Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(4):1027-1041. doi:10.7150/thno.22414 This issue Cite
Research Paper
Targeting ALDH2 for Therapeutic Interventions in Chronic Pain-Related Myocardial Ischemic Susceptibility
Chen Li1,2*, Wanqing Sun3*, Chunhu Gu4*, Zheng Yang1,2, Nanhu Quan5, Jingrun Yang2, Zhaoling Shi2, Lu Yu6 ![]() , Heng Ma1,2
, Heng Ma1,2 ![]()
1. Department of Pathophysiology, Fourth Military Medical University, Xi'an, 710032, China;
2. Department of Physiology, Fourth Military Medical University, Xi'an, 710032, China;
3. Fuwai Hospital, National Center of Cardiovascular Diseases, Beijing 100037, China;
4. Department of Cardiovascular Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, China;
5. Cardiovascular center, First Affiliated Hospital of Jilin University, Changchun, 130000, China;
6. Department of Pathology, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, China.
* These authors contributed equally to this work
Received 2017-8-16; Accepted 2017-10-9; Published 2018-1-1
Abstract

Clinical observations have demonstrated a link between chronic pain and increased ischemic heart disease mortality, but the mechanisms remain elusive. Reactive aldehydes have recently been confirmed as a new player in pain pathologies, while our previous study demonstrated that reactive aldehydes (4-HNE) induced carbonyl stress contributing to myocardial ischemic intolerance. The aim of this study was to explore whether chronic pain increases susceptibility to myocardial ischemia/reperfusion (MI/R) injury and to investigate the underlying mechanisms focusing on toxic aldehyde and carbonyl stress.
Methods: Chronic pain was induced by chronic compression of the dorsal root ganglion (CCD). After 2 weeks CCD, aldehyde dehydrogenase (ALDH2) KO or wild-type (WT) littermate mice were then subjected to in vivo MI/R.
Results: In CCD-WT mice, heightened nociception paralleled circulating aldehyde (4-HNE) accumulation and cardiac protein carbonylation. Mechanistically, CCD-induced 4-HNE overload provoked cardiac Sirtuin 1 (SIRT1) carbonylative inactivation and inhibited Liver kinase B1 (LKB1) - AMP-activated protein kinase (LKB1-AMPK) interaction, which resulted in exacerbated MI/R injury and higher mortality compared with non-CCD WT mice. ALDH2 deficiency further aggravated CCD-induced susceptibility to MI/R injury. Exogenous 4-HNE exposure in peripheral tissue mimicked chronic pain-induced aldehyde overload, elicited sustained allodynia and increased MI/R injury. However, cardiac-specific ALDH2 upregulation by AAV9-cTNT-mediated gene delivery significantly ameliorated chronic pain-induced SIRT1 carbonylative inactivation and decreased MI/R injury (minor infarct size, less apoptosis, and improved cardiac function).
Conclusion: Collectively, chronic pain-enhanced carbonyl stress promotes myocardial ischemic intolerance by SIRT1 carbonylative inactivation and impairment of LKB1-AMPK interaction. ALDH2 activation and prevention of protein carbonylation may be a potential therapeutic target for myocardial ischemic vulnerability in chronic pain patients. Our results newly provided overlapping cellular mechanisms of chronic pain and myocardial dysfunction interplay.
Keywords: chronic pain, myocardial ischemic intolerance, ALDH2, 4-HNE, carbonylation.
Introduction
Pain is not an isolated pathological process. Chronic pain is one of the most inextricable clinic problems and living with pain is linked with poor health for many comorbid conditions [1]. A previous report has demonstrated that chronic pain is associated with increased morbidity and mortality, predominated by cardiovascular disease (CVD) and cancer [2]. Moreover, cardiovascular risk factors are more prevalent in people reporting chronic pain [3]. Ischemic heart disease contributes significantly to morbidity and mortality. Although transient peripheral nociception precondition has been shown to have cardioprotection against myocardial ischemia/reperfusion (MI/R) injury [4], clinical observations have revealed higher ischemic heart disease mortality in individuals with chronic pain [5]. The related mechanisms remain elusive. Different from acute nociceptive stimulation, chronic pain leads to long-term malignant stress, which is likely to be the pathophysiological basis of cardiovascular damage. Identifying the molecular basis that links chronic pain with increased susceptibility to MI/R injury is not only scientifically important but may also reveal new therapeutic targets against pain-related heart injury. Furthermore, non-steroidal anti-inflammatory pain medications (NSAID's) for pain treatment are accompanied by the risk of cardiovascular damage [6, 7]. Cyclooxygenase-2 (COX-2) inhibitors have limited efficacy and increase the risk of cardiovascular problems [8]. Therefore, a novel approach for cardiac-safe pain control is of scientific importance.
As a new player in eliciting pain and pain-sensitization [9], reactive aldehydes are common by-products of lipid peroxidation on mitochondrial and plasma membranes in response to inflammation and cellular injury. 4-hydroxy-2-nonenal (4-HNE), the most abundant and reactive carbonyl species, has been shown to cause in vivo cytotoxic effects [10] and carbonyl stress [11]. 4-HNE undergoes Michael-addition reactions at its C=C double bond through nucleophilic attacked on Cys, Lys or Arg amino acid residues of proteins, thereby forming protein carbonylation. Accumulation of carbonyl stress-induced cytotoxic products can severely impair cellular functions [12]. 4-HNE is a pain trigger in response to tissue injury. More importantly, it has been demonstrated that 4-HNE is also increased in both acute and inflammatory pain models of rodents [13]. Due to its high diffusion properties [14], 4-HNE may operate both short- and long-term effects on other organs (e.g., heart) far from the site of tissue injury. Our previous study demonstrated that 4-HNE induced carbonyl stress contributes to myocardial ischemic intolerance [11, 15] and cardiomyocyte senescence [16]. However, the pathophysiological role of carbonyl stress in possible relationships between chronic pain and MI/R injury is unclear.
Aldehyde dehydrogenase 2 (ALDH2), an abundantly expressed protein in heart and brain, plays a pivotal role in cardiac and neural aldehyde metabolism and detoxification [17]. A new understanding has been established that ALDH2 activation is necessary and sufficient for cardioprotection by reducing cardiac ischemic damage [12, 15]. 4-HNE is a substrate as well as a potent inhibitor of ALDH2. Excessive production of 4-HNE can be completely prevented by the presence of Alda-1, a selective activator of ALDH2 [18]. Our previous data confirmed that activation of ALDH2 indeed reduces MI/R injury [15], improves the cardiac autophagy [16] and prevents carbonylative inactivation of myocardial SIRT-1 [11]. However, it is still unknown whether reduction of aldehyde load by ALDH2 activation may improve cardiac anti-I/R injury capacity under such specific pathological condition of chronic pain.
Therefore, the purposes of the present study were to determine whether myocardial I/R injury is amplified in animals with chronic pain; if so, to investigate whether the enhanced I/R injury observed in these animals can be rescued by ALDH2 activation; and to identify possible mechanisms by which carbonyl stress confers chronic pain-induced myocardial ischemic vulnerability.
Results
Chronic pain sensitizes heart to MI/R injury
To determine the mechanical allodynia in mice, the ipsilateral hind paw withdrawal mechanical threshold (PWMT) was assessed in WT and ALDH2 knock out (KO) mice. There was no difference in the PWMT among WT and ALDH2 KO mice under non-CCD conditions. However, the PWMT was decreased in both WT and KO mice 1 day after CCD surgery and remained stable for more than 3 weeks (Figure 1A), indicating that CCD surgery caused allodynia effect. Meanwhile, PWMT was reduced more significantly in CCD-ALDH2 KO mice in comparison with CCD-WT mice (Figure 1B). Similar allodynia was observed when the nociceptive score [13] was estimated (Figure 1C).
Chronic pain sensitizes heart to MI/R injury. (A) Nociceptive threshold in WT and ALDH2 KO mice subjected to CCD operation at baseline and up to 3 weeks; *P < 0.05 vs. CCD-WT. (B) Ipsilateral nociceptive threshold in WT and ALDH2 KO mice 2 weeks post-CCD compared to the no-CCD control; *P < 0.05 vs. no-CCD WT control; #P < 0.05 vs. CCD-WT. (C) Ipsilateral nociceptive threshold was assessed with a scoring scale 2 weeks post-CCD in WT and ALDH2 KO mice; *P < 0.05 vs. CCD-WT. (D) Representative photographs and quantitative data for infarct size (INF) and area at risk (AAR) in hearts from WT and ALDH2 KO mice hearts 2 weeks post-CCD followed by in vivo MI/R (30 min ischemia/4 h reperfusion) compared to the no-CCD treatment control. (E) Cardiac CK activity relative levels from WT and ALDH2 KO mice hearts 2 weeks post-CCD followed by in vivo MI/R (30 min ischemia/4 h reperfusion) compared to no-CCD control. (F) left ventricular ejection fraction (LVEF) was assessed by echocardiography in WT and ALDH2 KO mice 2 weeks post-CCD or no-CCD control followed by in vivo MI/R (30 min ischemia/4 weeks reperfusion) or sham. (G) Survival curves of WT and ALDH2 KO mice 2 weeks post-CCD or no pain control subjected to 30 min cardiac ischemia and 8 weeks reperfusion or sham (n=30). *P < 0.05 vs. no-CCD WT control; #P < 0.05 vs. CCD-WT. Values are mean ± SEM, n = 6 per group.
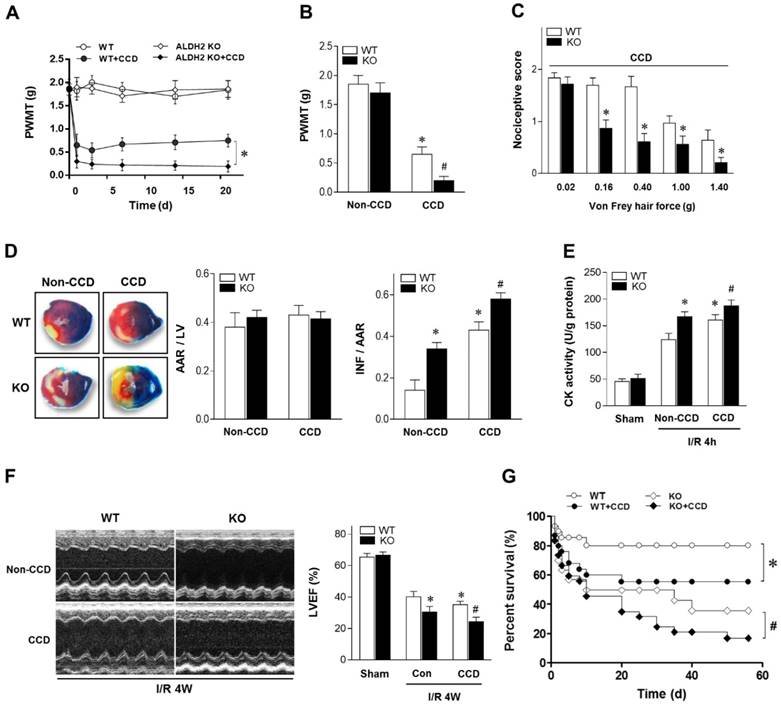
To verify the effect of chronic pain on the cardiac tolerance to ischemic stress, WT and ALDH2 KO mice with or without CCD were subjected to 30 min of ischemia followed by 4 h of reperfusion in vivo. Of interest, chronic pain increased susceptibility to MI/R injury. CCD-WT mice hearts manifested enlarged myocardial infarct size (Figure 1D), increased Creatine kinase MB (CK-MB) levels (Figure 1E) and depressed cardiac contractile function (Figure 1F) compared with non-CCD WT control hearts. Furthermore, the intolerance to MI/R injury became worse in ALDH2 deficient hearts. Ultimately, MI/R-induced mortality was increased in CCD-WT mice more than in non-CCD WT control mice. CCD-ALDH2 KO mice also showed even higher mortality (Figure 1G). These data demonstrate that chronic pain leads to enhanced susceptibility to MI/R injury, which is particularly evident in the ALDH2 deficient heart.
Impaired LKB1-AMPK interaction
Although the concept of AMPK as a cardioprotective molecule during ischemia and I/R conditions is well accepted, the question whether chronic pain could influence ischemic AMPK activation still remains unclear. Compared with non-CCD WT control hearts, post ischemic cardiac AMPK and its downstream target ACC phosphorylation were decreased in CCD-WT mice (Figure 2A).
Aldehydic overload in CCD mice exacerbated by ALDH2 deficiency resulting in cardiac SIRT1 inactivation and impaired LKB1-AMPK interaction. (A) Immunoblots of p-AMPKα (Thr 172) and p-ACC (Ser 79) from WT and ALDH2 KO mice hearts 2 weeks post-CCD. Bar graphs show the ratio of phosphorylated to total AMPK; *P < 0.05 vs. no-CCD WT control; #P < 0.05 vs. WT no pain MI. (B) Immunoblots of AMPKα from LKB1 immune-complexes during ischemic conditions in WT and ALDH2 KO mice 2 weeks post-CCD. (C) Representative co-immunoprecipitation (Co-IP) analysis of LKB1 acetylation in the cardiac tissue from WT and ALDH2 KO mice 2 weeks post-CCD. (D) Plasm 4-HNE levels at baseline and up to 3 weeks in WT and ALDH2 KO mice subjected to CCD operation; *P < 0.05 vs. no-CCD WT control; #P < 0.05 vs. CCD-WT. (E) Representative analysis of cardiac protein carbonyl level in WT and ALDH2 KO mice 2 weeks post-CCD; *P < 0.05 vs. no-CCD WT control; #P < 0.05 vs. CCD-WT. (F) Representative western blot analysis of cardiac ALDH2 expression in WT and ALDH2 KO mice 2 weeks post-CCD compared to the no pain control. (G) Cardiac ALDH2 activation in CCD-WT mice compared to the no-CCD WT control; *P < 0.05 vs. no-CCD WT control. (H) SIRT1 carbonylation and activity in WT and ALDH2 KO mice 2 weeks post-CCD; *P < 0.05 vs. no-CCD WT control; #P < 0.05 vs. CCD-WT. Values are mean ± SEM, n = 6 per group.
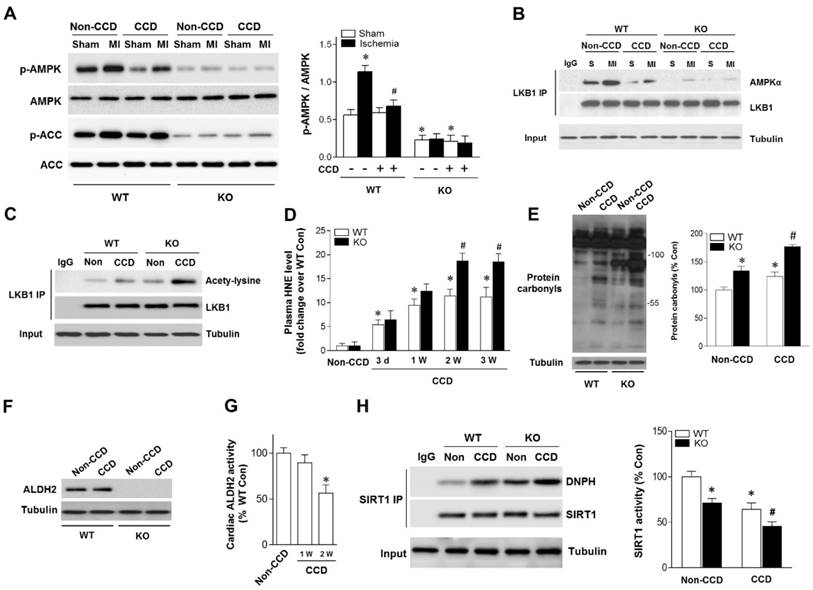
Because LKB1 is the major upstream kinase of AMPK in the heart [19, 20], we next evaluated the LKB1-AMPK interaction during ischemia in both WT and ALDH2 KO hearts. Notably, the LKB1 complex indicated a decreased LKB1-AMPK interaction during ischemia in CCD-WT hearts (Figure 2B), which demonstrates that this mechanism was inhibited by chronic pain. Moreover, ALDH2 deficiency further blocked LKB1-AMPK interaction and AMPK phosphorylation during ischemia in the CCD-ALDH2 KO hearts (Figure 2B). Most noticeably, we newly observed that LKB1 acetylation was higher in CCD-WT hearts compared to the no-CCD WT control, which was further augmented by ALDH2 KO (Figure 2C). Previous studies reported that the acetylation of LKB1 seriously affects its ability to activate AMPK [21]. Consistent with these notions, our results indicated that LKB1-AMPK interaction and ischemic AMPK activation were impaired under chronic pain conditions.
Aldehydic overload and cardiac SIRT1 carbonylative inactivation
Increasing evidence suggests that LKB1 deacetylation is regulated by SIRT1 [21]. Our previous research demonstrated that SIRT1 is an important carbonylation attack target [11, 16] and carbonyl modification leads to SIRT1 inactivation. We hypothesized that carbonyl stress may be involved in chronic pain and further explored the potential mechanisms of chronic pain affecting cardiac SIRT1 activity. In WT mice, the circulating 4-HNE levels were increased as early as 1 day post-CCD, becoming statistically significant 3 days post-CCD, peaking 2 weeks post-CCD, and remaining elevated thereafter. CCD-ALDH2 KO mice had higher plasm 4-HNE levels than CCD-WT mice at 2 weeks post-CCD (Figure 2D). We further found that CCD-induced circulating aldehyde overload was accompanied by cardiac carbonyl stress (Figure 2E). ALDH2 activity (not protein level) in heart was significantly decreased at 2 weeks post-CCD (Figure 2F,G), conforming to the significant increase of cardiac protein carbonyls compared with no pain controls (Figure 2E). Notably, ALDH2 deficiency markedly exacerbated the cardiac carbonyl stress after CCD surgery for 2 weeks (Figure 2D). These results suggest that reactive aldehydes produced by chronic pain were sufficient to mediate cardiac carbonyl stress. ALDH2 deficiency enhanced hyperalgesia and aldehydic overload. Furthermore, using co-immunoprecipitation, we found that chronic pain (2 weeks post-CCD) increased SIRT1 carbonylation and caused an ~40% decrease in SIRT1 activity in WT mice hearts (Figure 2H). The SIRT1 carbonylation and inactivation further deteriorated in ALDH2 KO hearts after CCD surgery (Figure 2H). These results indicate that chronic pain-induced cardiac SIRT1 carbonylation impairs ischemic AMPK activation via LKB1 hyperacetylation, which further deteriorated with ALDH2 deficiency.
4-HNE elicits sustained allodynia, SIRT1 carbonylation and increased MI/R injury
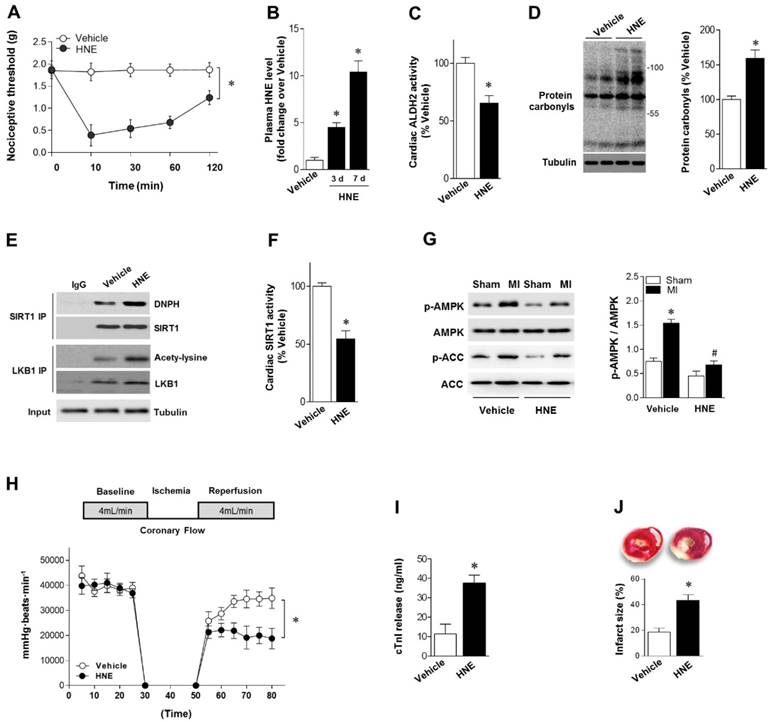
Our results showed that reactive aldehydes of 4-HNE are produced by chronic pain. We next determined whether exogenous 4-HNE could mimic pain-related intolerance to MI/R injury. Injection of 4-HNE (150 nmol in 50 µL per paw [13, 22]) into hind paws of mice induced a robust and sustained allodynia lasting for >120 min, as evidenced by the decreased nociceptive threshold (Figure 3A). WT mice were treated with 4-HNE for 7 days (one dose per day). Plasma 4-HNE levels were significantly increased (Figure 3B), approaching peak levels at 2 weeks post-CCD. 4-HNE treatment suppressed ALDH2 activity (Figure 3C) and generated cardiac protein carbonyls (Figure 3D). We also demonstrated that SIRT1 carbonylation and LKB1 acetylation were elevated in the heart after 4-HNE exposure (Figure 3E), which was consistent with decreased cardiac SIRT1 activity (Figure 3F). Isolated hearts from WT mice with or without 4-HNE treatment were also subjected to Langendorff heart perfusion (independent of neural and humoral factors) to further investigate whether exposure of 4-HNE could increase cardiac vulnerability to ischemic injury. Hearts were perfused for 20 min at basal conditions followed by 20 min of global no-flow ischemia, and were then reperfused for 30 min. Comparing with vehicle control hearts, ischemic AMPK phosphorylation was impaired in 4-HNE-exposed hearts (Figure 3G). Post-ischemic cardiac function in 4-HNE-exposed hearts was significantly reduced during reperfusion as evidenced by decreased heart rate-left ventricle (LV) pressure products (Figure 3H). There were no significant changes in heart rate between 4-HNE-treated and vehicle control groups (432±18 beats/min vs. 441±23 beats/min). I/R-induced cardiomyocyte injury, evidenced by the amount of cardiac-specific troponin-I (cTnI) released from cardiomyocytes (Figure 3I) and myocardial infarct size (Figure 3J), was significantly increased in 4-HNE-exposed hearts.
4-HNE elicits sustained allodynia and increased MI/R injury. (A) Injection of 4-HNE (150 nmol in 50 µL per paw) into hind paws of mice induced sustained allodynia lasting for 120 min. (B) Plasma 4-HNE levels in vehicle or 4-HNE-treated groups at 3 and 7 days. (C) Cardiac ALDH2 activity and (D) protein carbonyl levels in vehicle or 4-HNE-treated groups at 7 days. (E) Representative co-immunoprecipitation (Co-IP) analysis of carbonylation of SIRT1 and acetylation of LKB1 in the cardiac tissue from vehicle or 4-HNE-treated groups at 7 days. (F) Cardiac SIRT1 activity in vehicle or 4-HNE-treated groups at 7 days; *P < 0.05 vs. vehicle. (G) Hearts isolated from vehicle or 4-HNE-treated mice at 7 days were perfused ex vivo for 20 min with global no-flow ischemia, and then reperfused for 30 min. Immunoblots of ischemic p-AMPKα (Thr 172) and p-ACC (Ser 79) in hearts from vehicle or 4-HNE-treated groups. Bar graphs show the ratio of phosphorylated to total AMPK; *P < 0.05 vs. vehicle sham; #P < 0.05 vs. vehicle MI. (H) Cardiac function shown by heart rate-LV pressure products. (I) Cardiac cTnI release and (J) infarct size at the end of reperfusion in hearts from vehicle or 4-HNE-treated groups mice subjected to 20 min global no-flow ischemia and 2 h reperfusion; *P < 0.05 vs. vehicle group. Values are mean ± SEM, n = 6 per group.
ALDH2 activation mediates cardioprotection against MI/R injury in chronic pain mice
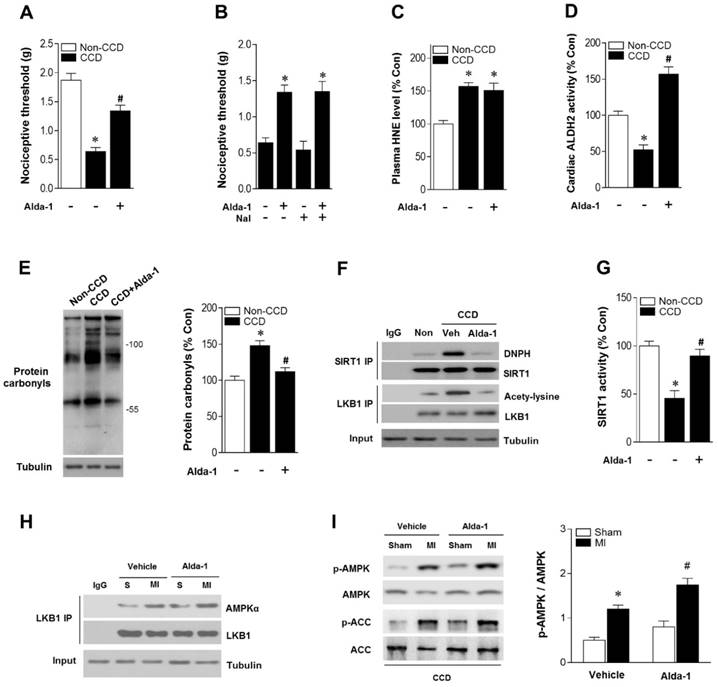
As ALDH2 metabolizes acetaldehyde, we hypothesized that activating ALDH2 would detoxify 4-HNE and protect the hearts from MI/R injury in individuals suffering from chronic pain. Consistent with our and others' previous observations [16, 23, 24], ALDH2 activator Alda-1 did not affect basal cardiac physiological state, suggesting that long-term Alda-1 treatment is cardiac-safe (Table S1). Administration of Alda-1 reversed nociceptive threshold in CCD WT mice (Figure 4A). Consistent with the previous report [13], we also found that naloxone did not block Alda-1-induced analgesic effect, suggesting that the anti-nociceptive effect of Alda-1 is opioid receptor signaling independent (Figure 4B). We found that Alda-1 treatment decreased circulating 4-HNE levels (Figure 4C), enhanced cardiac ALDH2 activity (Figure 4D) and inhibited cardiac protein carbonyl levels (Figure 4E). Alda-1 treatment effectively suppressed the chronic pain-induced SIRT1 carbonylation (Figure 4F) and inactivation (Figure 4G), which in turn alleviated LKB1 hyperacetylation (Figure 4F). On this basis, Alda-1 treatment markedly enhanced LKB1-AMPK interaction during ischemia (Figure 4H) and increased ischemic AMPK and ACC phosphorylation compared with that in untreated group (Figure 4I). Notably, ALDH2 activation significantly improved the tolerance to MI/R injury in CCD mice hearts. MI/R-induced infarct size was reduced (Figure 5A), CK-MB activity was decreased (Figure 5B), and cardiac function (Figure 5C,D) was improved in Alda-1-treated CCD mice. Finally, treatment with Alda-1 also improved the survival rate of post-MI/R CCD mice (Figure 5E). These data suggest that ALDH2 activation may represent a mechanism-based analgesic and cardioprotective therapeutic strategy for chronic pain-related vulnerability to MI/R injury.
ALDH2 activation reduces nociception and aldehydic load in CCD mice. (A) Nociceptive threshold in no-CCD WT control or WT mice 2 weeks post-CCD with or without Alda-1 (ALDH2 selective activator) treatment (3 mg/kg BW per injection, 3 doses per day for 2 weeks); *P < 0.05 vs. no-CCD WT control; #P < 0.05 vs. CCD alone. (B) Alda-1 treatment-induced anti-nociceptive effect is independent of opioid receptor pathway (Naloxone, opioid receptor antagonist, 1mg/kg per subcutaneous injection give 10 min prior to Alda-1 treatment); *P < 0.05 vs. CCD alone. (C) Plasma 4-HNE level. (D) Cardiac ALDH2 activity. (E) Cardiac protein carbonyl levels. (F) Representative co-immunoprecipitation analysis of carbonylation of SIRT1 and acetylation of LKB1 and (G) SIRT1 activity in the cardiac tissue from no pain WT control or 2 weeks post-CCD with or without Alda-1 treatment groups; *P < 0.05 vs. no pain control; #P < 0.05 vs. CCD alone. (H) Immunoblots of AMPKα from LKB1 immune-complexes and (I) immunoblots of p-AMPKα (Thr 172) and p-ACC (Ser 79) during sham or ischemic conditions (ischemia 10 min, in vivo) in no pain WT control or 2 weeks post-CCD with or without Alda-1 treatment groups. Bar graphs show the ratio of phosphorylated to total AMPK; *P < 0.05 vs. CCD vehicle sham; #P < 0.05 vs. CCD vehicle MI. Values are mean ± SEM, n = 6 per group.
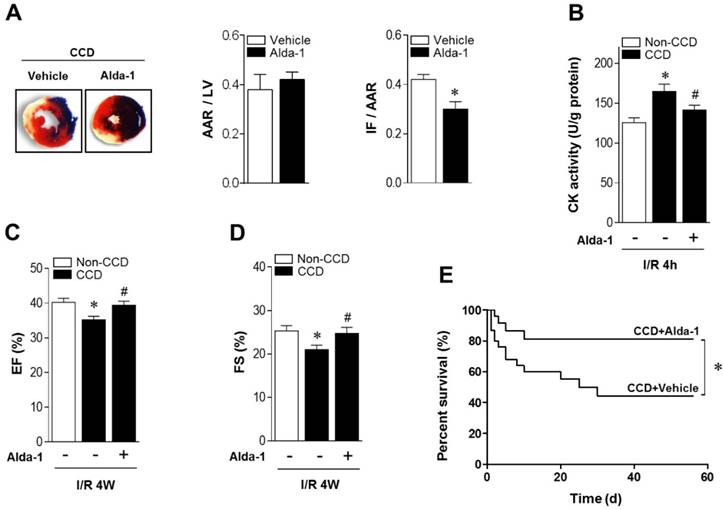
ALDH2 activation reduces MI/R injury in CCD mice. (A) Representative photographs and quantitative data for infarct size (INF) and area at risk (AAR) (*P < 0.05 vs. CCD vehicle) and (B) casapase-3 activity in hearts from Alda-1-treated or not treated hearts 2 weeks post-CCD followed by in vivo MI/R injury (30 min ischemia/4 h reperfusion); *P < 0.05 vs. no-CCD control; #P < 0.05 vs. CCD alone. (C) Average ejection fraction (EF) and fractional shortening (D) assessed by echocardiography in no pain control or Alda-1-treated or not treated CCD hearts after in vivo MI/R (30 min ischemia/4 weeks reperfusion); *P < 0.05 vs. no-CCD control; #P < 0.05 vs. CCD alone. (E) Survival curves of Alda-1-treated or not treated CCD mice subjected to 30 min cardiac ischemia/8 weeks reperfusion (n=30). *P < 0.05 vs. CCD vehicle. Values are mean ± SEM, n = 6 per group.
Cardiac-specific ALDH2 upregulation ameliorates MI/R injury in chronic pain mice
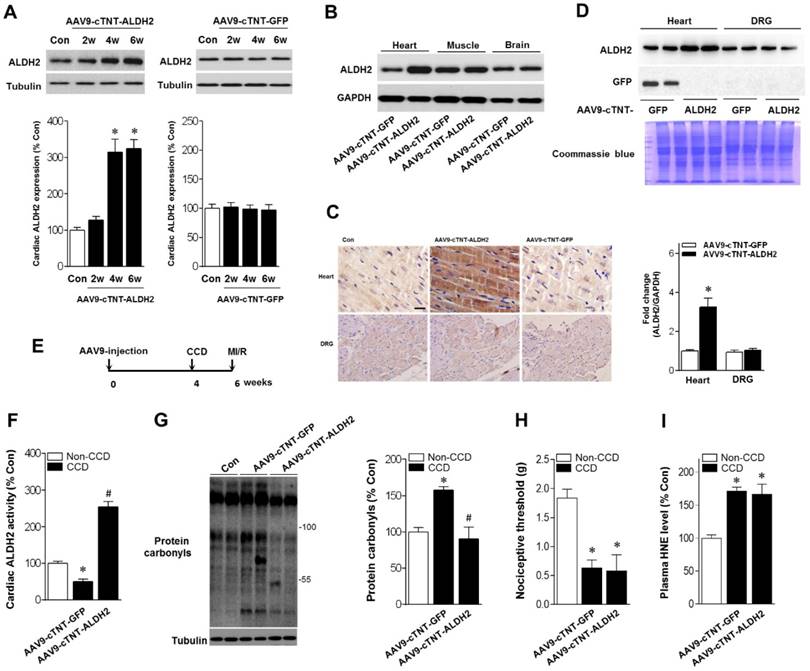
Above-mentioned results have shown that ALDH2 is a key factor in chronic pain-related susceptibility to MI/R injury. The concomitant problem is how to protect the heart in the presence of chronic pain. Cardiac gene transfer may serve as a novel therapeutic approach. We used an Adeno-associated virus (AAV) of serotype 9 expressing ALDH2 under the control of the cardiac-specific cTNT promoter (AAV9-cTNT-ALDH2) [25, 26]. In vivo gene transfer of AAV9-cTNT-ALDH2 or Green fluorescent protein (GFP, negative control) were performed via intra-ventricular cavity injections [27]. Twenty-eight days after control AAV9-cTNT-GFP injection, GFP expression in mouse ventricular myocytes was documented by fluorescence microscopy and a transduction rate of 81±3.2% was observed with a dose of 3×1011 vg/mouse. AAV9-cTNT-ALDH2 exposure elevated ALDH2 in the heart (Figure 6A), but not in the brain, muscle or dorsal root ganglion (DRG) (Figure 6B-D), indicating cardiac-specific ALDH2 overexpression. CCD surgery was performed in AAV9-cTNT-ALDH2 (or GFP) injected mice (Figure 6E). Our results demonstrated that cardiac-specific ALDH2 upregulation in CCD mice markedly enhanced ALDH2 activation (Figure 6F) and decreased cardiac protein carbonylation (Figure 6G). However, the nociceptive threshold (Figure 6H) and plasm HNE level (Figure 6I) were not changed compared to the control. Consistent with the decline in cardiac carbonyl stress, AAV9-cTNT-ALDH2-injected CCD mice hearts exhibited reduced SIRT1 carbonylation (Figure 7A), increased SIRT1 activity (Figure 7B) and suppressed LKB1 acetylation (Figure 7A) compared with GFP-CCD mice hearts. These data indicated that ALDH2 upregulation in cardiomyocytes improved cardiac SIRT1 activity during chronic pain. The LKB1-AMPK interaction during ischemia (Figure 7C) and ischemic AMPK phosphorylation (Figure 7D) in the AAV9-ALDH2-injected CCD mice hearts was also significantly enhanced compared with GFP-control CCD mice hearts. Langendorff heart perfusion system showed that cardiac ALDH2 upregulation directly reduced the susceptibility to I/R injury in CCD mice hearts, evidenced by improved post-ischemic left-ventricular function (Figure 7E), decreased cTnI release (Figure 7F) and decreased infarct size (Figure 7G). Thus, we provide the first evidence, to our knowledge, that cardiac-specific upregulation of ALDH2 through gene transfer with AAV9 vectors bearing the cardiac troponin T promoter results in a functional benefit and enables cardioprotection against MI/R injury in animals with chronic pain.
Cardiac-specific ALDH2 upregulation in vivo enhances ALDH2 activation and ameliorates cardiac protein carbonylation. (A) Representative western blot analysis and quantification of protein levels of cardiac ALDH2 expression in AAV9-cTNT-ALDH2-injected mice or negative controls injected with AAV9-cTNT-GFP; *P < 0.05 vs. no-AAV control (B) Immunoblot results show ALDH2 was specifically upregulated in the mouse heart after AAV9-cTNT-ALDH2 treatment. (C) Immunohistochemical analysis was performed to confirm the cardiac-specific ALDH2 upregulation vs. DRG (scale bar, 20 μm). (D) Representative immunoblots and quantification of protein levels of ALDH2 expression in the heart and DRG from AAV9-cTNT-ALDH2- and AAV9-cTNT-GFP-exposed mice; *P < 0.05 vs. AAV9-cTNT-GFP-injected heart. (E) Protocol for ALDH2 gene therapy in mouse model. CCD surgery was performed, followed 2 weeks later by ex vivo I/R by Langendroff perfusion in AAV9-cTNT-ALDH2-injected mice or AAV9-cTNT-GFP-injected mice. (F) Cardiac ALDH2 activation. (G) Representative analysis of cardiac protein carbonyl level. (H) Ipsilateral nociceptive threshold and (I) plasma 4-HNE levels in rAAV9-ALDH2-injected CCD mice or rAAV9-LacZ-injected CCD mice compared to the no pain control mice; *P < 0.05 vs.no-AAV9 control; #P < 0.05 vs. AAV9-cTNT-GFP-injected heart. Values are mean ± SEM, n = 6 per group.
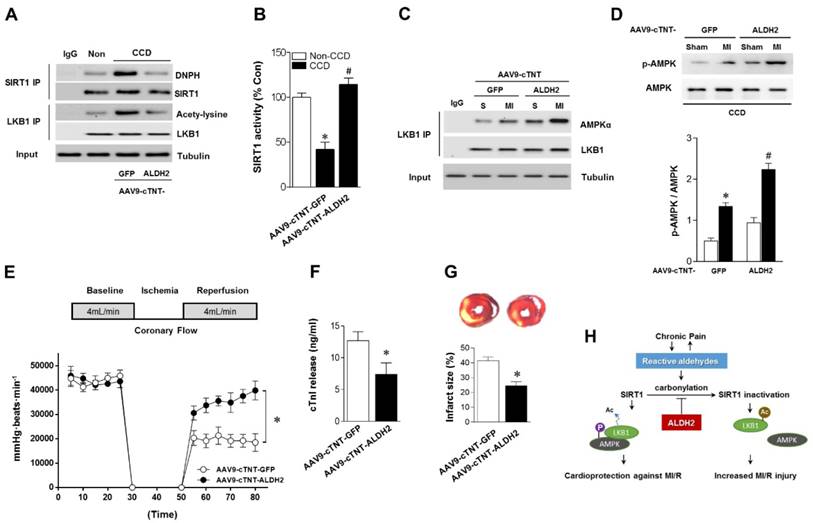
Cardiac-specific ALDH2 upregulation in vivo ameliorates I/R injury in CCD mice. (A) Representative co-immunoprecipitation analysis of SIRT1carbonylation and LKB1 acetylation as well as (B) SIRT1 activity in the cardiac tissue from AAV9-cTNT-ALDH2-injected CCD mice or AAV9-cTNT-GFP-injected CCD mice compared to the no pain control mice; *P < 0.05 vs. no CCD control; #P < 0.05 vs. AAV9-cTNT-GFP-injected CCD mice. (C) Immunoblots of AMPKα from LKB1 immune-complexes and (D) immunoblots of p-AMPKα (Thr 172) during sham or ischemic conditions. Bar graphs show the ratio of phosphorylated to total AMPK; *P < 0.05 vs. AAV9-cTNT-GFP CCD sham; #P < 0.05 vs. AAV9-cTNT-GFP CCD ischemia. (E) Cardiac function shown by heart rate-LV pressure products and (F) Cardiac caspase-3 activity or (G) infarct size from AAV9-cTNT-ALDH2-injected CCD mice or AAV9-cTNT-GFP-injected CCD mice hearts subjected to 20 min global no-flow ischemia and 2 hours reperfusion; *P < 0.05 vs. AAV9-cTNT-GFP-injected CCD group. Values are mean ± SEM, n = 6 per group. (H) Summary of working hypothesis.
Discussion
The relationship between chronic pain and cardiac damage is potentially of pathophysiological and clinical interest, but it is poorly understood. In the present study, we showed that CCD-induced chronic pain caused vulnerability of mice hearts to MI/R injury and resultant higher mortality. Reactive aldehydes (i.e., 4-HNE), which produced and induced pain hypersensitivity, had a direct adverse impact on ischemic hearts. We also identified the cardiac SIRT1 as an important target of 4-HNE-induced carbonyl stress. 4-HNE-induced SIRT1 carbonylation may be associated with impairment of LKB1-AMPK interaction resulting in decreased ischemic AMPK activation, which ultimately increased mortality. This represents that aldehydic overload is, at least in part, a critical pathological basis contributing to chronic neuropathic pain and increased susceptibility to MI/R injury (Graphical abstract). Pharmacological ALDH2 activation by Alda-1 treatment protected hearts against MI/R injury and helped ameliorate pain in mice suffering from chronic pain. More importantly, we, for the first time, established cardiac-specific upregulation of ALDH2 by AAV9-cTNT-mediated gene transfer, which prevented chronic pain-induced cardiac SIRT1 carbonylative inactivation and enabled long-term cardioprotection against MI/R. Our results newly provided overlapping cellular mechanisms by which chronic pain and myocardial dysfunction interplay. Therefore, it can be put forward accordingly as a targeted therapeutic for the comorbidity of chronic pain and cardiac damage.
The interaction between pain and cardiovascular dysfunction is complex, with previous studies reporting conflicting results. It was discovered that transient acute peripheral nociceptive stimulation has cardioprotective effect, which is considered as preconditioning against MI/R injury [4, 28]. However, recent clinical and epidemiological data reported higher cardiovascular mortality in patients with chronic pain [1, 2], especially IHD mortality at nearly three-fold [5]. These results suggest that chronic pain-induced pathophysiological conditions and abnormal stress may lead to cardiovascular vulnerability but the mechanism is unknown. Therefore, we hope to explore the mechanism link between chronic pain and MI/R injury. Here we provide new evidence in CCD mice (a widely used and clinically relevant animal model for study of chronic neuropathic pain and hyperalgesia [29-31]) that chronic pain significantly enhances post-ischemic myocardial injury, increases infarct size, and worsens cardiac functional recovery during in vivo regional MI/R. By contrast, the effective analgesia possesses cardioprotective properties. These results suggest that mechanism-based pain therapeutic approaches may have broad clinical applications in a variety of cardiovascular diseases, such as the chronic pain-related cardiovascular vulnerability.
A recent study demonstrated that aldehyde is closely related to pain [32]. Aldehydes are ubiquitously present in the environment and are also generated during numerous endogenous physiological processes. Aldehydes as diffusible and highly reactive agents in cells that can form adducts with lipids, proteins and DNA, which in turn leads to inactivation of intracellular signaling. Therefore, accumulation of cytotoxic and reactive aldehydes can severely impair cellular functions [12]. 4-HNE may also be generated by the nociception processes. The Mochly-Rosen laboratory recently demonstrated that acute inflammatory pain increased 4-HNE accumulation in the site of injury [13]. Previous studies suggested that leukocytes may be responsible for generating 4-HNE, malondialdehyde and acetaldehyde at sources of injury [33]. Under chronic pain stimulation, nerve endings could produce reactive aldehydes and release them into the circulation [34]. Here we present the first in vivo evidence that a significant peak in reactive aldehyde overload is observable after 2 weeks chronic neuropathic pain presence, and this overload signal remains for further days. More importantly, we also observed a significant positive correlation between increased circulating 4-HNE and cardiac protein carbonyl levels, suggesting that chronic pain-induced aldehyde overload is potentially responsible for cardiac carbonyl stress. The evidence of administering 4-HNE in peripheral tissue for mimetic in vivo aldehyde overload further demonstrated that reactive aldehyde is responsible for cardiac carbonylative protein accumulation in post-CCD hearts. By using an in vivo MI/R model and an ex vivo heart perfusion model, our data clearly showed that chronic pain produces reactive aldehydes and exacerbates MI/R injury. On the contrary, preventing 4-HNE accumulation by ALDH2 activation during chronic pain presence markedly reduced subsequent MI/R injury. Therefore, this is a valuable approach to target efficient detoxification of 4-HNE and prevent cardiac aldehyde overload in therapies of cardiovascular diseases. The catalytic activity of ALDH2 on 4-HNE metabolism has emerged as a key role in cardioprotection. The cardioprotective effects of ALDH2 during MI/R have been demonstrated by ALDH2 transgenic model [12, 15, 17]. Furthermore, reducing ALDH2 activity contributes to peripheral neuropathy, whereas ALDH2 activators may help ameliorate pain [13]. As much as ~36-45% of the East Asians population carries the ALDH2*2 mutant allele, which results in a dramatic reduction in the enzymatic activity. Clinically, comparative studies show that Asian populations are more sensitive to painful stimuli than other ethnicities. Individual carriers of the ALDH2*2 mutation are more susceptible to developing peripheral neuropathy [35]. Moreover, epidemiological studies demonstrated that individuals carrying ALDH2*2 mutation are more susceptible to CVD and to the morbidity of CVD [36]. Collectively, these evidences suggest that ALDH2 activation during chronic pain presence not only reduces pain but also causes cardioprotection against MI/R injury, providing a cardiac-beneficial pain therapeutic target. Our pharmacological (ALDH2 activator) and genetic (cardiac-specific ALDH2 upregulation and ALDH2 KO) evidence demonstrate that lack of ALDH2 in CCD mice increases cardiac carbonyl stress and thus renders cardiomyocytes more susceptible to I/R injury. Cardiac-specific ALDH2 upregulation or ALDH2 activation treatment was sufficient to protect hearts against MI/R injury and help ameliorate pain in mice suffering from chronic pain.
Another significant finding of the current study is that the carbonylative inactivation of SIRT1, the crucial kinase responsible for LKB1-AMPK activation, is responsible for chronic pain-sensitized MI/R injury. Cardiac SIRT1 has been found to resist oxidative stress and suppress cardiomyocyte apoptosis, which both are the main factors responsible for I/R injury. Consistently, cardiac-specific deletion of SIRT1 does not show overt phenotype under basal condition but results in a more vulnerable phenotype during MI/R [37], indicating that SIRT1 activation is important for heart adaptation to I/R stress. Our group [11, 16] and others [38] have reported that SIRT1 activity is regulated by carbonylation, which significantly destroys the myocardial anti-ischemic injury ability. The present study demonstrated that preventing carbonylative SIRT1 inactivation during chronic pain exhibits benefits to MI/R injury. AMPK has been shown to have substantial cardioprotective capabilities against MI/R injury [39]. AMPK activation during ischemia produces effects that have been demonstrated to be beneficial to the myocardium [40]. We provide evidence that ischemic AMPK phosphorylation was significantly impaired in hearts from CCD mice. ALDH2 activation or cardiac-specific ALDH2 upregulation augment ischemic AMPK phosphorylation by facilitating SIRT1 activity. Moreover, LKB1 has been characterized as a major upstream kinase of AMPK in the heart [41]. It has been reported [19] that LKB1-AMPK interaction during ischemia triggers AMPK phosphorylation in the heart. More importantly, the acetylation and deacetylation of LKB1 are mediated by SIRT1, and they can affect its activity secondary to that of AMPK [21, 42]. The protective activities of SIRT1 may be achieved at least in part by fine tuning the acetylation/deacetylation status and stabilities of LKB1 protein [42]. However, the contribution of these molecular events to increased vulnerability for MI/R injury during chronic pain presence remains unclear. In the present study, the ischemia-induced LKB1-AMPK interaction was significantly blunted in the hearts from CCD mice. We found that ALDH2 therapy (Alda-1 treatment or cardiac-specific ALDH2 upregulated by AAV9-cTNT-mediated gene delivery) prevented SIRT1 carbonylative inactivation, which subsequently reduced LKB1 acetylation and enhanced LKB1-AMPK interaction, thus extenuating MI/R injury in chronic pain mice.
In summary, the novel findings in the present study suggest that elevated reactive aldehyde concentration during chronic pain sensitizes MI/R injury via SIRT1 carbonylative inactivation and impairment of LKB1-mediated AMPK activation (Figure 7H). The data further demonstrate that ALDH2 activation or cardiac-specific ALDH2 upregulation blocks cardiac carbonyl stress and attenuates ischemic vulnerability in individuals with chronic pain. The current experimental results have broad clinical implications, indicating that ALDH2 activation and prevention of protein carbonylation might be a potential therapeutic target for chronic pain-induced myocardial ischemic susceptibility.
Materials and Methods
Animals
Male wild-type (WT) mice and ALDH2 KO mice (all C57BL/6 background), 3 mo of age, were used. ALDH2 KO mice were backcrossed into the C57BL/6 background (generation N10) at the animal resource center. The ALDH2 KO mice were originally provided by Dr. H. Zhang (Fuwai Hospital, National Center for Cardiovascular Disease, China). All experimental procedures were reviewed and approved by the Animal Use and Care Committee for Research and Education of the Fourth Military Medical University (Xi'an, P. R. China), and were in accordance with the guidelines provided by the National Institute of Health and the International Association for the Study of Pain.
CCD Surgery
CCD operations were performed as previously described [29-31, 43]. Under 2% isoflurane anesthesia, the intervertebral foramina of L3 and L4 were exposed. An L-shaped steel rod, 2 mm in length and 0.3 mm in diameter, was implanted into each foramen to compress each DRG. No pain control group of mice received no surgical treatment. After CCD surgery the operated mice were singly housed. Food and an automated water supply were available ad lib. The housing facility maintained an air temperature of 22.2±1.1°C, humidity of 50% (± 20%), and a 2:12 light dark cycle. The health and welfare of the animals were checked daily.
Nociception assessment
To quantify the mechanical sensitivity of the hindpaw, mice were placed in individual plastic boxes and allowed to acclimate for 30 min. A series of calibrated von Frey filaments (Stoelting, Kiel, WI, USA) were applied to the plantar surface of the hindpaw (ranging 0.02 g to 2.0 g for mouse) with a sufficient force to bend the filaments for 5 seconds or until paw withdraw. In the presence of a response, the filament of the next lower force was applied. In the absence of a response, the filament of the next greater force was applied. A positive response was indicated by a sharp withdrawal of the paw. Each filament was applied 10 times and the minimal value that caused at least 6 responses was recorded as the paw withdrawal threshold. All behavioral studies were performed under blind conditions [44].
Alda-1 and 4-HNE administration
CCD WT mice were treated with Alda-1 (selective ALDH2 activator; Calbiochem #126920) for 2 weeks. Alda-1 dose (3 mg/kg BW per injection, dissolved in 50% polyethylene glycol and 50% dimethyl sulfoxide by volume) was established in preliminary studies and given in multiple doses (3 doses per day)[13, 16]. Alda-1 was injected subcutaneously to the dorsal side of the neck. One subset of CCD-WT animals received the vehicle alone and served as the control. For 4-HNE administration, mice were injected intraplantarly (i.pl.) with HNE (150 nmol in 50 µL per paw) or vehicle (0.9% saline) was used to induced nociceptive response as previously described [13, 22]. Immediately afterward, the paw threshold was assessed at 10, 30, 60 and 120 min, using von Frey test as previously described [13].
Measurement of 4-HNE levels
Circulating 4-HNE levels were measured using OxiSelectTM HNE-His Adduct ELISA Kit (Cell Biolabs, San Diego, USA) according to the manufacturer's instructions. The plasma 4-HNE concentrations were calculated according to the standard curve [45, 46].
ALDH2 enzymatic activity
ALDH2 enzymatic activity was determined by measuring the conversion of NAD+ to NADH by absorbance of 340 nm, as previously described [15, 47]. ALDH2 activity was expressed as nmol NADH/min per mg protein. The ALDH2 activity of the control group was set as 100%.
Total protein and SIRT1 carbonylation assessment
The protein carbonyl content of tissue was determined as described previously [11, 16]. The carbonyl groups in the protein side chains were derivatized to 2,4-dinitrophenylhydrazone (DNPhydrazone) by reaction with 2,4-dinitro phenylhydrazine (DNPH). The DNP-derivatized protein samples were separated by polyacrylamide gel electrophoresis followed by immunoblotting. Additional studies were performed to detect covalent modification of SIRT1 by carbonylation. SIRT1 was immunoprecipitated using whole-cell extracts in accordance with published methods [38, 48]. To determine the carbonylation of SIRT1, blots were probed first with anti-SIRT1 antibody. After stripping, membranes were equilibrated with 20% (v/v) methanol, 80% Tris-buffered saline for 5 min. Then they were incubated with 0.5 mM 2,4-DNPH for 30 min at room temperature. The membranes were washed and then incubated overnight in anti-DNPH antibody (Abcam), as described previously [11].
SIRT1 activity assay
SIRT1 deacetylase activity was evaluated in crude nuclear extracts from heart samples. Trichostain A (0.2 mM; Sigma-Aldrich, St. Louis, MO, USA), components of Fluor de Lys SIRT1 Fluorescent Activity Assay/Drug Discovery Kit (Enzo Life Sciences, Farmingdale, NY, USA), and 170 μM NAD+ were mixed at 37°C for 1 h, followed by incubation in developer for 15 min at room temperature according to the manufacturer's instructions. Fluorescence intensity was measured using a Fluoroskan Ascent® microplate fluorometer (Thermo Electron Corp., Milford, MA, USA). No-enzyme and time 0 negative controls were generated by incubating developer II solution with 2 mM NAM before mixing the substrates with or without samples. SIRT1 activity was calculated with the corrected arbitrary fluorescence units of the tested samples and expressed as fluorescence units relative to the control [48].
In vivo regional ischemia and assessment of myocardial Infarction
Mice were anaesthetized with 2% isoflurane, intubated, and ventilated with oxygen (Rodent Ventilator, Harvard Apparatus, Millis, MA, USA), and core temperature was maintained at 37°C with a heating pad. After left lateral thoracotomy, the left anterior descending coronary artery was occluded for 30 min with an 8-0 nylon suture and polyethylene tube to prevent arterial injury and then reperfused for 4 h. An electrocardiogram (ECG) and blanching of the LV confirmed ischemic repolarization changes (ST-segment elevation) during coronary occlusion (ADInstruments, Colorado Springs, CO, USA). At different time points, the left ventricle (LV) was isolated before freeze-clamping in liquid nitrogen. Freeze-clamped LV was stored at -80°C for further immunoblotting analysis. The hearts were then excised and stained with 2,3,5-triphenyltetrazolium (TTC) and Evans blue dye to delineate the extent of myocardial necrosis as a percent of the ischemic area at risk (AAR). Hearts were then fixed, sectioned, and photographed with a Leica microscope and analyzed with the ImageJ Software (U.S. National Institutes of Health, Bethesda, MD, USA), as previously described [15, 39].
Echocardiography
Mice were anesthetized using 12 μL/g BW of 2.5% avertin (Sigma-Aldrich), and cardiac function was determined using echocardiography (VisualSonics VeVo 770) as previously described [16].
Creatine kinase (CK) activity assay
Blood samples for CK activity measurement were collected 4 h after reperfusion from mice subjected to I/R and determined spectrophotometrically at 340 nm as described previously [11].
Isolated heart perfusions
Mice were anaesthetized with 2% isoflurane, and hearts were excised and perfused in a Langendorff fashion with a Krebs-Henseleit bicarbonate (KHB) buffer containing 7 mM glucose, 0.4 mM oleate, 1% bovine serum albumin, and 10 mU/mL insulin. Hearts were perfused for 20 min at a flow of 4 mL/min, followed by 20 min of global, no-flow ischemia and 30 min of reperfusion. A fluid-inflated balloon connected to the Chart5 system from ADInstruments was inserted into the LV to measure heart rate and left ventricular developed pressure (LVDP). The balloon was filled to achieve a baseline LV end-diastolic pressure of 5 mmHg, which was kept constant during ischemia and reperfusion [15, 19]. On reperfusion, the perfusate was collected into a beaker on ice. A 1 mL aliquot of the total perfusate was used for detection of cardiac troponin-I (cTnI) using the High Sensitivity Mouse Cardiac Troponin-I ELISA Kit (Life Diagnostics, West Chester, PA, USA) [19].
In vivo Gene therapy
Recombinant adeno-associated virus (serotype 9) vectors carrying mouse ALDH2 (accession number BC005476) with a c-TNT promoter (AAV9-cTNT-ALDH2) (Hanbio Inc, Shanghai, China) were used. AAV9-cTNT-GFP was used as a negative control. Mice underwent gene transfer via aortic cross-clamping as previously described [27, 49, 50]. After dissection of the aorta and pulmonary artery, AAV9-cTNT-ALDH2 [3×1011 vector genomes (vg) per mouse] or AAV9-cTNT-GFP was injected into the left ventricular cavity through a 27G catheter while the aorta and pulmonary artery were cross-clamped for 30 s. This procedure allows the solution that contains the AAV to circulate down the coronary arteries and perfuse the heart without direct manipulation of the coronaries [51]. After cross-clamping was released, the chest was closed. Animals were extubated on spontaneous breathing and observed until fully awake. Myocardial ALDH2 expression was analyzed 2, 4 and 6 weeks after in vivo gene transfer.
Immunoblotting analysis
Immunoblots and immunoprecipitation were performed as previously described [16, 19]. Rabbit antibodies p-AMPKα (Thr172), AMPKα, Tubulin, acetyl-lysine, LKB1 and horseradish peroxidase-linked secondary antibodies were obtained from Cell Signaling (Danvers, MA, USA). ALDH2 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The Zeba™ Micro Spin Desalting Columns 7K MWCO (Thermo Fisher, #89877) and Pierce Co-Immunoprecipitation (Co-IP) Kit (Thermo Fisher, #26149) were used for Co-IP experiments.
Statistical analysis
Values are mean ± SEM. Data analysis was performed using GraphPad Prism 5.0 software (GraphPad Software, La Jolla, CA, USA). Significance was determined by either a 2-tailed, unpaired Student's t test or ANOVA using Tukey's post-test. Data from the isolated heart perfusion experiments comparing cardiac function over time were analyzed by 2-way repeated-measures ANOVA. Log-rank testing was used to evaluate equality of survival curves. A value of P<0.05 was considered statistically significant.
Abbreviations
MI/R: myocardial ischemia/reperfusion; CCD: chronic compression of the dorsal root ganglion; DRG: dorsal root ganglion; ALDH2: aldehyde dehydrogenase 2; 4-HNE: 4-hydroxy-2-nonenal; DNPH: 4-dinitro phenylhydrazine; LKB1: liver kinase B1; AMPK: adenosine 5'-monophosphate (AMP)-activated protein kinase; ACC: Acetyl-CoA carboxylase; PWMT : paw withdrawal mechanical threshold; IF: infarct size; AAR: area at risk; LVEF: left ventricular ejection fraction; FS: Fraction shortening; LVDP: left ventricular developed pressure; CK: Creatine kinase; KHB buffer: Krebs-Henseleit bicarbonate buffer; AAV9: adeno-associated virus (serotype 9); GFP: green fluorescent protein; cTnI: cardiac troponin I; cTnT: cardiac troponin T.
Supplementary Material
Supplementary Table 1.
Acknowledgements
This work was supported by the following grants: National Natural Science Foundation of China No.81322004 and 31671424 (to Dr. Ma); No. 31571413 and 31201037 (to Dr. Yu); No.81500264 (to Dr. Sun); No.81370352 (to Dr. Gu). The Science and Technology Research and Development Program of Shaanxi Province, China (No. 2011KJXX66 and No. 2015KW-050 to Dr. Ma). The Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry No. HG4601 (to Dr. Ma). The State Scholarship Fund, China (No. 201403170453 to Dr. Ma). The innovation team grant of Shaanxi province (No. 2014KCT-20 to Dr. Ma).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sibille KT, Steingrimsdottir OA, Fillingim RB, Stubhaug A, Schirmer H, Chen H. et al. Investigating the Burden of Chronic Pain: An Inflammatory and Metabolic Composite. Pain Res Manag. 2016;2016:7657329
2. McBeth J, Symmons DP, Silman AJ, Allison T, Webb R, Brammah T. et al. Musculoskeletal pain is associated with a long-term increased risk of cancer and cardiovascular-related mortality. Rheumatology (Oxford). 2009;48:74-7
3. Goodson NJ, Smith BH, Hocking LJ, McGilchrist MM, Dominiczak AF, Morris A. et al. Cardiovascular risk factors associated with the metabolic syndrome are more prevalent in people reporting chronic pain: results from a cross-sectional general population study. Pain. 2013;154:1595-602
4. Jones WK, Fan GC, Liao S, Zhang JM, Wang Y, Weintraub NL. et al. Peripheral nociception associated with surgical incision elicits remote nonischemic cardioprotection via neurogenic activation of protein kinase C signaling. Circulation. 2009;120:S1-9
5. Torrance N, Elliott AM, Lee AJ, Smith BH. Severe chronic pain is associated with increased 10 year mortality. A cohort record linkage study. Eur J Pain. 2010;14:380-6
6. Howes LG. Selective COX-2 inhibitors, NSAIDs and cardiovascular events - is celecoxib the safest choice? Ther Clin Risk Manag. 2007;3:831-45
7. Coxib traditional NTC, Bhala N Emberson J, Merhi A Abramson S. et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769-79
8. Howes LG, Krum H. Selective cyclo-oxygenase-2 inhibitors and myocardial infarction: how strong is the link? Drug Saf. 2002;25:829-35
9. Galer EL, Grace PM. Reactive aldehydes: a new player in inflammatory pain. Annals of translational medicine. 2015;3:S23
10. Leonarduzzi G, Robbesyn F, Poli G. Signaling kinases modulated by 4-hydroxynonenal. Free radical biology & medicine. 2004;37:1694-702
11. Gu C, Xing Y, Jiang L, Chen M, Xu M, Yin Y. et al. Impaired Cardiac SIRT1 Activity by Carbonyl Stress Contributes to Aging-Related Ischemic Intolerance. PloS one. 2013;8:e74050
12. Chen CH, Sun L, Mochly-Rosen D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovascular research. 2010;88:51-7
13. Zambelli VO, Gross ER, Chen CH, Gutierrez VP, Cury Y, Mochly-Rosen D. Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Science translational medicine. 2014;6:251ra118
14. Singh M, Kapoor A, Bhatnagar A. Oxidative and reductive metabolism of lipid-peroxidation derived carbonyls. Chemico-biological interactions. 2015;234:261-73
15. Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. European heart journal. 2011;32:1025-38
16. Wu B, Yu L, Wang Y, Wang H, Li C, Yin Y. et al. Aldehyde dehydrogenase 2 activation in aged heart improves the autophagy by reducing the carbonyl modification on SIRT1. Oncotarget. 2016;7:2175-88
17. Chen CH, Ferreira JC, Gross ER, Mochly-Rosen D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiological reviews. 2014;94:1-34
18. Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nature structural & molecular biology. 2010;17:159-64
19. Morrison A, Chen L, Wang J, Zhang M, Yang H, Ma Y. et al. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 2015;29:408-17
20. Sun W, Quan N, Wang L, Yang H, Chu D, Liu Q. et al. Cardiac-specific Deletion of the Pdha1 Gene Sensitizes Heart to Toxicological Actions of Ischemic Stress. Toxicol Sci. 2016;151:193-203
21. Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. The Journal of biological chemistry. 2008;283:27628-35
22. Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B. et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13519-24
23. Sun L, Ferreira JC, Mochly-Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Science translational medicine. 2011;3:107ra11
24. Gomes KM, Campos JC, Bechara LR, Queliconi B, Lima VM, Disatnik MH. et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovascular research. 2014;103:498-508
25. Lucas A, Mialet-Perez J, Daviaud D, Parini A, Marber MS, Sicard P. Gadd45gamma regulates cardiomyocyte death and post-myocardial infarction left ventricular remodelling. Cardiovascular research. 2015;108:254-67
26. Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18:43-52
27. Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E. et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601-5
28. Robbins N, Koch SE, Rubinstein J. Targeting TRPV1 and TRPV2 for potential therapeutic interventions in cardiovascular disease. Transl Res. 2013;161:469-76
29. Song XJ, Hu SJ, Greenquist KW, Zhang JM, LaMotte RH. Mechanical and thermal hyperalgesia and ectopic neuronal discharge after chronic compression of dorsal root ganglia. Journal of neurophysiology. 1999;82:3347-58
30. Wang T, Hurwitz O, Shimada SG, Qu L, Fu K, Zhang P. et al. Chronic Compression of the Dorsal Root Ganglion Enhances Mechanically Evoked Pain Behavior and the Activity of Cutaneous Nociceptors in Mice. PloS one. 2015;10:e0137512
31. Fan N, Donnelly DF, LaMotte RH. Chronic compression of mouse dorsal root ganglion alters voltage-gated sodium and potassium currents in medium-sized dorsal root ganglion neurons. Journal of neurophysiology. 2011;106:3067-72
32. Zambelli VO, Chen CH, Gross ER. Reactive aldehydes: an initial path to develop precision medicine for pain control. Annals of translational medicine. 2015;3:258
33. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxidants & redox signaling. 2014;20:1126-67
34. Guedes RP, Araujo AS, Janner D, Bello-Klein A, Ribeiro MF, Partata WA. Increase in reactive oxygen species and activation of Akt signaling pathway in neuropathic pain. Cellular and molecular neurobiology. 2008;28:1049-56
35. Suzuki Y, Taniyama M, Muramatsu T, Higuchi S, Ohta S, Atsumi Y. et al. ALDH2/ADH2 polymorphism associated with vasculopathy and neuropathy in type 2 diabetes. Alcoholism, clinical and experimental research. 2004;28:111S-6S
36. Jo SA, Kim EK, Park MH, Han C, Park HY, Jang Y. et al. A Glu487Lys polymorphism in the gene for mitochondrial aldehyde dehydrogenase 2 is associated with myocardial infarction in elderly Korean men. Clinica chimica acta; international journal of clinical chemistry. 2007;382:43-7
37. Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N. et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170-82
38. Caito S, Rajendrasozhan S, Cook S, Chung S, Yao H, Friedman AE. et al. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. Faseb J. 2010;24:3145-59
39. Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W. et al. Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010;122:282-92
40. Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R. et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578-82
41. Sakamoto K, Zarrinpashneh E, Budas GR, Pouleur AC, Dutta A, Prescott AR. et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. American journal of physiology Endocrinology and metabolism. 2006;290:E780-8
42. Zu Y, Liu L, Lee MY, Xu C, Liang Y, Man RY. et al. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circulation research. 2010;106:1384-93
43. Hu SJ, Xing JL. An experimental model for chronic compression of dorsal root ganglion produced by intervertebral foramen stenosis in the rat. Pain. 1998;77:15-23
44. Kou ZZ, Li CY, Tang J, Hu JC, Qu J, Liao YH. et al. Down-regulation of insulin signaling is involved in painful diabetic neuropathy in type 2 diabetes. Pain physician. 2013;16:E71-83
45. Guo JM, Liu AJ, Zang P, Dong WZ, Ying L, Wang W. et al. ALDH2 protects against stroke by clearing 4-HNE. Cell research. 2013;23:915-30
46. Lee WC, Wong HY, Chai YY, Shi CW, Amino N, Kikuchi S. et al. Lipid peroxidation dysregulation in ischemic stroke: plasma 4-HNE as a potential biomarker? Biochemical and biophysical research communications. 2012;425:842-7
47. Li SY, Gomelsky M, Duan J, Zhang Z, Gomelsky L, Zhang X. et al. Overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene prevents acetaldehyde-induced cell injury in human umbilical vein endothelial cells: role of ERK and p38 mitogen-activated protein kinase. The Journal of biological chemistry. 2004;279:11244-52
48. Tong C, Morrison A, Mattison S, Qian S, Bryniarski M, Rankin B. et al. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress. Faseb J. 2013;27:4332-42
49. Cutler MJ, Wan X, Plummer BN, Liu H, Deschenes I, Laurita KR. et al. Targeted sarcoplasmic reticulum Ca2+ ATPase 2a gene delivery to restore electrical stability in the failing heart. Circulation. 2012;126:2095-104
50. Sun A, Cheng Y, Zhang Y, Zhang Q, Wang S, Tian S. et al. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. Journal of molecular and cellular cardiology. 2014;71:92-104
51. Quan N, Sun W, Wang L, Chen X, Bogan JS, Zhou X. et al. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. Faseb J. 2017;31:4153-4167
Author contact
![]() Corresponding authors: Heng Ma, M.D., Ph.D., Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fourth Military Medical University, Xi'an 710032, China. E-mail: hengmaedu.cn or Lu Yu, M.D., Ph.D. Department of Pathology, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, China. E-mail: yuluedu.cn
Corresponding authors: Heng Ma, M.D., Ph.D., Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fourth Military Medical University, Xi'an 710032, China. E-mail: hengmaedu.cn or Lu Yu, M.D., Ph.D. Department of Pathology, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, China. E-mail: yuluedu.cn