Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(14):3824-3840. doi:10.7150/thno.25308 This issue Cite
Research Paper
Targeting S1PR1/STAT3 loop abrogates desmoplasia and chemosensitizes pancreatic cancer to gemcitabine
Manendra Babu Lankadasari1,2, Jayasekharan S. Aparna1, Sabira Mohammed1,2, Shirley James1, Kazunori Aoki3, Valsalakumari S. Binu4, Sindhu Nair5, Kuzhuvelil B. Harikumar1 ![]()
1. Cancer Research Program, Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram, Kerala State, India 695014
2. Ph.D scholar, Manipal Academy of Higher Education (MAHE), Manipal, India
3. Division of Molecular and Cellular Medicine, National Cancer Center Research Institute, Tsukiji 5-1-1, Chuo-ku, Tokyo 104-0045, Japan
4. Department of Biostatistics, National Institute of Mental and Neurosciences, Bengaluru, India 560029
5. Department of Pathology, Regional Cancer Centre, Thiruvananthapuram, India 695025
Received 2018-2-2; Accepted 2018-4-19; Published 2018-6-13
Abstract

Rationale: Pancreatic cancer is associated with poor prognosis with a 5-year survival rate of less than 6%. Approximately 90% of pancreatic cancer patients harbor somatic mutations in the KRAS gene. Multiple lines of evidence suggest a persistent activation of STAT3 in KRAS-driven oncogenesis contributing to desmoplasia and gemcitabine resistance. Sphingosine 1-phosphate receptor 1 (S1PR1) is an integral component of tumor progression and maintains an activated state of STAT3. FTY720 is an approved drug for multiple sclerosis and acts as a functional antagonist for S1PR1. Here we explored the potential utility of FTY720 to target S1PR1/STAT3 and other major signaling pathways in pancreatic cancer, and sought proof-of-principle for repurposing FTY720 for the treatment of pancreatic cancer.
Methods: We examined the activity of FTY720 in the proliferation, apoptosis, and cell cycle assays in human and mouse pancreatic cancer model systems. Further, we studied the efficacy of using a combination of FTY720 and gemcitabine as opposed to individual agents in vitro as well as in vivo
Results: Treatment of human and mouse pancreatic cancer cells with FTY720 resulted in inhibition of growth, increased apoptosis, and cell cycle arrest. FTY720 in combination with gemcitabine breached the mitochondrial membrane potential, altered the S1PR1-STAT3 loop, and inhibited epithelial to mesenchymal (EMT) transition. Data from murine models exhibited a marked reduction in the tumor size, increased apoptosis, inhibited NF-κB, S1PR1/STAT3, Shh signaling and desmoplasia, modulated the expression of gemcitabine-metabolizing transport enzymes, and restored the expression of tumor suppressor gene PP2A.
Conclusion: Taken together, our results established FTY720 as a propitious molecule, which increases the efficacy of gemcitabine and represents a promising agent in the management of pancreatic cancer.
Keywords: pancreatic cancer, FTY720, S1P receptor 1, PP2A, metastasis, gemcitabine, STAT3
Introduction
Pancreatic cancer is currently the fourth leading cause of cancer-associated deaths in the United States [1]. According to the recent report by Pancreatic Cancer Action Network, by 2030 pancreatic cancer is projected to become the second leading cause of cancer-related deaths surpassing breast, prostate, and colorectal cancers [2]. The average survival rate remains at 5% despite the intense efforts over the past several decades. Approximately 90% of pancreatic cancer patients harbor somatic mutations of the KRAS gene. Over several decades with marginal effects on patient survival, gemcitabine remains the primary drug of choice for pancreatic cancer therapy [1].
Sphingosine 1-Phosphate (S1P), a pleiotropic lipid molecule present inside the cells, produced by phosphorylation of sphingosine by Sphingosine Kinases (SphK1 and 2) regulates numerous cellular events such as cell growth, migration, lymphocyte egress, and vascular integrity [3]. S1P acts either intracellularly or through the cell surface G protein-coupled receptors (S1P receptor, S1PR 1-5) [4]. It was shown that pancreatic stellate cells were activated by S1P derived from pancreatic cancer and resulted in increased cell growth and invasion [5]. SphK1 is involved in cell proliferation, gemcitabine resistance, and peritoneal metastasis of pancreatic cancer cells, and SphK1-/- mice show reduced tumor burden compared to wild-type mice [6]. S1PR1 has been reported as one of the key factors responsible for persistent STAT3 activation in different tumor types [7, 8]. However, the precise role of S1PR1 in pancreatic cancer is not clear. Fingolimod (FTY720)is the first FDA-approved oral drug for the treatment of the relapsing form of multiple sclerosis [9]. FTY720 is phosphorylated by both sphingosine kinases 1& 2 and can bind to four of the five S1P receptors except for S1PR2 and acts as a functional antagonist to S1PR1 [10, 11]. Apart from its proven role in immunosuppression and preventing T cell egress from secondary lymphoid organs, FTY720 inhibited growth and induced apoptosis in different cancer cell lines in vitro as well as in vivo including multiple myeloma [12], renal cell carcinoma [13], and colorectal cancer [11]. There were studies demonstrating the in vitro efficacy of FTY720 in pancreatic cancer [14]. However, the underlying mechanism of action is still elusive.
In this study, we showed that S1P receptor modulator FTY720 inhibited the growth of pancreatic cancer in two pre-clinical mouse models, an immunodeficient and a syngeneic model with an intact immune system. In both models, FTY720 suppressed tumor growth by chemosensitizing cancer cells to gemcitabine, a currently approved drug for treating pancreatic cancer which inhibited desmoplasia and epithelial-to mesenchymal transition (EMT). Thus, we provided compelling in vitro and in vivo evidence to support the use of FTY720 as a propitious therapeutic agent for the treatment of pancreatic cancer.
Methods
Materials
FTY720 and gemcitabine were purchased from Selleckchem (Houston, TX). The Annexin-FITC kit was procured from Biotool (Houston, TX). Source of other chemicals, antibodies and kits are provided in Supplementary Material.
Cell lines
BxPC-3, AsPC-1 cells were acquired from the American Type Culture Collection (ATCC, Manassas, VA), MIA PaCa-2 and PANC-1 were from the National Centre for Cell Sciences (Pune, India). PAN 02, a C57BL/6-derived pancreatic cancer cell line was obtained from the National Cancer Institute (Frederick, MD), HDPE cell line was a kind gift from Dr. Florencia McAllister, UTMDACC (Houston, TX) and was grown in keratinocyte serum free medium with 5 ng/ml recombinant human EGF. Cells were cultured in Dulbecco's modified Eagle medium containing 10% fetal bovine serum (FBS) and the antibiotic mixture (penicillin, streptomycin and amphotericin). All cell lines were tested for the presence of mycoplasma and found to be negative.
Animals
NOD.CB17-Prkdcscid/J mice (7-8-week old, hereafter referred as NOD-SCID) and C57Bl/6 (8-10-week old) mice were obtained from Jackson laboratories and kept in individually ventilated cages (IVCs) with standard rodent chow, water ad libitum, and a 12 h light/dark cycle. The animal experiment protocol got prior approval from Institutional Animal Ethics Committee (IAEC) of Rajiv Gandhi Centre for Biotechnology and followed the rules and regulations prescribed by Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India.
Cell proliferation assay
Human and mouse pancreatic cancer cell lines were maintained in their appropriate media in the presence of antibiotic solution and FBS. To study the effect of FTY720 on the metabolic activity of cells, MTT assay was used. 5×104/0.2 mL cells were plated in 96-well plates and treated with different concentrations of FTY720 and incubated for 24 and 48 h. Subsequently the medium was removed and cells were washed with PBS. 20 µL of MTT (5 mg/mL) (Sigma, #M2128) was added to each well and incubated for 4 h. Then the medium was removed and the crystals were dissolved in DMSO and read at 595 nm in TECAN spectrophotometer.
Annexin V-FITC staining
Cells were plated in 6-well plates at a density of 3×105 and incubated for 12 h followed by incubation for 24 h with various concentrations of FTY720. Cells were trypsinized and washed with pre-chilled PBS and resuspended in 100 µL of 1x binding buffer with 5 µL of Annexin V-FITC and 5 µL of propidium iodide for 15 min in the dark. Subsequently, 400 µL of binding buffer was added and cells were filtered through cell strainer and analyzed using FACS scanner.
Cell cycle analysis
Cells were plated at a density of 3×105 in a 6-well plate and after 24 h cells were treated with FTY720 and incubated for 24 h. The cells were washed with PBS and fixed with ice cold 70% ethanol and incubated with RNase A (1 mg/mL) (Sigma, #R6513) for 30 min. 10 µL of propidium iodide (1 mg/mL) (Sigma, #P4170) was added followed by incubation for 15 min in the dark and analysis was carried out by BD FACS Aria system.
Cell migration assay
1×105 cells were plated in 12-well plates and allowed to reach 90% confluence. A wound was made using a 200 µL tip and the wells were washed with PBS to remove the floating cells. Subsequently, FTY720-containing medium was added and the cells were monitored for the rate of wound closure. 100× images were taken at 0 h, 12 h and 24 h using a phase contrast microscope.
Quantification of reactive oxygen species
3×105 cells were plated in 6-well plates and incubated overnight. The cells were treated with FTY720 for 24 h and then exposed to 1% FBS with 5 µM H2DCFDA (Thermo Fisher Scientific, #D399). The cells were then kept for 30 min in dark and analyzed in FACS system.
Assessment of loss of mitochondrial membrane potential
3×105 cells were plated in 6-well plates and incubated for 24 h. Then the cells were treated with FTY720 (10 µM), gemcitabine (1 µM), or the combination and incubated for 24 h. Subsequently, the cells were trypsinized and suspended in 0.1 µM DiOC6 (3) in serum-free medium and kept at 37 ºC for 15 min in dark. Cells were centrifuged at 130 g for 5 min, washed twice with PBS and analyzed in FACS within 15 min.
Clonogenic assay
1×105 cells were plated in 12-well plates and treated with FTY720, gemcitabine, or the combination of both and incubated for 24 h. Then the cells were counted and 2×104 cells were re-plated in 6-well plates and incubated for 7 days. The colonies were washed with PBS and fixed for 30 min in 10% formalin, and stained with crystal violet. The growth rate was determined by melting the colonies in 10% acetic acid and measuring the amount of crystal violet released using TECAN spectrophotometer at 595 nm.
Western blotting
Tissue samples were homogenized in protein lysis buffer and the samples were centrifuged at 12000 ×g for 10 min. The supernatant was collected and used for Western blotting. For the in vitro study, cancer cells were plated in 60 mm dishes and treated with the drug for 24 h. Cells were lysed in protein lysis buffer and used for further analysis. Proteins were separated on polyacrylamide gels and transferred to nitrocellulose membranes. After the transfer, membranes were blocked with 5% skimmed milk and subsequently incubated with either of the following primary antibodies; S1PR1 (ab23695, 1:3000) was obtained from Abcam. STAT3 (sc-482, 1:2000), c-MYC (sc-764, 1:3000), E-Cadherin (sc-7870, 1:1000), N-Cadherin (sc-7939, 1:2000), Cyclin-D1 (sc-753, 1:1000), COX-2 (sc-7951, 1:1000), ERK 1 (sc-93, 1:3000), and β-Tubulin (sc-9104, 1:2000) were procured from Santa Cruz Biotechnology. p-STAT3 (9145S, 1:1000), Vimentin (5741, 1:3000), p-ERK 1/2 (9106, 1:2000), and p-Akt (9271, 1:1000) were purchased from cell signaling Technology. HRP conjugated secondary antibody was added and the detection were performed using ECL solution.
Generation of luciferase-expressing stable cell lines
Luciferase-expressing pancreatic cancer cell lines were generated using pLenti CMV Puro LUC (w168-1) (Addgene #17477) [15] and transfection was carried out using lentiviral 3rd generation transfection system. Briefly, HEK293T cells were grown to 70% confluency and pRRE (gag/pol), pMD2G (VSVG), pRSV (Rev), and pLenti CMV Puro LUC plasmids were suspended in 0.25 M CaCl2, equilibrated with same volume of 2× HEPES solution, and entire solution was added to the wells. The medium was changed after 14 h and the viral particles were collected at 24 and 48 h. MIA PaCa-2 and PAN 02 cells were grown to 50% confluency and transfected with the viral particles. Luciferase-expressing cells were selected using 2 μg/mL of puromycin (Sigma, #P8833) starting from 48 h after infection till 7 passages. The presence of luciferase was confirmed by imaging the cells under IVIS.
Generation of orthotopic pancreatic cancer mice models
MIA PaCa-2 and PAN 02 pancreatic cancer cells (both harboring KRAS mutations) were used for generating the orthotopic model in NOD-SCID and C57Bl/6 mice, respectively, as previously described [16]. All procedures in mice were performed during light cycle. Animals were anesthetized using a mixture of ketamine-xylazine. A small incision was made on the right abdominal side and spleen was gently pulled out without causing injury to underlining organs. MIA PaCa-2-Luc cells (5×105 cells/50 µL volume) or PAN 02-Luc cells (5×104 cells/50 µL volume) were injected directly into pancreas using a 27-gauge needle. The wounds were closed with metallic clips. Animals were randomized into four groups after initial IVIS imaging (Xenogen Corp., Alameda, CA) on day 7 following tumor implantation for further experimentation: (a) untreated controls (vehicle only); (b) FTY720 alone (1 mg/kg body weight once daily p.o.); (c) gemcitabine alone (25 mg/kg body weight twice weekly by i.p. injection); and (d) combination of FTY720 (1 mg/kg body weight), once daily p.o., and gemcitabine (25 mg/kg body weight twice weekly by i.p. injection. Animals were imaged every week using IVIS system by injecting 100 µL of luciferin (1 mg/mL) through the i.p route. Animals were sacrificed on day 35 after tumor implantation. Primary tumors in the pancreas were excised and the final tumor volume was measured as V = (length × width2) / 2, where V is the volume of the tumor [17]. The distal macroscopic metastatic lesions were counted in liver, lungs, intestine and abdominal cavity. Part of the tumor was fixed in formalin and used for immunohistochemical analysis and the remaining portion was snap frozen in liquid nitrogen for protein and RNA extraction.
Immunohistochemical staining
The formalin fixed tissues were sliced to 5 µm and after xylene and graded ethanol treatment; antigen retrieval was carried out by using citrate buffer, pH 6. Peroxidase activity was blocked using 3% H2O2 in methanol followed by another blocking step using a commercial kit (Dako, #K500711). The primary antibody in antibody diluent was used with overnight incubation at 4 ºC. Slides were washed and incubated with the secondary antibody for 1 h and developed using DAB solution. Hematoxylin was used as the counter stain to detect the nucleus and the slides were mounted with DPX.
Sirius red staining
The nuclei were stained with hematoxylin for 8 min, followed by washing in distilled water. Picro-sirius red stain (Sigma, #365548) was overlaid on the tissue sections and incubated for 1 h. After washing with acidified water, the slides were dehydrated by placing them in xylene and mounted with DPX.
RT2 profiler PCR array
RNA was isolated by RNeasy Lipid Tissue mini kit (Qiagen, #74804) with a DNase treatment step included. RNA was quantified using Nanodrop spectrophotometer 2000. 1.5 µg of RNA was used for reverse transcription by High Capacity cDNA reverse transcription kit (ABI, #4368814). The cDNA was then mixed with SYBR green PCR master mix (ABI, #4367659) and 25 µL per well was added in RT² Profiler PCR Array (Qiagen, #PAHS-181Z) with , 96-well plate format. The data were then exported and uploaded into the RT2 profiler PCR array web portal (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php) for analysis.
Real-time PCR
RNA was isolated using RNeasy Lipid Tissue mini kit following vendor's protocol following which 1.5 µg RNA was converted to cDNA using High Capacity cDNA reverse transcription kit. The cDNA was then mixed with SYBR green PCR master mix and 20 µL was added to each well and the reaction was performed in ABI real time instrument. Subsequently, ΔΔCT was calculated after normalizing with the control gene.
PP2A activity assay
The PP2A activity in the tumor samples was quantified using a kit (Millipore, #17-313) following the manufacturer's protocol. Briefly, a standard curve was generated using Phosphate Standard -Solution C (Catalog # 20-103). The protein lysates were isolated from tissues using protein lysis buffer. PBS was replaced with TBS to avoid the background signal due to phosphate ions. 60 µg of protein lysate was incubated with 4 µg of Anti-PP2A antibody and 30 µL of Protein A agarose slurry followed by incubation for 1-2 h at 4 ºC with continuous shaking. Next, 60 µL of diluted phosphopeptide (750 µM) and 20 µL of assay buffer were added and incubated for 10 min at 30 °C in a shaking incubator. The mixture was centrifuged and 25 µL was transferred into each well of a 96-well plate and 100 µL of malachite green detection reagent was added and incubated for 10 min or till color developed and read at 650 nm.
Isolation of acinar cells
The pancreas of mice was dissected, minced, and washed with 0.01% trypsin inhibitor (Invitrogen, #17075029) in PBS followed by DMEM containing antibiotics and 5 mg/mL BSA fraction V (Thermo Fisher Scientific, #15260-037). 10 mL of digestive solution containing 1 mg/mL collagenase (Sigma, #C2139) and 0.2 mg/mL BSA fraction V was added and incubated for 30 min at 37 °C (120-140 cycles/min). The disrupted cells were grown in DMEM containing 10% FBS, 0.1 mg/mL trypsin inhibitor and 5 mg/mL BSA fraction V.
Statistical analysis
Various in vitro assays were performed at least three independent times with a minimum of 3-4 technical replicates in each treatment condition. Statistical analysis was done using either unpaired t-test or one-way ANOVA followed by post-hoc Tukey test using GraphPad 6.0 software. IHC quantification of (TUNEL, Ki-67, p65, cyclin D1, and β-catenin) was performed as follows. For the statistical analysis, we counted 2000 cells (40× magnification) from each group (except for vimentin which is 800 cells/group). α-SMA and Sirius red staining intensity were calculated using ImageJ software.
Results
FTY720 induced apoptosis in pancreatic cancer cell lines but not in primary pancreatic cells
In view of the FDA approval of FTY720, a functional S1PR1 antagonist for multiple sclerosis, we used a drug repurposing approach to study the potential benefits of FTY720 in pancreatic cancer. We initially performed a dose response study to test the viability of different human and mouse pancreatic cancer cell lines. Treatment of cells with increasing concentrations of FTY720 ranging from 0 to 20 µM significantly inhibited the growth of all pancreatic cancer cell lines tested (Figure 1A). Also, we did not observe any significant cytotoxicity of FTY720 in mouse primary acinar cells and HDPE cells (Figure S1A-B). The IC50 ranged from 5-10 µM. We selected MIA PaCa-2 and PAN 02 cells for subsequent analysis.
FTY720 inhibited the growth and induces apoptosis in pancreatic cancer cell lines. (A) Human and mouse pancreatic cancer cells were treated with indicating concentrations of FTY720 (for 24 and 48 h) and cell survival was analyzed using MTT assay. (B) MIA PaCa-2 cells were treated with FTYT720 (10µM and 15µM) and the rate of apoptosis was quantified after 24h using annexin V-FITC staining. Scatter plot from FACS (upper panel) and quantification of live and apoptotic population (lower panel).(C) The effect of FTY720 on cell cycle was quantified using propidium iodide staining after treating MIA PaCa-2 cells with 10µM of FTY720. (D) MIA PaCa-2 cells were treated with FTY720 alone and the increased ROS positive population was quantified using DCFDA staining. (E) The anti-migratory capability of FTY720 was checked using wound healing assay. Briefly, a scratch was made when the cells reach 90% confluence and treated with an increased dose of FTY720 and the closure of wound was monitored at 12h and 24 h of time intervals. Representative images (left panel) and quantification (right panel). Data is presented as mean ± S.D and representative data from at least 3 independents experiments is shown, Statistical significance was calculated using t-test * p≤0.001.
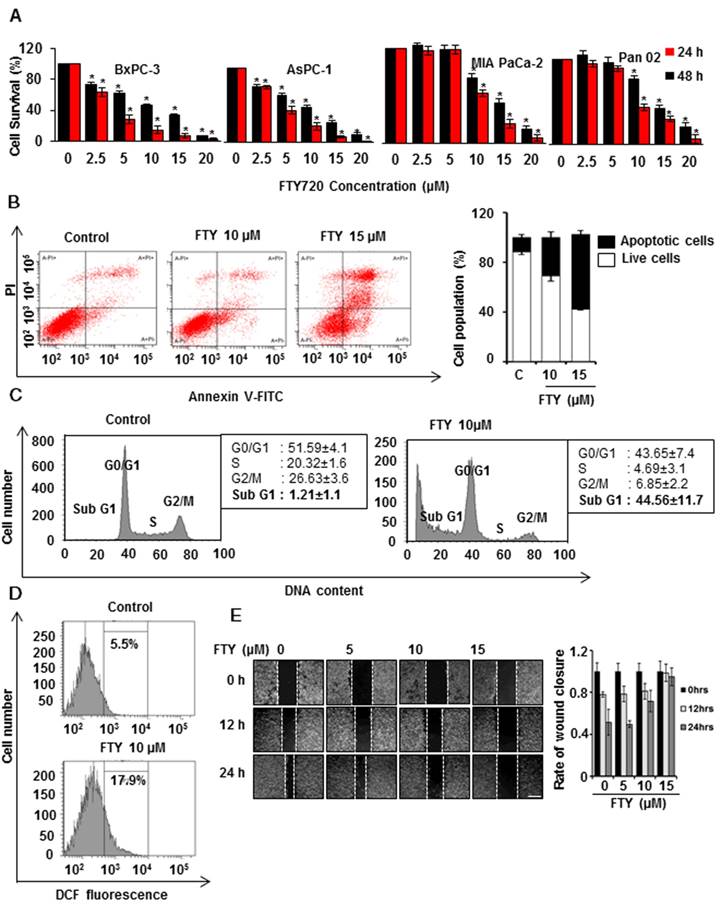
Next, we examined the possible mechanism of FTY720-mediated cell death in vitro. Annexin V5 staining was used to differentiate between apoptosis and necrosis and showed an increase in the apoptotic cell population after treatment with FTY720 (Figure 1B and Figure S1C) which also induced cell cycle arrest in both MIA PaCa-2 and PAN 02 cell lines in SubG1 phase (Figure 1C and Figure S1D). Further, we analyzed any involvement of reactive oxygen species (ROS) in FTY720 induced apoptosis and observed higher ROS levels after the treatment (Figure 1D). FTY720 is known to inhibit S1PR1 which plays an important role in cell migration. We, therefore, performed a wound healing assay to assess the effect of FTY720 on pancreatic cancer cell migration. We observed a marked inhibition of migration potential of FTY720-treated cells which was evident from the decrease in wound closure area (Figure 1E and Figure S2A)
FTY720 enhanced cell death induced by gemcitabine
Combination therapy is gaining more attention due to acquired drug resistance when treated with a single agent. We, therefore, investigated whether FTY720 could be used as a combinatorial agent with gemcitabine which is one of the major chemotherapy drugs for pancreatic cancer. The MTT assay using different concentrations of FTY720 and gemcitabine (Figure 2A) showed that 10 µM of the combination of both drugs induced maximum cell death; however, there was no synergistic effect of the doses tested. We performed DiOC6(3) staining to assess the loss of mitochondrial membrane potential (MMP) which is considered to be a marker for apoptosis. We found that both FTY720 and gemcitabine, either alone or in combination, promoted mitochondrial depolarization. As compared to control cells, the combination induced a statistically significant depolarization indicating the involvement of mitochondria in FTY720-driven apoptosis (Figure 2B and Figure S3A). Also, a colony formation assay to check the proliferative capacity of MIA PaCa-2 and PAN 02 cells after the treatment revealed that FTY720+Gemcitabine drastically inhibited the tumorigenic potential of MIA PaCa-2 cells (Figure 2C).
FTY720 enhanced the effects of gemcitabine in pancreatic cancer cell lines. (A) MIA PaCa-2 cells were treated with indicating concentrations of FTY720and gemcitabine (for 24h) and cell survival was analyzed using MTT assay. (B) MIA PaCa-2 cells were treated with FTY720 in combination with gemcitabine and the loss in mitochondrial membrane potential was quantified using DiCO6(3) staining after 24h. (C) The clonogenic potential of the cells was quantified after 24 h pre-treatment of cells with the drug alone and the combinations and incubating for 24h. Cells were collected and plated again as 1000 cells per well and allowed them to grow in drug free environment. Representative images were shown in left panel. Then the colonies were fixed with 10% neutral buffered formalin and stained with crystal violet. The quantification was performed using TECAN spectrophotometer at 590nm after melting the stained colonies (right panel). (D) MIA PaCa-2 cells were treated with FTY720 and in combination with gemcitabine for 24h and the expression of S1PR1, p-STAT3, STAT3, and c-Myc; EMT markers, E-cadherin, N-cadherin and Vimentin and proliferative markers CyclinD1 and Cox-2 expression were analyzed using western blotting.β-Tubulin served as internal control
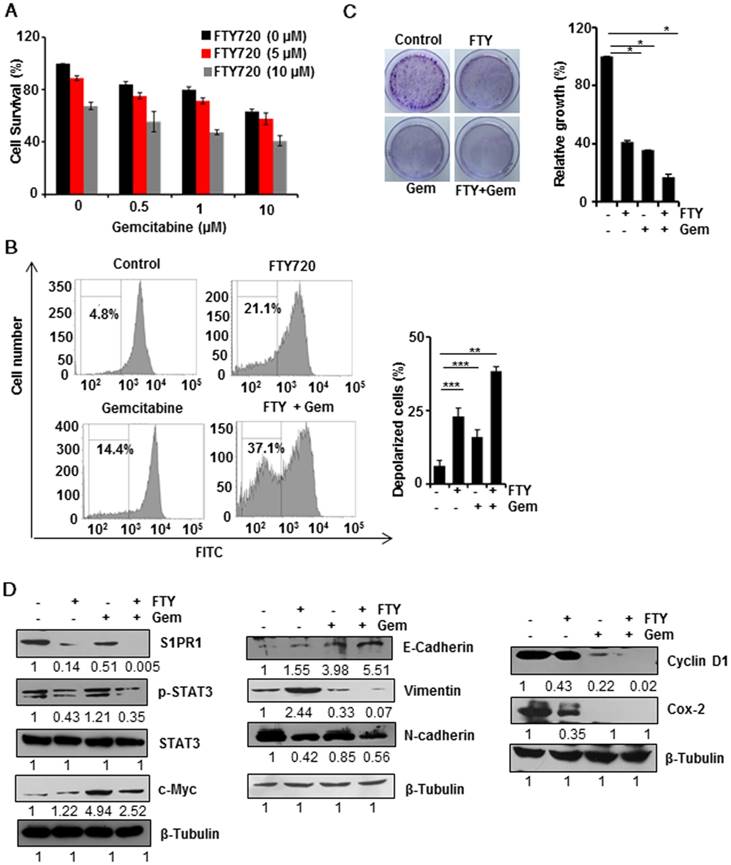
FTY720 and gemcitabine combination inhibited the S1PR1-STAT3 loop, EMT, and expression of cell proliferation markers in pancreatic cancer cells in vitro
We then analyzed the effect of FTY720 and gemcitabine combination on the expression of various signaling molecules using Western blotting. FTY720 either alone or in combination with gemcitabine reduced the expression of S1PR1 and phosphorylation of STAT3. However, the expression of a STAT3-dependent gene c-Myc was upregulated in the gemcitabine-treated group in the presence of activated STAT3 but downregulated with the combination treatment in pancreatic cancer cells. Together these results indicated that FTY720 +Gemcitabine interfered with persistent S1PR1/STAT3 loop in pancreatic cancer (Figure 2D). We then studied the individual and combined effect of FTY720 and gemcitabine in EMT phenotype and observed that combination regimen decreased the mesenchymal phenotype (Vimentin and N-cadherin) with a concomitant increase in the epithelial phenotype (E-cadherin) (Figure 2D). We next examined the effect of FTY720 on the expression of proliferation markers such as cyclin D1 and Cox-2 and detected a marked reduction in the expression of both proteins (Figure 2D). Collectively these results showed that FTY720 in combination with gemcitabine could reduce cell proliferation and effectively blocked mesenchymal transition in vitro.
FTY720 sensitized pancreatic cancer cells to gemcitabine in a preclinical model
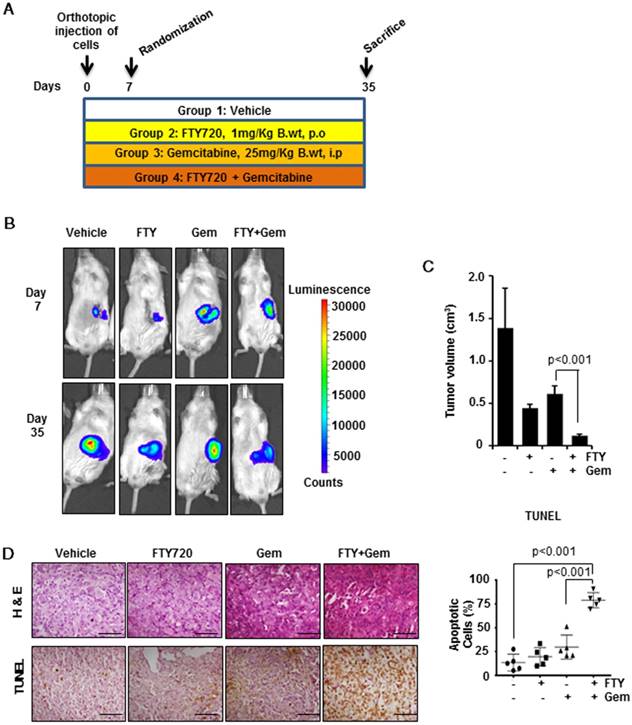
To confirm our in vitro findings, we developed a pre-clinical model of pancreatic cancer in immunodeficient SCID mice. The experimental scheme is shown in Figure 3A. Mice were injected orthotopically with luciferase-tagged MIA PaCa-2 cells. Mice were randomized on day 7 and treatment of FTY720 (1 mg/kg body weight, a therapeutically relevant dose) and gemcitabine was started and continued till mice were sacrificed. Animals were monitored for any adverse reactions to the compounds. The progression of tumors was monitored weekly by non-invasive bioluminescence imaging (Figure 3B and Figure S4A). Animals were sacrificed on day 35 and gross necropsy was performed (Figure S4B). Pancreatic tumors were excised from each animal and tumor volume was recorded. The tumor volume was statistically reduced in the combination group as compared to gemcitabine alone group (p<0.001) (Figure 3C). We also recorded the metastasis to major adjacent organs and abdominal cavity and the presence of ascites which is commonly seen in advanced metastatic pancreatic cancer patients (Table S1). There was dissemination of tumor cells to the abdominal cavity, liver, colon, and intestine which was significantly inhibited with FTY720 and gemcitabine combination. Severe ascites was seen in control and gemcitabine-treated animals that was markedly reduced (mild ascites) in the combination group of FTY720 and gemcitabine. These results collectively indicated the role of FTY720 in enhancing the effect of gemcitabine and reducing its side effects. The tissue sections were stained with hematoxylin-eosin and independently assessed by a pathologist. The vehicle-treated groups showed high grade adenocarcinoma. The FTY720-treated animals showed both apoptosis and necrosis whereas necrotic lesions were observed in gemcitabine-treated groups. In the FTY720 and gemcitabine combination group, we could observe a higher number of cells undergoing either apoptosis or necrosis (Figure 3D, upper panel).
FTY720 in combination with gemcitabine effectively reduced the tumor burden and increased apoptosis in an orthotopic model. (A) The schematic representation of the experimental procedure. 1×106 MIA PaCa2-luc cells were injected into the pancreas of NOD-SCID mice and after 7 days, they were grouped. The group1 received saline (100 µL orally, daily), group 2 FTY720 (1mg/kg B. wt. orally, daily), group 3 gemcitabine (25mg/kg B. wt., IP, twice a week) and group 4, a combination of FTY720 (1 mg/kg B. wt., orally, daily); gemcitabine (25 mg/kg B. wt., IP, twice a week). (B) Bioluminescence imaging of orthotopically implanted MIA PaCa-2 Luc cells in live mice (day 7 and Day 35). (C) The tumor volumes in mice measured on last day expressed in cm3. (D) The paraffin embedded sections were stained with hematoxylin and eosin (top panel) and the rate of apoptosis was measured in vivo using TUNEL assay (bottom panel). The quantification of TUNEL positive cells in each group (right panel). Statistical analysis was performed using one way ANOVA followed by post hoc Tukey test. (* p≤0.001)
FTY720 decreased cell proliferation and increased apoptosis in pancreatic cancer
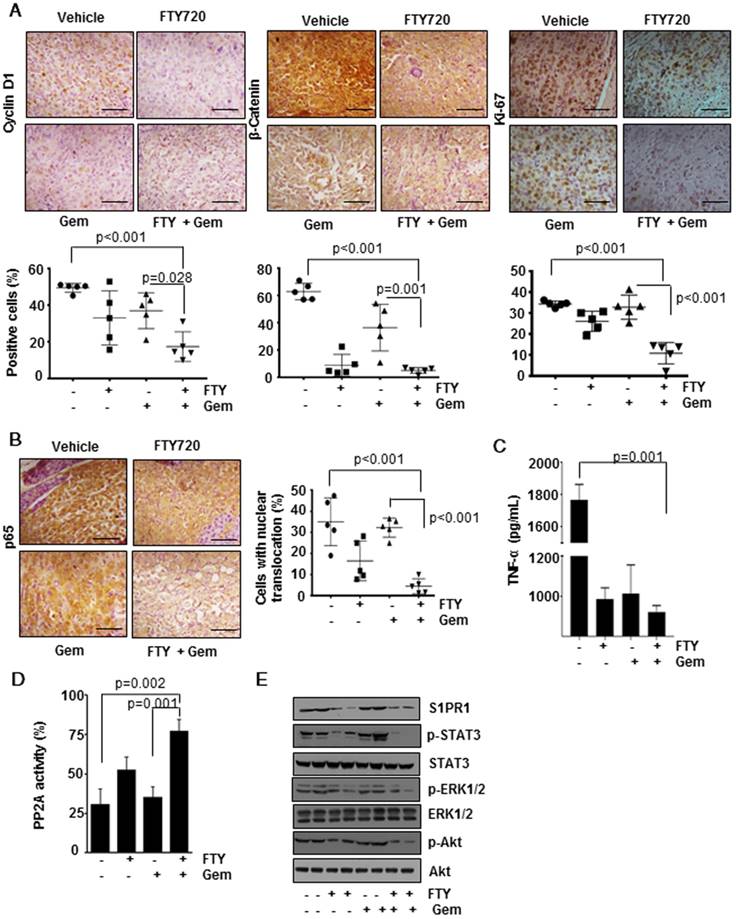
The process of tumorigenesis is a balancing act between cell proliferation and apoptosis. We analyzed apoptosis in vivo by the TUNEL assay (Figure 3D, lower panel) and found significantly greater number of TUNEL-positive cells in the combination group (p<0.001) indicating a higher rate of apoptosis. To verify the presence of highly proliferating cells, we performed immunohistochemical analysis of Cyclin D1 (Figure 4A, left panel), β-Catenin (Figure 4A, center panel) and Ki-67 (Figure 4A, right panel). We observed a significantly decreased cell proliferation (p<0.001) in FTY720 plus gemcitabine group. We manually counted positive cells and data were expressed as the % of positive cells out of total cells. Statistical analysis indicated that FTY720 combined with gemcitabine treatment was better than gemcitabine alone in decreasing the cell proliferation.
FTY720 in co-operation with gemcitabine inhibited cell proliferation and downregulated the activation of NF-kB and S1PR1/STAT3 pathways and PP2A activation in pancreatic cancer. (A) Immunohistochemical analysis of proliferative markers cyclin D1, β-catenin and Ki-67 in tissue samples (upper panel). The quantification positive cells are shown lower panel. (B) FTY treatment inhibited the activation of NF-κB and NF-κB dependent gene expression. Immunohistochemical analysis of nuclear translocation of p65, representative image (left panel) and quantification of nuclear positive cells (right panel). Statistical analysis was performed using one way ANOVA followed by post hoc Tukey test; * p≤0.001 vs control. (C) The circulatory TNF in serum was measured using ELISA. Statistical significance was calculated using t-test, * p≤0.001 vs control. (D) PP2A enzyme activity was measured using a commercially available kit. Statistical significance was calculated using t-test, ** p≤0.05 vs control. (E) FTY720 inhibited the S1PR1/STAT3 loop and downstream signaling in pancreatic cancer. Immunoblot analysis showing the expression of indicated proteins in tissue lysates of pancreatic cancer samples.
FTY720 inhibited NF-kB signaling and NF-kB-dependent gene expression
Next, we examined the effect of FTY720 treatment on NF-kB, a versatile transcription factor which regulates the expression of several genes involved in the process of inflammation and cancer [18]. Immunohistochemical detection of p65 translocation to nucleus revealed that in both vehicle and gemcitabine-treated groups the number of cells with positive nuclear expression of p65 was dramatically increased (Figure 4B) but was significantly decreased in the combination group (p<0.001 vs vehicle). Further, we measured the levels of TNF-α, a well-known target gene of NF-kB, in the sera of animals using an ELISA method and found that FTY720 either alone or in combination was effective in decreasing the TNF production (Figure 4C). We also profiled the expression of 84 genes (Table S2) using an RT2 profiler array. We selected the genes whose expression was regulated by NF-kB and found that combination treatment was able to downregulate the NF-kB-dependent gene expression further supporting our observations (Figure 5A).
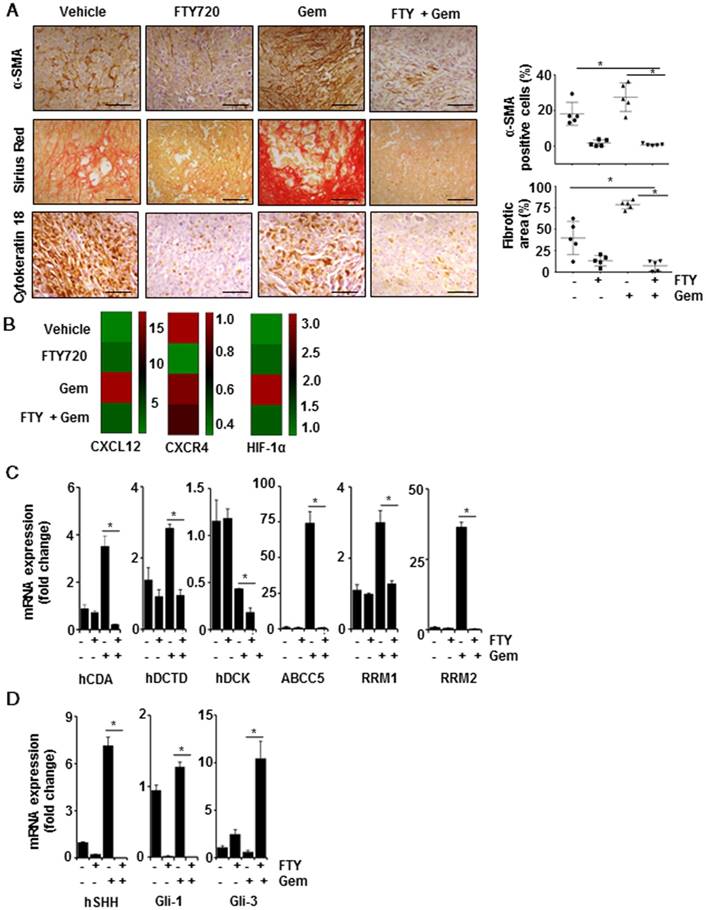
FTY720 sensitizes tumors towards gemcitabine through altered desmoplasia and reduced Shh signaling. (A) Representative images of tissue sections of α-SMA (top panel), quantification of positive cells (right panel), Sirius red (central panel), quantification of fibrotic area (right panel) and Cytokeratin-18 (lower panel). Right panels represent the quantification data using imageJ software. Statistical analysis was performed using one way ANOVA followed by post hoc Tukey test. (* p≤0.001). (B) RT2 Profiler PCR Array was used to measure the mRNA expression of CXCL12, CCR4, HIF1α and the data was represented in heat map. (C) Real time PCR data of major gemcitabine metabolizing genes,β-actin was served as housekeeping gene, Statistical significance was calculated using t-test, * p≤0.001 vs vehicle. (D) Real time PCR data of showing the expression of Shh, Gli-1 and Gli-3 in pancreatic cancer tissue samples. β-actin was served as housekeeping gene, Statistical significance was calculated using t-test, * p≤0.001 vs vehicle.
FTY720 reactivated PP2A in pancreatic cancer
Accumulating evidence demonstrated that PP2A is a tumor suppressor protein and its expression and activity is downregulated in many cancers [19]. FTY720 is a known activator of PP2A but whether the same mechanism is involved in the context of pancreatic cancer is not known. To verify this, western blotting performed to determine the expression of PP2A protein levels and it did not reveal any significant difference between the four groups (data not shown). Further, an enzymatic assay revealed a considerable decrease in PP2A activity in the gemcitabine-treated group. Treatment with FTY720 either alone or in combination with gemcitabine reactivated PP2A as seen from enhanced activity as compared to gemcitabine alone group (Figure 4D).
FTY720 treatment abrogated downstream signaling cascade through the S1PR1-STAT3 loop
Next, we sought to understand the role of the S1PR1-STAT3 loop which is one of the major signalling pathways in maintaining tumorigenicity in many cancers. Consistent with the available information in the literature, we observed that FTY720 treatment reduced the expression of S1PR1 in pancreatic cancer tissue samples. This was related to p-STAT3 levels whose expression was abolished by treatment with FTY720 and gemcitabine (Figure 4E). We also analyzed the expression of gene targets downstream of S1P/S1PR1 which were shown to be involved in tumor progression and metastasis such as p-ERK1/2, and p-Akt [20]. FTY720, either alone or in combination with gemcitabine, dramatically reduced the S1P/S1PR1-mediated signaling pathways. These results emphasize the role of FTY720 in inhibiting the activation of pathways mediated by S1PR1/STAT3 loop in pancreatic cancer.
FTY720 inhibited desmoplasia and myofibroblastic features
One of the fundamental characteristics of pancreatic cancer is the presence of abundant desmoplasia and myofibroblasts (the activated form of pancreatic stellate cells; PSCs) which are mainly responsible for collagenous stroma in pancreatic cancer [21, 22]. To understand the distribution of myofibroblasts, we stained the tissue samples with α-SMA, which is considered as an activation marker for PSCs, and found that majority of cells had a high level of α-SMA expression that was significantly reduced in FTY720 treated samples (Figure 5A, upper panel, p<0.001 vs Vehicle). Subsequently, we stained the tissue sections with Sirius red which detects the presence of collagen. As compared to the vehicle-treated group, animals treated with gemcitabine alone displayed a large area positive for collagen staining associated with fibrosis-like pathology. These pathological changes were largely alleviated in FTY720- treated groups (Figure 5A, center panel, p<0.001 vs vehicle). Myofibroblasts serve as the primary source for extracellular matrix (ECM) in desmoplasia. We further stained the tissue sections with an epithelial marker cytokeratin-18 and observed high level of expression in vehicle- and gemcitabine-treated groups that was significantly decreased in both FTY720- treated groups (Figure 5A, lower panel).
Three genes, CXCR4, CXCL12 and HIF1α, in the RT2 profiler array, have been shown to have a direct link to fibrotic changes. Both CXCR4 and CXCL12 were highly upregulated in gemcitabine alone-treated animals compared to the rest of the groups. Both genes were reported to be involved in the activation of myofibroblasts in different tissue types and CXCR4 agonists are currently in clinical trials for pancreatic cancer [23]. HIF-1α is yet another factor which regulates CXCR4, pro-fibrosis and ECM deposition [24]. Our array data demonstrated that HIF1α expression was elevated in gemcitabine groups and was significantly downregulated in FTY720-treated animals (Figure 5B). Taken together, our results confirmed that FTY720 was effective in preventing the desmoplasia, deposition of ECM and activation of PSCs, thereby reducing the adverse effects of gemcitabine.
FTY720 modulated the expression of enzymes in gemcitabine metabolism
We explored the metabolic status of gemcitabine in the tumor microenvironment after reduced desmoplasia. Gemcitabine inhibits DNA synthesis by incorporating into the DNA in its activated dFdCTP form. We performed real-time PCR to examine the expression of genes involved in this activation process. Primer sequences used for qPCR are given in Table S3. We detected reduced expression of deoxycytidine kinase (DCK) in the gemcitabine-treated group which showed the acquired gemcitabine resistance. Also, FTY720 effectively decreased the expression of deoxycytidine monophosphate deaminase (DCTD) and cytidine deaminase (CDA) (gemcitabine-inactivating enzymes). Almost 90% of the gemcitabine was converted to 2′-deoxy-2′,2′-difluorouridine (dFdU), an inactivated form in the presence of CDA [25], and FTY720 effectively decreased the CDA expression even in the presence of gemcitabine. Also, ribonucleotide reductases M1 and M2 (RRM1 and RRM2) act as biomarkers for gemcitabine resistance and their increased expression has been shown to lead to gemcitabine resistance [26]. FTY720 reduced the expression of both genes. One of the major reasons for acquired gemcitabine resistance is its efflux from the cells by various ABC transporters. Gemcitabine treatment increased ABCC5 expression which effluxes the drug outside the cells [27]. We found lowered expression of gemcitabine efflux pump, ABCC5, in the presence of the combination treatment with gemcitabine and FTY720 indicating the localization of gemcitabine inside the cells (Figure 5C). Together these results showed that in the presence of FTY720, the availability of the active form of gemcitabine within tumor microenvironment would be higher through inhibition of desmoplasia, gemcitabine-metabolizing enzymes, and transporter proteins leading to a decrease in gemcitabine resistance.
FTY720 inhibited S1PR1/STAT3 loop-mediated Sonic-hedgehog (Shh) signaling in pancreatic cancer
The results so far indicated that FTY720 could suppress the stromal resistance and desmoplasia through suppression of S1PR1/STAT3 loop. To further understand how S1PR1/STAT3 pathway regulates the desmoplasia, we evaluated the role played by Shh signaling. We observed that mRNA transcripts of both Shh and Gli-1 were significantly upregulated while Gli-3, a known suppressor of desmoplastic changes, was considerably downregulated in the group treated with gemcitabine alone (Figure 5D). The role of STAT3 in Shh activation and stromal reactions was previously established by other groups [28]. Collectively, these data suggested that inhibition of S1PR1/STAT3 loop resulted in inactivation of Shh pathway and desmoplasia, reduced resistance to gemcitabine, and improved the delivery of gemcitabine to tumor sites.
FTY720 inhibited EMT in pancreatic cancer
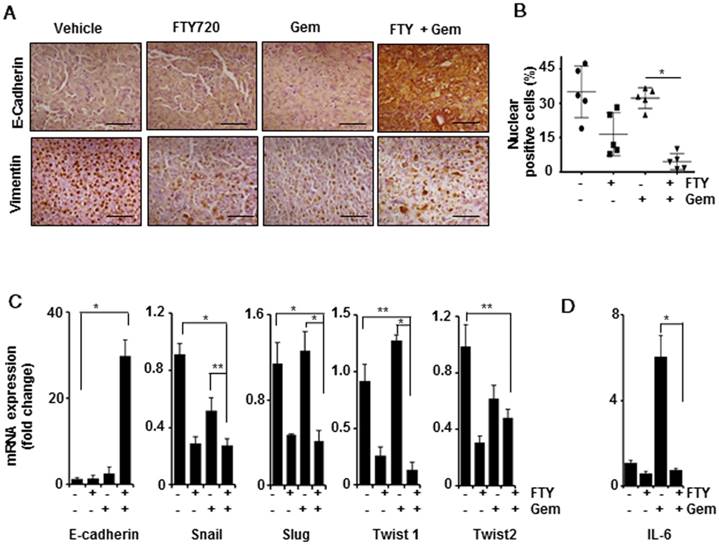
It has been reported that mesenchymal population showed higher resistance towards gemcitabine leading to chemoresistance in pancreatic cancer and inhibition of EMT resulted in enhanced sensitivity to gemcitabine [29]. We examined whether FTY720 treatment altered the EMT signature in pancreatic cancer. Upon staining the tissue sections with E-Cadherin and vimentin E-cadherin expression was found to be highly elevated in the combination group (Figure 6A). A limited number of positive cells were detected in the vehicle-treated group. In case of vimentin, we observed an abnormal nuclear localization in the vehicle-treated group. It was of note that the nuclear labelling of vimentin was reduced in all three treatment groups (* p≤0.001 vs control), however, cytoplasmic staining of vimentin could also be detected. We counted the cells with both cytoplasmic and nuclear vimentin and expressed the percentage of nuclear-positive vimentin in each group (Figure 6B). We then performed a quantitative PCR to study the mRNA expression levels of E-cadherin, Snail, Slug, Twist 1, and Twist 2. Primer sequences are presented in Table S3. The E-cadherin levels were statistically upregulated over 30-fold in the FTY720 + gemcitabine-treated group (Figure 6C). The remaining four EMT markers were also downregulated in the combination group as compared to gemcitabine alone group indicating that the combination treatment was more effective in inhibiting mesenchymal phenotype and thereby reversing the malignant nature of pancreatic cancer. IL-6, a STAT3-dependent gene, was shown to be involved in the process of mesenchymal transition [30]. We observed a significant upregulation of IL-6 mRNA transcript in the gemcitabine-treated group which may lead to chemoresistance (Figure 6D). Based on these observations, we concluded that FTY720 abrogated S1PR1/STAT3/IL-6 pathway and prevented the mesenchymal transition leading to improved drug sensitivity.
FTY720 in combination with gemcitabine inhibited the epithelial mesenchymal transition in pancreatic cancer. (A) Representative Immunohistochemical images of E-Cadherin (upper panel) and Vimentin (lower panel). (B) Quantification of nuclear positive Vimentin expression in all the groups. p≤0.001.(C) Real time PCR data of the various EMT markers, E-Cadherin, Snail, Slug, Twist1 and Twist2 (Left to right). β-actin was served as housekeeping gene, Statistical significance was calculated using t-test, * p≤0.001, **p≤0.01 vs vehicle
FTY720 regulated inflammation and immunity gene signature in pancreatic cancer
Inflammation and cancer are closely related and there is increasing evidence for the use of anti-inflammatory molecules in cancer management. FTY720 is an immunosuppressant drug and our in vitro data emphasized its role as an anti-inflammatory molecule. To determine anti-inflammatory properties of FTY720, we selected an 84-gene signature panel of genes primarily involved in inflammation. The genes with most significant changes in expression are shown in Supplementary Figure 5B-C. We observed that the combination treatment with FTY720 and gemcitabine was most effective as compared to either agent alone. Interestingly, there was a several-fold elevation in the expression of CCL4, PD1 (~4-fold), CXCR1, and IDO1 (~40-fold) in the gemcitabine-treated group as compared to the vehicle-treated animals. These genes were known to confer drug resistance [31, 32]. However, the expression of these genes was downregulated in the FTY720-treated group.
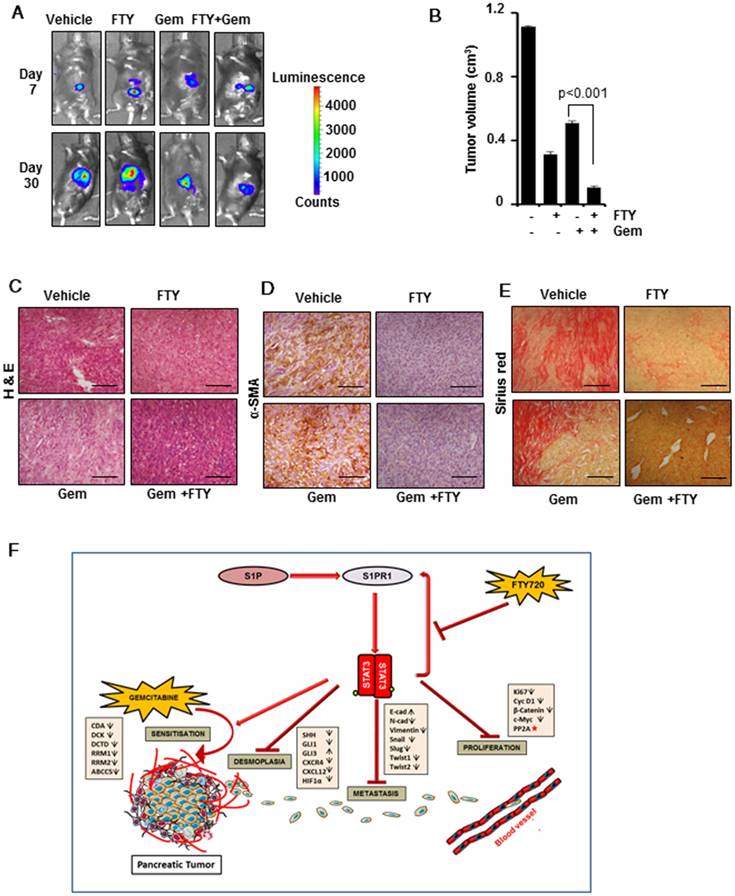
FTY720 acted in synergy with gemcitabine in a syngeneic model of pancreatic cancer
After elucidating the regulatory role of FTY720 in desmoplasia and EMT signature in pancreatic cancer, we next aimed to address the involvement of host immune system in FTY720-mediated tumor suppression and its co-operative action with gemcitabine in pancreatic cancer. We adopted a syngeneic model of pancreatic cancer in C57Bl/6 mice by injecting PAN 02 cells in the pancreas. We found that FTY720 either as a single agent or in combination with gemcitabine was effective in reducing the tumor volume (Figure 7A-C and Figure S6A-B) confirming that irrespective of the nature of immune system (intact or immunocompromised) FTY720 was able to reduce the tumor burden. We also stained for the markers of desmoplasia in PAN 02-derived tumor sections and found that gemcitabine increased the expression of α-SMA (Figure 7D and Figure S6C) and collagen. (Figure 7E and Figure S6D). The FTY720-treated animals showed reduced desmoplastic changes which further supported the beneficial effects of FTY720 in preventing desmoplasia in clinically relevant doses. A schematic illustration of pathways altered during the combination therapy is presented in Figure 7F.
FTY720 in combination with gemcitabine inhibited the tumor progression and desmoplasia in a syngeneic model of pancreatic cancer. (A) Representative bioluminescent images of the C57Bl/6 mouse implanted with PAN 02-luc (5×104 cells). (B) The tumor volumes in mice measured on last day expressed in cm3. (C) The paraffin embedded sections were stained with hematoxylin and eosin and representative H&E images of each group. (D) Representative images of tissue sections of α-SMA staining (left panel). (E) Representative images of tissue sections of Sirius red staining (right panel).Statistical analysis was performed using one way ANOVA followed by post hoc Tukey test; * p≤0.001 vs control. (F) Graphic illustration of mode of action of FTY720 in combination with gemcitabine in pancreatic cancer. FTY720 reduces the cell proliferation by decreasing the expression of certain proliferation markers and by activation of PP2A (red star). It also abrogates HIF1α/CXCL12/CXCL4 loop, Shh pathway resulting in reduced desmoplasia and metastasis. This reduction in desmoplastic content leads to increased accumulation of gemcitabine and thereby increased sensitivity through altering gemcitabine metabolizing genes.
Discussion
STAT3 is a major transcription factor which is under the control of various cytokines and growth factors and its activation provides survival advantage to cancer cells [11]. There is substantial evidence confirming the role of activated STAT3 in pancreatic cancer initiation, progression and metastasis. However, its role in inducing chemoresistance and the beneficial usage of its inhibitors in pancreatic cancer management were not fully elucidated. Targeting STAT3 in cancer through the proteins which have a regulatory role in STAT3 activation might represent a viable strategy. In this respect, S1PR1 appears to be an ideal candidate as blocking S1PR1 led to inhibition of STAT3 signaling in different tumor types [7, 11]. In the current study, we demonstrated that in vitro treatment of pancreatic cancer cells with FTY720, which indirectly blocks STAT3 activation, resulted in dose-dependent increased apoptosis. For possible successful clinical translation of the current findings, FTY720 was used in therapeutically relevant doses in pre-clinical models of pancreatic cancer [33]. One of the major obstacles is the development of resistance to the chemotherapeutic drug, gemcitabine, widely used in pancreatic cancer therapy [1]. Recent findings have shown an enhanced clinical activity of combination treatments with gemcitabine and other agents targeting signaling pathways associated with tumor microenvironment [34]. In the present study a statistically significant response in tumor regression was observed for the combination of gemcitabine and FTY720 in both models irrespective of host immune system.
NF-κB is another transcription factor which, together with STAT3, serves as a central hub for tumor progression and metastasis by regulating the expression of genes responsible for the production of various cytokines and chemokines associated with tumorigenesis [35]. The involvement of S1P in NF-κB signaling and production of pro-inflammatory cytokines was reported previously [36]. FTY720 inhibited the S1P/S1PR1 feed forward loop leading to decrease in STAT3 and NF-κB activation and pro-inflammatory cytokine production in colon cancer [11]. Our current study is the first report showing FTY720 as a potent inhibitor of NF-κB-STAT3 cross-talk in pancreatic cancer.
Desmoplasia has major clinical implications as it contributes to chemoresistance and shows a negative association between patient survival and ECM deposition [21]. In this study, we observed desmoplasia-associated changes in the gemcitabine-treated group leading to chemoresistance. One of the undesired side effects of gemcitabine therapy is the upregulation of CXCL12/CXCR4 axis in cancer cells providing the pro-survival advantage, aggressiveness, and conferring resistance to gemcitabine [37]. The gemcitabine-dependent activation of CXCR4 leads to accumulation of NF-kB and HIF1α in the nuclei of pancreatic cancer cells and NF-κB subsequently activates the transcription of both CXCR4 and HIF1 [37]. Interestingly, FTY720, alone and in combination with gemcitabine, was effective in reducing desmoplasia and expression of CXCR4, CXCL12, and HIF1α. FTY720 functioned as a CXCR4 agonist in our study for which the exact mechanism remains to be elucidated. Also, a direct correlation between enhanced expression of activated STAT3 and increased desmoplastic stroma was reported in human pancreatic cancer [28] and Shh was shown to be a direct target of STAT3 involved in the formation of desmoplasia [38]. We observed that S1PR1 was an important factor for persistent activation of STAT3 and FTY720 and gemcitabine combination was effective in down-regulating STAT3 expression leading to reduced Shh levels and in minimizing desmoplasia. Currently, stromal targeting in chemotherapeutic interventions for pancreatic cancer is gaining interest [34]. Our study, for the first time, reported the beneficial effects of clinically relevant doses of FTY720 in preventing desmoplasia. Furthermore, by inhibiting the desmoplastic changes, ECM deposition, and modulating the expression of gemcitabine-metabolizing enzymes, FTY720 reduced the resistance and improved the intra-tumor delivery of gemcitabine through stromal remodelling.
PP2A is considered as a tumor suppressor gene and negatively regulates many pathways involved in cell survival, apoptosis, and drug resistance in a wide array of cancer types including pancreatic cancer [39]. However, in most cancers, PP2A is either functionally deactivated or genetically altered. Farrell et al reported that PP2A activity was decreased in pancreatic cancer and restoring its activity reduced the tumorigenic potential of pancreatic cancer cells [39]. Our results showed that FTY720, either alone or in combination with gemcitabine, activated PP2A. Hence, regulating the activity of PP2A and modulating the downstream oncogenic signaling is another mechanism through which FTY720 exerts its anticancer activity.
EMT is another characteristic feature which contributes to early stage dissemination, invasion, metastasis, and drug resistance in pancreatic cancer [40]. Suppression of transition to mesenchymal phenotype leads to enhanced sensitivity to gemcitabine treatment associated with a decrease in cancer cell proliferation. The role of FTY720 in the reversal of EMT is still largely an unexplored area. In this study, we observed statistically significant downregulation of EMT markers in FTY720 + gemcitabine-treated animals. Also, E-cadherin was upregulated and the dissemination and peritoneal metastasis of tumor cells were significantly inhibited in the groups treated with the combination of drugs in both models tested. Collectively, the results indicated that FTY720 inhibited metastasis by reducing EMT and enhancing the activity of gemcitabine in mouse models of pancreatic cancer.
We profiled the major genes involved in inflammation and immunity in pancreatic cancer and detected several-fold upregulation of genes known to be involved in drug resistance in gemcitabine-treated animals. Indoleamine 2,3-dioxygenase-1 (IDO) was reported to be involved in tryptophan metabolism and its over-expression was associated with decreased overall disease-free survival and acquired immune response [32]. Currently, an IDO inhibitor (Indoximod) is in clinical trials together with gemcitabine for metastatic pancreatic cancer (NCT02077881). Here we observed that FTY720 and gemcitabine combination was very effective in downregulating IDO1 expression.
The dose of FTY720 used in this study was 1 mg/kg body weight which is equivalent to 5.6 mg/d in humans similar to that used in clinical trials for multiple sclerosis [9]. A further reduction in tumor volume can be predicted with increase in drug dose. However, for a successful clinical translation, we limited our study to clinically relevant dose of FTY720.
In summary, we demonstrated that FTY720 in combination with gemcitabine was effective in the pre-clinical model of pancreatic cancer. Possible mechanisms for the observed beneficial effects of FTY720 include (a) enhancing the activity of gemcitabine and inhibiting the proliferation of pancreatic cancer cells, (b) suppressing the S1PR1/STAT3 loop, Shh signaling, EMT and desmoplasia thus significantly down-regulating the major pathways responsible for drug resistance and metastasis, and (c) restoring the activity and function of the tumor suppressor PP2A. Furthermore, the combination of FTY720 and gemcitabine was found to have a better efficacy than either agent alone.
Therefore, it is worthwhile to consider repurposing of FTY720, an orally bioavailable FDA approved drug, either alone or in combination with gemcitabine for the treatment of pancreatic cancer.
Abbreviations
COX-2: cyclooxygenase-2; CXCL12: C-X-C motif chemokine ligand 12; CXCR4: C-X-C chemokine receptor type 4; DMSO: dimethyl sulfoxide; EGF: epidermal growth factor; EMT: epithelial-mesenchymal transition; ERK-1/2: phosphor-extracellular signal-regulated kinase-1/2; FACS: fluorescence-activated cell sorting; FDA: food and drug administration; FITC: fluorescein isothiocyanate; HIF-1α: hypoxia inducible factor-1 alpha, KRAS: Kirsten rat sarcoma; MTT: [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; PP2A: protein phosphatase 2A; S1P: sphingosine-1 phosphate; S1PR1: Sphingosine-1-phosphate receptor 1; Shh: sonic hedgehog.SphK1: sphingosine kinase 1; STAT3: signal transducer and activator of transcription 3; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labelling; α-SMA: alpha-smooth muscle actin.
Acknowledgements
Authors express their gratitude to Professor M. Radhakrishna Pillai, Director, RGCB for sharing resources and useful discussions. We are also grateful to the personnel of Flow cytometry core facility and Bio-imaging facility for their excellent technical assistance and Animal research facility for mouse colony maintenance. We also acknowledge Dr. Florencia McAllister, UT M. D. Anderson Cancer Center, Houston for providing HDPE cells Dr. S. Lakshmi, Regional Cancer Centre, Thiruvananthapuram and Dr. Ruby John Anto, RGCB for sharing reagents and members of K.B.H lab for critical reading of the manuscript. M.B.L and S.M acknowledge senior research fellowship from University Grant Commission and Indian Council of Medical Research respectively. M.B.L also acknowledges the members of Doctoral Advisory Committee for their suggestions. This work was supported by a research grant from Department of Biotechnology (No.BT/PR6965/BRB/10/1145/2012) and in part by faculty start-up grant and DBT-Ramalingaswami fellowship (No.BT/RLF/Re-entry/38/2011) to K.B.H.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73-85
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21
3. Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58-67
4. Yester JW, Tizazu E, Harikumar KB, Kordula T. Extracellular and intracellular sphingosine-1-phosphate in cancer. Cancer Metastasis Rev. 2011;30:577-97
5. Bi Y, Li J, Ji B, Kang N, Yang L, Simonetto DA. et al. Sphingosine-1-phosphate mediates a reciprocal signaling pathway between stellate cells and cancer cells that promotes pancreatic cancer growth. Am J Pathol. 2014;184:2791-802
6. Aoki H, Aoki M, Katsuta E, Ramanathan R, Idowu MO, Spiegel S. et al. Host sphingosine kinase 1 worsens pancreatic cancer peritoneal carcinomatosis. J Surg Res. 2016;205:510-7
7. Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A. et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nature Med. 2010;16:1421-8
8. Liu Y, Deng J, Wang L, Lee H, Armstrong B, Scuto A. et al. S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood. 2012;120:1458-65
9. Kappos L, Antel J, Comi G, Montalban X, O'Connor P, Polman CH. et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124-40
10. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V. et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355-60
11. Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC. et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107-20
12. Yasui H, Hideshima T, Raje N, Roccaro AM, Shiraishi N, Kumar S. et al. FTY720 induces apoptosis in multiple myeloma cells and overcomes drug resistance. Cancer Res. 2005;65:7478-84
13. Gstalder C, Ader I, Cuvillier O. FTY720 (Fingolimod) Inhibits HIF1 and HIF2 Signaling, Promotes Vascular Remodeling, and Chemosensitizes in Renal Cell Carcinoma Animal Model. MolCancer Ther. 2016;15:2465-74
14. Shen Y, Cai M, Xia W, Liu J, Zhang Q, Xie H. et al. FTY720, a synthetic compound from Isaria sinclairii, inhibits proliferation and induces apoptosis in pancreatic cancer cells. Cancer Lett. 2007;254:288-97
15. Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO. et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PloS one. 2009;4:e6529
16. Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, Pandey MK. et al. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int J Cancer. 2010;127:257-68
17. Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J. et al. De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 2017;77:5503-17
18. Sethi G, Sung B, Aggarwal BB. Nuclear factor-kappaB activation: from bench to bedside. Exp Biol Med (Maywood). 2008;233:21-31
19. Grech G, Baldacchino S, Saliba C, Grixti MP, Gauci R, Petroni V. et al. Deregulation of the protein phosphatase 2A, PP2A in cancer: complexity and therapeutic options. Tumour Biol. 2016;37:11691-700
20. Nagahashi M, Yamada A, Katsuta E, Aoyagi T, Huang WC, Terracina KP. et al. Targeting the SphK1/S1P/S1PR1 axis that links obesity, chronic inflammation and breast cancer metastasis. Cancer Res. 2018;78:1713-25
21. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C. et al. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res. 2015;21:3561-8
22. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579-96
23. Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31-82
24. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A. et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391-402
25. Ueno H, Kiyosawa K, Kaniwa N. Pharmacogenomics of gemcitabine: can genetic studies lead to tailor-made therapy? Br J Cancer. 2007;97:145-51
26. Minami K, Shinsato Y, Yamamoto M, Takahashi H, Zhang S, Nishizawa Y. et al. Ribonucleotide reductase is an effective target to overcome gemcitabine resistance in gemcitabine-resistant pancreatic cancer cells with dual resistant factors. J Pharmacol Sci. 2015;127:319-25
27. Oguri T, Achiwa H, Sato S, Bessho Y, Takano Y, Miyazaki M. et al. The determinants of sensitivity and acquired resistance to gemcitabine differ in non-small cell lung cancer: a role of ABCC5 in gemcitabine sensitivity. Molecular Cancer Ther. 2006;5:1800-6
28. Wormann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Gorgulu K. et al. Loss of P53 function activates JAK2-STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology. 2016;151:180-93 e12
29. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
30. Siddiqui I, Erreni M, Kamal MA, Porta C, Marchesi F, Pesce S. et al. Differential role of Interleukin-1 and Interleukin-6 in K-Ras-driven pancreatic carcinoma undergoing mesenchymal transition. Oncoimmunology. 2018;7:e1388485
31. Chen L, Fan J, Chen H, Meng Z, Chen Z, Wang P. et al. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci Rep. 2014;4:5911
32. Zhang T, Tan XL, Xu Y, Wang ZZ, Xiao CH, Liu R. Expression and Prognostic Value of Indoleamine 2,3-dioxygenase in Pancreatic Cancer. ChinMed J. 2017;130:710-6
33. Copland DA, Liu J, Schewitz-Bowers LP, Brinkmann V, Anderson K, Nicholson LB. et al. Therapeutic dosing of fingolimod (FTY720) prevents cell infiltration, rapidly suppresses ocular inflammation, and maintains the blood-ocular barrier. Am J Pathol. 2012;180:672-81
34. Carr RM, Fernandez-Zapico ME. Pancreatic cancer microenvironment, to target or not to target? EMBO Mol Med. 2016;8:80-2
35. Greten FR, Weber CK, Greten TF, Schneider G, Wagner M, Adler G. et al. Stat3 and NF-kappaB activation prevents apoptosis in pancreatic carcinogenesis. Gastroenterology. 2002;123:2052-63
36. Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY. et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084-8
37. Arora S, Bhardwaj A, Singh S, Srivastava SK, McClellan S, Nirodi CS. et al. An undesired effect of chemotherapy: gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factor kappaB- and hypoxia-inducible factor 1alpha-mediated up-regulation of CXCR4. J Biol Chem. 2013;288:21197-207
38. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457-61
39. Farrell AS, Allen-Petersen B, Daniel CJ, Wang X, Wang Z, Rodriguez S. et al. Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Mol Cancer Res. 2014;12:924-39
40. Elaskalani O, Razak NB, Falasca M, Metharom P. Epithelial-mesenchymal transition as a therapeutic target for overcoming chemoresistance in pancreatic cancer. World J Gastrointest Oncol. 2017;9:37-41
Author contact
![]() Corresponding author: K. B. Harikumar, Ph. D., Cancer Research Program, Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram, Kerala State, 695014, India. Ph: +91-471-2529-596; Fax: + 91-471-2346-333; E-Mail: harikumarres.in
Corresponding author: K. B. Harikumar, Ph. D., Cancer Research Program, Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram, Kerala State, 695014, India. Ph: +91-471-2529-596; Fax: + 91-471-2346-333; E-Mail: harikumarres.in