Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(17):4679-4694. doi:10.7150/thno.26643 This issue Cite
Research Paper
Dopaminergic precursors differentiated from human blood-derived induced neural stem cells improve symptoms of a mouse Parkinson's disease model
Yanpeng Yuan1,2,3, Xihe Tang1,2,3, Yun-Fei Bai4, Shuyan Wang1,2,3, Jing An1, Yanchuan Wu5, Zhi-Qing David Xu4, Y. Alex Zhang1,3 ![]() , Zhiguo Chen1,2,3
, Zhiguo Chen1,2,3 ![]()
1. Cell Therapy Center, Beijing Institute of Geriatrics, Xuanwu Hospital, Capital Medical University, and Key Laboratory of Neurodegeneration, Ministry of Education, Beijing 100053, China
2. Center of Neural Injury and Repair, Beijing Institute for Brain Disorders, Beijing, 100069, China
3. Center of Parkinson's Disease, Beijing Institute for Brain Disorders, Beijing, 100069, China
4. Department of Neurobiology, Beijing Key Laboratory of Major Brain Disorders, Beijing Institute of Brain Disorders, Capital Medical University, Beijing, 100069, China.
5. Central Laboratory, Beijing Institute of Geriatrics, Xuanwu Hospital, Capital Medical University, and Key Laboratory of Neurodegeneration, Ministry of Education, Beijing 100053, China
Received 2018-4-12; Accepted 2018-8-9; Published 2018-9-9
Abstract

Autologous neural stem cells (NSCs) may offer a promising source for deriving dopaminergic (DA) cells for treatment of Parkinson's disease (PD).
Methods: By using Sendai virus, human peripheral blood mononuclear cells (PBMNCs) were reprogrammed to induced NSCs (iNSCs), which were then differentiated to dopaminergic neurons in vitro. Whole-genome deep sequencing was performed to search for mutations that had accumulated during the reprogramming and expansion processes. To find the optimal differentiation stage of cells for transplantation, DA precursors obtained at various differentiation time points were tested by engraftment into brains of naïve immunodeficient mice. At last, the safety and efficacy of iNSC-derived DA precursors were tested by transplantation into the striatum of immunodeficient PD mouse models.
Results: PBMNC-derived iNSCs showed similar characteristics to fetal NSCs, and were able to specifically differentiate to DA neurons with high efficiency in vitro. The sequencing data proved that no harmful SNVs, Indels and CNVs were generated during the reprogramming and expansion processes. DA precursors obtained between differentiation day 10 to 13 in vitro were most suitable for transplantation when a balanced graft survival and maturation were taken into account. Two weeks after transplantation of DA precursors into mouse PD models, the motor functions of PD mice started to improve, and continued to improve until the end of the experiments. No graft overgrowth or tumor was observed, and a significant number of A9-specific midbrain DA neurons were surviving in the striatum.
Conclusion: This study confirmed the efficacy of iNSC-derived DA precursors in a mouse PD model, and emphasized the necessity of genomic sequencing and vigorous safety assessment before any clinical translation using iNSCs.
Keywords: induced neural stem cells, Parkinson's disease, genomic sequencing, cell therapy, reprogramming
Introduction
Parkinson's disease (PD) is caused by progressive degeneration of the dopaminergic neurons located at the substantia nigra (SN) of the midbrain, and afflicts more than 1% of people more than 60 years old [1, 2]. Because the neurodegenerative pathology in PD is restricted in neuronal subtypes—only dopamine (DA) neurons are lost and in space—the damaged neurons are localized in midbrain and project to striatum—cellular therapy is considered a promising strategy for treatment of PD [3, 4]. Fetal ventral mesencephalon (fVM) tissue transplantation has shown marked efficacy in some PD patients, but did not meet the primary end point in two double-blind placebo-controlled clinical trials [5-7]. Retrospective analysis revealed several possible reasons for the mild clinical results that included: 1) immune recognition of incoming grafts [6, 8, 9]; 2) heterogeneity of grafts [6, 8, 9]; and 3) niche problems in host brains [9-11].
Reprogramming technologies may provide a solution for the first two issues. Our lab and other groups have shown that autologous induced pluripotent stem cells (iPSCs)-derived DA precursors show efficacy in a nonhuman primate PD model [12, 13]. An alternative cellular source for autologous transplantation is induced neural stem cells (iNSCs), which have limited plasticity—only to neurons and glia—and theoretically pose less tumorigenic risk. We have previously reported generation of iNSCs from human peripheral blood mononuclear cells (PBMNCs) by using a non-integrative episomal vector system [14]. Nevertheless, the potential of blood-derived iNSCs in treating PD has not been explored yet. In addition, clinical translation of this strategy requires a careful safety assessment on the mutations that may have accumulated during the reprogramming process, and a timely derivation and differentiation method that can better meet the clinical needs. Inactivation of transgenes expressed by episomal vectors necessitates serial dilutions of vectors through cell divisions, which is a lengthy process. Sendai virus, an RNA virus that replicates in the form of negative-sense single stranded RNA in the cytoplasm of infected cells, does not go through a DNA phase nor integrate into the host genome, and therefore offers an efficient non-integrative vehicle for reprogramming [15-17]. Moreover, Sendai viruses can be rapidly removed by increasing the culture temperature to 39 ℃ [15, 16].
In the current study, we reprogrammed human PBMNCs to iNSCs by using a Sendai virus system, analyzed the mutations that had been acquired through the conversion and expansion processes, and tested the efficacy of iNSC-derived DA precursors in a PD model.
Methods
Isolation and expansion of PBMNCs
The isolation and expansion of PBMNCs was carried out as previously described [14]. 10 mL of donor's peripheral venous blood was collected with written informed consent, and from it around 9×106 mononuclear cells were isolated by using Ficoll-Paque PLUS (GE, Fairfield, USA). PBMNCs were expanded for two weeks in the medium containing 100 ng/mL human recombinant SCF, 10 ng/mL human interleukin 3 (IL-3), 40 ng/mL human insulin-like growth factor 1(IGF-1), 2 U/mL erythropoietin (EPO) (all from Life Technologies, Carlsbad, USA), 1 μM dexamethasone, and 100 μg/mL human transferrin (both from Sigma-Aldrich, St. Louis, USA). After two weeks, around 2×107 MNCs were obtained.
Reprogramming of PBMNCs to iNSCs by Sendai virus infection
Three million PBMNCs were suspended in SCF medium and then infected by Sendai virus (Life Technologies, Carlsbad, USA) encoding OCT3/4, SOX2, KLF4, and c-MYC (OSKM) (MOI=10). Two days later, these cells were plated at a density of 2×105/well on poly-D-lysine/laminin-coated 12-well plates in NSC medium, consisting of DMEM/F12:Neurobasal (1:1), 1×N2, 1×B27, 2 mM GlutaMAX, 1% NEAA (Life Technologies), 10 ng/mL recombinant human leukemia inhibitory factor (rhLIF, Millpore, Billerica, USA), 3 μM CHIR99021 and 2 μM SB431542 (both from Gene Operation, Michigan, USA). The medium was changed every other day. Ten days later, epithelium-like clones appeared in the culture. On days 32 to 35, the clones were large enough to be picked up and transferred onto PDL/laminin-coated 96-well plates for expansion. To inactivate residual Sendai virus, the incubator temperature was raised from 37 °C to 39 °C for one week when iNSCs were at passage number three. Culture for one week at higher temperature resulted in death of around half of the iNSCs, and the surviving cells were passaged in PDL/laminin-coated 24-well plates, and then onto coated 6-well plates when the cell population was sufficiently large.
Isolation and expansion of fetal NSCs
Aborted fetal brain tissue was obtained with written informed consent in accordance with the Declaration of Helsinki. The research protocol was reviewed and approved by the Institutional Review Committee of Xuanwu Hospital, Capital Medical University. The isolation and expansion of fetal brain NSCs was done as described previously [14]. Briefly, cerebral cortical tissue was gently dissected into small pieces (about 1 mm3), triturated gently with a Pasteur pipet in a 15 mL centrifuge tube (Corning, New York, USA) and pelleted by gravity for 5 min. Then the supernatant was filtrated with a 50-μm cell strainer and the cells were plated in NSC medium consisting of DMEM/F12, 2% B27, 1% penicillin-streptomycin (Life Technologies), 20 ng/mL basic fibroblast growth factor (bFGF) and 20 ng/mL EGF (PeproTech, Rocky Hill, USA).
Characterization of iNSCs in vitro
The identity of iNSCs was examined by immunostaining for NSC markers and by differentiation assays. To test the expression of NSC markers, iNSCs (passage No. 10, 20, 30, 40, and 50), and fetal NSCs were seeded at a density of 1×104 cells per 12 mm glass coverslip that had been coated with PDL/laminin in proliferation medium. NSC markers SOX1, SOX2, OLIG2, PAX6, NESTIN, GFAP and KI67 were tested by immunofluorescent staining. For differentiation assays, cells were seeded at a density of 5×103 cells per 12 mm glass coverslip that had been coated with PDL/laminin in NSC differentiation medium (DMEM/F12, 1×N2, 1×B27, 1×NEAA, 1×GlutaMAX) for 4 weeks for spontaneous differentiation. By spontaneous differentiation, no oligodendrocytes were generated. To derive oligodendrocytes specifically, iNSCs were seeded at a density of 2×104 cells per 12 mm glass coverslip coated with PDL /laminin and cultured in differentiation medium supplemented with 1 μM RA (Sigma-Aldrich), 20 ng/mL PDGF-AB (PeproTech), 10 ng/mL bFGF (PeproTech), and 1 μM SAG1 (Enzo) for the first 2 weeks. After that, RA and bFGF were replaced with 60 ng/mL T3, 1 mM cAMP (both from Sigma-Aldrich), 10 ng/mL insulin-like growth factor 1 (IGF-1), and 10 ng/mL NT3 (both from PeproTech) and differentiated for another 6 weeks.
Karyotype analysis
Karyotype analysis was carried out as described before [14]. Briefly, iNSCs were cultured in NSC medium for 2 days and then the cells were incubated with 100 ng/mL colcemid (Life Technologies) for 90 min, after which they were fixed with an ice-cold methanol:glacial acetic acid (3:1) solution 3 times. Chromosomes were stained with Hoechst (Life Technologies) and counted using 630× magnification.
RNA isolation and RT-PCR
RNA isolation was performed using the RNeasy plus Mini Kit (Qiagen, Hilden, Germany). Briefly, cells were collected and re-suspended in lysis buffer. RNA was eluted in 30 μL distilled water. Reverse transcription was carried out using the GoScriptTM Reverse Transcription System (Promega, Madison, USA). RT-PCR analyses for detection of transgenes, neural stem cell genes and Sendai virus genes were performed using the primer pairs listed in Table S1. The PCR program is as follows: 94 °C 2 min, 94 °C 30 s, 52-60 °C 30 s, 72 °C 45 s, 72 °C 7 min. Steps 2-4 were repeated 35 times. The temperature of Step 3 depended on the Tm value of the primers used.
Whole-genome sequencing and analysis
Genomic DNA was prepared from 2 million cells of episomal iNSCs p43 (reported in our previous paper), iNSC1 p27, iNSC2 p22, and parental PBMNCs for whole-genome sequencing by using Qiagen DNeasy Blood and Tissue Kit (catalog No. 69504). Next generation sequencing was performed by Illumina Hiseq X Ten. The variants from each derived iNSC line were then compared to the parental PBMNCs. The flow chart for data analysis is presented in Figure S1.
Differentiation of iNSCs specifically to DA neurons
A two-step differentiation method was used to derive DA cells from iNSCs. In the first stage, iNSCs were seeded at a density of 5×103 cells per 12 mm glass coverslip that had been coated with PDL/laminin in NSC differentiation medium (DMEM/F12, 1×N2, 1×B27, 1×NEAA) supplemented with 1 μM SAG1 and 100 ng/mL FGF8b for 10 days. For stage two, SAG1 and FGF8b in the medium were replaced with 0.2 mM ascorbic acid, 0.5 mM cAMP, 10 μM DAPT (all from Sigma-Aldrich), 10 ng/mL GDNF, 10 ng/mL BDNF and 1 ng/mL TGF-βIII (all from PeproTech) and cultured for another 2 weeks. The differentiated cells were fixed by 4% paraformaldehyde on days 10, 13, 15, 18 and 24 for analysis.
Dopamine release assay
iNSCs were differentiated on PDL/Laminin-coated 12-well plates. On day 13, 15, 18 and 24 of differentiation, the cells were washed twice with warm HBSS solution and then incubated with 550 μL HPLC medium for 24 h. 500 μL supernatant was collected and mixed with 7 μL perchloric acid (7 N, Sigma-Aldrich) before being stored at -80 °C for future use. For HPLC assay, the supernatant was centrifuged at 15,000 ×g at 4 °C for 25 min, and 100 μL supernatant was used for detection of DA, DOPAC, HVA, 5-HIAA and 5-HT using a reverse-phase column (Agilent, Santa Clara, CA) and an electrochemical detector system (5600A, Thermo Fisher Scientific, Waltham, MA). Three biological replicates were included for each group at every differentiation time point and the experiments were repeated twice.
Electrophysiology
Whole-cell patch clamp recording was applied on iNSC-derived mDA neurons on day 35 of differentiation. DA neurons were bathed in artificial cerebrospinal fluid that included 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1.25 mM NaH2PO4, 1 mM MgSO4, 25 mM glucose, and 26 mM NaHCO3. The internal solution consisted of 143 mM KCl, 8 mM NaCl, 10 mM HEPES, 1 mM MgCl2, 2% Biocytin, 2 mM NaATP and 0.4 mM NaGTP [18]. Cells were visualized on an IX71 Olympus system. The electrode resistance was 3-5 MΩ. Action potentials were induced in a current clamp mode using a HEKA EPC-10 patch-clamp amplifier (HEKA Electronic Inc., Lambrecht, Germany). Series resistance was constantly monitored. Data acquisition and analysis were performed using PatchMaster 2.71 (HEKA).
Animals and cell transplantation
All the mouse experiments were done according to the guidelines for the Care and Use of Laboratory Animals established by Beijing Association for Laboratory Animal Science. Adult male SCID-beige (SPF grade) mice weighing 20-25 g were used for tumor formation test, survival test and 6-OHDA (H4381, Sigma-Aldrich) lesioned PD models. Adult C57BL/6 mouse PD models were also used for transplantation experiments to confirm the results obtained with SCID-beige mice. To generate unilateral PD models, 6-OHDA was injected into the striatum of the right hemisphere (A/P +0.5 mm, M/L -2.1 mm, D/V -3.2 mm). A total of 10 μg 6-OHDA was injected per mouse in 2 μL of saline with 0.02% ascorbic acid. For engraftment experiments, 2×105 DA precursors suspended in 4 μL transplantation buffers (5 g/L glucose in HBSS) were injected into the right side of the striatum (A/P +0.5 mm, M/L -2.1 mm, D/V -3.4 mm). For the behavioral test, the mice received a subcutaneous injection of 0.5 mg/kg apomorphine (A4393, Sigma-Aldrich), and the number of contralateral rotations per min was scored. Mice with stable lesions (>7 rpm/min) were selected for transplantation studies [19]. The stable SCID-beige PD mice were randomly divided into 2 groups for transplantation experiments: 6-OHDA+cells group (n = 10) and 6-OHDA+buffer group (n = 8). The stable C57BL/6 PD models were randomly divided into 4 groups: 6-OHDA+iNSC1 group (n = 10), 6-OHDA+iNSC2 group (n = 6), 6-OHDA+iNSC3 group (n = 6), and 6-OHDA+buffer group (n = 6). For engrafted C57BL/6 PD models, cyclosporine was administered two days before through 4 weeks after transplantation. The apomorphine-induced contralateral rotation test was performed one week before and 2, 4, 6, 8 and 12 weeks after transplantation for SCID-beige mice, and 2 and 4 weeks after transplantation for C57BL/6 mice. Twelve weeks after transplantation, the SCID-beige mice were perfused for histological analysis.
Histological analysis
Immunostaining was performed as previously described [20]. Briefly, cells were blocked by 3% donkey serum for 2 h at room temperature. All primary antibodies and working dilution information are listed in Table S2. The secondary antibodies were Cy3-conjugated donkey anti-sheep/rabbit/goat/mouse/rat (1:400), FITC-conjugated donkey anti-sheep/rabbit/goat/mouse/rat (1:200), Cy5-conjugated donkey anti-rabbit/mouse/goat/rat (1:200, all from Jackson ImmunoResearch).
The brains were sliced at 40 μm thickness by using a Leica 2000R (Leica, Mannheim, Germany). The slices were stained using a similar method as for staining cultured cells. Pictures were captured using a confocal laser scanning microscope (TCS SP5, Leica).
Statistical analysis
The data are expressed as mean ± SEM. The statistical analyses and statistical graphing were carried out using Graphpad Prism 5 (Graphpad software, La Jolla, CA). Three or more groups were compared by using two-way ANOVA with Dunnett's multiple comparisons test. The differences were considered to be statistically significant when the p value was less than 0.05. The numbers of positive cells were calculated using ImageJ software (NIH, USA). For quantification of surviving grafted cells in each mouse, one out of every three serial sections (40 μm thickness per section) throughout the graft sites were counted. The behavioral test of animal rotations was recorded using a HUAWEI G7 camera.
The whole-genome sequencing data have been deposited in ENA's Sequence Read Archive with the accession number ERP106583.
Results
Generation of iNSCs from PBMNCs by using Sendai virus
PBMNCs were isolated from the peripheral venous blood of a healthy 33-year-old male donor. The PBMNCs were negative for pluripotent and early neural markers as previously described [14]. PBMNCs were transfected by non-integrative Sendai viral vectors encoding OCT4, SOX2, KLF4, and c-MYC (OSKM) for two days. Following transfection, the cells were cultured in NSC medium containing human recombinant leukemia inhibitor factor (hrLIF), SB431542 and CHIR99021 on PDL/laminin-coated plates. After about two weeks, epithelium-like clones appeared and could be selected manually (Figure 1A-B). CHIR99021 is a small molecule inhibitor of glycogen syntheses kinase 3 (GSK3) and could activate canonical WNT signaling pathway [21]. SB431542 is a small molecule inhibitor of transforming growth factor β (TGF-β) and Activin receptors, and has been implicated in mesenchymal to epithelial transition and reprogramming [22, 23]. These iNSCs could stably self-renew in the presence of hrLIF, SB and CHIR, either as spheres or in a monolayer form (Figure 1B). The iNSCs showed a good proliferative capacity and had been passaged more than 50 times in the lab as of this writing. We plotted the growth curve of cells of passage number 10, 20, and 30, and all three exhibited similar proliferation rates (Figure 1C). The iNSCs also showed a normal karyotype (Figure 1D).
PBMNCs are induced into neural stem cells by using Sendai virus. (A) A schematic representation of the neural stem cells induction procedure. PBMNCs: peripheral blood mononuclear cells. (B) Phase-contrast images of the PBMNCs, an iNSC clone, iNSCs in sphere and iNSCs in monolayer culture. Scale bars, 200 μm. (C) The growth curve of iNSCs P10, P20 and P30. (n=3 independent experiments). (D) Karyotype analysis of iNSCs. (E) Detection of remaining Sendai virus by PCR and immunocytochemistry staining. Scale bars, 50 μm. (F) Expression of transgenes (SOX2, OCT4, KLF4, cMYC) and GAPDH reference gene at different passages of iNSCs. The values represent mean ± SEM in this figure and all the others in this article.
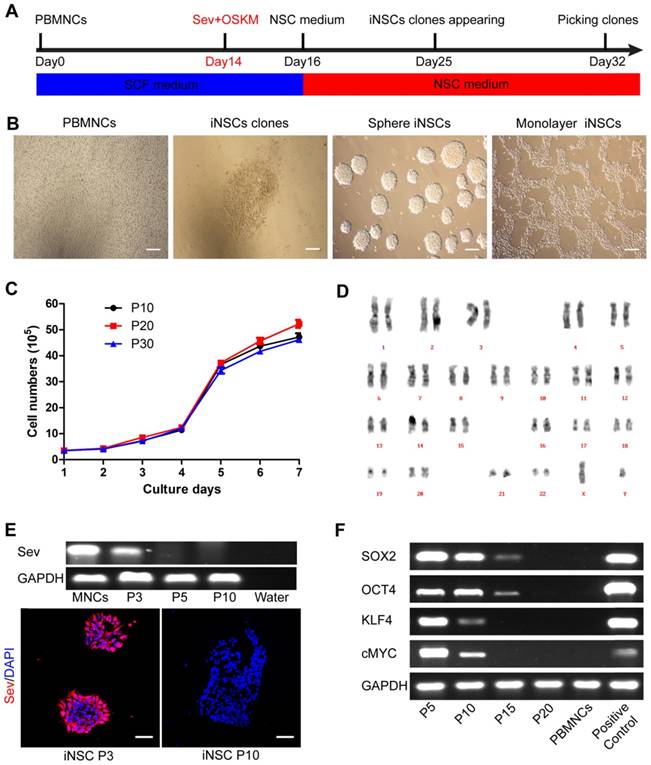
The Sendai virus used in the study was the third generation virus that was sensitive to temperature change. When the derived iNSCs had been passaged three times, the incubator temperature was raised to 39 ℃ for one week to inactivate the virus. The residual virus could be detected by PCR and immunostaining of the viral protein Sev, which was positive at passage three but disappeared at passage 10 (Figure 1E; primer sequences are listed in Table S1). After virus inactivation, the transgene expression (OSKM) was gradually diluted and lost over cell divisions (Figure 1F). All the four transgenes were lowered to an undetectable level by using PCR at passage 20 (Figure 1F; primer sequences are listed in Table S1).
Peripheral blood-derived iNSCs are similar to fetal brain NSCs
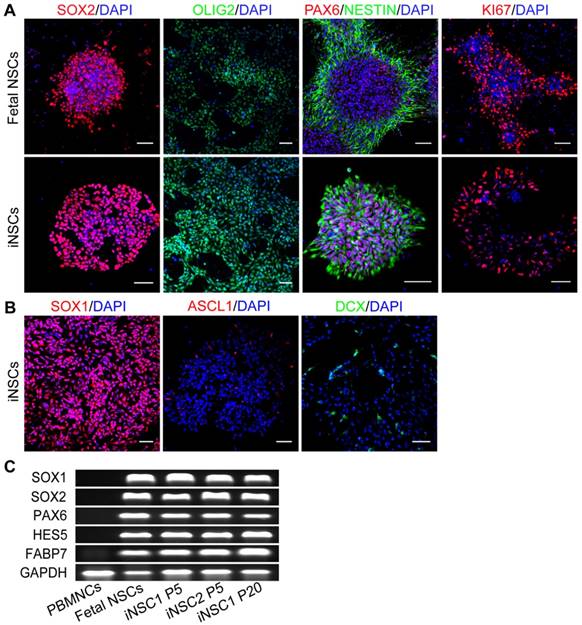
The identity of the induced cells was examined by comparison with fetal brain NSCs derived from aborted fetus 12 weeks of gestation. Both fetal NSCs and iNSCs (passage No. 30) were positive for neural stem cell markers SOX2, OLIG2, PAX6 and NESTIN, and the marker for proliferative potential, KI67 (Figure 2A). iNSCs were also positive for SOX1 (Figure 2B). Even cultured in proliferation conditions, a small percent of iNSCs and fetal NSCs would assume spontaneous neuronal differentiation and become ASCL1 and Doublecortin (DCX) positive (Figure 2B). iNSCs of early through late passages were all positive for SOX1, SOX2, NESTIN and GFAP (Figure S2). The PCR results showed that both iNSCs and fetal NSCs expressed SOX1, SOX2, HES5, PAX6 and FABP7—all neural stem cell genes (Figure 2C; primer sequences in Table S1).
iNSCs are similar to fetal NSCs. (A) Immunostaining for SOX2, OLIG2, PAX6, NESTIN, and KI67—neural stem cell markers of iNSCs (P30) and fetal NSCs. Scale bars, 50 μm. (B) Immunostaining for SOX1, ASCL1 and DCX. Scale bars, 50 μm. DCX: Doublecortin. (C) Expression of SOX1, SOX2, PAX6, HES5, FABP7 and GAPDH in iNSCs and fetal NSCs. P30: passage No. 30. Also see Figure S2.
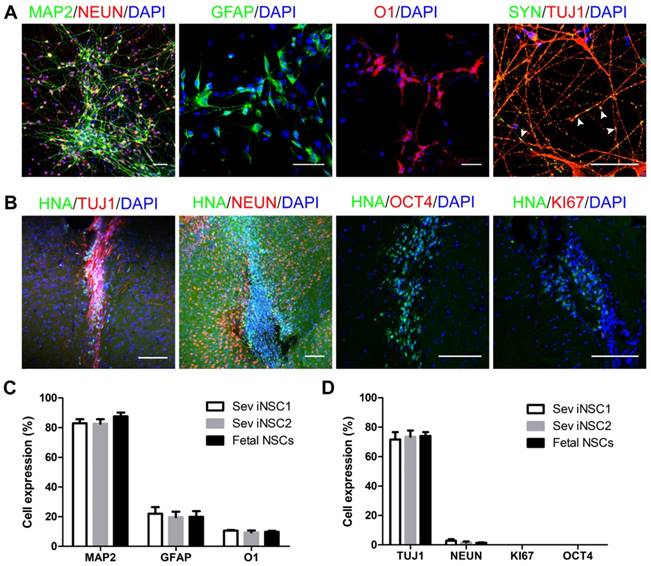
The differentiation ability of iNSCs was analyzed by subjecting the cells to a differentiation basal medium on PDL/laminin-coated coverslips. After 6 weeks, a majority of the cells turned into MAP2+ mature neurons (82.89%), which were co-labeled with NEUN. Synapsin (SYN) staining indicated that the differentiated neurons might have established synaptic connections (Figure 3A, C). Under this condition, a small number of induced cells differentiated into GFAP+ astrocytes (21.92%, Figure 3A, C), but no oligodendrocyte was observed (data not shown). When iNSCs were subjected to a glial differentiation condition and continuously cultured for 7-8 weeks, O1+ oligodendrocytes emerged (10.57%), similar to that from fetal NSCs (Figure 3A, C). The safety of iNSCs was examined by transplanting the induced and fetal cells into the striatum of immunodeficient SCID-beige mice. Eight weeks following engraftment, no overgrowth of tumor formation was observed. All the transplanted cells were negative for OCT4 and KI67, and 73.43% of the grafts were positive for tubulin beta III (TUJ-1) and 1.67% for NeuN (Figures 3B, D).
Differentiation of iNSCs in vitro and in vivo. (A) Immunofluorescent staining of MAP2, GFAP, O1 and SYN on iNSC (P30)-differentiated cells. Scale bars, 50 μm. SYN: Synapsin. White arrowheads represent SYN/TUJ1-double positive cells. (B) Immunofluorescent staining of HNA, TUJ1, NEUN, KI67 and OCT4 in SCID-beige mouse brain one month after iNSC (P30) transplantation. Scale bars, 50 μm. HNA: human nuclei antibody. (C) Differentiation efficiency of iNSC1, iNSC2 and fetal NSCs (n=3 independent experiments). (D) Differentiation efficiency of iNSCs following transplantation into SCID-beige mice (n=3). P30: passage No. 30.
Whole-genome sequencing analysis
It is of great clinical significance to know whether during the reprogramming and expansion processes the obtained cells have accumulated any harmful mutation(s) [24, 25]. The parental blood mononuclear cells and three iNSCs lines were subjected to whole-genome deep sequencing. The three iNSC lines included one iNSC line reprogramed using episomal vectors (reported in our previous study [14]), and two Sendai virus-reprogrammed lines (iNSC1 and iNSC2, two separate clones). For all three iNSC lines, >1×109 reads per sample were generated and analyzed. Table 1 shows a summary of the sequence analyses of the three iNSC lines. We obtained alignable DNA sequences of >140 Gb for each iNSC line and PBMNCs. The sequencing was done at an average read depth of 41-fold, with 98.70% of the autosomal genome covered by at least 10 reads. With reference to the parental PBMNCs, single-nucleotide variants (SNVs), insertions and deletions (Indels), and copy number variants (CNVs) were analyzed for the three iNSC lines (Figure S1) [26, 27]. When the sequences were directly compared, 648 to 715 likely SNVs were identified between each iNSC line and its parental PBMNCs sequenced in pairs. We chose 16 SNVs among the three daughter iNSCs for verification by PCR and Sanger sequencing, and 14 of them (88%) were correct (Table S3). When the variants were screened by using a condition of Reads ≥ 35 and VAF (variant allele fraction, mutation reads divided by total reads for the same site) ≥ 0.40, each of the three iNSC lines harbored 13-29 high-confidence SNVs (Table S4, Supplementary Spreadsheet SNVs and Table S5) and 0-1 Indel (Table S4, Supplementary Spreadsheet Indels and Table S6) mutations compared with the parental PBMNCs, with the sev iNSC2 showing fewer high-confidence mutations than the other iNSC lines. To investigate the functional relevance of SNVs found in iNSCs after induction and expansion, we next focused on SNVs in exons. High-quality sequencing revealed 23 SNVs in episomal iNSCs residing within the coding regions; 20 of them were non-synonymous (NS) and three of them were synonymous (S). Fifteen SNVs in sev iNSC1 were found in the coding regions; 9 of them were non-synonymous and 6 were synonymous. The paired sequencing data revealed 9 SNVs in coding regions in sev iNSC2, with 5 of them being non-synonymous and 4 synonymous. The NS:S ratio was 6.67 for episomal iNSC line, and 1.50 and 1.25 for sev iNSC1 and iNSC2, respectively (Table 1). The two sev iNSC lines showed similar NS:S ratios.
Whole-genome sequencing data summary.
| Features/iNSCs lines | Episomal iNSC | Sev iNSC1 | Sev iNSC2 |
|---|---|---|---|
| Total nucleotides sequenced | 151 Gb | 151 Gb | 146 Gb |
| Alignable nucleotides | 145 Gb | 145 Gb | 140 Gb |
| Genome coverage (fold) | 41.75 × | 41.94 × | 41.75 × |
| Total SNVs (compared to hg19 database) | 3283047 | 3299641 | 3292380 |
| Total SNVs (compared to parental cells) | 692 | 648 | 715 |
| High quality, filtered SNVs a | 29 | 27 | 13 |
| SNVs in coding regions | 23 | 15 | 9 |
| Synonymous (S) | 3 | 6 | 4 |
| Non-synonymous (NS) | 20 | 9 | 5 |
| NS:S ratio | 6.67 | 1.5 | 1.25 |
| Oncogenes in coding regions b | 1 | 0 | 0 |
| Suppressor genes in coding regions c | 1 | 0 | 0 |
| NSC related genes in coding regions d | 0 | 0 | 0 |
| Total Indels (compared to hg19 database) | 906589 | 908051 | 906913 |
| Total Indels (compared to parental cells) | 89 | 80 | 112 |
| Indels in coding regions | 1 | 0 | 0 |
| Total CNVs (compared to hg19 database) e | 45 | 17 | 14 |
| Total CNVs (compared to parental cells) f | 190 | 0 | 0 |
| CNVs in coding regions g | 16 | 0 | 0 |
| CNVs in coding region (pathogenic class) | 9 | 0 | 0 |
a. Screen condition: reads ≥ 35; daughter iNSC lines VAF ≥ 0.4.
b-d. Screen condition: reads ≥ 35; daughter iNSC lines VAF ≥ 0.4, non-synonymous.
e-f. Screen condition: parental PBMNCs are wildtype; CNV classes are pathogenic and likely pathogenic; CNV size ≥ 500Kb.
g. Screen condition: parental PBMNCs are wildtype; CNV classes are pathogenic and likely pathogenic; CNV size ≥ 500Kb; variants reside in exons.
CNV class: 1, pathogenic; 2, likely pathogenic; 3, of uncertain significance; 4, likely benign; 5, benign.
Among the coding region mutations, we were particularly concerned with those that had occurred in oncogenes, tumor suppressor genes, and NSC-related genes, which are relevant to tumorigenic risk (Table 1). In the two sev iNSC lines, zero non-synonymous mutations in the coding region were found that belonged to oncogenes, tumor suppressor genes, or NSC-related genes. In the episomal iNSC line, one oncogene (KLF4) and one tumor suppressor gene (YARS) showed non-synonymous mutation in the coding region. Regarding Indel mutation, zero high-quality filtered mutations in the coding region were found in either of the sev iNSC lines, and one Indel mutation (KLF4) was detected in the episomal iNSC line, causing a heterozygous truncation in exon 2. None of the exonic SNVs and Indels found in the three iNSC lines occurred in the conserved regions. To learn about the possible impact of the mutations, we looked into the Provean/ SIFT/ Polyphen2_HDIV/ Polyphen2_HVAR and mutationtaster databases, ACMG clinical database, Chinese population database, east Asian population database (EXAC), ESP database, HGMD, Clinvar, OMIM and SWISS databases. The collective information obtained by referring to these databases will lead to an overall assessment of the mutation: either negative or positive. “Negative” means a person harboring this particular mutation would not likely be diagnosed as a genetic disease patient caused by this mutation. “Positive” means the opposite. All of the SNVs and Indels found in the three iNSC lines gave a “negative” result, suggesting that the SNVs and Indels overall represented a benign nature of mutations.
Compared to the hg19 database, there were 14-45 filtered CNVs in the three iNSC lines (Table 1). With reference to the parental PBMNCs, the numbers of CNVs in episomal iNSC, sev iNSC1, and sev iNSC2 were 190, zero, and zero, respectively. The screen conditions were: 1) parental PBMNCs were wildtype; 2) CNV classes were pathogenic and likely pathogenic; 3) CNV size ≥ 500 kb. With the current WGS technology, the reads and library fragments only span several hundred base pairs, and the read depths are randomly distributed. These limitations render the current WGS technology not very reliable in accurately detecting CNVs, particularly small fragment CNVs. We therefore set the screening condition for CNV as “size ≥ 500 kb”. The impact of individual CNV was rated as 1, pathogenic; 2, likely pathogenic; 3, of uncertain significance; 4, likely benign; and 5, benign, based on the collective information obtained by visiting various databases (DECIPHER, ClinGen, meMAF, DGV databases). CNVs rated as “pathogenic” and “likely pathogenic” that occurred in coding regions were screened; there were 16, zero, and zero such CNVs in episomal iNSCs, sev iNSC1, and sev iNSC2, respectively (Supplementary Spreadsheet CNVs and Table S7). When the screening condition was set more strictly as “pathogenic”, the numbers of CNVs changed to nine, zero, and zero, respectively (Table 1).
iNSCs can be differentiated to mature midbrain DA neurons with high efficiency in vitro
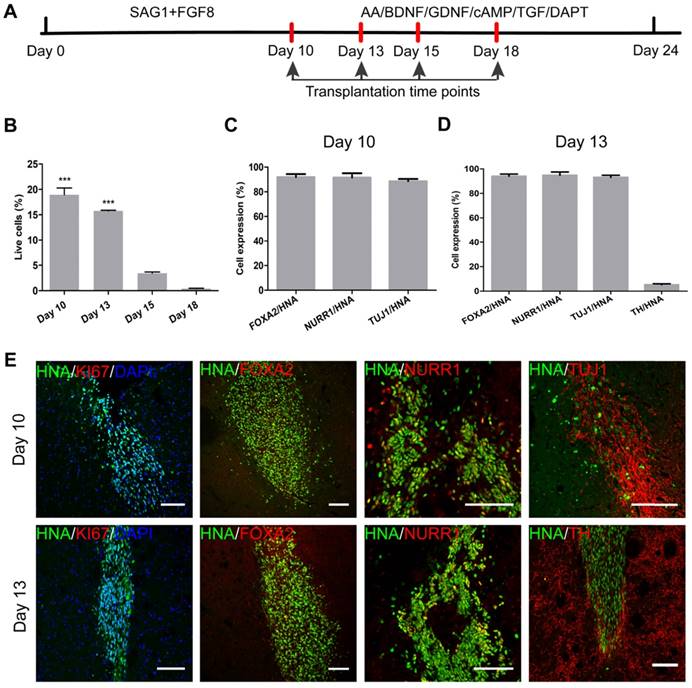
iNSCs generated this way resembled primitive NSCs that are LIF-responsive [28, 29]. Therefore, we tested the capacity of iNSCs to specifically give rise to DA neurons. The differentiation scheme included two stages (Figure 4A). During stage one, iNSCs were treated with SAG1 (a small molecule to activate sonic hedgehog (SHH)) and FGF8b, for 10 days, to drive iNSCs towards a floor plate fate [30-33]. During stage two (after day 10), SAG1 and FGF8b were withdrawn and replaced by ascorbic acid (AA), brain derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), cyclic adenosine monophosphate (cAMP), transforming growth factor βIII (TGF-βIII) and DAPT, to facilitate the maturation of DA neurons [12, 13].
iNSCs differentiate into midbrain dopamine neurons in vitro. (A) A schematic representation of the iNSC differentiation procedure in vitro. AA: ascorbic acid; BDNF: brain-derived neurotrophic factor; GDNF: glial cell line-derived neurotrophic factor; cAMP: cyclic adenosine monophosphate; TGF: transforming growth factor. (B) Immunofluorescent staining for FOXA2, NURR1, and KI67 on day 10, FOXA2, NURR1, GIRK2 and TH on days 18 and 24, on cells derived from P30 iNSC1. Scale bars, 50 μm. (C) Efficiency of differentiation from P30 iNSC1 on days 10, 18 and 24 (n=3 independent experiments). (D) Percentages of TH/FOXA2, TH/NURR1, and GIRK2/TH on differentiation day 24. (E) DA, DOPAC and HVA levels secreted by iNSC-derived midbrain dopamine neurons tested by HPLC on day 24. DA: dopamine; DOPAC: 3,4-dihydroxyphenylacetic acid; HVA: homovanillic acid. (F) DA secretion on differentiation days 0 (control), 13, 15, 18 and 24. ***p < 0.001 by two-way ANOVA with Dunnett's multiple comparison test (n=6). (G) DOPAC secretion on differentiation days 0 (control), 13, 15, 18 and 24. **p < 0.01, ***p < 0.001 by two-way ANOVA with Dunnett's multiple comparison test (n=6). Also see Figures S3-5.
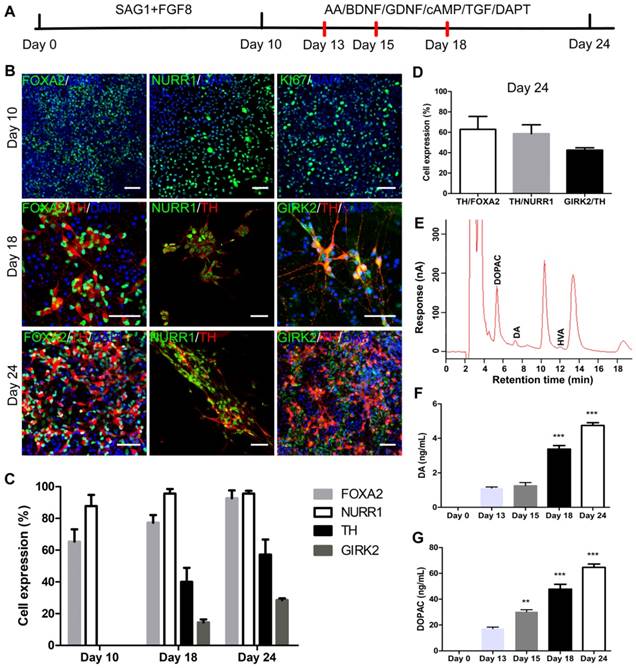
We examined the expression of markers forkhead box A2 (FOXA2), orphan nuclear receptor (NURR1), G-protein-coupled inward rectifier potassium (GIRK2), and Tyrosine hydroxylase (TH) on days 10, 13, 15, 18 and 24 (Figure 4B). FOXA2 is a transcription factor that is expressed in the floor plate of neural tube and then widely in the ventral midbrain [32, 34]. NURR1 is a marker for DA neural cells that are going through the last few cell cycles or just exit cell cycles. A previous study has shown that NURR1 is an ideal marker for selection of DA cells that can survive transplantation and are committed and can only mature to DA neurons [35]. GIRK2 is an A9 region-specific midbrain DA neuron marker [36, 37]. On day 10 of differentiation, on average, 65.33% and 87.76% cells were positive for FOXA2 and NURR1, respectively (Figure 4B-C and Figure S3A-B), but few TH+ or GIRK2+ cells were observed. At this time point, about 50% of cells were positive for KI67, in agreement with the observation that cell culture expanded through stage one. Along the course of differentiation, the proportion of FOXA2+ cells increased from 65.33% on day 10, to 77.33% on day 18 and 92.73% on day 24; and the proportion of NURR1+ cells increased from 87.76% on day 10, to 95.58% on day 24 (Figure 4B-C and Figure S3A-B). The percentage of TH+ cells reached 40.07% on day 18, and 57.23% on day 24; and the proportion of GIRK2+ cells was 14.45% on day 18 and increased to 28.55% on day 24 (Figure 4B-C and Figure S3C-D). On day 24, 62.87% of FOXA2+ cells were co-labeled with TH, and 58.69% of NURR1+ cells were co-labeled with TH. On average, 42.51% of TH+ cells also stained positive for GIRK2 (Figure 4D). To examine whether iNSCs of early (P10) vs. late (P50) passages show differential capacity in DA neuron differentiation, iNSCs were differentiated for 24 days and stained for FOXA2, NURR1, TH, and GIRK2 (Figure S4A). There were no statistically significant differences between the proportions of the above markers (Figure S4B).
The supernatant of cultures on day 13, 15, 18 and 24 was collected and tested for levels of DA and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA). Both DA and DOPAC were detected on day 13, 15, 18 and 24, and HVA was only detected on day 24 (Figure 4E and Figure S5). Compared to those on day 13, the levels of DA and DOPAC on day 18 and 24 were significantly higher (Figure 4F-G). DA is metabolized to DOPAC by monoamine oxidase (MAO), and DOPAC to HVA by catechol-O-methyltransferase (COMT). That DA and DOPAC appeared earlier than HVA suggested that the cells in culture had matured step by step. No 5-hydroxyindoleacetic acid (5-HIAA) or 5-hydroxytryptamine (5-HT) was detected in the supernatant at any timepoint by high-performance liquid chromatography (HPLC) (Figure 4E and Figure S5), indicating that the iNSCs did not differentiate to serotonergic neurons under the current condition.
Electrophysiological properties of mDA neurons derived from iNSCs
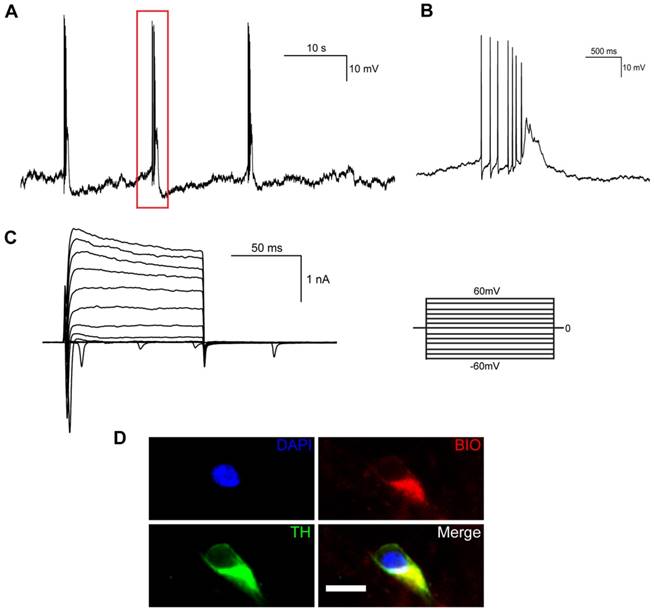
We also examined the electrophysiological properties of iNSC-derived mDA neurons by whole-cell patch clamp recording. When differentiated for 35 days, spontaneous action potentials could be detected in mature neurons (Figure 5A), which were spaced by groups of bursting spikes (a series of 2-10 spikes with decreasing amplitudes, Figure 5B). Both voltage-dependent outward potassium currents and inward sodium currents were observed by voltage clamp recording (Figure 5C). The mature neurons that had been recorded to release spontaneous action potentials were confirmed to be TH-positive (Figure 5D).
Electrophysiological properties of midbrain dopaminergic neurons differentiated from human iNSCs in vitro. (A) Spontaneous action potentials of an mDA neuron. Red rectangle represents the magnified section in (B) (n=3). (B) A group of bursting firing in (A), which included 7 bursting spikes of decreasing amplitudes. (C) Outward K+ current and inward Na+ current by voltage clamp recording (n=9). (D) Immunofluorescent staining of mDA neurons that had released spontaneous action potentials. BIO: biocytin; TH: tyrosine hydroxylase. Scale bars, 10 μm. (n=3).
Searching the optimal time window of DA cell differentiation for transplantation in vivo
Along the course of DA cell differentiation, the optimal stage of cells for transplantation should balance the ability to survive and to maturate. The more mature in vitro, the more vulnerable are the cells, and the more difficult for them to survive replating and transplantation. On the other hand, a higher degree of stemness corresponds to a better chance of survival, but a lower possibility of specification into terminally mature DA neurons. To find the optimal stage of cells for transplantation, we transplanted cells of differentiation day 10, 13, 15, and 18 into the striatum of naïve immunodeficient SCID-beige mice (Figure 6A). For each time point, 1×105 single cells in suspension were injected into the right side of the striatum (n = 4) to test the safety and differentiation capacity of cells. One month later, the SCID-beige mice were sacrificed by perfusion and the brains sliced for histological analysis.
Transplantation of mDA precursors into naïve mice in vivo. (A) A schematic representation of the experimental procedure of transplantation. (B) One month after transplantation into naïve SCID-beige mice, the percentages of surviving cells from differentiation day 10, day 13, day 15, and day 18 groups. ***p < 0.001 by two-way ANOVA with Dunnett's multiple comparison test (n=6). (C) One month after transplantation of differentiation day 10 cells, the percentages of surviving cells (HNA+) that co-expressed FOXA2, NURR1, and TUJ1 (n=4). HNA: human nuclei antibody. (D) One month after transplantation of differentiation day 13 cells, the percentages of surviving cells (HNA+) that co-expressed FOXA2, NURR1, and TUJ1 (n=4). HNA: human nuclei antibody. (E) One month after transplantation of differentiation day 10 and day 13 cells, representative images of immunofluorescent staining for FOXA2, NURR1, KI67, TUJ1 and TH are shown. Scale bars, 100 μm. Also see Figure S6.
Staining with human nuclei antibodies (HNA) was employed to identify engrafted cells. The surviving HNA+ cells were calculated, and were estimated to be 18,940 in the day 10 cell engraftment group; but, this number was reduced to only 411 in the day 18 engraftment group. The survival rates of the engrafted cells are presented in Figure 6B. One month after transplantation into naïve mouse brains, only a small number of day 15 and day 18 cells survived and the vast majority of grafts had died (Figure S6). As revealed from the in vitro differentiation data, the proportions of FOXA2+ and NURR1+ cells had already reached around 65.33% and 87.76% by day 10, and gradually increased over time. On day 13 in vitro, TH+ mature DA neurons started to show up (6.13%), and reached 25.50% and 40.07% by day 15 and day 18, respectively (Figure S3C). GIRK2+ cells appeared by day 15 (5.33%) and reached 14.45% by day 18 in vitro (Figure S3D). It seemed that by day 15 and day 18 in vitro, the cells had reached a state that was too mature to survive the transplantation procedure.
We examined the cells of differentiation day 10 one month after engraftment. About 91.97% and 91.63% of the surviving cells were positive for FOXA2 and NURR1, respectively (Figure 6C, E). About 88.63% were positive for TUJ-1, but few TH+ cells were observed. Compared to day 10 cells, day 13 cells gave rise to a higher percentage of TUJ-1+ cells (93.13%), and some TH+ cells (5.30%) were also observed one month after transplantation, at the expense of a slightly reduced overall survival rate (Figure 6B, D-E). The results showed that the optimal time window of cells for transplantation should be between day 10 and day 13 of differentiation. We then decided to use a mixture of cells from day 10 and day 13 for transplantation in a PD mouse model.
iNSC-derived mDA precursors are safe and show efficacy in PD models
Next, we tested the safety and efficacy of human iNSC-derived DA precursors in a mouse PD model. The mice (SCID-beige and C57BL/6) were lesioned unilaterally on the right side of the striatum by injecting 6-OHDA to generate PD models. Only those mice with more than 7 contralateral rotations per min were considered successful PD models. Five weeks later, 2×105 DA precursors in 4 μL buffer that included day 10 and day 13 cells mixed at a ratio of 1:7 were injected into the lesioned sites of SCID-beige PD models (n = 10). SCID-beige mice in the sham surgery PD group received buffer only (n = 8), and naïve SCID-beige mice without 6-OHDA injection or cellular transplantation were used as controls (n = 3). Apomorphine-induced rotation tests were performed 1 week prior to, and 2, 4, 6, 8 and 12 weeks after cell transplantation (Figure 7A). All SCID-beige mice survived after transplantation and were sacrificed with perfusion following the last behavior test. Two weeks after transplantation, the 6-OHDA + cells group already showed significantly improved behavioral performance compared to that of the 6-OHDA + buffer group (Figure 7B). The motor performance remained stable after week 2 post-transplantation through week 6, when another period of improvement started. The engrafted mice continued to improve from week 6 until the end of the experiments—week 12 post-transplantation (Figure 7B). The behavioral data of individual mice are presented in Figure S7. All the 10 engrafted mice showed significant improvement by the end of the experiment (Figure S7A), which was not seen in the 6-OHDA + buffer mice (Figure S7B). To exclude the possibility that the improvement in motor function might be specific to the SCID-beige genetic background, we employed additional PD mouse models of C57BL/6 background, and engrafted three sev iNSC-derived DA precursors or buffer into the lesioned sites (n = 6-10 for each condition). C57BL/6 PD mice also showed significantly improved behavioral performance 4 weeks after transplantation (Figure S8).
Transplantation of iNSC-derived mDA precursors into PD mouse models. (A) A schematic representation of the cell transplantation and behavioral test procedure. (B) The results of apomorphine-induced contralateral rotations at different time points from the 6-OHDA+buffer group (n=8), 6-OHDA+cells group (n=10), and control group (n=3). ***p < 0.001 by two-way ANOVA with Dunnett's multiple comparison test. (C) TH immunofluorescent staining for TH on an unlesioned control, 6-OHDA-leisoned hemisphere and the hemisphere with 6-OHDA+cells, at levels of striatum, MFB, and SN. Scale bars, 100 μm. MFB: medial forebrain bundle; SN: substantia nigra. (D) The percentages of FOXA2/TH, NURR1/TH, GIRK2/TH, TH/HNA in PD mice 12 weeks after transplantation (n=10). HNA: human nuclei antibody. (E) Immunofluorescent staining for TH, KI67, OCT4, FOXA2, NURR1 and GIRK2 in PD mice 12 weeks after transplantation. Scale bars, 50 μm. Also see Figures S7 and S8.
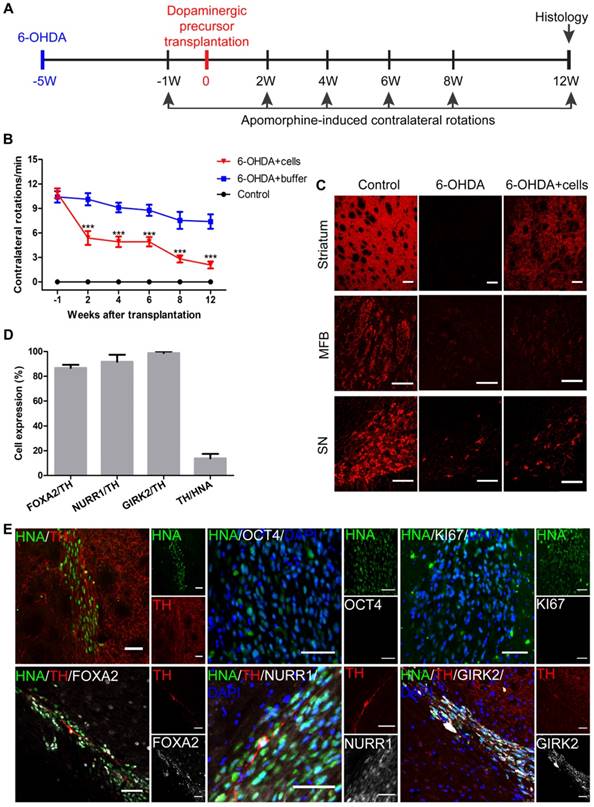
Immunofluorescent staining results revealed that TH-positive signals were almost absent at the striatum and medial forebrain bundle (MFB) in the 6-OHDA-lesioned SCID-beige mice that received no grafts (Figure 7C). The signals were also markedly reduced at substantia nigra pars compacta (SNpc). In contrast, in the engrafted SCID-beige mice, TH+ signals were greatly recovered in the striatum, and mildly but significantly increased at SNpc (Figure 7C).
Twelve weeks after transplantation, there were, on average, about 21,700 surviving cells in each SCID-beige mouse, as indicated by the human nuclei staining. Among the surviving cells, 13.84% were TH-positive DA neurons (Figures 7D). About 86.78% and 91.72% of the TH-positive cells were co-labeled with FOXA2 and NURR1, respectively (Figure 7D-E). In addition, about 98.77% of the TH-positive cells were positive for GIRK2, an A9 DA neuronal marker specifically expressed at the SNpc (Figure 7D-E). No overgrowth of grafts, or any KI67- or OCT4-positive transplanted cells were observed (Figure 7E).
Discussion
Compared to allogeneic DA precursors (fetal ventral mesencephalon tissue or ES-derived DA neural cells), autologous DA cells may be advantageous since they may circumvent immune recognition-associated problems [12, 13, 38, 39]. Takahashi and colleagues have also shown that human iPSC-derived DA cells can improve the symptoms of PD monkey models and the cells seem to be safe [38], and the group plans to conduct human clinical trials for PD intervention soon [40]. An alternative source for autologous DA neurons is induced neural stem cells [14]. NSCs belong to adult tissue stem cells and are generally considered to pose less tumorigenic risk than pluripotent stem cells do. In addition, differentiation to DA cells from NSCs would take a much shorter period of time compared with iPSCs, which normally need to first go through a differentiation stage to become neural stem cells [41]. Nevertheless, native neural stem cells that exist in the subgranular zone of the hippocampus and the subventricular zone in rodent brain show limited plasticity and can only differentiate to glutamatergic and GABAergic neurons [42-44], but not to dopaminergic neurons—the neuronal subtype relevant in PD. Compared to native neural stem cells, the iNSCs obtained in the current study seemed to be more primitive. The iNSCs possessed a higher degree of plasticity and were able to become dopaminergic, cholinergic, glutamatergic and GABAergic neurons [14]. The iNSCs were also double positive for SOX1 and SOX2. During embryo development, SOX1 is the earliest specific marker for NSCs [45, 46]. SOX1/SOX2 double positivity indicated a primitive state of NSCs. In our study, we also showed that the iNSCs could differentiate into A9-specific midbrain DA neurons, which were positive for GIRK2, in addition to NURR1 and TH.
However, a greater degree of plasticity normally corresponds to a higher level of stemness, and accordingly, tumorigenic risk. Therefore, the safety of these iNSCs needs to be scrutinized. We transplanted iNSC-derived DA cells into immunodeficient mice for as long as three months, and did not observe any graft overgrowth or tumor development, suggesting that the iNSCs were quite safe. For possible future clinical trials, patient-specific iNSC-DA precursors might need to be quality-assured in immunodeficient mouse brains for a longer period of follow-up time, for example, 6 months.
Another safety concern is whether iNSCs have accumulated harmful mutations during the reprogramming and expansion processes. In the experimental setting used in this study, the parental cells, peripheral blood mononuclear cells, represent a heterogeneous population, consisting of T, B, NK cells, monocytes, and a small percentage of dendritic and blood progenitor cells. Each of the three iNSC lines was derived from a single cell in the heterogeneous PBMN cells. That particular single cell first went through a reprogramming process to become an induced neural stem cell, and then expanded into a colony by a series of cell divisions. Mutations could happen during either the reprogramming and/or the expansion stage(s). Theoretically, if a mutation takes place during the reprogramming process, all the daughter cells in the colony should harbor that particular mutation, unless another corrective mutation occurs in some of the daughter cells during the expansion stage, which is highly unlikely. In this case (a mutation happens in the reprogramming stage), a heterozygous mutation would give rise to a VAF value of around 0.5 in the daughter cells, and a homozygous mutation would be around 1.0 in the daughter cells. If a mutation occurs during the expansion stage, the VAF value would be less than 0.5 for a heterozygous mutation; and a mutation that occurred during the first several cell divisions would lead to a relatively greater VAF value than that of the mutation derived from cell divisions happening later.
Genome replication does not have 100% fidelity and it is estimated that 3-30 mutations are acquired per haploid genome per mitotic division for a somatic cell [47, 48]. Considering that it takes 46-47 cell divisions for a fertilized egg to give rise to fully differentiated somatic cells in a human being, each somatic cell is expected to harbor 138-1410 spontaneous mutations. Therefore, the PBMN cells used as parental cells in this study would actually be genetically heterogeneous harboring mosaic mutations in individual cells. To distinguish mutations that had possibly happened during the reprogramming stage from those from the expansion stage, we used VAF = 0 as a cut ratio for parental cells and 0.4 for daughter cells. Only one homozygous mutation (KLF4) was found in all the 3 daughter iNSC lines, so we focused the analysis on the heterozygous mutations. Reads ≥ 35 was set as a condition to screen high-confidence mutations. Mutations with VAF = 0 in parental cells and VAF ≥ 0.4 in daughter iNSCs were considered mutations that had probably occurred during the reprogramming stage, although we cannot exclude the possibility that a mutation that confers proliferative advantage was obtained during the expansion stage, which led to increased VAF values over cell passages. That VAF > 0 in parental cells, and VAF ≥ 0.4 in daughter iNSCs suggests two possibilities. One was that the daughter iNSCs originated from the rare population in the parental cells that already had this mutation; the other possibility was that the mutation was acquired during the reprogramming process. That VAF = 0 in parental cells, and VAF < 0.4 in daughter iNSCs suggests that the mutation was probably acquired not during the reprogramming process, but during the expansion stage. That VAF > 0 in parental cells, and VAF < 0.4 in daughter cells suggests that the iNSCs probably did not originate from a rare population already harboring that mutation, and the mutation was acquired during the expansion stage. The results showed that 13-29 SNVs and 0-1 Indels were probably acquired during the reprogramming process. Interestingly, the 13 probably reprogramming stage-acquired SNVs in sev iNSC2 were all included in the 27 SNVs in sev iNSC1, but none of these 13 overlapped with the 29 SNVs acquired during reprogramming in episomal iNSCs. This suggests that these 13 SNVs are possibly hot spots often associated with Sendai virus-mediated reprogramming method using the four factors (OCT4, SOX2, KLF4 and c-MYC).
In this study, 283-336 SNVs and 28-70 Indels were acquired probably during the expansion stage. It takes around 10 cell divisions for a founder iNSC to form a colony (around 1000 cells), followed by a series of passages. Passage numbers 43, 27, and 22 were used for sequencing analysis for episomal iNSCs, sev iNSC1, and sev iNSC2, respectively. From these data, it is estimated that 9-13 mutations (SNVs and Indels) were acquired per cell cycle for the iNSC lines. 64 SNVs and 5 Indels were found commonly shared by all three iNSC lines, suggesting that these are probably hot spot mutations favored in the culture condition used in the current study. Nevertheless, all the SNVs and Indels detected in the three iNSC lines were predicted to have an overall harmless nature.
There were 9 pathogenic CNVs detected in episomal iNSCs. In contrast, no CNV was detected in sev iNSC1, or sev iNSC2. It is unclear whether it was the higher passage number of episomal iNSCs or the reprogramming method that had made the difference. Further studies are needed to address this issue.
For a cell source to be considered as donor cells in a clinical application, two minimal requirements need to be met. First, the tumorigenic risk should be minimal. The two sev iNSC lines did not show any SNVs or Indels in the coding regions of oncogenes, tumor suppressor genes, or neural stem cell genes. Engraftment into the striatum of immunodeficient mice for three months did not show any graft overgrowth. All these results suggest that the iNSCs obtained by Sendai virus method are quite safe. Second, the mutations should not affect the functions of transplanted cells pertaining to PD treatment, for example, the differentiation to DA neurons, the efficiency of differentiation, the secretion of dopamine from derived DA neurons, appropriate marker expression patterns on iNSCs and derived DA cells, and functional restoration in PD mouse models, etc. Two weeks after transplantation of iNSC-derived DA precursors, PD mouse models already showed significant improvement in symptoms. Another marked period of observed improvement was from 6 to 12 weeks post-transplantation. The curves in behavioral improvement may reflect the dynamic changes of implanted cells, and the collective output from the survival, differentiation, maturation, migration, and synaptic connection of those cells.
This study emphasizes the necessity of deep genomic sequencing before application of personalized therapy using reprogrammed stem cells. A thorough safety and efficacy assessment should be performed before any clinical application. Blood cell-derived, Sendai virus-mediated iNSCs can differentiate to DA precursors and improve symptoms in a mouse PD model. The cells seem to be safe in spite of some mutations accumulated from the reprogramming and expansion processes.
Abbreviations
AA: ascorbic acid; BDNF: brain derived neurotrophic factor; cAMP: cyclic adenosine monophosphate; CNVs: copy number variants; COMT: catechol-O-methyltransferase; DA: dopamine; DOPAC: 3,4-dihydroxyphenylacetic acid; FGF: fibroblast growth factor; FOXA2: forkhead box A2; GIRK2: G-protein-coupled inward rectifier potassium; HPLC: high-performance liquid chromatography; hrLIF: human recombinant leukemia inhibitor factor; HVA: homovanillic acid; Indels: insertions and deletions; MAO: monoamine oxidase; NSC: neural stem cells; NURR1: orphan nuclear receptor; PBMNCs: peripheral blood mononuclear cells; PD: Parkinson's disease; SHH: sonic hedgehog; SNVs: single-nucleotide variants; TGF-βIII: transforming growth factor βIII; TH: tyrosine hydroxylase; VAF: variant allele fraction.
Supplementary Material
Supplementary tables 1-4 and figures.
Supplementary table 5.
Supplementary table 6.
Supplementary table 7.
Acknowledgements
This work was supported by the Stem Cell and Translation National Key Project (2016YFA0101403), National Basic Research Program of China (2011CB965103, 2012CBA01307), National Natural Science Foundation of China (81661130160, 81422014, 81561138004, 3140090027), Natural Science Foundation of Beijing Municipal (5142005), Beijing Talents Foundation (2017000021223TD03), Support Project of High-level Teachers in Beijing Municipal Universities in the Period of 13th Five-year Plan (CIT&TCD20180333), Beijing Medical System High Level Talent Award (2015-3-063), the Royal Society-Newton Advanced Fellowship (NA150482), Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201706), and Natural Science Foundation of Shandong Province (2016ZDJS07A09).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Buttery PC, Barker RA. Treating Parkinson's disease in the 21st century: can stem cell transplantation compete? J Comp Neurol. 2014;522:2802-16
2. Williams-Gray CH, Evans JR, Goris A, Foltynie T, Ban M, Robbins TW. et al. The distinct cognitive syndromes of Parkinson's disease: 5 year follow-up of the CamPaIGN cohort. Brain. 2009;132:2958-69
3. Chen Z. Cell therapy for Parkinson's disease: New hope from reprogramming technologies. Aging Dis. 2015;6:499-503
4. Wijeyekoon R, Barker RA. Cell replacement therapy for Parkinson's disease. Biochim Biophys Acta. 2009;1792:688-702
5. Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R. et al. Transplantation of embryonic dopamine neurons for severe Parkinson's disease. N Engl J Med. 2001;344:710-9
6. Olanow CW, Goetz CG, Kordower JH, Stoessl AJ, Sossi V, Brin MF. et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson's disease. Ann Neurol. 2003;54:403-14
7. Lindvall O, Brundin P, Widner H, Rehncrona S, Gustavii B, Frackowiak R. et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson's disease. Science. 1990;247:574-7
8. Chen Z, Palmer TD. Cellular repair of CNS disorders: an immunological perspective. Hum Mol Genet. 2008;17:R84-92
9. Olanow CW, Kordower JH, Lang AE, Obeso JA. Dopaminergic transplantation for Parkinson's disease: current status and future prospects. Ann Neurol. 2009;66:591-6
10. Petit GH, Olsson TT, Brundin P. The future of cell therapies and brain repair: Parkinson's disease leads the way. Neuropathol Appl Neurobiol. 2014;40:60-70
11. Shin E, Tronci E, Carta M. Role of serotonin neurons in L-DOPA- and Graft-Induced Dyskinesia in a rat model of Parkinson's disease. Parkinsons Dis. 2012;2012:370190
12. Hallett PJ, Deleidi M, Astradsson A, Smith GA, Cooper O, Osborn TM. et al. Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson's disease. Cell Stem Cell. 2015;16:269-74
13. Wang S, Zou C, Fu L, Wang B, An J, Song G. et al. Autologous iPSC-derived dopamine neuron transplantation in a nonhuman primate Parkinson's disease model. Cell Discov. 2015;1:15012
14. Tang XH, Wang SY, Bai YF, Wu JY, Fu LL, Li M. et al. Conversion of adult human peripheral blood mononuclear cells into induced neural stem cell by using episomal vectors. Stem Cell Res. 2016;16:236-42
15. Sochacki J, Devalle S, Reis M, Mattos P, Rehen S. Generation of urine iPS cell lines from patients with Attention Deficit Hyperactivity Disorder (ADHD) using a non-integrative method. Stem Cell Res. 2016;17:102-6
16. Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348-62
17. Lu J, Liu H, Huang CT, Chen H, Du Z, Liu Y. et al. Generation of integration-free and region-specific neural progenitors from primate fibroblasts. Cell Rep. 2013;3:1580-91
18. Jiang H, Xu Z, Zhong P, Ren Y, Liang G, Schilling HA. et al. Cell cycle and p53 gate the direct conversion of human fibroblasts to dopaminergic neurons. Nat Commun. 2015;6:10100
19. da Conceicao FS, Ngo-Abdalla S, Houzel JC, Rehen SK. Murine model for Parkinson's disease: from 6-OH dopamine lesion to behavioral test. J Vis Exp. 2010;35:1376
20. Wu J, Sheng C, Liu Z, Jia W, Wang B, Li M. et al. Lmx1a enhances the effect of iNSCs in a PD model. Stem Cell Res. 2015;14:1-9
21. Zheng J, Choi KA, Bang PJ, Hyeon S, Kwon S, Moon JH. et al. A combination of small molecules directly reprograms mouse fibroblasts into neural stem cells. Biochem Bioph Res Co. 2016;476:42-8
22. Li W, Sun W, Zhang Y, Wei W, Ambasudhan R, Xia P. et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci U S A. 2011;108:8299-304
23. Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275-80
24. Cai J, Miao X, Li Y, Smith C, Tsang K, Cheng L. et al. Whole-genome sequencing identifies genetic variances in culture-expanded human mesenchymal stem cells. Stem Cell Reports. 2014;3:227-33
25. Bhutani K, Nazor KL, Williams R, Tran H, Dai H, Dzakula Z. et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat Commun. 2016;7:10536
26. Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C. et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15:12-3
27. Cheng L, Hansen NF, Zhao L, Du Y, Zou C, Donovan FX. et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell. 2012;10:337-44
28. Bauer S, Patterson PH. Leukemia inhibitory factor promotes neural stem cell self-renewal in the adult brain. J Neurosci. 2006;26:12089-99
29. Hitoshi S, Seaberg RM, Koscik C, Alexson T, Kusunoki S, Kanazawa I. et al. Primitive neural stem cells from the mammalian epiblast differentiate to definitive neural stem cells under the control of Notch signaling. Genes Dev. 2004;18:1806-11
30. Doi D, Samata B, Katsukawa M, Kikuchi T, Morizane A, Ono Y. et al. Isolation of human induced pluripotent stem cell-derived dopaminergic progenitors by cell sorting for successful transplantation. Stem Cell Reports. 2014;2:337-50
31. Chung S, Moon JI, Leung A, Aldrich D, Lukianov S, Kitayama Y. et al. ES cell-derived renewable and functional midbrain dopaminergic progenitors. Proc Natl Acad Sci U S A. 2011;108:9703-8
32. Fasano CA, Chambers SM, Lee G, Tomishima MJ, Studer L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell. 2010;6:336-47
33. Xi J, Liu Y, Liu H, Chen H, Emborg ME, Zhang SC. Specification of midbrain dopamine neurons from primate pluripotent stem cells. Stem Cells. 2012;30:1655-63
34. Oh SM, Chang MY, Song JJ, Rhee YH, Joe EH, Lee HS. et al. Combined Nurr1 and Foxa2 roles in the therapy of Parkinson's disease. EMBO Mol Med. 2015;7:510-25
35. Ganat YM, Calder EL, Kriks S, Nelander J, Tu EY, Jia F. et al. Identification of embryonic stem cell-derived midbrain dopaminergic neurons for engraftment. J Clin Invest. 2012;122:2928-39
36. Reyes S, Fu Y, Double K, Thompson L, Kirik D, Paxinos G. et al. GIRK2 expression in dopamine neurons of the substantia nigra and ventral tegmental area. J Comp Neurol. 2012;520:2591-607
37. Thompson L, Barraud P, Andersson E, Kirik D, Bjorklund A. Identification of dopaminergic neurons of nigral and ventral tegmental area subtypes in grafts of fetal ventral mesencephalon based on cell morphology, protein expression, and efferent projections. J Neurosci. 2005;25:6467-77
38. Kikuchi T, Morizane A, Doi D, Magotani H, Onoe H, Hayashi T. et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson's disease model. Nature. 2017;548:592-6
39. Chen Z, Phillips LK, Gould E, Campisi J, Lee SW, Ormerod BK. et al. MHC mismatch inhibits neurogenesis and neuron maturation in stem cell allografts. PLoS One. 2011;6:e14787
40. Ewen C. https://www.nature.com/news/reprogrammed-cells-relieve-parkinson-s-symptoms-in-trials-1.22531
41. Kumar M, Bagchi B, Gupta SK, Meena AS, Gressens P, Mani S. Neurospheres derived from human embryoid bodies treated with retinoic acid show an increase in nestin and Ngn2 expression that correlates with the proportion of tyrosine hydroxylase-positive cells. Stem Cells Dev. 2007;16:667-81
42. Sequerra EB, Miyakoshi LM, Froes MM, Menezes JRL, Hedin-Pereira C. Generation of glutamatergic neurons from postnatal and adult subventricular zone with pyramidal-like morphology. Cereb Cortex. 2010;20:2583-91
43. Azim K, Fischer B, Hurtado-Chong A, Draganova K, Cantu C, Zemke M. et al. Persistent Wnt/beta-catenin signaling determines dorsalization of the postnatal subventricular zone and neural stem cell specification into oligodendrocytes and glutamatergic neurons. Stem Cells. 2014;32:1301-12
44. Babu H, Cheung G, Kettenmann H, Palmer TD, Kempermann G. Enriched monolayer precursor cell cultures from micro-dissected adult mouse dentate gyrus yield functional granule cell-like neurons. Plos One. 2007;2:e388
45. Aubert J, Stavridis MP, Tweedie S, O'Reilly M, Vierlinger K, Li M. et al. Screening for mammalian neural genes via fluorescence-activated cell sorter purification of neural precursors from Sox1-gfp knock-in mice. Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11836-41
46. Wood HB, Episkopou V. Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech Develop. 1999;86:197-201
47. Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R. et al. A quantitative measurement of the human somatic mutation rate. Cancer Res. 2005;65:8111-7
48. Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26:345-52
Author contact
![]() Corresponding authors: Zhiguo Chen (chenzhiguoorg) or Y. Alex Zhang (yazorg)
Corresponding authors: Zhiguo Chen (chenzhiguoorg) or Y. Alex Zhang (yazorg)