Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(14):3737-3750. doi:10.7150/thno.25487 This issue Cite
Research Paper
Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma
Chuan Xing Wu1 ![]() , Xiao Qi Wang1,2
, Xiao Qi Wang1,2 ![]() , Siu Ho Chok1, Kwan Man1, Simon Hing Yin Tsang1, Albert Chi Yan Chan1, Ka Wing Ma1, Wei Xia1, Tan To Cheung1
, Siu Ho Chok1, Kwan Man1, Simon Hing Yin Tsang1, Albert Chi Yan Chan1, Ka Wing Ma1, Wei Xia1, Tan To Cheung1
1. Department of Surgery, The University of Hong Kong, Hong Kong.
2. Department of Medicine, The University of Hong Kong, Hong Kong.
Received 2018-2-10; Accepted 2018-5-20; Published 2018-6-13
Abstract

Rationale: Hepatocellular carcinoma (HCC) is an aggressive malignant solid tumor wherein CDK1/PDK1/β-Catenin is activated, suggesting that inhibition of this pathway may have therapeutic potential.
Methods: CDK1 overexpression and clinicopathological parameters were analyzed. HCC patient-derived xenograft (PDX) tumor models were treated with RO3306 (4 mg/kg) or sorafenib (30 mg/kg), alone or in combination. The relevant signaling of CDK1/PDK1/β-Catenin was measured by western blot. Silencing of CDK1 with shRNA and corresponding inhibitors was performed for mechanism and functional studies.
Results: We found that CDK1 was frequently augmented in up to 46% (18/39) of HCC tissues, which was significantly associated with poor overall survival (p=0.008). CDK1 inhibitor RO3306 in combination with sorafenib treatment significantly decreased tumor growth in PDX tumor models. Furthermore, the combinatorial treatment could overcome sorafenib resistance in the HCC case #10 PDX model. Western blot results demonstrated the combined administration resulted in synergistic down-regulation of CDK1, PDK1 and β-Catenin as well as concurrent decreases of pluripotency proteins Oct4, Sox2 and Nanog. Decreased CDK1/PDK1/β-Catenin was associated with suppression of epithelial mesenchymal transition (EMT). In addition, a low dose of RO3306 and sorafenib combination could inhibit 97H CSC growth via decreasing the S phase and promoting cells to enter into a Sub-G1 phase. Mechanistic and functional studies silencing CDK1 with shRNA and RO3306 combined with sorafenib abolished oncogenic function via downregulating CDK1, with downstream PDK1 and β-Catenin inactivation.
Conclusion: Anti-CDK1 treatment can boost sorafenib antitumor responses in PDX tumor models, providing a rational combined treatment to increase sorafenib efficacy in the clinic.
Keywords: cancer stem cells, hepatocellular carcinoma, PDX models, CDK1 inhibitor, RO3306, sorafenib
Introduction
Hepatocellular carcinoma (HCC) is one of the most prevalent and common malignant tumors globally. It is reported that only 10~20% of patients can have surgery when the tumors are found [1-3]. In terms of etiology, it is associated with HBV, HCV, alcohol, aflatoxin and hemochromatosis. Moreover, autoimmune hepatitis, obesity and diabetes were also recently confirmed to be related to the development of HCC [4-6]. Inactivation of tumor suppressor genes and overexpression of oncogenes contribute to HCC tumorigenesis and progression, as do genetic and epigenetic mutations [7, 8]. Although a large number of gene alterations have been identified in the past decades, no main driving genes could be universally targeted, which may be due to frequent alterations of the cancer cells following Darwinian laws [9].
Tumor heterogeneity is the main obstacle for various cancer therapies. Cancer stem cells, microenvironment alterations, and epigenetic and genetic mutations all can contribute to cancer heterogeneity [10-13]. CSCs lead to tumor heterogeneity due to their capability of differentiating into heterogenous cancer cells. The inappropriate government of the cell cycle may contribute to the generation of CSCs, which currently are thought to be partially responsible for tumorigenicity and tumor relapse [14]. Furthermore, dysfunctional cell cycle regulation, such as the expression of cyclin-dependent kinases (CDKs) in CSCs, may promote tumorigenesis and the failure of traditional chemotherapeutics to eradicate solid cancer. Hence, CDK inhibitors may be suitable candidates for HCC therapy, and some have entered clinical trials [15-18]. In this study, we found that HCC expresses high levels of the cyclin-dependent kinase CDK1, which is associated with poor overall survival. Previous findings suggested that CDK1 governed the phosphorylation of factor 4E-binding protein 1 (4E-BP1), which is important in tumorigenesis [19]. CDK1 interaction with pluripotency transcription factor Oct4 (octamer-binding transcripttion factor 4) plays an important role in embryonic stem cell stemness [20, 21]. However, targeted CDK inhibitors in a defined cancer stem cell population have not yet been elucidated.
We explored the CDK1 inhibitor RO3306 alone or in combination with sorafenib to see the efficacy of treatment in patient-derived xenograft (PDX) tumor models. PDX models have unique advantages for conducting preclinical trials as they mostly mimic molecular, histologic, and genomic characteristics of the primary tumor. This tendency reveals an increased acceptance for its predictive value and a high degree of representation of the clinical outcome, which could increase the overall success of transitioning to a new clinically approved treatment and delivery to patients [22-25]. They are a precious approach to integrating preclinical and clinical drug development [26, 27]. A CDK1 inhibitor (RO3306) in combination with sorafenib acts on hepatocellular carcinoma with a synergistic antitumor growth effect on PDX tumor models, which may be due to its effects on decreasing the liver cancer stem cell stemness via the CDK1/PDK1/β-Catenin signaling pathway. The anticancer effect was confirmed in CSC-derived orthotopic tumor models. Furthermore, with the down-regulation of β-Catenin, EMT was also suppressed, which shows anti-metastasis potential.
Methods
HCC specimens and cell lines
The collection and application of fresh tumor tissues was approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority of Hong Kong (UW05-3597/I022). Specimens were collected from three HCC patients who had not undergone any radiation or chemotherapy before liver resection. HCC tissues and normal liver samples (N21 and N23) were acquired from the Tissue Bank at the Department of Surgery at Queen Mary Hospital. The human HCC cell line MHCC97H (97H) was from the Cell bank of Shanghai, Chinese Academy of Sciences, and the normal hepatocyte cell line LO2 from ATCC was cultured in 10% FBS in DMEM:F12 (Life Technologies). Additionally, 97H liver cancer stem cells were isolated from 97H monolayer cells through the single sphere formation method. The second and third generation of 97H spheres from the dissected single sphere cells were used in our experiments [28]. The 97 liver CSCs were cultured in DMEM:F12 sphere medium without FBS.
Generation of PDX tumor models, CSC-derived orthotopic tumors and drug treatment
The fresh tumor tissues from Queen Mary Hospital were minced into a size of 1 mm × 1 mm. The tissues were immediately transplanted into the flank of NOD-SCID mice subcutaneously. This was considered the PDX F0 tumor. Once the F0 tumor volume reached ~2 cm3, we propagated another generation of NOD-SCID mice or nude mice for the experiment (F1). A CSC-derived orthotopic tumor model and lung metastasis model were generated following the procedures reported previously [28]. F1 tumor-bearing mice were randomly and blindly divided into four groups: (1) vehicle; (2) RO3306 4 mg/kg every 2 days, oral gavage administration; (3) sorafenib 30 mg/kg, twice a day, oral gavage administration; and (4) combinatorial treatment RO3306 4 mg/kg plus sorafenib 30 mg/kg. When most F1 tumors were larger than 20 mm3, we started the treatments and the tumor volumes (length × width2 / 2) and body weights were monitored approximately 3 times per week. The total treatment lasted for two weeks' time, and the mice were killed after another 2 weeks.
Immunoblots and coimmunoprecipitation (Co-IP)
Cell pellets and tumor tissue (50-75 mg) were put in protein lysis buffer (20 mM Tris pH 7.4, 250 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF) for 30 min. The protein concentration measurement followed the standard procedure of the PierceTM BCA Protein Assay Kit (Thermo Scientific). The cytoplasmic and nuclear protein were extracted by ReadyPrep Protein Extraction kit (Bio-Rad Laboratories. Inc.CA). The relevant proteins were transferred to the PVDF membrane and incubated with primary antibody against β-actin (Sigma-Aldrich, MO), CDK1, PDK1, β-Catenin, phospho-Akt (T308, S473), total Akt, phospho-Stat3 (T705, S727), total Stat3, Nanog, Oct4, Sox2, phospho-GSK3 β (S9) and cleaved Notch1 (all from Cell Signaling Technology), and E-cadherin and Snail1 (Santa Cruz Biotechnology). The high or low-level protein expressions were measured by ImageJ software, which has been described in our previous article [28, 29]. We defined CDK1/ PDK1-related HCC cases as cases where the ratio of the two proteins (CDK1 vs. PDK1) was 0.5 to 1 and HCC cases unrelated to CDK1/PDK1 as cases where the ratio was less than 0.5 or larger than 1 [28]. For protein interaction using primary antibodies against CDK1 and PDK1, 150 µg of protein was incubated with 10 µL of protein G sepharose bead slurry (Amersham Biosciences, NJ) at 4 ℃, rotating for 30 min. The supernatant was kept and primary anti-CDK1 antibody was added and incubated with gentle rocking overnight in a cold room. 40 µL protein G sepharose bead slurry was added and incubated with rotation for 3 h at 4 ℃. Finally, the immune-complexes were analyzed by immunoblotting using anti-PDK1 antibody.
RNA extraction and qPCR
Total RNA extraction rigidly followed the protocol of the RNeasy kit (Qiagen). gDNA treatment containing 300-500 µg RNA, 10x RO DNase buffer (1.8 µL), DNase-free RO (0.8 µL), and RNA-free water to a total volume of 18 µL was incubated at 37 ℃ for 10 min, followed by the addition of 1.8 µL of RO DNase stop solution, and then incubated at 65 ℃ for another 10 min. The reverse-transcription followed the procedure in the Transcriptor First Strand cDNA Synthesis Kit (Roche):10x buffer (3 µL), universal primer (2 µL), dNTP (2 µL), inhibitor (0.8 µL), RTase (1.2 µL), and RNA-free water (3 µL), with the procedure as follows: 25 ℃ for 10 min, 37 ℃ for 2 h, 85 ℃ for 5 min, store at 4 ℃. Quantitative PCR was performed using the Selected SYBR Green master mix (Life Technologies) on an ABI 7900HT Detection System. cDNA (1 µL), SYBR Green master mix (7.5 µL), target primer (2.5 µL), and RNA-free water was added to a total volume of 15 µL. The assay was performed on an ABI 7900HT Detection System with the following procedure: stage 1, 50 ℃ 2 min, 95 ℃ 10 min; stage 2, (95 ℃ 0.15 min, 60 ℃ 1 min) ×40 cycles; and stage 3, 95 ℃ 0.15 min, 60 ℃ 1 min. The primers are listed in Table S1.
H&E staining, IHC staining, PI staining and flow cytometry assay
Sections of approximately 4-6 µm were cut and mounted on a pre-coated glass slide, following the procedure we reported previously [28]. Fixed cells were incubated in 1 mL 70% ethanol at -20 ℃ for at least 1 h. The cells were centrifuged at 500 ×g for 5 min, and suspended in 500-1000 µL PI staining buffer containing PBS, 50 µg/mL PI and 100 µg/mL RNAse A. For the CSC marker measurements, the cells were labeled with antibodies against CD133-PE, CD90-APC, EpCAM-PE-Cy7 (eBioscience), and the corresponding isotype antibody as a control to exclude nonspecific background staining. They were then incubated in the dark for 20-30 min and analyzed by FACSCalibur (Becton Dickinson, San Jos, CA).
Sphere formation and tumorigenicity assays
The CSCs were seeded in 96 well plates at one cell per well for single sphere formation assay to measure the self-renewal ability. The number of CSCs was maintained around 600-800 per well in 96 plates for the overall colony formation assay. The tumor cells were pretreated with a low dose of RO3306 (2 mM) and sorafenib (2.5 mM) for 2 days, and then implanted into mice subcutaneously to trace the tumor growth by luciferase [28].
Lentivirus transduction to knockdown CDK1
To study the function of CDK1, the plasmids expressing shRNAs specifically targeting human CDK1 (shCDC2 and shCDC22) were built using the pEco-Lenti-H1-shRNA (GFP) kit (GenTarget Inc., San Diego, CA, USA). The oligos for shRNA were constructed by Life Technologies RNAi Designer and the Gene Link shRNA design program. The scramble control shRNA cloned in the vector was used as a control. These are listed in Table S1.
Statistical analysis
Statistical analysis among the different groups was conducted using SPSS 21 (IBM Corp., Armonk, NY, USA). All the data are presented as the mean ± SD of three independent experiments. Pearson correlation analysis was conducted to measure the correlation between the protein levels (calculated by ImageJ) in clinical tissues [28, 29]. Student's paired and independent t-tests were used. The Pearson's chi-square test was used to calculate the correlation of CDK1 related or unrelated to other proteins with clinicopathologic parameters. The Kaplan-Meier method and log-rank test were applied for the survival analysis. Univariable and multivariable analysis, and Cox hazard regression tests were conducted to analyze the independent prognostic factors. P<0.05 was considered statistically significant.
Results
The potential associated expression of CDK1/PDK1/β-Catenin and its clinical significance in HCC
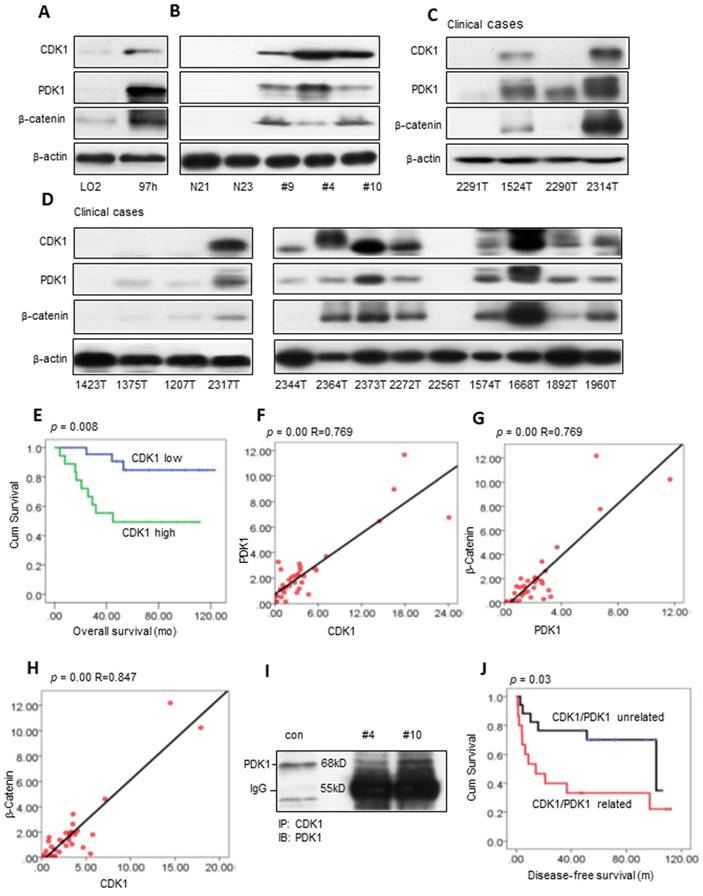
We evaluated CDK1, PDK1 and β-Catenin activity in cell lines and fresh human HCC tissues (Figure 1A-B). CDK1 was high in the 97H cells and liver cancers compared with the immortalized liver cell LO2 cells and normal human liver tissues. The high expression level of CDK1 correlates with increased PDK1 and β-Catenin. Western blot analysis showed that 46% (18/39) of clinical cases had a high expression level of CDK1 analyzed by ImageJ [28, 29] (Figure 1C-D and Figure S1A-C). What's more, the high activity level of CDK1 was associated with poor overall survival (log-rank=7.094, p=0.008, Figure 1E), which is consistent with the findings in ovarian cancer [30]. Clinically, the high activity level of CDK1 was significantly associated with one- and five-year tumor recurrence (p=0.013 and p=0.017, Pearson chi-square test; Table S2). Furthermore, there exists a correlation between CDK1 and PDK1 (p=0.00, R=0.769, linear regression analysis; 92.3% 36/39 cases), PDK1 and β-Catenin (p=0.00, R=0.769, linear regression analysis; 89.7% 35/39 cases), and CDK1 and β-Catenin (p=0.00, R=0.847, linear regression analysis; 87.2% 34 / 39 cases) (Figure 1F-H). Co-IP confirmed the interaction between CDK1 and PDK1 (Figure 1I), but there was no detectable association between PDK1 and β-Catenin or CDK1 and β-Catenin (data not shown). PDK1 is expressed in many cancers and plays an important role in tumor metastasis [31-34]. The CDK1 and PDK1 association revealed a poor disease-free survival compared with the CDK1 and PDK1 unrelated cases (log-rank=4.719, p=0.03, Figure 1J). CDK1 and PDK1 relation is associated with a five-year recurrence (p=0.025, Pearson chi-square test; Table S3). The univariate analysis displayed that CDK1 (high or low) or PDK1 (high or low) are the factors associated with overall survival. CDK1 related to PDK1 is a significant independent prognostic factor for disease-free survival based on multivariate analysis (p=0.009, Table S4), consistent with the Kaplan-Meier analysis (Figure 1J).
Potential clinical association of CDK1, PDK1 and β-catenin in HCC. (A-B) Western blot measurements of CDK1, PDK1 and β-catenin in the immortalized hepatocyte cell line LO2 and 97H liver cancer cell line, normal human liver tissue (N21, N23) and fresh HCC samples. (C-D) The expression levels of CDK1, PDK1 and β-catenin in clinical HCC tumors. (E) The overall survival of CDK1 high compared with CDK1 low HCC patients. (F-H) The correlation of CDK1 and PDK1, PDK1 and β-catenin, CDK1 and β-catenin. (I) The interaction between CDK1 and PDK1 detected by Co-IP. (J) The disease-free survival of the CDK1/PDK1 unrelated group compared with the CDK1/PDK1 related group. p < 0.05 was considered a significant difference.
The efficacy of sorafenib and RO3306 on PDX models
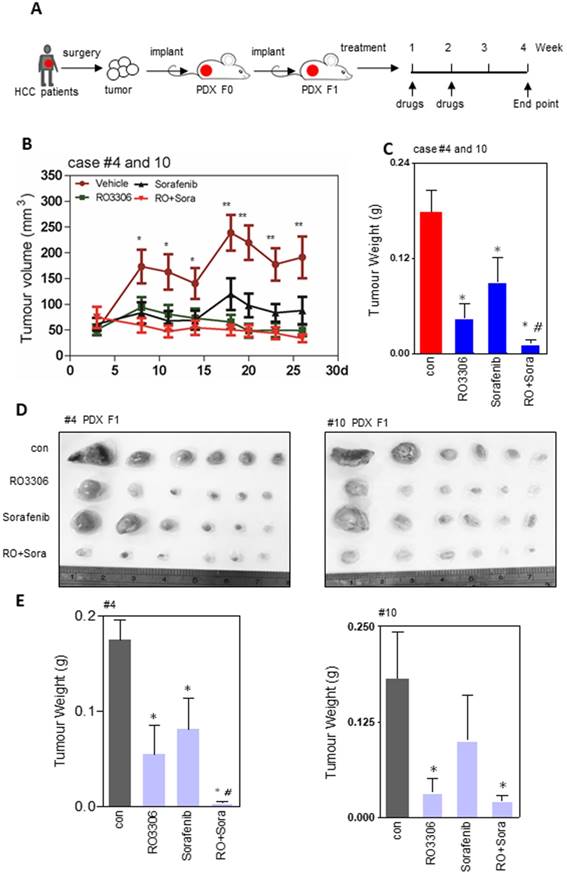
The PDX tumor models mostly resemble their original tumor, which is in line with our western blot and immunohistochemistry staining (Figure S1D-G). The activities of CDK1, PDK1 and β-Catenin are specific similar between the clinical tissues and paired PDX models (Figure S1E). To explore the direct effect of CDK1 inhibitor alone or combined with sorafenib, we applied them to F1 generation PDX tumor models in vivo (Figure 2A). The results showed that tumor growth was suppressed by 75%, 49% and 92% in RO3306, sorafenib, and combinatorial treatment groups. Particularly, in the RO3306 combined with sorafenib group, a synergistic anticancer effect on the overall PDX tumor models was revealed (Figure 2B-C). The HCC case #4 PDX model illustrated a dramatically synergistic reduction in tumor growth in line with the overall PDX tumors. HCC case #10 PDX model displayed tumor resistance to sorafenib treatment, whereas combinatorial treatment showed a significant enhanced antitumor effect, overcoming the tumor resistance (Figure 2D-E). Both PDX case #4 and case #10 were consistent with their tumor volume growth curves (Figure S2A-B). Taken together, the CDK1-targeted inhibitor can enhance the efficacy of sorafenib treatment, which would be applied to overcome its limited effect and improve the outcome of advanced HCC patients. In addition, the HCC case #9 PDX model indicated a dramatic resistance to RO3306, sorafenib, and combinatorial treatment, which we presumed is due to low tumor progression and growth (Figure S2C-D). Slow PDX tumor growth was also observed in aggressive triple negative breast cancer (HCI-004 of supplementary Figure 2 in that paper) [35], which makes it different than effective suppression. Moreover, with under treatment alone or in combination, the mouse body weights of various treatment groups had no significant difference, indicating the drug tolerance at the beginning of administration (Figure S2E).
Efficacy of treatments on tumor growth. (A) The diagram of the PDX tumor model (F0 and F1 generation) establishment and targeted drug administration schedule. (B) Growth curves of total tumor nodules of the PDX models were derived from HCC case #4 and case #10 from day 3 to day 26 in various treatment groups. (C) The tumor weights of the four indicated groups at the 4 week endpoint. (D) The tumor images of HCC case #4 and #10 PDX tumors at the endpoint. Each indicated treatment group included 6 mouse tumors per PDX cases. (E-F) The tumor weight of HCC case #4 and #10 PDX tumors corresponding to (D). *, p < 0.05; **, p < 0.01 compared to the control. #p < 0.05 compared to single agent treatment.
The synergistic effects of RO3306 and sorafenib targeting liver CSCs in vivo
Histologically, PDX tumors were paired to original HCC (Figure 3A), which may predict the clinical treatments. The activity levels of CDK1, PDK1 and β-Catenin illustrate synchronous downregulation with the combinatorial treatment, which may be the reason for the dramatic antitumor growth compared with single treatment groups (Figure 3B). The CDK1 activity level is higher in PDX case #4 than in case #10, and the decreased CDK1 level is 1.6-fold compared to 0.6-fold in the case #10 based on ImageJ analysis, which may explain the reason for the more dramatic inhibition of PDX case #4 (Figure 3B, upper panel). Wnt/β-Catenin signaling activation is pivotal in the maintenance of HCC stemness [36]. Flow cytometry analysis shows that the CD133+ and CD90+ CSC subpopulation decreased in the combinatorial treatment group compared with other groups (Figure 3C).
Antitumor effect is associated with downregulation of CDK1/PDK1/β-catenin and targeting the CSC population in PDX models. (A) H&E staining of HCC tissue and paired PDX tumors of the F1 generation (PDX F1). Scale bar represent 10 μm. (B) The expression levels of CDK1, PDK1 and β-catenin detected by western blot. (C) The CD133 and CD90 CSC population measured by flow cytometry. (D) Western blot analysis of the activation of stemness-related proteins Oct4, Sox2 and Nanog. (E) HCC PDX F1 case #10 had low activity level of pErk1/2. (F) Activation levels of AKT and pStat3 in the indicated groups of HCC PDX cases #4 and #10.
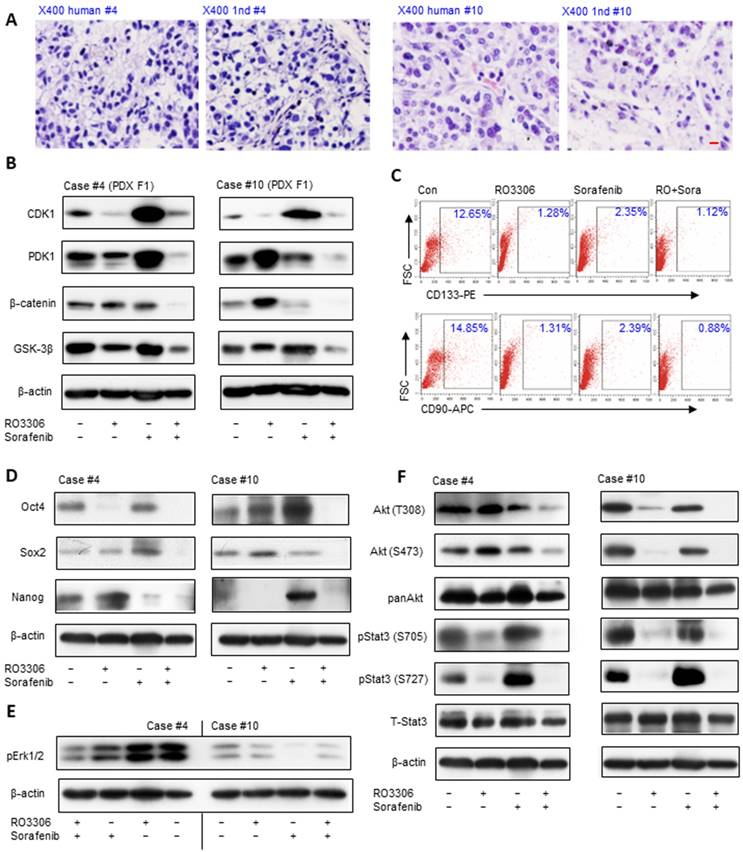
The reduction of CSCs was further verified by western blot results, which demonstrated downregulation of the stemness proteins Oct4, Sox2 and Nanog (Figure 3D). Specifically, RO3306 has a more prominent targeted inhibition effect on CSCs than sorafenib (Figure 3C), in line with the dramatically decreased level of the stemness-related proteins Oct4, Sox2 and Nanog in the RO3306 treatment (Figure 3D). Moreover, the upregulated level of Oct4 and Nanog may contribute to the resistance of the case #10 PDX model tumor following sorafenib treatment [37-39] (Figure 3D, right), Erk1/2 activity is considered a marker of sorafenib treatment in HCC [40, 41] and sorafenib resistance was confirmed by the low level of pErk1/2 in case #10 PDX tumor model (Figure 3E). The serine/threonine kinases Akt and Stat3 maintain the stemness of the stem cells and CSCs [28, 42], and the western blot results showed synergistic inhibition on the activity of Akt, especially for pStat3 (Figure 3F). In addition, the decreased levels of Snail 1 and Snail 2 with the upregulated level of E-cadherin in F1 PDX models illustrated the anti-metastatic potential (Figure S3). Taken together, the down-regulated expression of CDK1, PDK1 and β-Catenin with decreased CSCs may contribute to the antitumor effect on the F1 generation PDX tumor models.
RO3306 and sorafenib inhibited HCC CSC proliferation and self-renewal
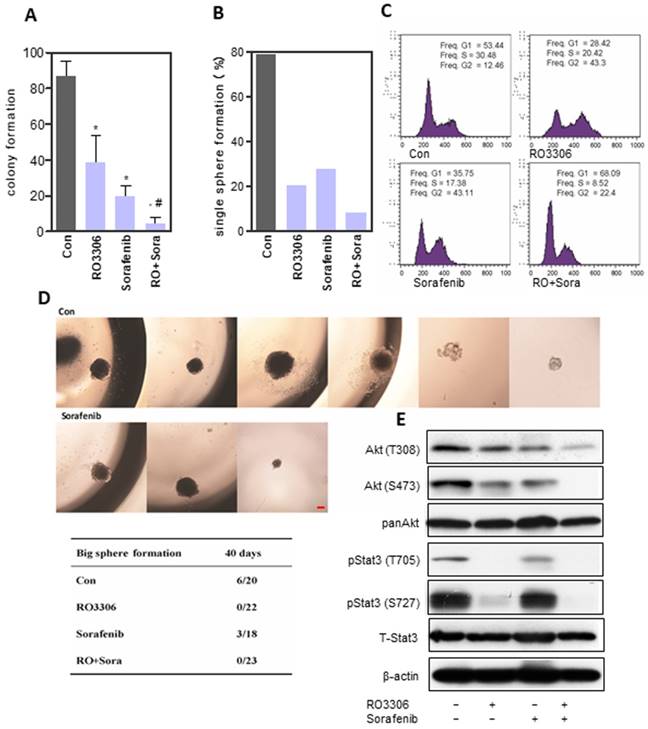
CSCs can be enriched in anchorage-independent sphere cells [28, 43]. The sphere formation assay showed a low dose of RO3306 (38.3 ± 15.2), sorafenib (19.8 ± 5.9) or combinatorial treatment (4.4 ± 3.7), which could significantly suppress cancer proliferation, particularly the combined therapy, which illustrated a synergistic effect (Figure 4A and Figure S4A). The self-renewal ability was detected by single-cell sphere formation and demonstrated similar results (Figure 4B). RO3306 and sorafenib combinatorial treatment decreased the CD90+ and EpCAM+ CSC subpopulations, as well as the expression of stemness-related genes Nanog and Sox2 (Figure S4B-C). Low-dose combinatorial inhibition decreased S-phase and increased cell arrest in the G2/M phase for 24 h (Figure 4C), whereas the upregulated Sub-G1 population indicated more cell apoptosis after a 72h exposure duration (Figure S4D). Next, to further evaluate self-renewal, single cells per well in 96 well plates were cultured for 40 days to detect huge sphere formation (≥1 mm). Surprisingly, RO3306 and the combinatorial treatment had no huge sphere formation compared with 27% (6/22) in the vehicle-treated and 17% (3/18) in the sorafenib groups (Figure 4D). RO3306 may have more suppression effects on CSC stemness compared with sorafenib, which shows the rational application of the synergistic combinatorial treatment. This was further confirmed by the synergistic inhibition on the activation of Akt and Stat3 (Figure 4E). Furthermore, the agent treatments, especially the combinatorial inhibition, could reverse the process of EMT and decrease the drug resistance gene ABCB1 (Figure S5A-G). Overall, RO3303 and sorafenib combination illustrated synergistic inhibition of CSC stemness.
Synergistic anticancer effect of RO3306 and sorafenib on liver CSC stemness. (A) 97H sphere cells were treated with DMSO, RO3306 (4 mM), sorafenib (5 mM), and RO3306 (4 mM) combined with sorafenib (5 mM). Colony formation was counted on day 7 (86.9 ± 8.3, 38.3 ± 15.2, 19.8 ± 5.9 and 4.4 ± 3.7 in DMSO, RO3306, sorafenib and combinatorial group). The data are presented from two independent experiments, each with eight replicates. An independent t-test was used for statistical comparison. *, p < 0.01 compared to the control. #, p < 0.05 compared to single agent treatments. (B) The 97H CSCs were serially diluted into single cells (one cell/well) followed by low-dose RO3306 (2 mM), sorafenib (2.5 mM), and RO3306 (2 mM) combined with sorafenib (2.5 mM) treatment for two days. The sphere formation of more than 40 cells were counted. Single-cell formation (%) was evaluated by the sphere formation number divided by the single cell seeding number in a 96 well plate. Single sphere formation was 78.7%, 20.3%, 27.8% and 8.4% in DMSO, RO3306, sorafenib and combinatorial group, separately. (C) PI staining analysis of cell cycle in the indicated groups. The data represent two independent experiments, each in triplicate. (D) Single-cell sphere formation following 2 days of treatment observed for 40 days to calculate the number of huge spheres formed (sphere size >1 mm). Huge single-cell formation was evaluated by the huge sphere formation number divided by the single cell seeding number in a 96-well plate. Scale bar represent 500 μm. (E) 97H sphere cells were treated with DMSO, RO3306 (4 mM), sorafenib (5 mM), and RO3306 (4 mM) combined with sorafenib (5 mM) for 48 h. Then, an analysis of the activation level of pStat3 and Akt was performed.
RO3306 increased the efficacy of sorafenib on CSC tumorigenicity
To further evaluate the oncogenic function, CSCs pre-treated with the indicated drugs were assessed in an in vitro single-cell sphere formation assay and for their tumorigenicity in vivo (Figure 5A). Functional assays showed 81.5%, 25.8%, 29.4% and 6.1% single-cell colony formation in the control, RO3306, sorafenib and combinatorial treatment groups (Figure 5B). Next, we confirmed the combination synergistic effect in vivo by a tumorigenicity assay (Figure S6A and Figure 5C-D). At the four-week endpoint, the tumor incidence was 100% (6/6) in the vehicle-treated groups compared with 83% (5/6), 33% (2/6) and 0% (0/6) in the RO3306, sorafenib and combinatorial treatment groups, respectively (Figure 5E). Moreover, the average tumor weight and volume were much lower in the indicated treatment groups than in the control vehicle-treated group (Figure 5F-G).
The combined effect on tumorigenicity and tumor growth. (A) The diagram of tumorigenicity in vivo and single-cell sphere formation assay. First, cells were pretreated with low-dose DMSO, RO3306 (2 mM), sorafenib (2.5 mM), and RO3306 (2 mM) combined with sorafenib (2.5 mM) for two days. (B) The comparison of single-cell sphere formation in indicated groups after pretreatment without further treatment. (C-D) The bioluminescence signal represented the tumor incidence and was confirmed by the tumor image, all at 4 weeks' time. (E) Statistical comparison of tumor incidence (%) in various groups after injection of pretreated cells without further treatment for 4 weeks. (F-G) Statistical comparison of the tumor size and volume based on the image in (D). (H-I) Antitumor and antimetastasis effect of the combinatorial treatment on 97H CSC-derived orthotropic tumor models. *, p < 0.01 compared to control. #, p < 0.05 compared to single agent treatments.
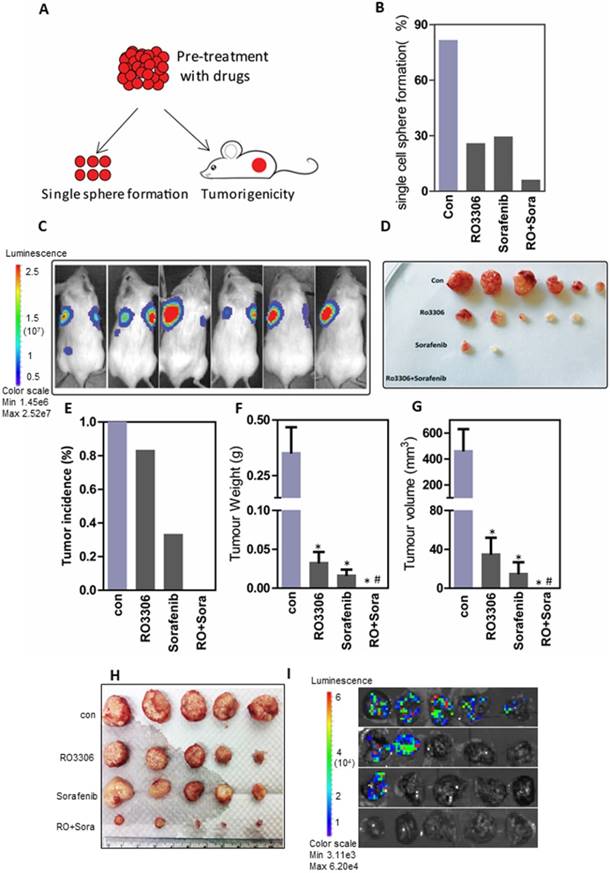
In addition, 100 days after total tumor resection, tumor recurrence occurred in three mice of the vehicle-treated groups with a huge tumor size; two mice in the RO3306-treated group with one new emerging tumor, all with small size; one new emerging tumor in the sorafenib group; and one new emerging tumor in the combinatorial group (Figure S6B). Overall, the tumor incidence was 100% in the vehicle-treated group with 3 recurring tumors, and 100% in the RO3306-treated group with 2 recurring tumors. However, the average tumor size was smaller in the RO3306-treated group compared to the vehicle-treated group (data not shown). The tumor incidence in the sorafenib-treated group was 50% (3/6), while that of the combinatorial treatment group was only 17% (1/6), both without tumor recurrence (Figure S6C). Together, this suggests that combinatorial treatment could synergistically decrease CSC tumorigenicity.
From the 97H CSCs-derived orthotopic tumor models, we confirmed the synergistic antitumor effect of the combinatorial treatment (tumor weight: 1.87 ± 0.31g, 0.56 ± 0.38g, 0.55 ± 0.32g vs 0.03 ± 0.03g) (Figure 5H and Figure S6D), which was in line with the PDX models of HCC case #4 and #10. Further, the combination antitumor growth effect along with the antimetastasis shown in our lung metastasis investigation 100%, 40%, 20% lung metastasis compared with 0% in the combinatorial treatment group. (Figure 5I and Figure S6E).
CDK1/PDK1/β-Catenin signaling in CSCs
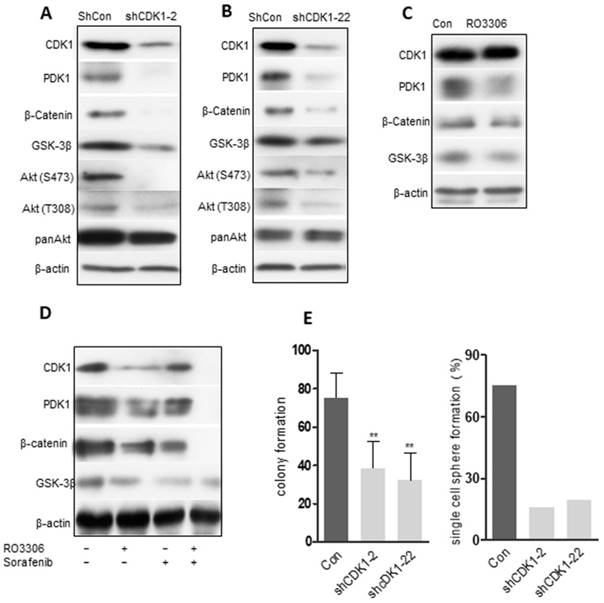
The clinical correlation among CDK1, PDK1 and β-Catenin, and the corresponding decrease in the expression levels of CDK1, PDK1 and β-Catenin demonstrated a significant, synergistic anticancer effect following combinatorial treatment in the PDX models. To further recapitulate the observation above, we evaluated the oncogenic function of CDK1 in vitro. Two shRNAs (shCDK1-2 and shCDK1-22) specifically targeting CDK1 were transfected into MHCC 97H sphere cells to avoid off-target effects. The knockdown effect was confirmed by western blot analysis and the results revealed that both shRNAs could specifically decrease the expression of CDK1, accompanied by a downregulation of PDK1, β-Catenin and AKT (Figure 6A-B). The PDK1 and AKT were believed to interact with each other, which was verified by our Co-IP analysis (Figure S7A). Pharmacological inhibition of MHCC 97H sphere cells could decrease PDK1 and β-Catenin without obvious down-regulation of CDK1 in 24 h (Figure 6C) (due to inhibition of the kinase activity of CDK1, but degradation of the protein CDK1 needs longer than 24 h). CDK1, PDK1 and β-Catenin expression levels were decreased by RO3306 and sorafenib alone or in combination after 48 h (Figure 6D). Functional assays showed that CDK1 silencing could dramatically suppress the clonogenic phenotype analyzed by reducing the colony formation and inhibiting the self-renewal ability measured by the single sphere formation assay (Figure 6E). Downregulation of CDK1 reverses the process of EMT (Figure S7B-E). The CD90+ CSC subpopulation also decreased after transfection silencing (Figure S7F). Taken together, the abolished tumor growth occurred through inhibiting the CDK1/PDK1/β-Catenin signal pathway in HCC.
The CDK1/PDK1/β-catenin axis in CSCs. (A-B) 97H CSCs were treated with scrambled shRNA (shCon) or shRNA (shCDK1-2 and shCDK1-22) against CDK1. Western blot results confirmed the decreased expression levels of CDK1 in 97H CSCs, with a downregulation of PDK1, β-catenin and AKT. (C) Pharmacological inhibition of CDK1 with RO3306 for 24 h. Western blot analysis of the expression levels of CDK1, PDK1 and β-catenin. (D) Inhibition of CDK1 combined with sorafenib or alone for 48 h. Western blot detected the indicated protein levels. (E) shCDK1 knockdown, followed by a colony formation assay. (F) shCDK1 knockdown group compared with the scrambled group for the single-cell sphere formation assay in a 96-well plate. The data represent three independent experiments, each in triplicate. **, p < 0.01 compared to control.
Discussion
HCC is one of the most aggressive cancers with very low survival and a high probability of metastasis. We illustrated that CDK1 is associated with poor HCC overall survival (p=0.008). High CDK1 expression is a risk factor of one- and five-year tumor recurrence (p=0.013 and p=0.017), indicating that CDK1 might play essential oncogenic roles in HCC progression. CDK1 is a member of the Ser/Thr protein kinase, which is essential for cell G1/S and G2/M phase transitions [44, 45]. The dysfunction of the cell cycle could lead to the generation of CSCs that currently are thought to partially contribute to oncogenicity [14, 46]. The abnormal cell cycle in CSCs might enhance tumorigenesis and lead to the failure of traditional chemotherapy. Exploring the CDK inhibitors in CSCs may permit the development of novel strategies to treat cancer [47]. Currently, sorafenib is the only FDA-approved targeted therapy for HCC, but its effect only extends 3 months in advanced HCC. There is an urgent need to identify new effective therapies for HCC patients.
One breakthrough has been achieved for palbociclib (a reversible CDK4/6 inhibitor) in combination with letrozole (aromatase inhibitor) for estrogen receptor-positive breast cancer treatment. The enhanced combination antitumor effects led to palbociclib being approved by the FDA in 2015 [48, 49]. Apart from the CDK4/6 inhibitor palbociclib, the combination of CDK1 inhibitors with other targeted therapies, including sorafenib, may also be conceivable. In our research, we illustrated that the CDK1 inhibitor RO3306 enhanced sorafenib treatment in PDX tumor models and the combination effect was confirmed in CSC-derived orthotopic tumor models. The PDX models are currently the most accepted tumor model to predict clinical outcomes. In the F1 generation, PDX tumor models showed synergistic anticancer growth effects with targeting of CSCs, accompanied by the downregulation of AKT and pStat3 activation. It also promoted the inhibition of self-renewal and proliferation in 97H CSCs in vitro. The in vivo tumorigenicity analysis showed the synergistic effect of the combination of RO3306 and sorafenib on 97H CSC-derived xenograft models. Notably, in our research we demonstrated that the adverse effects of RO3306 plus sorafenib are well tolerated, resemble the single sorafenib treatment and that RO3306 is a bearable drug with a similar body weight curve to the vehicle-treated group (Figure S2E).
Based on the anticancer evidence of the PDX model of case #4 and case #10 (Figure 2E), and with the corresponding results of the western blot analysis (Figure 3B), high expression activation level of CDK1 has a more dramatic response to combinatorial treatment compared to the corresponding controls. Moreover, the high activity of CDK1 showed a downregulation of CDK1, PDK1 and β-Catenin in PDX case #4 following treatment with the CDK1 inhibitor (Figure 3B, left). The higher expression levels of CDK1, Oct4 and Nanog after sorafenib treatment in case #10 would explain the reason for sorafenib resistance in the clinic (Figure 2E, right and Figure 3B, D) and was confirmed by the low level of pErk1/2 in case #10 PDX tumor model (Figure 3E). Intriguingly, whether the CDK1 levels were high or low, the decreased CDK1, PDK1 and β-Catenin levels occurred or didn't occur following single treatment, but the synergistic decline in the activity of CDK1, PDK1 and β-Catenin synchronized with the combinatorial treatment (Figure 3B). This was consistent with the result of the decreased stemness-related proteins Oct4, Sox2 and Nanog, as well as Akt and pStat3 activation. Taken together, the preclinical findings illustrate the possibility for using CDK1 inhibitors in complement with sorafenib for the treatment of specific CDK1-aberrant HCC tumors.
RO3306 has a more dramatic inhibition compared with the sorafenib treatment of case #10 (Figure 2E, right) and 97H CSCs (Figure 4D), which is in line with the western blot analysis showing the downregulation of the activation level of pStat3 (Figure 3F and Figure 4E). pStat3 is important to CSC maintenance and cancer progression [28, 50]. Based on the evidence that CDK1 plays an essential role in cancer progression and embryonic stem cells (ESC) through the association with stemness marker Oct4 [20, 21], the effect of CDK1 in a defined CSC population was investigated in our study. Dysregulation in cell cycle could lead to developmental abnormalities and cancer proliferation. Focusing on the defects of cell cycle regulation in cell division could provide new approaches for designing new and novel targeted drug therapies. Here, we confirmed the hypothesis of the essential role of CDK1 in CSCs and that pharmacological inhibition could suppress liver CSC progression via the cell cycle. The combinatorial treatment could decrease the S phase in 24 h treatment and promote cells to enter into a Sub-G1 phase during 48 h exposure (Figure 4C and Figure S4D).
We attempted to explore the molecular mechanism of the oncogenic role of CDK1. We reported that CDK1 interaction with another oncogenic molecule, PDK1, is in line with the previous findings in embryonic stem cells (39). PDK1 has been shown to play an important role in metastasis, moreover liver metastasis from breast cancer, which also has a high expression level of PDK1 (31, 32). Furthermore, we demonstrated that CDK1 and PDK1 association was related to tumor metastasis (p=0.025) and poor disease-free survival (p=0.03). Consistently, a Cox proportional hazard regression analysis revealed that CDK1 in association with PDK1 is an independent prognostic factor of disease-free survival (p=0.009). The CDK1 inhibitor RO3306 alone or in combination with sorafenib could decrease CDK1, PDK1 and β-Catenin in 97H CSCs and the PDX models in vivo. Furthermore, down-regulation of CDK1 by shRNAs and pharmacological inhibition of CDK1 by RO3306 showed a consequent decrease in PDK1 and β-Catenin, with tumor suppression on single-cell sphere formation and colony formation. The aberrant β-Catenin- related chromosome 8p losses were associated with large tumor size [51]. It is reported that human β-catenin alterations account for 26% of human liver tumors. The dysfunctional β-catenin pathway leads to carcinogenesis and is closely associated with mouse and human HCC [52]. Wnt/β-catenin signaling acts through inducing EMT, which is important to maintaining CSC stemness [53-57]. In our study, RO3306 could enhance sorafenib to decrease the CSC population in vivo and vitro, which may occur through the reversal of EMT with down-regulation of the mesenchymal markers Snail 1 and Snail 2, and upregulation of the epithelial marker E-cadherin, suggesting anti-metastatic potential. Reversing EMT is associated with the suppression of CDK1, PDK1 and β-Catenin activation.
In summary, we report that CDK1 was frequently overexpressed in HCC and associated with tumor progression via the CDK1, PDK1 and β-Catenin pathways (Figure S8 and Figure S9). RO3306 could enhance sorafenib treatment in F1 PDX tumor models by targeting CSC stemness. Together, this evidence supports a personalized therapeutic option for a novel molecular subgroup of patients with CDK1-aberrant HCC receiving a CDK1 inhibitor combined with sorafenib treatment.
Abbreviations
CDK1: cyclin-dependent kinase 1; Co-IP: Co-immunoprecipitation; CSCs: cancer stem cells; EMT: epithelial-mesenchymal transition; HCC: hepatocellular carcinoma; Nanog: homeobox protein NANOG; Oct4: octamer-binding transcription factor 4; PDK1: phosphoinositide-dependent kinase; PDX: patient-derived tumor xenograft model; Sox2: sex determining region Y-box2; Stat3: signal transducer and activator of transcription 3.
Acknowledgements
This study was supported by Healthy and Medical Research Fund, Research Council of Hong Kong (HMRF 03143396 to XQW).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245-1255
2. Nagasue N, Kohno H, Chang YC, Taniura H, Yamanoi A, Uchida M, Kimoto T. et al. Liver resection for hepatocellular carcinoma. Results of 229 consecutive patients during 11 years. Ann Surg. 1993;217:375-384
3. Huynh H, Chow PK, Tai WM, Choo SP, Chung AY, Ong HS, Soo KC. et al. Dovitinib demonstrates antitumor and antimetastatic activities in xenograft models of hepatocellular carcinoma. J Hepatol. 2012;56:595-601
4. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-2576
5. Bruix J, Sherman M, American Association for the Study of Liver D. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020-1022
6. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118-1127
7. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789-799
8. Liu M, Jiang L, Guan XY. The genetic and epigenetic alterations in human hepatocellular carcinoma: a recent update. Protein Cell. 2014;5:673-691
9. Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A. et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017;17:254-268
10. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726-734
11. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23-28
12. Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30-38
13. Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320-329
14. Cheng T. Cell cycle inhibitors in normal and tumor stem cells. Oncogene. 2004;23:7256-7266
15. McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, MacKenzie M. et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int J Cancer. 2002;102:463-468
16. Fischer PM, Gianella-Borradori A. CDK inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2003;12:955-970
17. Senderowicz AM. Novel small molecule cyclin-dependent kinases modulators in human clinical trials. Cancer Biol Ther. 2003;2:S84-95
18. Ambrosini G, Seelman SL, Qin LX, Schwartz GK. The cyclin-dependent kinase inhibitor flavopiridol potentiates the effects of topoisomerase I poisons by suppressing Rad51 expression in a p53-dependent manner. Cancer Res. 2008;68:2312-2320
19. Velasquez C, Cheng E, Shuda M, Lee-Oesterreich PJ, Pogge von Strandmann L, Gritsenko MA, Jacobs JM. et al. Mitotic protein kinase CDK1 phosphorylation of mRNA translation regulator 4E-BP1 Ser83 may contribute to cell transformation. Proc Natl Acad Sci U S A. 2016;113:8466-8471
20. Li L, Wang J, Hou J, Wu Z, Zhuang Y, Lu M, Zhang Y. et al. Cdk1 interplays with Oct4 to repress differentiation of embryonic stem cells into trophectoderm. FEBS Lett. 2012;586:4100-4107
21. Zhao R, Deibler RW, Lerou PH, Ballabeni A, Heffner GC, Cahan P, Unternaehrer JJ. et al. A nontranscriptional role for Oct4 in the regulation of mitotic entry. Proc Natl Acad Sci U S A. 2014;111:15768-15773
22. Keysar SB, Astling DP, Anderson RT, Vogler BW, Bowles DW, Morton JJ, Paylor JJ. et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol Oncol. 2013;7:776-790
23. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Cora D. et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508-523
24. Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D. et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res. 2012;18:2515-2525
25. Sivanand S, Pena-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, Yamasaki T. et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med. 2012;4:137ra175
26. Garrido-Laguna I, Tan AC, Uson M, Angenendt M, Ma WW, Villaroel MC, Zhao M. et al. Integrated preclinical and clinical development of mTOR inhibitors in pancreatic cancer. Br J Cancer. 2010;103:649-655
27. Jimeno A, Amador ML, Kulesza P, Wang X, Rubio-Viqueira B, Zhang X, Chan A. et al. Assessment of celecoxib pharmacodynamics in pancreatic cancer. Mol Cancer Ther. 2006;5:3240-3247
28. Wu CX, Xu A, Zhang CC, Olson P, Chen L, Lee TK, Cheung TT. et al. Notch inhibitor PF-03084014 inhibits hepatocellular carcinoma growth and metastasis via suppression of cancer stemness due to reduced activation of Notch1-Stat3. Mol Cancer Ther. 2017;16:1531-1543
29. Wang XQ, Zhang W, Lui EL, Zhu Y, Lu P, Yu X, Sun J. et al. Notch1-Snail1-E-cadherin pathway in metastatic hepatocellular carcinoma. Int J Cancer. 2012;131:E163-172
30. Yang W, Cho H, Shin HY, Chung JY, Kang ES, Lee EJ, Kim JH. Accumulation of cytoplasmic Cdk1 is associated with cancer growth and survival rate in epithelial ovarian cancer. Oncotarget. 2016;7:49481-49497
31. Du J, Yang M, Chen S, Li D, Chang Z, Dong Z. PDK1 promotes tumor growth and metastasis in a spontaneous breast cancer model. Oncogene. 2016;35:3314-3323
32. Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, Omeroglu A. et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015;22:577-589
33. Scortegagna M, Ruller C, Feng Y, Lazova R, Kluger H, Li JL, De SK. et al. Genetic inactivation or pharmacological inhibition of Pdk1 delays development and inhibits metastasis of Braf(V600E)::Pten(-/-) melanoma. Oncogene. 2014;33:4330-4339
34. Lian S, Shao Y, Liu H, He J, Lu W, Zhang Y, Jiang Y. et al. PDK1 induces JunB, EMT, cell migration and invasion in human gallbladder cancer. Oncotarget. 2015;6:29076-29086
35. DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R. et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17:1514-1520
36. Takai A, Fako V, Dang H, Forgues M, Yu Z, Budhu A, Wang XW. Three-dimensional organotypic culture models of human hepatocellular carcinoma. Sci Rep. 2016;6:21174
37. Wu G, Wilson G, Zhou G, Hebbard L, George J, Qiao L. Oct4 is a reliable marker of liver tumor propagating cells in hepatocellular carcinoma. Discov Med. 2015;20:219-229
38. Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, Xu Y. et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology. 2012;56:1004-1014
39. Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, Hess S. et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016;23:206-219
40. Caraglia M, Giuberti G, Marra M, Addeo R, Montella L, Murolo M, Sperlongano P. et al. Oxidative stress and ERK1/2 phosphorylation as predictors of outcome in hepatocellular carcinoma patients treated with sorafenib plus octreotide LAR. Cell Death Dis. 2011;2:e150
41. Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S. et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851-11858
42. Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA. et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823-826
43. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313-323
44. Ohashi R, Gao C, Miyazaki M, Hamazaki K, Tsuji T, Inoue Y, Uemura T. et al. Enhanced expression of cyclin E and cyclin A in human hepatocellular carcinomas. Anticancer Res. 2001;21:657-662
45. Chao Y, Shih YL, Chiu JH, Chau GY, Lui WY, Yang WK, Lee SD. et al. Overexpression of cyclin A but not Skp 2 correlates with the tumor relapse of human hepatocellular carcinoma. Cancer Res. 1998;58:985-990
46. Quesenberry PJ, Colvin GA, Lambert JF. The chiaroscuro stem cell: a unified stem cell theory. Blood. 2002;100:4266-4271
47. Boyer MJ, Cheng T. The CDK inhibitors: potential targets for therapeutic stem cell manipulations? Gene Ther. 2008;15:117-125
48. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016;6:353-367
49. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417-430
50. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9:50-63
51. Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, Monges G. et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763-1773
52. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M. et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847-8851
53. Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512-1520
54. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB. et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468-476
55. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-715
56. Bessede E, Staedel C, Acuna Amador LA, Nguyen PH, Chambonnier L, Hatakeyama M, Belleannee G. et al. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene. 2014;33:4123-4131
57. Shuang ZY, Wu WC, Xu J, Lin G, Liu YC, Lao XM, Zheng L. et al. Transforming growth factor-beta1-induced epithelial-mesenchymal transition generates ALDH-positive cells with stem cell properties in cholangiocarcinoma. Cancer Lett. 2014;354:320-328
Author contact
![]() Corresponding authors: Chuan Xing Wu, PhD and Xiao Qi Wang, PhD, Department of Surgery, The University of Hong Kong, 21 Sassoon Road, Hong Kong, Telephone: 852-39179616, Fax: 852-39179634, Email: u3003812hk and xqwanghk
Corresponding authors: Chuan Xing Wu, PhD and Xiao Qi Wang, PhD, Department of Surgery, The University of Hong Kong, 21 Sassoon Road, Hong Kong, Telephone: 852-39179616, Fax: 852-39179634, Email: u3003812hk and xqwanghk