Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(4):1180-1194. doi:10.7150/thno.20846 This issue Cite
Research Paper
LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d
Haihai Liang1,2#, Zhenwei Pan1#, Xiaoguang Zhao1,2, Li Liu1,2, Jian Sun1,2, Xiaomin Su1,2, Chaoqian Xu1, Yuhong Zhou1,2, Dandan Zhao1, Bozhi Xu1, Xuelian Li1, Baofeng Yang1,2, Yanjie Lu1,2, ![]() , Hongli Shan1,2,
, Hongli Shan1,2, ![]()
1. Department of Pharmacology (State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), College of Pharmacy, Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China
2. Northern Translational Medicine Research and Cooperation Center, Heilongjiang Academy of Medical Sciences, Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China.
#Authors with equal contribution to this study
Received 2017-5-3; Accepted 2017-11-6; Published 2018-2-2
Abstract

Rationale: Cardiac fibrosis is associated with various cardiovascular diseases and can eventually lead to heart failure. Dysregulation of long non-coding RNAs (lncRNAs) has recently been recognized as one of the key mechanisms involved in cardiac diseases. However, the potential roles and underlying mechanisms of lncRNAs in cardiac fibrosis have not been explicitly delineated.
Methods and Results: Using a combination of in vitro and in vivo studies, we identified a lncRNA NONMMUT022555, which is designated as a pro-fibrotic lncRNA (PFL), and revealed that PFL is up-regulated in the hearts of mice in response to myocardial infarction (MI) as well as in the fibrotic cardiac fibroblasts (CFs). We found that knockdown of PFL by adenoviruses carrying shRNA attenuated cardiac interstitial fibrosis and improved ejection fraction (EF) and fractional shortening (FS) in MI mice. Further study showed that forced expression of PFL promoted proliferation, fibroblast-myofibroblast transition and fibrogenesis in mice CFs by regulating let-7d, whereas silencing PFL mitigated TGF-β1-induced myofibroblast generation and fibrogenesis. More importantly, PFL acted as a competitive endogenous RNA (ceRNA) of let-7d, as forced expression of PFL reduced the expression and activity of let-7d. Moreover, let-7d levels were decreased in the MI mice and in fibrotic CFs. Inhibition of let-7d resulted in fibrogenesis in CFs, whereas forced expression of let-7d abated fibrogenesis through targeting platelet-activating factor receptor (Ptafr). Furthermore, overexpression of let-7d by adenoviruses carrying let-7d precursor impeded cardiac fibrosis and improved cardiac function in MI mice.
Conclusion: Taken together, our study elucidated the role and mechanism of PFL in cardiac fibrosis, indicating the potential role of PFL inhibition as a novel therapy for cardiac fibrosis.
Keywords: cardiac fibrosis, lncRNA PFL, let-7d, Ptafr
Introduction
Cardiovascular diseases, the leading cause of death in the world, are accompanied by cardiac interstitial fibrosis. Cardiac fibrosis is characterized by the aberrant proliferation of fibroblasts and deposition of extracellular matrix (ECM) in the heart, and it contributes to heart failure (HF)[1-4]. However, effective treatment of cardiac fibrosis is not currently available, partly due to our incomplete knowledge of the mechanisms underlying fibrogenesis. Therefore, a better understanding of the molecular mechanisms of cardiac fibrosis is essential for the development of novel anti-fibrotic strategies for cardiac disease.
Non-coding RNAs (ncRNAs) are a class of RNAs without protein-coding function and play a pivotal role in cellular physiology and pathology[5]. A large number of studies have shown that microRNAs (miRNAs), such as miR-101, miR-133 and miR-21, are involved in the process of cardiac fibrosis[6-8]. The let-7 family has been shown to participate in the generation and development of multiple cardiovascular diseases, including arrhythmia[9], myocardial infarction[10] and cardiac hypertrophy[11]. However, the contribution of let-7 in cardiac remodeling is still controversial, and it remains to be determined whether let-7 is involved in myocardial fibrosis[12, 13]. If so, the molecular and signaling mechanisms underlying the function of let-7 should be investigated. In addition, the upstream factors that regulate let-7 during cardiac fibrosis need to be elucidated.
Recently, long non-coding RNAs (lncRNAs) have been recognized as novel factors that regulate the initiation and progression of cardiac diseases by modulating chromatin remodeling, via transcriptional and posttranscriptional control and by competitively binding miRNAs[5, 14, 15]. However, insights into the biological roles of lncRNAs in cardiac fibrosis are only just beginning to emerge, and several studies have reported the involvement of lncRNAs in myocardial fibrosis[16-18]. Our previous work reported the differential expression of lncRNAs in MI mice using bioinformatics analysis based on microarray data and verified changes in 14 lncRNAs by qRT-PCR[19]. However, the detailed roles and mechanisms of these differentially expressed lncRNAs in the process of myocardial fibrosis have not been well understood.
In this study, we reported that overexpression of lncRNA NONMMUT022555, which has been named pro-fibrotic lncRNA (PFL), led to fibrogenesis through increasing cell viability and promoting fibroblast-myofibroblast transition by competitively binding let-7d, which inhibited the expression of platelet-activating factor receptor (Ptafr). The silencing of PFL by adenoviruses in vivo mitigated cardiac fibrosis and improved heart function in MI mice. This study indicated that manipulating the expression of PFL represents a promising therapeutic strategy for cardiac fibrosis.
Materials and Methods
Myocardial infarction (MI) models
The procedures for the use of animals (8-week-old C57BL/6 mice; 20-30 g) in this work were in accordance with the regulations of the Ethic Committees of Harbin Medical University and conformed to the NRC Guide for the Care and Use of Laboratory Animals (2011, 8th ed.). Mice anesthetized with pentobarbital (40 mg/kg, i.v.) were randomly divided into two groups: the sham-operated control group and the MI group. The trachea was cannulated and ventilated with a small animal ventilator at a tidal volume of 0.5 mL. A standard limb lead II electrocardiogram (ECG) was continuously recorded by an ECG recorder (BL 420, ChengDu TME Technology Co., Ltd., ChengDu, China). Body temperature was maintained at 37°C by placing the animal on a heating pad. The chest was opened by a left thoracotomy between the 3rd and 4th ribs. A segment of saline-soaked 7-0 sutures was looped around the left anterior descending coronary artery (LAD), near its origin from the left coronary artery. The LAD was occluded, and then, the chest was closed. All surgical procedures were performed under sterile conditions. Successful occlusion was confirmed by elevation of the S-T segment in lead II. After 28 days, the mice were anesthetized and sacrificed, and their hearts were quickly isolated. The portion of the heart bordering the MI was prepared for Masson staining and other experiments. Sham animals underwent the chest opening procedures without LAD occlusion.
Isolation and culture of cardiac fibroblasts (CFs) and cardiomyocytes (CMs) from neonatal mice
Neonatal mouse CFs were prepared via the following procedures: mouse hearts from 1- to 3-day-old C57BL/6 mice were finely minced and placed together in 0.25% trypsin. Pooled cell suspensions were centrifuged and then resuspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin. The suspension was plated onto culture flasks for 90 min to allow for preferential attachment of fibroblasts to the bottom of the culture flasks. Non-adherent and weakly attached cells were considered to be CMs and were transferred to new culture flasks for subsequent studies. CFs were grown to confluence and subsequently passaged using trypsin. Both CFs and CMs were incubated at 37°C with 5% CO2. Studies were conducted on CFs (passage 2-3) that were grown to subconfluence in serum-containing media. After starvation in serum-free medium for 24 h, CFs were administered recombinant human TGF-β1 (10 ng/mL, Sigma-Aldrich Co., LLC, USA) for 24 h.
Masson's trichrome staining
The hearts of MI mice or sham mice were quickly dissected and immersed in 4% neutral buffered formalin for 24 h and stained with Masson's trichrome. Fibrotic tissue was quantified with Image-Pro Plus 6.0. Three areas were analyzed for each slide, and each area was divided into 100 squares. Collagen staining (blue stain) in each square was scored as 1 (present) or 0 (absent). The results are shown as the percentage of area occupied by fibrosis to the total area.
In vitro translation assay
The full-length of PFL was acquired using PCR and inserted into pcDNA3.1 vector. Then the in vitro translation assay was performed according to the manufacturer's instructions (Cat#AM1200, Invitrogen). pcDNA3.1-C/EBPβ was used as a positive control.
Transfection procedures
For transfection, cells were first washed once with serum-free medium and then incubated in 4 mL of serum-free medium for 4-6 h. miRNA mimic (100 nM) or other constructs (1 µg/mL for PFL and shRNA-PFL) and lipofectamine 2000 (Invitrogen, Carlsbad, CA) were separately mixed with 500 μL of Opti-MEM ® I Reduced Serum Medium (Gibco, Grand Island, NY) for 5 min. Then, the two mixtures were combined and incubated at room temperature for 20 min. The lipofectamine:miRNA (or plasmid) mixture was added to the cells and incubated in 6-well culture plates at 37°C for 6 h. Let-7d mimic was purchased from RiboBio Co., Ltd. (Guangzhou, China). Subsequently, 5 mL of fresh medium containing 10% FBS was added to the flasks and the cells were maintained in the culture medium for 48 h until the subsequent experiments.
MTT cell viability assay
CFs were seeded in 96-well culture plates at 1×104 cells/well and incubated at 37°C with 5% CO2. After treatment, the MTT assay (Amresco, Solon, USA) was performed. Briefly, 20 μL of MTT solution (5 mg/mL) was added to each well and incubated for 4 h. Formazan crystals were then dissolved in 150 μL of DMSO. The optical density of the wells was measured at 490 nm with a microplate reader (BioTek, Richmond, USA).
EdU fluorescence staining
5-ethynyl-2′-deoxyuridine (EdU) fluorescence staining was used to detect newly synthesized DNA in CFs after the indicated treatment. All steps were performed according to the manufacturer′s instructions for the Cell-Light EdU DNA cell proliferation kit (RiboBio, Guangzhou, China).
Western blotting
For western blot analysis, total protein samples were extracted from tissues or cells as previously described[20]. Briefly, tissues or cells were lysed with RIPA lysis buffer (Beyotime, Jiangsu, China). A 60 μg protein sample was fractionated on a 10% or 15% SDS-polyacrylamide gel. After electrophoretic transfer to a nitrocellulose blotting membrane (Pall Life Science), the blots were probed with primary antibodies, with GAPDH (anti-GAPDH antibody from Kangchen, Shanghai, China, 1:2 000) as an internal control. Mouse and rabbit polyclonal antibodies for α-SMA (ab7817, 1:500) and Ptafr (ab104162, 1:200), respectively, were purchased from Abcam (Abcam Inc., USA). Antibodies against collagen 1 (14695-1-AP, 1:500) and FN1 (15613-AP, 1:500) were purchased from Proteintech (Rosemont, IL, USA). The rabbit polyclonal antibody against CTGF (sc-25440, 1:200) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The immunoreactivity was detected using an Odyssey Infrared Imaging System. The bands were quantified by measuring the band intensity for each group.
Immunofluorescence staining
Cultured CFs on sterile glass cover slips were washed briefly with cold PBS 3 times and fixed with 4% paraformaldehyde for 15 min. Then, the cell membrane was permeabilized by 0.4% Triton X-100 for 1 h and blocked by normal goat serum for 1 h, at 37°C. The cells were incubated with anti-α-SMA antibody (Abcam Inc., USA, ab7817, 1:100) and anti-vimentin antibody (Cell Signal Tech, #5741, 1:100) overnight at 4°C and subsequently incubated with a FITC-conjugated goat anti-mouse antibody for 1.5 h. The cells were then washed with PBS, and the nuclei were stained with DAPI (Roche Molecular Biochemicals) for 5 min at room temperature. Immunofluorescence was analyzed under a fluorescence microscope (Nikon 80i, Japan).
Ultrasound imaging measurements
Four weeks after MI, mice were anesthetized, and the prethoracic fur was removed using NairTM depilatory cream (Church & Dwight Co., Inc., Princeton, NJ, USA). Ultrasound imaging measurements were acquired using a Vevo®2100 High-Resolution Imaging system (Visual Sonics, Toronto, ON, Canada). Mice were positioned on a MicePad (part of the VisualSonics Vevo Integrated Rail System II) equipped with an integrated heater. Body temperature was maintained at 37°C. Pre-warmed Aquasonic Clear®Ultrasound Gel (Parker Laboratories, Inc., Fairfield, NJ, USA) was used as a coupling agent between the ultrasound scan-head and skin. Left ventricular systolic diameter (LVSd), left ventricular diastolic diameter (LVDd) and LV internal diameter (LVID) were measured in at least three consecutive cardiac cycles. Ejection fraction (EF) and fractional shortening (FS) were calculated using Vevo®2100 High-Resolution Imaging system (Visual Sonics).
Quantitative RT-PCR
Total RNA samples were extracted from the heart tissues of mice or cultured cells using TRIzol (Invitrogen, Carlsbad, CA). As described in our previous study[21], the relative expression levels of mRNAs and miRNAs were quantified using the mirVana qRT-PCR miRNA Detection Kit with SYBR Green I (Applied Biosystems, Foster City, CA). After a 40-cycle reaction, the threshold cycle (Ct) was determined and relative mRNA and miRNA levels were calculated based on the Ct values and normalized to the GAPDH or U6 level in each sample. The data were analyzed using the 2-ΔΔCT method. The primer sequences are shown in Table S1.
Luciferase reporter assays
Ptafr 3'UTR containing the conserved let-7d binding sites and mutated sequences were synthesized by Invitrogen and amplified by PCR. The PCR fragment was subcloned into the SacI and HindIII sites downstream the luciferase gene in the pMIR-Report plasmid (Promega). The let-7d sensor reporter was constructed according to the method previously described[22]. Briefly, the mouse genomic sequence (200 bp) flanking pre-let-7d was reversely inserted into the pGL3 vector downstream of the luciferase gene coding region.
PFL was amplified by PCR. The forward primer was 5'- CCGCTCGAGTCTTTTTAGCTACTCTCTACTTTC -3'. The reverse primer was 5'- ATAAGAATGCGGCCGC GACAAGAATCCTGACTTTGGG -3'. To produce mutated PFL devoid of let-7d binding site, the mutations were generated using QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). Wild type and mutated PFL were subcloned into the pGL3 vector (Promega) immediately downstream the coding region of luciferase gene. The primers as following:
Mut1 F: 5'- CATGTGGCCAGCAAACCACAGGGATTTCTTCATTCTTGACATCCAACAATTACAAACATG -3'; R: 5'- CATGTTTGTAATTGTTGGATGTCAAGAATGAAGAAATCCCTGTGGTTTGCTGGCCACATG -3'; Mut2 F: 5'- CACTAATGCTCATGAGGCAAGCACTTCGTCGCTCTATCCATCTCCTTAGCCAACACTTG -3'; R: 5'- CAAGTGTTGGCTAAGGAGATGGATAGAGCGACGAAGTGCTTGCCTCATGAGCATTAGTG -3'.
Next, the luciferase vector (0.1 μg) was cotransfected with let-7d mimics or PFL into HEK-293 cells or CFs using lipofectamine 2000 (Invitrogen, Carlsbad, CA). As an internal control, 10 ng of the renilla luciferase reporter was also included. After transfecting for 48 h, the cells were collected, and the dual-luciferase activities were measured by a luminometer according to the manufacturer's instructions (Promega, USA).
Pull-down assay with biotinylated miRNA
CFs were transfected with biotinylated miRNA (100 nM) as previously described[22] and harvested 48 h after transfection. The cells were washed with PBS followed by a brief vortex and incubated in lysis buffer on ice for 10 min. The lysates were incubated with M-280 streptavidin magnetic beads (Sigma). The beads were incubated at 4°C for 3 h and washed twice with ice-cold lysis buffer, three times with the low-salt buffer and once with high-salt buffer. The bound RNAs were purified using TRIzol for the analysis.
Construction of the plasmid expressing short-hairpin RNA (shRNA) and the adenovirus vector
The mmu-let-7d precursor DNA and shRNAs for mouse Ptafr and PFL were constructed using plasmid pAdv-shRNA ExpressionVector (Biowit Technologies, Shenzhen, China). The shRNA sequences are shown in Table S2. These different shRNA sequences were each inserted into the pAdv-CMV plasmid or pAdv-shRNA plasmid. A scrambled RNA was used as a negative control. The Adv harboring these shRNA fragments were generated by Biowit Technologies (Shenzhen, China). Adv was amplified in HEK293 cells.
In vivo adenovirus infection
Three days after surgery, the mice were injected with Adv-sh-PFL (or Adv-sh-Scr) and Adv-let-7d (or Adv-NC) via the tail vein (1x1010 plaque-forming units at a volume of 200 μL). Mice in the sham and MI groups underwent the same procedures but received 200 μL of saline.
Statistical analysis
Data are presented as the mean ± SEM. One-way analysis of variance (ANOVA) followed by Bonferroni or Dunnett's post hoc test was used for multiple group comparisons. A two-tailed p value < 0.05 was considered as a statistically significant difference. Data were analyzed using GraphPad Prism 5.0 and SPSS 14.0.
Results
LncRNA PFL as a regulator of cardiac fibrosis
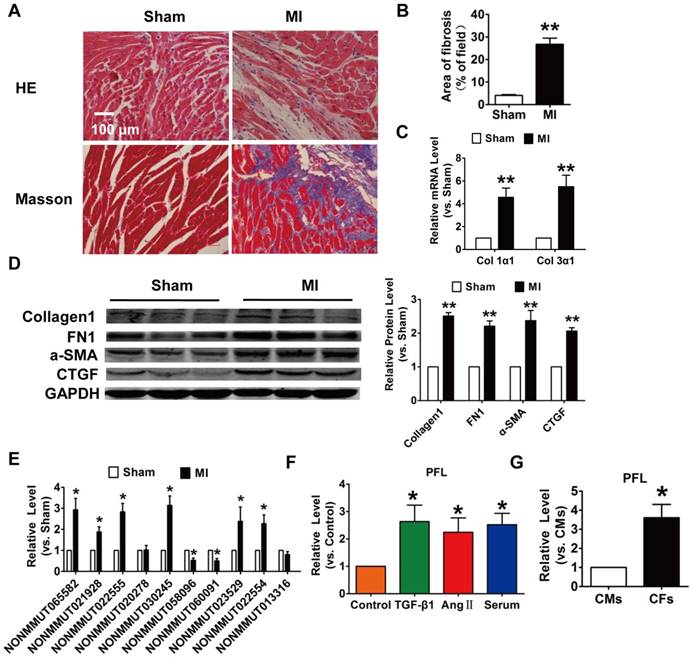
To explore the role of lncRNAs in cardiac fibrosis, we first developed an MI-induced cardiac fibrosis mouse model. 28 days after surgery, Masson's staining revealed marked collagen deposition in MI mice compared with sham mice (Fig. 1A-B). Meanwhile, the expression of collagen 1α1, collagen 3α1 and fibrosis-related genes, such as alpha smooth muscle actin (α-SMA), connective tissue growth factor (CTGF) and fibronectin 1 (FN1), were significantly increased in the MI mice (Fig. 1C-D). We then performed qRT-PCR to detect the expression of lncRNAs that were found to be differentially expressed in MI mice in our previous work. Eight lncRNAs were significantly dysregulated in the mice after MI (Fig. 1E). Among the eight lncRNAs, NONMMUT 022555 (Fig. S1) was markedly increased in the MI mice and was positively correlated with fibrosis-related proteins, such as Col1a2, Col4a1 and Fn1, which are known to be involved in the process of fibrosis[19]. Meanwhile, we performed in vitro translation followed by Coomassie stained PAA Gel, and found that NONMMUT 022555 is really a non-coding RNA without protein-coding function (Fig. S2). Thus, we focused on examining NONMMUT 022555 in our subsequent experiments and herein refer to this lncRNA as pro-fibrotic lncRNA (PFL) for convenience. Consistent with the results from the mouse model, PFL expression was also increased in cardiac fibroblasts (CFs) that were treated with transforming growth factor (TGF)-β1 (10 ng/mL), angiotensin II (Ang II) (100 nM) or 20% serum for 24 h (Fig. 1F). Intriguingly, qRT-PCR showed that PFL was enriched in CFs compared to cardiomyocytes (CMs) (Fig. 1G), indicating that PFL play a more important role in the pathological process of CFs than CMs after MI.
Increased expression of lncRNA NONMMUT022555 (PFL) in the hearts of mice subjected to myocardial infarction (MI) and in fibrotic cardiac fibroblasts (CFs). (A) Representative images of HE and Masson's trichrome staining of the LV sections of mouse hearts. (B) Quantification of the total fibrotic area using Image-Pro Plus. (C) mRNA expression of collagen 1α1 and collagen 3α1 was measured by qRT-PCR; GAPDH mRNA served as an internal control. n=4-6 mice per group. (D) Protein levels of fibrosis-related factors in the infarcted hearts were assessed by western blot analysis. (E) qRT-PCR analysis showing dysregulated lncRNAs in the hearts of mice after MI. (F) PFL was increased in CFs after treatment with TGF-β1 (10 ng/mL), Ang II (100 nM) or 20% serum for 24 h. (G) qRT-PCR detection of PFL expression in CMs and CFs. n=5-6 independent cell cultures. *p<0.05, **p<0.01 vs. sham, control or cardiomyocytes (CMs).
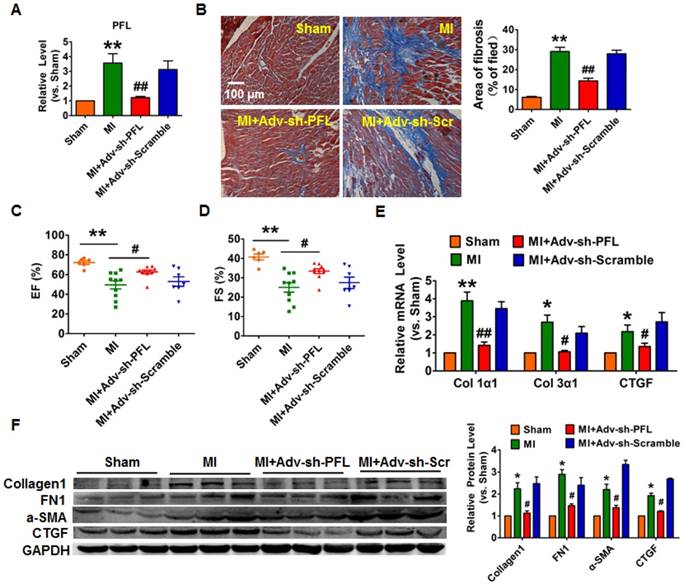
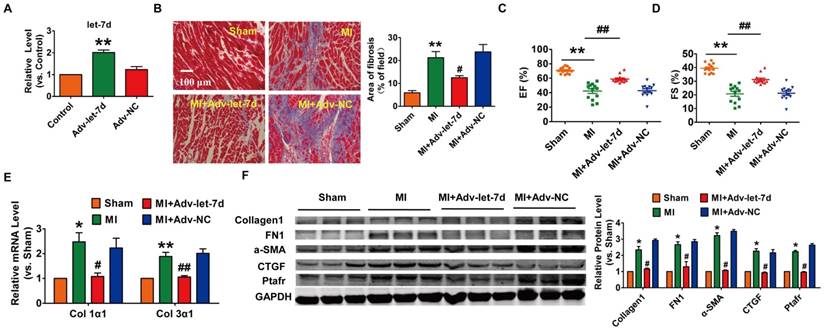
To evaluate the potential therapeutic action of PFL inhibition on cardiac fibrosis after MI in mice, we knocked down PFL in mice via a tail vein injection of adenoviruses carrying a PFL-specific short-hairpin RNA (Adv-sh-PFL) (Fig. S3A). Silencing PFL in normal mice had no effect on collagen production and heart function (Fig. S3B-D). However, knockdown of PFL in MI mice significantly abated the up-regulation of PFL (Fig. 2A), markedly reduced collagen deposition (Fig. 2B) and improved heart function in MI mice (Fig. 2C-D). Moreover, PFL silencing attenuated the expression of several fibrosis-related genes at both mRNA and protein levels and dampened cardiac fibrosis in MI mice (Fig. 2E-F).
Silencing of PFL mitigated cardiac remodeling after MI. (A) qRT-PCR analysis showing that Adv-sh-PFL reversed the up-regulation of PFL in MI mice; GAPDH mRNA served as an internal control, and Adv-sh-scramble served as an additional control. (B) Representative images of Masson's trichrome staining show that knockdown of PFL suppressed collagen deposition in MI mice; quantification of the total fibrotic area using Image-Pro Plus. Four weeks after MI, echocardiographic imaging showed that the silencing of PFL improved ejection fraction (EF) (C) and fraction shortening (FS) (D). (E-F) Western blot and qRT-PCR analysis revealed that knockdown of PFL alleviated cardiac fibrosis and fibrotic protein expression in MI mice; GAPDH served as a loading control, and Adv-sh-scramble served as a negative control. n=5-10 mice per group. *p<0.05 and **p<0.01 vs. sham mice; #p<0.05 and ##p<0.01 vs. MI mice.
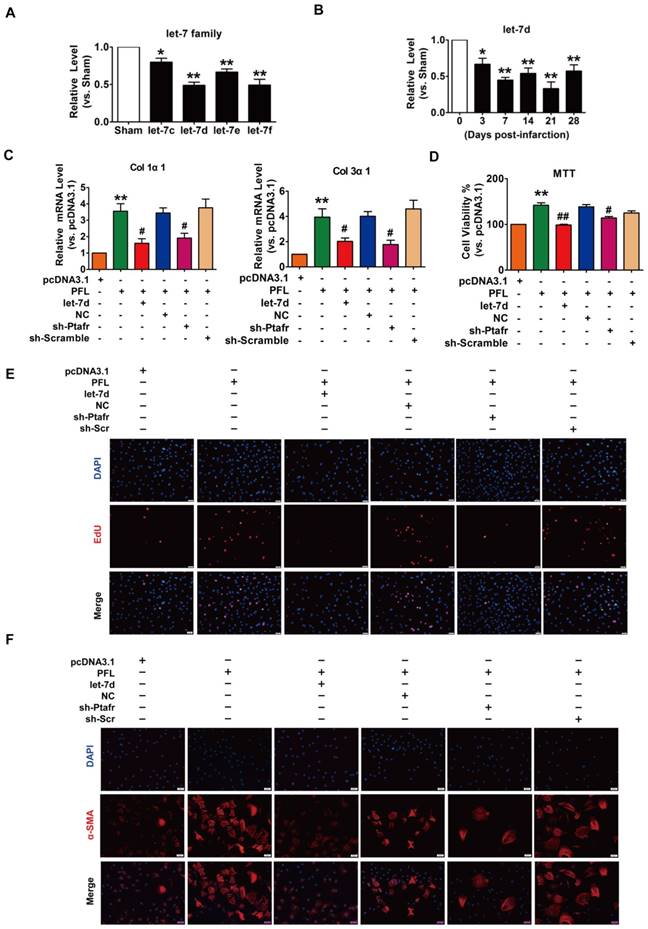
PFL acts as a ceRNA for let-7d to promote cardiac fibrosis
Accumulating evidence has shown that lncRNAs participate in the genesis and progression of cardiovascular diseases, such as myocardial ischemia-reperfusion injury and cardiac hypertrophy, by competitively binding to and decoying miRNAs[14, 22]. To explore whether miRNAs play a role in mediating the function of PFL in our models, we first performed a bioinformatics analysis using the RegRNA 2.0 database (http://regrna.mbc.nctu.edu.tw/html/prediction.html)[23] to identify miRNAs that contain potential binding sequences for PFL and are differentially expressed in MI mice (Table S3). Using this method, we determined that let-7 miRNA family expression levels were decreased in mice after MI and that the let-7 miRNA contained binding sites for PFL (Fig. 3A). Among the let-7 family, let-7d contains two binding sites for PFL and was down- regulated from day 3 to day 28 after coronary artery ligation (Fig. 3B). We then examined whether let-7d mediated the regulatory effect of PFL on cardiac fibrosis. qRT-PCR showed that overexpression of PFL by the pcDNA3.1 plasmid carrying the PFL gene (Fig. S4A) up-regulated the expression of collagen 1α1, collagen 3α1 and CTGF in CFs, which was attenuated by let-7d (Fig. 3C, Fig. S4B). To further explore the mechanism of PFL in fibrosis, we detected the effect of PFL on cell proliferation and fibroblast-myofibroblast transition in CFs. As shown in Fig. 3D-E, forced expression of PFL increased cell viability and promoted CFs proliferation, which were abrogated by let-7d. Moreover, enhanced expression of PFL promoted fibroblast-myofibroblast transition in CFs by regulating let-7d (Fig. 3F).
PFL acted as a ceRNA for let-7d to promote cardiac fibrosis. (A) Decreased expression of the let-7 family in the infarcted heart. (B) Decreased expression of let-7d in the peri-infarct area at varying time points, as evaluated by qRT-PCR; GAPDH served as an internal control. (C) qRT-PCR analysis revealed that forced expression of PFL (1 μg/mL) in CFs increased mRNA expression levels of collagen 1α1 and collagen 3α1, which were reversed by let-7d overexpression; GAPDH mRNA served as an internal control. MTT assay (D) and EdU staining (E) for the assessment of cell viability and proliferation in CFs overexpressing PFL in the presence or absence of let-7d mimics. (F) Immunostaining of α-SMA in CFs demonstrated that enhanced expression of PFL promoted the fibroblast-myofibroblast transition. *p<0.05 and **p<0.01 vs. Sham mice or pcDNA3.1; #p<0.05 and ##p<0.01 vs. PFL.
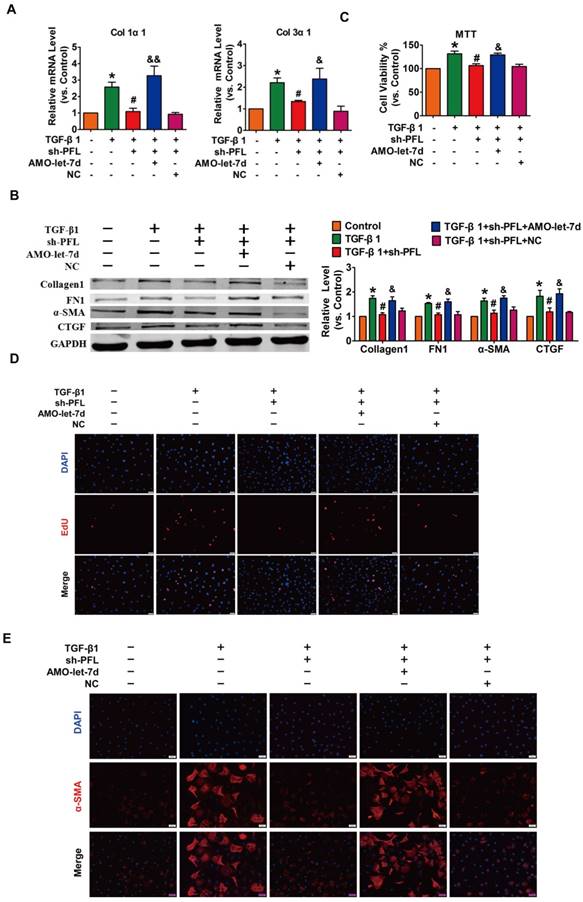
Next, TGF-β1 was used to promote fibrogenesis in CFs to further evaluate the anti-fibrotic effect of PFL. As illustrated in Fig. S4C, while all three shRNAs could repress PFL expression in the CFs compared with the scrambled shRNA, shRNA #2 showed the most potent inhibitory effect. Therefore, shRNA #2 was used for all subsequent experiments and was designated as sh-PFL. As illustrated in Fig. 4A-E and Fig. S4D, TGF-β1 increased cell viability and collagen production as well as promoted cell proliferation and fibroblast-myofibroblast transition in CFs, whereas silencing of PFL by shRNA alleviated the TGF-β1-induced fibrogenesis in CFs. More importantly, the effects of PFL inhibition on fibrogenesis were abated by silencing let-7d (Fig. 4A-E).
Silencing PFL alleviated TGF-β1-induced fibrogenesis in CFs. (A) Suppression of PFL attenuated the increase in collagen 1α1 and collagen 3α1 expression induced by TGF-β1, as measured by qRT-PCR; GAPDH mRNA served as an internal control. (B) Western blot analysis showing that knockdown of PFL alleviated TGF-β1-induced fibrotic protein expression (Collagen I, FN1, CTGF and α-SMA); GAPDH served as a loading control. (C-D) MTT assay and EdU staining for the assessment of proliferation, showing that the silencing of PFL inhibited TGF-β1-induced cell viability and proliferation. (E) Representative images of immunofluorescence staining showing that knockdown of PFL abated the TGF-β1-induced fibroblast-myofibroblast transition. n=4-6 independent cell cultures. *p<0.05 and **p<0.01 vs. control; #p<0.05 and ##p<0.01 vs. TGF-β1; and &p<0.05 and &&p<0.01 vs. TGF-β1+sh-PFL
Next, we used gain- and loss-of-function experiments to reveal the relationship between PFL and let-7d. Overexpression of PFL could significantly eliminate let-7d in CFs (Fig. 5A), whereas knockdown of PFL by shRNA produced the opposite effect (Fig. 5B). Coincidently, we found that knockdown of PFL in normal mice also resulted in the up-regulation of let-7d (Fig. 5C). To acquire more direct evidence of the interaction between PFL and let-7d, we constructed a let-7d sensor luciferase vector containing a perfect let-7d target site which was incorporated into the 3'UTR of the luciferase gene, a strategy that has been widely used in similar studies[14, 22]. As shown in Fig. 5D, the luciferase activity of the let-7d sensor was increased in CFs transfected with PFL, indicating that PFL bound let-7d to limit its inhibitory effect on luciferase activity. In contrast, the silencing of PFL by shRNA inhibited the luciferase activity of the let-7d sensor (Fig. 5D). Moreover, overexpression of let-7d inhibited the luciferase activity of its sensor, whereas co-transfection of PFL alleviated the inhibitory effect of let-7d on its sensor (Fig. 5E). Additional evidence was generated in experiments using a luciferase vector carrying a PFL fragment containing the binding sites or mutated binding sites for let-7d (Fig. 5F). As illustrated in Fig. 5G, let-7d suppressed the luciferase activity from the wild-type PFL or mutated PFL (mutation in either of the two let-7d binding sites) vector in HEK293 cells. However, the mutated let-7d failed to do so. Moreover, the inhibitory effect of let-7d was lost when the double-mutated PFL was used (mutation of both binding sites for let-7d).
Interaction between PFL and let-7d. (A) Forced expression of PFL with a PFL expression plasmid inhibited the expression of let-7d in CFs. **p<0.01 vs. pcDNA3.1. (B) Knockdown of PFL by its shRNA increased the expression of let-7d in CFs. **p<0.01 vs. sh-Scramble. (C) Knockdown of PFL increased the expression of let-7d in normal mice. *p<0.05 vs. control. GAPDH served as an internal control; pcDNA3.1 and sh-Scramble empty vector served as additional controls. (D-E) PFL binds to let-7d and regulates its activity. CFs were co-transfected with the let-7d sensor and PFL or sh-PFL and its corresponding scrambled form, and luciferase activity was detected. *p<0.05 and **p<0.01 vs. the let-7d sensor; ##p<0.01 vs. the let-7d sensor+let-7d. (F) The predicted binding sites of PFL and let-7d. Mut-1, mutated binding site 1; Mut-2, mutated binding site 2; and Mut-3, mutated binding sites 1 and 2. (G) Luciferase reporter activities of chimeric vectors carrying the luciferase gene and a fragment of PFL RNA containing wild-type or mutated let-7d binding sites. (H) The wild-type and mutated sequences of let-7d were shown. (I) PFL was associated with let-7d. CFs were transfected with biotinylated wild-type let-7d (Bio-let-7d-WT) or biotinylated mutant let-7d (Bio-let-7d-Mut). A biotinylated miRNA that is not complementary to PFL was used as a negative control (Bio-NC). Forty-eight hours after transfection, the cells were harvested for a biotin-based pull-down assay. PFL expression levels were analyzed by qRT-PCR. **p<0.01 vs. Bio-NC. n=6 independent cell cultures.
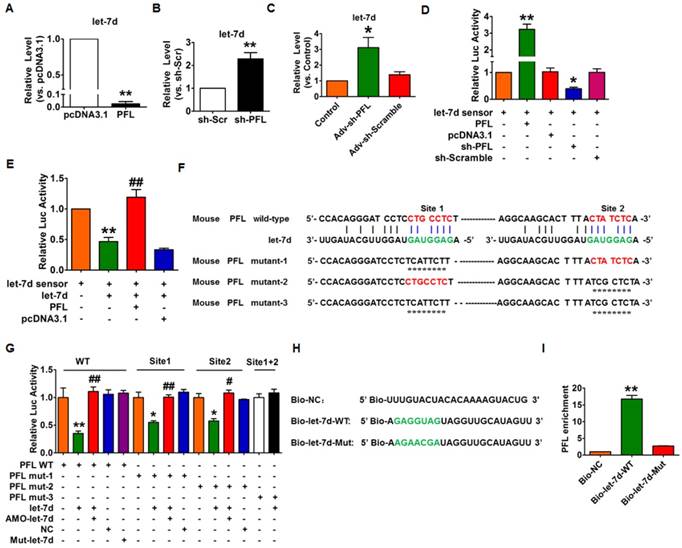
Next, we applied a biotin-avidin pull-down system to examine the direct binding between let-7d and PFL. CFs were transfected with biotinylated wild-type let-7d (Bio-let-7d-WT) or mutated let-7d (Bio-let-7d-Mut) (Fig. 5H), and then biotin-based pull-down and qRT-PCR assay were performed to test whether let-7d could pull down PFL. As shown in Fig. 5I, the introduction of let-7d caused the enrichment of PFL. However, the mutated let-7d, in which the base pairing between PFL and let-7d was disrupted, was unable to bind PFL, indicating that PFL can directly bind to let-7d in a sequence-specific manner.
The cardio-protective role of let-7d in cardiac fibrosis during MI
It has been reported that let-7d is involved in the process of lung fibrosis[24]; however, the role of let-7 in cardiac fibrosis is still under debate[12, 13]. Then, we injected adenoviruses carrying the let-7d precursor into mice and estimated the potential cardio-protective effect of let-7d on cardiac fibrosis during MI (Fig. 6A). As shown in Fig. 6B-D, forced expression of let-7d mitigated the area of interstitial fibrosis and improved heart function in MI mice. Meanwhile, overexpression of let-7d attenuated collagen deposition in MI mice, which was also accompanied by the inhibited expression of various fibrosis relevant proteins (collagen1, α-SMA, FN1 and CTGF) as well (Fig. 6E-F).
Forced expression of let-7d ameliorated cardiac fibrosis in the mice after MI. (A) Tail vein injection of Adv-let-7d increased let-7d expression in normal mice, as measured by qRT-PCR; GAPDH served as an internal control, and Adv-NC served as a negative control. (B) Representative images of Masson-stained hearts of MI mice after injection of Adv-let-7d; quantification of the total fibrotic area using Image-Pro Plus. Forced expression of let-7d restored the impaired cardiac function in infarcted hearts (C-D), ameliorated collagen production (E) and reversed the induction of fibrosis-related protein expression (F). n=13 mice per group for the heart function evaluation; and n=6 mice for the qRT-PCR and western blot assays. *p<0.05 and **p<0.01 vs. control or sham mice; #p<0.05 and ##p<0.01 vs. MI mice.
To reveal the action of let-7d in cardiac fibrosis, we first detected the changes in let-7d expression within fibrogenic CFs. As illustrated in Fig. 7A, let-7d was reduced in CFs exposed to TGF-β1, Ang II or serum. More importantly, qRT-PCR revealed that the expression of let-7d was significantly higher in CFs than that in CMs (Fig. 7B), indicating that let-7d may play important roles in the regulation of CFs. Notably, transfection of AMO-let-7d into CFs to inhibit the expression of endogenous let-7d resulted in the up-regulation of collagen 1α1, collagen 3α1 and CTGF at the mRNA level (Fig. 7C) as well as increased the protein expression levels of collagen1, α-SMA, FN1, and CTGF (Fig. 7D). Furthermore, knockdown of let-7d promoted fibroblast-myofibroblast transition in cultured CFs (Fig. 7E).
Overexpression of let-7d abrogated fibrogenesis in CFs. (A) Decreased expression of let-7d in CFs pretreated with TGF-β1, Ang II or serum. *p<0.05 and **p<0.01 vs. control; (B) Expression of let-7d in CMs and CFs. *p<0.05 vs. CMs. Inhibition of let-7d increased collagen 1α1 and collagen 3α1 mRNA expression (C) and promoted fibrogenesis (D) in CFs. *p<0.05 and **p<0.01 vs. NC. (E) Representative immunofluorescence images showing that suppression of let-7d promoted the transition of fibroblasts into myofibroblasts. Overexpression of let-7d mitigated TGF-β1-induced cell proliferation (F), collagen production (G), fibrogenesis (H) and the fibroblast-myofibroblast transition (I). *p<0.05 and **p<0.01 vs. control; #p<0.05 and ##p<0.01 vs. TGF-β1. Forced expression of let-7d alleviated 20% serum-driven proliferation (J) and fibrogenesis (K) in CFs. *p<0.05 and **p<0.01 vs. control; #p<0.05 and ##p<0.01 vs. serum. n=5-6 independent cell cultures.
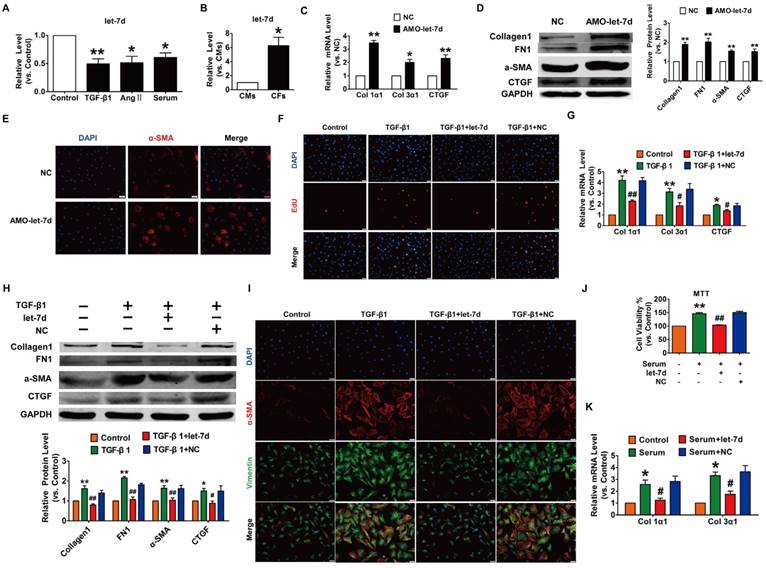
We further determined the anti-fibrotic effect of let-7d under various stimuli. As shown in Fig. 7F-H, forced expression of let-7d markedly attenuated TGF-β1-induced cell proliferation, collagen production and collagen 1α1, collagen 3α1 and CTGF expression as well as some other up-regulated fibrosis-related genes in cultured CFs. Meanwhile, transfection of let-7d suppressed the TGF-β1-induced transition of fibroblasts into myofibroblasts driven by TGF-β1 (Fig. 7I). In agreement with these results, we also found that let-7d mitigated proliferation, collagen production and fibrogenesis in CFs cultured in 20% serum (Fig. 7J-K).
Ptafr mediates the anti-fibrotic effects of let-7d
Next, we used the TargetScan database to search for candidate targets that have a potential let-7d binding site and mediate the anti-fibrotic action of let-7d. We found two conserved binding sites for let-7d in the 3'UTR of Ptafr that have been reported to be involved in the process of fibrosis. Our subsequent experiments validated the regulatory relationship between let-7d and Ptafr. As depicted in Fig. 8A, we constructed a luciferase expression vector carrying the targeting fragment containing the let-7d binding sites or mutated sequences (nucleotide replacement). The luciferase assay showed that let-7d inhibited the activities of the wild-type Ptafr luciferase vector or the vector carrying mutations in either of the binding sites (Fig. 8B). However, let-7d failed to diminish the luciferase activity of the double-mutated construct. These results indicated that both sites are necessary for let-7d to regulate Ptafr expression. More importantly, forced expression of let-7d inhibited the expression of Ptafr at both mRNA and protein levels, whereas knockdown of let-7d displayed the opposite effect (Fig. 8C-E).
Ptafr is a direct target of let-7d and mediates the anti-fibrotic function of let-7d. (A) Sequence alignment showing let-7d:PTAFR complementarity in the mouse, rat and human genes. The matched base pairs in the seed region are outlined by red and green. Mut-1, mutated binding site 1; Mut-2, mutated binding site 2; and Mut-3, mutated binding sites 1 and 2. (B) Luciferase reporter activities of chimeric vectors carrying the luciferase gene and a fragment of the Ptafr 3′-UTR containing the wild-type or mutant let-7d binding sites. (C-E) Enhanced expression of let-7d repressed, whereas inhibition of let-7d increased, the expression of Ptafr at the protein and mRNA levels. Overexpression of let-7d mitigated PAF-induced collagen production (F) and fibrogenesis (G) and blocked fibroblast-myofibroblast transition (H). (I) Proposed model for the mechanism of PFL and let-7d in cardiac fibrosis. Cardiac stress inhibits the expression of let-7d by increasing the expression of PFL, which results in the derepression of Ptafr and promotes cell proliferation and fibroblast-myofibroblast transition, leading to remodeling and cardiac dysfunction. n=5-6 independent cell cultures. *p<0.05 and **p<0.01 vs. control or NC; #p<0.05 vs. PAF.
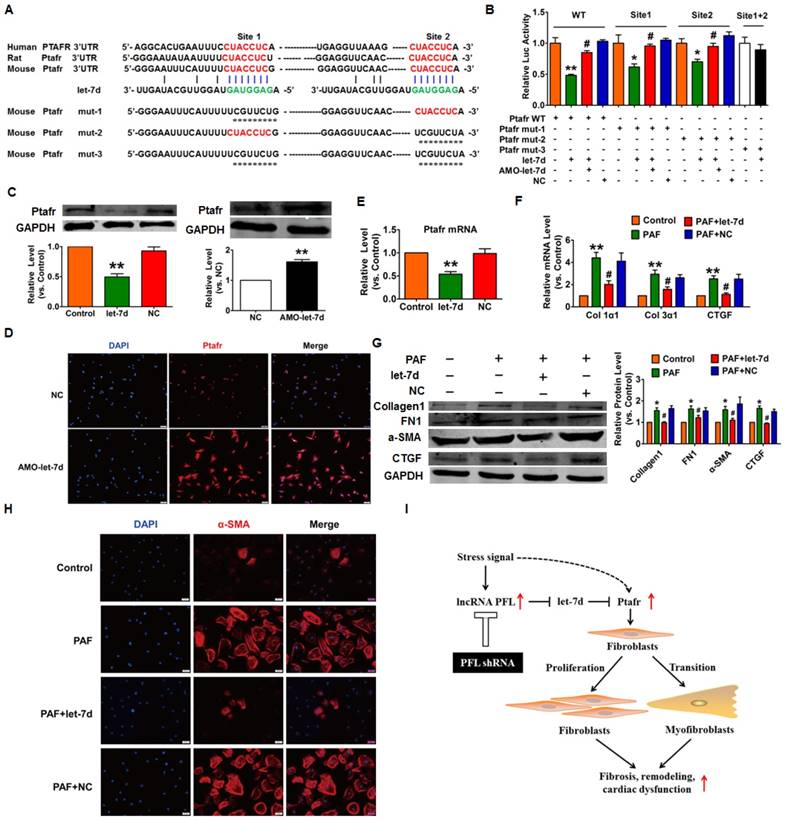
To examine whether Ptafr mediates the anti-fibrotic effect of let-7d, CFs were transfected with let-7d and then treated with PAF. Moreover, overexpression of let-7d inhibited PAF-driven collagen deposition (Fig. 8F), fibrogenesis (Fig. 8G) and myofibroblast generation in cultured CFs (Fig. 8H). Furthermore, similar to the effect of let-7d overexpression, we found that knockdown of Ptafr mitigated the effect of PFL on cell viability, collagen production and myofibroblast formation (Fig. 3C-F). These results indicated that Ptafr mediates the actions of PFL and let-7d during cardiac fibrogenesis.
Discussion
In the present study, we characterized the pro-fibrotic property of PFL in an MI model and elucidated the molecular events underlying the fine regulation of PFL in cardiac fibrosis. We found that PFL was remarkably up-regulated in MI mice and that this up-regulation promoted cardiac fibrogenesis in CFs. The pro-fibrotic action of PFL was mediated by the down-regulation and functional inactivation of let-7d. In contrast, knockdown of PFL alleviated cardiac fibrosis and improved heart function in MI mice. Furthermore, let-7d was identified as an anti-fibrotic miRNA, but it is down-regulated in MI, and this down-regulation relieves the repression of Ptafr and leads to its up-regulation. Decreased expression of let-7d is partially attributable to the miRNA-sponge action of the PFL. These new findings allowed us to conclude that PFL is a novel pro-fibrotic lncRNA that absorbs the anti-fibrotic miRNA let-7d, which in turn relieves the repression of the pro-fibrotic protein Ptafr to exaggerate cardiac remodeling and cardiac dysfunction (Fig. 8I).
As the first powerful bioactive lipid, PAF has been previously implicated in the initiation and progression of fibrotic diseases through activation of a G protein-coupled receptor, PTAFR[25-27]. Correa-Costa et al. found that knockout of Ptafr prevented renal fibrosis in mice[26]. Some studies have reported the up-regulation of PAF in HF[28, 29], indicating the potential role of PAF/PTAFR in cardiac remodeling. However, the upstream regulator of PTAFR in cardiac fibrosis is not completely known. In this study, we found that Ptafr is one of the direct targets for let-7d and mediates the anti-fibrotic effect of let-7d. The formation of fibroblastic foci and excessive deposition of ECM are the main pathologic features of myocardial fibrosis. Myofibroblasts, which have features of both fibroblasts and smooth muscle cells, are regarded as the major effector cells for the extensive synthesis and secretion of ECM. Our study revealed that let-7d inhibits fibroblast proliferation and fibroblast-myofibroblast transition by targeting Ptafr, which ultimately inhibits collagen production and cardiac fibrosis.
A variety of studies, including ours, have provided strong evidence that miRNAs play essential roles in cardiac fibrosis[4, 6, 30]. Increasing evidences showed that dysregulation of the let-7 family is involved in cardiac fibrosis[12, 13]. However, there is controversy about the actual role of the let-7 family in cardiac fibrosis. Tolonen et al. found that let-7c was decreased in the hearts of mice following MI[13]. Further study showed that inhibition of let-7c reduced cardiac fibrosis and prevented the deterioration of systolic function after MI. In contrast, Wang et al. observed the down-regulation of let-7i in CFs and in the hearts of mice exposed to Ang II; they also found that inhibition of let-7i exacerbated Ang II-induced cardiac fibrosis, whereas overexpression of let-7i reduced Ang II-induced cardiac fibrosis in mice[12]. In this study, we were the first to reveal that let-7d is down-regulated in mice after MI. Furthermore, we found that inhibition of let-7d caused cell proliferation, fibroblast-myofibroblast transition and then induced collagen deposition in CFs. In contrast, enhanced expression of let-7d prevented TGF-β1-induced or PAF-driven fibrogenesis by inhibiting Ptafr in CFs. More importantly, we found that forced expression of let-7d attenuates cardiac fibrosis and improves cardiac function after MI. Our study elucidated the anti-fibrotic effect and mechanisms of let-7d in cardiac fibrosis, and indicated that the exogenous application of let-7d may be a novel therapeutic strategy for the treatment of myocardial fibrosis.
A recent study from Dhahri et al. observed the reduced expression of let-7f in HUVECs treated with cigarette smoke extracts (CSE) and in the ischemic muscles of mice exposing to cigarette smoke, and they found that forced expression of let-7f promoted angiogenesis in HUVECs exposed to CSE by targeting TGF-βRI[31]. In this study, we also found the down-regulation of let-7f in the MI mice, which contains two potential binding sites for PFL. Thus, let-7f might play a protective role in the process of cardiac remodeling after MI through promoting angiogenesis and inhibiting fibrosis by regulating TGF-βRI, which warrants our further detailed study.
LncRNA has been shown to be involved in a wide range of diseases, such as cancer, cardiovascular diseases and nervous system diseases[5, 32, 33]. However, the role and mechanism of lncRNAs in the process of myocardial fibrosis are less known. A recent study from Viereck et al. found that the lncRNA Chast (cardiac hypertrophy-associated transcript) promoted cardiac hypertrophy and cardiac fibrosis in vitro and in vivo[5]. In this study, we found that PFL, one of the lncRNAs that was found to be differentially expressed in MI mice in our previous work[19], was significantly up-regulated in the hearts of mice after MI and in the CFs treated with TGF-β1, Ang II or serum. Moreover, our data showed that forced expression of PFL induced, whereas knockdown of PFL attenuated, fibrogenesis in CFs. More importantly, silencing PFL alleviated cardiac fibrosis and improved heart function in mice subjected to MI.
CeRNAs, which are RNA transcripts that have the same specific miRNAs binding sites, are considered to be natural decoys in miRNAs activity by competing for common miRNAs[34]. Recent studies have shown lncRNAs can function as ceRNA with the target in the process of various diseases, including organ fibrosis [4, 35-37] . For example, lncRNA H19 acts as a ceRNA to mediate CTGF expression by sponging miR-455 in cardiac fibrosis[38]. Yu et al. found that the lncRNA Growth Arrest-specific Transcript 5 (GAS5) inhibited liver fibrosis by competitively binding to miR-222. In addition, lncRNA could act as natural miRNA sponges to adsorb and to inhibit the function of miRNAs[39]. In this study, we found that both PFL and Ptafr contain the binding site of let-7d and revealed that PFL can directly bind to let-7d in a sequence-specific manner. So, we speculate that the up-regulated PFL during cardiac fibrosis competitively binds to let-7d and releases the inhibitory effects of let-7d on Ptafr, which results in the up-regulation of Ptafr and promotes fibrogenesis.
Moreover, we found other differentially expressed lncRNAs in MI mice, such as NONMMUT021928 and NONMMUT065582, which contain let-7d binding sites. In addition, those deregulated lncRNAs have binding sites of other miRNAs involved in fibrosis. As we know, one miRNA may regulate multiple targets and one target could be co-regulated by multiple miRNAs. Additionally, one lncRNA may regulate multiple miRNAs and many lncRNAs form a complex lncRNA-miRNA-target regulatory network to participate in the progression of cardiac fibrosis. In the future, the lncRNA-miRNA-target regulatory network should be systematically investigated to reveal the mechanism of cardiac fibrosis.
In addition to acting as ceRNA, lncRNAs has been reported to be involved in cardiovascular diseases through interacting with DNA or proteins, modulating their epigenetics by guiding chromatin-modifying complexes to the target genomic DNA loci[40]. Han et al. found that Mhrt, a cardiac-specific lncRNA, inhibits cardiac hypertrophy and failure by sequestering Brg1 and then preventing chromatin remodeling[15]. Wang et al. found that lncRNA Chaer is necessary for the development of cardiac hypertrophy through interacting with PRC2 and then inhibiting histone H3 lysine 27 methylation at the promoter regions of genes involved during cardiac hypertrophy[41]. Thus, we hypothesize that PFL may regulate methylation and histone modification or promote the interaction between DNA and proteins, and this results in cardiac fibrosis. In our future work, we will investigate the effect of PFL on epigenetics and explore the roles of dysregulation of PFL during cardiac remodeling.
Although tens of thousands of lncRNAs have been found in humans, lots of lncRNAs do not show the high interspecies conservation typical of protein-coding genes[42]. Similar to many other lncRNAs, we failed to find a homologous sequence of PFL in the human genome. However, in addition to sequence conservation, many studies have suggested other aspects of lncRNA conservation, such as their structure, function, and expression syntenic loci[43-45]. More in-depth study should be performed to seek the human homologue for PFL from different conservation dimensions to reveal its implications for clinical therapeutics.
In short, the present work revealed PFL as a critical pro-fibrotic lncRNA that promotes cardiac fibrosis by competitively binding let-7d. In a series of in vitro and in vivo experiments, we revealed the roles of PFL and let-7d in cardiac fibrosis and illuminated the regulatory relationship between them. These findings indicate that interfering with PFL expression may be considered as a novel strategy for the prevention and treatment of cardiac fibrosis and associated pathological processes.
Abbreviations
Ptafr: platelet-activating factor receptor; MI: myocardial infarction; CFs: cardiac fibroblasts; CMs: cardiomyocytes; lncRNA: long non-coding RNA; PFL: pro-fibrotic lncRNA; miRNAs: microRNAs; ECM: extracellular matrix; HF: heart failure; shRNA: short hairpin RNA; α-SMA: alpha smooth muscle actin; CTGF: connective tissue growth factor; FN1: fibronectin 1; Ang II: angiotensin II.
Supplementary Material
Figure S1. The sequence of lncRNA NONMMUT022555 (PFL). Figure S2. In vitro translation assay confirmed the PFL is really a non-coding RNA without protein-coding function. Figure S3. Tail vein injection of Adv-sh-PFL in normal mice have no effect on collagen deposition and heart function. Figure S4. Knockdown of PFL attenuated fibrogenesis in CFs. Table S1. Sequences of qRT-PCR primers. Table S2. Sequences of shRNA. Table S3. Predicted miRNAs that contain potential binding sequences for PFL.
Acknowledgements
This study was supported by the Major Program of National Natural Science Foundation of China (81530010); the National Key R&D Program of China (2017YFC1307403 to Baofeng Yang); the National Natural Science Foundation of China (81770284, 31671187, 81670207); and the Outstanding Youth Science Fund Project of Heilongjiang Province (JC201411).
Author Contributions
H.-L.S., Y.-J.L. and B.-F.Y. designed the research; H.-H.L., Z.-W.P., X.-G.Z., L.L., J.-S., X.-M.S., D.-D.Z., and X.-L.L. performed cellular experiments; H.-H.L., C.-Q.X., Y.-H.Z., B.-Z.X. and J.S. conducted animal experiments; H.-H.L. and H.-L.S. wrote the manuscript.
Competing Interests
The authors declared no conflicts of interest.
References
1. Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov. 2016;15:620-638
2. Zhong C, Wang K, Liu Y, Lv D, Zheng B, Zhou Q. et al. miR-19b controls cardiac fibroblast proliferation and migration. J Cell Mol Med. 2016;20:1191-1197
3. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC Cardiac Fibrosis. The Fibroblast Awakens. Circ Res. 2016;118:1021-1040
4. Piccoli MT, Bar C, Thum T. Non-coding RNAs as modulators of the cardiac fibroblast phenotype. J Mol Cell Cardiol. 2016;92:75-81
5. Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M. et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. 2016;8:326ra322
6. Liang H, Zhang C, Ban T, Liu Y, Mei L, Piao X. et al. A novel reciprocal loop between microRNA-21 and TGFbetaRIII is involved in cardiac fibrosis. Int J Biochem Cell Biol. 2012;44:2152-2160
7. Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J. et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-beta1 pathway. Circulation. 2012;126:840-850
8. Shan H, Zhang Y, Lu Y, Pan Z, Cai B, Wang N. et al. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res. 2009;83:465-472
9. Li X, Wang B, Cui H, Du Y, Song Y, Yang L. et al. let-7e replacement yields potent anti-arrhythmic efficacy via targeting beta 1-adrenergic receptor in rat heart. J Cell Mol Med. 2014;18:1334-1343
10. Seeger T, Xu QF, Muhly-Reinholz M, Fischer A, Kremp EM, Zeiher AM. et al. Inhibition of let-7 augments the recruitment of epicardial cells and improves cardiac function after myocardial infarction. J Mol Cell Cardiol. 2016;94:145-152
11. Gray C, Li M, Patel R, Reynolds CM, Vickers MH. Let-7 miRNA profiles are associated with the reversal of left ventricular hypertrophy and hypertension in adult male offspring from mothers undernourished during pregnancy after preweaning growth hormone treatment. Endocrinology. 2014;155:4808-4817
12. Wang X, Wang HX, Li YL, Zhang CC, Zhou CY, Wang L. et al. MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension. 2015;66:776-785
13. Tolonen AM, Magga J, Szabo Z, Viitala P, Gao E, Moilanen AM. et al. Inhibition of Let-7 microRNA attenuates myocardial remodeling and improves cardiac function postinfarction in mice. Pharmacol Res Perspect. 2014;2:e00056
14. Wang JX, Zhang XJ, Li Q, Wang K, Wang Y, Jiao JQ. et al. MicroRNA-103/107 Regulate Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury Through Targeting FADD. Circ Res. 2015;117:352-363
15. Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST. et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102-106
16. Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting CC, Alexanian M. et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med. 2017:9
17. Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL. et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ Res. 2017;121:575-583
18. Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S. et al. MIAT Is a Pro-fibrotic Long Non-coding RNA Governing Cardiac Fibrosis in Post-infarct Myocardium. Sci Rep. 2017;7:42657
19. Qu X, Song X, Yuan W, Shu Y, Wang Y, Zhao X. et al. Expression signature of lncRNAs and their potential roles in cardiac fibrosis of post-infarct mice. Biosci Rep. 2016:36
20. Liang H, Xu C, Pan Z, Zhang Y, Xu Z, Chen Y. et al. The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol Ther. 2014;22:1122-1133
21. Liang H, Gu Y, Li T, Zhang Y, Huangfu L, Hu M. et al. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis. 2014;5:e1238
22. Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y. et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377-1388
23. Chang TH, Huang HY, Hsu JB, Weng SL, Horng JT, Huang HD. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinformatics. 2013;14(Suppl 2):S4
24. Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF. et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220-229
25. Latchoumycandane C, Hanouneh M, Nagy LE, McIntyre TM. Inflammatory PAF Receptor Signaling Initiates Hedgehog Signaling and Kidney Fibrogenesis During Ethanol Consumption. PLoS One. 2015;10:e0145691
26. Correa-Costa M, Andrade-Oliveira V, Braga TT, Castoldi A, Aguiar CF, Origassa CS. et al. Activation of platelet-activating factor receptor exacerbates renal inflammation and promotes fibrosis. Lab Invest. 2014;94:455-466
27. Zhang H, Yang Y, Takeda A, Yoshimura T, Oshima Y, Sonoda KH. et al. A Novel Platelet-Activating Factor Receptor Antagonist Inhibits Choroidal Neovascularization and Subretinal Fibrosis. PLoS One. 2013;8:e68173
28. Detopoulou P, Nomikos T, Fragopoulou E, Chrysohoou C, Antonopoulou S Platelet activating factor in heart failure. potential role in disease progression and novel target for therapy. Curr Heart Fail Rep. 2013;10:122-129
29. Detopoulou P, Nomikos T, Fragopoulou E, Antonopoulou S, Kotroyiannis I, Vassiliadou C. et al. Platelet activating factor (PAF) and activity of its biosynthetic and catabolic enzymes in blood and leukocytes of male patients with newly diagnosed heart failure. Clin Biochem. 2009;42:44-49
30. Creemers EE, van Rooij E. Function and Therapeutic Potential of Noncoding RNAs in Cardiac Fibrosis. Circ Res. 2016;118:108-118
31. Dhahri W, Dussault S, Haddad P, Turgeon J, Tremblay S, Rolland K. et al. Reduced expression of let-7f activates TGF-beta/ALK5 pathway and leads to impaired ischaemia-induced neovascularization after cigarette smoke exposure. J Cell Mol Med. 2017;21:2211-2222
32. Leucci E, Vendramin R, Spinazzi M, Laurette P, Fiers M, Wouters J. et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016;531:518-522
33. Hossein-Nezhad A, Fatemi RP, Ahmad R, Peskind ER, Zabetian CP, Hu SC. et al. Transcriptomic Profiling of Extracellular RNAs Present in Cerebrospinal Fluid Identifies Differentially Expressed Transcripts in Parkinson's Disease. J Parkinsons Dis. 2016;6:109-117
34. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP A ceRNA hypothesis. the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353-358
35. Teng KY, Ghoshal K. Role of Noncoding RNAs as Biomarker and Therapeutic Targets for Liver Fibrosis. Gene Expr. 2015;16:155-162
36. Booton R, Lindsay MA. Emerging role of MicroRNAs and long noncoding RNAs in respiratory disease. Chest. 2014;146:193-204
37. Yu F, Zheng J, Mao Y, Dong P, Lu Z, Li G. et al. Long Non-coding RNA Growth Arrest-specific Transcript 5 (GAS5) Inhibits Liver Fibrogenesis through a Mechanism of Competing Endogenous RNA. J Biol Chem. 2015;290:28286-28298
38. Huang ZW, Tian LH, Yang B, Guo RM. Long Noncoding RNA H19 Acts as a Competing Endogenous RNA to Mediate CTGF Expression by Sponging miR-455 in Cardiac Fibrosis. DNA Cell Biol. 2017;36:759-766
39. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344-352
40. Bayoumi AS, Sayed A, Broskova Z, Teoh JP, Wilson J, Su H. et al. Crosstalk between Long Noncoding RNAs and MicroRNAs in Health and Disease. Int J Mol Sci. 2016;17:356
41. Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J. et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. 2016;22:1131-1139
42. Johnsson P, Lipovich L, Grander D, Morris KV. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta. 2014;1840:1063-1071
43. Rogoyski OM, Pueyo JI, Couso JP, Newbury SF. Functions of long non-coding RNAs in human disease and their conservation in Drosophila development. Biochem Soc Trans. 2017;45:895-904
44. Jenkins AM, Waterhouse RM, Muskavitch MA. Long non-coding RNA discovery across the genus anopheles reveals conserved secondary structures within and beyond the Gambiae complex. BMC Genomics. 2015;16:337
45. Diederichs S. The four dimensions of noncoding RNA conservation. Trends Genet. 2014;30:121-123
Author contact
![]() Corresponding author: Prof. Hongli Shan (e-mail: shanhonglihrbmu.edu.cn) and Prof. Yanjie Lu (e-mail: yjlu2008com). Baojian Road 157, Harbin, Heilongjiang 150081, P. R. China. Fax: 86 451 86667511; Tel: 86 451 86671354
Corresponding author: Prof. Hongli Shan (e-mail: shanhonglihrbmu.edu.cn) and Prof. Yanjie Lu (e-mail: yjlu2008com). Baojian Road 157, Harbin, Heilongjiang 150081, P. R. China. Fax: 86 451 86667511; Tel: 86 451 86671354