.J.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Boissier MC, Semerano L, Challal S, Saidenberg-Kermanac'h N, Falgarone G. Rheumatoid arthritis: from autoimmunity to synovitis and joint destruction. J Autoimmun. 2012;39:222-8
2. Antiga E, Volpi W, Cardilicchia E, Maggi L, Fili L, Manuelli C. et al. Etanercept downregulates the Th17 pathway and decreases the IL-17+/IL-10+ cell ratio in patients with psoriasis vulgaris. J Clin Immunol. 2012;32:1221-32
3. Nemoto Y, Kanai T, Takahara M, Oshima S, Okamoto R, Tsuchiya K. et al. Th1/Th17-mediated interstitial pneumonia in chronic colitis mice independent of intestinal microbiota. J Immunol. 2013;190:6616-25
4. Haines CJ, Chen Y, Blumenschein WM, Jain R, Chang C, Joyce-Shaikh B. et al. Autoimmune Memory T Helper 17 Cell Function and Expansion Are Dependent on Interleukin-23. Cell Rep. 2013;5:1378-88
5. Beurel E, Kaidanovich-Beilin O, Yeh WI, Song L, Palomo V, Michalek SM. et al. Regulation of Th1 Cells and Experimental Autoimmune Encephalomyelitis by Glycogen Synthase Kinase-3. J Immunol. 2013
6. Rostami A, Ciric B. Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J Neurol Sci. 2013;1-2:76-82
7. Ishigame H, Zenewicz LA, Sanjabi S, Licona-Limon P, Nakayama M, Leonard WJ. et al. Excessive Th1 responses due to the absence of TGF-beta signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc Natl Acad Sci U S A. 2013;110:6961-6
8. DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol. 2016;16:149-63
9. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725-9
10. Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345-50
11. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311-28
12. Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB. et al. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell. 2015;163:1413-27
13. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C. et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell. 2015;163:1400-12
14. Meiler F, Zumkehr J, Klunker S, Ruckert B, Akdis CA, Akdis M. In vivo switch to IL-10-secreting T regulatory cells in high dose allergen exposure. J Exp Med. 2008;205:2887-98
15. Djouad F, Bouffi C, Ghannam S, Noel D, Jorgensen C. Mesenchymal stem cells: innovative therapeutic tools for rheumatic diseases. Nat Rev Rheumatol. 2009;5:392-9
16. Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S. et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30:42-8
17. Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J. et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102:3837-44
18. Ren G, Su J, Zhao X, Zhang L, Zhang J, Roberts AI. et al. Apoptotic cells induce immunosuppression through dendritic cells: critical roles of IFN-gamma and nitric oxide. J Immunol. 2008;181:3277-84
19. Hegyi B, Kudlik G, Monostori E, Uher F. Activated T-cells and pro-inflammatory cytokines differentially regulate prostaglandin E2 secretion by mesenchymal stem cells. Biochem Biophys Res Commun. 2012;419:215-20
20. Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K. et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42-9
21. Sullivan C, Murphy JM, Griffin MD, Porter RM, Evans CH, O'Flatharta C. et al. Genetic mismatch affects the immunosuppressive properties of mesenchymal stem cells in vitro and their ability to influence the course of collagen-induced arthritis. Arthritis Res Ther. 2012;14:R167
22. Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, Cancedda R. et al. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol. 2005;35:1482-90
23. Bouffi C, Bony C, Courties G, Jorgensen C, Noel D. IL-6-dependent PGE2 secretion by mesenchymal stem cells inhibits local inflammation in experimental arthritis. PLoS One. 2010;5:e14247
24. Luz-Crawford P, Noel D, Fernandez X, Khoury M, Figueroa F, Carrion F. et al. Mesenchymal stem cells repress Th17 molecular program through the PD-1 pathway. PLoS One. 2012;7:e45272
25. Selleri S, Dieng MM, Nicoletti S, Louis I, Beausejour C, Le Deist F. et al. Cord-blood-derived mesenchymal stromal cells downmodulate CD4+ T-cell activation by inducing IL-10-producing Th1 cells. Stem Cells Dev. 2013;22:1063-75
26. Luz-Crawford P, Tejedor G, Mausset-Bonnefont AL, Beaulieu E, Morand EF, Jorgensen C. et al. Glucocorticoid-induced leucine zipper governs the therapeutic potential of mesenchymal stem cells by inducing a switch from pathogenic to regulatory Th17 cells in a mouse model of collagen-induced arthritis. Arthritis Rheumatol. 2015;67:1514-24
27. Beaulieu E, Morand EF. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat Rev Rheumatol. 2011;7:340-8
28. Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R. et al. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest. 2007;117:1605-15
29. Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol. 2002;22:7929-41
30. Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V. et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood. 2006;107:2037-44
31. Hamdi H, Godot V, Maillot MC, Prejean MV, Cohen N, Krzysiek R. et al. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood. 2007;110:211-9
32. Yang N, Baban B, Isales CM, Shi XM. Crosstalk between bone marrow-derived mesenchymal stem cells and regulatory T cells through a glucocorticoid-induced leucine zipper/developmental endothelial locus-1-dependent mechanism. FASEB J. 2015;29:3954-63
33. Robson NC, Phillips DJ, McAlpine T, Shin A, Svobodova S, Toy T. et al. Activin-A: a novel dendritic cell-derived cytokine that potently attenuates CD40 ligand-specific cytokine and chemokine production. Blood. 2008;111:2733-43
34. Semitekolou M, Alissafi T, Aggelakopoulou M, Kourepini E, Kariyawasam HH, Kay AB. et al. Activin-A induces regulatory T cells that suppress T helper cell immune responses and protect from allergic airway disease. J Exp Med. 2009;206:1769-85
35. Ihn HJ, Kim DH, Oh SS, Moon C, Chung JW, Song H. et al. Identification of Acvr2a as a Th17 cell-specific gene induced during Th17 differentiation. Biosci Biotechnol Biochem. 2011;75:2138-41
36. Luz-Crawford P, Djouad F, Toupet K, Bony C, Franquesa M, Hoogduijn MJ. et al. Mesenchymal Stem Cell-Derived Interleukin 1 Receptor Antagonist Promotes Macrophage Polarization and Inhibits B Cell Differentiation. Stem Cells. 2016;34:483-92
37. Luz-Crawford P, Ipseiz N, Espinosa-Carrasco G, Caicedo A, Tejedor G, Toupet K. et al. PPARbeta/delta directs the therapeutic potential of mesenchymal stem cells in arthritis. Ann Rheum Dis. 2016
38. Luz-Crawford P, Kurte M, Bravo-Alegria J, Contreras R, Nova-Lamperti E, Tejedor G. et al. Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res Ther. 2013;4:65
39. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI. et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141-50
40. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2009;37:D26-31
41. Medina-Rivera A, Defrance M, Sand O, Herrmann C, Castro-Mondragon JA, Delerce J. et al. RSAT 2015: Regulatory Sequence Analysis Tools. Nucleic Acids Res. 2015;43:W50-6
42. Huber S, Stahl FR, Schrader J, Luth S, Presser K, Carambia A. et al. Activin a promotes the TGF-beta-induced conversion of CD4+CD25- T cells into Foxp3+ induced regulatory T cells. J Immunol. 2009;182:4633-40
43. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO. et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92-107
44. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641-9
45. Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O'Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010;21:425-34
46. Ngo D, Beaulieu E, Gu R, Leaney A, Santos L, Fan H. et al. Divergent effects of endogenous and exogenous glucocorticoid-induced leucine zipper in animal models of inflammation and arthritis. Arthritis Rheum. 2013;65:1203-12
47. Scott B, Liblau R, Degermann S, Marconi LA, Ogata L, Caton AJ. et al. A role for non-MHC genetic polymorphism in susceptibility to spontaneous autoimmunity. Immunity. 1994;1:73-83
Author contact
![]() Corresponding author: Farida Djouad, Inserm U1183, IRMB, 34295 Montpellier cedex 5, France Tel: 33 (0) 4 67 33 04 75- Fax: 33 (0) 4 67 33 01 13 - E-mail: farida.djouadfr
Corresponding author: Farida Djouad, Inserm U1183, IRMB, 34295 Montpellier cedex 5, France Tel: 33 (0) 4 67 33 04 75- Fax: 33 (0) 4 67 33 01 13 - E-mail: farida.djouadfr