Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(17):4574-4590. doi:10.7150/thno.26758 This issue Cite
Research Paper
Combination of NIR therapy and regulatory T cell modulation using layer-by-layer hybrid nanoparticles for effective cancer photoimmunotherapy
Wenquan Ou1, Liyuan Jiang1, Raj Kumar Thapa1, Zar Chi Soe1, Kishwor Poudel1, Jae-Hoon Chang1, Sae Kwang Ku2, Han-Gon Choi3, Chul Soon Yong1, Jong Oh Kim1, ![]()
1. College of Pharmacy, Yeungnam University, 214-1, Dae-Dong, Gyeongsan 712-749, South Korea
2. Department of Anatomy and Histology, College of Korean Medicine, Daegu Haany University, Gyeongsan 712-715, South Korea
3. College of Pharmacy, Institute of Pharmaceutical Science and Technology, Hanyang University, 55, Hanyangdaehak-ro, Sangnok-gu, Ansan 426-791, South Korea
Received 2018-4-18; Accepted 2018-7-28; Published 2018-8-10
Abstract

The efficacy of combined near-infrared (NIR) and immune therapies for inhibiting tumor growth and recurrence has gained increasing research attention. Regulatory T cells in the tumor microenvironment constitute a major obstacle in achieving robust CD8+ T cell antitumor immunotherapy. In the present study, we designed a photoimmunotherapy-based strategy involving a combination of photothermal and photodynamic therapies, followed by Treg cell suppression, for eliciting an immune response with IR-780- and imatinib-loaded layer-by-layer hybrid nanoparticles.
Methods: The layer-by-layer hybrid nanoparticles were prepared through electrostatic interactions. Their photothermal effect, photodynamic effect as well as their effect on inhibiting Treg cells' suppressive function were investigated in vitro and in vivo. Their antitumor effect was evaluated using B16/BL6 and MC-38 tumor-bearing mice.
Results: The layer-by-layer hybrid nanoparticles, which were pH-sensitive, enabled the release of IR-780 dye for NIR-induced photothermal and photodynamic effects, and the release of imatinib-loaded glucocorticoid-induced TNF receptor family-related protein/poly(lactic-co-glycolic acid) (GITR-PLGA) nanoparticles to initiate antitumor immunotherapy. The photothermal and photodynamic effects caused by IR-780 under NIR exposure resulted in direct tumor apoptosis/necrosis and the production of tumor-associated antigen, promoted dendritic cell maturation, and enhanced the presentation of tumor-associated antigen to T cells, while the imatinib-loaded GITR-PLGA cores reduced the suppressive function of Treg cells, and consequently activated effective CD8+ T cells towards tumors.
Conclusion: With the significant photothermal, photodynamic and immunotherapies, the system successfully eradicated tumor growth, diminished tumor recurrence, and improved survival in vivo. The proposed nanoparticles provide a novel and versatile approach to boost antitumor photoimmunotherapy.
Keywords: imatinib, immunotherapy, IR-780, layer by layer, photodynamic therapy, photothermal therapy, Treg cell
Introduction
Photo-induced cancer therapy, which exploits non-toxic photosensitizers to efficiently generate photodynamic therapy (PDT) and photothermal therapy (PTT), has emerged as an attractive strategy for in vivo tumor ablation [1]. Upon exposure to near-infrared (NIR) irradiation, the photosensitizer converts the absorbed light energy into thermal energy and generates singlet oxygen (1O2) from tissue oxygen, ultimately resulting in local hyperthermia and tumor damage (necrosis and/or apoptosis) [2]. NIR-triggered tumor ablation has particular advantages over conventional therapeutic methods, including high and precise local temperature, preservation of surrounding tissues, short recovery time, destruction of tumor vessels, induction of acute inflammatory responses, and deprivation of oxygen and nutrients in tumor areas [3, 4]. However, their limited accumulation in tumor, water-insoluble characteristics, and instability restrict the application of photosensitizers. To overcome these drawbacks, chemical modifications of photosensitizer dye [5], polymer-conjugated photosensitizers [6], or photosensitizer-loaded nano-delivery systems have been developed to enhance the accumulation of photosensitizers in tumor [7].
During NIR exposure, molecular oxygen can be catalyzed to a range of reactive oxygen species (ROS). The ROS can directly induce tumor necrosis or apoptosis and the accumulation of dendritic cells (DCs) and neutrophils, which promote an antitumor immune response [8, 9]. It was demonstrated that PEGylated copper nanowires significantly elevated high-mobility group box 1 (HMGB1) protein release when used in combination with NIR irradiation [10] and play a crucial role in initializing the subsequent immune response against tumor [11]. HMGB1 belongs to the damage-associated molecular patterns (DAMPs), which can activate DCs to present the tumor-antigen to T cells. However, it has been suggested that the tumor microenvironment becomes so immunosuppressive that NIR exposure treatment alone may not be sufficient for tumor ablation and even has some immunosuppressive effects [12]. The recruitment and expansion of CD4+ CD25+ Foxp3+ Treg cells in the tumor microenvironment mostly contribute to the severe immunosuppression [13]. In this light, the integration of NIR irradiation and the inhibition of intratumoral Treg cells might induce tumor eradication and facilitate durable antitumor immunity. Imatinib (IMT), initially developed as an inhibitor of tyrosine kinase, has been widely used for treating leukemia and gastrointestinal stromal tumors [14]. Studies have shown that IMT reduces the activation of transcription factors STAT3 and STAT5 in Treg cells, inhibits Foxp3 expression, and impairs Treg immunosuppressive functions in vitro and in vivo [14-16]. However, its strong cytotoxicity to normal cells, and poor targeting effect and solubility restrict its further use.
In this study, we report the incorporation of IR-780 and IMT in layer-by-layer hybrid nanoparticles (LBL hNPs) to achieve PTT and PDT antitumor efficacy as well as intratumoral Treg cell downregulation function (Scheme 1). In this system, the hydrophobic drug IMT was loaded inside glucocorticoid-induced TNF receptor family-related protein (GITR) antibody-modified poly(lactic-co-glycolic acid) (PLGA) core, in which the GITR antibody functioned to target Treg cells [17]. IR-780 iodide, a lipophilic cationic NIR dye used as the photosensitizer was incorporated on the outside of GITR-PLGA core via electrostatic interactions. To protect the photosensitizer and GITR-PLGA core from degradation and to achieve pH sensitivity, poly-L-histidine (PLH) and poly(ethylene glycol)-block-poly(L-glutamic acid) (PEG-b-PLG) were used as polycationic and polyanionic coating layers. The PLH and PEG-b-PLG layers not only provide stimulus-responsive behavior triggered by the acidic tumor microenvironment, but also enable extended blood circulation and improved biocompatibility of the delivered payloads [18]. Particularly, owing to the amphoteric nature of the outside coating layers, after accumulating in the tumor area via the enhanced permeability and retention (EPR) effect [19], the LBL hNPs underwent protonation in the acidic tumor microenvironment, which would result in release of the IR-780 photosensitizer and the GITR-PLGA core. Subsequently, the released IMT-loaded GITR-PLGA core can be successfully targeted to the Treg cells within the tumor microenvironment to abrogate their immunosuppressive function. Furthermore, NIR-induced PTT and PDT can initiate rapid and irreversible damage to the cancer cell membrane, resulting in swelling and bursting, and release of intracellular components due to influx of water inside the cell. This process also induces relocation of calreticulin, Hsp70, and Hsp90 to the cell surface, along with the extracellular release of HMGB1 and ATP, which provides immunogenic signals to active dendritic cells [11]. Therefore, the interaction of NIR therapy and Treg cell modulation using LBL hNPs was expected to improve tumor ablation efficacy.
Schematic illustration of NIR therapy and regulatory T cell modulation using layer-by-layer hybrid nanoparticles (LBL hNPs) with mutual PTT, PDT, and immune-anticancer therapeutic effects.
Methods
Materials
Poly(D,L-lactide-co-glycolide) (PLGA; La:Ga=50:50, MW 7,000-17,000 Da), polyvinyl alcohol (PVA, MW 89,000-98,000), 1,3-diphenyl-2-benzofuran, 2,5-diphenyl-3,4-benzofuran (DPBF), IR-780 iodide, lipopolysaccharide (LPS), 2-(N-Morpholino) ethanesulfonic acid hydrate (MES) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Poly(D,L-lactide-co-glycolide)-N-hydroxysuccinimide endcap (PLGA-NHS; La:Ga=50:50, MW 20,000-45,000 Da) was purchased from PolySciTech (Akina, Inc. USA). Poly-L-histidine (PLH) (MW: 5,000-25,000) and poly (ethylene glycol)-block-poly(L-glutamic acid) (PEG-b-PLG) (PEG repeating units: 22 (MW=1,000 Da), PLG repeating units: 50, (MW=7,500 Da)) were obtained from Alamanda Polymers (Huntsville, AL, USA). Imatinib base (IMT) was acquired from LC Laboratories (Woburn, MA, USA). PerCP/Cy5.5 anti-mouse CD8a antibody, FITC-conjugated anti-mouse CD4, APC anti-mouse IFN-γ antibody, PE-conjugated anti-mouse CD25, APC-conjugated anti-mouse Foxp3, PE-cyanine7-conjugated anti-mouse B220, anti-mouse CD3 antibody, anti-mouse IL-4 antibody, anti-mouse CD28 antibody, Rat IgG2a isotype control antibody, TGF-β, IFN-γ, and IL-2 were purchased from Biolegend (San Diego, CA, USA). DiD solid dye, PE-granzyme B monoclonal antibody (NGZB), TGFβ-1 mouse ELISA kit and IFN-γ mouse ELISA kit were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals were of reagent grade and used directly as received.
B16/BL6 cells were obtained from the Korean Cell Line Bank (Seoul, South Korea). Cells were cultured in DMEM (Hyclone, Logan, UT) supplemented with 10% (v/v) fetal bovine serum (Hyclone, Logan, UT) and 1% (v/v) penicillin-streptomycin solution (Hyclone) in a humidified incubator (Thermo Fisher Scientific, USA) at 37 °C, under 5% CO2 condition. Animal care and use were in accordance with the guidelines for care and use of laboratory animals at the animal center of Yeungnam University, Republic of Korea.
Fabrication of LBL hNPs
PLGA NPs were prepared using a modified o/w single emulsion method [20]. Briefly, 5 mg PLGA/PLGA-NHS (at 25 % w/w) and 1 mg IMT were dissolved in 1 mL of acetonitrile (ACN). The solution was added dropwise into 1 mL of deionized water with 0.5% PVP under magnetic stirring at a speed of 360 rpm. The mixture was then added dropwise into 4 mL of deionized water (containing 0.5% PVP) to adjust a final concentration of 1 mg/mL, and stirred for 6 h at room temperature to remove the organic solvent. For the covalent attachment of GITR antibody onto the PLGA NP surface, 2.0 mg/mL GITR antibody (molar excess) was mixed with the PLGA NPs under 0.1 M MES buffer (pH=7.0) and stirred at room temperature for 4 h [21, 22]. Excess MES was removed by dialysis (MWCO 3500-Da dialysis bag, Spectrum Chemical, USA) against distilled water for 72 h. After centrifugation at 13,000 ×g, 4 °C for 10 min and washing with PBS, the resulting pellets was re-dispersed in 1 mL PBS and keep at 4 °C. The successful synthesis and graft ratio of antibody were analyzed by BCA Kit (Thermo Fisher Scientific, USA), where BSA was used as a standard protein.
Before LBL self-assembly, IR-780 with increasing input from 3% to 11% was encapsulated outside the PLGA NP cores by electrostatic interactions. Then, a saturating amount of polymer (1 mg/mL PLH and PEG-b-PLG) solution of opposite charge was added dropwise under continuous stirring. Typically, after adding each layer, mixing was facilitated by immediate sonication for 5 min [23]. The exact amount of polymer required for each layer was determined by titration [24]. Free reagents in the solution were removed by dialysis as described above.
Characterization of LBL hNPs
Hydrodynamic diameter, PDI, and ζ-potential of LBL hNPs were measured by dynamic light scattering (DLS) using a Nano-S90 ZetaSizer (Malvern Instruments, UK). After each drop (20 µL) (layer) of polymer titrated, the ζ-potential of the media was checked. LBL hNP morphology was determined by transmission electron microscopy (TEM, H7600; Hitachi, Tokyo, Japan), with 2% w/v phospho-tungstic acid prestaining treatment.
IMT was isolated from NPs by centrifugation using Amicon® Ultra Centrifugal Filters (MWCO 3500 Da, Merck Millipore, USA) at 3000 ×g for 30 min. IMT content in the supernatant was analyzed by HPLC using a C18 column (250 × 4.6 mm, 5 µm; GL Science, USA). The absorbance of IMT at 266 nm was detected under a 1 mL/min flow rate using a 60/40 ratio of 0.02 M KH2PO4/acetonitrile mobile phase. Drug EE and drug LC were calculated as follows: EE = (total weight of IMT-weight of IMT in supernatant /total weight of IMT) × 100%; LC = (total weight of IMT - weight of IMT in supernatant /total weight of NP) × 100%. IR-780 was isolated from LBL hNPs in the same way as IMT. For analysis, IR-780 was determined by using a UV/visible spectrophotometer (PerkinElmer U-2800; Hitachi, Tokyo, Japan).
The stability of LBL hNPs in complete medium and PBS solution was measured at a constant temperature of 37 °C under gentle shaking (100 rpm). Changes in hydrodynamic diameter and PDI were identified in triplicate by DLS at predetermined time intervals. The photostability of free IR-780 and IR-780 hNPs exposed to daylight at different time points were determined by analyzing the absorbance using a UV/visible spectrophotometer.
Drug release profiles of IR-780 and IMT from LBL hNPs were generated by dialysis. Briefly, 1 mL of LBL hNP solution was dialyzed against 50 mL PBS buffer (pH 5.0, 6.5 or pH 7.4) in a dialysis bag (MW=3500 Da, Spectrum, USA) under gentle shaking (100 rpm) at 37 °C. At predetermined time intervals, 50 μL sample was taken out and analyzed by HPLC as described above. The interference of IR-780 on this HPLC-based method was excluded by comparing the HPLC peaks of baseline, IMT standard sample (50.0 µg/mL), IR-780 standard sample (50.0 µg/mL), and the sample from drug release study (Figure S1).
Photothermal effect and singlet oxygen generation capacity
To investigate the photothermal effect of LBL hNPs, 1 mL of LBL hNP solution with IR-780 at concentrations of 1, 5, 10, and 20 µg/mL were exposed to 808 nm NIR irradiation at 1.0 W/cm2 for 3 min. During the irradiation period, a thermos-camera (Therm-app, Opgal Optronic Industries, Israel) was utilized to monitor and record the temperature variation. The absorbance of IR-780 at 780 nm was detected using a UV/visible spectrophotometer. The photothermal effect was compared with that of free IR-780 at concentrations of 1, 5, 10, 20 µg/mL.
Singlet oxygen generation after treatments with 2.0, 5.0, 10.0 µg/mL free IR-780 or IR-780-loaded LBL hNPs for various times under NIR irradiation (808 nm, 1.0 W/cm2) was determined by photooxidation of DPBF[25], and the variation in absorbance at 415 nm was measured using a UV/visible spectrophotometer (PerkinElmer U-2800; Hitachi, Tokyo, Japan).
In vitro cytotoxicity and uptake of LBL hNPs
In vitro cell toxicity of blank LBL hNPs, IR-780, and IR-780-loaded LBL hNPs (with and without NIR irradiation) was determined by MTT assay. Briefly, 3×104 B16 cells were seeded in a 96-well plate and after 24 h, they were incubated with free IMT, free IR-780, IMT plus IR-780 or LBL hNPs at different concentrations for 24 h. Then, 20 µL of MTT solution (5 mg/mL) was added into each well, and the cells incubated for another 4 h. After the addition of 100 µL of DMSO into each well, the absorbance at 570 nm was measured using a microreader (Thermo Fisher Scientific, USA). Cell viability was calculated using the following formula: Cell viability (%) = (A570(treated group) - A0) / (A570(without treatment) - A0) × 100%. A570(treated group) represents cells treated with blank hNPs, free IR-780, IMT, free IR-780, IMT plus IR-780 and IR-780-loaded LBL hNPs (with or without NIR irradiation), A570(without treatment) refers to the untreated group, while A0 refers to medium only. For NIR irradiation, after 6-h incubation with LBL hNPs, the medium was replaced with fresh medium and the cells were subjected to 808 nm NIR irradiation at 1.0 W/cm2 for 1 min per well, followed by incubation for a predetermined time.
In vitro cytotoxicity of different concentrations of IMT, IMT-loaded GITR-PLGA cores and LBL hNPs on CD8+ T and Treg cells were also evaluated using Annexin V and PI Kit (BD Biosciences, USA). The detailed method has been described in Supplementary Material.
The intracellular uptake of IR-780 and GITR-NPs in B16 cells was analyzed by flow cytometry (FACSCalibur; BD Biosciences, USA). To measure dose-dependent uptake, the media were replaced with 1, 3, and 5 µg/mL of IR-780-loaded LBL hNPs. After incubation for 3 h, uptake of IR-780 was measured by flow cytometry. Similarly, time-dependent cellular uptake of IR-780 and APC-GITR-PLGA core in B16 cells was analyzed by incubating cells with 3 µg/mL of IR-780-loaded LBL hNPs. After 3, 6, 9 h, cells were harvested and analyzed by flow cytometry.
To visualize the intracellular distribution of IR-780, 3 µg/mL of free IR-780 or IR-780-loaded LBL hNPs was added to B16 cells seeded on a glass slide. After 3 h, the cells were washed with PBS and fixed with 4% paraformaldehyde. After staining with LysoTracker Green and DAPI, cells were observed by confocal laser scanning microscopy (CLSM, K1-Fluo; Nanoscope, South Korea). The quantitative intracellular uptake of free IR-780 and IR-780-loaded LBL hNPs was further quantitatively confirmed using flow cytometry.
In vitro assay of apoptosis, necrosis and photothermal effect induced by LBL hNPs with NIR irradiation
To observe apoptosis and necrosis induction by IR-780-loaded hNPs plus NIR irradiation, B16 cells were incubated with 3 µg/mL of free IR-780 or IR-780-loaded hNPs and exposed to NIR irradiation at 1.0 W/cm2 for 1 min per well, and then incubated for 24 h. Then, the cells were stained with FITC-Annexin V and PI (BD Biosciences, USA) according to the manufacturer's protocol and analyzed by flow cytometry. Furthermore, temperature changes of cells after following NIR irradiation were measured using a thermal camera. Additionally, live and dead cells were evaluated by live and dead staining using AO (1 mg/mL) and PI (0.1 mg/mL) for 30 min and fluorescence microscopy (Eclipse Ti, Nikon Instruments, Melville, NY, USA).
In vitro assay of ROS generation
To examine intracellular ROS generation by LBL hNPs plus NIR irradiation, B16 cells were stained with DCFDA (1 µM) for 30 min. Then, the cells were treated with free IR-780 (3 µg/mL) or IR-780-loaded hNPs for 6 h. After NIR irradiation at 1.0 W/cm2 for 1 min per well, the cells were harvested and analyzed by flow cytometry.
In vitro assay of DC maturation and antigen presentation
Tumor antigen derived from LBL hNP-treated cells (with or without NIR irradiation) was collected from the growth media [26]. For the identification of enhancing DAMPs generation after NIR irradiation, western blotting with GAPDH and HMGB1 antibody was used to demonstrate higher expression of HMBG1 [27].
DCs were isolated from femurs of C57BL/6 mice and stimulated with 20 ng/mL GM-CSF and 10 ng/mL IL-4 for 7 days [28]. The harvested immature DCs were seeded in a 96-well plate (U bottom) at a density of 4×104 cells/mL and treated with PBS, LPS (positive control, 100 ng/mL), and tumor antigen with or without NIR irradiation. After 24 h, the cells were harvested and stained with PE-antimouse CD11c (Biolegend), PerCP/Cy5.5-antimouse CD40 (Biolegend), APC-antimouse CD80 (Biolegend) and FITC-CD86 (Biolegend). DC maturation was evaluated by flow cytometry using FlowJo software for analysis.
To confirm the enhancement of antigen presentation by DCs, tumor antigen (with or without NIR irradiation)- or LPS-treated DCs were cocultured with CD8+ T cells at the ratio of 1:20 [29]. After 3 days, the cells were collected and labeled with APC-CD69, FITC-CD8, and PE-IFN-γ. Effect on CD8+ T cells was analyzed using flow cytometry.
In vitro assay of downregulation of Treg cell differentiation
To generate Treg cells, single-cell suspensions were isolated from spleen and lymph nodes of 6-week-old C57BL/6 mice. CD4+ T cells were purified with CD4 MicroBeads (Miltenyi Biotec) and seeded at the density of 2×105 in a flat-bottom 96-well plate coated with anti-CD28 (1 μg/mL; eBioscience) and anti-CD3 (5 μg/mL; Biolegend), together with anti-IL-4 (5 μg/mL; Biolegend), anti- IFN-γ (5 μg/mL; Biolegend), IL-2 (10 ng/mL; Biolegend), TGF-β1 (10 ng/mL; Biolegend), and IMT-loaded LBL hNPs. The targeting effect of LBL hNPs on Treg cells was assessed by using APC-labeled GITR and the effect on Treg cell differentiation was identified by treatment with 2.5, 5, 10, 20 µM IMT-loaded LBL hNPs. After 4 days, Treg cells were characterized by surface staining using the following antibodies: FITC-conjugated anti-CD4 (Biolegend), PE-cyanine 7-conjugated anti-B220 (Biolegend), PE-anti-CD25 (Biolegend), and intracellular staining using APC-anti-Foxp3 (eBioscience). During the staining procedure, fixation and permeabilization buffer (eBioscience) were used according to the manufacturer's instructions. Finally, the percentage of differentiated Treg cells was determined by flow cytometry using FlowJo software for analysis.
A coculture system was further used to identify the effect of LBL hNPs on Treg cells in a tumor environment. In this experiment, 2×105 Treg cells were cocultured with 3×104 B16 tumor cells and incubated with LBL hNPs (IR-780: 5 µg/mL; IMT: 5 µM). Competitive cellular uptake between these two cell lines was assessed. Similarly, Treg cell differentiation under tumor condition was also verified by flow cytometry as described above. The detailed method has been described in Supplementary Material.
For the determination of STAT3, p-STAT3, STAT5, and p-STAT5, Treg cells were purified and induced with PBS, free IMT, LBL hNPs, and LBL hNPs with GITR antibody pretreatment. At day 3, protein samples were collected and the expression of the above proteins was visualized by western blotting.
Proliferation of Treg cells after treatment with LBL hNPs at different IMT concentrations was confirmed using CFSE (Thermo Fisher Scientific, USA) according to the manufacturer's recommendations. The effects of free IMT, GITR-PLGA cores from LBL hNPs, and GITR pretreatment on Treg cell proliferation were determined using the same procedure.
In vivo biodistribution of LBL hNPs
The biodistribution of LBL hNPs in vivo was assessed in mice bearing B16/BL6 tumors of ≥200 mm3. IR-780 (8 mg/kg)-loaded LBL hNPs were intravenously administered to the tumor-bearing mice. The distribution of IR-780 fluorescence was recorded at 0, 8, and 24 h using an in vivo animal imaging system (FOBI, NeoScience, Korea). After the last time point, the accumulation of IR-780 fluorescence in the heart, liver, spleen, lungs, and kidneys was measured in the sacrificed mice.
In vivo evaluation of antitumor efficacy
To assess the in vivo therapeutic effect, 5-week-old C57BL/6 mice were transplanted with B16/BL6 cancer cells (1×106 per mice). When the tumor volume reached 100 mm3 in size, mice were randomly assigned into 5 treatment groups (n=6), including: PBS, NIR irradiation, free IMT (10.0 mg/kg), free IR-780 (8.0 mg/kg, with NIR irradiation), LBL hNPs (with NIR irradiation). The corresponding doses were injected intravenously into the mice. Eight hours later, the tumor sites were exposed to 808 nm, 1.0 W/cm2 NIR irradiation for 3 min. Temperature elevation during NIR treatment was detected using the Thermos-app camera. The injections were administered every two days and during this period, body weight and tumor volume were recorded every other day. The tumor volume was defined as Volume (mm3) = length (mm) × width2 (mm2)/2.
Throughout the treatment, blood was taken from the eyes of the mice every two days and the TGF-β level in the blood was identified by ELISA (Thermo Fisher Scientific, USA). At day 20, heart, liver, lungs, kidneys, and tumors were isolated from sacrificed mice and stained with hematoxylin and eosin (H&E). Additionally, in vivo intra-tumoral CD4+ (helper T), CD8+ (cytotoxic T) infiltrations, cell proliferation marker (Ki-67) expression, and tumor angiogenesis marker (CD31) expression were evaluated using primary antisera. CD4+- and CD8+-immunolabeled cell numbers (cells/mm2 of tumor mass), mean percentages of Ki-67- and CD31-immunolabeled cells (%/mm2 of tumor mass), and tumor cell volumes (%/mm2 of mass) were analyzed on tumor histological specimens by histomorphometry.
To evaluate the fate of Treg and CD8+ T cells during treatment, spleens and tumors were isolated at day 20 and were dissociated to generate single-cell suspensions using collagenase and DNase. Lymphocytes were enriched by Percoll gradient (GE Healthcare Life Sciences, Utah, USA) and stained with FITC-conjugated anti-CD4 (Biolegend), PE-cyanine 7-conjugated anti-B220 (Biolegend), APC anti-mouse IFN-γ Antibody (Biolegend), PE-Granzyme B monoclonal antibody (NGZB) (eBioscience), PE-anti-CD25 (Biolegend), PerCP/Cy5.5 anti-mouse CD8a antibody (Biolegend), and APC-anti-Foxp3 (eBioscience). The percentages of Treg cells, granzyme B+CD8+ T cells, and IFN-γ+ CD8+ T cells were determined by flow cytometry.
Statistical analysis
All experimental data are presented as the mean ± SD. Differences among groups were determined using one-way ANOVA analysis followed by Tukey's test. p* < 0.05 and p**<0.01 were considered statistically significant.
Results and Discussion
Preparation and determination of LBL hNPs
To generate LBL hNPs consisting of a GITR antibody-modified PLGA core and outer polymer layers, nanoparticles of different PLGA concentrations were optimized according to hydrodynamic diameter, zeta potential, and polydispersity index (PDI) as shown in Figure S2. Then, the cores were loaded with IMT, and the maximum loading capacity (LC) was found to be 13% (Figure S3A). Subsequently, GITR antibody was successfully conjugated to the PLGA cores, with a conjugation efficiency of 78.2 ± 3.8% (GITR-PLGA). Next, IR-780 was incorporated at different percentages of the total weight in the surface of GITR-PLGA nanoparticles by electrostatic interactions. The maximum loading efficacy (LE) and loading capacity (LC) of IR-780 in LBL hNPs were 85% and 10%, respectively (Figure S3B).
To yield self-assembling LBL hNPs, alternate polymers (PLH and PEG-b-PLG) were coated on the surface of the GITR-PLGA core. The amounts of PLH and PEG-b-PLG polymers were determined by checking zeta potential changes. After the addition of six layers, LBL hNPs suffered from a slight increase in hydrodynamic diameter (Figure S4A), probably attributable to the strong ionic electrostatic interaction between PLH and PEG-b-PLG and the formation of a dense polymer mesh outside the PLGA core [30, 31]. Results showed that the addition of five or six layers significantly (p < 0.01) increased the hydrodynamic diameter, from 142.6 nm (four layers) to 183.0 nm (five layers). Furthermore, the PLGA cores coated with four or six layers successfully protected IR-780 from degradation during the study period. However, in the group coated with two layers, fluorescence of IR-780 was stably maintained for 8 h, following which it started to decrease (Figure S4B). We selected four layers for further study to generate a stable drug delivery system with the potential to penetrate the tumor microenvironment via the EPR effect. Although the amount of polymers cannot be measured during the addition of alternate layers, the reversed surface charges can be used to determine successful formation of a layer-by-layer architecture (Figure 1A and Figure S4C). An increase in the PLGA surface charge (from -27.0 ± 1.2 to 7.9 ± 1.7 mV) was measured after the addition of PLH polymer. Similarly, reversal of charge appeared after the addition of the PEG-b-PLG second layer. LBL hNPs can be successfully generated via the reversed electrostatic interaction of different layers [24]. After the incorporation of IR-780 and outer four layers to IMT-loaded GITR-PLGA cores, the hydrodynamic diameter slightly increased from 134.8 ± 2.4 nm to 144.9 ± 3.0 nm (Figure 1B). However, loss of IR-780 was not observed before or after coating with outer layers, (Figure S4D), suggesting that LBL hNPs had better drug loading capacity. The generation of a layer covering the GITR-PLGA core and the spherical shape of nanoparticles were further confirmed by TEM image (Figure 1C). The LBL hNPs kept stable in PBS at pH 7.4 and in RPMI complete media at least 24 h, with slight changes in size and PDI (Figure S5A). Additionally, compared with free IR-780 dye, the absorbance of IR-780 in LBL hNPs under daylight exposure remained unchanged within 24 h (Figure S5B), suggesting the excellent photostability of LBL hNPs. Consequently, LBL hNPs enhance the stability of IR-780 for achieving NIR effects.
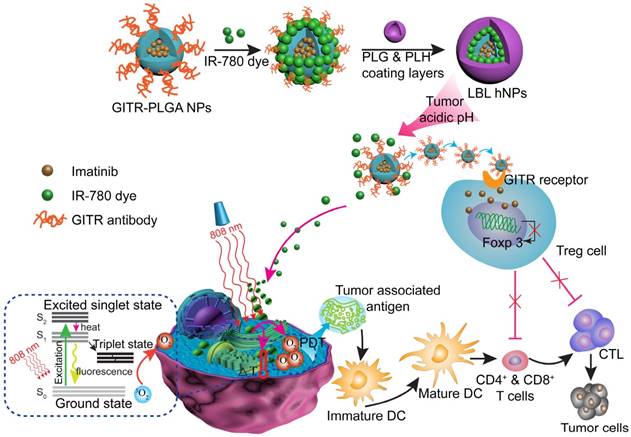
Characterization, NIR-induced photothermal effect, and in vitro drug release profile of LBL hNPs. (A) Changes in zeta potential after coating of GITR-PLGA cores with alternating PLH and PEG-b-PLG layers (n=3). (B) Comparison of hydrodynamic diameter before and after the addition of 4 coating layers. (C) TEM image of LBL hNPs. (D) Temperature elevation of LBL hNPs in the presence of NIR irradiation (808 nm, 1.0 W/cm2), with IR-780 concentrations ranging from 1 µg/mL to 20 µg/mL. (E) IR thermography of LBL hNPs within different IR-780 concentrations. (F) Temperature increase of PBS, free IR-780, and LBL hNPs under NIR exposure (808 nm, 1.0 W/cm2); the concentration of IR-780 was 5 µg/mL. (G) UV absorbance of free IR-780 and LBL hNPs at 780 nm during the treatment period. (H) In vitro release profile of LBL hNPs at pH 5.0, 6.5 and pH 7.4. (I) Hydrodynamic diameter at pH 5.0, 6.5, and pH 7.4.
In vitro photothermal (PTT) effect
Photothermal conversion capability was evaluated by measuring the temperature elevation of free IR-780 or IR-780-loaded LBL hNPs at various concentrations under 808 nm, 1.0 W/cm2 NIR irradiation. Both free IR-780 (Figure S6A) and LBL hNPs (Figure 1D-E) exhibited concentration-dependent temperature increases. However, in free IR-780, the temperature reached a plateau at approximately 53 °C in 80 s, whereas IR-780-loaded LBL hNPs heated up to 67 °C in 80 s (Figure 1F), primarily resulting from the protection from degradation and non-radiative relaxation of IR-780 in LBL hNPs [32]. Studies have shown occurrence of irreversible cell damage when the hyperthermia temperature generated from PTT exceeded 42°C [33]. Therefore, in this study, 5 µg/mL IR-780-loaded LBL hNPs were chosen for later experiments. In addition, compared with free IR-780 (5 µg/mL) with NIR irradiation treatment, LBL hNPs exhibited a high increase in temperature and protection from degradation under NIR exposure (Figure 1G), suggesting a strengthened PTT effect when using LBL hNPs.
To further explore the capacity of singlet oxygen generation in LBL hNPs, 1.3-diphenyliso-benzofuran (DPBF) probe was utilized to investigate the singlet oxygen production. Remarkably, compared to free IR-780, which generated very little singlet oxygen, concentration-dependent singlet oxygen production was detected in IR-780-loaded LBL hNPs at increasing concentrations of 2.0, 5.0, 10.0 µg/mL within the NIR irradiation period (Figure S6B), probably owing to the protective photostability and reduced self-quenching of singlet oxygen in LBL hNPs [34].
In vitro cumulative release
In vitro drug release of IR-780 and IMT from LBL hNPs was evaluated at pH 5.0, 6.5 (mimicking tumor microenvironment pH) and pH 7.4. Remarkably, both IR-780 and IMT were more strongly released at pH 5.0 and 6.5 than at pH 7.4 (Figure 1H). This can be explained by enhancement of the proton effect at lower pH, which promotes proton entry into the coating layer, thus accelerating the disruption of LBL hNPs. At pH 7.4, IR-780 and IMT release reached a plateau at 33.1% and 29.7% at 24 h, respectively. Furthermore, the hydrodynamic diameter increased from 151 nm at pH 7.4 to 187 nm at pH 6.5, and to 882 nm at pH 5.0 (Figure 1I), indicating accelerated collapse of LBL hNPs at lower pH, suggesting accelerated drug release in the acidic tumor microenvironment.
In vitro cell cytotoxicity and cellular uptake of LBL hNPs
The cytotoxicity of IMT, IMT-loaded GITR-PLGA core and LBL hNPs toward Treg and CD8+ T cells was investigated using Annexin V/PI staining. At concentrations ≤ 5.0 µM, both the GITR-PLGA core and LBL hNPs had no significant cytotoxic effect on CD8+ T or Treg cells, showing more than 90% viable cells (Figure S7A-B). Next, in vitro cell viability of B16BL/6 tumor cells after photothermal damage by LBL hNPs was investigated. After 24 h, NIR irradiation treatment with LBL hNPs induced stronger cytotoxicity than no-NIR treatment or free IR-780 (with or without NIR irradiation), with an IC50 of 2.5 µg/mL, while blank LBL hNPs exhibited no cytotoxicity (Figure 2A). Consequently, the application of LBL hNPs might improve the therapeutic effect of IR-780 against B16BL/6 cancer cells.
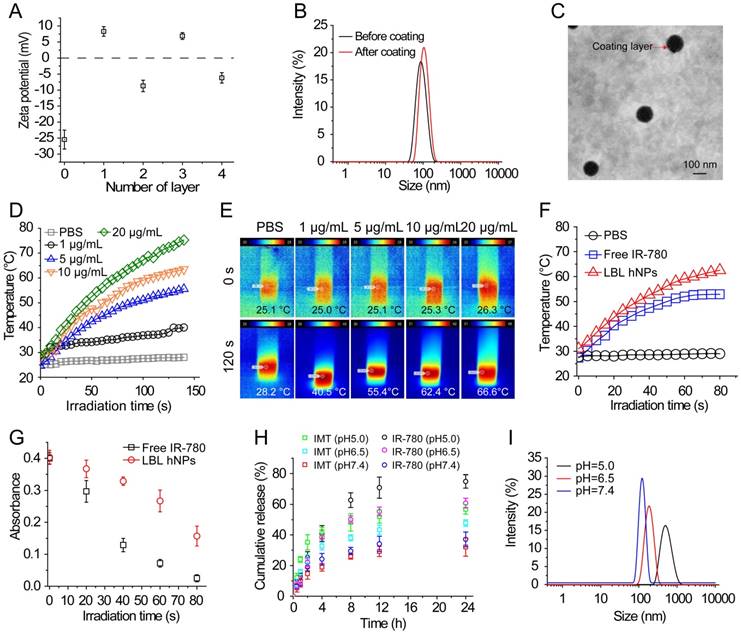
In vitro cytotoxicity and intracellular localization of IR-780. (A) In vitro cytotoxicity against B16BL/6 cancer cells of blank LBL hNPs, free IMT, IMT+IR-780, free IR-780, free IR-780 (NIR, 808 nm, 1.0 W/cm2, 1 min), LBL hNPs, and LBL hNPs (NIR) at indicated concentrations (n=6). (B) In vitro cellular uptake of IR-780 in B16BL/6 cells treated with IR-780-loaded LBL for 3, 6, or 9 h. The concentration of IR-780 was 3 µg/mL (n=3). (C) In vitro cellular uptake of GITR-PLGA cores from LBL hNPs in B16BL/6 cells exposed to IR-780-loaded LBL after incubation for 3, 6, or 9 h (n=3). (D) Confocal images of B16BL/6 cells exposed to PBS, free IR-780 or LBL hNPs. The concentration of IR-780 was 3 µg/mL.
The cellular uptake of IR-780-loaded LBL hNPs was investigated in cell culture media adjusted to pH 6.5 to mimic the tumor microenvironment. After 3-h incubation, IR-780-loaded LBL hNPs were taken up by B16BL/6 cells in a dose-dependent manner (Figure S8A). As quantitatively indicated by flow-cytometry, IR-780 from LBL hNP-treated B16BL/6 cells showed time-dependent uptake within 9 h (Figure 2B), whereas uptake of free IR-780 increased during the first 3 h, but then saturated, indicating high cellular internalization when LBL hNPs were used (Figure S8B). Notably, limited uptake of GITR-PLGA nanoparticles was observed in B16BL/6 cells, which was ascribed to low GITR receptor expression (Figure 2C). Confocal laser microscopy of cellular internalization confirmed stronger IR-780 uptake in cells exposed to LBL hNPs than in those incubated in the presence of free IR-780 or PBS (Figure 2D), suggesting enhanced uptake of IR-780 encapsulated in LBL hNPs. Furthermore, flow cytometric analysis demonstrated higher intracellular uptake of IR-780 in the group treated with LBL hNPs than in those treated with free IR-780 (Figure S8C). Hence, these results confirmed that loading of IR-780 dye in LBL hNPs could increase its cellular uptake in tumor cells.
In addition, after incubation in the acidic pH-mimicking environment of tumors, in vitro specific binding of the GITR-PLGA cores from LBL hNPS to Treg cells showed that GITR-PLGA cores can be successfully incorporated by Treg cells in a dose-dependent manner; up to 97.7% Treg cells incorporated the GITR-PLGA NPs, whereas their IgG isotype-modified PLGA NPs without GITR antibody modification showed less Treg uptake (Figure S9A-B). However, after pretreatment with 1.0 µg/mL GITR antibody, the uptake of GITR-PLGA cores was reversed to low levels, indicating the importance of GITR antibody in guiding the specific uptake of GITR-PLGA cores.
In vitro photodynamic effect and immune cell activation
To investigate photo-induced apoptosis and necrosis, B16BL/6 cells were incubated with LBL hNPs for 9 h, followed by 1 min of NIR irradiation (808 nm, 1.0 W/cm2) and further incubation until 24 h. In the absence of NIR irradiation, both free IR-780- and LBL hNP-treated cells exhibited negligible apoptosis/necrosis compared with the control group (Figure 3A). However, under NIR irradiation, LBL hNPs induced more late apoptosis (48.87%) and necrosis (50.2%) than free IR-780 (p < 0.01). The high level of necrosis in the LBL hNPs plus NIR group may result from physical or chemical damage, especially the production of heat during NIR exposure [35], whereas the increase in cell apoptosis may result from the involvement of Bcl-2 family members or NF-κβ and release of cytochrome C after PDT [36]. As the same photosensitizer and irradiation conditions result in both photothermal and photodynamic effects, their outcomes cannot be distinguished to be direct, indirect, or working in conjunction with other factors without further investigation. Cytotoxicity was further demonstrated by live and dead staining using fluorescence microscopy. In the LBL hNP-treated group under NIR irradiation, most cells were stained with PI, indicating the dead cells after treatment (Figure 3B). Potentially, the photo-induced cytotoxicity originated from a synergy of the photothermal effect and ROS generation. The temperature changes of cells after NIR irradiation revealed that the temperature elevation in the LBL hNP group during 1 min NIR irradiation was approximately 45°C, whereas it was only 37°C in the free IR-780 group, suggesting that more cell damage occurred during the photothermal effect in the LBL hNP group (Figure S10). However, the temperature of the group with free NIR irradiation treatment did not increase. Furthermore, no significant ROS production was observed in groups without NIR irradiation (Figure 3C-D). However, after NIR treatment, significant ROS generation was detected in LBL hNP-treated B16/BL6 tumor cells (Figure 3E-F), confirming a strong photodynamic effect.
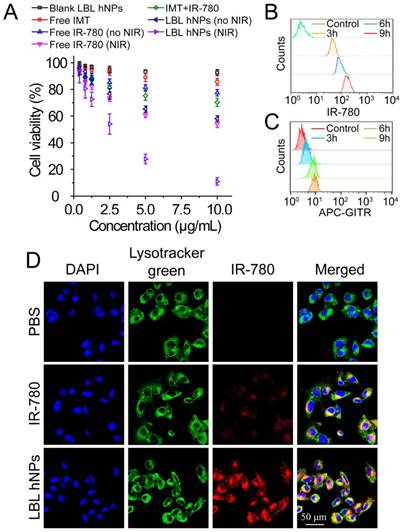
In vitro apoptosis/necrosis, ROS generation, activation of BMDCs and CD8+ T cells induced by LBL hNPs plus NIR irradiation. (A) Apoptosis/necrosis levels of B16BL/6 cells exposed to PBS, free IR-780, or LBL hNPs without or with NIR exposure (808 nm, 1.0 W/cm2, 1 min) as assessed by flow cytometry using Annexin V-FITC/PI staining (n=3). (B) Live and dead cell assay of B16BL/6 cells exposed to PBS, free IR-780, or LBL hNPs without or with NIR exposure (808 nm, 1.0 W/cm2, 1 min) using AO and PI staining. (C-D) In vitro ROS generation and statistical ROS levels in B16BL/6 cells exposed to PBS, free IR-780, or LBL hNPs without NIR irradiation (n=3). (E-F) In vitro ROS generation and ROS levels in B16BL/6 cells exposed to PBS, free IR-780, or LBL hNPs plus NIR irradiation (808 nm, 1.0 W/cm2, 1 min). n=3, **p < 0.01, one-way ANOVA. (G) HMGB1 content in B16BL/6 cells exposed to LBL hNPs in the presence or absence of NIR exposure. (H) In vitro maturation of non-matured BMDCs exposed to tumor-associated antigen derived from B16BL/6 cells treated with PBS or LBL hNPs (without or with NIR, 808 nm, 1.0 W/cm2, 1 min). The concentration of IR-780 was 3 µg/mL. LPS was used as a positive control. (I-J) CD69 and IFN-γ levels on CD8+ T cells after coculture with tumor-associated antigen-presenting BMDCs. The CD69 level was detected by flow cytometry while the IFN-γ level was measured by ELISA. **p < 0.01, one-way ANOVA.
After photothermal and photodynamic effects induced by NIR irradiation, necrotic cells generate a range of DAMP molecules, such as calreticulin, ATP, and HMGB1 [37, 38]. These molecules play crucial roles in cancer immunotherapy. Particularly, secretion of HMGB1 promotes immune cell recruitment and DC maturation [39]. LBL hNP-treated cells, with or without NIR irradiation, were evaluated for DAMP expression by western blotting. Remarkably, HMGB1 protein content was markedly higher in the NIR treatment group than in the non-exposed group (Figure 3G). The maturation of bone-derived (BM-) DCs was investigated after incubation with tumor-specific antigen derived from LBL hNP-treated B16BL/6 cells. Up to 82.9% of DCs became mature upon treatment with LBL hNPs in the presence of NIR irradiation (Figure 3H), a level close to that of LPS-treated cells, which acted as a positive control. In the groups treated with PBS or without NIR irradiation, only 24.2% or 25.4% of DCs were mature, respectively. This significant difference was further confirmed using other maturation-specific markers such as CD40 and CD80 (Figure S11), indicating that the specific tumor antigen induced by NIR exposure interacted with DCs and greatly promoted DC maturation.
The interaction of immature DCs with tumor-associated antigen enables them to transform into mature DCs and to deliver the major histocompatibility complex to T cells, thus promoting the proliferation and differentiation of effective T cells [40]. To establish whether DCs that captured tumor antigen can present it to CD8+ T cells, we cocultured them with CD8+ T cells. After 3 days, CD69 and IFN-γ were highly detected in cells exposed to tumor antigen derived from LBL hNPs plus NIR irradiation, with 59.8% CD8+CD69+ T cells and 150 pg/mL IFN-γ (Figure 3I-J). Therefore, after exposure to NIR irradiation, LBL hNPs enhanced the exposure of tumor-associated antigen, promoted DC maturation and downstream activation of effective T cells.
In vitro downregulation of Treg cells
To evaluate the effect of IMT on Treg cell differentiation, CD4+ T cells from spleen and lymph nodes were cultured with IMT at various concentrations. Flow cytometry demonstrated a dose-dependent effect of IMT on Treg differentiation (Figure 4A). Notably, 5 µM IMT induced a more than 2-fold reduction in Treg cell differentiation (23.2 ± 2.3% vs. 50.2 ± 1.7% at 0 µM). Next, the Treg cell-suppressive effects of free IMT (5 µM), GITR-PLGA cores from IMT-loaded LBL hNPs and IMT-loaded LBL hNPs with GITR antibody pretreatment were compared. The GITR-PLGA cores from LBL hNPs significantly reduced Treg cell differentiation to 12.0 ± 3.0%, while CD25+ Foxp3+ T cells in the free IMT group accounted for 25.1 ± 1.4%, suggesting an enhancement in Treg-inhibition function by applying LBL hNPs (Figure 4B). However, this inhibitory effect on Treg cells was reversed by GITR antibody pretreatment, confirming a successful targeting impact of GITR on Treg cells.
In vitro Treg cell-suppressive effect of GITR-PLGA cores from LBL hNPs. (A) Effect of IMT on Treg cell differentiation. The IMT concentration varied from 2.5 to 20 μM. The Treg cell level in the cytokine-non-treated group was regarded as the normal level in healthy mice (n=3). (B) Effect of free IMT (5 µΜ), GITR-PLGA cores from LBL hNPs and LBL hNPs with excessive GITR antibody pretreatment on Treg cell differentiation, with the non-IMT-treated group used as a control (n=3). (C) Schematic illustration of in vitro Treg cell and B16BL/6 cell Transwell coculture system. (D-E) Competitive cellular uptake of GITR-PLGA cores from LBL hNPs between Treg cells (D) and B16BL6 cells (E) in the coculture system. (F) Effect of free IMT, GITR-PLGA cores from LBL hNPs and LBL hNPs with excessive GITR antibody pretreatment on Treg cell differentiation in the presence of Treg cells and B16BL/6 cells coculture system (n=3). **p < 0.01, one-way ANOVA. (G) Western blot assay of STAT3, p-STAT3, STAT5, and p-STAT5 expression in Treg cells exposed to free IMT, LBL hNPs, or LBL hNPs with excessive GITR antibody pretreatment. The concentration of IMT was 5 µM. (H) Effect of IMT-loaded GITR-PLGA cores from LBL hNPs on Treg cell proliferation. The concentration of IMT ranged from 0 to 20 μM (n=3). (I) In vitro determination of Treg cell proliferation using CellTraceTM CFSE probe after exposure to free IMT, LBL hNPs, and LBL hNPs with GITR pretreatment. The concentration of IMT was 5 μM.
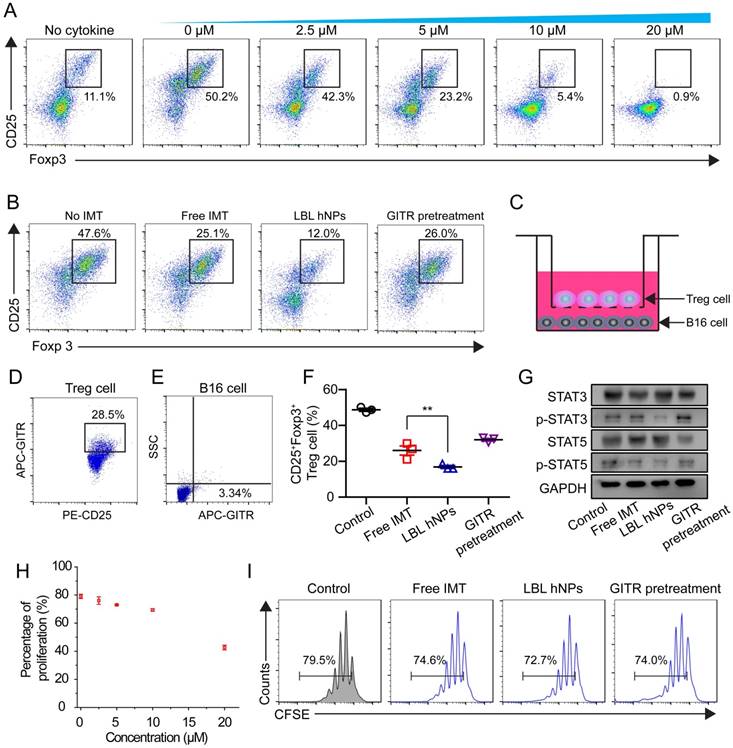
Next, we aimed to clarify the Treg cell-inhibitory effect of GITR-PLGA cores from LBL hNPs under coculture of B16BL/6 and Treg cells. To identify whether the GITR-PLGA cores from LBL hNPs can successfully target Treg cells in the presence of B16BL/6 tumor cells, competitive cellular uptake was measured using the transwell coculture system (Figure 4C). Compared to the IgG isotype-modified GITR-PLGA cores, which exhibited lesser uptake by Treg and B16BL/6 cells (Figure S12), GITR-PLGA cores were clearly observed in CD25+ Treg cells (28.5 ± 4.7%), whereas the percentage in B16BL/6 cells was only 3.3 ± 1.4% (Figure 4D-E). The accelerated cellular uptake of GITR-PLGA core in Treg cells was largely derived from the high expression of GITRs on Treg cells [17]. Subsequently, remarkable downregulation of Treg cells (19.7%, p<0.01) was detected in LBL hNP compared with free IMT (30.8%) treatment groups (Figure 4F). A reverse effect was also observed following GITR antibody pretreatment. Altogether, the application of LBL hNPs resulted in significant inhibition of Treg cell differentiation.
To better understand the mechanism of IMT-induced Treg-suppressive function, STAT3, p-STAT3, STAT5, and p-STAT5 were quantified in Treg cells by western blotting. It has been demonstrated that the upregulation of Foxp3 in Treg cells may relate to binding of STAT3 and STAT5 to a highly conserved STAT-binding site located in the first intron of the Foxp3 gene [41, 42]. After incubation with LBL hNPs, STAT3 and STAT5 phosphorylation was decreased, while higher levels of p-STAT3 and p-STAT5 were maintained in free IMT or GITR antibody pretreatment groups (Figure 4G). Further evaluation of Treg cell proliferation proved that ≤10 µM IMT-loaded LBL hNPs had nearly no effect on Treg cell proliferation (Figure 4H). Additionally, there was no remarkable difference in proliferation between the free IMT or GITR pretreatment group and the LBL hNP-treated group (Figure 4I). Therefore, the suppressive effect of LBL hNPs on Treg cells was excluded to be exerted via inhibition of proliferation; instead, the reduction in STAT3 and STAT5 phosphorylation may account for the suppressive function of Treg cells.
In vivo biodistribution of LBL hNPs
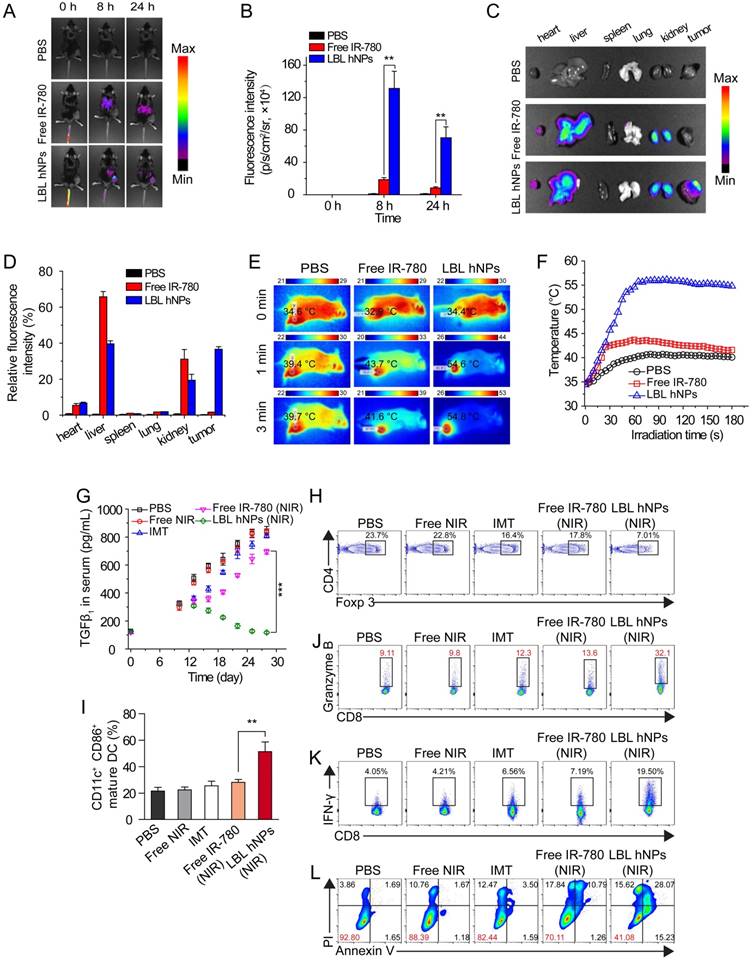
To determine the in vivo distribution of IR-780-loaded LBL hNPs, C57BL/6 mice with B16BL/6 cell xenografted tumors (200 mm3) were intravenously injected with free IR-780 (8 mg/kg) or IR-780 loaded-LBL hNPs (8 mg/kg IR-780). After 8 h, strong fluorescence was observed throughout the liver and kidneys in the free IR-780 group (Figure 5A-B). However, in the LBL hNP-treated group, clear fluorescence signal was observed specifically in the tumor and gradually attenuated during 24 h, indicating successful tumor targeting by the LBL hNPs. After the mice were killed, strong fluorescence accumulation in tumor was verified in LBL hNP-treated animals, while in the free IR-780 treatment group, fluorescence was intensely accumulated in the liver and kidneys (Figure 5C-D). This accumulation and retention of nanoparticles in tumors is considered to be induced by the EPR effect [43].
In vivo distribution, PTT effect, and immune-antitumor effect of LBL hNPs. (A) Fluorescence distribution in B16BL/6 tumor-bearing mice at 0, 8, and 24 h after intravenous administration of PBS, free IR-780 and LBL hNPs. (B) Mean fluorescence intensity in tumors in PBS, free IR-780 (8 mg/kg) and LBL hNPs treatment groups (n=3). **p < 0.01, one-way ANOVA. (C) Fluorescence distribution in the heart, liver, spleen, lungs, kidneys, and tumors after 24-h treatment with PBS, free IR-780 or LBL hNPs. (D) Relative fluorescence intensity percentage of these tissues after treatment with PBS, free IR-780 or LBL hNPs (n=3). (E-F) In vivo IR thermography and corresponding temperature increase at the tumors of mice injected with PBS, free IR-780, or LBL hNPs plus 3 min NIR irradiation (808 nm, 1.0 W/cm2). (G) Mean serum TGF-β levels in mice treated with PBS, NIR, IMT, free IR-780 (NIR), or LBL hNPs (NIR), at different days (n=6). ***p < 0.001, one-way ANOVA. (H) Intratumoral Treg cell percentages in mice treated with PBS, free NIR, IMT, free IR-780 (NIR), or LBL hNPs (NIR) at day 20 (n=6). (I) Intratumoral maturation of DCs in the indicated treatment groups (n=6). **p < 0.01, one-way ANOVA. (J-K) Secretion of antitumor cytokine Granzyme B and IFN-γ from intratumoral CD8+ T cells in the indicated treatment groups (n=6). (L) In vivo tumor cell apoptosis and necrosis in mice treated with PBS, NIR, IMT, free IR-780 (NIR), or LBL hNPs (NIR) (n=6).
The in vivo uptake of GITR-PLGA cores from LBL hNPs by intratumoral Treg cells was further confirmed using DiD-labeled LBL hNPs. At predetermined time points, single cells were obtained from the tumor and uptake of DiD-labeled GITR-PLGA cores were detected as CD3+CD4+Foxp3+DiD+ T cells (Figure S13A). LBL hNPs (GITR+) displayed significant (p < 0.01) time-dependent intratumoral Treg cell uptake of GITR-PLGA cores, whereas the uptake percentage was lower (p < 0.001) in the LBL hNPs (GITR-)-treated group (Figure S13B), confirming the successful uptake of GITR-PLGA core by intratumoral Treg cells in vivo. The relatively high cellular internalization of GITR-PLGA cores from LBL hNPs may result from the higher expression of GITR in Treg cells than in other T cells, as BioGPS data showed significantly higher (p < 0.0001) GITR expression in Treg cells than in CD8+ T cells or NK cells (Figure S13C).
In vivo photothermal effect and antitumor immunotherapy
In vivo generation of hyperthermia at the tumor site was investigated by intravenously injecting PBS, free IR-780, and IR-780-loaded LBL hNPs. After 8 h, the mice were exposed to NIR irradiation (808 nm, 1.0 W/cm2) for 3 min. The temperature elevation was monitored throughout the treatment using an infrared thermograph. Within 60 s, LBL hNPs significantly elevated the temperature of the tumor up to 55 °C (Figure 5E-F), confirming the preferential tumor accumulation and good photothermal effect of LBL hNPs. Free IR-780 heated the tumor surface up to only 44 °C during the first 60 s, and the temperature decreased thereafter. PBS as a control under NIR irradiation resulted in a mild temperature elevation, suggesting that NIR irradiation alone failed to increase the tumor temperature.
To examine the in vivo therapeutic effect, C57BL/6 mice bearing B16BL/6 cancer cells were intravenously administered the indicated formulations. During the treatment period, LBL hNPs plus NIR irradiation moderately increased serum TGF-β (Figure 5G), which finally reverted to the normal level (120 pg/mL), while in other treatment groups, the levels continued to increase. Increased serum TGF-β abrogates cancer immunotherapy by promoting Treg cell proliferation and differentiation [44, 45]. Thus, the reduced TGF-β level following LBL hNP treatment suggests successful targeting and suppression of Treg cells by IMT-loaded GITR-PLGA cores from LBL hNPs. To further evaluate the effect of IMT-loaded GITR-PLGA cores from LBL hNPs on Treg cells, the frequency of intratumoral Treg cells isolated from tumor after 3 dosages was analyzed by flow cytometry. The Treg-targeting IMT-loaded GITR cores inside the LBL hNPs resulted in a reduction of intratumoral Treg cells to 7.01% (Figure 5H), while other groups had more than 20% intratumoral Treg cells, suggesting Treg targeting and downregulation are successfully achieved by LBL hNPs in vivo. Thus, LBL hNPs are suitable to downregulate Treg cell function in vivo and pave the way for promoting antitumor immune cells.
Substantial DC maturation was observed in LBL hNP-treated cells under NIR exposure, indicating successful generation and presentation of tumor antigen to DCs under LBL hNP treatment (Figure 5I). Subsequently, mature DCs were capable of presenting tumor antigen to CD8+ T cells, leading to the generation of tumor antigen-specific effective T cells. Notably, LBL hNP-treated cells showed a 3-fold increase in granzyme B and a 5-fold increase in IFN-γ secretion in intratumoral CD8+ T cells after 3 dosages, while CD8+ T cells in the other groups had less granzyme B and IFN-γ secretion (Figure 5J-K). These observations indicated an enhancement of the cytolytic effect on tumors by activated cytotoxic T cells in the LBL hNP-treated group under NIR exposure. Both IFN-γ and granzyme B secreted by effective CD8+ T cells exert key roles in eliminating tumors [46]. Our results demonstrate that tumor-specific antigen generated under the stimulus of LBL hNPs plus NIR treatment can be captured and presented by DCs, leading to the enhanced proliferation of cytotoxic T cells, which are effective against tumor in vivo.
Free IMT or NIR treatment generated negligible amounts of ROS in vivo, whereas LBL hNPs plus NIR promoted ROS production at the tumor sites in vivo (Figure S14) as compared to free IR-780, free IMT, NIR alone, and PBS, confirming that LBL hNPs under NIR treatment induce adequate PDT against tumor in vivo. Approximately 59% of tumor cells were apoptotic in LBL hNP-treated animals, while only 7.2%, 11.61%, 17.56%, and 30% were apoptotic in mice exposed to PBS, NIR alone, free IMT, and free IR-780 (with NIR), respectively (Figure 5L). This tumor damage primarily resulted from enhanced effect of PTT and PDT after NIR irradiation.
The antitumor study in vivo is illustrated in Figure 6A. After treatment for 18 days, PBS as well as NIR allowed severe tumor growth (up to 2000 mm3) (Figure 6B-C). Free IMT and free IR-780 plus NIR irradiation effectively inhibited tumor growth at the early stage, but tumor growth took off at a later stage, primarily due to the low drug concentration at the tumor sites. Thus, the effects of NIR on tumor and of IMT on immune cells are not sufficient. However, in mice treated with LBL hNPs plus NIR irradiation, total tumor ablation was observed, and no tumor regrowth occurred after 9 days of treatment. Crucially, this treatment had a superior capacity to extend survival (Figure 6D), while short survival was detected in other groups.
In vivo tumor ablation and histopathological investigation. (A) In vivo antitumor therapy schedule for LBL hNPs administration. (B) Images of B16BL/6 tumor-bearing C57BL/6 mice taken at 28 days. (C) Average tumor growth curves for mice after indicated treatments. The intravenous injection began at day 10 after random group division (n=6). ***p < 0.001, one-way ANOVA. (D) Survival curves of mice after treatment with PBS, NIR, IMT, free IR-780 (NIR), or LBL hNPs (NIR) (n=5). Log-rank test. (E) Histopathological and immunohistochemical evaluation of intra-tumoral CD4+ T (Th) cells, CD8+ T cells, and tumor necrosis. Each tumor section was obtained from B16BL/6 tumor-bearing mice treated with PBS, NIR, IMT, free IR-780 (NIR), or LBL hNPs (NIR).
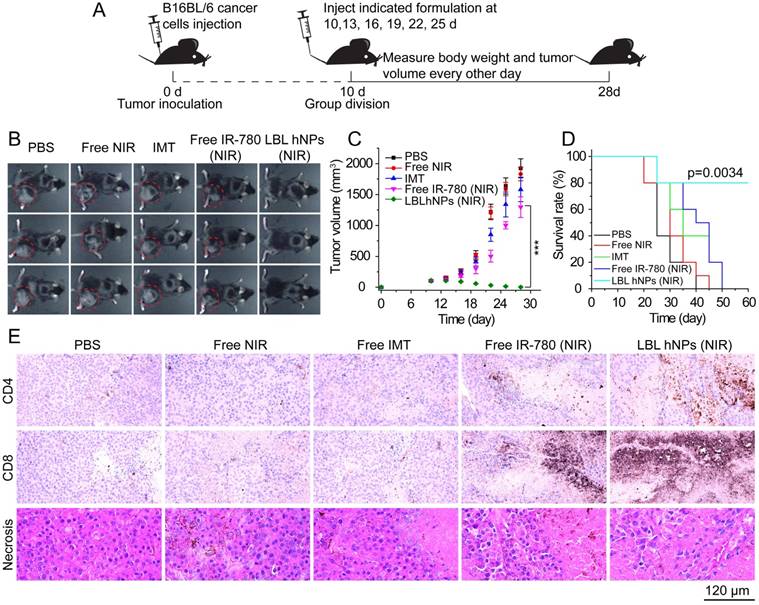
Histopathological investigation of mice after 3 dosages revealed remarkable levels of intratumoral CD4+ T cells (helper T cells), intratumoral CD8+ T cells, and more necrotic tumor cells in LBL hNP plus NIR-treated animals (Figure 6E) as compared to animals treated with PBS, NIR, and free IMT, which showed no significant differences in these features. In the free IR-780 (NIR) group, CD4+ T and CD8+ T cells were more abundant than in the PBS group, suggesting a subtle IR-780-induced NIR effect, but the levels were not near those in the LBL hNP (NIR) group. These results were in accordance with those of flow-cytometric analysis. Meanwhile, reductions in Ki-67 (a tumor proliferation marker) and CD31 (an angiogenesis marker) expression (Figure S15A) in LBL hNP plus NIR-treated animals confirmed the abrogation of tumor cell proliferation and intratumoral vessels. Immunohistochemical indexes exhibited significant differences between the LBL hNP plus NIR-treated and the other groups (Table S1). Moreover, treatment with LBL hNPs induced no pathological changes in main organs (Figure S15B).
To further demonstrate the efficacy of LBL hNPs in tumor eradication, the MC-38 tumor model was used for in vivo antitumor experiments (Figure S16). Consistent with the results shown in Figure 6, LBL hNPs under NIR irradiation showed superior antitumor effect than any other treatment group (Figure S16A-B). Furthermore, the serum level of cytokine TGFβ, which is a measure of Treg cell function, was also measured. The groups treated with LBL hNPs plus NIR showed significant reduction in serum TGFβ levels compared to other treated groups (Figure S16C). In addition, the serum levels of IFN-γ and TNF-α, which act as inflammatory factors associated with PDT-induced antitumor immunity, were also estimated. Significantly higher IFN-γ and TNF-α levels were observed in mice after LBL hNP plus NIR treatment (Figure S16D-E), indicating that our system successfully initiated the cellular immune response. The CD8+/Treg cell ratio acted as an important index for predicting in vivo antitumor immunity [47]. Further investigation of the intratumoral CD8+/Treg cell ratio demonstrated that the strategy of combining inhibition of Treg cells with NIR treatment (LBL hNPs plus NIR) generated higher CD8+ effector T cells against tumors compared to Treg cell inhibition or NIR treatment alone (Figure S16F).
Currently, studies related to combination photoimmunotherapy use both NIR-induced PDT effect and immune-checkpoint blockade (anti-PD-L1 and anti-CTLA4 antibody) to trigger anticancer photoimmunotherapy [48-50]. Similarly, our system also utilized the NIR dye IR-780 to achieve PDT/PTT-induced tumor eradication and promote maturation of dendritic cells, followed by induction of CD8 T cell-guided antitumor immunotherapy. However, instead of using the immune-checkpoint inhibitor, we utilized LBL hNPs, with IMT-loaded GITR-modified PLGA nanoparticles as the core, to specifically inhibit the suppressive function of Treg cells in the tumor microenvironment. The application of layer-by-layer coating on the nanoparticles facilitated the release of IR-780 and GITR-modified PLGA cores at lower pH in the tumor microenvironment. The modification of GITR antibody on the surface ensured accurate target effect of nanoparticles in the tumor microenvironment because of the high GITR receptor level on Treg cells. In the tumor microenvironment, Treg cells inhibit activation and proliferation of CD8+ effector T cells and promote tumor progression. In our system, NIR irradiation directly eradicated the tumor and facilitated the production of more tumor-associated antigens. The IMT-loaded nanoparticles inhibited the suppressive function of Treg cells both in vitro and in vivo, contributing to the activation and proliferation of more antitumor effector T cells. In summary, the combination of NIR treatment and Treg cell inhibition by LBL hNPs can initiate potent antitumor activity.
Conclusion
In conclusion, a photothermal, photodynamic anticancer effect and concomitant immuno-anticancer effect can be achieved using IR-780 dye and IMT-loaded LBL hNPs. The LBL hNPs not only possess the capacity to enhance PTT and PDT effects under NIR irradiation, but are also capable of releasing IR-780 and Treg-targeting GITR-PLGA cores at tumor pH. This makes them suitable for inhibiting the Treg cell suppressive function, promoting DC maturation and triggering downstream CD8+ T cell activation against tumor. In vivo experiments in two different tumor models demonstrated that LBL hNPs under NIR exposure eradicate tumor growth and recurrence by hyperthermia, generation of ROS, inhibition of tumor proliferation, and shutdown of tumor vessels. Simultaneously, the downregulation of Treg cell suppressive function by IMT-loaded GITR-PLGA cores in vivo efficiently paved the way for DC maturation and presentation of tumor-associated antigen. This strongly promoted the activation of effective CD8+ T cells, which elicited higher granzyme B and IFN-γ levels that inhibit tumor growth. Thus, the proposed nanoparticles may provide a new strategy to eliminate tumors using photoimmunotherapy.
Abbreviations
BMDCs: bone-derived dendritic cells; DAMP: damage-associated molecular pattern; DCs: dendritic cells; EPR: enhanced permeability and retention; GITR: glucocorticoid-induced TNF receptor; HMGB1: high-mobility group box 1; IMT: imatinib; LBL hNPs: layer by layer hybrid nanoparticles; LC: loading capacity; LE: loading efficiency; NIR: near-infrared; PDT: photodynamic therapy; PTT: photothermal therapy; ROS: reactive oxygen species.
Supplementary Material
Supplementary methods, figures and table.
Acknowledgements
This research was supported by National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIP) (No. 2018R1A2A2A05021143) and by the Medical Research Center Program (2015R1A5A2009124) through the NRF funded by MSIP.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liang C, Xu L, Song G, Liu Z. Emerging nanomedicine approaches fighting tumor metastasis: animal models, metastasis-targeted drug delivery, phototherapy, and immunotherapy. Chem Soc Rev. 2016;45:6250-69
2. Lucky SS, Soo KC, Zhang Y. Nanoparticles in photodynamic therapy. Chem Rev. 2015;115:1990-2042
3. Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer. 2006;6:535-45
4. Thapa RK, Byeon JH, Ku SK, Yong CS, Kim JO. Easy on-demand self-assembly of lateral nanodimensional hybrid graphene oxide flakes for near-infrared-induced chemothermal therapy. NPG Asia Mater. 2017;9:e416
5. Yuan A, Qiu X, Tang X, Liu W, Wu J, Hu Y. Self-assembled PEG-IR-780-C13 micelle as a targeting, safe and highly-effective photothermal agent for in vivo imaging and cancer therapy. Biomaterials. 2015;51:184-93
6. Shon S-M, Choi Y, Kim J-Y, Lee DK, Park J-Y, Schellingerhout D. et al. Photodynamic therapy using a protease-mediated theranostic agent reduces cathepsin-B activity in mouse atheromata in vivo. Arterioscler Thromb Vasc Biol. 2013;33:1360-5
7. Yan F, Duan W, Li Y, Wu H, Zhou Y, Pan M. et al. NIR-laser-controlled drug release from DOX/IR-780-loaded temperature-sensitive-liposomes for chemo-photothermal synergistic tumor therapy. Theranostics. 2016;6:2337-51
8. Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part two—cellular signaling, cell metabolism and modes of cell death. Photodiagnosis Photodyn Ther. 2005;2:1-23
9. Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: Part three—photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagnosis Photodyn Ther. 2005;2:91-106
10. Li K-C, Chu H-C, Lin Y, Tuan H-Y, Hu Y-C. PEGylated copper nanowires as a novel photothermal therapy agent. ACS Appl Mater Interfaces. 2016;8:12082-90
11. Ogawa M, Tomita Y, Nakamura Y, Lee M-J, Lee S, Tomita S. et al. Immunogenic cancer cell death selectively induced by near infrared photoimmunotherapy initiates host tumor immunity. Oncotarget. 2017;8:10425-36
12. Mroz P, Hamblin MR. The immunosuppressive side of PDT. Photochem Photobiol Sci. 2011;10:751-8
13. Liu C, Workman CJ, Vignali DAA. Targeting regulatory T cells in tumors. FEBS J. 2016;283:2731-48
14. Larmonier N, Janikashvili N, LaCasse CJ, Larmonier CB, Cantrell J, Situ E. et al. Imatinib mesylate inhibits CD4+CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol. 2008;181:6955-63
15. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H. et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17:1094
16. Park K. Combined therapy of imatinib and an anti-CTLA4 immune-checkpoint inhibitor. J Control Release. 2018;281:196
17. Sacchetti C, Rapini N, Magrini A, Cirelli E, Bellucci S, Mattei M. et al. In vivo targeting of intratumor regulatory T cells using PEG-modified single-walled carbon nanotubes. Bioconjug Chem. 2013;24:852-8
18. Dang X, Gu L, Qi J, Correa S, Zhang G, Belcher AM. et al. Layer-by-layer assembled fluorescent probes in the second near-infrared window for systemic delivery and detection of ovarian cancer. Proc Natl Acad Sci U S A. 2016;113:5179-84
19. Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136-51
20. McCall RL, Sirianni RW. PLGA nanoparticles formed by single- or double-emulsion with Vitamin E-TPGS. J Vis Exp. 2013:51015
21. Chen C, Mei H, Shi W, Deng J, Zhang B, Guo T. et al. EGFP-EGF1-conjugated PLGA nanoparticles for targeted delivery of siRNA into injured brain microvascular endothelial cells for efficient RNA interference. PLOS ONE. 2013;8:e60860
22. Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Control Release. 2007;120:18-26
23. Poon Z, Chang D, Zhao X, Hammond PT. Layer-by-layer nanoparticles with a pH-sheddable layer for in vivo targeting of tumor hypoxia. ACS Nano. 2011;5:4284-92
24. Ramasamy T, Haidar ZS, Tran TH, Choi JY, Jeong J-H, Shin BS. et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10:5116-27
25. Li L, Zhao J-F, Won N, Jin H, Kim S, Chen J-Y. Quantum dot-aluminum phthalocyanine conjugates perform photodynamic reactions to kill cancer cells via fluorescence resonance energy transfer. Nanoscale Res Lett. 2012;7:386
26. Min Y, Roche KC, Tian S, Eblan MJ, McKinnon KP, Caster JM. et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat Nano. 2017 [Epub ahead of print]
27. Fang RH, Hu C-MJ, Luk BT, Gao W, Copp JA, Tai Y. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Letters. 2014;14:2181-8
28. Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml Barbara U. et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+MHCII+ macrophages and dendritic cells. Immunity. 2015;42:1197-211
29. Klechevsky E, Flamar A-L, Cao Y, Blanck J-P, Liu M, O'Bar A. et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood. 2010;116:1685-97
30. Haidar ZS, Hamdy RC, Tabrizian M. Protein release kinetics for core-shell hybrid nanoparticles based on the layer-by-layer assembly of alginate and chitosan on liposomes. Biomaterials. 2008;29:1207-15
31. Yoon H-J, Lim TG, Kim J-H, Cho YM, Kim YS, Chung US. et al. Fabrication of multifunctional layer-by-layer nanocapsules toward the design of theragnostic nanoplatform. Biomacromolecules. 2014;15:1382-9
32. Yang T, Wang Y, Ke H, Wang Q, Lv X, Wu H. et al. Protein-nanoreactor-assisted synthesis of semiconductor nanocrystals for efficient cancer theranostics. Adv Mater. 2016;28:5923-30
33. Cobley CM, Au L, Chen J, Xia Y. Targeting gold nanocages to cancer cells for photothermal destruction and drug delivery. Expert Opin Drug Deliv. 2010;7:577-87
34. Guo Z, Zou Y, He H, Rao J, Ji S, Cui X. et al. Bifunctional platinated nanoparticles for photoinduced tumor ablation. Adv Mater. 2016;28:10155-64
35. Kitsis RN, Molkentin JD. Apoptotic cell death “Nixed” by an ER-mitochondrial necrotic pathway. Proc Natl Acad Sci U S A. 2010;107:9031-2
36. Mroz P, Yaroslavsky A, Kharkwal GB, Hamblin MR. Cell death pathways in photodynamic therapy of cancer. Cancers. 2011;3:2516-39
37. Bai J, Liu Y, Jiang X. Multifunctional PEG-GO/CuS nanocomposites for near-infrared chemo-photothermal therapy. Biomaterials. 2014;35:5805-13
38. Bezu L, Gomes-da-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R. et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187
39. Gougeon ML, Melki MT, Saidi H. HMGB1, an alarmin promoting HIV dissemination and latency in dendritic cells. Cell Death Differ. 2012;19:96-106
40. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543-55
41. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D. et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571-9
42. Larmonier N, Janikashvili N, LaCasse CJ, Larmonier CB, Cantrell J, Situ E. et al. Imatinib mesylate inhibits CD4+CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL(negative) tumors. J Immunol. 2008;181:6955-63
43. Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Cancer Nanotechnol. 2010:25-37
44. Wan YY, Flavell RA. TGF-β and regulatory T cell in immunity and autoimmunity. J Clin Immunol. 2008;28:647-59
45. Reginato E, Mroz P, Chung H, Kawakubo M, Wolf P, Hamblin MR. Photodynamic therapy plus regulatory T-cell depletion produces immunity against a mouse tumour that expresses a self-antigen. Br J Cancer. 2013;109:2167-74
46. Cai SF, Fehniger TA, Cao X, Mayer JC, Brune JD, French AR. et al. Differential expression of granzyme B and C in murine cytotoxic lymphocytes. J Immunol. 2009;182:6287-97
47. Ou W, Thapa RK, Jiang L, Soe ZC, Gautam M, Chang J-H. et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J Control Release. 2018;281:84-96
48. He C, Duan X, Guo N, Chan C, Poon C, Weichselbaum RR. et al. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat Commun. 2016;7:12499
49. Yang G, Xu L, Chao Y, Xu J, Sun X, Wu Y. et al. Hollow MnO2 as a tumor-microenvironment-responsive biodegradable nano-platform for combination therapy favoring antitumor immune responses. Nat Commun. 2017;8:902
50. Xu J, Xu L, Wang C, Yang R, Zhuang Q, Han X. et al. Near-infrared-triggered photodynamic therapy with multitasking upconversion nanoparticles in combination with checkpoint blockade for immunotherapy of colorectal cancer. ACS Nano. 2017;11:4463-74
Author contact
![]() Corresponding author: Prof. Jong Oh Kim, Ph.D. Tel: +82-53-810-2813; Fax: +82-53-810-4654; E-mail: jongohkimac.kr
Corresponding author: Prof. Jong Oh Kim, Ph.D. Tel: +82-53-810-2813; Fax: +82-53-810-4654; E-mail: jongohkimac.kr