Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Author Biography
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(17):4633-4648. doi:10.7150/thno.26687 This issue Cite
Research Paper
Aphthous ulcer drug inhibits prostate tumor metastasis by targeting IKKɛ/TBK1/NF-κB signaling
Chaping Cheng1,2,#, Zhongzhong Ji1,2,#, Yaru Sheng1, Jinming Wang1, Yujiao Sun2, Huifang Zhao1, Xiaoxia Li1, Xue Wang1,2, Yuman He1, Jufang Yao1, Li Wang1, Chenlu Zhang1, Yanjing Guo1, Jianming Zhang4, Wei-Qiang Gao1,2, ![]() , Helen He Zhu1,3,
, Helen He Zhu1,3, ![]()
1. State Key Laboratory of Oncogenes and Related Genes, Renji-Med-X Stem Cell Research Center, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China
2. School of Biomedical Engineering & Med-X Research Institute, Shanghai Jiao Tong University, Shanghai 200030, China
3. Department of Urology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China
4. National Research Center for Translational Medicine, Shanghai State Key Laboratory of Medical Genomics, Rui-Jin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China.
# These authors contribute equally to this paper.
Received 2018-4-15; Accepted 2018-7-12; Published 2018-9-9
Abstract

Tumor metastasis is the major cause of death for prostate cancer (PCa) patients. However, the treatment options for metastatic PCa are very limited. Epithelial-mesenchymal transition (EMT) has been reported to be an indispensable step for tumor metastasis and is suggested to associate with acquisition of cancer stem cell (CSC) attributes. We propose that small-molecule compounds that can reverse EMT or induce mesenchymal-epithelial transition (MET) of PCa cells may serve as drug candidates for anti-metastasis therapy.
Methods: The promoters of CDH1 and VIM genes were sub-cloned to drive the expression of firefly and renilla luciferase reporter in a lentiviral vector. Mesenchymal-like PCa cells were infected with the luciferase reporter lentivirus and subjected to drug screening from a 1274 approved small-molecule drug library for the identification of agents to reverse EMT. The dosage-dependent effect of candidate compounds was confirmed by luciferase reporter assay and immunoblotting. Wound-healing assay, sphere formation, transwell migration assay, and in vivo intracardiac and orthotopic tumor xenograft experiments were used to evaluate the mobility, metastasis and tumor initiating capacity of PCa cells upon treatment. Possible downstream signaling pathways affected by the candidate compound treatment were analyzed by RNA sequencing and immunoblotting.
Results: Drug screening identified Amlexanox, a drug used for recurrent aphthous ulcers, as a strong agent to reverse EMT. Amlexanox induced significant suppression of cell mobility, invasion, serial sphere formation and in vivo metastasis and tumor initiating capacity of PCa cells. Amlexanox treatment led to downregulation of the IKK-ɛ/ TBK1/ NF-κB signaling pathway. The effect of Amlexanox on EMT reversion and cell mobility inhibition can be mimicked by other IKK-ɛ/TBK1 inhibitors and rescued by reconstitution of dominant active NF-κB.
Conclusions: Amlexanox can sufficiently suppress PCa metastasis by reversing EMT through downregulating the IKK-ɛ/TBK1/NF-κB signaling axis.
Keywords: Amlexanox, prostate cancer, metastasis, IKK-ɛ/TBK1, mesenchymal-epithelial transition
Introduction
According to 2017 cancer statistics, prostate cancer (PCa) is one of most common cancers and highest causes of cancer-related mortality in men worldwide [1]. Tumor metastasis, which remains incurable, is the major contributor to PCa-related death [2, 3]. Therefore, treatment approaches to target prostate tumor metastases are in urgent need. Epithelial-mesenchymal transition (EMT), a biological process during which cells lose epithelial cell-specific characteristics such as cell polarity and cell-cell junction, and meanwhile acquire mesenchymal cell attributes, is a critical step in the metastasis of various types of tumors including PCa [4]. During the initiation of metastasis, tumor cells that undergo EMT acquire enhanced mobility and invasiveness to disseminate from the primary tumor and enter the circulation. Importantly, it has been reported recently by other groups and us that EMT endows tumor cells with cancer stem cell (CSC)-like traits including the ability to regenerate tumor and develop therapeutic resistance [5]. Upregulation of master EMT transcriptional regulators Zeb1 and Snail has been shown to be essential for tumor metastasis and enables a transition from non-CSC state to CSC state to promote tumorigenicity [6-9]. In addition, we demonstrated in a previous study that Zeb1 confers castration resistance on PCa cells through induction of stem cell-like properties [10]. Therefore, we proposed that identification of viable agents to reverse EMT or induce MET for the development of targeted therapy against tumor metastases and CSC activities is of great value and clinical importance.
The transcriptional factor NF-κB is an important regulator of many cellular processes in inflammatory immune response, cell proliferation, survival and invasion [11]. There are five NF-κB family members in mammals: p65 (Rel A), c-Rel, RelB, p50/p105 and p52/p100. Nuclear localization and activation of NF-κB are blocked by its association with IκB proteins. In response to stimuli, IκB kinase (IKK) activation results in phosphorylation and subsequent degradation of IκB proteins. Released NF-κB subunits form heterodimers or homodimers to exert a DNA binding effect and transactivate target genes [12, 13]. NF-κB is recently reported to play a major role in cancer. Constitutive activation of NF-κB has been found in a variety of malignancies including PCa, pancreatic cancer, glioma, breast cancer, head and neck cancer, acute myeloid leukemia, etc [14]. In addition to its well-studied role in tumor-related inflammation, activation of NF-κB has been demonstrated to enhance metastatogenesis via EMT [14-17]. The EMT inducer TNFα relies on activation of NF-κB and downstream upregulation of Twist1 for its tumor metastasis-promoting effect [18, 19]. Inactivation of IKKα, an important upstream component of the NF-κB signal pathway, inhibits the metastatogenesis of PCa in TRAMP mice [20].
IκB kinase ɛ (IKKɛ) and the IκB kinase-related TANK-binding kinase I (TBK1) are non-canonical IKKs that activate the NF-κB signal pathway in a way distinct from canonical IKKs such as IKKα and IKKβ. Several NF-κB effectors including IκBα, IKKβ, p65 and Crel can be modulated by IKKɛ and TBK1 upon stimuli, which facilitates the nuclear translocation of NF-κB and transcriptional upregulation of downstream genes [21-23]. Recent studies uncovered that IKBKE acts as an oncogene, amplification and overexpression of which lead to a constitutive activation of the NF-κB signaling pathway in breast cancer [24]. Deregulated expression of IKKɛ has also been reported in various types of cancer [25-30]. In addition, IKKɛ is found to promote tumor cell invasion and tumor metastasis by elevating EMT [26, 31]. Therefore, targeting the IKKɛ/TBK1 and NF-κB signaling axis may serve as a feasible way to suppress tumor metastasis.
In this study, using a novel high-throughput system for small-molecule drug screening, we identify Amlexanox, a commonly used clinical drug to treat recurrent aphthous ulcers, as a potent agent to reverse EMT. Amlexanox administration effectively represses PCa cell migration and tumor metastasis in vitro and in vivo by inhibition of the NF-κB signal pathway through specifically targeting IKKɛ and TBK1.
Results
Establishment of a high-throughput drug screening system for the discovery of agents to reverse EMT
To reflect and monitor the epithelial or mesenchymal status of cancer cells, we established lentiviral reporter systems utilizing mCherry or eGFP driven by promoter of CDH1 [32] or VIM [33] (Figure S1A). The CDH1 gene encodes E-cadherin, an essential component in adherent junctions and a frequently used epithelial cell marker. The VIM gene encodes vimentin, a type III intermediate filament protein specifically expressed in mesenchymal cells. A PCa cell line PC3 was infected with either E-cadherin-mCherry or vimentin-eGFP reporter viruses and selected with puromycin or hygromycin for generation of stable transfected cell lines. qRT-PCR using flow cytometry-sorted eGFP or mCherry positive or negative PC3 cells confirmed that the fluorescence intensities were well associated with the E-cadherin or vimentin expression levels, indicating that the reporter driven by promoter of CDH1 or VIM can faithfully reflect the endogenous gene expression (Figure S1B).
In order to perform high-throughput screening to identify potential agents to reverse EMT, we constructed a lentivirus plasmid containing the CDH1 promoter-driven firefly luciferase and the VIM promoter-driven renilla luciferase (Figure 1A). PC3 was infected with the dual-luciferase reporter lentivirus and selected with puromycin for a stable transfectant. The dual-luciferase reporter was validated by a significant decrease in the ratio of E-cadherin-firefly to vimentin-renilla upon treatment with a known EMT inducer, TGF-β (Figure 1B).
High-throughput drug screening from the approved drug library identifies Amlexanox as a potent compound to reverse EMT. (A) Map of the lentiviral dual-luciferase EMT reporter plasmid in which firefly luciferase expression is driven by the CDH1 gene promoter, while renilla luciferase is driven by the VIM promoter. (B) The ratio of E-cadherin-firefly to vimentin-renilla luciferase intensities in dual-luciferase reporter lentivirus-infected PC3 cells significantly decreases in response to the potent EMT inducer TGF-β (n=24). (C) Selection of single-cell-derived PC3 clones with higher mesenchymal properties. (D) Compared to parental PC3 cells, PC3-clone 4 expresses lower levels of epithelial markers E-cadherin and ZO-1, and higher levels of mesenchymal makers vimentin, and N-cadherin and EMT-inducing transcription factor Zeb1. E-cad: E-cadherin; N-cad: N-cadherin; Vim: vimentin. (E) Screening of a small-molecule compound library containing 1274 approved drugs on PC3-clone 4 cells identifies 110 compounds that are able to induce a higher expression of CDH1 promoter-driven luciferase. The Y axis in (A-C) is calculated by dividing individual normalized luciferase values by that of vehicle control. (F) Four compounds with greatest effect on EMT reversion from the first drug screening were selected for a dosage dependence test. Amlexanox displays a nice dosage-dependent effect on promoting CDH1-firefly luciferase expression. Compound 1: Betamethasone; Compound 2: Aminacrine; Compound 3: Lansoprazole; Compound 4: Amlexanox. (G) PC3-clone 4 cells acquire a more flat and polygonal epithelial shape after Amlexanox treatment. Scale bar = 50 μm. (H) Amlexanox treatment leads to dosage-dependent downregulation of EMT-inducing transcriptional factors Zeb1 and N-cadherin, and upregulation of epithelial-specific protein E-cadherin both in PC3 cells and DU145 cells. Unpaired t-test was used for the statistical analysis. ***, P<0.001. Data are presented as mean ± SEM.
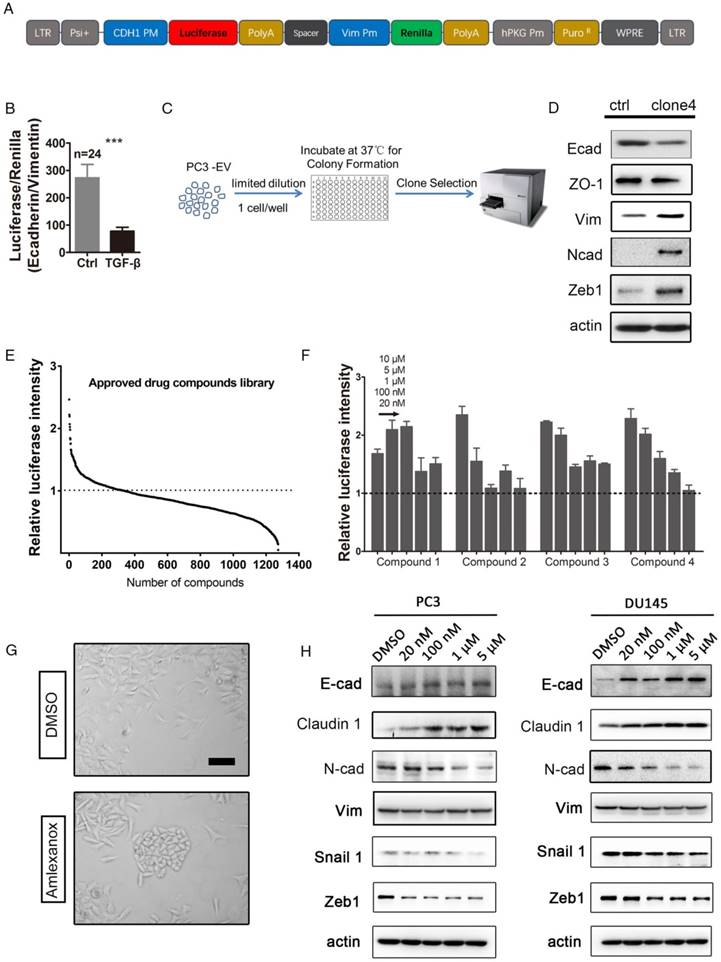
Although our previous study revealed that both mesenchymal and epithelial cell markers are expressed in PC3 cells [10], great heterogeneity existed in the cell line. Using limiting dilution, we generated single-cell derived clones of luciferase reporter PC3 cells in 96-well plates (Figure 1C). Clones containing elongated spindle-shaped mesenchymal-like cells were picked up and examined for their epithelial or mesenchymal status by evaluating the intensity of firefly or renilla luciferase. PC3-clone 4 was selected for subsequent screening for agents to reverse EMT due to its lower expression of epithelial-specific proteins E-cadherin and ZO-1, and higher expression levels of mesenchymal markers vimentin, N-cadherin and Zeb1 (Figure 1D).
Screening from an approved drug library identifies Amlexanox as a potent compound to reverse EMT
We next performed high-throughput screening for agents to reverse EMT from an approved drug library containing 1274 small molecules on the PC3-clone 4 reporter cells. After a 7-day drug treatment at a concentration of 10 μM, PC3-clone 4 cells were collected and tested for firefly luciferase activity. To rule out the effect of drug treatment on cell viability and proliferation, we normalized the luciferase intensity to the cell number determined by the cell counting kit-8 assay. We found that 110 out of 1274 small molecules were able to induce a higher expression of CDH1 promoter-driven luciferase compared to the vehicle control (Figure 1E). In the secondary screening, we used five drug concentrations at 10 μM, 5 μM, 1 μM, 100 nM and 20 nM of the top 4 potent candidates for further validation (Figure 1F and Table S2). Amlexanox, a drug used for recurrent aphthous ulcers, displayed a nice dosage-dependent effect on promoting firefly luciferase expression (compound 4 in Figure 1F). We then focused on whether Amlexanox can be used as an agent to reverse EMT in preventing cancer metastasis.
Interestingly, we observed that PC3-clone 4 cells acquired a more flat, polygonal epithelial-like shape after a 7-day treatment of Amlexanox at 5 μM compared to the mesenchymal phenotype of the vehicle-treated group (Figure 1G). We conducted real-time quantitative PCR to evaluate the mRNA levels of mesenchymal markers, adhesion molecules and integrins upon Amlexanox treatment. As shown in Figure S2A, Amlexanox treatment of PC3 cells lead to upregulated transcription of adhesion molecules (EpCAM, DSP, Claudin1, ZO1 and E-cadherin) and suppression of mesenchymal genes and integrin α5. Immunoblotting experiments further demonstrated a dose-dependent effect of Amlexanox on induction of epithelial markers E-cadherin and Claudin1 expression and on suppression of mesenchymal marker N-cadherin as well as the core transcriptional factor Zeb1 and Snail for EMT (Figure 1H). Suppression of vimentin expression by Amlexanox was more obviously detected in DU145 than in PC3. Taken together, these observations suggested that Amlexanox was a promising agent to reverse EMT.
Amlexanox suppresses mobility and migration of PCa cells
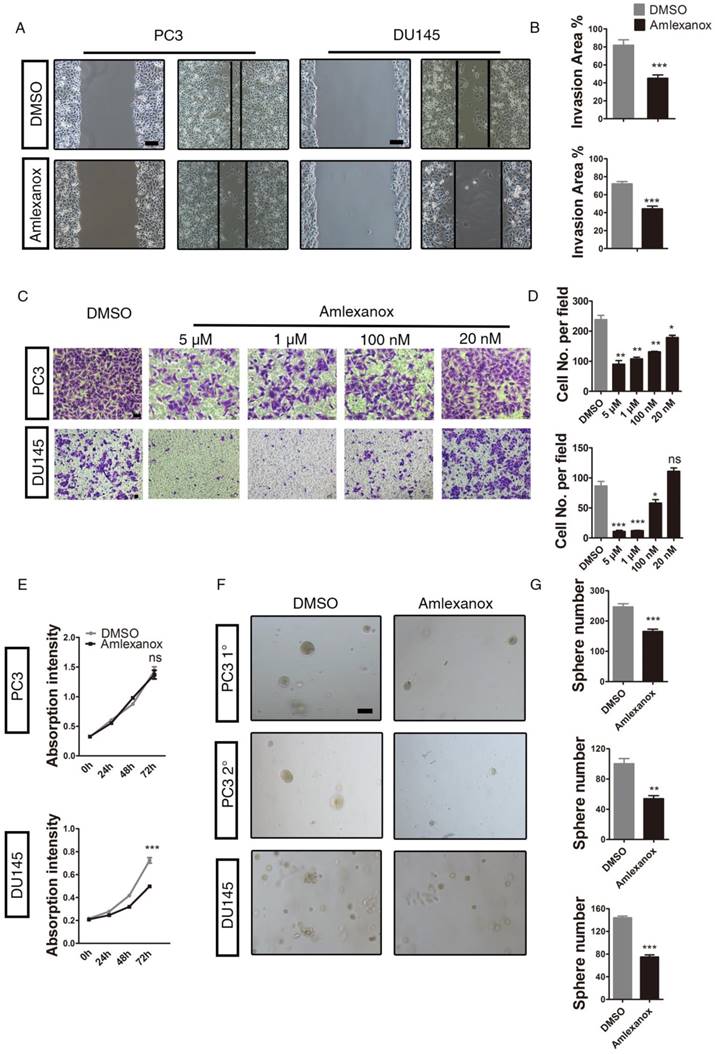
It has been demonstrated previously by different research groups that EMT is an essential step in tumor cell invasion and migration [34]. We next determined whether Amlexanox would exhibit an inhibitory role on cancer cell mobility and migration. As shown in Figure 2A-B, Amlexanox treatment led to a marked reduction in mobility of PC3 and DU145 cells, as characterized by a wound-healing assay. Transwell migration experiments further indicated an inhibitory effect of Amlexanox on PC3 and DU145 cell migration in a dose-dependent manner (Figure 2C-D). We further assessed the impact of Amlexanox treatment on cell proliferation utilizing the cell counting kit-8 assay. As shown in Figure 2E, Amlexanox did not affect the doubling time of PC3 cells, but exhibited a moderate but significant inhibition on DU145 cell proliferation. EMT was reported to associate with acquisition of tumor-initiating cell (TIC) or CSC characteristics [4, 5, 35]. Thus, reversal of EMT may compromise the TIC traits of cancer cells. We found that treatment of PC3-clone 4 with Amlexanox resulted in a striking loss of sphere-forming capacity, as determined by a serial sphere assay, a frequently used method to evaluate TIC capacity in vitro. Similar effects were observed in DU145 and VCaP cells (Figure 2F-G and Figure S3A-C).
Amlexanox suppresses mobility and migration of PCa cells in vitro.(A-B) Amlexanox represses the mobility of PC3 and DU145 cells, as characterized by a wound healing assay (n=18). Scale bar = 100 μm. (C-D) Amlexanox treatment causes dose-dependent suppression of PC3 and DU145 cell transwell migration in the Boyden chamber assay (n=3). Scale bar = 50 μm. (E) Moderate effect of Amlexanox on PC3 and DU145 cell proliferation via cell counting kit-8 (n=6). (F-G) The serial sphere generation capacities of PC3 and the sphere formation ability of DU145 cells were markedly inhibited by Amlexanox treatment (n=3). Scale bar = 100 μm. Unpaired t-test was used for the statistical analysis. *, P<0.05; **, P<0.01; ***, P<0.001. Data are presented as mean ± SEM.
Amlexanox significantly inhibits the metastasis and tumor-initiating capacity of PCa cells in vivo
To establish a metastatic xenograft tumor model, PC3 cells were labeled by infection with a lentivirus constitutively expressing a firefly luciferase gene and a tomato-Red gene. Stable transfected cells were intracardially injected into nude mice and subjected to serial in vivo selection of a metastatic subpopulation. PC3 cells isolated from the bone marrow of recipient mice by FACS sorting were termed PC3-M. We then tested whether the reversion of EMT by Amlexanox represses tumor cell metastasis in vivo. The PC3-M cells were first treated with Amlexanox at 5 μM or vehicle for 7 days to inhibit EMT and induce a more epithelial state.
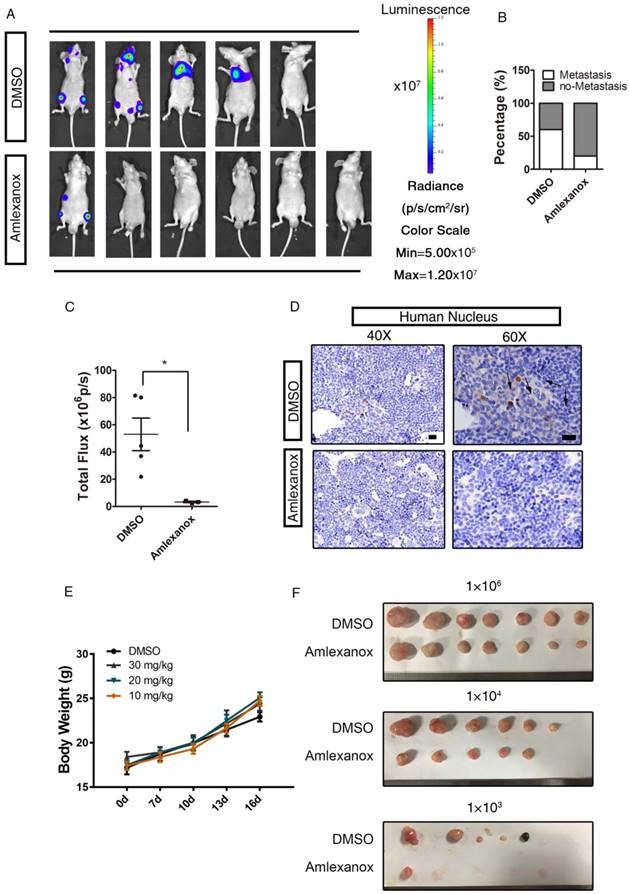
Nude mice transplanted with pretreated PC3-M cells continued to receive 30 mg/kg Amlexanox or vehicle treatment via intraperitoneal injection twice a week for 2 weeks. As indicated in Figure 3A-B, the incidence of metastatic tumor formation in Amlexanox-treated mice (1/6) was far lower than in the control group (4/5). Quantitative analysis of in vivo bioluminescence imaging data also showed a marked reduction of metastatic activity in the Amlexanox-treated group (Figure 3C). Immunochemical staining of the human nucleus further substantiated that metastasis of PC3 cells could be readily detected in femurs with specific human nucleus staining from vehicle-treated mice but not in xenograft recipients receiving Amlexanox (Figure 3D). Of note, this anti-metastatic effect of Amlexanox is not due to toxicity, as we did not observe body weight loss or major organ weight change in mice treated with 30 mg/kg Amlexanox (Figure 3E and Table 1). We next asked whether Amlexanox may exert a similar inhibitory impact on PC3-M cell metastasis in vivo without pretreatment in vitro. For that purpose, untreated PC3-M cells were implanted into nude mice via intracardiac injection. Amlexanox was given to recipient mice at 30 mg/kg via i.p. injection twice a week for 2 weeks starting from one week after the inoculation. As shown in Figure S4B-C, the incidence of tumor metastasis development was lower in mice that received Amlexanox (1/5) compared to the control group (3/5).
Amlexanox represses the metastasis of PCa cells in vivo. (A-C) Pretreatment with Amlexanox significantly suppresses the metastases formation of PC3-m cells (Amlexanox group: n=6; DMSO group: n=5). Quantitative analysis of the total photon flux is shown in Figure 3C (mean of each metastasis). (D) Immunochemical staining of human nuclei in sections of mouse femurs. Human nuclei were stained by the anti-nuclei antibody (Merck MAB1281, clone 235-1) that only recognizes the human-specific nucleus to identify the human prostate cancer cells in the mouse xenograft models. Positively stained cells are indicated by arrowheads. Scale bar = 20 μm. (E) Amlexanox does not exert toxicity when administered systemically to mice even at a high dosage of 30 mg/kg. Different doses of Amlexanox were injected intra-peritoneally to nude mice twice a week for 2 weeks to see toxicity. Alterations in body weight was not detected upon Amlexanox treatment. (F) Amlexanox treatment resulted in a decrease of the in vivo tumor-initiating ability of PC3 cells by the limited dilution assay. Unpaired t-test was used for the statistical analysis. *, P<0.05. Data are presented as mean ± SEM.
Tissue weight of major organs upon Amlexanox treatment.
| Tissue Weight (g) | Control | Amlexanox (30 mg/kg) | P-Value |
|---|---|---|---|
| Liver | 1.6510 ± 0.0649 | 1.5990 ± 0.0915 | 0.6630 |
| Lung | 0.1563 ± 0.0099 | 0.1850 ± 0.0053 | 0.0634 |
| Heart | 0.1695 ± 0.0049 | 0.1732 ± 0.0285 | 0.9045 |
| Spleen | 0.1890 ± 0.0487 | 0.1315 ± 0.0193 | 0.3343 |
| Kidney | 0.5067 ± 0.0472 | 0.4608 ± 0.0064 | 0.3903 |
| Testis | 0.1801 ± 0.0136 | 0.1784 ± 0.0025 | 0.9064 |
| Prostate & Seminal Vesical | 0.1875 ± 0.0705 | 0.1173 ± 0.0215 | 0.3946 |
The intracardiac injection of PC3-M cells allowed efficient and quick generation of tumor metastasis in vivo. However, in this model, two major steps of metastasis, which include invasion of the cancer cells into the stroma and penetration into circulation, were skipped. We therefore determined to assess the effect of Amlexanox on tumor metastasis utilizing an orthotopic xenograft model. Tomato-Red reporter-expressing PC3-M cells were injected into the left anterior lobe of nude mouse prostates. Amlexanox or vehicle treatment was given to recipients twice a week for 3 weeks from one week after the inoculation. Examination of the tomato-Red signal from the right anterior lobe of prostates enabled detection of metastatic PCa cells. As shown in Figure S4D, the mice receiving Amlexanox treatment displayed a lower frequency in metastasis formation (1/5) compared to the control groups (4/5). Taken together, these data suggested that Amlexanox achieved great anti-metastatic efficacy in PCa xenograft mouse models.
To further test the postulation that Amlexanox may negatively regulate the tumor-initiating capacity of PCa cells in vivo, we utilized a limiting dilution xenograft assay in which 1×103, 1×104 or 1×106 PC3 cells were implanted subcutaneously into nude mice. As shown in Figure 3F and Table 2, Amlexanox treatment led to a significant suppression on tumor formation incidence with the most striking effect observed in the 1000 cell xenograft group. Considering that we did not detect a significant effect of Amlexanox on PC3 cell proliferation in vitro, we proposed that the different tumor-forming incidence and tumor size of vehicle- and Amlexanox-treated groups were caused by suppression of tumor-initiating cells upon Amlexanox treatment. However, we could not exclude the possibility that Amlexanox may affect tumor microenvironment and inhibit PC3 cell growth in vivo indirectly.
Tumor initiating frequency in the limited dilution assay.
| Cells transplanted | Treatment | Tumors |
|---|---|---|
| 1×106 | Vehicle i.p. | 7/7 |
| Amlexanox i.p. | 7/7 | |
| 1×104 | Vehicle i.p. | 6/7 |
| Amlexanox i.p. | 5/7 | |
| 1×103 | Vehicle i.p. | 5/7 |
| Amlexanox i.p. | 1/7 |
The anti-metastasis effect of Amlexanox is dependent on the IKKɛ/TBK1/ NF-κB signaling axis
To uncover the mechanism underlying the inhibitory effect of Amlexanox on metastasis, we performed RNA sequencing to compare the transcriptional difference between Amlexanox- and vehicle-treated PCa cells. Consistent with our observations, gene set enrichment analysis (GSEA) of the RNA-seq data indicated a suppression of CDH1 targets and metastasis-related gene expression upon Amlexanox treatment (Figure 4A). Amlexanox was previously reported to act as a specific inhibitor of IKKɛ and TBK1, two non-canonical IKKs in the NF-κB pathway, by competitively binding to their ATP binding domain [36]. Meta-analysis of sequencing or microarray data sets from the cBioportal and Oncomine databases showed amplification and transcriptional upregulation of IKBKE and TBK1 in human prostate cancer samples (Figure S5A-B). In addition, components of NF-κB pathway including NFKB1, NFKB2, Rel A and Rel B were found to be significantly co-expressed with IKBKE and TBK1 in PCa patient samples (Figure S5C). Kyoto Encyclopedia of Genes and Genomes (KEGG) and GSEA analysis of our RNA-seq data revealed that the NF-κB pathway was the most significantly affected signaling pathway by Amlexanox treatment (Figure 4B-C). We therefore hypothesized that Amlexanox may reverse EMT through inhibition of the IKKɛ/TBK1/NF-κB signaling axis.
The anti-metastasis effect of Amlexanox acts through the IKKɛ/TBK1/NF-κB signaling axis. (A-B) GSEA analysis of RNA-seq data indicates downregulation of metastasis-associated signature genes and increased expression of genes in the NF-κB signaling pathway in Amlexanox-treated PC3 cells. (C) KEGG analysis of RNA-seq data from PC3 cells treated with Amlexanox or vehicle. (D) PC3-clone 4 displays a higher activation of IKKɛ/TBK1/NF-κB pathway. (E) Amlexanox treatment inhibits activation of the NF-κB pathway and induces MET.
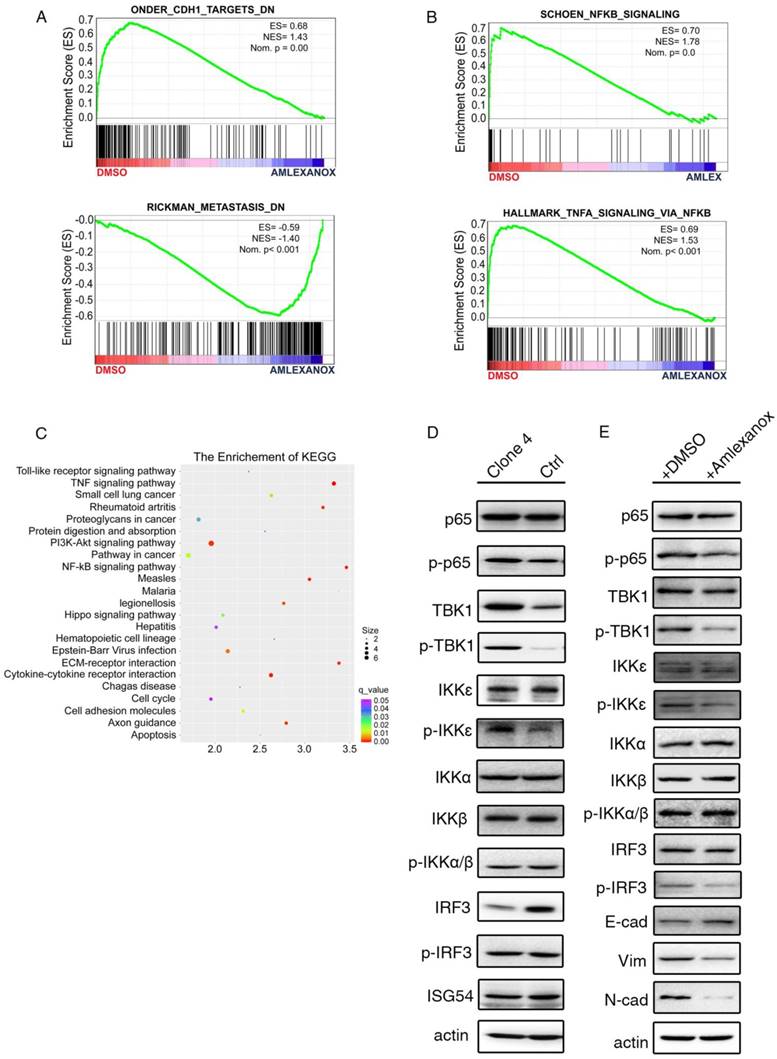
We first assessed the expression and activation of components of the IKKɛ/TBK1/NF-κB signaling pathway and other IKKs in the mesenchymal-like PC3-clone 4 and its parental cells. Interestingly, we found that TBK1 and the phosphorylated levels of IKKɛ and TBK1 were notably higher in PC3-clone 4 compared to its parental cell line (Figure 4D). Two previously reported targets of IKKɛ/TBK1 activation [37], phosphorylated-IRF3 and ISG54, were comparable between PC3-clone 4 cell line and its parental cell line, while the total IRF3 level was downregulated in PC3-clone 4 cells (Figure 4D). Instead, we detected a marked increase of phosphorylated-NF-κB p65 subunit, which can be activated downstream of IKKɛ and TBK1, in PC3-clone 4 cells (Figure 4D). IKKα and IKKβ, two canonical regulators of NF-κB signaling pathway, however, were expressed and phosphorylated at similar levels between PC3-clone 4 cell line and its parental cell line, suggesting the upregulation of NF-κB activation was caused by IKKɛ/TBK1 but not IKKα and IKKβ (Figure 4D). Taken together, these data indicated a constitutive upregulation of IKKɛ/TBK1/NF-κB signaling in the PCa cells that underwent EMT.
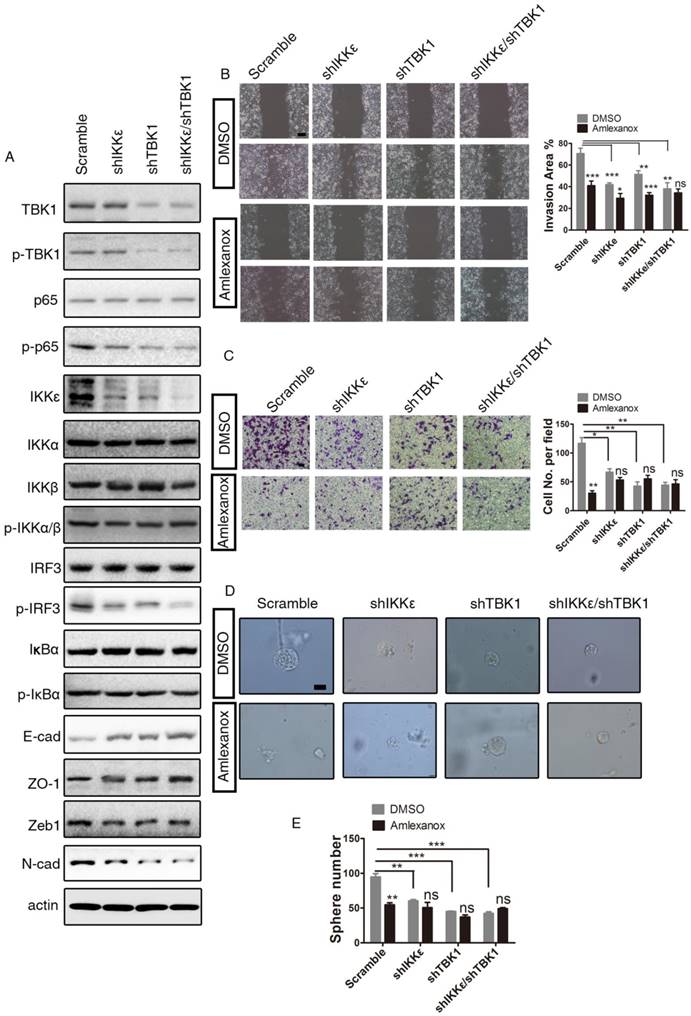
We then investigated the impact of Amlexanox treatment on IKKɛ/TBK1/NF-κB signaling. As shown in (Figure 4E), Amlexanox downregulated the phosphorylation levels of IRF and p65, two common IKKɛ/TBK1 targets in PC3-clone 4 cells. In contrast, the phosphorylation of IKKα and IKKβ were not affected. To determine whether inhibition of IKKɛ/TBK1 was responsible for the metastasis-suppressing effect of Amlexanox, we tested the impact of IKKɛ and/or TBK1 knockdown on EMT and cell migration. PC3-clone 4 cells stably transfected with shIKKɛ and/or shTBK1 displayed decreased p65 phosphorylation and expression of N-cadherin and Zeb1 and increased expression of epithelial markers E-cadherin and ZO1. A synergistic effect of the combination of shIKKe/shTBK1 was detected on N-cadherin and ZO-1 expressions, but not so obviously on E-cadherin (Figure 5A). Furthermore, similar to Amlexanox, IKKɛ and/or TBK1 knockdown or treatment with two other IKKɛ/TBK1 antagonists, In-1 or 67307, displayed strong reversion of EMT, cell mobility, transwell migration and sphere forming of PC3-clone 4 (Figure 5B-E and Figure S6). Collectively, these observations indicated that Amlexanox can inhibit IKKɛ/TBK1/NF-κB signaling pathway, which plays an essential role in promoting EMT and prostate tumor cell mobility.
IKKɛ/TBK1 axis plays a positive role in inducing EMT. (A) IKKɛ, TBK1 or IKKɛ/TBK1 knock-down leads to downregulation of the NF-κB signaling pathway, mesenchymal marker N-cadherin and EMT transcriptional factor ZEB1 and to upregulation of epithelial markers E-cadherin and ZO-1 in PC3 cells. (B) Amlexanox represses the mobility of PC3 cells characterized by the wound healing assay (n=9). Scale bar = 100 μm. (C) IKKɛ, TBK1 or IKKɛ/TBK1 knock-down suppresses PC3 cell transwell migration in the Boyden chamber assay (n=3). Scale bar = 50 μm. (D-E) Sphere generation capacities were inhibited by IKKɛ, TBK1 or IKKɛ/TBK1 knock-down (n=3). Scale bar = 50 μm. Unpaired t-test was used for the statistical analysis. *, P<0.05; **, P<0.01; ***, P<0.001. Data are presented as mean ± SEM.
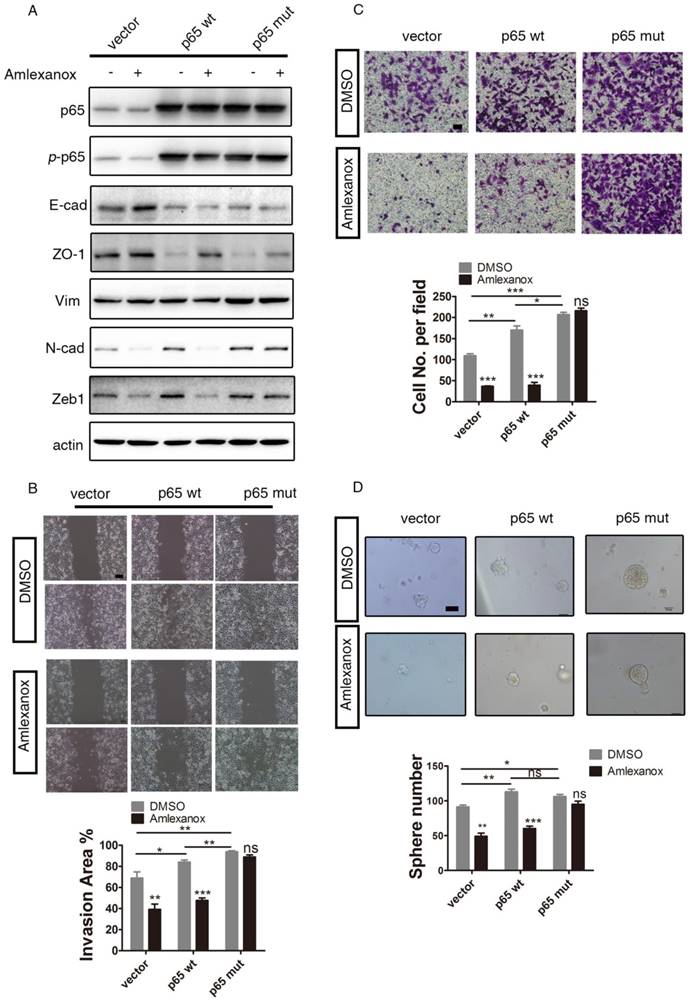
We next performed additional rescue experiments. As shown in Figure 6A, introducing ectopic expression of a dominant active form of p65 (phosphorylation mimic mutation at Ser536 of p65 [38]) abrogated the EMT-reversing effects of Amlexanox. The inhibition of cell migration, wound healing and sphere formation by Amlexanox could also be overridden by p65S536D expression (Figure 6B-D). These data further substantiated our conclusion that Amlexanox reversed EMT by targeting the NF-κB signaling axis.
The dominant active form of p65 abrogates the reversion of EMT and inhibition of cell mobility of PCa cells by Amlexanox. (A) Ectopic expression of dominant active p65 on PC3 cells abrogates the EMT-suppressing effects of Amlexanox. (B) p65S536D expression overrides the inhibitory impact of Amlexanox on PCa cell migration in the Boyden chamber assay (n=3). (C) p65S536D expression abrogates the suppression of Amlexanox on PCa cell mobility by the wound healing assay (n=9). Scale bar = 100 μm. (D) The inhibitory effect of Amlexanox on the sphere forming ability of PCa cells is overridden by the ectopic expression of dominant active p65 (n=3). Scale bar = 50 μm. Unpaired t-test was used for the statistical analysis. *, P<0.05; **, P<0.01; ***, P<0.001. Data are presented as mean ± SEM.
Discussion
In the current study, we designed and established a luciferase reporter system utilizing the cis-regulatory elements of CDH1 and VIM genes to drive the expression of firefly or renilla luciferase reporter to successfully monitor the epithelial or mesenchymal status of PCa cells. Based on this system, high-throughput screening from an approved small-molecule drug library lead to the discovery of Amlexanox as a strong agent to reverse EMT. We find that Amlexanox exerts a profound inhibitory effect on serial sphere formation, invasion and migration of PCa cells. Systemic administration of Amlexanox markedly suppressed the metastasis and tumor initiating capacity of PCa in vivo. Mechanistically, we further demonstrate that the EMT-reversing and tumor metastasis-suppressing effect of Amlexanox relies on inhibition of the IKKɛ/TBK1/NF-κB signaling axis.
Due to the significance of EMT in tumor metastasis and cancer stem cells, other research groups have also made efforts in search of EMT inhibitors or MET inducers. However, small-molecule drug candidates with favorable in vivo tolerance and good pharmacodynamics are still lacking. Dr. Robert A. Weinberg's group recently used a luciferase-based system to screen possible small molecules to induce endogenous expression of CDH1 from a 400 compound library. They found two candidate compounds, forskolin and cholera toxin, that acted to increase the levels of adenosine 3′,5′-monophosphate (cAMP) and subsequently to activate protein kinase A (PKA), and can induce MET in mesenchymal-like mammary epithelial cells [39]. Despite the good in vitro effects of forskolin and cholera toxin on suppression of mesenchymal characteristics, cell mobility and on regain of epithelial cell traits, in vivo usage of those compounds was infeasible because of their toxicity or poor pharmacodynamics. On the other hand, in medicinal chemistry, repositioning of current clinical drugs or those chemical compounds that are used in clinical trials may serve as an attractive approach for anti-EMT drug discovery owing to their good in vivo safety and known pharmacokinetics and pharmacodynamics [40].
In the present study, we screened an existing drug library and identified Amlexanox, which is used for recurrent aphthous ulcers treatment, as a potent agent to reverse EMT in PCa cells. Amlexanox treatment achieves great anti-metastasis and anti-tumor initiating effect without obvious toxicity in mouse xenograft models. These results suggest a promising repositioning of Amlexanox for the treatment of metastatic PCa. However, although work in our lab and other groups demonstrated that EMT promotes tumor-initiating cell activities, tumor metastasis and development of castration resistance [10, 41-43], other studies also suggested that EMT may not be required for tumor metastasis but is essential for tumor chemoresistance in lung and pancreatic cancers [44, 45]. Therefore, the potential clinical application of Amlexanox on tumor metastasis in broader tumor types in addition to prostate cancer needs to be further evaluated.
We demonstrated via biochemical analysis that IKKε/TBK1/NF-κB signaling is upregulated in PC3 cells that have undergone EMT. Constitutive activation of NF-κB signaling was reported to be required for EMT and metastatogenesis [46-48]. We show here that the EMT reversion and anti-metastasis effects of Amlexanox are exerted through targeting IKKε/TBK1, which subsequently leads to repression of NF-κB signaling. Interestingly, IKKε has been identified as an oncogene in breast cancer [24]. Gene amplification or overexpression of IKKε and deregulation of downstream NF-κB has been detected in human breast cancers. IKKε knockdown resulted in severe suppression of breast cancer cell survival and proliferation. Consistently, we observe a moderate but significant increase of DU145 cell doubling time upon treatment with Amlexanox. In addition, the positive role of NF-κB in tumor-related inflammation has been shown [12, 17]. Both tumor-promoting and suppressive roles of inflammation were reported in various tumor types [49-51]. The impact of Amlexanox on inflammatory tumor microenvironment and on tumor progression in immunocompetent mouse models awaits further investigation.
In conclusion, we demonstrate in this study that the TBK1/IKKε inhibitor Amlexanox can be used as a potent agent to reverse EMT. The reversion of mesenchymal-like PCa cells back to the epithelial state by Amlexanox is accompanied by a loss of in vivo metastatic and tumor-initiating ability. These results highlight the great potential of Amlexanox as an anti-metastatic agent or in combinational therapies with other interventional approaches for the treatment of advanced PCa.
Methods
Cell culture and treatment
PC3 and DU145 cell lines were purchased from ATCC. VCaP cells were kindly provided as a gift by Dr. Dong Gao at Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. The cell lines have been recently authenticated by short Tandem Repeat (STR) profiling at the Shanghai Biowing Applied Biotechnology Company. Both cell lines were cultured in Dulbecco's modified Eagle's medium (Gibco) containing 10% fetal bovine serum and 80 U/mL penicillin and streptomycin. TGFβ (PeproTech, 100-21c) was used to induce EMT at 100 ng/mL. In the drug screening or confirmation experiments, cells were treated with indicated concentrations of Amlexanox (TargetMol, T1639), DMSO or IKKɛ/TBK1 inhibitors MRT67307 (termed 67307, Medchemexpress, HY-13018) or IKK-IN-1 (termed In-1, Medchemexpress, HY-13873) for 7 days. Drug-containing medium was refreshed every 3 days.
Construction of lentiviral plasmids
For the construction of the dual-luciferase reporter lentiviral plasmids, we first synthesized a scaffold DNA fragment that contained elements of XhoI-HindIII-XbaI-SV40 late polyA-Spacer-ClaI-NheI-AvrII-SV40 late polyA-XhoI via chemosynthesis. The scaffold DNA fragment was then cloned into the pBlunt-Zero Vector (Transgene) according to the manufacturer's instruction to generate a vector plasmid. The coding sequences of luciferase and renilla, flanked with restriction sites HindIII/XbaI and NheI/AvrII respectively, were obtained by PCR amplification using the high-fidelity DNA polymerase Q5TM (NEB). Vectors pGL3-basic and pRL-CMV were used as templates to clone Luciferase and Renilla. The human VIM promoter and CDH1 promoter were amplified by genomic PCR and cloned into the pBlunt-Zero vector flanked with restriction sites of ClaI/NheI and XhoI/HindIII respectively. The dual-fluorescence reporter lentiviral plasmid was re-constructed from the dual-luciferase reporter lentiviral plasmid by replacing the luciferase and renilla with mCherry- and eGFP-coding sequences respectively.
The sequences of IKKɛ and TBK1 shRNA (IKKɛ: GAGCTATCTCACCAGCTCC; TBK1: GACAGAAGTTGTGATCACA) were cloned to the pSUPER vector, which was a gift from Dr. Tom Maniatis (# 26210, Addgene plasmid) to knock down the expression of IKKɛ or TBK1 or IKKɛ/TBK1. P65 wt and p65S536D were constructed following previously reported methods [38].
Validation of the EMT-indicator system
To evaluate whether the EMT fluorescent or luciferase reporter can reflect the endogenous E-cadherin or vimentin gene expression level, PC3 cells were infected with the EMT fluorescent or luciferase reporter lentiviruses and were subjected to selection for a stably transfected cell line. mCherry+ or GFP+ cells from PC3 transfected with the EMT fluorescent reporter virus were FACS sorted for RNA extraction. Real-time quantitative PCR was used to evaluate the correlation of E-cadherin or vimentin expression levels with mCherry or GFP levels in sorted cells. To validate the EMT luciferase reporter, TGFβ (PeproTech, 100-21c) was used to induce EMT of the EMT luciferase reporter virus-transfected PC3 cells at 100 ng/mL for 7 days. Cells were lysed and subjected to luciferase activity measurement according to the manufacturer's instruction (Dual-Glo, Promega, E2920). Ratiometric measurement was calculated by dividing the intensity of firefly luciferase by the intensity of renilla luciferase.
Drug screening
For the high-throughput drug screening, 1000 PC3-clone 4 cells were seeded into 96-well plates. Twenty-four hours later, cells were treated with individual compounds at a concentration of 10 μM. Drug-containing medium was refreshed every 3 days. Seven days later, cell number in each well was quantified by the cell proliferation assay as described in detail below. Cell proliferation assay was carried out using the cell counting kit-8 according to the manufacturer's instruction (Dojindo, CK04). The plates were then washed with phosphate buffered saline (PBS) twice, and subsequently subjected to the luciferase activity test following the manufacturer's instructions (ONE-Glo, Promega, E6110). The approved drug library (L1000) was purchased from TargetMol. All cell proliferation and luciferase assays were performed in triplicate. Luciferase intensity was measured using the GloMax Discover System (Promega).
Immunoblotting and immunofluorescent staining
Immunoblotting and immunofluorescent staining were performed using conventional methods as previously reported [52]. The primary and secondary antibodies used in the study are list in Table S1.
Wound healing assay and Transwell migration experiment
For the wound healing assay, cells (pretreated with Amlexanox or DMSO for 7 days) were seeded into 6-well culture plates (Corning). When the cells reached confluence, the culture medium was replaced with DMEM medium without serum to minimize cell proliferation. A pipette tip was used to make a straight scratch. The cell scratch was examined and photographed under a light microscope at 0 h, 12 h and 24 h. Cell-free area were quantified by ImageJ software. For the transwell migration experiment, cells (pretreated with Amlexanox or DMSO for 7 days) were seeded into the upper chamber of 24-well transwell plates (6.5 mm insert, 8.0 μm pores, costar 3422) with 100 μL serum-free medium. 500 μL 10% FBS-supplemented medium was added to the lower chamber. Amlexanox or DMSO were added to the culture medium at indicated concentrations. Cells were removed from the upper surface of the chamber using a cotton swab after 24 to 48 h. The migrated cells on the chamber bottom were fixed with 4% PFA and stained with crystal violent for visualization. Three fields per chamber were photographed for migrated cell quantification.
Sphere formation assay
Single PCa cells were resuspended in sphere culture medium (DMEM/F12 medium supplemented with 2% B27 (Gibco, 17504044), 1% N2 (Gibco, 17502001), 20 ng/mL fibroblast growth factor (PeproTech, 100-18B) and 20 ng/mL epidermal growth factor (PeproTech, AF-100-15-100), mixed at a 1:1 ratio with Matrigel (BD, 356234), and then seeded in 24-well culture dishes (Costar) at 1000 cells/well in a volume of 200 μL. Spheres formed were examined and photographed under a light microscope after 10 days. For serial sphere formation assay, spheres were harvested by centrifugation and digested with TrypLE for 5 min to obtain single cell suspensions. Secondary spheres were generated as described above.
Animal studies
Animal experiments were conducted following protocols approved by Ren Ji Hospital's committee on animal care. For the limited dilution assay, PCa cells were implanted subcutaneously at indicated cell numbers. Amlexanox (30 mg/kg) or vehicle was administered by intraperitoneal injection twice a week starting from 1 week after implantation. Tumors were harvested and imaged one month later. For intracardiac injection of PCa cells, cells were washed twice with DMEM, re-suspended in 100 μL PBS at 1×107/mL and injected into the left ventricle of 8-week-old male nude mice (Shanghai SLAC Laboratory Animal) anesthetized by tribromoethyl alcohol (Sigma, T48402). In orthotopic tumor xenograft models, 1×106 cells were suspended in 20 μL 50% Matrigel and injected into the left anterior lobe of anesthetized 8-week-old male nude mice. Mice received Amlexanox (30 mg/kg) or vehicle by intraperitoneal injection twice a week starting from 1 week after tumor implantation. Three weeks later, the mice were administrated 150 mg/kg D-luciferin (ThermoFisher, L2916) via intraperitoneal injection, then anesthetized and imaged using a Caliper IVIS bioluminescence system (Caliper LifeScience. USA).
RNA sequencing and analysis
Total RNA were extracted from PC3 cells treated with Amlexanox or vehicle using the Quick RNA MicroPrep kit (Zymo research, USA). Libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, USA) according to the manufacturer's instructions. The sequencing of libraries was performed using the Illumina HiSeq platform to generate 150 bp paired-end reads. After removal of adaptor sequences and poor quality sequences, clean reads were analyzed utilizing the TopHat-Cufflinks-Cuffmerge-Cuffdiff pipeline with default parameters. TopHat v2.0.13 was used to map the reads to the homo sapiens GRCh38 reference genome. Significance of differential gene expressions were defined by fold change between two groups of more than 1 and p-values less than 0.05. We further used the GO functional enrichment and KEGG analysis to annotate data with the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 (https://david.ncifcrf.gov/). Significance was calculated using a modified Fisher's exact test. The RNA-seq dataset was deposited to the GEO database with the accession number GSE110206.
Statistical analysis
The statistical analyses in this manuscript were performed between two groups of independently selected and identically distributed samples; therefore, we used unpaired Student's t-test and considered p-values <0.05 as statistically significant. All statistical analyses were performed using the Graphpad Prism 5 software. Data are presented as mean ± SEM (n≥3)
Data and materials availability
The RNA-seq dataset can be found in the GEO database with the accession number GSE110206.
Abbreviations
cAMP: adenosine 3',5'-monophosphate; CSC: cancer stem cell; DAVID: database for annotation, visualization and integrated discovery; EMT: epithelial-mesenchymal transition; GEO: gene expression omnibus; GSEA: gene set enrichment analysis; In-1: IKK-IN-1; IKK: IκB kinase; IKKɛ: IκB kinase ɛ; IκB: inhibitor of NF-κB; KEGG: kyoto encyclopedia of genes and genomes; MET: mesenchymal-epithelial transition; PBS: phosphate buffered saline; PCa: prostate cancer; PKA: protein kinase A; STR: short tandem repeat; TANK: TRAF family member-associated NF-kappa-B activator; TBK1: TANK-binding kinase I; TIC: tumor-initiating cell; TRAF: tumor necrosis factor receptor-associated factor.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The study is supported by funds to W-Q Gao from the Chinese Ministry of Science and Technology (2017YFA0102900), the National Natural Science Foundation of China (NSFC, 81372189 and 81630073), the Science and Technology Commission of Shanghai Municipality (16JC1405700), Shanghai Eastern Hospital (Pudong) Stem Cell Research Base Fund, and the KC Wong foundation, and by funds to H.H. Zhu from the NSFC (81772743), Shanghai Rising-Star Program (17QA1402100), the Shanghai Youth Talent Support Program, Shanghai Institutions of Higher Learning (The Program for Professor of Special Appointment (Young Eastern Scholar)), School of Medicine, Shanghai Jiao Tong University (Excellent Youth Scholar Initiation Grant 16XJ11003), and Ren Ji Hospital (Seed Project RJZZ14-010).
Author contributions
Experiments were designed by HHZ and WQG; CC performed drug screening, in vitro and in vivo functional studies; ZJ conducted the plasmid construction, immunoblotting and RNA-seq analysis; HHZ, WQG and CC analyzed the data and wrote the manuscript; YS, XW, HZ, YH and LW provided support for high-throughput drug screening and in vitro experiments; JW, YS, JY, XL, YG and LZ assisted in animal experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Irshad S, Abate-Shen C. Modeling prostate cancer in mice: something old, something new, something premalignant, something metastatic. Cancer Metastasis Rev. 2013;32:109-22
3. Suva LJ, Washam C, Nicholas RW, Griffin RJ. Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol. 2011;7:208-18
4. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21-45
5. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611-29
6. Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H. et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017;19:518-29
7. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A. et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487-95
8. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F. et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154:61-74
9. Ni T, Li XY, Lu N, An T, Liu ZP, Fu R. et al. Snail1-dependent p53 repression regulates expansion and activity of tumour-initiating cells in breast cancer. Nat Cell Biol. 2016;18:1221-32
10. Li P, Wang J, Chu M, Zhang K, Yang R, Gao WQ. Zeb1 promotes androgen independence of prostate cancer via induction of stem cell-like properties. Exp Biol Med. 2014;239:813-22
11. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705-16
12. Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301-10
13. Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H. et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569-81
14. Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336:25-37
15. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651
16. Hayden MS, Ghosh S. NF- B, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203-34
17. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-44
18. Bates RC, Mercurio AM. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell. 2003;14:1790-800
19. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL. et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72:1290-300
20. Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL. et al. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature. 2007;446:690-4
21. Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999;18:6694-704
22. Tojima Y, Fujimoto A, Delhase M, Chen Y, Hatakeyama S, Nakayama K. et al. NAK is an IkappaB kinase-activating kinase. Nature. 2000;404:778-82
23. Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30:631-41
24. Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF. et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065-79
25. Hildebrandt MA, Tan W, Tamboli P, Huang M, Ye Y, Lin J. et al. Kinome expression profiling identifies IKBKE as a predictor of overall survival in clear cell renal cell carcinoma patients. Carcinogenesis. 2012;33:799-803
26. Colas E, Perez C, Cabrera S, Pedrola N, Monge M, Castellvi J. et al. Molecular markers of endometrial carcinoma detected in uterine aspirates. Int J Cancer. 2011;129:2435-44
27. Guo JP, Shu SK, He L, Lee YC, Kruk PA, Grenman S. et al. Deregulation of IKBKE is associated with tumor progression, poor prognosis, and cisplatin resistance in ovarian cancer. Am J Pathol. 2009;175:324-33
28. Lu J, Yang Y, Guo G, Liu Y, Zhang Z, Dong S. et al. IKBKE regulates cell proliferation and epithelial-mesenchymal transition of human malignant glioma via the Hippo pathway. Oncotarget. 2017;8:49502-14
29. Peant B, Gilbert S, Le Page C, Poisson A, L'Ecuyer E, Boudhraa Z. et al. IkappaB-Kinase-epsilon (IKKepsilon) over-expression promotes the growth of prostate cancer through the C/EBP-beta dependent activation of IL-6 gene expression. Oncotarget. 2017;8:14487-501
30. Geng B, Zhang C, Wang C, Che Y, Mu X, Pan J. et al. IkappaB-kinase-epsilon in the tumor microenvironment is essential for the progression of gastric cancer. Oncotarget. 2017;8:75298-307
31. Hsu S, Kim M, Hernandez L, Grajales V, Noonan A, Anver M. et al. IKK-epsilon coordinates invasion and metastasis of ovarian cancer. Cancer Res. 2012;72:5494-504
32. Li LC, Chui RM, Sasaki M, Nakajima K, Perinchery G, Au HC. et al. A single nucleotide polymorphism in the E-cadherin gene promoter alters transcriptional activities. Cancer Res. 2000;60:873-6
33. Rittling SR, Baserga R. Functional analysis and growth factor regulation of the human vimentin promoter. Mol Cell Biol. 1987;7:3908-15
34. Li P, Yang R, Gao WQ. Contributions of epithelial-mesenchymal transition and cancer stem cells to the development of castration resistance of prostate cancer. Mol Cancer. 2014;13:55
35. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124-34
36. Reilly SM, Chiang S-H, Decker SJ, Chang L, Uhm M, Larsen MJ. et al. An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19:313-21
37. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT. et al. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491-6
38. Adli M, Baldwin AS. IKK-i/IKKϵ Controls Constitutive, Cancer Cell-associated NF-κB Activity via Regulation of Ser-536 p65/RelA Phosphorylation. J Biol Chem. 2006;281:26976-84
39. Pattabiraman DR, Bierie B, Kober KI, Thiru P, Krall JA, Zill C. et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science. 2016;351:aad3680
40. Jin G, Wong ST. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov Today. 2014;19:637-44
41. Zhang K, Zhao H, Ji Z, Zhang C, Zhou P, Wang L. et al. Shp2 promotes metastasis of prostate cancer by attenuating the PAR3/PAR6/aPKC polarity protein complex and enhancing epithelial-to-mesenchymal transition. Oncogene. 2016;35:1271-82
42. Park JJ, Park MH, Oh EH, Soung NK, Lee SJ, Jung JK. et al. The p21-activated kinase 4-Slug transcription factor axis promotes epithelial-mesenchymal transition and worsens prognosis in prostate cancer. Oncogene. 2018 doi.org/10.1038/s41388-018-0327-8
43. Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol. 2011;8:428-39
44. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
45. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
46. Ren D, Yang Q, Dai Y, Guo W, Du H, Song L. et al. Oncogenic miR-210-3p promotes prostate cancer cell EMT and bone metastasis via NF-kappaB signaling pathway. Mol Cancer. 2017;16:117
47. Verzella D, Fischietti M, Capece D, Vecchiotti D, Del Vecchio F, Cicciarelli G. et al. Targeting the NF-kappaB pathway in prostate cancer: a promising therapeutic approach? Curr Drug Targets. 2016;17:311-20
48. McCall P, Bennett L, Ahmad I, Mackenzie LM, Forbes IW, Leung HY. et al. NFkappaB signalling is upregulated in a subset of castrate-resistant prostate cancer patients and correlates with disease progression. Br J Cancer. 2012;107:1554-63
49. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y. et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. 2016;7:13720
50. Gunn L, Ding C, Liu M, Ma Y, Qi C, Cai Y. et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. 2012;189:2985-94
51. Jing H, Kase J, Dorr JR, Milanovic M, Lenze D, Grau M. et al. Opposing roles of NF-kappaB in anti-cancer treatment outcome unveiled by cross-species investigations. Genes Dev. 2011;25:2137-46
52. Guo Y, Zhang K, Cheng C, Ji Z, Wang X, Wang M. et al. Numb(-/low) Enriches a Castration-Resistant Prostate Cancer Cell Subpopulation Associated with Enhanced Notch and Hedgehog Signaling. Clin Cancer Res. 2017;23:6744-56
Author Biography
 Prof. Wei-Qiang Gao received his PhD from Columbia University in 1989 and did his post-doctoral research at Columbia University and the Rockefeller University. From 1993-2010 he was a Scientist, Senior Scientist and Group/Project Leader at Genentech, Inc. He moved to China in 2010 and started to work as a Professor at Shanghai Jiao Tong University. He is now serving as Dean of the School of Biomedical Engineering, Director of State Key Laboratory of Oncogenes and Related Genes, and Director of Renji Hospital Stem Cell Research Center in Shanghai Jiao Tong University. Dr. Gao has made important contributions to the fields of neuroscience, stem cells and tumorigenesis. He has published more than 80 papers as either corresponding or first author, including in Nature, Cell, Science, Neuron, Nature Neuroscience, Nature Communications, Gastroenterology, PNAS, etc. and has been granted 48 US patents. He is a scholar of the national “Thousand-Talents Program”, the Chief Scientist of 2 program projects from the Ministry of Science and Technology of China and 2 key grants from the National Natural Science Foundation of China. He has served as a reviewer for grant proposals of Wellcome Trust in UK, NIH in US, and NSFC and 36 journals including Nature, Nature Medicine, Nature Cell Biology, Nature Reviews Oncology, Nature Communications, Cancer Cells, Cell Reports, PNAS, etc. His research interests are in cancer research and cancer stem cells, stem cells and tissue repair/regeneration, hearing loss and inner ear hair cell regeneration.
Prof. Wei-Qiang Gao received his PhD from Columbia University in 1989 and did his post-doctoral research at Columbia University and the Rockefeller University. From 1993-2010 he was a Scientist, Senior Scientist and Group/Project Leader at Genentech, Inc. He moved to China in 2010 and started to work as a Professor at Shanghai Jiao Tong University. He is now serving as Dean of the School of Biomedical Engineering, Director of State Key Laboratory of Oncogenes and Related Genes, and Director of Renji Hospital Stem Cell Research Center in Shanghai Jiao Tong University. Dr. Gao has made important contributions to the fields of neuroscience, stem cells and tumorigenesis. He has published more than 80 papers as either corresponding or first author, including in Nature, Cell, Science, Neuron, Nature Neuroscience, Nature Communications, Gastroenterology, PNAS, etc. and has been granted 48 US patents. He is a scholar of the national “Thousand-Talents Program”, the Chief Scientist of 2 program projects from the Ministry of Science and Technology of China and 2 key grants from the National Natural Science Foundation of China. He has served as a reviewer for grant proposals of Wellcome Trust in UK, NIH in US, and NSFC and 36 journals including Nature, Nature Medicine, Nature Cell Biology, Nature Reviews Oncology, Nature Communications, Cancer Cells, Cell Reports, PNAS, etc. His research interests are in cancer research and cancer stem cells, stem cells and tissue repair/regeneration, hearing loss and inner ear hair cell regeneration.
 Prof. Helen He Zhu received her Bachelor's Degree from Fudan University, China. She earned her PhD in Molecular Pathology at the School of Medicine, University of California-San Diego in 2011 followed by postdoctoral training in the Department of Biology, University of California-San Diego. In 2012, Dr. Zhu relocated back to China and started to work as Associate Professor then Professor of the School of Medicine, Shanghai Jiao Tong University. Her publications include first author and corresponding author papers in PNAS, Blood, Gastroenterology, Clinical Cancer Research, Cancer Research, etc. Her current work focuses on 1) development of novel anti-metastasis drugs, 2) molecular and cellular mechanisms for castration resistance in prostate cancer, 3) identification of adult prostate stem cells and prostate cancer stem cells.
Prof. Helen He Zhu received her Bachelor's Degree from Fudan University, China. She earned her PhD in Molecular Pathology at the School of Medicine, University of California-San Diego in 2011 followed by postdoctoral training in the Department of Biology, University of California-San Diego. In 2012, Dr. Zhu relocated back to China and started to work as Associate Professor then Professor of the School of Medicine, Shanghai Jiao Tong University. Her publications include first author and corresponding author papers in PNAS, Blood, Gastroenterology, Clinical Cancer Research, Cancer Research, etc. Her current work focuses on 1) development of novel anti-metastasis drugs, 2) molecular and cellular mechanisms for castration resistance in prostate cancer, 3) identification of adult prostate stem cells and prostate cancer stem cells.
![]() Corresponding authors: Wei-Qiang Gao (gao.weiqiangedu.cn) or Helen He Zhu (zhuhecraneedu.cn). Tel: 86-21-68383917, Fax: 86-21-68383916. Address: Stem Cell Research Center, Ren Ji Hospital, 160 Pujian Rd., School of Medicine, Shanghai Jiao Tong University, Shanghai, 200127, China.
Corresponding authors: Wei-Qiang Gao (gao.weiqiangedu.cn) or Helen He Zhu (zhuhecraneedu.cn). Tel: 86-21-68383917, Fax: 86-21-68383916. Address: Stem Cell Research Center, Ren Ji Hospital, 160 Pujian Rd., School of Medicine, Shanghai Jiao Tong University, Shanghai, 200127, China.