Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Methods
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
Author biography
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(16):4509-4519. doi:10.7150/thno.27428 This issue Cite
Research Paper
CD28null pro-atherogenic CD4 T-cells explain the link between CMV infection and an increased risk of cardiovascular death
Alejandra Pera1,2, ![]() , Stefano Caserta3,†, Fabio Albanese1, Pinar Blowers1, George Morrow1, Nadia Terrazzini4, Helen E Smith5, Chakravarthi Rajkumar1, Bernhard Reus6, James R Msonda6,7, Murielle Verboom8, Michael Hallensleben8, Rainer Blasczyk8, Kevin A Davies1, Florian Kern1
, Stefano Caserta3,†, Fabio Albanese1, Pinar Blowers1, George Morrow1, Nadia Terrazzini4, Helen E Smith5, Chakravarthi Rajkumar1, Bernhard Reus6, James R Msonda6,7, Murielle Verboom8, Michael Hallensleben8, Rainer Blasczyk8, Kevin A Davies1, Florian Kern1
1. Department of Clinical and Experimental Medicine, Brighton and Sussex Medical School, Brighton, United Kingdom.
2. Immunology and allergy GC-01 group Maimonides Biomedicine Institute of Cordoba (IMIBIC), Reina Sofia Hospital, University of Cordoba, Cordoba, Spain.
3. Department of Global Health and Infectious Diseases, Brighton and Sussex Medical School, Brighton, United Kingdom.
4. School of Pharmacy and Biomolecular Sciences, University of Brighton.
5. Family Medicine and Primary Care, Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore.
6. School of Engineering and Informatics, University of Sussex, Brighton, UK.
7. University of Malawi, The Polytechnic, Blantyre, Malawi.
8. Institute for Transfusion Medicine, Hannover Medical School, Hannover, Germany.
†School of Life Sciences University of Hull, Hull, UK.
Received 2018-5-23; Accepted 2018-7-18; Published 2018-8-7
Abstract

An increased risk of cardiovascular death in Cytomegalovirus (CMV)-infected individuals remains unexplained, although it might partly result from the fact that CMV infection is closely associated with the accumulation of CD28null T-cells, in particular CD28null CD4 T-cells. These cells can directly damage endothelium and precipitate cardiovascular events. However, the current paradigm holds that the accumulation of CD28null T-cells is a normal consequence of aging, whereas the link between these T-cell populations and CMV infection is explained by the increased prevalence of this infection in older people. Resolving whether CMV infection or aging triggers CD28null T-cell expansions is of critical importance because, unlike aging, CMV infection can be treated.
Methods: We used multi-color flow-cytometry, antigen-specific activation assays, and HLA-typing to dissect the contributions of CMV infection and aging to the accumulation of CD28null CD4 and CD8 T-cells in CMV+ and CMV- individuals aged 19 to 94 years. Linear/logistic regression was used to test the effect of sex, age, CMV infection, and HLA-type on CD28null T-cell frequencies.
Results: The median frequencies of CD28null CD4 T-cells and CD28null CD8 T-cells were >12-fold (p=0.000) but only approximately 2-fold higher (p=0.000), respectively, in CMV+ (n=136) compared with CMV- individuals (n=106). The effect of CMV infection on these T-cell subsets was confirmed by linear regression. Unexpectedly, aging contributed only marginally to an increase in CD28null T-cell frequencies, and only in CMV+ individuals. Interestingly, the presence of HLA-DRB1*0301 led to an approximately 9-fold reduction of the risk of having CD28null CD4 T-cell expansions (OR=0.108, p=0.003). Over 75% of CMV-reactive CD4 T-cells were CD28null.
Conclusion: CMV infection and HLA type are major risk factors for CD28null CD4 T-cell-associated cardiovascular pathology. Increased numbers of CD28null CD8 T-cells are also associated with CMV infection, but to a lesser extent. Aging, however, makes only a negligible contribution to the expansion of these T-cell subsets, and only in the presence of CMV infection. Our results open up new avenues for risk assessment, prevention, and treatment.
Keywords: CD28null CD4 T-cells, CD28null CD8 T-cells, cardiovascular disease, aging, Cytomegalovirus, coronary complications
Introduction
A recent publication shows that Cytomegalovirus (CMV) infection increases the risk of cardiovascular death by over 20% [1] but no specific mechanisms explaining this effect were identified. CMV infection, however, is notorious for promoting large expansions of terminally differentiated effector T-cells, including CD4 T-cells. This is particularly observable in older people [2, 3]. Moreover, there is good evidence that terminally differentiated T-cells may cause vascular damage, to the extent that therapies specifically targeting T-cells in advanced atherosclerosis are being developed [4-7]. Among activated CD4 T-cells, cardiologists are particularly interested in CD28null CD4 T-cells [8-10]. These terminally differentiated effector cells do not express CD28, a co-stimulatory receptor molecule, which antigen-presenting cells engage during early T-cell activation. CD28null CD4 T-cells were initially discovered in rheumatoid arthritis (RA) [11-14], but later associated with unstable angina and coronary artery plaque instability (CAD) [15]. Multiple links between these cells and cardiovascular complications have since been reported [16-20]. Down-regulation of CD28 on CD4 T-cells is thought to be triggered by continuous/repetitive antigen exposure [21], which could be the result of a persistent viral infection, for example with CMV. CD28null CD8 T-cells, which frequently express the surface marker, CD57, were also mentioned in the context of cardiovascular and autoimmune disease but seem to be associated with a broader range of conditions [22].
CD28null CD4 T-cells accumulate in older people and show reduced proliferative capacity among many other signs of cellular senescence. Large frequencies of these cells are, therefore, primarily attributed to normal (immune system) aging [9, 21, 23]. While an association of CMV infection with increased numbers of CD28null CD4 T-cells was repeatedly reported in the literature [10, 23-27], this link is generally considered to be indirect and explained by the fact that older people are more likely to be CMV infected [23, 28]. Nobody has yet studied CD28null CD4 or CD8 T-cells in a large enough number of CMV-seronegative (CMV-) older people to resolve this issue. However, several smaller studies in the fields of autoimmune and cardiovascular disease offer some insight. Following participant stratification by CMV infection status they reported frequencies of CD28null CD4 T-cells of 5-10% or more in CMV-seropositive (CMV+) but not exceeding 1.5% in CMV- participants [29-32]. These results suggest that clinically relevant expansions of CD28null CD4 T-cells are effectively limited to CMV+ individuals, because the frequencies of CD28null CD4 T-cells that were associated with clinical effects were generally on the order of several percent [10, 33].
It was not the purpose of our work to show that CD28null CD4 T-cells are associated with cardiovascular (CV) morbidity or mortality, since there is overwhelming evidence for this association in the literature [8-10]. Instead, we examined the frequencies of CD28null CD4 T-cells in n=93 CMV- and n=122 CMV+ generally healthy older people and a corresponding cohort of young people; CD28null CD8 T-cells were evaluated in parallel. Our investigation was focused on the intriguing possibility that, independently of aging, CMV infection is a major risk factor for the expansion of the highly pro-atherogenic CD28null CD4 T-cell subset [8, 9]. This would help explain the significant effect of CMV infection on CV mortality [1] and open up interesting new avenues for research, including future therapeutic interventions.
Results
Frequencies of CD28null CD4 T-cells are an order of magnitude higher in CMV+ compared to CMV- individuals
CD4 T-cells were divided into CD28+ and CD28- ('null') subsets. These were further subdivided by their expression of the differentiation marker, CD27 [26, 34] (Figure 1A and Figure S1). Frequencies of CD28null CD4 T-cells (in percent of CD4 T-cells) ranged from barely detectable to 70% and were log10 transformed for improved visualization and normality of data distribution [35]. Across all individuals (CMV+ and CMV-), CD28null CD4 T-cell frequencies displayed a bimodal distribution (Figure 1B, top). Plotting CMV- and CMV+ individuals separately provided two very similar, near-normal distributions (Figure 1B, middle and bottom); however, in CMV+ participants the median was significantly higher (by a factor of 12.7) (Figure 1C). This difference was explained by CD28null CD4 T-cells lacking CD27 (p = 0.000). Those expressing CD27 made no contribution (Figure 1D). ROC analysis based on CD28null CD4 T-cell frequencies for discriminating CMV- and CMV+ individuals revealed an AUC of >0.910 (p=0.000) for all CD28null CD4 T-cells (and an even higher AUC for CD28nullCD27- CD4 T-cells). Note that a value >1.5% of CD28null CD4 T-cells identified a CMV+ individual with >95% probability (Figure 1E).
CD28null CD4 T-cell numbers are by an order of magnitude higher in CMV+ compared to CMV- people. (A) Dotplots showing CD27/CD28 expression on CD4 T-cells in CMV- and CMV+ individuals. CD28null cells are indicated (circle). (B) The CD28null CD4 T-cell frequency distribution (log10-transformed CD4 T-cell fractions) is shown for the whole cohort (top, n=242), CMV- (middle, n=106) and CMV+ individuals (bottom, n=136). (C) Scatterplots show log10-transformed fractions of CD28null CD4 T-cells. The UQ+1.5×IQR for CMV- individuals is indicated (dotted line). (D) Scatterplots show the log10-transformed fractions of the CD27+ and CD27- subsets of CD28null CD4 T-cells. Error bars in scatterplots show median, upper, and lower quartiles. (E) The ROC curve shows the separation of CMV- and CMV+ populations by CD28null CD4 T-cell frequencies.
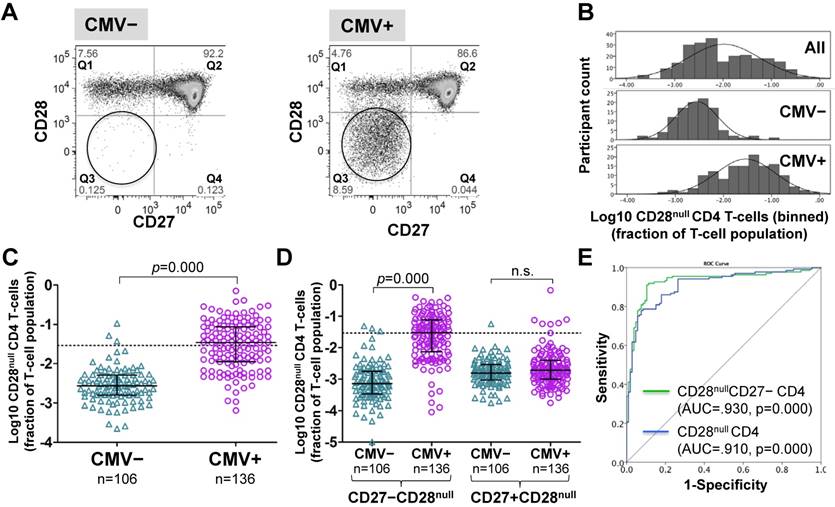
As no clear definition of 'T-cell expansion' exists in the literature, in analogy to our own previous work [35], we defined expansions of CD28null CD4 T-cells as frequencies above a non-parametric, upper outlier limit (upper quartile + 1.5×interquartile range). Only CMV- people were used as 'normal' reference group because of the observed effects of CMV infection on CD28null CD4 T-cell frequencies (Figure 1B). CD28null CD4 T-cell expansions were, therefore, defined as frequencies exceeding 2.9% of CD4 T-cells (Figure 1C, dotted line). According to this definition, <3% (3/106) of CMV- but >55% (76/136) of CMV+ individuals had expansions.
CMV infection is associated with a smaller increase of CD28null CD8 T-cells than CD28null CD4 T-cells
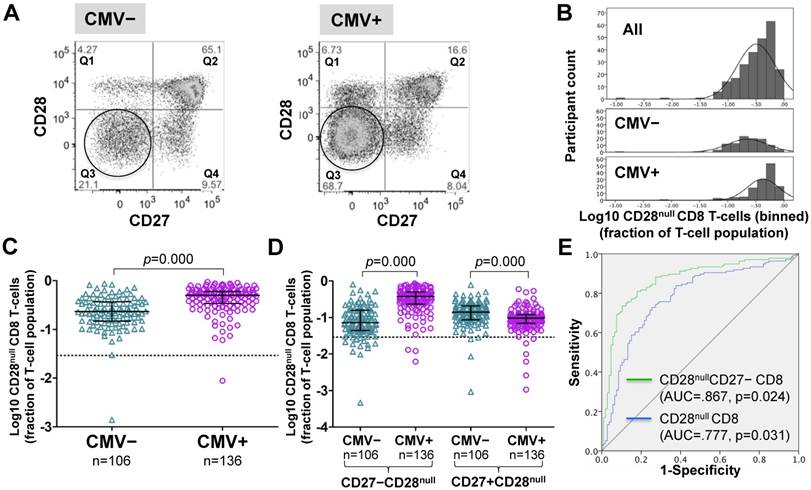
Frequencies of CD28null CD8 T-cells (in percent of CD8 T-cells) were generally higher in CMV+ individuals (Figure 2A). However, unlike for CD28null CD4 T-cells, no clear bimodal distribution was visible in the whole cohort (Figure 2B, top). Nevertheless, separate analysis of CMV- and CMV+ individuals revealed an approximately 2.2-fold median difference between the two populations (Figure 2B, middle and bottom, and Figure 2C). In analogy to CD28null CD4 T-cells, this difference was explained by the subset lacking CD27 expression (p=0.000). Interestingly, the subset expressing CD27 was significantly reduced in CMV+ individuals (Figure 2D). As with CD28null CD4 T-cells, ROC analysis provided good discrimination between CMV- and CMV+ individuals based on all CD28null CD8 T-cells (AUC=0.777; p=0.000) but even better discrimination when only CD28nullCD27- CD8 T-cells were taken into account (Figure 2E). On the whole, the overlap between CMV- and CMV+ populations was bigger with respect to CD28null CD8 than CD28null CD4 T-cell frequencies.
In CMV+ people, CD28null CD8 T-cell numbers are about twice as high as in CMV- people. (A) Dotplots show CD27 versus CD28 expression on CD8 T-cells in CMV- and CMV+ individuals. CD28null cells are indicated (circle). (B) The CD28null CD8 T-cell frequency distribution (log10-transformed CD8 T-cell fractions) is shown in the whole cohort (top, n=242), CMV- (middle, n=106) and CMV+ individuals (bottom, n=136). (C) Scatterplots show log10-transformed fractions of CD28null CD8 T-cells. (D) Scatterplots show log10-transformed fractions of the CD27+ and CD27- subsets of CD28null CD8 T-cells. Error bars in scatterplots show median, upper, and lower quartiles. (E) The ROC curve shows the separation of CMV- and CMV+ populations by CD28null CD8 T-cell frequencies.
The effect of age on CD28null CD4 and CD8 T-cells is marginal
To quantify the effects of CMV infection and age on CD28null CD4 T-cell frequencies, we used a hierarchical multiple regression model including sex as a possible confounder. Models based on sex alone or sex and age together explained only 0.2% and 1.8% of the variance of CD28null CD4 T-cell frequencies, respectively. After adding CMV infection status, the model explained 47.8% of that variance. While both CMV infection and age had statistically significant effects, becoming CMV+ was equivalent to the effect of 253 years of aging (Table 1). The corresponding model for CD8 T-cells showed an even smaller effect of age and a much smaller effect of CMV infection on the variance of CD28null CD8 T-cell frequencies, with only 17.8% of that variance being explained by CMV status (Table 2). Without CMV status, the model explained only 2.1% of this variance.
Effect of CMV infection status, age and sex on the size of the CD28null CD4 T-cell subset.a
| Variable | Parameter estimate | Standard error | 95% CI for parameter | t | P-value | R2 b | |
|---|---|---|---|---|---|---|---|
| lower bound | upper bound | ||||||
| Intercept | -2.857 | 0.183 | -3.218 | -2.497 | -15.614 | 0.000 | 47.8% c |
| Sex | 0.020 | 0.069 | -0.117 | 0.156 | 0.287 | 0.774 | |
| Age | 0.004 | 0.002 | 7.168×10-5 | 0.008 | 2.005 | 0.046 | |
| CMV infection | 1.012 | 0.070 | 0.874 | 1.149 | 14.499 | 0.000 | |
a The table shows the final step of the hierarchical multiple regression model.
b Percent variability of dependent variable explained.
c R2 for models including sex alone or sex and age were 0.2 and 1.8%, respectively.
Effect of CMV infection status, age and sex on the size of the CD28null CD8 T-cell subset.a
| Variable | Parameter estimate | Standard error | 95% CI for parameter | t | P-value | R2 b | |
|---|---|---|---|---|---|---|---|
| lower bound | upper bound | ||||||
| Intercept | -0.777 | 0.106 | -0.986 | -0.567 | -7.313 | 0.000 | 17.8% c |
| Sex | 0.026 | 0.040 | -0.105 | 0.054 | -0.639 | 0.524 | |
| Age | 0.003 | 0.001 | 0.000 | 0.005 | 2.102 | 0.037 | |
| CMV infection | 0.273 | 0.041 | 0.193 | 0.352 | 6.733 | 0.000 | |
a The table shows the final step of the hierarchical multiple regression model.
b Percent variability of dependent variable explained.
c R2 for models including sex alone or sex and age were 0.1% and 2.1 %, respectively.
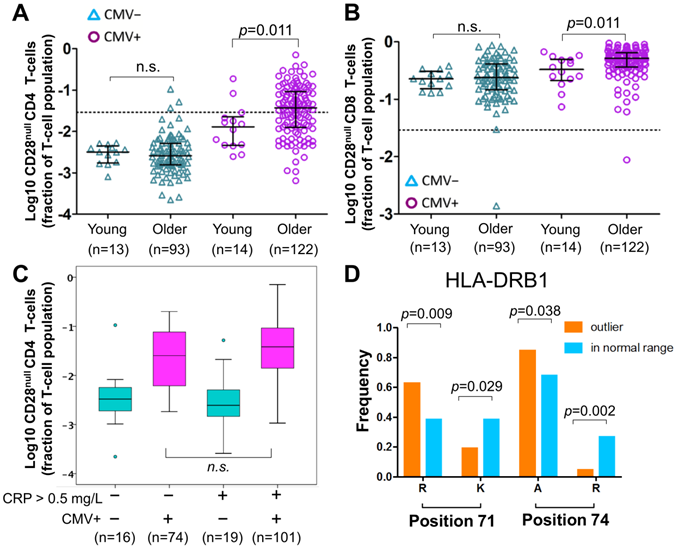
Of note, CD28null CD4 T-cell frequencies were not significantly different between young and older CMV- individuals, whereas among CMV+ individuals a small, statistically significant difference between the age groups was observed (Figure 3A). The latter corresponded to the small but significant effect of aging found in the regression models, which take the whole population (CMV- and CMV+) into account. The same applied to CD28null CD8 T-cells (Figure 3B).
Effect of age, HLA and CRP on CD28null CD4 T-cells. (A-B) Scatterplots show differences in CD28null CD4 (A) and CD8 (B) T-cell frequencies between young and older CMV- and CMV+ people (n.s.= not significant). CD28null T-cell fractions were log10-transformed for better visualization. (C) Box-plots show the levels of CD28null CD4 T-cell frequencies in four subgroups based on CMV infection status (CMV- green, CMV+ red) and CRP levels (≤ 5 mg/L or >5mg/L). (D) Bar-charts show significant differences in the frequencies of arginine (R), lysine (K), and alanine (A) at positions 71 and 74 of exon 2 of HLA-DRB1 in individuals with and without expansions. Significance levels are indicated.
CRP only has a small effect on CD28null CD4 T-cell frequencies
In analogy to a historic study on CMV and CAD [36], we compared the frequencies of CD28null CD4 T-cells between 4 subgroups based on CMV infection status (CMV+ or CMV-) and normal versus raised CRP levels (≤ 5 mg/L or > 5 mg/L). CRP was available in a total of n=210 older participants. The highest frequencies of CD28null CD4 T-cells (median) were found in CMV+ people with CRP levels > 5 mg/L (Figure 3C), the same group that was previously found to have the highest CAD prevalence. However, the difference in CD28null CD4 T-cells frequencies between CMV+ individuals with CRP-levels ≤ 5 mg/L or > 5 mg/L was statistically not significant; thus, CRP was clearly not a critical factor for their accumulation. The same trend was observed for CD28null CD8 T-cells but differences were not statistically significant either (not shown).
HLA-type significantly affects CD28null CD4 T-cell expansions
The frequencies of CD28null CD4 T-cells were significantly higher in CMV+ than in CMV- participants but varied by about a factor of 1000 in both groups, suggesting they were affected by additional parameters (Figure 1B). We previously observed an effect of HLA type on the frequencies of CMV-specific T-cells [35] and now wished to explore if this was also true for CD28null CD4 T-cells. Since the effect of CMV infection on the CD28null CD8 T-cell subset was small and the frequencies of these cells were less variable, we focused on CD28null CD4 T-cells for the remainder of the analysis.
Since CD4 T-cells are class-II MHC restricted, high-resolution HLA-DRB1 and DQB1 typing was performed in a subgroup of 64 CMV+ individuals (no materials for HLA-typing were available for the remaining participants). We used data reduction analysis to identify the most relevant alleles/allele groups in regard to CD28null CD4 T-cell frequencies and then built a linear regression model including the selected alleles/allele groups along with age and sex as independent variables (sex was considered to be a possible confounder). This model explained 22.8% of the variation of CD28null CD4 T-cell frequencies in CMV+ individuals, and showed that the effects of both age and sex were minor and statistically insignificant (Table 3).
Effect of sex, age, and HLA-type on CD28null CD4 T-cell frequencies.a
| Hierarchical multiple regression levels | Variables considered | P b | R2 c |
|---|---|---|---|
| (1) | Sex | 0.136 | 0.035 (3.5%) |
| (2) | Sex, Age | 0.093 | 0.075 (7.5%) |
| (3) | Sex, Age DRB1*03:01P, DQB1*02:01P, DQB1*03:02P | 0.009 | 0.228 (22.8%) |
aHierarchical multiple regression was used to analyze the effect of HLA-type on the frequencies of CD28null CD4 T-cells among CMV+ individuals.
b Level of significance for the model.
c Percent variability of dependent variable explained.
The model did not, however, directly answer if the presence or absence of certain HLA-alleles/allele groups made the occurrence of expansions of CD28null CD4 T-cells more or less likely (i.e., frequencies exceeding 2.9% of CD4 T-cells, see above). For that purpose, we used binary logistic regression where the binary outcome was 'expansion' or 'no expansion'. The initial binary model was based on the same independent HLA variables as the above linear model. All models also included age and sex as independent variables; HLA variables not making significant contributions were removed during model optimization. Our optimized model included only HLA-DRB1*03:01P, age, and sex. It showed that HLA-DRB1*03:01P had a strongly negative effect on CD28null CD4 T-cell expansions and provided correct classification in 73.4% of cases with a good model fit (Table S3). To potentially improve classification, we built alternative models (always including age and sex) based on the amino acid sequence of the HLA-DRB1 binding groove (its exon 2 sequence) rather than allele group/allele names. Amino acids in certain positions of the binding groove determine if a peptide can be presented or not [37], and certain combinations of amino acids in positions 11, 71 and 74 ('haplotypes') were previously linked to disease severity and, interestingly, cardiovascular events in RA (Table S4) [38, 39]. An optimized model based on these haplotypes provided correct classification in 75% of cases and, similar to the effect of HLA-DRB1*03:01P in the previous model, the haplotype Ser11-Lys71-Arg74 had a strongly negative effect on expansion (Table S5). Interestingly, both HLA-DRB1*03:01P and Ser11-Lys71-Arg74 are associated with a mild course of RA [38, 39]. In our cohort, HLA-DRB1*03:01P was the only allele group expressing Ser11-Lys71-Arg74, i.e., the models confirmed each other. We finally investigated if classification could be further improved by directly using amino acids in certain positions of exon 2 as model variables (rather than the presence/absence of set haplotypes). Interestingly, significant differences between groups existed with respect to positions 71 (Lys71, Arg71) and 74 (Ala74, Arg74) but not position 11 (Figure 3D). Our final, optimized model was based on Arg71 and Arg74, and provided correct classification in 79.7% of cases with an excellent fit (Table 4). Note that the effect of age and sex in all models was minor.
Arginine in position 74 of HLA-DRB1 protects from CD28null CD4 T-cell expansion.
| Variable | Parameter estimate | Standard error | p-value | OR | 95% CI for OR | Pseudo-R2 (model fit) b |
|---|---|---|---|---|---|---|
| Sex (m) | -1.063 | 0.640 | 0.097 | 0.345 | 0.098 - 1.211 | |
| age | 0.055 | 0.023 | 0.019 | 1.056 | 1.009 - 1.106 | |
| Arg71 | 1.748 | 0.871 | 0.045 | 5.742 | 1.041 - 31.680 | 0.351 - 0.468 |
| Arg74 | -2.166 | 0.807 | 0.007 | 0.115 | 0.024 - 0.550 | |
| constant | -4.029 | 1.872 | 0.031 | 0.018 | n.a. |
a A binary logistic regression model was constructed based on sex (step 1), age (step 2), and Arg71 as well as Arg74 (step 3). The table shows step 3 of the model, which provided correct classification in 79.4% of cases.
b The model fit is shown as the range between Cox & Snell and Nagelkerke pseudo-R2.
CD28null CD4 T-cell accumulations seem to affect blood pressure
The average number of anti-hypertensives, an indirect indicator of the severity of hypertension, was significantly higher in participants with CD28null CD4 T-cell expansions. This was true when beta-blockers and diuretics were included (p=0.021) but also when they were excluded (p=0.027). Moreover, 45/143 (31%) of participants without CD28null CD4 T-cell expansions but 33/72 (46%) of those with expansions were taking one or more anti-hypertensives (p=0.05, Chi-square test) when diuretics and beta-blockers were included; the figures were 38/143 (27%) and 30/72 (42%), respectively, when diuretics and beta-blockers were excluded (p=0.03, Chi-square test). The percentages of participants with previous vascular complications (at least one of the following, TIA or stroke, coronary graft, or stent) were not significantly different between these groups. However, individuals with complications had on average higher levels of CD28null CD4 T-cells (not statistically significant). With respect to lipid profiles (total cholesterol, HDL-cholesterol, LDL-cholesterol, and triglycerides), no significant differences between CMV- and CMV+ individuals (Table S6) or between those with and without expansions of CD28null CD4 T-cells were found (n=56 and n=112, respectively).
Significant proportions of CD28null CD4 T-cells are CMV specific
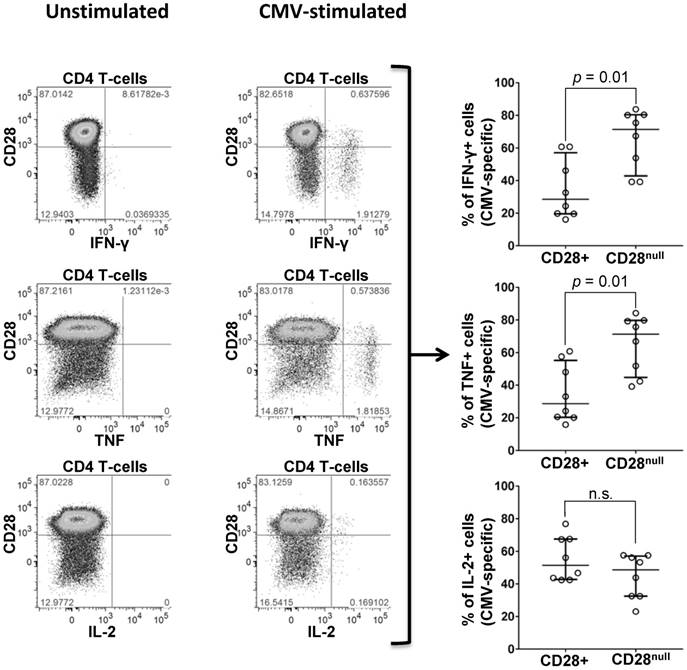
To quantify the proportion of CD28null cells among CMV-specific CD4 T-cells, PBMC from 7 CMV+ individuals (Table S7) were stimulated with peptide pools, each representing one complete CMV protein. CMV-specific CD4 T-cells were identified by intracellular IFN-ɣ, TNF, or IL-2; the distributions of activated cells into CD28null and CD28+ subsets were analyzed with respect to each cytokine (Figure 4). IFN-ɣ and TNF-producing CMV-specific CD4 T-cells were predominantly CD28null (median >70%). Very few cells expressed IL-2 and no significant difference between the CD28null and CD28+ subsets was observed. Between 3% and 18% (median 7%) of CD28null CD4 T-cells responded to single, selected CMV peptide pools. In agreement with the literature [29], all CD28null CD4 T-cells strongly expressed the vascular adhesion marker, CD49d. This was, moreover, true for all CMV-specific T-cells (Figure S2 and Figure S3).
Expression of CD28 by CMV-specific cells. PBMCs from a representative CMV+ individual stimulated with CMV-peptide (UL83). Plots show unstimulated (left) and stimulated CD4 T-cells (middle). CD28 expression is shown versus IFN-ɣ (top), TNF (middle), and IL-2 (bottom), each identifying responding cells. SEB stimulation (positive control) strongly induced all three cytokines (not shown). Individual scatter graphs (right) show CD28 expression in responding cells (proteins UL55, UL83, or UL86, n=7). At least 40% and up to almost 90% (median 70%) of IFN-ɣ and TNF-producing CMV-specific T-cells are CD28null. Error bars show median and interquartile range.
Methods
Ethics Statement
The study was approved by the UK National Research Ethics Service (NRES) (09/H1102/84 and 13/LO/1270). Written informed consent was obtained from all participants. The study was conducted in agreement with the Declaration of Helsinki.
Participants and samples
Twenty-seven healthy, young volunteers (19-32 years) were recruited from university (students/staff), 215 generally healthy, older volunteers (60-94 years) were recruited by general medical practitioners. Some of the older volunteers had previously experienced vascular events, including TIA, stroke, or CV complications treated with coronary stent implantation or grafts. Such individuals were not excluded if generally well with normal physical activity. Individuals with significant CV morbidity, however, were excluded (please see Supplementary Material for more details).
CMV serology
CMV immunoglobulin G (IgG) serology (Architect CMV IgG, Abbot, Maidenhead, UK) was performed at the Brighton and Sussex University Hospital Trust (BSUHT) virology laboratory.
Whole blood antibody staining for CD28null CD4 T-cells
Fresh whole blood was stained with fluorophore-labeled monoclonal antibodies: CD45, CD3, CD4, CD8, CD28 and CD27 in a 'lyse and wash' protocol (see Supplementary Material).
Additional phenotypic characterization of CD28null CD4 T-cells
Frozen PBMC from 8 old CMV+ individuals were used to characterize the expression of CD49d on CD28null CD4 T-cells (see Supplementary Material).
CMV peptides
CMV proteins for stimulation were chosen based on CD4 T-cell responses previously studied in the same individuals (see Supplementary Material).
CMV reactivity of CD28null CD4 T-cells
PBMCs from CMV+ individuals were activated with overlapping peptide pools, stained and acquired by flow-cytometry (see Supplementary Material).
CRP and lipid profiles
CRP and lipid profiles were determined at the Brighton and Sussex University Hospital Trust (BSUHT) pathology laboratory (Cobas analyzer platform, Roche, Burgess Hill, UK). Non-HDL-cholesterol was calculated as 'total cholesterol - HDL-cholesterol'. The LDL/HDL cholesterol ratio was calculated as 'LDL-cholesterol/HDL-cholesterol'.
HLA-typing and sequence analysis
HLA-typing was performed at the Institute for Transfusion Medicine, Hannover Medical School, Hannover/Germany (see Supplementary Material).
Data acquisition and analysis
Samples were acquired on an LSR II flow-cytometer (BD). FlowJo v9.x software (Tree Star Inc., Ashland, OR) was used for data analysis. Gating strategies are described in Figure S1-3.
Statistical analysis
GraphPad Prism 7 (GraphPad Software, Inc., San Diego, California, USA) and SPSS v23/v24 software (IBM, London, UK) were used for visualization and statistical analysis. Population frequencies were log10-transformed to improve normality and visualization. Hierarchical/binary logistic regression models were run in SPSS. For additional details please see Supplementary Material.
Discussion
Our results show that CMV infection is significantly associated with the accumulation of CD28null CD4 T-cells, which are known to trigger cardiovascular complications and even stroke [9]. Our data further suggest that CMV may directly drive this subset with a significant proportion of these cells recognizing CMV-antigens. The frequencies of CD28null CD4 T-cells were an order of magnitude (>12-fold) higher in CMV+ compared to CMV- individuals but only marginally affected by age (note that in older CMV- individuals, frequencies were below 1.5% in >95% of all cases). A small effect of age was noted in CMV+ individuals only and might be related to the duration of CMV infection. These observations seem to refute the idea that accumulation of CD28null CD4 T-cells is a result of normal (immune-system) aging [40].
The fact that only CMV-infected people can account for high frequencies of CD28null CD4 T-cells sheds new light on numerous published reports. For illustration, in 2007, Liuzzo et al. [18] reported CD28null CD4 T-cell frequencies in 3 patient groups with increasing severity of CAD. Thirty-five percent of those with the mildest disease displayed CD28null CD4 T-cell frequencies >4%. Such frequencies, however, were displayed by 50% and 75%, respectively, of the groups with greater and the greatest severity of CAD. While the CMV infection status of these individuals was unknown, our results suggest that at least 95% of the individuals with CD28null CD4 T-cell frequencies >4% were CMV+. It would appear that in Liuzzo's study, the proportion of CMV-infected individuals increased with increasing disease severity. Others made similar observations regarding high frequencies of CD28null CD4 T-cells in cohorts whose CMV infection status was unknown [10, 33]. Our results strongly argue that these observations reflect CMV-associated immunopathology, rather than normal (immune) aging [23]. Clearly, CMV infection triggers mechanisms that result in the accumulation of clinically relevant frequencies of these cells. The reason we are reiterating this is that, unlike aging, the effects of CMV infection may respond to appropriate treatment. Our work might be of particular relevance for CVD in the context of autoimmunity, in particular RA, where CD28null CD4 T-cell expansions are rife and were originally discovered [10]. CVD and autoimmunity are strongly associated with each other [41], and CMV-driven expansions of CD28null CD4 T-cells are one plausible link between these pathologies [33].
We also explored the effects of CMV infection and age on CD28null CD8 T-cells. As with CD28null CD4 T-cells, the frequencies of their CD8 counterparts were only marginally affected by age and any such effect was limited to CMV+ individuals. As a result, age per se is not an independent contributor to CD28null T-cell expansion in the CD8 compartment either. Of note, the median frequencies of CD28null CD8 T-cells in CMV- participants were on the order of 25%, i.e., about two orders of magnitude higher than the median CD28null CD4 T-cell frequencies. These constitutionally high levels suggest that CD28null CD8 T-cells are involved in the recognition of multiple antigens, which might explain their functional heterogeneity [22]. In CMV+ individuals, their frequencies were 'only' about twice as high as those of CMV- individuals. The effect of CMV on CD28null T-cell accumulation in the CD8 T-cell compartment was, therefore, much smaller than in the CD4 T-cell compartment.
Recently, the identification of CMV as the most likely, main driver of premature heart disease in HIV+ individuals caused a significant paradigm shift [42, 43]. Robustly testing the effect of CMV infection in this situation required a large number of CMV- individuals. This was also true for understanding the role of CMV infection in the accumulation of CD28null CD4 T-cells in our study. Widespread overestimation of CMV prevalence in the aging population in the US and Western Europe might explain why the effect of CMV on CD28null CD4 T-cells was not more robustly investigated before [23].
Regarding other factors influencing the frequencies of CD28null CD4 T-cells, the discovery of a protective effect of HLA-DRB1*0301P against expansion of these cells was exciting, because a protective effect of HLA-DRB1*0301P was previously described with respect to severe disease in RA [39, 44]. In RA, CD28null CD4 T-cells were shown to correlate with disease severity and the extent of extra-articular damage [12, 27]. HLA-DRB1*0301P might present peptides inducing regulatory T-cells that subsequently inhibit CD28null CD4 T-cell expansions. General inflammation levels, however, seemed to have only a small effect on CD28null CD4 T-cells. In CMV+ individuals with increased CRP levels (> 5 mg/L), CD28null CD4 T-cell frequencies were just slightly higher than in those with normal levels, whereas in CMV- individuals CRP had no noticeable effect.
In agreement with previous studies [26, 27], our results demonstrate that many CD28null CD4 T-cells recognize CMV antigens. Since we tested responses against select CMV proteins only, the sizes of the measured responses (median 7%, maximum of 18%) will have grossly underestimated the true proportion of CMV-specific cells among CD28null CD4 T-cells. This is because CMV+ individuals with a large response to one CMV protein usually recognize multiple additional ones [35]. Due to the fact that a majority of CMV-reactive CD4 T-cells are CD28null, each additional response will account for additional CD28null CD4 T-cells. No published report conclusively shows that these cells recognize antigens other than CMV. However, cross-reactivity between CMV-antigens and the stress-induced protein, heat-shock protein 60 [45] exists at the antibody level. Cross-reactivity with stress-induced proteins, for example, should be explored for T-cells as well. In any event, the majority of CMV-specific CD4 T-cells have the aggressive, CD28null phenotype and high expression of the vascular adhesion molecule, CD49d, which probably makes them an exemplar of pro-atherogenic T-cells [29].
Conclusions
Our study explains, at least in part, why CMV infection increases the risk of CVD-related death [1]. By inducing pro-atherogenic CD28null T-cells, as a direct response to CMV infection (CMV-specific CD28null CD4 T-cells), or indirectly, by mechanisms yet to be discovered, CMV drives immune cell-mediated vascular damage. This 'collateral' damage of CMV infection has been the (unrecognized) topic of many published papers dealing with CD28null CD4 T-cells and CVD and is a fascinating example of immunopathology. We must now start looking for ways to reduce CD28null CD4 T-cells in CMV-infected older people at risk of cardiovascular complications. Simply treating CMV might have real potential to achieve this. For example, aciclovir was shown to dramatically reduce the frequencies of CMV-specific CD4 T-cells in older humans [46] and valaciclovir was shown to reduce CMV-specific T-cells in mice [47]. Attempting to reduce the frequencies of CD28null CD4 T-cells with these drugs in individuals at risk could be the next decisive step forward. However, in the long run, we may need to develop effective, targeted interventions earlier in life. Knowing all relevant target antigens of CD28null CD4 T-cells and their HLA-associations will be particularly useful for that purpose.
Abbreviations
CMV: cytomegalovirus; CVD: cardiovascular disease; CAD: coronary artery disease; RA: rheumatoid disease; ROC: receiver-operator curve.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
We would like to thank the Nurses at the Clinical Investigation and Research Unit (CIRU) of the Brighton and Sussex University Hospital Trust (BSUHT) for their invaluable support. We would also like to thank all participating GP practices. We are particularly grateful to all participants.
Funding
This work was supported by The Dunhill Medical Trust, grant numbers R278/0213 and R107/0209.
Author contributions
Study design and supervision: FK, KAD, HES, CR. Experimental work: AP, GM, NT, FA, PB. Data-Analysis: AP, SC, FK, MV, MH, RB, PB, BR, JRM. Writing the manuscript: AP, FK, SC, KD, HES, MH, RB, BR.
Data and materials availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
FK holds a part-time position as Head of Immunology (R&D) at JPT Peptide Technologies, Berlin, Germany. FK and RB partly own a patent describing the use of protein-spanning peptide libraries for the antigen-specific stimulation of T-cells as described in the present work (WO 01/63286 A2). AP, SC, FA, PB, GM, NT, HES, CR, BR, JRM, MV, MH and KAD have nothing to disclose.
References
1. Wang H, Peng G, Bai J, He B, Huang K, Hu X. et al. Cytomegalovirus infection and relative risk of cardiovascular disease (ischemic heart disease, stroke, and cardiovascular death): A meta-analysis of prospective studies up to 2016. J Am Heart Assoc. 2017:6 (7). pii: e005025
2. Bajwa M, Vita S, Vescovini R, Larsen M, Sansoni P, Terrazzini N. et al. CMV-specific T-cell responses at older ages: Broad responses with a large central memory component may be key to long-term survival. J Infect Dis. 2017;215:1212-20
3. Fletcher JM, Vukmanovic-Stejic M, Dunne PJ, Birch KE, Cook JE, Jackson SE. et al. Cytomegalovirus-specific CD4+ T cells in healthy carriers are continuously driven to replicative exhaustion. J Immunol. 2005;175:8218-25
4. Hofmann U, Frantz S. Role of t-cells in myocardial infarction. Eur Heart J. 2016;37:873-9
5. Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G. et al. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail. 2017;10:e003688
6. Tracy RP, Doyle MF, Olson NC, Huber SA, Jenny NS, Sallam R. et al. T-helper type 1 bias in healthy people is associated with cytomegalovirus serology and atherosclerosis: The multi-ethnic study of atherosclerosis. J Am Heart Assoc. 2013;2:e000117
7. Bullenkamp J, Dinkla S, Kaski JC, Dumitriu IE. Targeting T cells to treat atherosclerosis: Odyssey from bench to bedside. Eur Heart J Cardiovasc Pharmacother. 2016;2:194-9
8. Dumitriu IE, Araguas ET, Baboonian C, Kaski JC. CD4+ CD28 null T cells in coronary artery disease: When helpers become killers. Cardiovasc Res. 2009;81:11-9
9. Dumitriu IE. The life (and death) of CD4+ CD28(null) T cells in inflammatory diseases. Immunology. 2015;146:185-93
10. Broadley I, Pera A, Morrow G, Davies KA, Kern F. Expansions of cytotoxic CD4+CD28- T cells drive excess cardiovascular mortality in rheumatoid arthritis and other chronic inflammatory conditions and are triggered by CMV infection. Front Immunol. 2017;8:195
11. Schmidt D, Martens PB, Weyand CM, Goronzy JJ. The repertoire of CD4+ CD28- T cells in rheumatoid arthritis. Mol Med. 1996;2:608-18
12. Martens PB, Goronzy JJ, Schaid D, Weyand CM. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40:1106-14
13. Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. 1996;97:2027-37
14. Namekawa T, Wagner UG, Goronzy JJ, Weyand CM. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998;41:2108-16
15. Liuzzo G, Goronzy JJ, Yang H, Kopecky SL, Holmes DR, Frye RL. et al. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation. 2000;101:2883-8
16. Rizzello V, Liuzzo G, Brugaletta S, Rebuzzi A, Biasucci LM, Crea F. Modulation of CD4(+)CD28null T lymphocytes by tumor necrosis factor-alpha blockade in patients with unstable angina. Circulation. 2006;113:2272-7
17. Brugaletta S, Biasucci LM, Pinnelli M, Biondi-Zoccai G, Di Giannuario G, Trotta G. et al. Novel anti-inflammatory effect of statins: Reduction of CD4+CD28null T lymphocyte frequency in patients with unstable angina. Heart. 2006;92:249-50
18. Liuzzo G, Biasucci LM, Trotta G, Brugaletta S, Pinnelli M, Digianuario G. et al. Unusual CD4+CD28null T lymphocytes and recurrence of acute coronary events. J Am Coll Cardiol. 2007;50:1450-8
19. Alber HF, Duftner C, Wanitschek M, Dorler J, Schirmer M, Suessenbacher A. et al. Neopterin, CD4+CD28- lymphocytes and the extent and severity of coronary artery disease. Int J Cardiol. 2009;135:27-35
20. Gerli R, Schillaci G, Giordano A, Bocci EB, Bistoni O, Vaudo G. et al. CD4+CD28- T lymphocytes contribute to early atherosclerotic damage in rheumatoid arthritis patients. Circulation. 2004;109:2744-8
21. Vallejo AN, Brandes JC, Weyand CM, Goronzy JJ. Modulation of CD28 expression: Distinct regulatory pathways during activation and replicative senescence. J Immunol. 1999;162:6572-9
22. Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology. 2011;134:17-32
23. Maly K, Schirmer M. The story of CD4+ CD28- T cells revisited: Solved or still ongoing? J Immunol Res. 2015;2015:348746
24. Hooper M, Kallas EG, Coffin D, Campbell D, Evans TG, Looney RJ. Cytomegalovirus seropositivity is associated with the expansion of CD4+CD28- and CD8+CD28- T cells in rheumatoid arthritis. J Rheumatol. 1999;26:1452-7
25. Looney RJ, Falsey A, Campbell D, Torres A, Kolassa J, Brower C. et al. Role of cytomegalovirus in the T cell changes seen in elderly individuals. Clin Immunol. 1999;90:213-9
26. van Leeuwen EM, Remmerswaal EB, Vossen MT, Rowshani AT, Wertheim-van Dillen PM, van Lier RA. et al. Emergence of a CD4+CD28- granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J Immunol. 2004;173:1834-41
27. Fasth AE, Snir O, Johansson AA, Nordmark B, Rahbar A, Af Klint E. et al. Skewed distribution of proinflammatory CD4+CD28null T cells in rheumatoid arthritis. Arthritis Res Ther. 2007;9:R87
28. Hecker M, Qiu D, Marquardt K, Bein G, Hackstein H. Continuous cytomegalovirus seroconversion in a large group of healthy blood donors. Vox Sang. 2004;86:41-4
29. Thewissen M, Somers V, Hellings N, Fraussen J, Damoiseaux J, Stinissen P. CD4+CD28null T cells in autoimmune disease: Pathogenic features and decreased susceptibility to immunoregulation. J Immunol. 2007;179:6514-23
30. Morgan MD, Pachnio A, Begum J, Roberts D, Rasmussen N, Neil DA. et al. CD4+CD28- T cell expansion in granulomatosis with polyangiitis (Wegener's) is driven by latent cytomegalovirus infection and is associated with an increased risk of infection and mortality. Arthritis Rheum. 2011;63:2127-37
31. Pierer M, Rothe K, Quandt D, Schulz A, Rossol M, Scholz R. et al. Association of anticytomegalovirus seropositivity with more severe joint destruction and more frequent joint surgery in rheumatoid arthritis. Arthritis Rheum. 2012;64:1740-9
32. Jonasson L, Tompa A, Wikby A. Expansion of peripheral CD8+ T cells in patients with coronary artery disease: Relation to cytomegalovirus infection. Journal of Internal Medicine. 2003;254:472-8
33. Pera A, Broadley I, Davies KA, Kern F. Cytomegalovirus as a driver of excess cardiovascular mortality in rheumatoid arthritis: A red herring or a smoking gun? Circ Res. 2017;120:274-7
34. Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: Consensus and issues. Cytometry A. 2008;73:975-83
35. Sylwester A, Nambiar KZ, Caserta S, Klenerman P, Picker LJ, Kern F. A new perspective of the structural complexity of HCMV-specific T-cell responses. Mech Ageing Dev. 2016;158:14-22
36. Zhu J, Quyyumi AA, Norman JE, Csako G, Epstein SE. Cytomegalovirus in the pathogenesis of atherosclerosis: The role of inflammation as reflected by elevated C-reactive protein levels. J Am Coll Cardiol. 1999;34:1738-43
37. Stanevicha V, Eglite J, Sochnevs A, Gardovska D, Zavadska D, Shantere R. HLA class II associations with rheumatic heart disease among clinically homogeneous patients in children in latvia. Arthritis Res Ther. 2003;5:R340-6
38. Ling SF, Viatte S, Lunt M, Van Sijl AM, Silva-Fernandez L, Symmons DP. et al. HLA-DRB1 amino acid positions 11/13, 71, and 74 are associated with inflammation level, disease activity, and the health assessment questionnaire score in patients with inflammatory polyarthritis. Arthritis Rheumatol. 2016;68:2618-28
39. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X. et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291-6
40. Vallejo AN, Weyand CM, Goronzy JJ. T-cell senescence: A culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med. 2004;10:119-24
41. Prasad M, Hermann J, Gabriel SE, Weyand CM, Mulvagh S, Mankad R. et al. Cardiorheumatology: Cardiac involvement in systemic rheumatic disease. Nat Rev Cardiol. 2015;12:168-76
42. Aiello AE, Simanek AM. Cytomegalovirus and immunological aging: The real driver of HIV and heart disease? J Infect Dis. 2012;205:1772-4
43. Lichtner M, Cicconi P, Vita S, Cozzi-Lepri A, Galli M, Lo Caputo S. et al. Cytomegalovirus coinfection is associated with an increased risk of severe non-aids-defining events in a large cohort of HIV-infected patients. J Infect Dis. 2015;211:178-86
44. Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH. et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature. 2017;545:243-7
45. Bason C, Corrocher R, Lunardi C, Puccetti P, Olivieri O, Girelli D. et al. Interaction of antibodies against cytomegalovirus with heat-shock protein 60 in pathogenesis of atherosclerosis. Lancet. 2003;362:1971-7
46. Pachnio A, Begum J, Fox A, Moss P. Acyclovir therapy reduces the CD4+ T cell response against the immunodominant pp65 protein from cytomegalovirus in immune competent individuals. PLoS One. 2015;10:e0125287
47. Beswick M, Pachnio A, Lauder SN, Sweet C, Moss PA. Antiviral therapy can reverse the development of immune senescence in elderly mice with latent cytomegalovirus infection. J Virol. 2013;87:779-89
Author biography
 Dr. Alejandra Pera is a postdoctoral fellow in immunology at Cordoba University. She is first or senior author on 9 publications and coauthor on 16 further publications in high quality journals including, for example, PNAS, Curr Opin Immunol, Cancer Immunology and Immunotherapy, and Frontiers in Immunology. Dr. Pera has won several highly competitive Spanish national research contracts. Current research interests in Dr Pera's group include: (1) immunosenescence and CMV infection; (2) NK cell immunotherapy; and (3) immunopathology of cardiovascular disease.
Dr. Alejandra Pera is a postdoctoral fellow in immunology at Cordoba University. She is first or senior author on 9 publications and coauthor on 16 further publications in high quality journals including, for example, PNAS, Curr Opin Immunol, Cancer Immunology and Immunotherapy, and Frontiers in Immunology. Dr. Pera has won several highly competitive Spanish national research contracts. Current research interests in Dr Pera's group include: (1) immunosenescence and CMV infection; (2) NK cell immunotherapy; and (3) immunopathology of cardiovascular disease.
![]() Corresponding author: Alejandra Pera, PhD, Maimonides Institute for Biomedical Research (IMIBIC), Edificio IMIBIC, avda Menedez Pidal s/n Córdoba, Spain
Corresponding author: Alejandra Pera, PhD, Maimonides Institute for Biomedical Research (IMIBIC), Edificio IMIBIC, avda Menedez Pidal s/n Córdoba, Spain