Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(7):1869-1878. doi:10.7150/thno.20524 This issue Cite
Research Paper
Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide
Adam Vandergriff1,2*, Ke Huang2*, Deliang Shen3 ![]() *, Shiqi Hu1,2, Michael Taylor Hensley2, Thomas G. Caranasos4, Li Qian5, Ke Cheng1,2
*, Shiqi Hu1,2, Michael Taylor Hensley2, Thomas G. Caranasos4, Li Qian5, Ke Cheng1,2 ![]()
1. Joint Department of Biomedical Engineering at University of North Carolina at Chapel Hill and North Carolina State University,
2. Department of Molecular and Biomedical Sciences and Comparative Medicine Institute, North Carolina State University, Raleigh, North Carolina
3. Department of Cardiovascular Medicine, First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
4. Department of Cardiothoracic Surgery, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina
5. Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina
* Equal Contributions
Received 2017-4-11; Accepted 2018-1-23; Published 2018-2-14
Abstract

Rationale: Cardiac stem cell-derived exosomes have been demonstrated to promote cardiac regeneration following myocardial infarction in preclinical studies. Recent studies have used intramyocardial injection in order to concentrate exosomes in the infarct. Though effective in a research setting, this method is not clinically appealing due to its invasive nature. We propose the use of a targeting peptide, cardiac homing peptide (CHP), to target intravenously-infused exosomes to the infarcted heart.
Methods: Exosomes were conjugated with CHP through a DOPE-NHS linker. Ex vivo targeting was analyzed by incubating organ sections with the CHP exosomes and analyzing with fluorescence microscopy. In vitro assays were performed on neonatal rat cardiomyocytes and H9C2 cells. For the animal study, we utilized an ischemia/reperfusion rat model. Animals were treated with either saline, scramble peptide exosomes, or CHP exosomes 24 h after surgery. Echocardiography was performed 4 h after surgery and 21 d after surgery. At 21 d, animals were sacrificed, and organs were collected for analysis.
Results: By conjugating the exosomes with CHP, we demonstrate increased retention of the exosomes within heart sections ex vivo and in vitro with neonatal rat cardiomyocytes. In vitro studies showed improved viability, reduced apoptosis and increased exosome uptake when using CHP-XOs. Using an animal model of ischemia/reperfusion injury, we measured the heart function, infarct size, cellular proliferation, and angiogenesis, with improved outcomes with the CHP exosomes.
Conclusions: Our results demonstrate a novel method for increasing delivery of for treatment of myocardial infarction. By targeting exosomes to the infarcted heart, there was a significant improvement in outcomes with reduced fibrosis and scar size, and increased cellular proliferation and angiogenesis.
Keywords: Targeting, exosomes, cardiac regeneration, heart disease, myocardial infarction
Introduction
Due to the limited regeneration of cardiomyocytes [1], stem cells have been the premier choice for promoting myocardial regeneration. Numerous clinical trials—such as APOLLO [2], C-Cure [3], CADUCEUS [4], and SCIPIO [5]—have demonstrated the cardiac regenerative potential of various stem cells. Subsequent studies demonstrated the lack of long term stem cell engraftment and differentiation [6-8]. The majority of the beneficial effects of stem cells come from the secretome [9], which includes extracellular microvesicles (EMVs). There are three known types of EMVs: apoptotic bodies, microvesicles, and exosomes. All are composed of a lipid bilayer, yet exosomes are differentiated by being formed within endosomes [10]. Acting as mediators of paracrine signals, exosomes protect and carry proteins and miRNA between cells [11-16].
Exosomes produced by cardiosphere-derived stem cells (CDCs) have been proven to induce myocardial regeneration via transportation of miRNA to the myocardium [17-19]. Despite the efficacy of exosomes, the methods for delivery to the heart are less than ideal. Previous studies have used both intracoronary and intramyocardial injections, with intramyocardial delivery being the more effective of the two [20]. While intramyocardial injections are acceptable in animal studies, in a clinical setting it is a much more serious procedure requiring a physician to perform the catheterization procedure [21]. Ideally, exosomes would be delivered intravenously, but it has been shown that the majority of intravenous injected exosomes are absorbed within the liver [22-24]. To offset the non-specific delivery, in our previous study [25] we utilized a dosage that was approximately ten times greater than what was used for intramyocardial studies [18] as shown in Table 1. In this study, we create infarct-targeting exosomes, through the use of a cardiac homing peptide (CHP) [26,27], to increase the efficacy and decrease the effective dose of intravenously delivered exosomes.
Comparison of exosome dosage administered in previous studies. * indicates weight was not listed in cited paper but was extrapolated from our own measurements of this strain of animal.
| Study | Delivery Method | Animal | Weight (kg) | Exosomes Injected | Dosage (XO/kg) | Relative Dosage |
|---|---|---|---|---|---|---|
| Vandergriff et al. 2015 [25] | IV | SCID Mice | 0.03 | 3.0×1010 | 1.0×1012 | 5000% |
| Ibrahim et al. 2014 [18] | IM | SCID Mice | 0.03* | 2.8×109 | 9.3×1010 | 467% |
| Gallet et al. 2016 [20] | IC | Yucatan Pig | 80† | 3.3×1012 | 4.1×1010 | 206% |
| Gallet et al. 2016 [20] | IM | Yucatan Pig | 80† | 1.65×1012 | 2.1×1010 | 103% |
| This study | IV | SD Rats | 0.3 | 6.0×109 | 2.0×1010 | 100% |
† indicates weight was not listed in paper but was acquired from other sources [79].
Methods
Exosome isolation
Exosomes were isolated as previously described [25,28] using ultrafiltration. Briefly, cells were grown to confluency in fetal bovine serum (FBS; Corning; Corning, NY)-supplemented Iscove's Modified Dulbecco's Medium (Gibco; Waltham, MA) then the media was changed to media without FBS. The media was conditioned on the cells for an extended period of time: 14 days for cardiosphere-derived cells (CDCs) [18,20], 5 days for HT1080 [29] (MilliporeSigma; St. Louis, MO). The HT1080-XOs were only used for the ex vivo targeting experiment; all other experiments used CDC-XOs. The conditioned media was filtered through a 0.22 μm sterilization filter to remove any cell particulates or apoptotic bodies, then concentrated and buffer exchanged to PBS using a 100 kDa ultrafiltration column (EMD Millipore; Billerica, MA). Sizes and concentrations of the isolated exosomes were ascertained using nanoparticle tracking analysis (NTA; NanoSight, Malvern, Worcestershire, United Kingdom). For electron microscopy, exosomes were stained using uranyl oxalate following a previously described protocol [30]. SDS-PAGE and Western blotting was performed using 5 μg of protein on 4-15% gradient gels (Bio-rad; Hercules, CA) and transferred using a wet transfer method.
Exosome tagging
The exosomes were tagged with a peptide shown to target the infarcted heart known as Cardiac Homing Peptide (CHP; CSTSMLKAC) or a Scramble peptide (Scr; CSKTALSMC) that is chemically identical but has a randomized internal sequence [26]. Both peptides were synthesized by GenScript (Piscataway, NJ). The peptide was conjugated to the exosomes using modifications of previously described methods [31]. DOPE-NHS (dioleoylphosphatidylethanolamine N-hydroxysuccinimide; COATSOME® FE-8181SU5, NOF America; White Plains, NY) and the peptide were combined with a 100-fold molar excess of peptide and allowed to react for 1 h to form the DOPE-peptide. The DOPE-peptide was then incubated with the exosomes with a lipid:exosome ratio of 6000:1 (i.e., 6000 molecules of DOPE-peptide for each exosome; see Supplementary Methods for rationale). An illustration of the conjugation reactions is shown in Figure 1A. Following labeling with the lipophilic dyes DiI (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindocarbocyanine Perchlorate; ThermoFisher Scientific; Waltham, MA) or DiR (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindotricarbocyanine Iodide; ThermoFisher Scientific; Waltham, MA), excess reagents were then removed through the use of a 100 kDa ultrafiltration spin filter and adjusted to the final concentration using PBS.
Study overview. (A) Myocardium-targeting exosomes were produced by reacting DOPE-NHS to Cardiac Homing Peptide (CHP). The lipophilic tails of the DOPE-CHP then spontaneously insert into the exosomal membrane, coating the exosome in CHP peptide. The exosomes were then intravenously injected into rats following I/R injury. (B) Western blot for PCNA verifies absence of cell particulates in purified exosomes and CD-81 shows presence of exosomes. (C) Nanoparticle tracking analysis shows that tagging the exosomes with CHP resulted in no significant changes in exosome size, with a modal exosome size of ~95 nm. (D) Transmission electron microscopy confirms the exosome structure. (E-F) Ex vivo labelling of infarcted rat heart sections showed increased retention of both CHP-tagged exosomes compared to Scr-tagged exosomes (DAPI in blue and DiI-labeled exosomes in red). Scale bars: D = 50 nm, E = 1 mm.
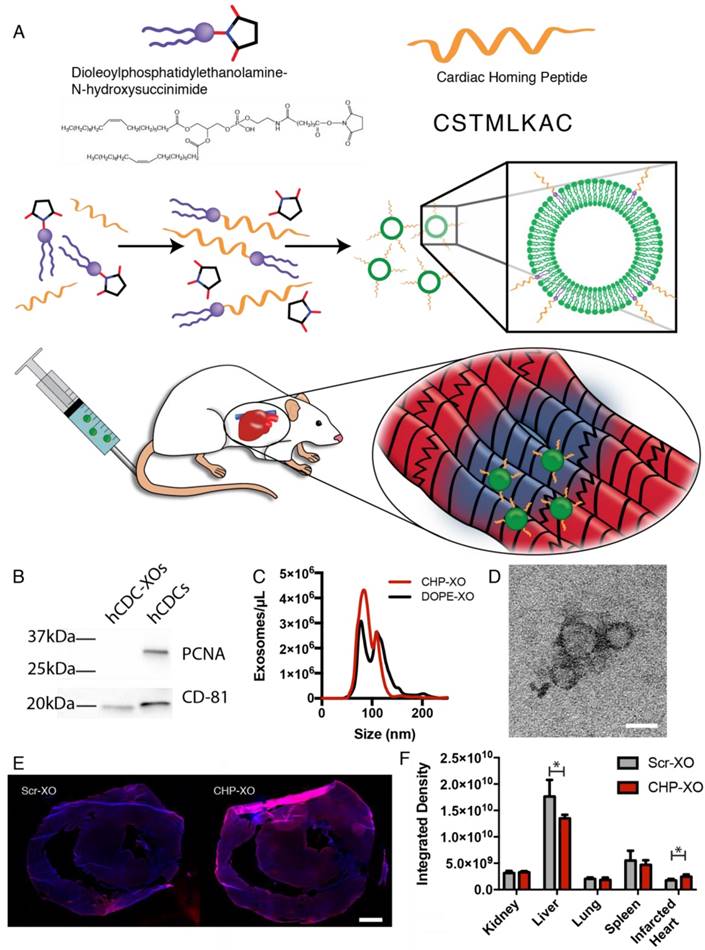
Ex vivo targeting studies
Exosomes derived from HT1080 cells were tagged with CHP peptide then labelled with DiI. Exosomes were incubated with fixed cryosections of rat organs for 1 h, followed by 3×5 min washes with PBS and DAPI labeling. To visualize the entire organs, images were stitched using ImageJ [32]. To avoid animal-to-animal variation on exosome binding, the infarcted heart sections and the other organs were acquired from a single rat for the ex vivo targeting assay. All images were acquired using the same exposure time to allow for the use of quantitative measurements.
In vitro study
Fresh neonatal rat cardiomyocytes (NRCMs) were isolated as previously described [33,34]. The cells were cultured for 2 days in 4-well culture slides. To stress the cells, serum-free medium containing either 2.50×108 CHP-XOs, Scr-XOs, or PBS was added, and cells were incubated for 24 h prior to fixation for analysis. Cells were stained for alpha-Sarcomeric Actinin (α-SA) to label the muscle fibers of the cardiomyocytes. To determine viability, cells that were positive for α-SA, contained DAPI-labelled nuclei, and were rounded up were counted as viable cells. To simulate ischemic injury, H9C2 cells (Sigma Aldrich; St. Louis, MO) were incubated with 500 μM H2O2 for 2 h followed by 24 h of treatment with either Scr-XOs or CHP-XOs. Cell apoptosis was assessed by TUNEL staining.
Animal housing and care
All procedures performed for this study were approved by the North Carolina State University Institutional Animal Care and Use Committee and were conducted within AAALAC International-accredited facilities. 5-7 week-old female SD rats (Charles River Laboratories; Worcester, MA) were utilized. All of the rats were clinically healthy prior to the study and were free of recognized pathogens (rat coronavirus, rat parvovirus, rat minute virus, Kilham rat virus, Toolan's H-1 virus, rat theilovirus, Sendai virus, pneumonia virus of mice, mycoplasma pulmonis, pneumocistis carinii). Rats were housed in static microisolator cages on direct-contact corn cob bedding (The Andersons, Inc; Maumee, OH). The room was maintained in controlled environmental conditions with a 12:12 h light:dark cycle and temperature between 68-72 °F. Rats were provided with standard rodent chow (Formula 5001; LabDiet, St. Louis, MO) ad libitum and provided continuous access to water using suspended water bottles.
Surgical procedures
Thirty female SD rats were anesthetized through intraperitoneal administration of 0.8-0.9 μL/g anesthetic combination of a 2:1 mixture of ketamine and xylazine. The depth of anesthesia was tested by performing a withdrawal reflex test of at least 2 toes, with absence of reflex confirming sufficient anesthesia. Under artificial ventilation with a rodent ventilator (SAR-1000 Small Animal Ventilator, CWE, Inc.; Ardmore, PA), an ischemia/reperfusion surgical operation was performed. The left coronary artery (LAD) was ligated using a 10 cm length of 7-0 silk suture through a 15 mm opening at the 4th intercostal space. A plain knot was tied and left in-situ for 30 min. Ischemia was confirmed in all rats by the appearance of discoloration of the heart surface. After 30 min, the ligature was released and reperfusion was verified by reddening of the previously discolored area of the heart muscle. The chest was closed under negative pressure. Rats were then extubated and observed for approximately 10 min until they were able to move.
Echocardiography
Echocardiograms were acquired at 4 h and 21 days after ischemia/reperfusion surgery with a Philips CX30 ultrasound system and L15-7io transducer (Philips; Amsterdam, Netherlands). The individual who performed the echocardiography and analysis was blinded to the treatment each animal received. Animals were anesthetized with 1.5% isoflurane during echocardiography. Ejection fraction was measured using Simpson's method using B-mode images to measure ventricular volume.
Administration of exosomes and animal randomization
Animals were transferred into cages following an observational period. The rats were treated with PBS (n=6), scramble peptide exosomes (Scr-XO; n=7), or CHP targeted exosomes (CHP-XO; n=7) via tail vein injection. In all three groups, the injection volume was 200 μL. In both the Scr-XO- and CHP-XO-treated groups, the treatments were 6×109 Scr-XO or CHP-XO in 200 μL PBS, respectively. Animals from each group were distributed equally between all cages. For randomization, the sequence of treatments was predetermined in a cyclical manner so as to keep group sizes the same. Animals were randomly drawn from cages and assigned to one of the three treatment groups. As predicted based on our previous studies, 6 animals died due to complications during or shortly after surgery. No animals died following delivery of treatment. Due to several deaths of animals assigned to one group, the predetermined sequence was adjusted towards the end of the study to rebalance the groups. The group sequence was determined by an individual whom was not present for surgery nor the echocardiography. For ex vivo imaging, 4 animals were sacrificed at 24 h following injection. The animal mortality rate was 20% for this study.
Histology and microscopy
All rat specimens were sacrificed at day 21. Hearts were excised, transversely bisected across the infarct area, and equilibrated with increasing sucrose solutions up to 30% overnight. Hearts were then embedded in Optimal Cutting Temperature compound (OCT; Tissue-Tek; Torrance, CA), snap-frozen in liquid nitrogen, and then cryosectioned with a thickness of 5 μm for Masson's Trichrome staining and 10 μm for haematoxylin and eosin staining (H&E) and immunohistochemistry (IHC). Masson's Trichrome staining was performed using HT15 Trichrome Staining (Masson) Kit (Sigma Aldrich; St. Louis, MO). H&E staining was performed using traditional methods and chemicals (Sigma-Aldrich; St. Louis, MO).
For immunohistochemistry (IHC), heart sections were then fixed in 4% paraformaldehyde solution, permeabilized with 0.01% saponin (Sigma-Aldrich, St. Louis, MO), and blocked using Protein Block solution (Dako; Glostrup, Denmark). Primary antibodies were incubated overnight at 4 °C, and subsequently secondary antibodies at room temperature for 1.5 h. Samples were then treated with DAPI (LifeTech, Carlsbad, CA) and mounted in Prolong Gold Mounting Media (LifeTech, Carlsbad, CA). Images were acquired using an Olympus IX81 fluorescence microscope and Zeiss LSM710 confocal microscope. Image processing and analysis was performed using ImageJ. For analyzing DiI uptake, nuclear staining was subtracted prior to measurement to avoid detection of Ki67 labelling.
Statistics
All figures display the mean with the standard deviation as the error bar. Statistical analysis was performed using GraphPad Prism (GraphPad Software, Inc.; La Jolla, CA). For all analyses, two-way ANOVA with Tukey's multiple comparison test was used to analyze the data. A p<0.05 was considered significant and is indicated on all figures with an asterisk. Technical replicates were acquired for each portion as follows. For echocardiograms, three technical replicates (image sequences) were acquired and analyzed. For microscopy, at least two sections were imaged for each specimen, and for high magnification images (not whole heart) at least three images were acquired and analyzed.
Results
Peptide labeling of exosomes increases retention to infarcted heart sections
To determine proper isolation of hCDC-XOs without cellular contamination, exosomes were characterized using both Western blotting, NTA, and TEM. Western blots confirmed exosome presence with the exosome marker CD81, and a lack of cellular contamination was verified using the nuclear protein PCNA (Figure 1B). Exosome size was confirmed to be 95 nm via NTA and TEM imaging confirmed the round morphology of exosomes (Figure 1C-D). It was not possible to directly determine the exosome labelling; NTA was not able to differentiate between labelled and unlabeled exosomes (Figure 1C). Instead we used indirect methods to test whether the addition of a targeting peptide would increase retention of exosomes. To do so, exosomes were incubated on slide sections of rat kidney, liver, lung, spleen, and infarcted heart. Exosomes were tracked and quantified by DiI labelling. It has been shown that exosomes demonstrate innate homing abilities [35]. Based upon the fluorescence integrated density taken from a series of images, CHP-tagged exosomes exhibited improved targeting to the infarcted heart (Figure 1E-F). In the other organs, the liver showed a decreased signal of CHP-XOs when compared to Scr-XOs. These data indicate that CHP conjugation is able to increase on-target and decreases off-target delivery of systemically administered exosomes.
CHP-XOs and Scr-XOs increase viability of NRCMs via increased uptake
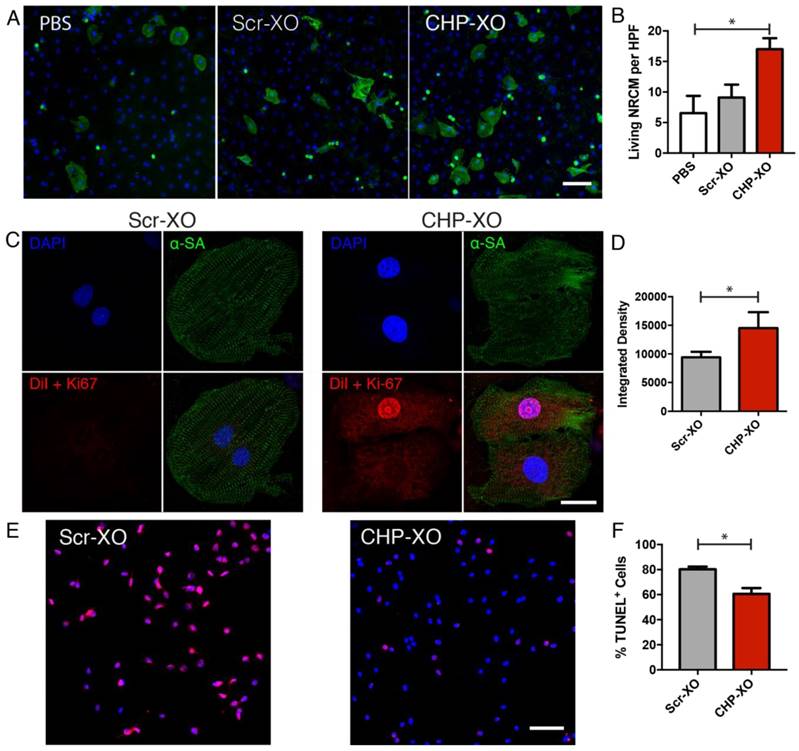
Prior to carrying out studies in an animal model, the targeted exosomes were tested on NRCMs cultured in vitro. In order to stress the NRCMs, the medium was switched to serum-free medium, and 2.50×108 exosomes were administered to each well of a 4-well culture slide. Overall viability of NRCMs were analyzed by counting viable α-sarcomeric actinin-positive cells. The results of counting indicated that the CHP-XO group had increased viable NRCMs compared to the PBS group (Figure 2A-B). Qualitative analysis of DiI-labelled exosomes showed uptake of exosomes whether tagged with the scramble or CHP peptides, though the CHP-tagged group had a significant increase in retention. This also confirmed that the CHP peptides were nontoxic to cardiomyocytes. Exposing H9C2 cardiac cells to a simulated infarction with hydrogen peroxide demonstrated that CHP-XOs reduced the number of apoptotic cells when compared to the Scr-XOs.
In vitro analysis of targeted exosomes. Following stress by serum starvation, treatment with CHP-XOs increased cardiomyocyte viability (A-B; n=3). CHP-XOs also demonstrated a significant increase in uptake in cardiomyocytes compared to Scr-XOs as shown with confocal microscopy (C-D; n=6). In H9C2 cardiac cells, treatment with CHP-XOs showed a significant reduction in TUNEL-positive cells (E-F). Scale bars A-E = 100 μm. Scale bar C = 20 μm.
CHP-XOs treatment increases cardiac function and reduces cardiac fibrosis following ischemia/reperfusion injury
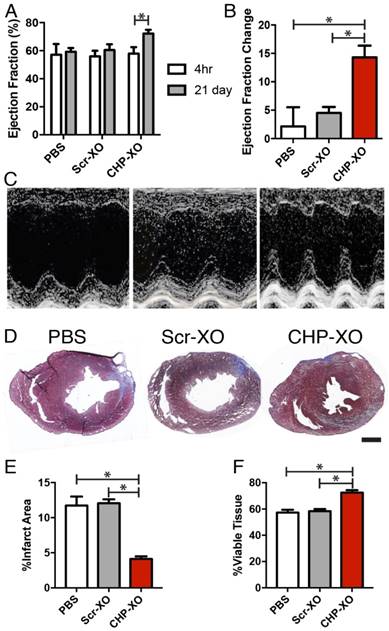
After a 4 h recovery period following the ischemia/reperfusion injury, the ejection fraction of all animals was indistinguishable in all groups, indicating a similar degree of initial injury. The animals were given 21 days for the treatments to take effect and repair the myocardial damage. Heart function was again measured by echocardiogram and tissues were collected for histological analysis. Echocardiography results demonstrated that CHP-XO treatment resulted in increased heart function over both other groups (Figure 3A-C). As cardiac fibrosis contributes to the reduction of cardiac function, Masson's Trichrome staining was used to measure the size of the fibrotic region. Upon measuring the images, injection of CHP-XOs led to a decrease in the infarcted tissue area and consequently an increase in viable tissue in the at-risk region (Figure 3D-F).
Heart function and morphology. Echocardiography results showed that after 21 days, animals receiving CHP-XOs showed an increase in ejection fraction much greater than both the PBS and Scr-XO groups (A-C; PBS n=6, Scr-XO and CHP-XO n=7). Data from graphs A-B are based upon measurements via Simpson's method using B-mode images. M-mode images in C are only for illustrative and qualitative purposes. Morphological analysis of the heart performed using Masson's Trichrome staining showed a significant reduction of fibrotic lesions (blue) in specimens from the CHP-XO group (D-F; n=3). Scale bar = 1 mm.
CHP-XOs improve cellular survival and vascular growth in myocardium
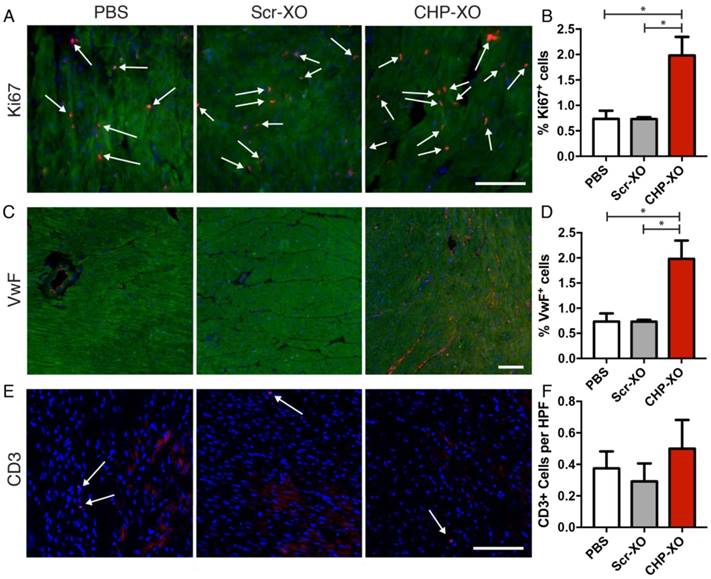
In order to determine the cellular response to the CHP-XO therapy, immunohistochemistry was performed with several targets. To distinguish between cardiomyocytes and other cells, α-SA was used as a co-stain for immunofluorescence. Imaging of Ki67 showed that there was increased cardiomyocyte proliferation following CHP-XO treatment (Figure 4A-B). Angiogenesis was measured by imaging of Von Willebrand factor (VwF) and showed significant increases with CHP-XO treatment (Figure 4C-D). As the exosomes were from human origin, negative effects by an immune response were possible. To test for this, T-cells were stained for CD3 and there was no difference between the groups (Figure 4E-F).
Myocardial proliferation and angiogenesis. Cardiomyocyte proliferation was measured using Ki67 (A; red, indicated with arrows), and angiogenesis was measured with VwF (B; red). Co-staining with α-SA (green) was used to differentiate myocardium from other cardiac tissues. When analyzed quantitatively, CHP-XOs promoted both cellular proliferation (B) and angiogenesis (D), both key factors of myocardial regeneration. Despite being of human origin, the exosomes did not illicit an immune response, as verified by CD3 staining for T-cells (E-F). Scale bars = 100 μm.
Discussion
After a long period of dormancy since the discovery of exosomes in the 1980's [36-40], researchers have begun to understand the value of exosomes and their importance in cell-to-cell signaling. Exosomes secreted by cardiosphere-derived stem cells have been shown to promote cardiac regeneration [18,20,41]. It has been indicated that the beneficial effects of exosomes are due to its ability to shuttle miRNA between cells [11-16]. CDC-XOs contain numerous miRNAs, in particular miR21 and miR146a [18], both of which have been shown to have beneficial effects on the injured myocardium [42-46]. miR21 reduces myocardial apoptosis by modulating expression of programmed cell death 4 (PDCD4), FasL, and AKT pathways [47-49]. miR146a represses interleukin-1 receptor-associated kinase1 (IRAK1) and tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), reducing activation of nuclear factor κΒ (NF-κΒ) [18,44]. The effects of the miRNAs do not appear to be the result of any single miRNA, but rather the effects are dependent upon multiple miRNAs working in tandem [18,49]. When combined, miR21 and miR146a result in significant reduction of p38 mitogen-associated kinase phosphorylation (p-p38 MAPK) [49]. As miRNAs do not require an exact match to the target mRNA, miRNAs can repress many different proteins [50-56]. When co-expressed or inhibited, this leads to multiple interactions and alterations as overlapping portions of pathways are modulated [50,57-59]. Of the many possible outcomes of the miRNA modulation, we demonstrate that CHP-XOs promote cardiac repair through reduced scar size (Figure 3D-E), increased cardiac proliferation (Figure 4A-B), and increased angiogenesis (Figure 4C-D).
The nano-scale size of exosomes has a dichotomous effect on the therapeutic potential of exosomes. Their small size allows exosomes to traverse capillary beds in the lungs and other tissues, which can trap cells or larger particles, but simultaneously reduces the rate of extravasation of the exosomes to the hypothesized target tissue [60]. When delivered intravenously, large amounts of exosomes have been shown to accumulate in off target organs such as the liver [22-24]. Thus far, no studies have definitively shown any side effects of excess exosome injection, though one study did report death of one animal when receiving large doses of intravenous exosomes, possibly due to pulmonary embolism [60]. Drug targeting has primarily been studied as a means to reduce the negative off-target effects of chemotherapeutic drugs [61-63], although there has been an increased interest in drug targeting for cardiac regeneration [64-73]. Following myocardial infarction, the myocardium undergoes several changes that can be exploited for targeting. One such change is an increase in vascular permeability, analogous to the enhanced permeability and retention (EPR) effect in cancer, allowing for increased extravasation of liposomes [74]. Changes in protein expression as well as exposure of normally intracellular proteins such as myosin have been utilized for targeting. Due to their high specificity, antibodies and antibody fragments have been the targeting molecule of choice [75]. Recent studies also showed that platelet membranes could be utilized to target cells to the infarct heart [76]. Recent developments in phage display have yielded many new targeting molecules such as the CHP [26,27]. Though the exact mechanism by which CHP interacts with the myocardium is unknown, by conjugating it to exosomes it amplifies the regenerative effects of the exosomes. A limitation of our study is that the mechanisms underlying the regenerative properties of exosomes were not fully elucidated. Nevertheless, recent studies from our group and others have demonstrated pro-angiogenic, pro-myogeneic, anti-apoptotic, and anti-inflammatory roles of stem cell secretome [77, 78].
Conclusions
Our results demonstrate an effective method for increasing the potency of CDC-XOs for cardiac regeneration through the use of Cardiac Homing Peptide. The use of DOPE-NHS allows for peptides or other molecules to be conjugated to exosomes and possibly other membrane-based nanoparticles. Targeting the exosomes resulted in increased uptake in cardiomyocytes in vitro, as well as increased functional recovery in an animal model by reducing fibrosis, inducing cardiomyocyte proliferation and increasing angiogenesis. The use of cardiac-targeted exosomes represents a promising method for the treatment of myocardial infarction.
Abbreviations
CHP: cardiac homing peptide; CDC: cardiosphere-derived cell; DOPE: dioleoylphosphatidylethanolamine; EMV: extracellular microvesicle; I/R: ischemia/reperfusion; NHS: N-hydroxysuccinimide; NRCM: neonatal rat cardiomyocyte; XO: exosome.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work is supported by National Natural Science Foundation of China (81370216), NSFC-Henan Talent Training Fund (U1404802), Health and Family Planning Commission of Henan Province scientific and technological innovation talents 51282 project - Leading Talent (100) and 5451 project (2015002), US National Institute of Health (HL123920 and HL137093), NC State University Chancellor's Faculty Excellence Program, NC State Chancellor's Innovation Fund, University of North Carolina General Assembly Research Opportunities Initiative grant, and American Heart Association (16PRE30130010).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J. Evidence for Cardiomyocyte Renewal in Humans. Science. 2009;324:98-102
2. Houtgraaf JH, Den Dekker WK, Van Dalen BM, Springeling T, De Jong R, Van Geuns RJ, Geleijnse ML, Fernandez-Aviles F, Zijlsta F, Serruys PW, Duckers HJ. First experience in humans using adipose tissue-derived regenerative cells in the treatment of patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2012;59:539-540
3. Bartunek J, Behfar A, Dolatabadi D, Vanderheyden M, Ostojic M, Dens J, El Nakadi B, Banovic M, Beleslin B, Vrolix M, Legrand V, Vrints C, Vanoverschelde JL, Crespo-Diaz R, Homsy C. et al. Cardiopoietic stem cell therapy in heart failure: The C-CURE (cardiopoietic stem cell therapy in heart failURE) multicenter randomized trial with lineage-specified biologics. J Am Coll Cardiol. 2013;61:2329-2338
4. Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LEJ, Berman D, Czer LSC, Marbán L, Mendizabal A, Johnston P V, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marbán E. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895-904
5. Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK. et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): Initial results of a randomised phase 1 trial. Lancet. 2011;378:1847-1857
6. Yau TM, Kim C, Ng D, Li G, Zhang Y, Weisel RD, Li RK. Increasing transplanted cell survival with cell-based angiogenic gene therapy. Ann Thorac Surg. 2005;80:1779-1786
7. Terrovitis J V, Smith RR, Marbán E. Assessment and optimization of cell engraftment after transplantation into the heart. Circ Res. 2010;106:479-494
8. Bonios M, Terrovitis J, Chang CY, Engles JM, Higuchi T, Lautamäki R, Yu J, Fox J, Pomper M, Wahl RL, Tsui BM, O'Rourke B, Bengel FM, Marbán E, Abraham MR. Myocardial substrate and route of administration determine acute cardiac retention and lung biodistribution of cardiosphere-derived cells. J Nucl Cardiol. 2011;18:443-450
9. Stastna M, Van Eyk JE. Investigating the secretome lessons about the cells that comprise the heart. Circ Cardiovasc Genet. 2012;5:o8-o18
10. Kowal J, Tkach M, Théry C. Biogenesis and secretion of exosomes Introduction: the discovery of exosomes. Curr Opin Cell Biol. 2014;29:116-125
11. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654-659
12. Simons M, Raposo G. Exosomes - vesicular carriers for intercellular communication. Curr Opin Cell Biol. 2009;21:575-581
13. Mittelbrunn M, Gutiérrez-Vázquez C, Villarroya-Beltri C, González S, Sánchez-Cabo F, González MÁ, Bernad A, Sánchez-Madrid F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2:282
14. Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, Paris L, Abalsamo L, Colone M, Molinari A, Podo F, Rivoltini L, Ramoni C, Fais S. Immune Surveillance Properties of Human NK Cell-Derived Exosomes. J Immunol. 2012;189:2833-2842
15. Yellon DM, Davidson SM. Exosomes: nanoparticles involved in cardioprotection? Circ Res. 2014;114:325-332
16. Vyas N, Dhawan J. Exosomes: mobile platforms for targeted and synergistic signaling across cell boundaries. Cell Mol Life Sci. 2017;74:1567-1576
17. Gray WD, French KM, Ghosh-Choudhary S, Maxwell JT, Brown ME, Platt MO, Searles CD, Davis ME. Identification of therapeutic covariant microRNA clusters in hypoxia-treated cardiac progenitor cell exosomes using systems biology. Circ Res. 2015;116:255-263
18. Ibrahim AGE, Cheng K, Marbán E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports. 2014;2:606-619
19. Chen L, Wang Y, Pan Y, Zhang L, Shen C, Qin G, Ashraf M, Weintraub N, Ma G, Tang Y. Cardiac progenitor-derived exosomes protect ischemic myocardium from acute ischemia/reperfusion injury. Biochem Biophys Res Commun. 2013;431:566-571
20. Gallet R, Dawkins J, Valle J, Simsolo E, de Couto G, Middleton R, Tseliou E, Luthringer D, Kreke M, Smith RR, Marbán L, Ghaleh B, Marbán E. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J. 2016;38:201-211
21. Sherman W, Martens TP, Viles-Gonzalez JF, Siminiak T. Catheter-based delivery of cells to the heart. Nat Clin Pract Cardiovasc Med. 2006;3(Suppl 1):S57-64
22. Takahashi Y, Nishikawa M, Shinotsuka H, Matsui Y, Ohara S, Imai T, Takakura Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol. 2013;165:77-84
23. Bala S, Csak T, Momen-Heravi F, Lippai D, Kodys K, Catalano D, Satishchandran A, Ambros V, Szabo G. Biodistribution and function of extracellular miRNA-155 in mice. Sci Rep. 2015;5:10721
24. Tseliou E, Fouad J, Reich H, Slipczuk L, Couto G de, Aminzadeh M, Middleton R, Valle J, Weixin L, Marbán E. Exosomes from cardiac stem cells amplify their own bioactivity by converting fibroblasts to therapeutic cells. J Am Coll Cardiol. 2015;66:599-11
25. Vandergriff AC, de Andrade JBM, Tang J, Hensley MT, Piedrahita JA, Caranasos TG, Cheng K. Intravenous Cardiac Stem Cell-Derived Exosomes Ameliorate Cardiac Dysfunction in Doxorubicin Induced Dilated Cardiomyopathy. Stem Cells Int. 2015:960926
26. Kanki S, Jaalouk DE, Lee S, Yu AYC, Gannon J, Lee RT. Identification of targeting peptides for ischemic myocardium by in vivo phage display. J Mol Cell Cardiol. 2011;50:841-848
27. Won YW, McGinn AN, Lee M, Bull DA, Kim SW. Targeted gene delivery to ischemic myocardium by homing peptide-guided polymeric carrier. Mol Pharm. 2013;10:378-385
28. Cappione A, Gutierrez S, Mabuchi M, Smith J, Strug I, Nadler T. A Centrifugal Ultrafiltration-Based Method for Enrichment of Microvesicles. Danvers, MA. 2014
29. Shtam TA, Kovalev RA, Varfolomeeva EY, Makarov EM, Kil Y V, Filatov M V. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun Signal. 2013:11
30. Théry C, Aled C, Sebastian A, Graça R. Isolation and Characterization of Exosomes from Cell Culture Supernatants. Curr Protoc Cell Biol. 2006;3:1-29
31. Kajimoto T, Okada T, Miya S, Zhang L, Nakamura S. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat Commun. 2013;4:2712
32. Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463-1465
33. Vandergriff AC, Hensley MT, Cheng K. Isolation and Cryopreservation of Neonatal Rat Cardiomyocytes. J Vis Exp. 2015:1-7
34. Vandergriff AC, Hensley MT, Cheng K. Cryopreservation of Neonatal Cardiomyocytes. Methods Mol Biol. 2015;1299:153-160
35. Lai RC, Yeo RWY, Tan KH, Lim SK. Exosomes for drug delivery - a novel application for the mesenchymal stem cell. Biotechnol Adv. 2013;31:543-551
36. Pan BT, Johnstone RM. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell. 1983;33:967-978
37. Harding C, Stahl P. Transferrin recycling in reticulocytes: pH and iron are important determinants of ligand binding and processing. Biochem Biophys Res Commun. 1983;113:650-658
38. Pan B-T, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol. 1985;101:942-948
39. Johnstone RM. Revisiting the road to the discovery of exosomes. Blood Cells, Mol Dis. 2005;34:214-219
40. Harding C V, Heuser JE, Stahl PD. Exosomes: Looking back three decades and into the future. J Cell Biol. 2013;200:367-371
41. Barile L, Lionetti V, Cervio E, Matteucci M, Gherghiceanu M, Popescu LM, Torre T, Siclari F, Moccetti T, Vassalli G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res. 2014;103:530-541
42. Zhu H, Fan G-C. Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am J Cardiovasc Dis. 2011;1:138-149
43. Eulalio A, Mano M, Ferro MD, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376-381
44. Wang X, Ha T, Liu L, Zou J, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C. Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2013;97:432-442
45. Palomer X, Capdevila-Busquets E, Botteri G, Davidson MM, Rodríguez C, Martínez-González J, Vidal F, Barroso E, Chan TO, Feldman AM, Vázquez-Carrera M. miR-146a targets Fos expression in human cardiac cells. Dis Model Mech. 2015;8:1081-1091
46. Wang J, Liew OW, Richards AM, Chen YT. Overview of microRNAs in cardiac hypertrophy, fibrosis, and apoptosis. Int J Mol Sci. 2016;17:1-21
47. Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, Abdellatif M. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of fas ligand. J Biol Chem. 2010;285:20281-20290
48. Tu Y, Wan L, Fan Y, Wang K, Bu L, Huang T, Cheng Z, Shen B. Ischemic Postconditioning-Mediated miRNA-21 Protects against Cardiac ischemia/reperfusion Injury via PTEN/Akt Pathway. PLoS One. 2013:8
49. Huang W, Tian SS, Hang PZ, Sun C, Guo J, Du ZM. Combination of microRNA-21 and microRNA-146a Attenuates Cardiac Dysfunction and Apoptosis During Acute Myocardial Infarction in Mice. Mol Ther Acids. 2016;5:e296
50. Kasinski a L, Kelnar K, Stahlhut C, Orellana E, Zhao J, Shimer E, Dysart S, Chen X, Bader AG, Slack FJ. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene. 2014;34:1-9
51. Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell. 2009;136:215-233
52. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15-20
53. Lewis BP, Shih I-H, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of Mammalian MicroRNA Targets. Cell. 2003;115:787-798
54. Farh KK-H, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The Widespread Impact of Mammalian MicroRNAs on mRNA Repression and Evolution. Science. 2005;310:1817-1822
55. Grimson A, Farh KKH, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol Cell. 2007;27:91-105
56. Mazière P, Enright AJ. Prediction of microRNA targets. Drug Discov Today. 2007;12:452-458
57. Dong CG, Wu WKK, Feng SY, Wang XJ, Shao JF, Qiao J. Co-inhibition of microRNA-10b and microRNA-21 exerts synergistic inhibition on the proliferation and invasion of human glioma cells. Int J Oncol. 2012;41:1005-1012
58. Noguchi S, Yasui Y, Iwasaki J, Kumazaki M, Yamada N, Naito S, Akao Y. Replacement treatment with microRNA-143 and -145 induces synergistic inhibition of the growth of human bladder cancer cells by regulating PI3K/Akt and MAPK signaling pathways. Cancer Lett. 2013;328:353-361
59. Pisano F, Altomare C, Cervio E, Barile L, Rocchetti M, Ciuffreda MC, Malpasso G, Copes F, Mura M, Danieli P, Viarengo G, Zaza A, Gnecchi M. Combination of miRNA499 and miRNA133 exerts a synergic effect on cardiac differentiation. Stem Cells. 2015;33:1187-1199
60. Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015;199:145-155
61. Yang T, Choi M-K, Cui F-D, Kim JS, Chung S-J, Shim C-K, Kim D-D. Preparation and evaluation of paclitaxel-loaded PEGylated immunoliposome. J Control Release. 2007;120:169-177
62. Kang D Il, Lee S, Lee JT, Sung BJ, Yoon J-Y, Kim J-K, Chung J, Lim S-J. Preparation and in vitro evaluation of anti-VCAM-1-Fab'-conjugated liposomes for the targeted delivery of the poorly water-soluble drug celecoxib. J Microencapsul. 2011;28:220-227
63. She W, Li N, Luo K, Guo C, Wang G, Geng Y, Gu Z. Dendronized heparin-doxorubicin conjugate based nanoparticle as pH-responsive drug delivery system for cancer therapy. Biomaterials. 2013;34:2252-2264
64. Klibanov AL, Khaw B-A, Nossiff N, O'Donnell SM, Huang L, Slinkin MA, Torchilin VP. Targeting of macromolecular carriers and liposomes by antibodies to myosin heavy chain. Am J Physiol. 1991;261:60-65
65. Khaw B-A, Torchilin V, Vural I, Narula J. Plug and seal: Prevention of hypoxic cardiocyte death by sealing membrane lesions with antimyosin-liposomes. Nat Med. 1995;1:1195-1198
66. Khaw B-A, Khudairi T. Dose-response to cytoskeletal-antigen specific immunoliposome therapy for preservation of myocardial viability and function in langendorff instrumented rat hearts. J Liposome Res. 2007;17:63-77
67. Khaw B-A, DaSilva J, Hartner WC. Cytoskeletal-antigen specific immunoliposome-targeted in vivo preservation of myocardial viability. J Control Release Off J Control Release Soc. 2007;120:35-40
68. Ko YT, Hartner WC, Kale A, Torchilin VP. Gene delivery into ischemic myocardium by double-targeted lipoplexes with anti-myosin antibody and TAT peptide. Gene Ther. 2009;16:52-59
69. Scott RC, Crabbe D, Krynska B, Ansari R, Kiani MF. Aiming for the heart: targeted delivery of drugs to diseased cardiac tissue. Expert Opin Drug Deliv. 2008;5:459-470
70. Dvir T, Bauer M, Schroeder A, Tsui JH, Anderson DG, Langer R, Liao R, Kohane DS. Nanoparticles for targeting the infarcted heart. Nano Lett. 2011;11:4411-4414
71. Zhang B, Green J V, Murthy SK, Radisic M. Label-free enrichment of functional cardiomyocytes using microfluidic deterministic lateral flow displacement. PLoS One. 2012;7:e37619
72. Nguyen MM, Carlini AS, Chien MP, Sonnenberg S, Luo C, Braden RL, Osborn KG, Li Y, Gianneschi NC, Christman KL. Enzyme-Responsive Nanoparticles for Targeted Accumulation and Prolonged Retention in Heart Tissue after Myocardial Infarction. Adv Mater. 2015;27:5547-5552
73. Kamps JA, Krenning G. Micromanaging cardiac regeneration: Targeted delivery of microRNAs for cardiac repair and regeneration. World J Cardiol. 2016;8:163
74. Weis SM. Vascular permeability in cardiovascular disease and cancer. Curr Opin Hematol. 2008;15:243-249
75. Liu M, Li M, Sun S, Li B, Du D, Sun J, Cao F, Li H, Jia F, Wang T, Chang N, Yu H, Wang Q, Peng H. The use of antibody modified liposomes loaded with AMO-1 to deliver oligonucleotides to ischemic myocardium for arrhythmia therapy. Biomaterials. 2014;35:3697-3707
76. Tang J, Su T, Huang K, Dinh PU, Wang Z, Vandergriff AC, Hensley MT, Cores J, Allen T, Li TS, Sproul E, Mihalko E, Lobo LJ, Ruterbories L, Lynch A, Brown AC, Caranasos TG, Shen D, Stouffer GA, Gu Z, Zhang J, Cheng K. Targeted repair of heart injury by stem cells fused with platelet nanovesicles. Nature Biomedical Engineering. 2018;2:17-26
77. Tang J, Shen D, Caranasos TG, Wang Z, Allen TA, Vandergriff A, Hensley MT, Dinh PU, Cores J, Li TS, Zhang J, Kan Q, Cheng K. Therapeutic microparticles functionalized with biomimetic cardiac stem cell membranes and secretome. Nature Communications. 2017;8:13724
78. Luo L, Tang J, Nishi K, Yan C, Dinh PU, Cores J, Kudo T, Zhang J, Li TS, Cheng K. Fabrication of synthetic mesenchymal stem cells for hear repair. Circulation Research. 2017;120:1768-1775
79. Kim H, Song KD, Kim HJ, Park WC, Kim J, Lee T, Shin DH, Kwak W, Kwon YJ, Sung S, Moon S, Lee KT, Kim N, Hong JK, Eo KY. et al. Exploring the genetic signature of body size in Yucatan miniature pig. PLoS One. 2015;10:1-16
Author contact
![]() Corresponding author: Ke Cheng: ke_chengedu or Deliang Shen: shendeliang1com
Corresponding author: Ke Cheng: ke_chengedu or Deliang Shen: shendeliang1com