Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Methods
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(16):4409-4428. doi:10.7150/thno.26467 This issue Cite
Research Paper
Suppression of metastasis through inhibition of chitinase 3-like 1 expression by miR-125a-3p-mediated up-regulation of USF1
Ki Cheon Kim1#, Jaesuk Yun2#, Dong Ju Son1, Ji Young Kim1, Jae-Kyung Jung1, Jeong Soon Choi1, Yu Ri Kim1, Ju Kyung Song1, Sun Young Kim1, Sin Kook Kang1, Dae Hwan Shin1, Yoon-Seok Roh1, Sang-Bae Han1, ![]() , Jin Tae Hong1,
, Jin Tae Hong1, ![]()
1. College of Pharmacy and Medical Research Center, Chungbuk National University, Osongsaengmyeong 1-ro 194-21, Osong-eup, Heungduk-gu, Cheongju, Chungbuk, 28160, Republic of Korea
2. Department of Pharmacy, Wonkwang University, 460 Iksandae-ro, Iksan, Jeonbuk, 54538, Republic of Korea
#These authors contributed equally to this work.
Received 2018-4-3; Accepted 2018-7-15; Published 2018-8-7
Abstract

Rationale: Chitinase 3-like 1 (Chi3L1) protein is up-regulated in various diseases including solid cancers. According to Genome-Wide Association Study (GWAS)/Online Mendelian Inheritance in Man (OMIM)/Differentially Expressed Gene (DEG) analyses, Chi3L1 is associated with 38 cancers, and more highly associated with cancer compared to other oncogenes such as EGFR, TNFα, etc. However, the mechanisms and pathways by which Chi3L1 is associated with cancer are not clear. In current study, we investigated the role of Chi3L1 in lung metastasis.
Methods: We performed the differentially expressed gene analysis to explore the genes which are associated with Chi3L1 using the web-based platform from Biomart. We investigated the metastases in lung tissues of C57BL/6 mice injected with B16F10 melanoma following treatment with Ad-shChi3L1. We also investigated the expression of USF1 and Chi3L1 in Chi3L1 KD mice lung tissues by Western blotting and IHC. We also analyzed lung cancer cells metastases induced by Chi3L1 using migration and cell proliferation assay in human lung cancer cell lines. The involvement of miR-125a-3p in Chi3L1 regulation was determined by miRNA qPCR and luciferase reporter assay.
Results: We showed that melanoma metastasis in lung tissues was significantly reduced in Chi3L1 knock-down mice, accompanied by down-regulation of MMP-9, MMP-13, VEGF, and PCNA in Chi3L1 knock-down mice lung tissue, as well as in human lung cancer cell lines. We also found that USF1 was conversely expressed against Chi3L1. USF1 was increased by knock-down of Chi3L1 in mice lung tissues, as well as in human lung cancer cell lines. In addition, knock-down of USF1 increased Chi3L1 levels in addition to augmenting metastasis cell migration and proliferation in mice model, as well as in human cancer cell lines. Moreover, in human lung tumor tissues, the expression of Chi3L1 was increased but USF1 was decreased in a stage-dependent manner. Finally, Chi3L1 expression was strongly regulated by the indirect translational suppressing activity of USF1 through induction of miR-125a-3p, a target of Chi3L1.
Conclusion: Metastases in mice lung tissues and human lung cancer cell lines were decreased by KD of Chi3L1. USF1 bound to the Chi3L1 promoter, however, Chi3L1 expression was decreased by USF1, despite USF1 enhancing the transcriptional activity of Chi3L1. We found that USF1 induced miR-125a-3p levels which suppressed Chi3L1 expression. Ultimately, our results suggest that lung metastasis is suppressed by knock-down of Chi3L1 through miR-125a-3p-mediated up-regulation of USF1.
Keywords: Chi3L1, lung cancer, metastasis, USF1, miRNA 125a-3p
Introduction
For decades, metastasis has been considered the final stage of the deterioration process in tumor development [1]. The mechanisms underlying invasion and metastasis were largely an enigma, until now. It was evident that as carcinomas arising from epithelial tissues progressed to higher pathological grades of malignancy, reflected by local invasion and distant metastasis, the associated cancer cells typically developed alterations in their shape, as well as in their attachment to other cells and the extracellular matrix [2]. Lung cancer is the most commonly diagnosed malignant tumor and accounts for approximately 20% of all cancer-related deaths worldwide [3]. Its propensity to metastasis undoubtedly contributes to its low survival and poor outcome in patients with this disease. Even cancer cells lacking the ability to metastasize can be transformed into metastasizable cells, by both the intrinsic properties of the tumor tissues and factors or genes in the tumor microenvironments, under certain circumstances [4]. Signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa B (NF-κB) are known to be significant factors regulating lung cancer cell proliferation, survival, cancer angiogenesis, and metastasis [5, 6]. Several cytokines, such as interleukin-1 beta and tumor necrosis factor alpha (TNF-α), are also significant for lung cancer metastasis [7]. In addition, epidermal growth factor receptor (EGFR), which is known to activate STAT3, is critical for lung cancer metastasis [8]. It has been reported that activation of both Toll-like receptor 2 complex and Toll-like receptor 6 complex with TNF-α enhances Lewis lung carcinoma metastatic growth [9]. Most recently, it was reported that metastasis of colorectal cancer was triggered via inhibition of growth factor independent 1 transcriptional repressor, which can suppress the targeted inflammatory genes [10]. Even though many target genes have been suggested as key factors in the regulation of metastasis, several other genes have been identified as risk factors for cancer metastasis in cancer patients [11]. Thus, multiple key factors could contribute to lung metastasis.
Chitinase 3-like 1 (Chi3L1; also known as YKL-40, 40 kDa) is a glycoprotein expressed and secreted by various types of cells [12]. Chi3L1 has been associated with many diseases, such as rheumatoid arthritis, osteoarthritis, liver fibrosis, inflammatory bowel disease, bacterial septicemia, neurological diseases, and atherosclerotic cardiovascular disease [13-15]. Moreover, Chi3L1 is also a significant factor in cancer development. The levels of circulating Chi3L1 and Chi3L1 expression are elevated in various cancers, including lung, prostate, colon, rectum, ovary, kidney, breast, glioblastomas, and malignant melanoma [16-18]. A high level of serum Chi3L1 also reflects metastasis of cancer [19]. Chi3L1 could be associated with colorectal and cervical angiogenesis, as well as pulmonary melanoma and breast metastasis [20, 21]. In patients with metastatic non-small cell lung cancer (NSCLC) and melanoma, the serum Chi3L1 level was identified as an independent prognostic biomarker [22]. Although a higher expression of Chi3L1 in cancer cells than normal cells has been reported, and a lot of studies demonstrated that Chi3L1 could be associated with metastasis, the regulatory mechanism of Chi3L1 in lung metastasis and the related factor of Chi3L1 expression are unclear. Therefore, we decided to focus on the effects of Chi3L1 on metastasis, as well as the regulating factors for Chi3L1 in lung metastasis.
The Genome-Wide Association Study (GWAS), Online Mendelian Inheritance in Man (OMIM), and differentially expressed gene (DEG) analyses indicated that Chi3L1 was associated with 38 cancers. In prior studies, metastatic lung carcinoma was significantly associated with Chi3L1 compared to other cancers [23-25]. It is also known that the Chi3L1 promoter sequence contains binding sites, such as specific binding sites for nuclear SPI1 (spleen focus forming virus proviral integration oncogene 1), specificity protein 1, SP3 (specificity protein 3), acute myeloid leukemia 1, CCAAT/enhancer-binding protein, and upstream stimulatory factor 1 (USF1) [26]. Using gene identifier mapping through expression profile data with Biomart and Gene Expression Omnibus (GEO) analysis of several genes [27], we found that USF1 was significantly and primarily associated with Chi3L1 (Figure S1). USF1 is a member of the basic helix-loop-helix (bHLH) leucine zipper family and can function as a cellular transcription factor [28]. USF1 can activate the transcription of genes containing pyrimidine-rich initiator elements and enhance-box (E-box) motifs, which are found in Chi3L1 [29]. It has been stated that single nucleotide polymorphisms of USF1 regulate papillary thyroid cancer and hepatocellular carcinoma [30, 31]. USF1 and USF2 are associated with postoperative metastatic recurrence in patients with hepatocellular carcinoma [32]. USF1 was found to increase the expression of major vault protein, which mediates drug resistance through the inhibition of transport processes in colorectal and renal cell adenocarcinoma cell lines [33]. USF1 inhibits human telomerase reverse transcriptase expression, leading to telomerase reactivation and oral carcinogenesis [34]. It was also mentioned that USF2-null mice displayed marked prostate hyperplasia [35]. However, the involvement of USF1 in the Chi3L1-mediated lung tumor metastasis has not yet been documented.
Therefore, in this study, we investigated the role of Chi3L1 in lung cancer metastasis and USF1-mediated lung metastasis. Additionally, we studied the mechanisms by which USF1 and Chi3Li regulate lung metastasis, using a Chi3L1 knock-down (KD) mice model, as well as cultured lung cancer and melanoma cell lines.
Results
Metastasis of B16F10 mouse melanoma to lung was decreased by injection of an adenoviral vector encoding mouse Chi3L1 short hairpin (sh) RNA (Ad-shChi3L1)
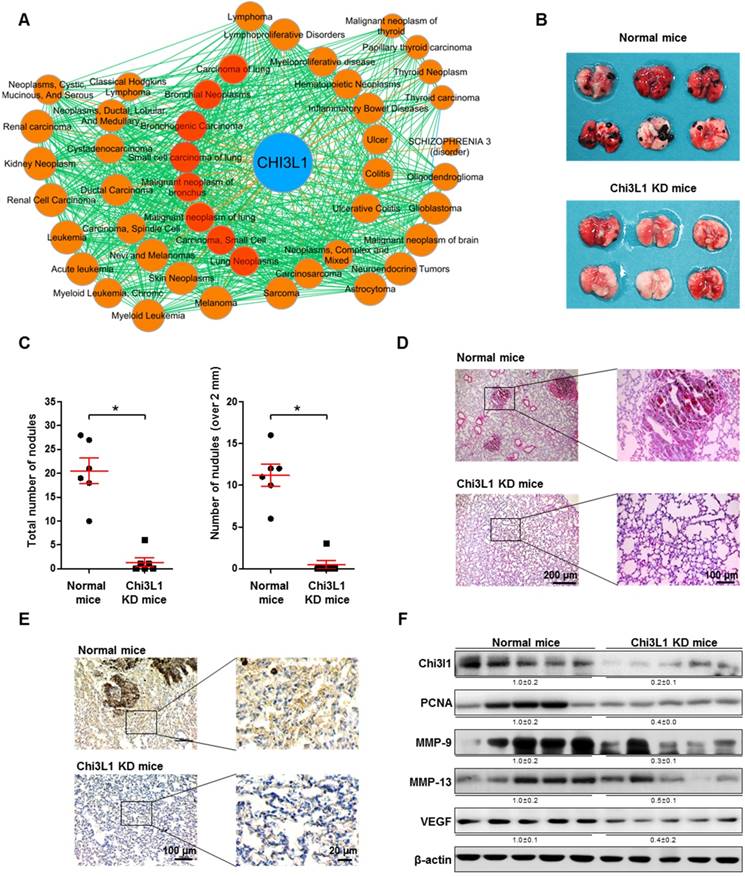
Chi3L1 is known as a metastatic biomarker in patients with malignant tumors [21, 36]. Through GWAS/OMIM/DEG analysis, we found that Chi3L1 is significantly associated with 38 cancers. Among these cancers, eight (red circles) are related to lung cancer (Figure 1A). With the same GWAS/OMIM/DEG analysis, we found that Chi3L1 is the most significant factor in the development of cancer and is more highly associated with cancer compared to EGFR, which was associated with 27 cancers (Figure S2). To investigate the association between Chi3L1 and cancerous metastasis, C57BL/6 mice were injected with B16F10 melanoma following treatment of Ad-shChi3L1. B16F10 cells were selected to research lung metastasis in C57BL/6 mice because B16F10 is a cell line visible with the naked eyes in various tissues and the cells have no immune rejection responses in C57BL/6 mice. Furthermore, the B16F10 cell line is an ideal model for lung metastasis research because metastasized cancer grows aggressively in the lung. Previously, we reported that B16F10 cells were inoculated to mice before the Chi3L1 knock-down (data not shown); however, the accumulation of B16F10 cells in the lung was slightly decreased by the knock-down of Chi3L1, but no significant difference was observed within 3 weeks between control and Chi3L1 knocked down group. We judged this experiment to be not proper because the observing term was too short and cell number was too great to confirm the metastatic inhibitory effect. Furthermore, mice injected with fewer B16F10 cells returned to a healthy state without cancer by protective mechanisms, such as immune systems. We first tested the efficiency of Ad-shRNA infection using immunohistofluorescence analysis. Lung tissues were transduced at a confluency of about 60% with Ad-shRNA-Chi3L1 (Figure S3). We found that metastatic melanomas were significantly reduced in the lung tissues of the Chi3L1 KD mice, as shown in Figure 1B. The number of melanoma nodules was significantly reduced, from an average of 20.5 in the lung tissues of the normal mice to an average of 1.3 in the lung tissues of the Chi3L1 KD mice. The number of melanoma nodules larger than 2 mm in diameter was also reduced compared to the normal mice, from an average of 11.2 to 0.5 (Figure 1C). Similarly, melanoma tumor development was significantly lower in the Chi3L1 KD mice than normal mice (Figure 1D-E). We also found Chi3L1 expression was also significantly decreased by Ad-shChi3L1 treatment (Figure 1E-F). Matrix metalloproteinases (MMPs), vascular endothelial growth factor (VEGF), and proliferating cell nuclear antigen (PCNA) are crucial malignant tumor invasion/migration, angiogenesis, and growth markers, respectively [37-39]. Thus, we chose these indicators as tumor markers and investigated their expression. Expression of MMP-9, MMP-13, and PCNA, as well as VEGF, was significantly suppressed in the lung tissues of Chi3L1 KD mice (Figure 1F). We also determined the phosphorylation of ERK and STAT3 since ERK and STAT3 pathways are well-known signals of various cancer metastases [40-42]. Phosphorylation of extracellular regulated kinase (ERK) and STAT3, both malignant metastasis signal markers, was also decreased in the lung tumor tissues of Chi3L1 KD mice (Figure S4). Thus, these data indicate that ERK and STAT3 activation may be the downstream signal pathway of Chi3L1 that enables the progression of cancer metastasis and invasion.
Knock-down (KD) of Chi3L1 inhibits lung metastasis of B16F10 melanoma in C57BL/6 mice model. (A) Gene (Chi3L1)-disease network was analyzed based on the GWAS/OMIM/DEG records (p<10-6). (B) Image of lung tumor tissues of C57BL/6 mice injected with B16F10 mouse melanoma. Black colonies are B16F10 melanoma colonies on the lung tumor tissue surface. (C) Counts of total and >2 mm diameter nodules in each lung tumor tissue. *, p<0.05. Error bars represent standard deviation. Normal mice, n=6; Chi3L1 KD mice, n=6. (D) Image of lung tumor tissue section stained with hematoxylin and eosin. (E) Immunohistochemistry to detect Chi3L1-positive cells in mice lung tissues (blue denotes nuclei; brown denotes Chi3L1 expression). (F) Expressions of Chi3L1, PCNA, MMP-9, MMP-13, and VEGF in C57BL/6 mice lung tissues (n=5 mice). Values below the Western blot bands indicate densitometry analysis using ImageJ software.
Cell migration and growth of human lung cancer cells were decreased by treatment with small interfering Chi3L1 RNA (siChi3L1)
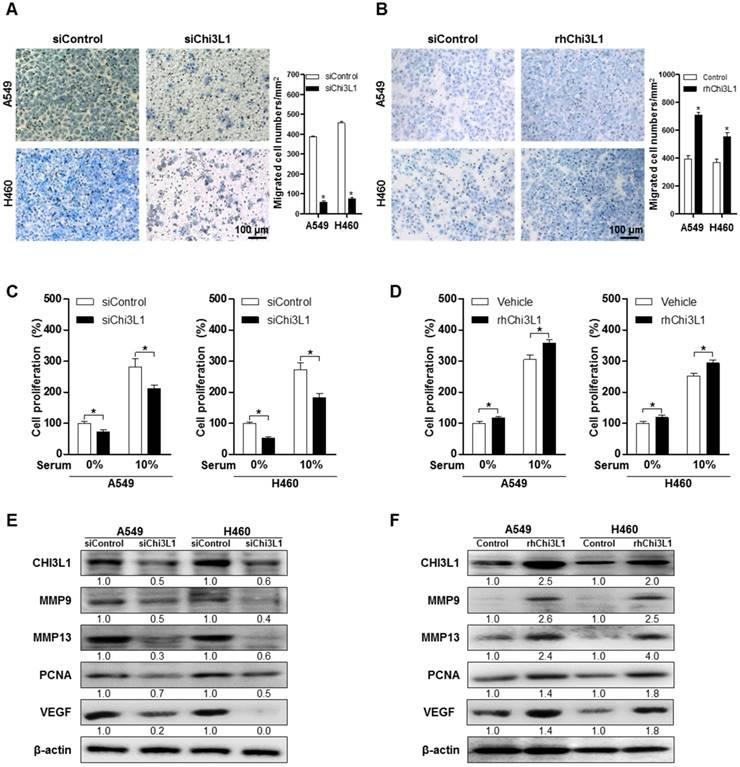
To study the effect of Chi3L1 on metastasis of lung cancer cell lines, we measured the migration of human lung cancer in the siChi3L1-treated A549 and H460 cancer cell lines using the trans-well migration assay. As a result, cell migration was significantly decreased by treatment with siChi3L1 in both A549 and H460 cell lines (Figure 2A). Conversely, the number of migrant cells was significantly increased by treatment with 100 ng/mL of recombinant human Chi3L1 (rhChi3L1) protein (Figure 2B). We also investigated migration ability using a wound healing method, which confirmed that siChi3L1 treatment suppressed cell migration (Figure S5A). Additionally, cell proliferation was decreased by siChi3L1 treatment, regardless of the presence of serum (Figure 2C). In contrast, cell proliferation was increased by treatment with rhChi3L1 (100 ng/mL; Figure 2D). Additionally, we assessed the cell proliferation of B16F10 cells with either KD of Chi3L1 using siChi3L1 RNA or recombinant mouse Chi3L1 treatment in vitro. The cell proliferation of B16F10 cells was increased by recombinant mouse Chi3L1 treatment but decreased by Chi3L1 KD regardless of the presence or absence of serum (Figure S5B). To investigate the effects of Chi3L1 on metastatic gene expression, we measured the expression of the cell migrating makers MMP-9 and -13, the cell proliferating marker PCNA, as well as the angiogenesis marker VEGF using Western blotting. Expression of MMP-9 and -13, PCNA, and VEGF was reduced by siChi3L1 treatment (Figure 2E) in A549 and H460 cells, but the expression was increased by rhChi3L1 treatment (Figure 2F).
Knock-down of Chi3L1 inhibits cell migration and proliferation in human lung cancer cell lines A549 and H460. (A) Cancer cell migration was measured by trans-well migration assays of A549 and H460 cells treated with siChi3L1 or siControl. *, p<0.05. (B) Trans-well migration assays of A549 and H460 cells treated with rhChi3L1 protein. *, p<0.05. (C) Cell proliferation was measured by MTT assay of A549 and H460 cells treated with siChi3L1. *, p<0.05. (D) Cell proliferation was measured by MTT assay of A549 and H460 cells treated with rhChi3L1. *, p<0.05. (E) Expressions of Chi3L1, MMP-9, MMP-13, PCNA, and VEGF after siChi3L1 treatment were analyzed by Western blotting. (F) Expressions of Chi3L1, MMP-9, MMP-13, PCNA, and VEGF after rhChi3L1 treatment were analyzed by Western blotting. All experiments were prepared in duplicate and repeated three times. Values below the Western blot indicate densitometry analysis using ImageJ software.
USF1 was associated with Chi3L1 in lung metastasis
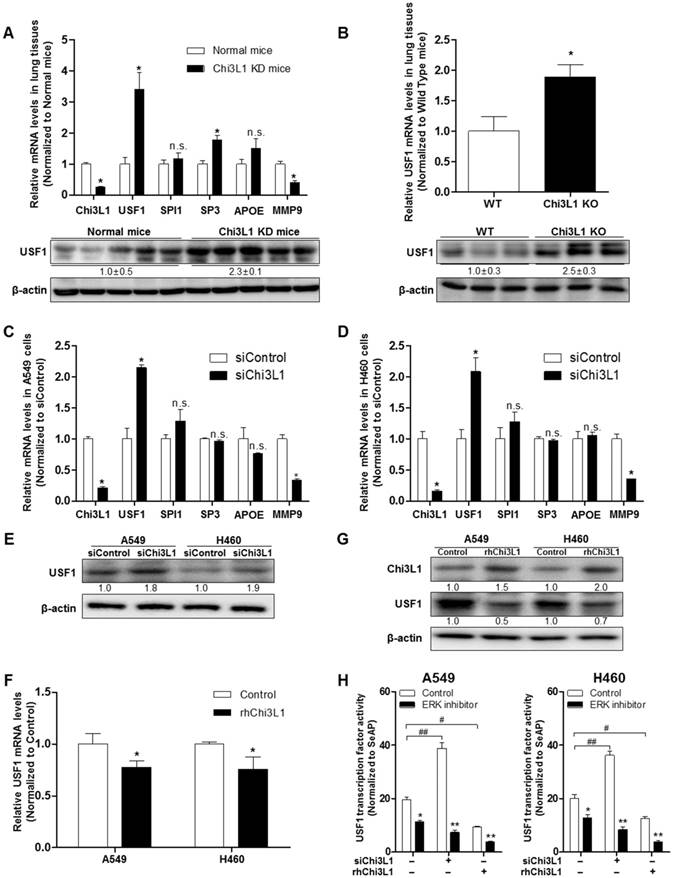
To identify genes regulating Chi3L1, we used Biomart and GEO for gene identifier mapping with profile data (Figure S1). Following the analysis, 20 genes were predicted to be associated with Chi3L1. Among them, four genes (USF1, SPI1, SP3, apolipoprotein E (APOE)) were significantly related to the Chi3L1 pathway, and one gene (MMP-9) was co-expressed with Chi3L1. We focused on these five genes which are related to the phenomenon induced by Chi3L1, particularly, lung cancer metastasis. To identify the genes affected by KD of Chi3L1, we performed quantitative real-time polymerase chain reaction (qPCR) for the five genes selected. It was observed that USF1 and SP3 expression was largely increased in the lung tumor tissue of Chi3L1 KD mice, whereas MMP-9 was decreased (Figure 3A). Additionally, we investigated the expression of USF1 protein in the lung tumor tissues of Chi3L1 KD mice. Expression of USF1 protein was significantly increased in the lung tumor tissues of Chi3L1 KD mice (Figure 3A). We also confirmed the up-regulation of USF1 mRNA levels and protein expression in the lung tumor tissues of Chi3L1 knock-out (KO) mice compared to wild-type (WT) mice (Figure 3B). To confirm these signal pathways, we conducted qPCR for the above five genes in the human lung cancer cell lines A549 and H460 after treatment with human Chi3L1 siRNA. Similar to the in vivo results, the USF1 gene was significantly increased by treatment with siChi3L1, whereas MMP-9 was significantly decreased in both cell lines (Figure 3C-D). Furthermore, expression of the USF1 protein was significantly increased in both siChi3L1-treated A549 and H460 cell lines (Figure 3E). To confirm the effects of Chi3L1 on USF1 expression, USF1 mRNA and protein levels were investigated after treatment of A549 and H460 cell lines with rhChi3L1. The mRNA level (Figure 3F) and protein expression (Figure 3G) of USF1 were significantly decreased by treatment with rhChi3L1 in both cell lines. It was also significant to see whether other pathways could be involved in Chi3L1 expression. In this regard, it is noteworthy that the production of Chi3L1 was decreased by the activation of the retinoic acid-inducible gene (RIG)-like helicase pathway [43]. Thus, we confirmed the expression of RIG-like helicase and downstream proteins. In USF1 KD lung cancer cell lines, mitochondrial antiviral-signaling protein (MAVS) mRNA levels were not changed but RIG-I and melanoma differentiation-associated protein 5 (MDA-5) mRNA levels were significantly increased (Figure S6). Based on these results, we speculated that the inhibition of Chi3L1 by USF1 was not dependent on the RIG-like helicase pathway because USF1 KD unexpectedly increased RIG-I and MDA-5 mRNA. It was also previously reported that USF1 is regulated by the ERK pathway [44, 45]. To elucidate the signal pathway of USF1 regulated by Chi3L1, USF1 transcriptional activity was investigated by using USF1 transcriptional response element (TRE) containing luciferase activity vector after Chi3L1 KD or treatment of rhChi3L1 in the presence of ERK inhibitor (U0126; 10 μM). USF1 transcriptional activity was significantly decreased by treatment with ERK inhibitor; however, the inhibition was extended in the treatment of siRNA or rhChi3L1 (Figure 3H). These data indicated that USF1 expression was affected by Chi3L1 in an ERK pathway-dependent manner but not in a RIG-I helicase-dependent manner in human lung cancer cell lines.
Up-regulated USF1 expression via knock-down (KD) of Chi3L1 in vivo and in vitro. (A) Relative expressions of Chi3L1, USF1, SPI1, SP3, APOE, and MMP-9 mRNA levels in Chi3L1 KD mice compared to normal mice. *, p<0.05. Lower panel shows expression of USF1 protein in lung tumor tissues of Chi3L1 KD mice. (B) Expression of USF1 mRNA (upper panel) and protein (lower panel) in lung tumor tissues of Chi3L1 KO mice (n=3 in each). Quantification of protein expression was measured and analyzed using ImageJ software. *, p<0.05. (C) Relative expressions of Chi3L1, USF1, SPI1, SP3, APOE, and MMP-9 mRNA in A549 cells by siChi3L1 treatment compared to siControl. *, p<0.05. (D) Relative expressions of Chi3L1, USF1, SPI1, SP3, APOE, and MMP-9 mRNA in H460 cells following siChi3L1 treatment compared to siControl. *, p<0.05. (E) Expression of USF1 protein with siChi3L1 treatment in A549 and H460 cell lines. (F) Relative expressions of USF1 mRNA following rhChi3L1 treatment. *, p<0.05. (G) Expressions of Chi3L1 and USF1 after rhChi3L1 treatment in A549 and H460 cell lines. (H) USF1 transcriptional activity following rhChi3L1 treatment or siChi3L1 treatment in A549 and H460 cell lines. ERK inhibitor (U0126 10 μM). *, p<0.05; **, p<0.01; #, p<0.05; ##, p<0.01. All in vitro experiments were prepared in duplicate and repeated three times. Values below the Western blot indicate densitometry analysis using ImageJ software.
Effect of USF1 on Chi3L1 expression, melanoma metastasis, and cancer cell growth and migration
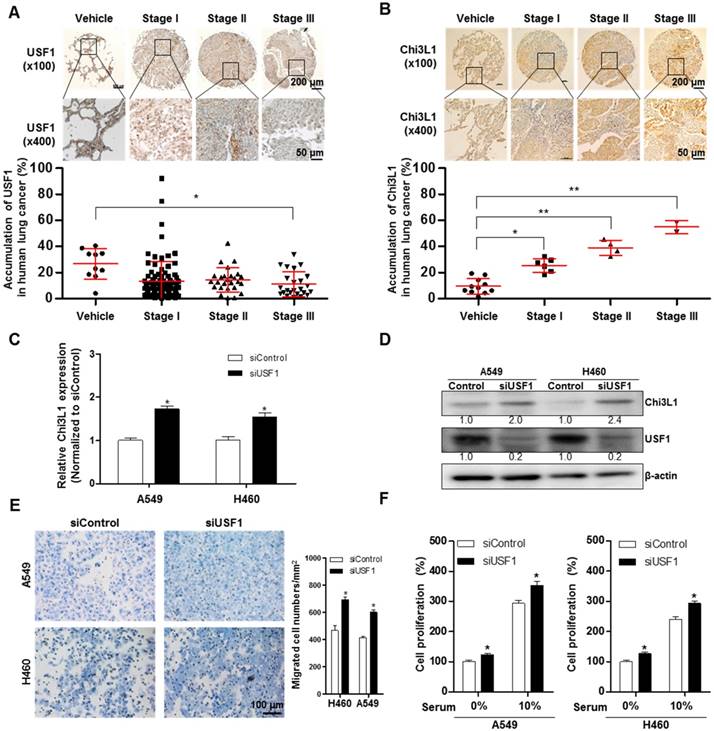
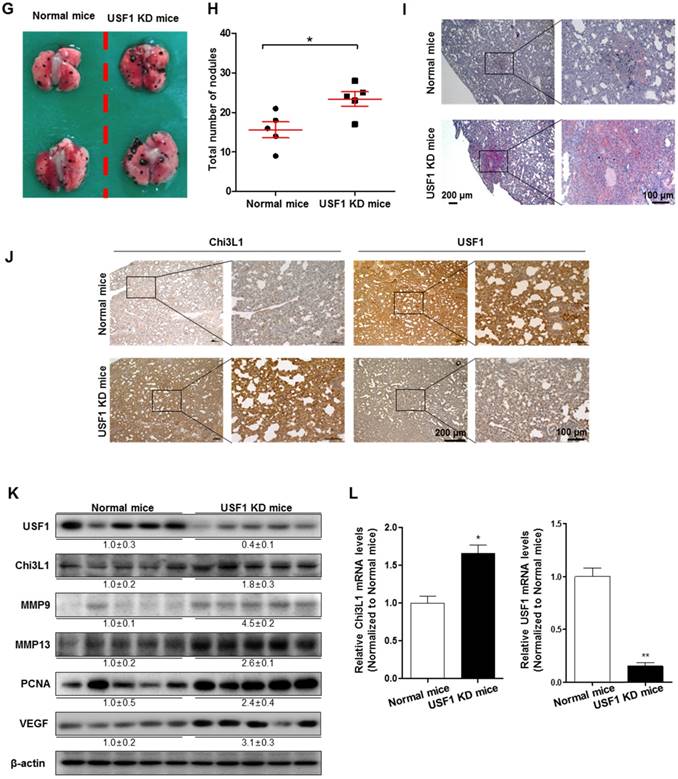
To further investigate the role of USF1 in Chi3L1-mediated cancer cell growth and metastasis inhibition, we first checked USF1 expression in human lung tumor tissues. As indicated (Figure 4A), USF1 expression was suppressed in human lung tumor tissue; however, Chi3L1 expression was increased in a stage-dependent manner (Figure 4B). We also confirmed these phenomena using lung cancer patients (n=15). To prove the expression of Chi3L1 and USF1 in lung cancer patients, normal or tumor lung tissues were obtained from hospitalized lung cancer patients in Keimyung University Dongsan Medical Center, Chonnam National University Hospital, and Chonbuk National University Hospital, under the respective Institutional Review Board (IRB) approval from each hospital. The results showed that Chi3L1 expression was increased, but USF1 expression was decreased in lung tumor tissues compared to normal lung tissues (Figure S7A). Moreover, USF1 expression decreased inversely with the increase of Chi3L1 expression in 15 lung tumor patients in a tumor stage-dependent manner (Figure S7B). To confirm the reciprocal effect of USF1 and Chi3L1 on their expression, we investigated the Chi3L1 mRNA level and protein expression in both A549 and H460 cell lines after siUSF1 treatment. The Chi3L1 mRNA level and protein expression were significantly increased by siUSF1 in both cell lines (Figure 4C-D). In addition, treatment with siUSF1 significantly induced A549 and H460 cell migration (Figure 4E) and cell proliferation (Figure 4F). To confirm the effect of USF1 on cancer metastasis in vivo, chemically modified siUSF1 (50 nmol per mouse) was injected through the tail vein of C57BL/6 mice injected with B16F10 melanoma, and then metastatic melanoma was assessed. Metastatic melanomas in the lung tissues of the mice were significantly increased in the siUSF1-transfected group (Figure 4G). Additionally, the number of melanoma nodules was significantly increased in the lung tissues of this group (Figure 4H), and the melanoma colony was largest in USF1 KD mice (Figure 4I). Immunohistochemistry (IHC) confirmed that Chi3L1 expression was increased and USF1 expression was decreased in the USF1 KD mice lung tissue section (Figure 4J). The expressions of Chi3L1, MMP-9, MMP-13, PCNA, and VEGF in mice lung tissues were also increased by siUSF1 treatment (Figure 4K). To confirm the reciprocal effects of USF1 on Chi3L1 mRNA, we performed qPCR. Accordingly, Chi3L1 mRNA level was increased, but USF1 mRNA level was decreased in USF1 KD lung tissues (Figure 4L). These results indicated that USF1 KD-induced cancer cell metastasis and proliferation occurred through the enhancement of Chi3L1 expression.
Effect of USF1 on Chi3L1 expression, melanoma metastasis, and cancer cell growth and migration. (A) Immunohistochemistry (IHC) analysis to detect USF1-positive cells in human lung cancer tissue microarray (blue denotes nuclei; brown denotes USF1). Lung cancer tissue array, including TNM, clinical stage, and pathology grade; 75 cases/150 cores. Quantification of USF1 gene expression was measured and analyzed using ImageJ software. *, p<0.05. (B) IHC analysis to detect Chi3L1-positive cells in the human lung cancer tissue microarray (blue denotes nuclei; brown denotes Chi3L1). Lung cancer tissue array, including TNM, clinical stage, and pathology grade; 12 cases/24 cores. *, p<0.05; **, p<0.01. (C) Relative expressions of Chi3L1 mRNA levels by siUSF1 treatment compared to siControl. *, p<0.05. (D) Chi3L1 and USF1 protein expressions following siUSF1 treatment compared to siControl in A549 and H460 cells. (E) Trans-well migration assay of A549 and H460 cells treated with siUSF1. *, p<0.05. (F) Cell proliferation was measured by MTT assay of A549 and H460 cells treated with siUSF1. *, p<0.05. (G) Metastases on the surface of the lung in the B16F10 mouse melanoma-injected group (n=5). Black colony is B16F10 melanoma colonization on the surface of lung tissues of five grains of mice per group. (H) Count of total nodules in each lung tissue. *, p<0.05. (I) Image of B16F10 formation in lung tissues by hematoxylin and eosin. (J) IHC analysis to detect cells positive for Chi3L1 and USF1 in the lung tumor tissues of normal and USF1 KD mice (blue denotes nuclei; brown denotes Chi3L1). (K) Expressions of Chi3L1, PCNA, MMP-9, MMP-13, and VEGF proteins in the lung tumor tissues of normal and USF1 KD mice. Quantification of protein expression was measured and analyzed using ImageJ software. (L) Relative expressions of Chi3L1 mRNA by siUSF1 treatment compared to siControl. *, p<0.05; **, p<0.01. All in vitro experiments were prepared in duplicate and repeated three times. Values below the Western blot indicate densitometry analysis using ImageJ software.
Modulation of Chi3L1 expression by miR-125a-3p-mediated USF1
We next investigated how USF1 regulates Chi3L1 expression by first determining the transcriptional level of Chi3L1. An E-box motif (CTCGTG) is located 190 bp upstream of the Chi3L1 transcription start site (TSS) [26]; thus, Chi3L1 could be regulated by USF1. To confirm USF1 binding to the Chi3L1 promoter, we performed chromatin immunoprecipitation (ChIP) assay with KD of Chi3L1 and rhChi3L1 treatment. USF1 was bound to Chi3L1 under the normal condition (Figure 5A). However, the binding of USF1 to the Chi3L1 promoter was significantly increased by siChi3L1 treatment but was decreased by rhChi3L1 treatment in A549 and H460 cells (Figure 5A). Moreover, siUSF1 also inhibited the binding of USF1 to Chi3L1. These data indicated that Chi3L1 expression could be regulated by USF1 through binding to the Chi3L1 promoter. We designed a luciferase vector with a Chi3L1 promoter from 1,200 bp upstream to 48 bp downstream of the Chi3L1 TSS (Figure S8A) to see if USF1 could inhibit transcription of Chi3L1. Unexpectedly, we found that Chi3L1 transcriptional activity was decreased by siUSF1 treatment in A549 and H460 cells (Figure 5B). Protein expressions are regulated by transcription and translation. Micro-RNAs (miRNAs) are commonly known to interrupt the translation of various genes by binding at the 3' untranslated region (3'UTR) in mature messenger RNA (mRNA) and breaking mRNA. Given that miRNA is disrupting the target mRNA, regardless of the transcription of the target, we focused on the modified levels of miRNA in the lung tissues of Chi3L1 KD mice. Using a program for predicting miRNA binding sites on mRNA, we selected three miRNAs (miR-24, miR-342-3P, and miR-125a-3p) that could bind to the 3'UTR in the mouse Chi3L1 mRNA. To identify the downstream factor of Chi3L1, we examined miR-24, miR-342-3p, and miR-125a-3p levels in the lung tumor tissues of KD mice. Interestingly, miR-125a-3p and miR-24 were significantly increased in the lung tumor tissues of Chi3L1 KD mice (Figure 5C). To identify the effect of miRNAs as a target on Chi3L1, we examined the Chi3L1 mRNA level after treatment with a miR-24 or miR-125a-3p mimic in A549 and H460 cells.
Suppression of Chi3L1 expression by miR-125a-3p induction is associated with USF1. (A) ChIP analysis for USF1 and Chi3L1 promoter binding activity. Relative binding activity of USF1 and the Chi3L1 promoter was analyzed by qPCR. *, p<0.05; **, p<0.01. (B) Chi3L1 transcriptional activity was measured by luciferase expression using transfection of luciferase-expressing plasmid DNA vector. Luciferase activity was analyzed 12 h after siUSF1 treatment. *, p<0.05. (C) miR-24, -125a-3p, and -342-3p levels were measured in lung tumor tissues of Chi3L1 KD mice compared to normal mice. *, p<0.05. (D) Relative expression of Chi3L1 mRNA in miR-24- and miR-125a-3p mimic-treated A549 and H460 cell lines. *, p<0.01. (E) Relative expression of Chi3L1 mRNA in siUSF1-treated A549 and H460 cell lines with miR-24 and miR-125a-3p mimic. *, p<0.05; #, p<0.05. (F) Relative Chi3L1 mRNA levels by 3'UTR-contained luciferase vector in A549 and H460 cell lines. WT vector contains original Chi3L1 mRNA 3'UTR. MT vector contains miR-125a-3p binding site deleted Chi3L1 mRNA 3'UTR. * p<0.05. (G) Expression of miR-125a-3p in Chi3L1 KO mice. *, p<0.05. (H) Expression of miR-125a-3p by treatment with rhChi3L1 in A549 and H460 cells. *, p<0.05. (I) Expression of miR-125a-3p in siChi3L1 and siUSF1 co-treated A549 and H460 cell lines. *, p<0.05; #, p<0.05. (J) miR-125a-3p transcriptional activity in siUSF1 or 6×Myc-tagged USF1-transfected A549 and H460 cell lines. *, p<0.05; #, p<0.05; ##, p<0.01. (K) Expression of miR-125a-3p in USF1 KD mice. **, p<0.01. All experiments were repeated three times, and samples in each experiment were prepared in duplicate.
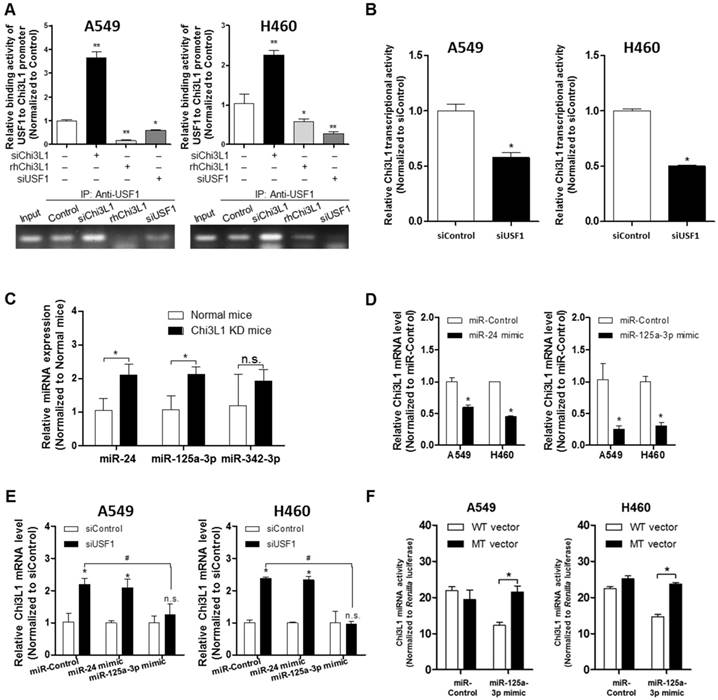
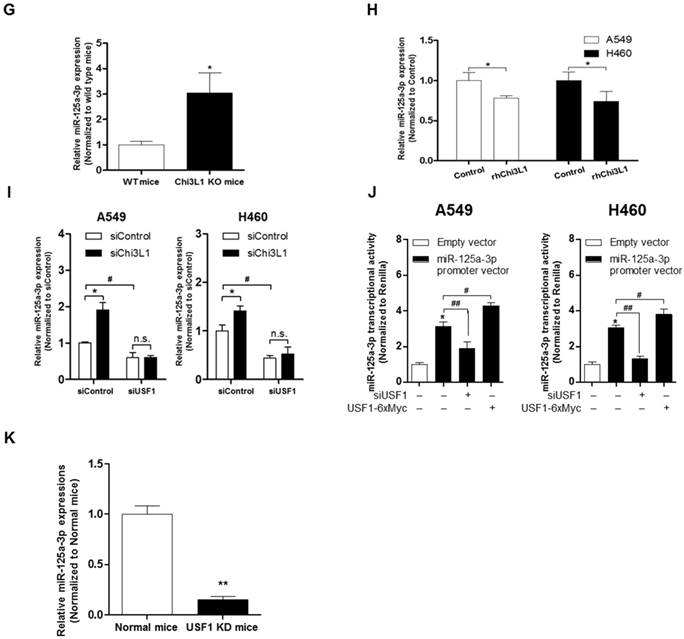
The results (Figure 5D) demonstrated that the Chi3L1 mRNA level was significantly decreased by miR-24 and miR-125a-3p; however, miR-125a-3p was more significantly decreased. Moreover, these inhibitory effects on Chi3L1 expression were observed under the USF1 KD condition only in miR-125a-3p mimic-treated cells (Figure 5E). Thus, we focused on deciphering the involvement of miR-125a-3p in Chi3L1 expression. Even though the 3'UTRs of Chi3L1 mRNA in mice and humans are very different, we noticed that the binding location of miR-125a-3p on the 3'UTR of Chi3L1 mRNA, predicted by following the miRNA support vector regression method [46], was similar between mice and humans (Figure S8B). Moreover, the mouse Chi3L1 protein has a motif identical to the human Chi3L1. To further confirm whether miR-125a-3p binds to the 3'UTR binding region of Chi3L1 mRNA, as the predicted region, the Chi3L1 mRNA level was detected by using Chi3L1 mRNA 3'UTR-tagged luciferase vector (Figure S8C). Luciferase activity containing the WT of Chi3L1 mRNA 3'UTR (WT Vector) was decreased by miR-125a-3p mimic treatment. However, the activity containing the mutant type of Chi3L1 mRNA 3'UTR (MT Vector), Δ(63-81) of Chi3L1 mRNA 3'UTR, was continued in human cancer cell lines, despite treatment with miR-125a-3p mimic (Figure 5F). Next, we investigated the levels of miR-125a-3p in Chi3L1 KO mice compared to WT mice. The level of miR-125a-3p was significantly elevated in the lung tissues of Chi3L1 KO mice relative to WT mice (Figure 5G). To confirm the effects of Chi3L1 on the level of miR-125a-3p, we analyzed the level of miR-125a-3p after rhChi3L1 treatment in A549 and H460 cells. The miR-125a-3p level was significantly decreased by treatment with rhChi3L1 in both cells (Figure 5H). However, the miR-125a-3p level was increased by KD of Chi3L1 (Figure 5I). To further confirm that the expression of miR-125a-3p by Chi3L1 could be associated with USF1, we determined miR-125a-3p in the double KD of Chi3L1 and USF1. Accordingly, miR-125a-3p expression was suppressed by USF1 KD, and the elevated level of miR-125a-3p by Chi3L1 KD was also decreased by USF1 KD in both A549 and H460 cell lines (Figure 5I). To confirm the effect of USF1 on the miR-125a-3p transcriptional activity, a miR-125a-3p promoter containing luciferase vector was designed (Figure S8D), and the luciferase activity in KD or overexpression of USF1 cells was determined. The miR-125a-3p transcription level was decreased by KD of USF1 but increased by USF1 overexpression (Figure 5J). Moreover, we confirmed that miR-125a-3p expression was significantly decreased in USF1 KD mice (Figure 5K). These data indicated that Chi3L1 expression was inhibited by miR-125a-3p-mediated USF1.
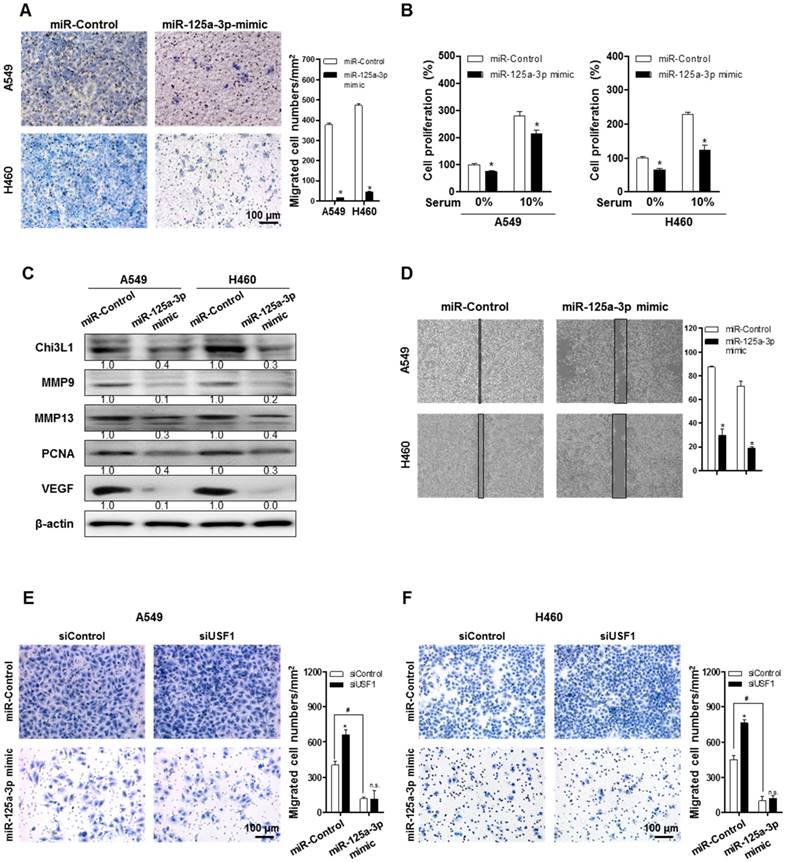
Inhibitory effect of miR-125a-3p on cancer migration and growth of lung cancer and Chi3L1 expression
To clarify the impact of miR-125a-3p on cancer metastasis and growth, cell migration and proliferation were investigated after treatment with a miR-125a-3p mimic in A549 and H460 cells. Pictures of the migrated cells were captured using a light microscope, and the number of cells was counted using ImageJ software. The migration of A549 and H460 cells was significantly decreased by treatment with the miR-125a-3p mimic (Figure 6A). Cell proliferation of A549 and H460 was also significantly reduced by miR-125a-3p mimic treatment, both in serum-free conditions and in the presence of 10% fetal bovine serum (FBS) (Figure 6B). To investigate the effects of miR-125a-3p on metastatic and proliferative genes, we analyzed the expressions of MMP-9, MMP-13, PCNA, and VEGF using Western blotting. The expressions of MMP-9, MMP-13, PCNA, and VEGF were significantly decreased by treatment with the miR-125a-3p mimic (Figure 6C). To investigate the inhibitory effect of miR-125a-3p on cell migration in the presence of Chi3L1 and USF1, we analyzed the cell migration in the KD of Chi3L1 or USF1 in the miR-125a-3p mimic-treated cells. We also confirmed that the ability to migrate was suppressed by treatment with the miR-125a-3p mimic using a migration assay through the wound healing method (Figure 6D). The data (Figure 6E-F) showed that USF1 KD increased cell migration but siUSF1-elevated cell migration was inhibited in A549 and H460 cell lines. The Chi3L1 mRNA level was elevated by the miR-125a-3p inhibitor, and this effect was not changed in USF1-overexpressing cells (Figure S9). These results indicated that miR-125a-3p-mediated USF1 suppressed Chi3L1 expression, which could be significantly associated with cancer cell migration.
miR-125a-3p mimic inhibits cell migration and proliferation by inhibiting Chi3L1 expression. (A) Trans-well migration assays of A549 and H460 cell lines treated with miR-125a-3p mimic. *, p<0.05. (B) Cell proliferation was measured by MTT assay of A549 and H460 cell treated with miR-125a-3p mimic. *, p<0.05. (C) Expressions of Chi3L1, MMP-9, MMP-13, PCNA, and VEGF with miR-125a-3p treatment were analyzed by Western blotting. (D) Wound healing migration assay of A549 and H460 cells treated with miR-125a-3p mimic. *, p<0.05. (E) Trans-well migration assays of miR-125a-3p mimic-treated A549 cell lines treated with siUSF1. *, p<0.05; #, p<0.05. (F) Trans-well migration assays of miR-125a-3p mimic-treated H460 cell lines treated with siUSF1. *, p<0.05; #, p<0.05. All experiments were repeated three times, and samples in each experiment were prepared in duplicate. Values below the Western blot indicate densitometry analysis using ImageJ software.
Methods
Adenoviral vector generation for KD of Chi3L1 by Ad-shChi3L1 injection and treatment of mice
Ad-shChi3L1 was used to KD Chi3L1 expression. Ad-shChi3L1 was constructed by Sirion Biotech (Martinsried, Germany). The U6-shRNA-SV40-pA region of the pO6A5 shuttle vector was transferred via recombination in a bacterial artificial chromosome vector, containing the genome of the replication Ad5-based vector deleted for the E1/E3 genes (involved in replication and immunomodulation, respectively). Adenoviral particles were produced by construction of the shRNA expression shuttle vector into HEK-293 cells. Cloning success for the resultant vector was verified by restriction analysis and DNA sequencing. To investigate the function of Chi3L1 on metastasis of melanoma in vivo, first, B16F10 cells were inoculated to mice before the Chi3L1 KD; however, the accumulation of B16F10 cells in the lung was slightly decreased by Chi3L1 KD, but no significant difference was observed within 3 weeks between control and Chi3L1 KD group. We assumed that this experiment was not proper because the observation duration was too short and cell number was too great to confirm the metastatic inhibitory effect. Furthermore, fewer B16F10 cell-injected mice returned to healthy condition without cancer by protective mechanisms, such as immune systems. Thus Ad-shChi3L1 or Ad-shControl were inoculated with 7.5×106 plaque-forming units (p.f.u.) per animal via the tail vein at 24 h before the injection of B16F10. The next day, C57BL/6 mice were injected with B16F10 mouse melanoma via the tail vein. The following day, either Ad-shChi3L1 or Ad-shControl were injected at 108 p.f.u. per animal via the tail vein, as a boosting shot. One week and 2 weeks after the boosting shot with Ad-shChi3L1, second and third injections were performed to inhibit Chi3L1 continually, despite degradation of the adenovirus by immune responses. All animal studies followed the guidelines of the Animal Care and Use Committee of Chungbuk National University (Cheongju, Chungbuk, Korea). All protocols involving mice in this study were reviewed and approved by the Chungbuk National University Institutional Animal Care and Use Committee (IACUC) and complied with the Korean National Institute of Health Guide for the Care and Use of Laboratory Animals (CBNUA-1073-17-01).
Human lung tumor tissue array
The human lung cancer tissue microarray, containing lung tumors from 140 patients and 10 samples of normal tissue, and the lung carcinoma tissue microarray, containing lung tumors from 12 patients and 12 samples of normal tissue, were purchased from US Biomax (Rockville, MD, USA). Tissue arrays were subjected to IHC analysis, as described below. Human lung cancer and normal lung tissues from 15 lung cancer patients were obtained from Keimyung University Dongsan Medical Center, Chonnam National University Hospital, and Chonbuk National University Hospital. All studies using human tissues were conducted in accordance with the Declaration of Helsinki and were approved by the Ethics Committee of Chungbuk National University Medical Centre (IRB No.: CBNU-IRB-2011-U01).
Plasmid vector and luciferase activity assay
Chi3L1 promoter dual-reporter (HPRM 12518-LvPG04) and negative control (NEG-LvPG04) lentiviral plasmid vectors were purchased from GeneCopoeia (Rockville, MD, USA). miR-125a-3p promoter dual-luciferase vectors and LvPG04[Gluc/minP] USF1 TRE, containing dual-luciferase vector, were cloned using lentiviral plasmid vector from Bionics (Seoul, Republic of Korea). pcDNA3.1+6×Myc-tagged Chi3L1 and USF1 were also cloned (Bionics). miTarget 3'UTR miRNA Target Clone (pEZX-MT06) was purchased from GeneCopoeia. Furthermore, Chi3L1 mRNA 3'UTR WT and miR-125a-3p binding predicted site deletion mutation were cloned (Bionics). To measure the transcriptional luciferase activity, A549 and H460 cells were plated in 12-well plates (1.5×105 cells/well) and transiently transfected with negative luciferase vector or target luciferase vector or miR target WT Vector or miR target MT Vector (1 μg/well/ea.) for 6 h, using a mixture of plasmid and Lipofectamine 3000 in Opti-MEM medium, according to the manufacturer's specification (Invitrogen, Carlsbad, CA, USA). For KD or overexpression of the target gene, siRNA or miRNA mimic or miRNA inhibitor (10 pmol/well/ea.) were treated following siRNA reverse transfection, according to the manufacturer's protocol. The medium was harvested to measure the luciferase activity after an appropriate time following the transfection of siRNA or overexpression vector, for analysis of dual-luciferase activity. Luciferase activity was measured by using the Secrete-Pair™ Dual Luminescence Assay kit (GeneCopoeia), and the results were read on a luminometer, as described by the manufacturer's specifications (WinGlow, Bad Wildbad, Germany). miRNA 3'UTR Chi3L1 mRNA luciferase activity was measured using the Luc-Pair™ Luciferase Assay kit (GeneCopoeia).
Cell culture
A549 and H460 human lung cancer cells and B16F10 mouse skin melanoma were obtained from the American Type Culture Collection (Manassas, VA, USA). RPMI1640 medium, Dulbecco's modified Eagle's medium (DMEM), penicillin, streptomycin, and FBS were purchased from Invitrogen. A549 and H460 cells were grown in RPMI1640 with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, at 37 °C in 5% CO2 humidified air. B16F10 cells were grown in DMEM with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, at 37 °C in 5% CO2 humidified air.
CRISPR/Cas9 technique-based Chi3L1 null mutation mice
8-month-old C57BL/6 mice purchased from Central Lab. Animal, Inc. (Seoul, Korea) were housed in standard cages in an Assessment and Accreditation of Laboratory Animal Care credited specific pathogen-free (SPF) animal facility on a 12 h light-12 h dark cycle. All animal protocols were approved by the Animal Care and Use Committee of the Model Animal Research Center in Korea Research Institute of Bioscience and Biotechnology. Single guide RNAs (sgRNAs) targeting a site of the genome corresponding to the N-terminal region of Chi3L1 were designed using ZiFiT (http://zifit.partners.org/ZiFiT/) program. The candidates of target nucleotides having a potential off-target with 1 or 2 base mismatch were avoided for the design (Figure S9). The two complimentary oligos of each sgRNA were annealed and cloned in pT7-gRNA vector, which is a vector designed for synthesis of sgRNA [47]. Microinjection was performed in fertilized eggs from C57BL/6 mice. The embryos were harvested in M2 medium and cultured in M16 medium for 2-3 h. The mixture of sgRNA (100 ng/µL) and Cas9 protein (80 ng/µL) was injected into the cytoplasm of the one-cell-stage embryos. Injected embryos were incubated overnight in the culture medium before embryo transfer into pseudo-pregnant female mice (ICR strain) [48]. After birth, genomic DNAs were extracted from tails of the progenies and subjected to PCR using the primer sets (Table S1). PCR amplicons were denatured and slowly reannealed to facilitate heteroduplex formation. The reannealing procedure comprised a 5-min denaturing step at 95 °C, followed by cooling to 85 °C at -2 °C/s, and further cooling to 25 °C at -0.1 °C/s. Reannealed amplicons were treated with five units of T7 endonuclease I (New England BioLabs) for 30 min at 37 °C and then analyzed by agarose gel electrophoresis. To check the potential off-target effects, the genomic regions encompassing the potential off-target sites with 1- or 2-base mismatch were PCR amplified and subjected to the T7E1 assay or sequencing analysis. For PCR genotyping assay, primer sets amplifying the region of exon 3 were designed as follows: forward: 5′-GAGTTTAGTATCCCATATCACC-3′, reverse: 5′-GGCCACATATTTTGTCACTCAT-3′. USF1 mRNA and protein expression between wild type and Chi3L1 KO mice, we applied the lung tissues of 3 mice per each group.
Disease-gene relationship analysis
To identify the character of a specific gene, we analyzed the relationship between the target gene(s) and various diseases, by utilizing the DiseaseConnect (http://disease-connect.org) resource. This web server combines comprehensive omics and literature data, including detailed lists and representations of disease-gene relations derived from various sources, such as GWAS, OMIM, DEG, Gene Reference into Function (GeneRIF) and GeneWays [25]. Chi3L1 was entered into the web server to search for all diseases related to Chi3L1 (p<1×10-8).
siRNA injection for KD of USF1 in vivo
For the examination of gene and miRNA expression and cellular function in vivo, In Vivo Ready siUSF1 or In Vivo Control siRNA were purchased from Ambion® by Life Technologies Inc. (Carlsbad, CA, USA; Table S3). These siRNAs included Ambion® In Vivo chemical modifications for superior serum stability for in vivo application. For the in vivo experiment, siRNA is mixed with Invivofectamine®3.0 Reagent (Life Technologies Inc.; Table S4), at least 1 h before use. The siRNA-Invivofectamine® mixture was injected into 8-week-old C57BL/6 male mice every 3 days for 2 weeks, by tail-vein injection [49]. B16F10 cells were collected and delivered to mice by tail-vein injection (2×105 cells per mouse), 1 day from the first injection of the siRNA-Invivofectamine® mixture.
Histological analysis
Lung tissues were dissected and immediately fixed in 4% formaldehyde solution, followed by dehydration in a graded ethanol series (70-100%) and embedding in paraffin. The tissues were then sectioned (4 μm thick) with a rotary microtome (Sakura Finetek Europe BV, Alphen aan den Rijn, The Netherlands), and stained with hematoxylin and eosin. Sections were viewed under a light microscope (Olympus, Tokyo, Japan), and photographed at ×200 magnification.
Immunohistochemistry (IHC)
Lung tissues metastasized with B16F10 melanoma specimens were fixed in formalin and paraffin-embedded for examination. Sections (4 µm thick) were used for IHC. The mouse lung sections were blocked with 3% normal horse serum diluted in phosphate-buffered saline (PBS) for 30 min; the sections were then blotted and incubated with rabbit polyclonal antibodies for Chi3L1 and mouse monoclonal antibodies for USF1 (Table S1) at the appropriate dilution (1:100 dilution) in blocking serum for 4 h at room temperature. The slides were washed in PBS, followed by the avidin-biotin-peroxidase complex (ABC; Vector Laboratories, Burlingame, CA, USA). The slides were washed, the peroxidase reaction was developed with diaminobenzidine and peroxide, and the slides were mounted in Aqua-Mount and evaluated under a light microscope (Olympus) at ×200 magnification. A negative control was performed by omitting the primary antibody.
Western blot analysis
Lung tissues were homogenized in lysis buffer containing a protease inhibitor cocktail tablet (Roche Diagnostics, Mannheim, Germany). The total protein concentration of each lysate was measured using the BCA Protein Assay Reagent (Pierce, Rockford, IL, USA). Proteins in the lysates were electrophoretically separated in 7.5-15.0% sodium dodecyl sulfate-polyacrylamide gel and then transferred to polyvinylidene difluoride membranes (GE Healthcare Life Sciences, Piscataway, NJ, USA). The membranes were blocked with 5% bovine serum albumin (Roche Diagnostics) overnight at 4 °C and then incubated overnight at 4 °C with the primary antibodies. Antibodies used in this study are listed in Table S1. The membranes were next incubated with horseradish peroxidase-conjugated secondary antibodies overnight at 4 °C. Bands were visualized with enhanced chemiluminescence (Amersham Pharmacia Biotech, Buckinghamshire, UK), and the intensities of the bands were quantified by NIH ImageJ software (University Health Network Research, Toronto, Canada).
qPCR and miRNA quantitative reverse-transcription PCR analysis
Total RNA was collected from mice lung tissues or lung cancer cell lines in 700 μL of QIAzol® and purified using the miRNeasy Mini kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer's protocol. Total RNA was reverse-transcribed into complementary DNA (cDNA) using a High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA), and then subjected to qPCR using QuantiFast® SYBR® Green PCR Master Mix (Qiagen) with custom-designed specific primers on a StepOnePlus™ Real-Time PCR System (Applied Biosystems). 18S was used as the housekeeping control. Primer sequences for this study are listed in Table S2. For the detection of miRNA, cDNA was prepared in a reverse transcription reaction using a miScript II RT kit (Qiagen). Mature miRNA expression was determined using a miRNA-specific miScript Primer Assay and a miScript SYBR® Green PCR kit (Qiagen) on a StepOnePlus™ Real-Time PCR System (Applied Biosystems). Specific mature and pre-mature miRNA primers were purchased from Qiagen. Relative fold changes in target gene expression were calculated using Rnu6B as an internal control. The qPCR experiments were performed as described previously [50]. The fold change between groups was determined for all targets using the 2ΔΔCt method.
Cell proliferation assay
A549 and H460 cells were plated in 96-well plates and subsequently treated with either siChi3L1 or miR-125a-3p and incubated for 6 h. After transfection, the culture medium was changed to 10% FBS-containing RPMI or serum-free RPMI and then incubated for 24 h. Cell viabilities were measured by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (Sigma-Aldrich, St. Louis, MO, USA), according to the manufacturer's instructions. Briefly, MTT (5 mg/mL) was added and the plates were incubated at 37 °C for 2 h before 100 μL dimethyl sulfoxide (DMSO) was added to each well. Finally, the absorbance of each well was read at a wavelength of 540 nm using a microplate reader.
Trans-well migration assay
Migration of human lung cancer cells, A549 and H460, was quantitatively measured on permeable inserts (8 μm pore trans-well; Corning Inc.). The siChi3L1- or miR-125a-3p- or siUSF1-treated A549 and H460 cells were plated at 2.0×104 cells per well and incubated at 37 °C, 5% CO2 in a humidified incubator for 17 h. After incubation, the cells were fixed with 3.7% formaldehyde for 2 min and then washed with 1×PBS twice. Next, the cells were permeated with 100% methanol for 15 min and stained with trypan blue for 20 min. Non-migrated cells on the inside of the wells were removed with a cotton swab, and the images, captured under a light microscope (Olympus) at ×200 magnification, were analyzed using NIH ImageJ software.
Treatment of siRNA and miRNA
SiChi3L1 RNA and control siRNA were purchased from OriGene Technologies (Rockville, MD, USA; Table S3). SiUSF1 and control siRNA were purchased from Santa Cruz Biotechnology (Dallas, TX, USA; Table S3). miR-125a-3p mimic and control miRNA were purchased from Thermo Fisher Scientific (Waltham, MA, USA; Supplementary Table S3). Human lung cancer cells A549 and H460 (2×104 cells/cm2) were plated in 6- or 24-well plates and transiently treated with siRNA or miRNA mimetic molecule using Lipofectamine® RNAiMAX Reagent, according to the manufacturer's protocol (Thermo Fisher Scientific; Table S4). The treated cells were examined by the cell proliferation and migration assay, and for other molecular mechanisms by qPCR, Western blotting, and ChIP assay.
ChIP assay
The ChIP assay was undertaken using a commercially available kit (Cell Signaling Technology, Danvers, MA, USA; Table S4). In brief, cells were cross-linked with 1% formaldehyde. After 10 min, the cross-linking reaction was stopped by the addition of glycine. The extracted chromatin was digested and fragmented into 150-900 bp, which was then immunoprecipitated by ChIP-grade antibodies against USF1 (Santa Cruz Biotechnology) or a normal immunoglobulin G using protein G agarose beads. Afterward, the DNA and transcriptional factor complexes were uncross-linked to obtain the pure DNA fragment. PCR was then performed using the primers spanning the putative USF1 binding site in the human Chi3L1 promoter. Primer sequences for this study are listed in Table S2. For normalization of cross-linked USF1 and Chi3L1 promoter binding, histone H3 antibody (Table S1) was used for immunoprecipitation.
Image analysis quantification
ImageJ (Windows version 1.5k; http://imagej.nih.gov/ij) was used for analysis of the Western blotting, IHC, and migrated cell images. Densitometry, cell counting, and IHC quantification were performed as described previously [51].
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad Software, La Jolla, CA, USA). Statistical comparisons of paired groups were determined by Student's t-tests. Results are expressed as group means ± standard deviation. Statistical significance was set at p<0.05 for all tests.
Discussion
The expression levels of circulating Chi3L1 and Chi3L1 are elevated in various cancers [16-18]. A high level of serum Chi3L1 reflects aggressiveness in metastatic breast cancer [19], colorectal, and cervical angiogenesis, as well as pulmonary melanoma metastasis [20, 21]. In patients with metastatic NSCLC and melanoma, serum Chi3L1 level was identified as an independent prognostic biomarker [22, 36]. Additionally, Chi3L1 KO BALB/c mice with allergic pulmonary cancer had decreased levels of myeloid-derived cells, pro-inflammatory mediators, cancer volume, and metastasis [52]. Similar to these previous data, the present study showed that KD of Chi3L1 using Ad-shChi3L1 inhibited cancer metastasis and metastatic gene expression in C57BL/6 mice lungs. We also found that lung metastasis was significantly inhibited in Chi3L1 KO mice (data not shown). The KD of Chi3L1, using siChi3L1, decreased cell migration and proliferation, as well as the expression of cancer growth and metastasis genes, such as MMP-9, MMP-13, PCNA, and VEGF, in A549 and H460 human lung cancer cell lines. Conversely, cell migration and proliferation, as well as the expressions of related genes, were significantly increased by treatment with rhChi3L1. In addition to these experimental data, our GWAS/OMIM/DEG analysis showed a significant association between Chi3L1 and metastatic lung cancer. These results indicate that Chi3L1 significantly promotes lung cancer metastasis [24, 25]. However, the underlying mechanisms and Chi3L1 signals associated with lung tumor metastasis were not completely elucidated.
To determine the targets or signals by which Chi3L1 induced lung metastasis, we identified several Chi3L1-mediated genes by using gene mapping and expression profile data analysis [27]. Among the genes, we identified USF1 as being primarily associated with Chi3L1. The expression of USF1 was significantly increased by KD of Chi3L1 in vivo and in vitro. Moreover, the expression of USF1 in Chi3L1 KO mice was significantly increased at both the mRNA and protein levels. In contrast, the expression of USF1 was significantly decreased by treatment with rhChi3L1. USF1 is a transcription factor of the bHLH leucine zipper family that can activate the transcription of genes containing the E-box motif in the promoter region [28, 29]. To confirm the effects of USF1 on metastatic melanoma, we examined Chi3L1 expression and metastatic melanoma in mice lung tissues using in vivo-modified siUSF1. Metastasis was increased, and the expression of metastatic and proliferative marker proteins was also induced in USF1 KD mice. Furthermore, the migration and proliferation of A549 and H460 cells were increased by siUSF1 treatment. The expression of Chi3L1 was also reduced by siUSF1 treatment in vivo and in vitro. We also found that the expression of Chi3L1 was up-regulated, but USF1 was down-regulated in a stage-dependent manner in both human lung tumor tissues and tumor tissues of lung cancer patients. USFs contribute to inflammatory and immune responses, as they can inhibit NF-κB/STAT3 signaling while also stimulating immunoglobulin light-chain genes, which are significant factors in tumorigenesis [53]. In this regard, we found that phosphorylation of STAT3 was inhibited in the lung tumor tissues of Chi3L1 KD mice. Pertinently, previous data suggests that USFs are closely related to colorectal, renal, and oral carcinogenesis [33, 34]. Furthermore, USF1 and USF2 have been associated with postoperative metastatic recurrence in patients with hepatocellular carcinoma [32]. Thus, our results suggest that USF1 overexpression could be significant for Chi3L1 KD-mediated inhibition of lung tumor metastasis.
We next studied how USF1 regulated Chi3L1. Recently, it was reported that Chi3L1 expression was stimulated by semaphorin 7a and integrin alpha v but inhibited by plexin C1 [21]. Conversely, the expressions of semaphorin 7a, integrin alpha v, and plexin C1 were not affected by the Chi3L1 expression. It was considered that USF1 regulated Chi3L1 expression independently of semaphorin 7a and integrin levels and plexin C1 pathway. Additionally, Chi3L1 expression was suppressed by RIG-like helicase, which stimulates the expressions of LIM Kinase 2, cofilin, phosphatase and tensin homolog, and B-Raf proto-oncogene. Notably, the production of Chi3L1 was decreased by the activation of the RIG-like helicase pathway. Thus, we confirmed the expression of RIG-like helicase and downstream proteins. In USF1 KD lung cancer cell lines, RIG-I and Mda-5 mRNA levels were significantly increased. These data indicated that the expression of Chi3L1 by USF1 was independent of the RIG-like helicase pathway. Chi3L1 is also regulated by interleukin (IL)-13, γ-interferon, and other cytokines, such as IL-1, IL-6, and TNF-α [54]. These cytokines could regulate Chi3L1 expression [55]. Notably, USF1 could regulate the expression of cytokines such as IL-10 [56] and TGF-β [57]. Moreover, USF1 is important for cytokine-induced gene expression. USF1 is also involved in cytokine-releasing UV-induced tyrosinase expression [58]. Thus, it is possible that USF-1 could be involved in Chi3L1 expression by gene expression changes induced by cytokines, such as IL-13 or γ-interferon. These data indicate that cytokine-induced USF1 expression could also be involved in USF1-mediated Chi3L1 expression.
We also experimentally showed that Chi3L1 expression was up-regulated by USF-1 siRNA, and Chi3L1 knock-down increased the expression of USF-1. The mean half-life of Chi3L1 mRNA was 13.4 h [59]; however, the mean full-translation time of general genes was reportedly approximately 7 min, indicating that Chi3L1 mRNA could be fully translated during the maintenance of mRNA [60]. However, in our present study, we found that Chi3L1 transcriptional activity did not reflect the full translation of Chi3L1 protein after USF1 KD by siUSF1 (Figure 4 and Figure 5). Thus, we considered that miRNA could inhibit the translation of mRNA. In this regard, we focused on miRNA as a target of Chi3L1 mRNA. Even though USF1 binds the Chi3L1 promoter at the transcriptional level, Chi3L1 expression was inhibited by USF1; thus, we speculated the translational regulation of Chi3L1 by USF1 occurs through miRNA. Relatedly, emerging evidence has shown that human miRNA genes are frequently located in genomic regions associated with cancer, and the disruption of miRNA expression patterns has been detected in many human cancers [61]. Previously, miR-125a-3p was predicted to be a target of both mouse and human Chi3L1 mRNAs [62]. Curiously, in our study, the expressions of both miR-125a-3p and USF1 were significantly increased by KD of Chi3L1 in vivo and in vitro. These data indicate that miR-125a-3p could be associated with USF1 in the regulation of Chi3L1. We also found that these two factors were associated with each other in the regulation of Chi3L1. We thus hypothesized that miR-125a-3p was regulated by the USF1 transcription factor. In support of this statement, we found that KD of USF1 reduced the expression of miR-125a-3p while USF1 overexpression had the opposite effect. Conversely, USF1 was increased by KD of Chi3L1 and by miR-125a-3p, a target of Chi3L1. In a functional study of miR-125a-3p on Chi3L1 expression and metastasis, we found that miR-125a-3p mimic treatment decreased the expression of Chi3L1, together with reducing cell migration and proliferation in A549 and H460 cells. Furthermore, we demonstrated that miR-125a-3p transcriptional activity was significantly decreased by siUSF1 but increased by USF1 overexpression. We also revealed that Chi3L1 levels were significantly reduced by the miR-125a-3p treatment, regardless of the presence or absence of USF1. Additionally, we found that the decrease of Chi3L1 mRNA level by USF1 overexpression was abolished by treatment with miR-125a-3p inhibitor. This result confirmed that USF1 could suppress Chi3L1 expression through activation of miR-125a-3p.
In previous studies, miR-125a-3p inhibited adhesion, proliferation, migration, and survival through the suppression of metastasis-associated factors, such as Fyn (a tyrosine kinase), breast cancer susceptibility gene 1, focal adhesion kinase, and paxillin, in prostate and breast cancer [63-65]. Additionally, miR-125a-3p inhibited NSCLC proliferation, migration, and invasion, by targeting metastasis-associated gene 1 [66]. It was also reported that the expression of miR-125a-3p was significantly decreased in most malignant glioma samples relative to normal brain tissues and glioma tissues of low malignancy [67]. It has been documented that the expression of Chi3L1 could be altered by other miRNAs. The expression of Chi3L1 in bone formation and differentiation was reduced by miR-24, and loss of Chi3L1 reduced bone formation and mineralization [68]. Hepatitis C virus (HCV) infection- and TNF-α-mediated Chi3L1 expression is inhibited by miR-449a, and HCV patients demonstrate up-regulation of Chi3L1 along with down-regulation of miR-449a [69]. In our present study, Chi3L1 expression could induce cancer cell proliferation and migration; however, miR-125a-3p suppressed Chi3L1 protein expression via inhibiting Chi3L1 mRNA. Therefore, regulation of miR-125a-3p could be a potential candidate for further development as an anti-cancer mechanism. Although miRNA therapeutics reportedly suppress gene expression more effectively than anti-sense oligonucleotide strategies, they have some limitations, such as identification of tissue-specific miRNA, biological instability, off-target effects, and delivery in the cell system. Up to a certain extent, these problems have been resolved by chemical modifications using cholesterol conjugation, morpholinos, cationic lipids, and cationic nanoparticles [70]. Still, more research is needed to understand the mechanism of action for better miRNA therapeutics. These data suggest that miR-125a-3p could regulate USF1 to suppress Chi3L1, and thus inhibit cancer metastasis (Figure S10). Collectively, the present study suggested that Chi3L1 could be a significant target of lung metastasis.
Abbreviations
3'UTR: 3' untranslated region; Ad-shChi3L1: adenoviral vector encoding mouse chitinase 3-like 1 short hairpin ribonucleic acid; Ad-shControl: adenoviral vector encoding mouse negative control short hairpin ribonucleic acid; APOE: apolipoprotein E; bHLH: basic helix-loop-helix; cDNA: complementary deoxyribonucleic acid; Chi3L1: chitinase 3-like 1; ChIP: chromatin immunoprecipitation; DEG: differentially expressed gene; E-box: enhance-box; EGFR: epidermal growth factor receptor; ERK: extracellular regulated kinase; FBS: fetal bovine serum; GWAS: genome-wide association Study; IHC: immunohistochemistry; IL: interleukin; KD: knock-down; KO: knock-out; MAVS: mitochondrial antiviral-signaling protein; MDA-5: melanoma differentiation-associated protein 5; miR-125a-3p: micro ribonucleic acid 125a-3p; miR-342-3p: micro ribonucleic acid 342-3p; miR-Control: micro ribonucleic acid negative control; MMP: matrix metalloproteinase; mRNA: messenger ribonucleic acid; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NSCLC: non-small cell lung cancer; OMIM: online mendelian inheritance in man; PCNA: proliferating cell nuclear antigen; qPCR: quantitative real-time polymerase chain reaction; rhChi3L1: recombinant human chitinase 3-like 1 protein; RIG: retinoic acid-inducible gene; rmChi3L1: recombinant mouse chitinase 3-like 1 protein; sgRNA: single guide ribonucleic acid; siChi3L1: small interference chitinase 3-like 1 ribonucleic acid; siControl: small interference negative control ribonucleic acid; siUSF1: small interference upstream stimulatory factor 1 ribonucleic acid; SP3: specificity protein 3; SPI1: spleen focus forming virus proviral integration oncogene 1; STAT3: signal transducer and activator of transcription 3; TNF-α: tumor necrosis factor alpha; TNM: tumor-node-metastasis; TRE: transcriptional response element; TSS: transcription start site; USF1: upstream stimulatory factor 1; VEGF: vascular endothelial growth factor; WT: wild type.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
This work was supported by a National Research Foundation of Korea [NRF] grant, funded by the Korean government (MSIP) (No. MRC2017R1A5A2015541).
Author Contributions
K.C.K. and J. Y. conducted most of the experiments, performed data analysis, generated most of the experimental mice, and were the primary writers of the manuscript. D.J.S contributed the developments of methodology. J.Y.K., J.K.J., J.S.C., Y-S.R., and S-B.H. provided advice throughout the project. Y.R.K., J.K.S., S.Y.K., S.K.K., and D.H.S. supported the experiments and data analysis. J.T.H. supervised the entire project and had a major role in the experimental design, data interpretation, and writing of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Klein CA. Cancer. The metastasis cascade. Science. 2008;321:1785-1787
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-674
3. Morales-Oyarvide V, Mino-Kenudson M. High-grade lung adenocarcinomas with micropapillary and/or solid patterns: a review. Curr Opin Pulm Med. 2014;20:317-323
4. Fidler IJ, Kripke ML. The challenge of targeting metastasis. Cancer Metastasis Rev. 2015;34:635-641
5. Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502-2512
6. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11-19
7. Apte RN, Dotan S, Elkabets M. et al. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25:387-408
8. Soon YY, Leong CN, Koh WY. et al. EGFR tyrosine kinase inhibitors versus cranial radiation therapy for EGFR mutant non-small cell lung cancer with brain metastases: a systematic review and meta-analysis. Radiother Oncol. 2015;114:167-172
9. Kim S, Takahashi H, Lin W. et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102-106
10. Xing W, Xiao Y, Lu X. et al. GFI1 downregulation promotes inflammation-linked metastasis of colorectal cancer. Cell Death Differ. 2017;24:929-943
11. Mantovani A. Cancer: inflaming metastasis. Nature. 2009;457:36-37
12. Rehli M, Krause SW, Andreesen R. Molecular characterization of the gene for human cartilage gp-39 (CHI3L1), a member of the chitinase protein family and marker for late stages of macrophage differentiation. Genomics. 1997;43:221-225
13. Coffman FD. Chitinase 3-Like-1 (CHI3L1): a putative disease marker at the interface of proteomics and glycomics. Crit Rev Clin Lab Sci. 2008;45:531-562
14. Bonneh-Barkay D, Wang G, Starkey A. et al. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J Neuroinflammation. 2010;7:34-2094 -7-34
15. Wu S, Hsu LA, Cheng ST. et al. Circulating YKL-40 level, but not CHI3L1 gene variants, is associated with atherosclerosis-related quantitative traits and the risk of peripheral artery disease. Int J Mol Sci. 2014;15:22421-22437
16. Johansen JS, Schultz NA, Jensen BV. Plasma YKL-40: a potential new cancer biomarker? Future Oncol. 2009;5:1065-1082
17. Choi IK, Kim YH, Kim JS. et al. High serum YKL-40 is a poor prognostic marker in patients with advanced non-small cell lung cancer. Acta Oncol. 2010;49:861-864
18. Lee CG, Da Silva CA, Dela Cruz CS. et al. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol. 2011;73:479-501
19. Jensen BV, Johansen JS, Price PA. High levels of serum HER-2/neu and YKL-40 independently reflect aggressiveness of metastatic breast cancer. Clin Cancer Res. 2003;9:4423-4434
20. Kawada M, Seno H, Kanda K. et al. Chitinase 3-like 1 promotes macrophage recruitment and angiogenesis in colorectal cancer. Oncogene. 2012;31:3111-3123
21. Ma B, Herzog EL, Lee CG. et al. Role of chitinase 3-like-1 and semaphorin 7a in pulmonary melanoma metastasis. Cancer Res. 2015;75:487-496
22. Thöm I, Andritzky B, Schuch G. et al. Elevated pretreatment serum concentration of YKL-40-An independent prognostic biomarker for poor survival in patients with metastatic nonsmall cell lung cancer. Cancer. 2010;116:4114-4121
23. Barrenas F, Chavali S, Holme P. et al. Network properties of complex human disease genes identified through genome-wide association studies. PLoS One. 2009;4:e8090
24. Rappaport N, Nativ N, Stelzer G. et al. MalaCards: an integrated compendium for diseases and their annotation. Database (Oxford). 2013;2013:bat018
25. Liu CC, Tseng YT, Li W. et al. DiseaseConnect: a comprehensive web server for mechanism-based disease-disease connections. Nucleic Acids Res. 2014;42:W137-W146
26. Rehli M, Niller HH, Ammon C. et al. Transcriptional regulation of CHI3L1, a marker gene for late stages of macrophage differentiation. J Biol Chem. 2003;278:44058-44067
27. Montojo J, Zuberi K, Rodriguez H. et al. GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Res. 2014;3:153
28. Hung CC, Kuo CW, Wang WH. et al. Transcriptional activation of Epstein-Barr virus BRLF1 by USF1 and Rta. J Gen Virol. 2015;96:2855-2866
29. Zeng Y, Li H, Zhang X. et al. Basal transcription of APOBEC3G is regulated by USF1 gene in hepatocyte. Biochem Biophys Res Commun. 2016;470:54-60
30. Zhao X, Wang T, Liu B. et al. Significant association between upstream transcription factor 1 rs2516839 polymorphism and hepatocellular carcinoma risk: a case-control study. Tumour Biol. 2015;36:2551-2558
31. Yuan Q, Bu Q, Li G. et al. Association between single nucleotide polymorphisms of upstream transcription factor 1 (USF1) and susceptibility to papillary thyroid cancer. Clin Endocrinol (Oxf). 2016;84:564-570
32. Chen B, Chen XP, Wu MS. et al. Expressions of heparanase and upstream stimulatory factor in hepatocellular carcinoma. Eur J Med Res. 2014;19:45
33. Ikeda R, Nishizawa Y, Tajitsu Y. et al. Regulation of major vault protein expression by upstream stimulating factor 1 in SW620 human colon cancer cells. Oncol Rep. 2014;31:197-201
34. Chang JT, Yang HT, Wang TC. et al. Upstream stimulatory factor (USF) as a transcriptional suppressor of human telomerase reverse transcriptase (hTERT) in oral cancer cells. Mol Carcinog. 2005;44:183-192
35. Chen N, Szentirmay MN, Pawar SA. et al. Tumor-suppression function of transcription factor USF2 in prostate carcinogenesis. Oncogene. 2006;25:579-587
36. Schmidt H, Johansen JS, Gehl J. et al. Elevated serum level of YKL-40 is an independent prognostic factor for poor survival in patients with metastatic melanoma. Cancer. 2006;106:1130-1139
37. Vihinen P, Kahari VM. Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer. 2002;99:157-166
38. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4-10
39. Stoimenov I, Helleday T. PCNA on the crossroad of cancer. Biochem Soc Trans. 2009;37:605-613
40. Yu Y, Luk F, Yang JL. et al. Ras/Raf/MEK/ERK pathway is associated with lung metastasis of osteosarcoma in an orthotopic mouse model. Anticancer Res. 2011;31:1147-1152
41. Ai X, Wu Y, Zhang W. et al. Targeting the ERK pathway reduces liver metastasis of Smad4-inactivated colorectal cancer. Cancer Biol Ther. 2013;14:1059-1067
42. Choi C, Helfman DM. The Ras-ERK pathway modulates cytoskeleton organization, cell motility and lung metastasis signature genes in MDA-MB-231 LM2. Oncogene. 2014;33:3668-3676
43. Ma B, Herzog EL, Moore M. et al. RIG-like Helicase Regulation of Chitinase 3-like 1 Axis and Pulmonary Metastasis. Sci Rep. 2016;6:26299
44. Renault MA, Jalvy S, Potier M. et al. UTP induces osteopontin expression through a coordinate action of NFkappaB, activator protein-1, and upstream stimulatory factor in arterial smooth muscle cells. J Biol Chem. 2005;280:2708-2713
45. Hu XT, Zhu BL, Zhao LG. et al. Histone deacetylase inhibitor apicidin increases expression of the alpha-secretase ADAM10 through transcription factor USF1-mediated mechanisms. FASEB J. 2017;31:1482-1493
46. Betel D, Koppal A, Agius P. et al. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90
47. Jao LE, Wente SR, Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci U S A. 2013;110:13904-13909
48. Ittner LM, Gotz J. Pronuclear injection for the production of transgenic mice. Nat Protoc. 2007;2:1206-1215
49. Gonzalez-Rodriguez A, Reibert B, Amann T. et al. In vivo siRNA delivery of Keap1 modulates death and survival signaling pathways and attenuates concanavalin-A-induced acute liver injury in mice. Dis Model Mech. 2014;7:1093-1100
50. Son DJ, Kumar S, Takabe W. et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat Commun. 2013;4:3000
51. Gu SM, Park MH, Yun HM. et al. CCR5 knockout suppresses experimental autoimmune encephalomyelitis in C57BL/6 mice. Oncotarget. 2016;7:15382-15393
52. Libreros S, Garcia-Areas R, Keating P. et al. Allergen induced pulmonary inflammation enhances mammary tumor growth and metastasis: Role of CHI3L1. J Leukoc Biol. 2015;97:929-940
53. Horbach T, Gotz C, Kietzmann T. et al. Protein kinases as switches for the function of upstream stimulatory factors: implications for tissue injury and cancer. Front Pharmacol. 2015;6:3
54. Johansen JS, Olee T, Price PA. et al. Regulation of YKL-40 production by human articular chondrocytes. Arthritis & Rheumatology. 2001;44:826-837
55. Singh SK, Bhardwaj R, Wilczynska KM. et al. A complex of nuclear factor I-X3 and STAT3 regulates astrocyte and glioma migration through the secreted glycoprotein YKL-40. J Biol Chem. 2011;286:39893-39903
56. Zhang L, Handel MV, Schartner JM. et al. Regulation of IL-10 expression by upstream stimulating factor (USF-1) in glioma-associated microglia. J Neuroimmunol. 2007;184:188-197
57. Weigert C, Brodbeck K, Sawadogo M. et al. Upstream stimulatory factor (USF) proteins induce human TGF-beta1 gene activation via the glucose-response element-1013/-1002 in mesangial cells: up-regulation of USF activity by the hexosamine biosynthetic pathway. J Biol Chem. 2004;279:15908-15915
58. Galibert MD, Carreira S, Goding CR. The Usf-1 transcription factor is a novel target for the stress-responsive p38 kinase and mediates UV-induced Tyrosinase expression. EMBO J. 2001;20:5022-5031
59. Sharova LV, Sharov AA, Nedorezov T. et al. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 2009;16:45-58
60. Vassilenko KS, Alekhina OM, Dmitriev SE. et al. Unidirectional constant rate motion of the ribosomal scanning particle during eukaryotic translation initiation. Nucleic Acids Res. 2011;39:5555-5567
61. Calin GA, Sevignani C, Dumitru CD. et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999-3004
62. Betel D, Wilson M, Gabow A. et al. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149-53
63. Ninio-Many L, Grossman H, Shomron N. et al. microRNA-125a-3p reduces cell proliferation and migration by targeting Fyn. J Cell Sci. 2013;126:2867-2876
64. Ninio-Many L, Grossman H, Levi M. et al. MicroRNA miR-125a-3p modulates molecular pathway of motility and migration in prostate cancer cells. Oncoscience. 2014;1:250-261
65. Xu X, Lv YG, Yan CY. et al. Enforced expression of hsa-miR-125a-3p in breast cancer cells potentiates docetaxel sensitivity via modulation of BRCA1 signaling. Biochem Biophys Res Commun. 2016;479:893-900
66. Zhang H, Zhu X, Li N. et al. miR-125a-3p targets MTA1 to suppress NSCLC cell proliferation, migration, and invasion. Acta Biochim Biophys Sin (Shanghai). 2015;47:496-503
67. Yin F, Zhang JN, Wang SW. et al. MiR-125a-3p regulates glioma apoptosis and invasion by regulating Nrg1. PLoS One. 2015;10:e0116759
68. Jin T, Lu Y, He QX. et al. The Role of MicroRNA, miR-24, and Its Target CHI3L1 in Osteomyelitis Caused by Staphylococcus aureus. J Cell Biochem. 2015;116:2804-2813
69. Sarma NJ, Tiriveedhi V, Subramanian V. et al. Hepatitis C virus mediated changes in miRNA-449a modulates inflammatory biomarker YKL40 through components of the NOTCH signaling pathway. PLoS One. 2012;7:e50826
70. Jain CK, Gupta A, Dogra N. et al. MicroRNA therapeutics: the emerging anticancer strategies. Recent Pat Anticancer Drug Discov. 2014;9:286-296
Author contact
![]() Corresponding authors: Sang-Bae Han, PhD, Professor, College of Pharmacy & Medical Research Center, Chungbuk National University, 194-31 Osongsaengmyeong 1-ro, Osong-Biocampus Building Room 303, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Korea. Phone: 82-43-261-2815; FAX: 82-43-268-2732; E-mail: shanac.kr and Jin Tae Hong, PhD, Professor, College of Pharmacy & Medical Research Center, Chungbuk National University, 194-31 Osongsaengmyeong 1-ro, Osong-Biocampus Building Room 303, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Korea. Phone: 82-43-261-2813; FAX: 82-43-268-2732; E-mail: jinthongac.kr
Corresponding authors: Sang-Bae Han, PhD, Professor, College of Pharmacy & Medical Research Center, Chungbuk National University, 194-31 Osongsaengmyeong 1-ro, Osong-Biocampus Building Room 303, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Korea. Phone: 82-43-261-2815; FAX: 82-43-268-2732; E-mail: shanac.kr and Jin Tae Hong, PhD, Professor, College of Pharmacy & Medical Research Center, Chungbuk National University, 194-31 Osongsaengmyeong 1-ro, Osong-Biocampus Building Room 303, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Korea. Phone: 82-43-261-2813; FAX: 82-43-268-2732; E-mail: jinthongac.kr