Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(5):1361-1375. doi:10.7150/thno.18299 This issue Cite
Research Paper
Sarcoma-Targeting Peptide-Decorated Polypeptide Nanogel Intracellularly Delivers Shikonin for Upregulated Osteosarcoma Necroptosis and Diminished Pulmonary Metastasis
Suoyuan Li1,4*, Tao Zhang1*, Weiguo Xu2*, Jianxun Ding2 ![]() , Fei Yin3, Jing Xu3, Wei Sun1, Hongsheng Wang3, Mengxiong Sun1, Zhengdong Cai1,3
, Fei Yin3, Jing Xu3, Wei Sun1, Hongsheng Wang3, Mengxiong Sun1, Zhengdong Cai1,3 ![]() , Yingqi Hua1,3
, Yingqi Hua1,3 ![]()
1. Department of Orthopedics, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080, P. R. China
2. Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China
3. Shanghai Bone Tumor Institution, Shanghai 201620, P. R. China
4. Department of Orthopedics, Suzhou Municipal Hospital, Nanjing Medical University, Suzhou 215000, P. R. China
*These authors contributed equally to this work.
Received 2016-11-10; Accepted 2017-12-18; Published 2018-2-2
Abstract

Purpose: Osteosarcoma is the most common primary bone cancer and is notorious for pulmonary metastasis, representing a major threat to pediatric patients. An effective drug targeting osteosarcoma and its lung metastasis is urgently needed.
Design: In this study, a sarcoma-targeting peptide-decorated disulfide-crosslinked polypeptide nanogel (STP-NG) was exploited for enhanced intracellular delivery of shikonin (SHK), an extract of a medicinal herb, to inhibit osteosarcoma progression with minimal systemic toxicity.
Results: The targeted, loaded nanogel, STP-NG/SHK, killed osteosarcoma cells by inducing RIP1- and RIP3-dependent necroptosis in vitro. Necroptosis is a novel cell death form that could be well adapted as an efficient antitumor strategy, the main obstacle of which is its high toxicity. After intravenous injection, STP-NG/SHK efficiently suppressed tumor growth and reduced pulmonary metastasis, offering greater tumor necrosis and higher RIP1 and RIP3 upregulation compared to free SHK or untargeted NG/SHK in vivo. Additionally, the treatment with NG/SHK or STP-NG/SHK showed minimal toxicity to normal organs, suggesting low systemic toxicity compared to free SHK. Conclusion: The STP-guided intracellular drug delivery system using the necroptosis mechanism showed profound anti-osteosarcoma activity, especially eliminated lung metastasis in vivo. This drug formulation may have great potential for treatment of osteosarcoma.
Keywords: Sarcoma-Targeting Peptide, Polypeptide Nanogel, Intracellular Drug Delivery, Necroptosis, Osteosarcoma Chemotherapy
Introduction
It is well known that apoptosis induced by chemotherapeutic agents is the most common tumor killing mechanism [1]. However, tumor cells eventually accumulate resistance to apoptosis after multiple treatments [2]. Drug resistance may result from disrupted apoptosis machinery and upregulated membrane transporters of drugs. Historically, necrosis was thought to be uncontrollable and an unregulated cell death pathway until the report of regulated necrotic cell death in 2004 [3]. Necroptosis, as it is called, is a recently reported programmed cell death mode and is distinct from apoptosis [4]. Necroptosis can be caused by tumor necrosis factor receptor (TNFR) superfamily, interferon receptors, T cell receptors, cellular metabolism, Toll-like receptors, genotoxic stresses, or various small molecule compounds. The core necroptosis pathways are receptor-interacting protein kinase 1 (RIP1), receptor-interacting protein 3 (RIP3), and mixed lineage kinase domain-like protein (MLKL) [5]. Furthermore, the necroptosis pathway, distinct from the apoptosis pathway, cannot be blocked by traditional barriers of apoptosis [6]. There are several experimental therapies that can rapidly kill cancer cells through necroptosis, such as TNF+Smac mimetics+zVAD and obatoclax [7, 8], which suggests that triggering necroptosis holds great potential as an advanced strategy for cancer therapy.
Shikonin (SHK) is an active naphthoquinone compound extracted from Lithospermum root with extensive pharmacological activity [9]. Our previous study indicated that SHK had rapid and profound antitumor effects on both primary and metastatic osteosarcomas by inducing RIP1- and RIP3-dependent necroptosis [10]. However, the poor solubility and severe systemic toxicity of SHK hinder its usage as a clinical antitumor drug [11], as demonstrated by emaciated mice at the end of SHK treatment [12].
Recently, polymeric nanoparticles have gained increasing attention for improving the water solubility and reducing side effects of antineoplastic drugs. In addition, compared with small molecule drugs, the polymeric nanocarrier-incorporated agents also have the following advantages: (1) ensuring drugs bypass biological barriers and extending the circulation time in vivo; (2) allowing drugs to selectively accumulate in tumors through the enhanced permeability and retention (EPR) effect; (3) releasing drugs in a controllable fashion; (4) extending drug retention time at the lesion site; and (5) exhibiting a functionalized surface for conjugating various targeting ligands to identify tumors [13]. All these properties support the use of polymeric nanoparticles as promising nanocarriers for controlled delivery of SHK.
Osteosarcoma is the most common primary bone malignancy, which is the second highest cause of cancer-related deaths during the second and third decades of life [14]. Currently, surgical resection and adjuvant/neoadjuvant chemotherapy are preferred treatment regimens, but more than 30% of patients lose their lives within five years when resistance to chemotherapy occurs [15]. Current chemotherapy techniques for osteosarcoma suffer from lack of specificity and have high systemic toxicity. Therefore, chemotherapy formulations with lower side effects, higher antitumor efficacy, and longer survival are desperately needed for the treatment of osteosarcoma.
In order to fulfill this requirement, based on a passive targeting strategy [16], a sarcoma-targeting peptide (STP)-modified reduction-responsive poly(ethylene glycol)-poly(L-phenylalanine-co-L-cystine) (STP-PEG-P(LP-co-LC)) nanogel was prepared for targeted intracellular delivery of SHK, which was abbreviated as STP-NG/SHK (Scheme 1). STP with an amino acid sequence of VNTANST can specifically recognize vimentin (VIM), which is overexpressed on the surface of cancer cells [17, 18], including osteosarcoma cells [19]. In detail, the selective internalization of STP-NG/SHK by cancer cells and subsequently enhanced cell necroptosis through RIP1 and RIP3 pathways were revealed in osteosarcoma 143B cells in vitro. In addition, STP-NG/SHK selectively accumulated in the human 143B osteosarcoma-xenografted nude mouse model and demonstrated enhanced inhibition of osteosarcoma growth and metastasis with reduced systemic toxicity.
Schematic illustration for preparation of STP-NG/SHK, and RIP1- and RIP3-dependent cell necroptosis in primary tumor and lung metastasis.
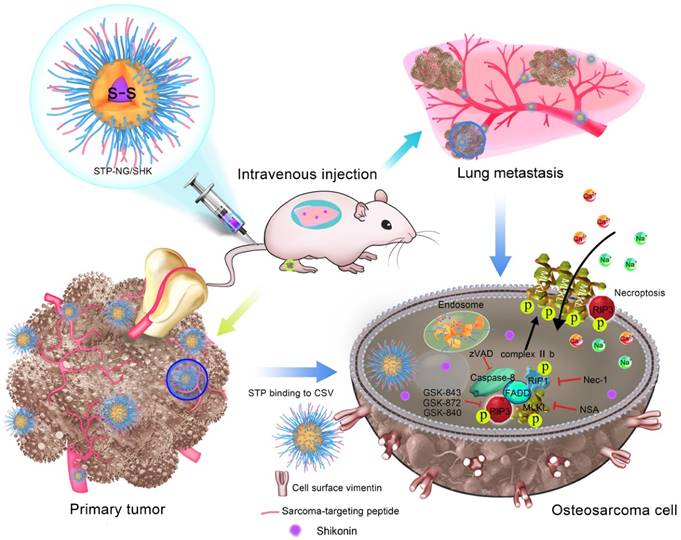
Materials and Methods
Materials
mPEG (number average molecular weight (Mn) = 5000 g mol-1), trifluoroacetic acid (TFA), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl), and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich (Shanghai, P. R. China). The amino-terminated mPEG (mPEG-NH2), L-phenylalanine N-carboxyanhydride (LP NCA), and L-cystine N-carboxyanhydride (LC NCA) were synthesized according to the previously reported protocol [20]. SHK (purity > 98%) was purchased from Shanghai Tauto Biotech Co., Ltd (Shanghai, P. R. China). Dimethyl sulfoxide (DMSO) was purchased from Sigma (Shanghai, P. R. China). High glucose Dulbecco's modified Eagle's medium (DMEM-H) and fetal bovine serum (FBS) were obtained from Thermo Scientific (Waltham, MA, USA). Hoechst 33342 was bought from Beyotime Co., Ltd (Shanghai, P. R. China). The peptide VNTANST-FITC (STP-FITC) was purchased from Sangon Biotech Co., Ltd (Shanghai, P. R. China).
Antibodies used for flow cytometry (FCM) and fluorescence microscopy were rabbit anti-VIM (Cell Signaling Technologies, Danvers, USA), rabbit anti-VIM 488 conjugate (Cell Signaling Technologies, Danvers, USA), and rabbit mAb IgG isotype control (Cell Signaling Technologies, Danvers, USA). Antibodies used for Western blot were rabbit anti-Actin (Santa Cruz, Dallas, USA), mouse anti-RIP1 (BD Biosciences, New Jersey, USA), rabbit anti-RIP3, rabbit anti-PARP, rabbit anti-caspase-3, and anti-mouse IgG HRP-linked antibody (Cell Signaling Technologies, Danvers, USA).
Synthesis of mPEG-P(LP-co-LC) and STP-PEG-P(LP-co-LC)
As shown in Scheme S1, mPEG-P(LP-co-LC) was first synthesized via the ring-opening polymerization (ROP) of LP NCA and LC NCA with mPEG-NH2 as a macroinitiator according to previously published protocol [21]. Second, tert-butoxycarbonyl-amino-modified PEG-NH2 (t-Boc-NH-PEG-NH2) was used as a macroinitiator to synthesize t-Boc-NH-PEG-P(LP-co-LC) by the same reaction procedure (Scheme S2). The t-Boc group was deprotected with TFA, and a maleimide group was conjugated onto the end of PEG by condensing carboxyl and amino groups with EDC·HCl and NHS as the condensing agents. The reactive maleimide-terminated polypeptide nanogel of MI-PEG-P(LP-co-LC) was synthesized. Finally, STP-PEG-P(LP-co-LC) was synthesized using a Michael addition reaction between STP and MI-PEG-P(LP-co-LC), as depicted in Scheme S3.
Preparation of NG/SHK and STP-NG/SHK
As mentioned previously, SHK was embedded into nanogels using a sequential dispersion and dialysis approach. Briefly, mPEG-P(LP-co-LC) (500.0 mg) was dispersed in 18.0 mL of N,N-dimethylformamide (DMF), and SHK (100.0 mg) was dissolved in 2.0 mL of DMF. Then 20.0 mL of MilliQ water was added dropwise into the above solution. The final solution was stirred for 12 h at room temperature, and then dialyzed against MilliQ water for 24 h (molecular weight cut-off (MWCO) = 3,500 Da). MilliQ water was replaced every 2 h. Subsequently, the SHK-loaded nanogel (NG/SHK) was collected by lyophilisation after filtration. STP-NG/SHK was prepared similarly as NG/SHK, but STP-PEG-P(LP-co-LC) replaced mPEG-P(LP-co-LC).
Drug loading content (DLC) and drug loading efficiency (DLE) were measured as follows: first, 1.0 mg of NG/SHK or STP-NG/SHK was dissolved in 10.0 mL of DMF and stirred for 12 h at room temperature. Then, SHK loaded in the nanogel was assayed using an ultraviolet-visible (UV-Vis) spectrophotometer (λabs = 520 nm). DLC and DLE of NG/SHK or STP-NG/SHK were calculated by Equations (1) and (2), respectively.
(1)
(2)
Characterization
The chemical structures of mPEG-P(LP-co-LC) and STP-PEG-P(LP-co-LC) were confirmed by both proton nuclear magnetic resonance (1H NMR) and Fourier-transform infrared (FT-IR) spectra, which are shown in Figures S1 and S2. The morphologies of NG/SHK and STP-NG/SHK were assessed using a JEM-1011 transmission electron microscopy (TEM; JEOL, Tokyo, Japan) with an accelerating voltage of 100 kV. A drop of loading nanogel solution (0.05 mg mL-1) in phosphate-buffered saline (PBS) was deposited onto a carbon-coated copper grid and dried at room temperature for more than one day before testing. Hydrodynamic radii (Rhs) were assessed by dynamic laser scattering (DLS) on a Wyatt QELS instrument (DAWN EOS, Wyatt Technology Corporation, Santa Barbara, CA, USA), and the scattering angle was set at 90°. For DLS measurements, the loading nanogels were dissolved in PBS at a concentration of 0.1 mg mL-1.
In Vitro SHK Release
SHK release from NG/SHK and STP-NG/SHK was measured in PBS at pH 7.4 without or with glutathione (GSH). The weighed freeze-dried nanogels were suspended in 10.0 mL of PBS with none or 10.0 mM GSH, and introduced into a dialysis bag (MWCO = 3,500 Da) to initiate release into 100.0 mL of PBS with none or 10.0 mM GSH at 37 °C, 75 rpm constant shaking. Next, 2.0 mL of release medium was removed and replenished with an equal volume of fresh medium. The release SHK was measured using a fluorescence UV-Vis spectrophotometer (λabs = 520 nm).
Cell Lines and Cell Culture
A human osteoblast hFOB1.19 cell line and a human osteosarcoma 143B cell line were obtained from American Type Culture Collection (ATCC; Rockville, MD, USA). hFOB1.19 cells was cultured in DMEM and F-12 (DMEM/F-12, 1:1, V/V; Thermo Scientific, Waltham, MA, USA) supplemented with 10% (V/V) FBS, 100.0 U mL-1 penicillin and 100.0 μg mL-1 streptomycin, 0.3 mg mL-1 G418 (Thermo Scientific, Waltham, MA, USA) in a humidified incubator at 34 °C in 5% (V/V) carbon dioxide (CO2). 143B cells were cultured in DMEM-H supplemented with 10% (V/V) FBS, 100.0 U mL-1 penicillin and 100.0 μg mL-1 streptomycin in a humidified incubator at 37 °C in 5% (V/V) CO2.
Confocal Laser Scanning Microscope (CLSM) Assessments
hFOB1.19 or 143B cells were seeded on coverslips in 24-well plates and incubated for 24 h. Then 0.1 mL of Hoechst 33342 was added to wells and incubated for 1 h at 37 °C. Cells were then rinsed with PBS three times. Cells in the anti-VIM 488 group were stained with anti-VIM conjugate 488 for 2 h at 37 °C; cells of the STP-FITC group were stained with STP-FITC for 2 h at 37 °C; anti-VIM+STP-FITC cells were stained with anti-VIM for 2 h at 37 °C, rinsed with PBS three times, and stained with STP-FITC for 2 h at 37 °C. All treated cells were rinsed with PBS three times and observed under a TCS SP8 confocal laser scanning platform (Leica Company, Ltd., Germany), and mean optical density (/pixel) of cytomembrane was calculated using ImageJ software (version 1.45s; Wayne Rasband, NIH, Bethesda, MD, USA).
Flow Cytometry Assays
hFOB1.19 or 143B cells were stained and then analyzed by FCM. PBS was a blank control and isotype IgG was the negative control. 143B cells plated in 6-well plates and synchronized with complete DMEM-H were incubated with blank DMEM-H as control, SHK (2.0 μΜ), NG/SHK (2.0 μΜ), or STP-NG/SHK (2.0 μΜ) for 24 h. Subsequently, the cells were washed twice with cold PBS. The cells used for cell cycle assessment were mixed with 200.0 μL of 1 × binding buffer (BD Biosciences, New Jersey, USA), and incubated at room temperature away from light for 15 min with propidium iodide (PI; Sigma, St. Louis, USA), NP-40, and RNaseA (BD Biosciences, New Jersey, USA). The cells used for cell death assessment were mixed in 200.0 μL of 1 × binding buffer (eBioscience, San Diego, USA), and incubated at room temperature away from light for 15 min with an annexin-V/7-aminoactinomycin-D (7-AAD) (eBioscience, San Diego, USA) double staining solution. Stained cells were analyzed using FCM. The cells at different cell cycle stages and necrosis levels were measured with BD Accuri C6 Software (BD Biosciences, New Jersey, USA).
Cytotoxicity Assays
Cells were seeded into 96-well plates, cultured overnight to adhere, and treated with SHK, NG/SHK, or STP-NG/SHK (0, 0.1, 0.2, 0.5, 1.0, 2.0, 5.0, or 10.0 μΜ SHK equivalent) for 24 h. Cells incubated with blank DMEM-H were a control. After 24 h incubation, supernatant was removed, and 190.0 μL of DMEM-H along with 10.0 μL cholecystokinin octapeptide (CCK-8) (Dojindo, Japan) was added to each well for 4 h incubation. Absorbance was measured with an ELX800 Micro Plate Reader (Bio-Tek Instruments, Canada) at 450 nm. Cell viability was calculated according to Equation 3 [22].
(3)
Western Blot Analyses
143B cells were treated with 2.0 μΜ SHK, NG/SHK, or STP-NG/SHK for 24 h. Cells were washed twice with PBS solution and lysed with radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Institute of Biotechnology, Beijing, P. R. China) and protease inhibitor (Thermo Scientific, Waltham, USA). Tumor tissues and lung metastases were retrieved from -80 °C storage and immersed in liquid nitrogen. The resulting powder was lysed with RIPA lysis buffer and protease inhibitor. Protein concentration was quantified with a Pierce BCA Protein Assay Kit (Thermo Scientific; Waltham, USA). Samples containing equal amounts of proteins (depending on antibody concentration, 15.0 - 50.0 μg) from lysates of cultured osteosarcoma cells and tumor tissue were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked in 5% (V/V) skim milk at room temperature for 1 h, and blots were probed overnight at 4 °C (1:1000, V/V) with diluted primary antibodies or β-actin to measure the protein of interest. After incubation, membranes were washed three times with washing buffer (PBS containing 0.1% Tween, V/V) for 10 min. Subsequently, membranes were incubated for 1 h with secondary antibodies (1:2000, V/V) at room temperature. Membranes were washed with Tween 20-PBS three times for 10 min and were developed using an Odyssey two color infrared laser imaging system. β-Actin was used as an internal control.
Establishment of Mouse Tumor Model
Animal experiments were performed on 4-week-old female BALB/c nude mice purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, P. R. China). 143B cells were digested, washed in cold PBS three times, and suspended in cold PBS at a final concentration of 1 × 108 cells mL-1. In this model, the 10.0 μL of cell suspension was injected into the medullary cavity of the tibia. All animal-related procedures were approved by the Animal Care and Use Committee of the Shanghai General Hospital, and the study was approved by the Science and Technology Commission of Shanghai Municipality (ID: SYXK 2007 - 0006) with a permit number of 2011-RES1.
Biodistribution Assays
143B osteosarcoma-bearing mice were randomly divided into three groups and treated with SHK, NG/SHK, or STP-NG/SHK at a free SHK dose of 2.0 mg per kg body weight (mg (kg BW)-1) via tail vein injection. At 8 and 72 h post-injection, mice were sacrificed, then the hearts, livers, spleens, lungs, kidneys, blood, and tumors were obtained for further assay. Blood samples were centrifuged (4,000 rcf, 10 min) to collect plasma. Tissues were washed with 10.0 mL of cold PBS before collection. Tissue samples were diluted in a solution of methanol and distilled water (1:1, V/V), and homogenized. The cleared supernatant was diluted with 0.1% (V/V) formic acid solution to measure free SHK with liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS; Agilent 6410 A; Agilent Technologies, Santa Clara, USA).
Therapeutic Efficiency and Systemic Toxicity Detections
Two weeks later, when tibial tumors were macroscopic (about 100 mm3 in volume), 143B osteosarcoma-bearing mice were randomly divided into four groups (n = 7), i.e., control, SHK, NG/SHK, or STP-NG/SHK at a SHK dose of 2.0 mg (kg BW)-1. Treatments were begun concomitantly and consisted of PBS, SHK (diluted in 5% DMSO, V/V), and various SHK formulations in PBS, injected into the tail vein every other day. Mice were weighed prior to treatment and were euthanized at two days after the last injection.
Mice were observed prior to tumor formation, and tumors were measured apically (AP) and longitudinally (L) with digital calipers. AP measurements were taken across the knee-cap, and L measurements were made in the front of the tibia, where tumor growth was maximal. Primary tumor size was estimated according to Equation 4 [23].
(4)
After all the treatments, posterior limbs with tumors were collected and weighed. Necrosis of primary tumor tissues were estimated using NIS-Elements imaging software (version 2.3; Nikon, Tokyo, Japan) after hematoxylin and eosin (H&E) staining. Briefly, three tumor nodes from each group were embedded, the largest section of tumor was sliced up for H&E staining, then five different visual fields were photographed under 100 × microscope, the necrosis area was quantified with NIS-Elements imaging software, and the tumor necrosis rate was normalized using the control group. Furthermore, the lungs were weighed and their weight recorded. Then the lung tumors were counted, and lung metastases were microscopically counted in the largest coronal sections. The main internal organs (i.e., the heart, liver, kidney, and spleen) were also histologically assessed.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism 5 (La Jolla, CA). For each experimental datum, standard error of the mean (SEM) from replicate experiments was calculated and appeared as error bars, which show SEM for at least three experiments. Means and SEMs were calculated for all groups and compared by one-way ANOVA and LSD multiple comparison test. *P < 0.05 was considered statistically significant, #P < 0.01 and &P < 0.001 were considered highly significant.
Results and Discussion
Preparations and Characterizations of Untargeted and Targeted SHK Nanoformulations
As shown in Figure 1 and Figures S1 and S2, the chemical structures of mPEG-P(LP-co-LC) and STP-PEG-P(LP-co-LC) were characterized by both 1H NMR and FT-IR spectroscopy. The degrees of polymerization (DPs) of LP and LC in both nanogels were determined to be 12 and 4, respectively, which were derived from the elemental analyses of carbon (C), hydrogen (H), nitrogen (N), and sulfur (S). NG/SHK and STP-NG/SHK had uniform spherical morphologies as measured by TEM (Figure 1A and 1B). The mean apparent diameters of NG/SHK and STP-NG/SHK were 65 and 75 nm, respectively. The Rhs of both nanogels measured with DLS were 70±3.5 nm and 85.9±5.5 nm, respectively. The zeta potentials of NG/SHK and STP-NG/SHK were -7.52 mV and -2.21 mV. These data confirmed that both NG/SHK and STP-NG/SHK exhibited a suitable size for selective accumulation in tumor tissues via the EPR effects [24].
Chemical structures and characterizations of untargeted and targeted SHK nanoformulations. Morphologies and Rhs of (A) NG/SHK and (B) STP-NG/SHK. (C) Release profiles of SHK from NG/SHK and STP-NG/SHK in PBS without or with 10.0 mM GSH.
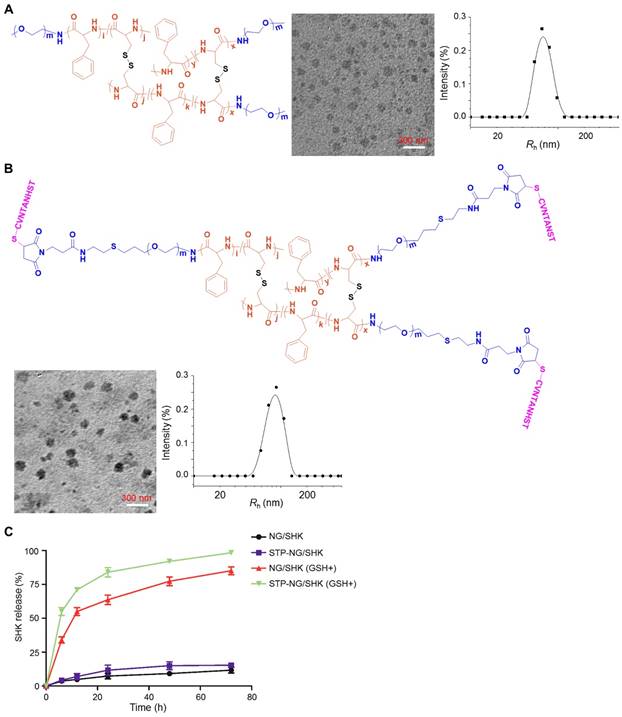
The DLC was 7.41% in NG/SHK and 7.49% in STP-NG/SHK, respectively. The release profiles of NG/SHK and STP-NG/SHK in vitro were conducted in PBS at pH 7.4 without and with 10.0 mM GSH, as depicted in Figure 1C. <17% of loaded SHK was released from the nanogel in PBS without GSH at 72 h, but the release contents of SHK from NG/SHK and STP-NG/SHK were accelerated up to 85.1% and 98.4% in the presence of GSH, respectively. A burst release in the presence of GSH may be attributed to the rapid dissolution of disulfide bond (S-S) in the core of the nanogel. The reduction-responsive profiles of NG/SHK and STP-NG/SHK may minimize the loss of SHK prior to arrival at the tumor, and subsequently enhance therapeutic efficacy and reduce side effects in vivo.
Necroptosis Induction via Specific Targeted Drug Delivery
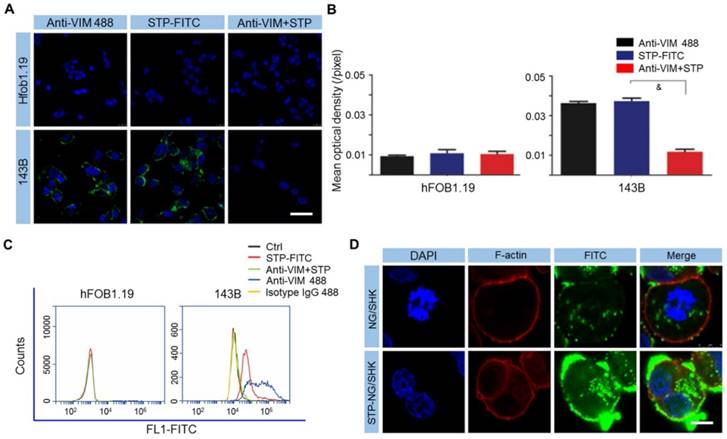
It is important to identify specific cell surface markers to facilitate nanoparticle targeting. In order to do so, the expression of VIM on the osteosarcoma surface was tested. As revealed by CLSM and FCM, 143B osteosarcoma cells showed higher cell surface vimentin (CSV) expression compared to the normal tissue counterpart osteoblast hFOB1.19 cells, which showed almost no expression (Figure 2A-C). STP was proved to bind to the osteosarcoma cell membrane, and this binding could be blocked if cells were pre-incubated with non-fluorescently labeled VIM antibody (Figure 2A-C). The data demonstrated that STP specifically binds to CSV in osteosarcoma, which could be further used as a targeting agent at the surface of polymeric nanocarriers. Moreover, neither STP nor anti-VIM antibody would bind to white blood cells (WBCs) in whole blood (Figure S3), which indicated ideal tumor specificity. CSV is associated with the epithelial-to-mesenchymal transition, upregulates tumor malignancy and metastasis, and is strongly expressed in osteosarcoma [19]. Due to the lack of CSV [19], STP did not bind to human osteoblast hFOB1.19 cells and blood cells. This discrepancy holds the hope of achieving ideal efficacy and minimal toxicity. Furthermore, STP-NG/SHK showed higher membrane binding due to its targeting effect, which then increased cellular uptake of NG/SHK (Figure 2D).
STP binds to osteosarcoma cells by CSV. (A) Cell-surface staining analysis for CSV in hFOB1.19 osteoblasts and 143B cells with CLSM. Scale bar = 25 μm. (B) Cells were stained with Anti-VIM conjugate 488 (green), STP-FITC (green), or Anti-VIM+ STP-FITC (green), and mean optical density (/pixel) was measured. Nuclei were stained with Hoechst 33342 (blue). Scale bar = 10 mm. (C) Immunologic assessment of STP binding to hFOB1.19 and 143B cells with FCM. Anti-VIM conjugate 488 was a positive control. Isotype controls were negative controls. Each set of data is represented as mean ± SEM (n = 3; &P < 0.001). (D) 143B cells were incubated with NG/SHK-FITC and STP-NG/SHK-FITC, and cellular uptake was measured by fluorecence imaging. Scale bar = 5 μm.
The cytotoxicity of SHK, NG/SHK, and STP-NG/SHK toward 143B cells in vitro was then evaluated by a CCK-8 assay. As shown in Figure 3A, the growth inhibitory effect was observed in all treatment groups. STP-NG/SHK showed the most profound cytotoxicity against 143B cells among all three drugs. The half-maximal inhibitory concentrations (IC50s) are depicted in Figure 3B. The IC50 at 24 h of treatment with STP-NG/SHK toward 143B cells was 1.15±0.09 μΜ, while those of NG/SHK and SHK were 1.52±0.01 μΜ and 2.29±0.12 μΜ. Cell death was also assessed in Figure 3C and Figure S4; this data was consistent with those of the cytotoxicity assays. The percentage of 7-AAD positive cells of STP-NG/SHK groups was 50.33% ± 2.60%, which was significantly higher than those of the NG/SHK group (41.67% ± 1.45%) and SHK group (30.33% ± 0.88%), (P < 0.001). In summary, NG/SHK and STP-NG/SHK showed higher cytotoxicity in vitro against osteosarcoma than naked SHK. Furthermore, the targeting peptide STP enhanced cytotoxicity compared to NG/SHK, possibly due to increased cellular uptake. To further confirm the mechanism of cell death, cell cycle was analyzed in each treatment group, and no difference within the groups was noted (Figure S5). This suggested that neither free SHK nor encapsulated SHK altered the cell cycle to render an anti-proliferation effect.
Cytotoxicity in vitro. (A) The cell viability of 143B cells was detected. (B) IC50s of SHK, NG/SHK, and STP-NG/SHK after 24 h. (C) Necroptosis in osteosarcoma 143B cells measured by FCM and Annexin V/7-AAD staining after treatments. Panel quadrants indicate populations of normal, early and late apoptotic, and necrotic cells. (D) RIP1, RIP3, PARP, caspase-3, and caspase-8 measurement via Western blot and (E) semiquantitative analyses. Each set of data is represented as mean ± SEM (n = 3; *P < 0.05, #P < 0.01, &P < 0.001).
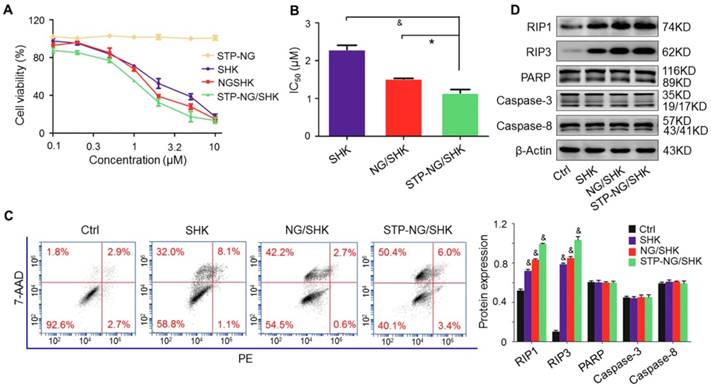
A panel of Western blot analyses was then performed to determine the form of induced cell death. Caspase-3, caspase-8, and PARP are indicators of apoptosis, and RIP1 and RIP3 are the markers of necroptosis. As shown in Figure 3D, the levels of RIP1 and RIP3 were elevated in 143B cells after the treatments with NG/SHK and STP-NG/SHK for 24 h compared with the control and SHK groups. However, PARP and caspase-3 were uniformly expressed with no difference in all groups. These results showed that nano-encapsulated NG/SHK and STP-NG/SHK induced cell death selectively through necroptosis to osteosarcoma cells in a greater extent than bare SHK, and STP-NG/SHK showed the greatest extent of necroptosis as revealed by densitometry (Figure 3E).
Elevated Intratumoral Accumulation of Loaded Nanogels
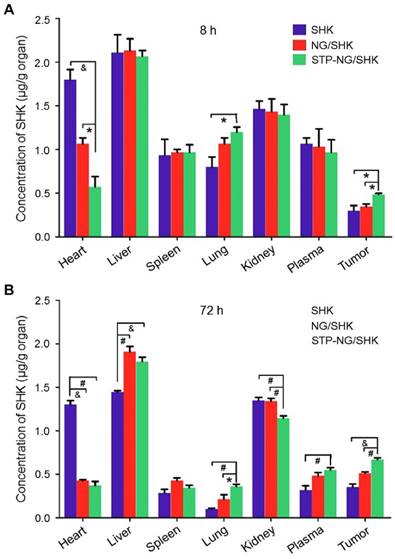
The tissue biodistribution of SHK was measured to confirm the enhanced intratumoral accumulation of SHK through the EPR effect. In theory, highly selective tissue biodistribution is related to increased efficacy and reduced side effects [25]. Figure 4 depicted the biodistribution of SHK at 8 and 72 h after injection. Across all groups, the highest SHK concentration was seen in the liver due to macrophage phagocytosis of the reticuloendothelial system (RES) [26]. NG/SHK and STP-NG/SHK had a higher hepatic storage than free SHK. High concentration of SHK in the livers and kidneys indicated that all the SHK formulations might be metabolized by the liver and/or kidney. At 72 h post-injection, the SHK concentrations decreased in normal organs while they increased in tumor issue (Figure 4B). Notably, more than three times higher drug concentration was found in the hearts of the free SHK group compared to those of the loaded nanogel groups. Of all toxicity concerns, the cardiotoxicity of SHK is the most worrisome, and this result may support a decrease in cardiotoxicity of STP-NG/SHK compared to free SHK and NG/SHK. More importantly, NG/SHK and STP-NG/SHK showed a higher drug concentration than free SHK in plasma and tumor tissues at 72 h post-injection. In particular, STP-NG/SHK exhibited significantly higher retention and release of SHK than NG/SHK. The evidence suggested that the STP does not guide the nanoparticle to the tumor. However, STP enhances nanoparticle-cell interaction once it reaches tumor site, and increases cellular uptake through EPR. This targeted effect might confer high drug activity as well as minimal systemic toxicity.
Biodistribution. Biodistribution of SHK in 143B osteosarcoma-bearing mice at (A) 8 h and (B) 72 h after injection of SHK, NG/SHK, or STP-NG/SHK at a dose of 2.0 mg (kg BW)-1, intravenous injection. Each set of data is represented as mean ± SEM (n = 3; *P < 0.05, #P < 0.01, &P < 0.001).
Upregulated Anti-Osteosarcoma Activities of NG/SHK and STP-NG/SHK In Vivo
The bioactivities of untargeted and targeted SHK-loaded nanogel were evaluated in vivo. Among all treatment groups, STP-NG/SHK showed superior anti-proliferation efficacy. In detail, the volume of the orthotopic tumor was measured after amputation. The three treatment groups showed that the growth of tumors was suppressed. After euthanization, the tumor volume was 363.6±24.0 mm3 in the STP-NG/SHK group, which was significant smaller than 660.1±59.3 mm3 in the NG/SHK group (P < 0.001), 754.0±56.6 mm3 in the SHK group (P < 0.001), and 1093.9±81.4 mm3 in the untreated control group (P < 0.001) (Figure 5A and 5B). Consistent with tumor volume, the average weight of the posterior limb containing the tumors was also the smallest in the STP-NG/SHK group (0.8±0.03 g) compared with those of the NG/SHK group (1.4±0.10 g) (P < 0.01), SHK group (1.8±0.09 g) (P < 0.001), and control group (2.2±0.09 g) (P < 0.001) (Figure 5C).
In vivo antitumor efficacy of STP-NG/SHK against primary osteosarcoma. (A) Tumor growth curves. (B) Posterior limb tumors. (C) Average weight of tibial primary osteosarcoma tumors. (D) Tumor necrosis calculated with NIS-Elements imaging software. (E) H&E of primary tumor. Scale bar = 100 μm. (F) RIP1, RIP3, PARP, caspase-3, and caspase-8 expression in primary tumor tissues per group measured with Western blot and (G) semiquantitative analyses. Each set of data is represented as mean ± SEM (for A, C and D, n = 7, for G n=3; #P < 0.01, &P < 0.001).
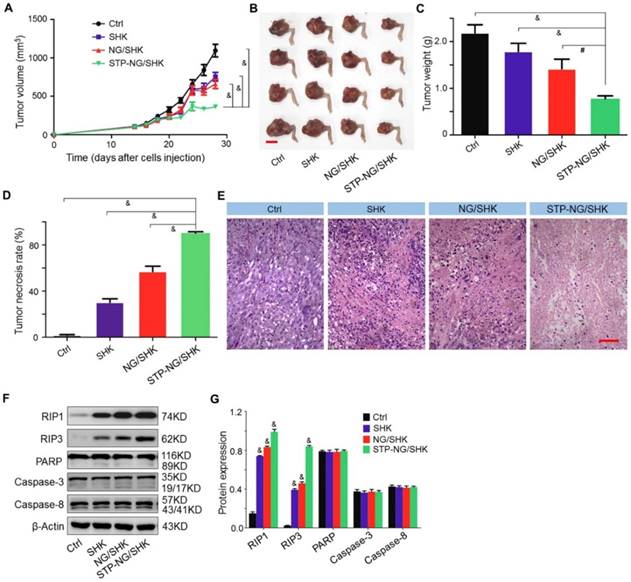
Moreover, pathological examinations were performed to calculate the tumor necrosis rate. The average tumor necrosis rate was calculated from three observation fields and analyzed by NIS-Elements imaging software (Nikon Instruments, Melville, NY, USA). Tumor necrosis areas of the STP-NG/SHK group were the highest (90.3% ± 0.7%) in comparison to those of the NG/SHK group (57.0% ± 2.6%) (P < 0.001), SHK group (30.3% ± 1.8%) (P < 0.001), and control group (1.8% ± 0.3%) (P < 0.001) (Figure 5D). Small area of necrosis in the untreated tumor samples may occur due to hypoxia within the center of the tumor. The representative H&E staining of treated tumor tissues indicated that large and deformed tumor cells were pyknotic or karyorrhexic (Figure 5E). These morphological changes were consistent with SHK-induced necroptosis.
To confirm the mechanism of cell death induced by SHK, the expressions of RIP1, RIP3, PARP, and caspase-3 in primary mouse tumor were assessed by Western blot and quantification analysis. As shown in Figure 5F and 5G, the expression of RIP1 and RIP3 was elevated after STP-NG/SHK treatment to a greater extent than with treatment by NG/SHK or SHK. However, the expression of PARP, caspase-3, and caspase-8 remained unchanged after treatment with all SHK formulations. Collectively, the above in vivo data showed a result consistent with the data of Figure 3D. Furthermore, all the results showed that the inhibition of cell proliferation induced by SHK formulations was through RIP1- and RIP3-dependent necroptosis. With the help of STP, STP-NG/SHK exhibited a higher antitumor efficacy than NG/SHK or SHK.
Enhanced Suppression of Osteosarcoma Pulmonary Metastasis of SHK Nanoformulations
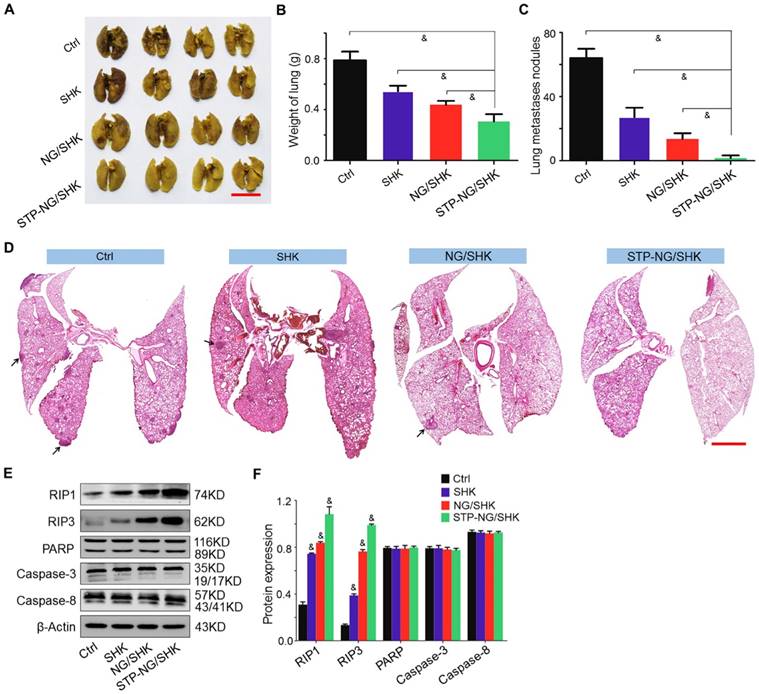
Osteosarcoma is frequently accompanied by pulmonary metastasis, which causes the death of patients. In this work, lung metastasis was assessed by apparent morphology, lung weight, lung metastatic nodules, and H&E staining of lung sections at the end of all treatments. Lung metastasis was more significantly reduced after STP-NG/SHK treatment compared with all other tested groups by gross morphology (Figure 6A). The average weight of lung was 0.3±0.03 g in the STP-NG/SHK group, which was lower than those of the NG/SHK group (0.4±0.01 g, P < 0.001), SHK group (0.5±0.02 g, P < 0.001), and control group (0.8±0.03 g, P < 0.001) (Figure 6B), indicating less solid tumor in the lung. Meanwhile, the number of lung metastases was significantly reduced by the treatment of STP-NG/SHK (0.8 ± 0.4) compared with those of the NG/SHK group (6.8 ± 0.8; P < 0.001), SHK group (13.6 ± 1.3, P < 0.001), and control group (32.0 ± 1.3; P < 0.001) (Figure 6C). The histopathological results of representative lung slides in all groups showed metastasis was mostly inhibited in the STP-NG/SHK group (Figures 6D and Figure S6). These data proved the profound inhibition of osteosarcoma pulmonary metastasis by STP-NG/SHK. This superior anti-metastasis activity should be attributed to tumor-targeting and intracellular drug release. The targeting of STP-NG/SHK to CSV was associated with the epithelial-to-mesenchymal transition, which is critical to metastasis [17]. Furthermore, as shown in Figure 6E and 6F, the expression of RIP1, RIP3, PARP, caspase-3, and caspase-8 in lung tissue was measured by Western blot analyses. Similar results are shown in Figure 5E and 5F, which indicated that the targeted loaded nanogel, STP-NG/SHK, killed osteosarcoma cells by inducing RIP1- and RIP3-dependent necroptosis in vivo.
In vivo antitumor efficiency of STP-NG/SHK against lung osteosarcoma metastasis. (A) Macroscopic appearance of lung metastatic osteosarcoma tumors per group. Scale bar = 1 cm. (B) Lung weight and (C) average microscopically (largest coronal sections) counted lung metastases. (D) H&E staining of metastases of osteosarcoma tumors. Arrows indicate lung metastases. Scale bar = 2 mm. (E) RIP1, RIP3, PARP, caspase-3, and caspase-8 expression in harvested lung metastases per group measured by Western blot and (F) semiquantitative analyses. Each set of data is represented as mean ± SEM (for B and C, n = 7; for E, n = 3; *P < 0.05, #P < 0.01, &P < 0.001).
Biosafety and Reduced Toxicity of SHK after Encapsulation
The biosafety of empty nanoparticle STP-NG was first tested. The empty nanoparticle showed no cytotoxicity toward 143B cells and hFOB1.19 cells (Figure S7A). Hepatic toxicity and systemic toxicity were also confirmed negative in vivo (Figure S7B and S7C). Being confident that the nanoparticle was free of toxicity, the toxicity of all SHK formulations was then tested in vivo. Body weight and histopathology of the heart, liver, kidney, and spleen were examined. As shown in Figure 7A, the body weight of osteosarcoma-bearing mice in the NG/SHK and STP-NG/SHK groups increased slightly over time by 1.5% and 9.8%, while the body weight in the free SHK and control groups dropped by 20.3% and 7.4%, respectively. Free SHK caused the most weight loss possibly due to high systemic toxicity. The decrease of body weight in the control group was caused by tumor burden-related cachexia [27]. The survival rates of 143B-bearing mice after the treatments with various SHK formulations and PBS as control are shown in Figure 7B. The earliest death occurred in the PBS group on day 42. With time, all the mice of the PBS group died after 72 days. The chemotherapy with all SHK formulations could prolong the survival times of mice. More fortunately, the groups of NG/SHK and STP-NG/SHK had encouraging results, and the 90-day survival rates were 28.6% and 71.4%, respectively, which are attributed to the upregulated tumor inhibition efficacies and decreased toxicities than the free SHK group. STP-NG/SHK with the targeting peptide STP increased the tumor accumulation and antitumor activity of encapsulated SHK, and prolonged the survival period.
Systemic toxicity of STP-NG/SHK against 143B osteosarcoma-bearing mice. (A) Weight changes. (B) Survival rates. (C) Mouse organs stained with H&E. Myocardia from mice in the SHK were damaged (★). No overt tissue damage was observed in STP-NG/SHK-treated mice. Scale bar = 100 μm (C). Each set of data is represented as mean ± SEM (for A, n = 7; *P < 0.05, #P < 0.01, &P < 0.001).
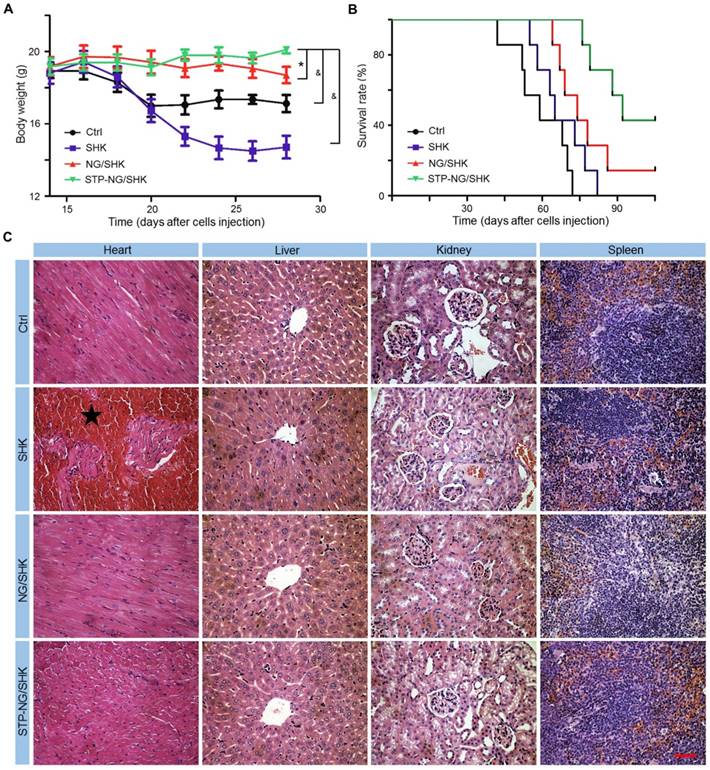
To further evaluate the systemic toxicity of all tested formulations, the main organs of all groups were isolated, sectioned, and stained by H&E. As shown in Figure 7C, the main toxicity by SHK is myocardial damage while minimal abnormality was observed in other organs. NG/SHK and STP-NG/SHK exhibited much less myocardial damage in the H&E slides compared to free SHK.
In summary, overall health conditions represented by body weights in the NG/SHK and STP-NG/SHK groups were better, and myocardial toxicity was reduced. These results suggested that lower systemic toxicity and higher safety could be achieved by nanogel encapsulation, in which STP may efficiently further augment tumor-specific affinity.
Discussion
Osteosarcoma patients develop resistance against conventional chemotherapeutic drugs during therapy, which ultimately leads to treatment failure [28]. It is of great clinical importance to develop advanced strategies against chemoresistance. Necroptosis holds great potential to overcome chemoresistance through the by-pass of apoptosis, in which RIP1 and RIP3 are crucial genes. Recently, the roles of RIP1 and RIP3 in necroptosis toward many cancers in response to therapeutic agents have been reported [29]. We have reported the profound antitumor effect of SHK against osteosarcoma [9]. Necroptosis is initiated by the formation of a complex, which includes caspase-8, Fas-associated death domain (FADD), RIP1, RIP3, and MLKL [30]. Apoptosis and necroptosis share substantial interplay between each other. Caspase-8 can inhibit the formation of necrosome by cleaving RIPK1. When caspase-8-dependent apoptosis is inhibited, necroptosis will be activated in a feedback loop. Therefore, an ideal modality to minimize chemoresistance is to include drugs inducing both apoptosis and necroptosis. However, the necroptosis-inducing drugs like SHK normally have rapid cytotoxicity causing severe systemic side effects. An approach to reduce toxicity and increase tumor accumulation is urgently needed, and nanocarrier systems are widely used to do so.
Recently, polymeric nanocarriers have been exploited for controlled drug delivery in treating osteosarcoma [31]. The drug-loaded polymer nanoplatforms selectively accumulated at the tumor site through passive and active targeting strategies, and further maintained the pharmacodynamics and reduced the side effects of small molecule antitumor drugs. As a typical example, Wang's group designed a PTX and etoposide (ETP) co-loaded PEGylated poly(lactide-co-glycolide) (PLGA) nanoparticle for osteosarcoma treatment [32]. The co-loaded PTX and ETP enhanced cell cycle arrest at the G2/M phase and upregulated cell apoptosis. Stimuli-responsive properties are additional important characteristics for passive targeting and controlled drug delivery. Moreover, the triggered release from polymeric nanocarriers has been mainly focused on pH, redox status, and enzyme activity for treating osteosarcoma. For instance, Kumar's group designed an acid-sensitive gemcitabine (GTB)-loaded core-shell nanocarrier to inhibit MG-63 osteosarcoma cells [33]. Several passive targeting platforms have been proposed to be effective, however, the influence of tumor microenvironment on nanocarriers often impairs drug efficacy. Active targeting is a powerful tool for enhancing intratumoral drug accumulation. For example, activated leukocyte cell adhesion molecule (ALCAM) was thought to be a biomarker of osteosarcoma, and the DOX-loaded anti-ALCAM diabody hybrid polymer liposomal nanoparticle (α-ALCAM-HPLN/DOX) offered more toxicity toward osteosarcoma cells than the non-targeted HPLN/DOX [34]. Although several osteosarcoma-targeting molecules have been described, specificity remains the main issue to be improved [35, 36].
In this study, the untargeted and targeted disulfide-core-cross-linked polypeptide nanogel was designed to controllably deliver SHK intracellularly, so the nanogel, especially STP-NG, delivered more drug to tumors when compared to free SHK in 72 h. Nanogels can be loaded with SHK and deliver it into tumor cells selectively and enhance bioactivity in vivo, representing an efficient drug delivery system for osteosarcoma therapy [37].
To increase specificity and efficacy, a binding structure was identified that allowed drug accumulation at the tumor surface. Cutrera's group used an in vivo reporter gene-mediated screening strategy to identify a tumor-targeting peptide, VNTANST (named STP in this work) [38], which possibly binds to VIM on the cell surface [39]. VIM is an intermediate filament protein, conventionally regarded as an intracellular structural protein in cells of mesenchymal origin, such as chondrocytes, fibroblasts, and macrophages [17]. High VIM expression has been reported for several tumors, including human prostate [40], gastrointestinal tract [41], hepatocellular [42], and breast tumors [43]. Upregulation of VIM is associated with the epithelial-to-mesenchymal transition, which is important for motility and tumor metastasis [44]. Sarcomas are mesenchymal malignancies and universally express VIM. In our study, STP (i.e., VNTANST) was first proved to specifically bind to CSV in osteosarcoma cells. The finding leads to our design of STP-NG/SHK with the purpose of targeting osteosarcoma. As expected, due to tumor specific targeting effect, STP-NG/SHK exhibited superior antitumor activity against osteosarcoma than SHK and NG/SHK. In vitro, NG/SHK and STP-NG/SHK showed similar efficacy, possibly due to direct exposure of the drug in cell culture. However, STP-NG/SHK showed significant higher activity in vivo. Most importantly, STP-NG/SHK was able to reduce pulmonary metastasis, which could eventually improve long-term survival. Meanwhile, systemic toxicity was minimal throughout the treatment with loaded nanogels, especially STP-NG/SHK, which is important since severe toxicity hinders the development of SHK as a therapeutic option for cancer. Our system offers active targeting to osteosarcoma cells by CSV, increased intracellular drug via endocytosis, and rapid tumor killing through necroptosis. Meanwhile, toxicity of bare SHK is minimized, which makes clinical safety viable.
Conclusions
For upregulated inhibition efficacy of osteosarcoma and pulmonary metastasis through drug-induced necroptosis, a STP-decorated disulfide-crosslinked polypeptide nanogel was developed for efficient encapsulation and targeted delivery of SHK. The targeting ability of STP toward 143B cells was confirmed, and the enhanced inhibition of cell proliferation by STP-NG/SHK was demonstrated through the RIP1- and RIP3-dependent necroptosis in vitro. More fascinatingly, STP-NG/SHK selectively accumulated in orthotopic 143B osteosarcoma through specifically recognizing VIM on the cell membrane. As a result, STP-NG/SHK exhibited excellent antitumor efficacy and inhibition of pulmonary metastasis through the RIP1- and RIP3-dependent necroptosis. Meanwhile, STP-NG/SHK showed low systemic toxicity. Thus, the SHK-loaded smart drug delivery system exhibited great potential for VIM-targeted malignancy chemotherapy via necroptosis.
Abbreviations
7-AAD: 7-aminoactinomycin-D; α-ALCAM-HPLN/DOX: DOX-loaded anti-ALCAM diabody hybrid polymer liposomal nanoparticle; ALCAM: activated leukocyte cell adhesion molecule; ATCC: American Type Culture Collection; t-Boc-NH-PEG-NH2: tert-butoxycarbonyl-amino-modified PEG-NH2; C: carbon; CCK-8: cholecystokinin octapeptide; CLSM: confocal laser scanning microscope; CO2: carbon dioxide; CSV: cell surface vimentin; DLC: drug loading content; DLE: drug loading efficiency; DLS: dynamic laser scattering; DMEM-H: high glucose Dulbecco's modified Eagle's medium; DMSO: dimethyl sulfoxide; DMF: N,N-dimethylformamide; DP: degree of polymerization; EDC·HCl: 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride; EPR effect: enhanced permeability and retention effect; ETP: etoposide; FADD: Fas-associated death domain; FBS: fetal bovine serum; FCM: flow cytometry; FT-IR spectrum: Fourier-transform infrared spectrum; GSH: glutathione; GTB: gemcitabine; H: hydrogen; 1H NMR spectrum: proton nuclear magnetic resonance spectrum; H&E: hematoxylin and eosin; IC50s: half-maximal inhibitory concentrations; LC-MS/MS: liquid chromatography coupled with tandem mass spectrometry; LC NCA: L-cystine N-carboxyanhydride; LP NCA: L-phenylalanine N-carboxyanhydride; mg (kg BW)-1: mg per kg body weight; MLKL: mixed lineage kinase domain-like protein; mPEG-NH2: amino-terminated mPEG; MWCO: molecular weight cut-off; N: nitrogen; NG/SHK: SHK-loaded nanogel; NHS: N-hydroxysuccinimide; PBS: phosphate-buffered saline; PLGA: poly(lactide-co-glycolide); PI: propidium iodide; PVDF: polyvinylidene difluoride; RES: reticuloendothelial system; RIP1: receptor-interacting protein kinase 1; RIP3: receptor-interacting protein 3; RIPA: radioimmunoprecipitation assay; ROP: ring-opening polymerization; S: sulfur; S-S: disulfide bond; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SEM: standard error of the mean; SHK: shikonin; STP: sarcoma-targeting peptide; STP-FITC: VNTANST-FITC; STP-PEG-P(LP-co-LC): STP-modified poly(ethylene glycol)poly(L-phenylalanine-co-L-cystine); TEM: transmission electron microscopy; TFA: trifluoroacetic acid; TNFR: tumor necrosis factor receptor; UV-Vis: ultraviolet-visible; VIM: vimentin; WBCs: white blood cells.
Supplementary Material
Supplementary schemes and figures for synthesis and characterization of nanogels. Supplementary figure for binding assay of STP-FITC and VIM antibody with peripheral WBCs. Supplementary figure for cytotoxicity of STP-NG/SHK in vitro. Supplementary figure for effect of STP-NG/SHK on the cell cycle in vitro. Supplementary figure for pulmonary metastasis counting.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos. 81202115, 51673190, 51603204, 51673187, and 51520105004), the Research Grant from Shanghai Hospital Development Center (No. SHDC12013107), and the Excellent Young Talent Program of Shanghai Municipal Commission of Health and Family Planning (No. XYQ2013108).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhu X, Sun Y, Chen D, Li J, Dong X, Wang J. et al. Mastocarcinoma therapy synergistically promoted by lysosome dependent apoptosis specifically evoked by 5-Fu@nanogel system with passive targeting and pH activatable dual function. J Control Release. 2017;254:107-18
2. Li S, Sun W, Wang H, Zuo D, Hua Y, Cai Z. Research progress on the multidrug resistance mechanisms of osteosarcoma chemotherapy and reversal. Tumour Biol. 2015;36:1329-38
3. Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272-82
4. Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014;5:e1312
5. Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol. 2014;76:129-50
6. Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C. et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350:328-34
7. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55-65
8. Philipp S, Sosna J, Adam D. Cancer and necroptosis: friend or foe? Cell Mol Life Sci. 2016;73:2183-93
9. Zhao Q, Assimopoulou AN, Klauck SM, Damianakos H, Chinou I, Kretschmer N. et al. Inhibition of c-MYC with involvement of ERK/JNK/MAPK and AKT pathways as a novel mechanism for shikonin and its derivatives in killing leukemia cells. Oncotarget. 2015;6:38934-51
10. Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z. et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013;13:580
11. Wiench B, Eichhorn T, Paulsen M, Efferth T. Shikonin directly targets mitochondria and causes mitochondrial dysfunction in cancer cells. Evid Based Compl Alt Med. 2012;2012:726025
12. Andújar I, Ríos JL, Giner RM, Recio MC. Pharmacological properties of shikonin - a review of literature since 2002. Planta Med. 2013;79:1685-97
13. Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Che Soc Rev. 2012;41:2971-3010
14. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64:83-103
15. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: Current treatment and a collaborative pathway to success. J Clin Oncol. 2015;33:3029-35
16. Li W, Li JF, Gao J, Li BH, Xia Y, Meng YC. et al. The fine-tuning of thermosensitive and degradable polymer micelles for enhancing intracellular uptake and drug release in tumors. Biomaterials. 2011;32:3832-44
17. Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033-46
18. Satelli A, Brownlee Z, Mitra A, Meng QH, Li S. Circulating tumor cell enumeration with a combination of epithelial cell adhesion molecule- and cell-surface vimentin-based methods for monitoring breast cancer therapeutic response. Clin Chem. 2015;61:259-66
19. Satelli A, Mitra A, Cutrera JJ, Devarie M, Xia X, Ingram DR. et al. Universal marker and detection tool for human sarcoma circulating tumor cells. Cancer Res. 2014;74:1645-50
20. Ding J, Shi F, Xiao C, Lin L, Li C, He C. et al. One-step preparation of reduction-responsive poly(ethylene glycol)-poly(amino acid)s nanogels as efficient intracellular drug delivery platforms. Polym Chem. 2011;2:2857-64
21. Huang K, Shi B, Xu W, Ding J, Yang Y, Liu H. et al. Reduction-responsive polypeptide nanogel delivers antitumor drug for improved efficacy and safety. Acta Biomater. 2015;27:179-93
22. Feng XR, Ding JX, Gref R, Chen XS. Poly (β-cyclodextrin)-mediated polylactide-cholesterol stereocomplex micelles for controlled drug delivery. Chinese J Polym Sci. 2017;35:693-9
23. Tan ML, Friedhuber AM, Dunstan DE, Choong PF, Dass CR. The performance of doxorubicin encapsulated in chitosan-dextran sulphate microparticles in an osteosarcoma model. Biomaterials. 2010;31:541-51
24. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81-9
25. Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172:782-94
26. Kangas L, Unkila M. Tissue selectivity of ospemifene: pharmacologic profile and clinical implications. Steroids. 2013;78:1273-80
27. Esteva-Font C, Jin BJ, Verkman AS. Aquaporin-1 gene deletion reduces breast tumor growth and lung metastasis in tumor-producing MMTV-PyVT mice. FASEB J. 2014;28:1446-53
28. Marina NM, Smeland S, Bielack SS, Bernstein M, Jovic G, Krailo MD. et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): An open-label, international, randomised controlled trial. Lancet Oncol. 2016;17:1396-408
29. Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689-97
30. Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347-53
31. Gu X, Ding J, Zhang Z, Li Q, Zhuang X, Chen X. Polymeric nanocarriers for drug delivery in osteosarcoma treatment. Curr Pharm Des. 2015;21:5187-97
32. Wang B, Yu X, Xu S, Xu M. Paclitaxel and etoposide co-loaded polymeric nanoparticles for the effective combination therapy against human osteosarcoma. J Nanobiotechnol. 2015;13:1-11
33. Chitkara D, Kumar N. BSA-PLGA-based core-shell nanoparticles as carrier system for water-soluble drugs. Pharm Res. 2013;30:2396-409
34. Federman N, Chan J, Nagy JO, Landaw EM, McCabe K, Wu AM. et al. Enhanced growth inhibition of osteosarcoma by cytotoxic polymerized liposomal nanoparticles targeting the alcam cell surface receptor. Sarcoma. 2012;2012:126906
35. Tiram G, Segal E, Krivitsky A, Shreberk-Hassidim R, Ferber S, Ofek P. et al. Identification of dormancy-associated microRNAs for the design of osteosarcoma-targeted dendritic polyglycerol nanopolyplexes. ACS Nano. 2016;10:2028-45
36. Gu W, Jia Z, Truong NP, Prasadam I, Xiao Y, Monteiro MJ. Polymer nanocarrier system for endosome escape and timed release of siRNA with complete gene silencing and cell death in cancer cells. Biomacromolecules. 2013;14:3386-9
37. Shi F, Ding J, Xiao C, Zhuang X, He C, Chen L. et al. Intracellular microenvironment responsive PEGylated polypeptide nanogels with ionizable cores for efficient doxorubicin loading and triggered release. J Mater Chem. 2012;22:14168-79
38. Craig R, Cutrera J, Zhu S, Xia X, Lee YH, Li S. Administering plasmid DNA encoding tumor vessel-anchored IFN-alpha for localizing gene product within or into tumors. Mol Ther. 2008;16:901-6
39. Cutrera J, Dibra D, Xia X, Hasan A, Reed S, Li S. Discovery of a linear peptide for improving tumor targeting of gene products and treatment of distal tumors by IL-12 gene therapy. Mol Ther. 2011;19:1468-77
40. Kolijn K, Verhoef EI, van Leenders GJ. Morphological and immunohistochemical identification of epithelial-to-mesenchymal transition in clinical prostate cancer. Oncotarget. 2015;6:24488-98
41. Stockman DL, Miettinen M, Suster S, Spagnolo D, Dominguez-Malagon H, Hornick JL. et al. Malignant gastrointestinal neuroectodermal tumor: clinicopathologic, immunohistochemical, ultrastructural, and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. Am J Surg Pathol. 2012;36:857-68
42. Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S. Cell-surface Vimentin: A mislocalized protein for isolating csVimentin(+) CD133(-) novel stem-like hepatocellular carcinoma cells expressing EMT markers. Int J Cancer. 2015;137:491-6
43. Azimi I, Beilby H, Davis FM, Marcial DL, Kenny PA, Thompson EW. et al. Altered purinergic receptor-Ca(2+) signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol Oncol. 2016;10:166-78
44. Dong Q, Zhu X, Dai C, Zhang X, Gao X, Wei J. et al. Osteopontin promotes epithelial-mesenchymal transition of hepatocellular carcinoma through regulating vimentin. Oncotarget. 2016;7:12997-3012
Author contact
![]() Corresponding author: Yingqi Hua, E-mail: yhuaedu.cn; Jianxun Ding, E-mail: jxdingac.cn; Zhengdong Cai, E-mail: czd856163.com.
Corresponding author: Yingqi Hua, E-mail: yhuaedu.cn; Jianxun Ding, E-mail: jxdingac.cn; Zhengdong Cai, E-mail: czd856163.com.