Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(17):4695-4709. doi:10.7150/thno.26203 This issue Cite
Research Paper
Gαi1 and Gαi3mediate VEGF-induced VEGFR2 endocytosis, signaling and angiogenesis
Jian Sun1#, Wei Huang2#, Shuo-fei Yang3#, Xiao-pei Zhang2#, Qing Yu2, Zhi-qing Zhang4, Jin Yao2 ![]() , Ke-ran Li2
, Ke-ran Li2 ![]() , Qin Jiang2
, Qin Jiang2 ![]() , Cong Cao2,5,4
, Cong Cao2,5,4 ![]()
1. Department of Anesthesiology, Huai'an Maternity and Child Healthcare Hospital, Yangzhou University Medical School, Huai'an, China
2. The Fourth School of Clinical Medicine, The Affiliated Eye Hospital, Nanjing Medical University, Nanjing 210029, China
3. Department of Vascular Surgery, Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China. Pujian Road 160, Shanghai, China
4. Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, Suzhou, China.
5. North District, The Municipal Hospital of Suzhou, Suzhou, China
# Co-first authors.
Received 2018-3-21; Accepted 2018-8-17; Published 2018-9-9
Abstract

VEGF binding to VEGFR2 leads to VEGFR2 endocytosis and downstream signaling activation to promote angiogenesis.
Methods: Using genetic strategies, we tested the requirement of α subunits of heterotrimeric G proteins (Gαi1/3) in the process.
Results: Gαi1/3 are located in the VEGFR2 endocytosis complex (VEGFR2-Ephrin-B2-Dab2-PAR-3), where they are required for VEGFR2 endocytosis and downstream signaling transduction. Gαi1/3 knockdown, knockout or dominant negative mutation inhibited VEGF-induced VEGFR2 endocytosis, and downstream Akt-mTOR and Erk-MAPK activation. Functional studies show that Gαi1/3 shRNA inhibited VEGF-induced proliferation, invasion, migration and vessel-like tube formation of HUVECs. In vivo, Gαi1/3 shRNA lentivirus inhibited alkali burn-induced neovascularization in mouse cornea. Further, oxygen-induced retinopathy (OIR)-induced retinal neovascularization was inhibited by intravitreal injection of Gαi1/3 shRNA lentivirus. Moreover, in vivo angiogenesis by alkali burn and OIR was significantly attenuated in Gαi1/3 double knockout mice. Significantly, Gαi1/3 proteins are upregulated in proliferative retinal tissues of proliferative diabetic retinopathy (PDR) patients.
Conclusion: These results provide mechanistic insights into the critical role played by Gαi1/3 proteins in VEGF-induced VEGFR2 endocytosis, signaling and angiogenesis.
Keywords: VEGFR2, Gαi1, Gαi3, endocytosis, angiogenesis, signaling
Introduction
Vascular endothelial growth factor (VEGF) is crucial for vascular development and stimulates the formation of blood vessels [1]. The over-production of VEGF is associated with pathologic angiogenesis supporting tumor growth, acute/chronic inflammation, and proliferative retinopathy [1]. VEGF promotes endothelial cell survival, proliferation, migration and invasion [2, 3], required for the formation, function and maintenance of the vasculature [2, 3].
Two receptor tyrosine kinase (RTK) receptors have been identified for VEGF (VEGFR1 and VEGFR2). The biological function of VEGF is primarily mediated through the VEGF receptor-2 (VEGFR-2, also known as KDR) [1]. VEGF receptor-1 (VEGFR1) has been hypothesized to act as a negative regulator of angiogenesis as it triggers weak signal transduction [1, 4]. VEGF binding to VEGFR2 leads to tyrosine auto-phosphorylation of VEGFR2 [5-7], followed by the recruitment and activation of several key adaptor proteins, including Shc, growth factor receptor-binding protein 2 (Grb2), Grb-2-associated binder 1 (Gab1), SH2-domain containing protein tyrosine phosphatase 1 (SHP1) and SHP2 [5-7]. Activation of the adaptor proteins stimulates key downstream signaling cascades, including PI3K (phosphoinositide-3 kinase)-Akt-mTOR (mammalian target of rapamycin), Erk-MAPK (mitogen-activated protein kinase) and phospholipase C γ (PLCγ) signaling pathways [5-7], to induce the transcription of multiple angiogenesis-associated genes [5-7].
Recent studies show that VEGFR2 endocytosis is indispensable for VEGFR2 signal transduction [8, 9]. VEGFR2 internalization is reported to be regulated by Ephrin-B2, and its interacting protein, Dab2 (clathrin-associated protein disabled 2) as well as the cell polarity protein PAR-3 [8, 9]. The formation of this complex facilitates clathrin-mediated VEGFR2 endocytosis, which is required for downstream signal transduction [8, 9]. Inhibition of the VEGFR2 endocytosis complex suppresses VEGF-induced signaling and angiogenesis [8, 9].
The inhibitory subunit of the heterotrimeric guanine nucleotide-binding proteins (G proteins), or the Gαi proteins, have three members, Gαi1, Gαi2, and Gαi3 [10]. Gαi proteins couple with GPCRs (G protein coupled receptors) to inhibit adenylate cyclase (AC), leading to decreased cyclic AMP (cAMP) production [10]. Our group reported an unconventional function of Gαi1 and Gαi3 in mediating signal transduction of the EGFR (epidermal growth factor receptor) [11]. In response to EGF, Gαi1 and Gαi3 are recruited to the EGFR, to mediate downstream PI3K-Akt-mTORC1 signaling transduction [11]. In further studies, we show that Gαi1/3 play a pivotal role in the signaling of other RTKs, including fibroblast growth factor (FGFR) [12], keratinocyte growth factor receptors (KGFR) [13-15] and brain derived neurotrophic factor (BDNF) [16]. The current study aims to understand the potential role of Gαi1/3 in VEGFR2-induced signaling. Our results demonstrate that Gαi1/3 are required for VEGF-induced VEGFR2 endocytosis, signaling and angiogenesis.
Results
Gαi1 and Gαi3 double knockout abolishes VEGF-induced Akt-mTORC1 and Erk activation in mouse embryonic fibroblasts
To examine the potential role of Gαi proteins (Gαi1/3) in VEGF-induced signaling, we utilized Gαi1 and Gαi3 double knockout (“DKO”) mouse embryonic fibroblasts (MEFs, an immortalized cell line) [11, 13, 16]. As shown in Figure S1A, Western blotting assay confirmed Gαi1 and Gαi3 deficiency in the DKO MEFs. Both WT and DKO MEFs were treated with VEGF (“-A”, 25 ng/mL). As shown, the level of VEGFR2 expression and VEGF-induced VEGFR2 phosphorylation (“p-”, at Tyr-1175) were equivalent between WT and DKO MEFs (Figure S1A, P>0.05 vs. WT MEFs). However, VEGF-induced downstream Akt activation was almost completely blocked in the DKO MEFs, tested using phospho-specific antibodies against Akt phosphorylation at both Ser-473 and Thr-308 residues (Figure S1B, P< 0.05 vs. WT MEFs). Verifying the above data, GSK3α/β phosphorylation, a well-known Akt downstream target, was largely inhibited as well (Figure S1B, P< 0.05 vs. WT MEFs). The expressions of total Akt1/2 and GSK3β in DKO MEFs were comparable to that of the WT MEFs (Figure S1B).
Similarly, VEGF-induced activation of mTOR complex 1 (mTORC1) was blocked in the DKO MEFs. As shown in Figure S1C, VEGF-induced phosphorylation of p70 S6 kinase 1(“S6K1”, at Thr-389), the primary substrate of mTORC1 [17], and S6 ribosomal protein (“S6”, at Ser-235/236) were significantly increased in the WT MEFs, but not in the DKO MEFs (P< 0.05 vs. WT MEFs). Total S6K1 and S6 protein levels were similar between WT and DKO MEFs (Figure S1C).
We next examined Erk-MAPK signaling, a keyVEGFR2 downstream signaling pathway [7]. VEGF-induced Erk activation, as detected using an antibody against p-Erk1/2 (Thr202/Tyr-204), was largely attenuated in the Gαi1 and Gαi3 DKO MEFs (Figure S1D, P< 0.05 vs. WT MEFs).
Gαi1 and Gαi3 are required for VEGF-induced Akt-mTORC1 and Erk activation in mouse embryonic fibroblasts
To study the individual roles of Gαi1 or Gαi3 in VEGF-induced signaling, Gαi1 or Gαi3 single knockout (SKO) MEFs were utilized [11-13, 16]. Results show that, in response to VEGF, the phosphorylation of Akt, S6 and Erk1/2 were reduced in the Gαi1 or Gαi3 SKO MEFs (Figure 1A, P< 0.05 vs. WT MEFs), but completely blocked in the DKO MEFs (Figure 1A, P< 0.05 vs. WT MEFs). Interestingly, in the MEFs, Gαi3 SKO resulted in a larger reduction of VEGF signaling compared to Gαi1 SKO (Figure 1A). In contrast to Gαi1 or Gαi3 SKO, Gαi2 SKO showed no significant effect on VEGF-induced signaling (Figure 1B). VEGF-induced phosphorylation of Akt, S6 and Erk1/2 were equivalent between WT and Gαi2 SKO MEFs (Figure 1B, P> 0.05 vs. WT MEFs).
Gαi1 and Gαi3 are required for VEGF-induced Akt-mTORC1 and Erk activation in mouse embryonic fibroblasts. (A-B) WT MEFs, Gαi1 or Gαi3 single knockout (SKO) MEFs, Gαi2 SKO MEFs, and Gαi1/3 double knockout (DKO) MEFs were treated with VEGF (“-A”, 25 ng/mL) for indicated times, and were tested by Western blotting assay of listed proteins. (C-D) DKO MEFs were transiently transfected with the vector encoding Ad-Gαi1, Ad-Gαi3 or empty vector (pDC315); VEGF (“-A”, 25 ng/mL)-induced signaling was tested. The same sets of lysate samples were run in sister gels to test different proteins (same for all figures). Indicated protein was quantified via ImageJ software (same for all figures). “n=5” means quantification from five replicate blot data (same for all figures). Bars stand for mean ± SD (same for all Figures).*P< 0.05 vs. WT MEFs. #P< 0.05 in (A). #P< 0.05 vs. Vector MEFs in (D).
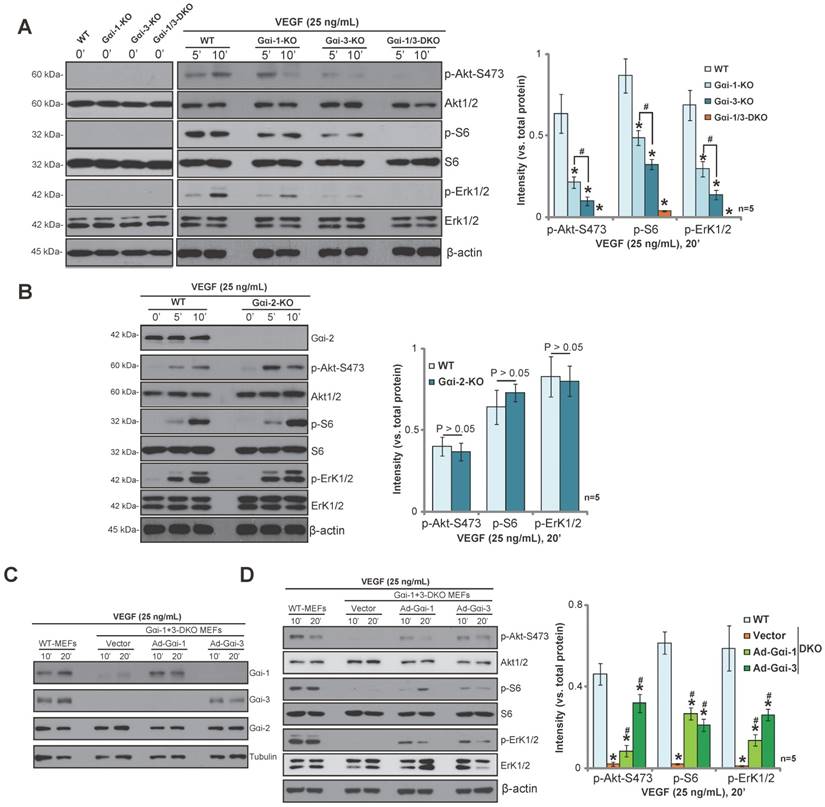
To further demonstrate the requirement for Gαi1 and Gαi3 in VEGF-induced signaling, adenovirus Gαi1 construct (“Ad-Gαi1”, no Tag) or adenovirus Gαi3 construct (“Ad-Gαi3”, no Tag) was exogenously expressed in the DKO MEFs. Western blotting confirmed the expression of exogenous Ad-Gαi1 and Ad-Gαi3 in the DKO MEFs (Figure 1C). After re-expression of Gαi1 or Gαi3, VEGF-induced Akt-mTORC1 and Erk1/2 activation were partially restored in DKO MEFs (Figure 1D). These results further demonstrate that Gαi1 and Gαi3 are required for VEGF-induced Akt-mTORC1 and Erk activation in MEFs.
Knockdown of Gαi1/3 inhibits VEGF-induced Akt-mTORC1 and Erk activation in mouse embryonic fibroblasts
We next used a RNA interference (RNAi) strategy to knockdown Gαi proteins in MEFs. Knockdown of Gαi1 or Gαi3 by targeted siRNA resulted in a partial decrease in phosphorylation of Akt (Ser-473), GSK3α/β, S6 and Erk1/2 in response to VEGF (Figure S2A). Gαi3 siRNA was more potent than Gαi1 siRNA in decreasing VEGF-induced signaling (Figure S2A, see quantification results). We then knocked down both Gαi1 and Gαi3 in MEFs, using a short hairpin RNA (shRNA) strategy. Gαi1 and Gαi3 shRNA lentivirus were added to WT MEFs, and stable MEFs were selected using puromycin [11, 13, 15, 16]. Western blotting results show a substantial knockdown of both Gαi1 (over 95%) and Gαi3 (over 90%) in stable MEFs with the lentivirus (Figure S2B). Consequently, VEGF-induced phosphorylation of Akt (Ser-473), GSK3α/β, S6 and Erk1/2 were significantly inhibited (Figure S2B, P< 0.05 vs. scramble control shRNA). Thus, siRNA/shRNA-mediated knockdown of Gαi1/3 inhibits VEGF-induced signaling in MEFs, further supporting a role of Gαi1/3 proteins in VEGFR2 signaling.
RTKs can be trans-activated by ligand-stimulated GPCRs [18]. To rule out this possibility, we treated MEFs with pertussis toxin (PTX), which ADP-ribosylates Gαi proteins to interfere with GPCR-dependent signaling. PTX had no effect on VEGF-induced phosphorylation of Akt (473S) and Erk1/2 (Figure S2C, P>0.05 vs. vehicle control).
Gαi1 and Gαi3 are required for VEGF-induced VEGFR2 endocytosis in mouse embryonic fibroblasts
Following VEGF binding, VEGFR2 is endocytosed via the clathrin-mediated pathway [8, 9, 19, 20], which is an indispensable step for downstream signal transduction [8, 9, 19, 20]. Activated VEGFR2 forms a multi-protein complex with Ephrin-B2 (a B-class ephrin subfamily transmembrane protein), the clathrin-associated protein disabled 2 (Dab2), and the cell polarity regulator PAR-3, resulting in clathrin-mediated VEGFR2 endocytosis [8, 9, 19, 20]. In line with these findings, we show that VEGFR2 co-immunoprecipitated with Ephrin-B2, Dab2 and PAR-3 in response to VEGF in WT MEFs (Figure 2A). Significantly, both Gαi1 and Gαi3 were located in the VEGFR2 endocytosis complex (Figure 2A). In response to VEGF, Gαi1/3 co-immunoprecipitated with the VEGFR2-Ephrin-B2-Dab2-PAR-3 complex (Figure 2B). Importantly, VEGF-activated VEGFR2 was unable to form a complex with Ephrin-B2-Dab2-PAR-3 when Gαi1/3 were absent (Figure 2C, P< 0.05 vs. WT cells). As a result, VEGF-induced VEGFR2 endocytosis, reflected by reduced VEGFR2 expression on the cell plasma membrane, was inhibited in DKO MEFs (Figure 2D). VEGFR endocytosis complex proteins, Ephrin-B2, Dab2 and PAR-3, were equivalent between WT and DKO MEFs (Figure 2D).
Gαi1 and Gαi3 are required for VEGF-induced VEGFR2 endocytosis in mouse embryonic fibroblasts. (A-B) WT MEFs were treated with VEGF (“-A”, 25 ng/mL) for indicated times; associations between VEGFR2, p-VEGFR2, Ephrin-B2, Dab2, PAR-3, Gαi1 and Gαi3 were tested by co-immunoprecipitation (“IP”) assay. (C) WT and DKO MEFs were treated with VEGF (“-A”, 25 ng/mL) for 5 min;VEGFR2, p-VEGFR2, Ephrin-B2, Dab2, PAR-3, Gαi1 and Gαi3 associations are shown. (D) The expressions of listed proteins in WT and DKO MEFs, treated with/out VEGF (“-A”, 25 ng/mL) for 5 min, were tested by Western blotting assay. (E) Stable WT-MEFs with empty vector (“Vec”) or dominant negative Gαi3 (G202T) (“DN-Gαi3”) were treated with VEGF (“-A”, 25 ng/mL) for 5 min; associations between VEGFR2, Ephrin-B2, Dab2, PAR-3 and Gαi3 were tested by co-immunoprecipitation (“IP”) assay. (F) The expressions of listed proteins in total cell lysates were also tested.*P< 0.05 vs. WT MEFs in (C). #P< 0.05 vs. untreated control (“C”) cells in (D). #P< 0.05 vs. “Vec” in (E-F).
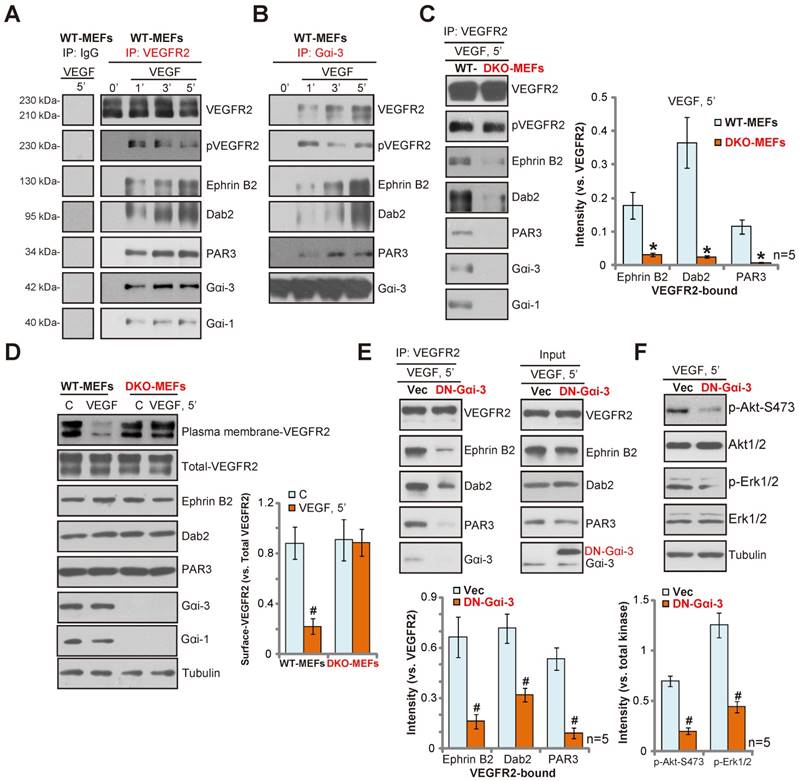
To further test the role of Gαi proteins in VEGFR endocytosis, an adenovirus dominant negative Gαi3 construct (“DN-Gαi3”, tagged with Flag), substituting a conserved Gly(G) residue with Thr (T) in the G3 box of Gαi3 [11, 13], was introduced into MEFs. This mutation prevents Gαi3 binding to adaptor/associated proteins [11, 21, 22]. We show that forced expression of the DN-Gαi3 disrupted the formation of the VEGFR2 endocytosis complex(VEGFR2-Ephrin-B2-Dab2-PAR-3) in WT MEFs (Figure 2E). Expression of the endocytosis complex proteins were unchanged by DN-Gαi3 expression (Figure 2E, “Input”). Consequently, VEGF-induced phosphorylation of Akt and Erk were also inhibited by DN-Gαi3 (Figure 2F, P< 0.05 vs. vector control). Overall, these results suggest that Gαi1/3 play a key role in the formation of the VEGFR2 endocytosis complex, required for VEGF-induced VEGFR2 endocytosis.
Gab1 lies downstream of Gαi1/3 in VEGFR2 signaling in mouse embryonic fibroblasts
After VEGF stimulation, endocytosed VEGFR2 binds to the key adaptor protein Grb-2-associated binder 1 (Gab1). Gab1-p85 association is required for PI3K-Akt-mTOR activation by VEGFR2 [23] and Gab1-SHP2 association is essential for Erk-MAPK activation [24]. As Gαi1/3 association with VEGFR2 is required for VEGFR2 endocytosis, we determined whether Gαi1/3 are also required for VEGF-induced Gab1 activation. Performing a titration and time course, we show that VEGF-induced Gab1 activation, tested by examining Tyr-627 phosphorylation, was almost blocked in Gαi1/3 DKO MEFs (Figure S3A-B). Similarly, knockdown of both Gαi1 and Gαi3 in WT MEFs attenuated Gab1 activation by VEGF (Figure S3C). Gαi1 or Gαi3 SKO resulted in partial inhibition of Gab1 activation by VEGF (Figure S3D), and Gαi2 SKO had no significant effect on VEGF-induced Gab1 phosphorylation (Figure S3E). Notably, VEGF (25 ng/mL)-induced phosphorylation of Akt, S6 and Erk1/2 were almost completely blocked in Gab1 knockout MEFs (Figure S3F). Abrogation of Gab1 signaling in Gαi1/3-depleted cells could explain the impairment of downstream Akt-mTOR and Erk-MAPK cascades in response to VEGF.
shRNA-mediated knockdown of Gαi1/3 inhibits VEGF-induced VEGFR2 endocytosis and downstream signaling activation in HUVECs
To study the role of Gαi1 or Gαi3 in VEGFR2 signaling in endothelial cells, a shRNA strategy was applied to knockdown Gαi1/3 in primary cultured human umbilical vein endothelial cells (HUVECs). Gαi1-shRNA and/or Gαi3-shRNA lentivirus were added to primary human HUVECs, and stable cells were selected with puromycin. As shown in Figure 3A, the applied shRNA lentivirus induced downregulation of Gαi1 (over 90%) and Gαi3 (over 99%) in HUVECs. VEGFR2 expression, Gαi2 expression, and VEGF-induced VEGFR2 phosphorylation were unaffected by Gαi1/3 shRNA (Figure 3A). VEGF-induced phosphorylation of Gab1, Akt, S6 and Erk1/2 were all decreased in stable HUVECs with Gαi1 or Gαi3 shRNA (Figure 3B, P< 0.05 vs. control shRNA). Gαi3 shRNA was more potent than Gαi1 shRNA in inhibiting VEGF-induced downstream signalings (Figure 3B, quantification results). The combined knockdown of Gαi1 and Gαi3 resulted in further inhibition of VEGFR2 signalings (Figure 3B).
shRNA-mediated knockdown of Gαi1/3 inhibits VEGF-induced VEGFR2 endocytosis and downstream signaling activation in HUVECs. (A-B) Stable HUVECs, expressing scramble control shRNA (“scr-shRNA”), Gαi1 shRNA, Gαi3 shRNA or Gαi1 plus Gαi3 shRNA, were treated with VEGF (“-A”, 25 ng/mL) for indicated times, and were tested by Western blotting assay of the listed proteins. (C) VEGFR2 associations with Ephrin-B2, Dab2 and PAR-3 as well as Gαi1 and Gαi3 were tested by co-immunoprecipitation assay. (D) VEGFR2 expression in cell plasma membrane and in total cell lysates, along with the VEGFR2 endocytosis complex protein in total cell lysates were also tested. (E) Stable HUVECs expressing WT-Gαi3 (Ad-Gαi3) or the empty vector (“pDC315”), were treated with VEGF (“-A”, 25 ng/mL) for indicated times, and were tested by Western blotting assay of the listed proteins.*P< 0.05 vs. HUVECs with “scr-shRNA” (B-C).# P< 0.05in (B). # P< 0.05 vs. “C” in (D).# P< 0.05 in (E).
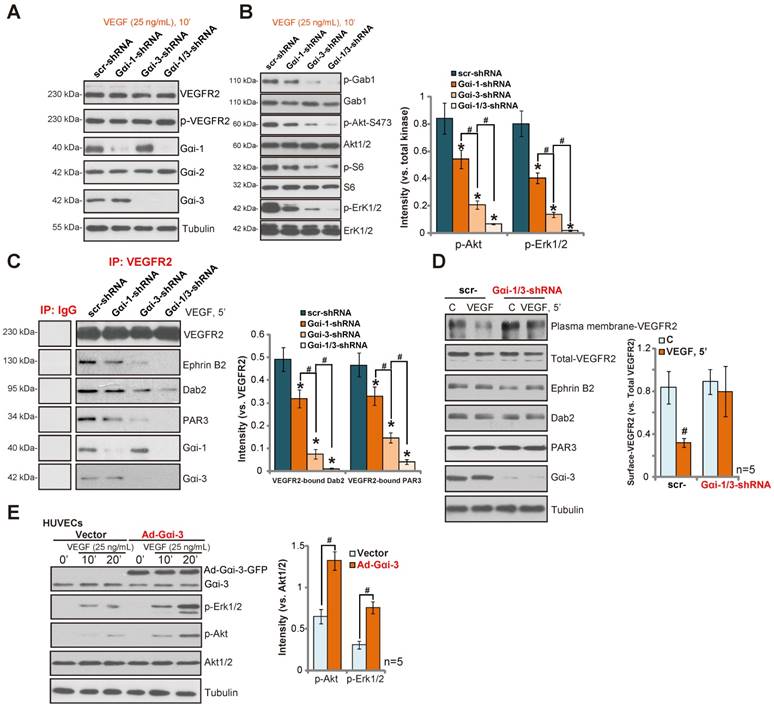
In HUVECs, VEGF-activated VEGFR2 forms an endocytosis complex, as shown by VEGF co-immunoprecipitation with Gαi1, Gαi3, Ephrin-B2, Dab2 and PAR-3 (Figure 3C). shRNA-mediated knockdown of both Gαi1 and Gαi3 disrupted the VEGFR2 endocytosis complex (VEGFR2-Ephrin-B2-Dab2-PAR-3) (Figure 3C). Correspondingly, VEGF-induced VEGFR2 endocytosis was inhibited, as the cell plasma membrane VEGFR2 level was not significantly reduced (Figure 3D). VEGFR2, Ephrin-B2, Dab2 and PAR-3 expressions were unaffected by Gαi1 and Gαi3 double knockdown (Figure 3D). Significantly, Gαi3 shRNA was more efficient in disrupting VEGF-induced VEGFR2 endocytosis complex (VEGFR2-Ephrin-B2-Dab2-PAR-3) formation than Gαi1 shRNA (Figure 3C). On the other hand, forced over-expression of Gαi3 (an adenovirus construct (“Ad-Gαi-3-Flag” [16]) in HUVECs increased the phosphorylation of Akt (Ser-473) and Erk1/2 in response to VEGF (Figure 3E, P< 0.05 vs. empty vector cells). Together, these results indicate that Gαi1/3 are required for VEGF-induced signaling in HUVECs.
shRNA-mediated knockdown of Gαi1/3 inhibits VEGF-induced proliferation, invasion, migration and vessel-like tube formation in HUVECs
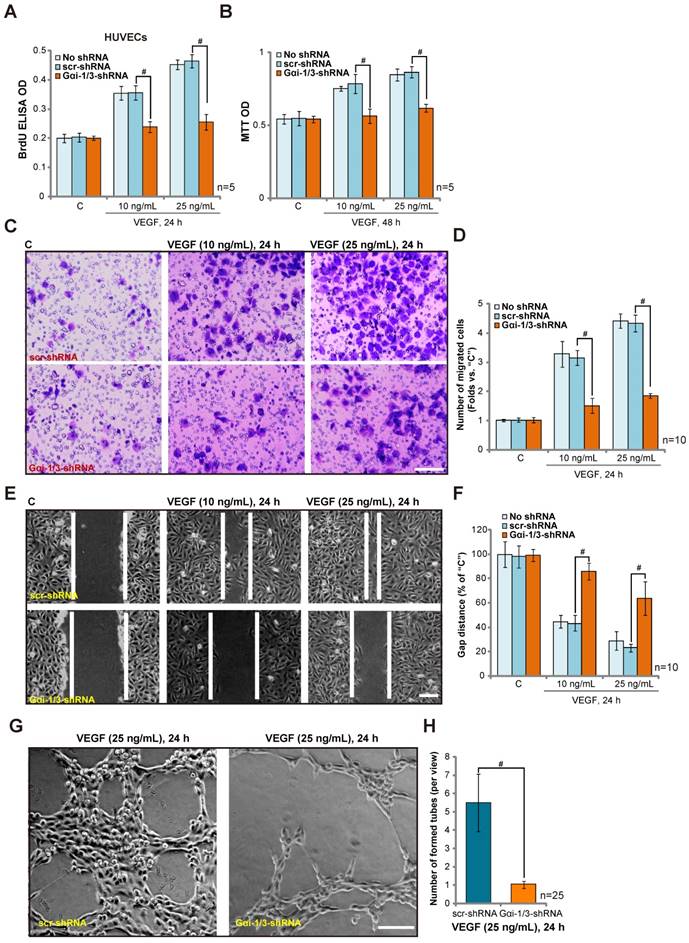
We next tested the potential role of Gαi1 and Gαi3 in endothelial cell function in vitro. Activation of VEGFR2 promotes endothelial cell invasion, migration, proliferation and angiogenesis [6]. Treatment with VEGF (10/25 ng/mL) promoted the proliferation of control HUVECs, reflected by an increase of BrdU ELISA optical density (OD) (Figure 4A) and MTT OD (Figure 4B). Significantly, the combined knockdown of Gαi1 and Gαi3 by targeted shRNA lentivirus (see Figure 3) inhibited VEGF-induced HUVEC proliferation (Figure 4A-B, P< 0.05 vs. control shRNA). In the process of neovascularization, newly-generated endothelial cells migrate to form vessel-like tube structures [25, 26]. As shown in Figure 4C, a Transwell invasion assay demonstrates that VEGF stimulation can promote the invasion of HUVECs, which was significantly inhibited by Gαi1 and Gαi3 shRNA (Figure 4C-D, P< 0.05 vs. control shRNA). Furthermore, results using a “scratch wound healing” assay, Figure 4E, show that Gαi1 and Gαi3 knockdown significantly decreased VEGF-induced HUVEC migration distance (Figure 4F, P< 0.05 vs. control shRNA). A scramble control shRNA (“scr-shRNA”) had no effect on HUVEC proliferation (Figure 4A-B) nor migration (Figure 4C-F). VEGF-induced HUVEC tube formation, tested by an in vitro Matrigel-based capillary-genesis assay, was dramatically inhibited by Gαi1 and Gαi3 shRNA (Figure 4G-H). Notably, Gαi3 shRNA was more potent than Gαi1 shRNA in inhibiting VEGF-induced cell proliferation (Figure S4A-B), migration (Figure S4C) and vessel-like tube formation (Figure S4D). Together, the functional studies show that Gαi1/3 shRNA inhibits VEGF-induced proliferation, invasion, migration and vessel-like tube formation in HUVECs.
shRNA-mediated knockdown of Gαi1/3 inhibits VEGF-induced proliferation, invasion, migration and vessel-like tube formation in HUVECs. Stable HUVECs, expressing scramble control shRNA (“scr-shRNA”) or lentiviral Gαi1 plus Gαi3 shRNA, were treated with/without VEGF (“-A”, 10/25 ng/mL) for indicated times. Cell proliferation was tested by BrdU ELISA assay (A) and MTT OD assay (B). Cell invasion and migration were tested by Transwell assay (C-D) and scratch wound healing assay (E-F), respectively. HUVEC vessel-like tube formation was tested by the Matrigel-based capillary-genesis assay (G-H). For the Transwell assay, ten random views of each condition were included to calculate the average number of invaded cells. For the scratch wound healing assay assay, ten random views of each condition were included to calculate the average migration distance. For the vessel-like tube formation assay, 25 random views were included to calculate the average number of formed vessels. #P< 0.05. Bar=100 μm.
Gαi1 and Gαi3 shRNA inhibits in vivo angiogenesis
Alkali injuries are common to the human cornea, resulting in angiogenic responses. An alkali burn will lead to over-production of VEGF, which induces endothelial cell proliferation and corneal neovascularization (“CoNV”) (see our previous study [27]). Seven days after alkali burn, a significant CoNV was detected in the burned mouse cornea (Figure 5A), which was largely inhibited by eyedrops of Gαi1 and Gαi3 shRNA lentivirus (Figure 5A-B) (P< 0.05 vs. control shRNA, n=10). We also examined the effects of Gαi1 and Gαi3 shRNA lentivirus on the oxygen-induced retinopathy (OIR) model, an in vivo retinal angiogenesis model [28]. Mice at P12 were exposed to 75% oxygen in a hyperoxic chamber for 5 days, which leads to inhibition of the vasculature in the central retina, causing a central avascular area. The return of mice to normoxic conditions (21% oxygen) results in a massive production of VEGF [28, 29]. This causes a retinal vasculature proliferative response, leading to revascularization of the central avascular area, reflected by the development of pathological neovascular tufts on the vitreal side of the internal limiting membrane.
Gαi1 and Gαi3 shRNA inhibits in vivo angiogenesis. Eight-week-old male mice who had received a corneal alkali burn were treated with Gαi1 and Gαi3 shRNA lentivirus or scramble control shRNA (scr-shRNA). On day 7, corneal neovascularization (CoNV) was observed by slit-lamp (A, bar=1 mm), and neovascularization was quantified (B). (C-D) Mice injected with Gαi1 and Gαi3 shRNA lentivirus at P12 in the OIR model following removal from a 75% oxygen chamber had a significantly increased retinal vaso-obliteration ratio (VO%) on P17 than littermates injected with scramble control shRNA (scr-shRNA) (Bar=500 μm). Gαi1 and Gαi3 shRNA injection inhibited normal retinal vascular development at P17 as well. (E-F) Expressions of listed proteins in the retinal tissues of listed groups are shown. “No shRNA” stands for no shRNA lentivirus injected. #P< 0.05 in (E-F).
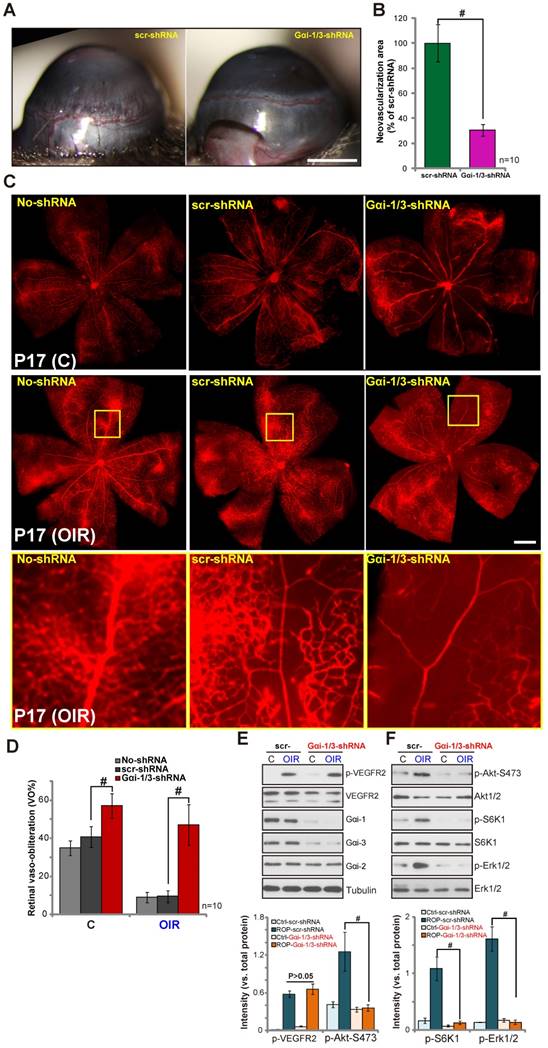
We observed a significant retinal neovasculation in mice exposed to OIR (Figure 5C). OIR-induced retinal angiogenesis was quantified via reduction of retinal vaso-obliteration (VO%) using the previously described method [29]. As shown, intravitreal injection of scramble shRNA only slightly inhibited normal retinal vascular development and OIR-induced retinal angiogenesis (P17, as compared to retinas without shRNA injection (“No shRNA”) (Figure 5C-D). Significantly, OIR-induced retinal angiogenesis was almost completely blocked by Gαi1 shRNA and Gαi3 shRNA (Figure 5C-D) (P< 0.05 vs. control shRNA, n=10). Gαi1 shRNA and Gαi3 shRNA injection also inhibited normal retinal vascular development (Figure 5C-D). The results are in line with the known function of VEGF signaling for the vascular development process [30, 31]. Together, these results indicate that Gαi1 and Gαi3 shRNA inhibits in vivo angiogenesis.
The retinal tissues of control (normoxia) and OIR model mice (four mice per group) were isolated and analyzed biochemically. As shown in Figure 5E, Gαi1 and Gαi3 shRNA caused over 95% reduction of Gαi1 and Gαi3 expression in the mouse retinas (as compared to the control shRNA retinas). Gαi2 expression was unchanged (Figure 5E). Gαi1 and Gαi3 shRNA had no effect on VEGFR2 expression and OIR-induced VEGFR2 phosphorylation (Figure 5E) but resulted in over 90% inhibition of downstream Akt, S6K1 and Erk1/2 phosphorylation (as compared to the control shRNA retinas, Figure 5F). Thus, Gαi1 and Gαi3 shRNA inhibited Akt and Erk1/2 activation in retinal tissues of OIR mice.
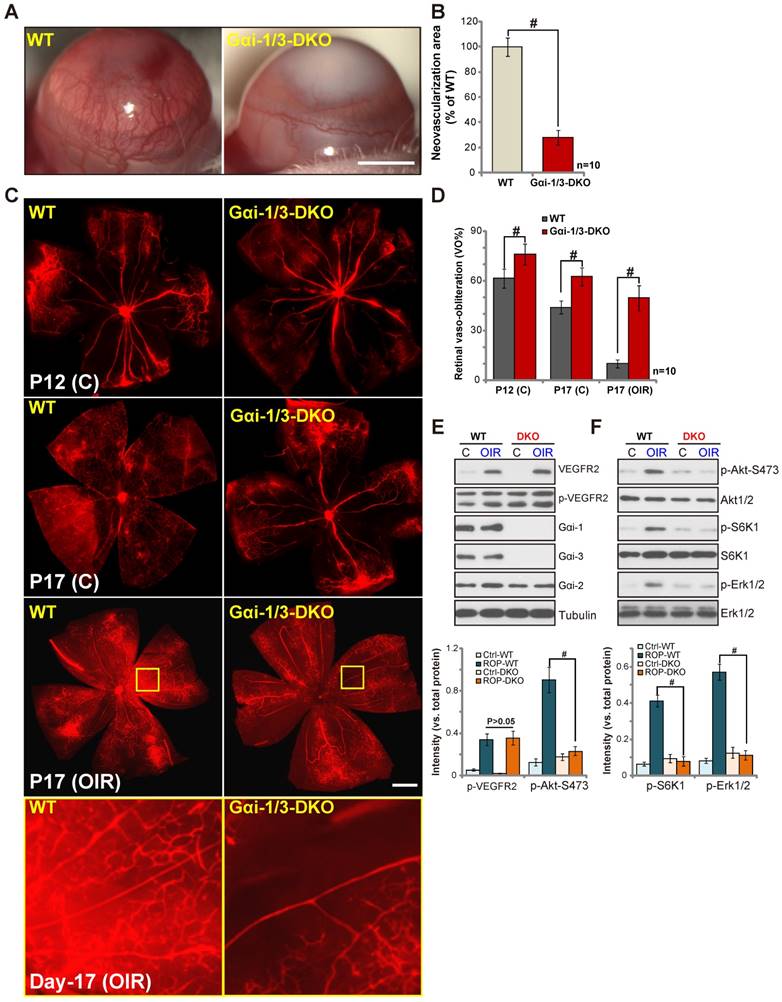
In vivo angiogenesis is impaired in Gαi1 and Gαi3 DKO mice
We generated Gαi1/Gαi3 DKO mice using the CRISPR-Cas-9 method (see our previous study [16]). The alkali burn induced significant CoNV in the wild type (“WT”) mice (Figure 6A), whereas neovascularization was largely attenuated in the DKO mice (Figure 6A-B, P< 0.05 vs. WT mice). Similarly, examining the OIR mice, we show that OIR-induced retinal neovasculation observed in the WT mice (Figure 6C), was almost completely blocked in the DKO mice (Figure 6C-D, P< 0.05 vs. WT mice). Further, normal retinal physiological angiogenesis was also inhibited in Gαi1 and Gαi3 DKO mice at P12 and P17 (Figure 6C-D). When analyzing signaling changes in the retinal tissues (four mice per group), we show that Gαi1/Gαi3 DKO (Figure 6E) blocked OIR-induced Akt, S6K1 and Erk1/2 phosphorylation in vivo (Figure 6F). VEGFR2 expression and OIR-induced VEGFR2 phosphorylation, as well as Gαi2 expression, were equivalent between WT and DKO mice (Figure 6E). The genetic evidence further confirm that Gαi1 and Gαi3 are required for angiogenesis and VEGF signaling in vivo.
In vivo angiogenesis is impaired in Gαi1 and Gαi3 DKO mice. Eight-week-old wild-type (“WT”) and Gαi1 and Gαi3 double knockout (“DKO”) mice received a corneal alkali burn. On day 7, corneal neovascularization (CoNV) was observed by slit-lamp (A, bar=1 mm), and neovascularization was quantified (B). (C-D) Mice were subjected to OIR; representative images are shown at P12/P17, and retinal vaso-obliteration ratios (VO%) were quantified. Normal retinal physiological angiogenesis was inhibited in the DKO mice at P12 and P17 (Bar=500 μm). (E-F) Expressions of listed proteins in the retinal tissues of listed groups are shown. #P< 0.05.
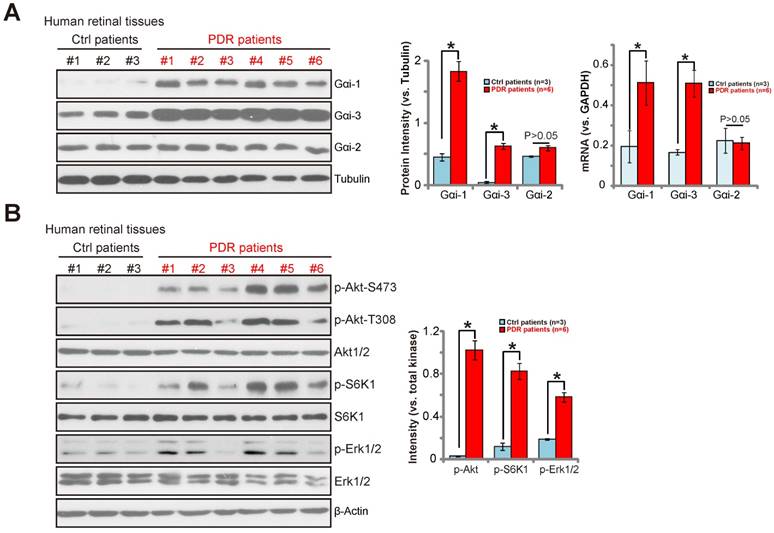
Gαi1 and Gαi3 upregulation in proliferative retinal tissues in PDR patients
VEGF is considered one primary factor involved in neovascularization in the proliferative diabetic retinopathy (PDR) [32]. Increased level of VEGF has been reported in the vitreous and fibro-vascular tissues from eyes with PDR [33]. We thus investigated whether Gαi1 and Gαi3 are dysregulated in the proliferative retinas of human patients. As described, a total of six retinal proliferative membrane tissues from PDR patients as well as three retinas from age-matched traumatic retinectomy patients were tested. Expression of Gαi1 and Gαi3 in human retinal tissues was tested by the Western blotting assay and quantitative real-time PCR assay (qRT-PCR) assay. Results in Figure 7A (the left panel) demonstrate that Gαi1 and Gαi3 proteins were clearly upregulated in the proliferative retinal tissues (P< 0.05, as compared to the retinas from control patients). Furthermore, Gαi1 and Gαi3 mRNA were also increased in proliferative retinal tissues of PDR patients (P< 0.05, as compared to the retinas from control patients) (Figure 7A, right panel). However, Gαi2 expression was equivalent between the two groups (Figure 7A). Further analysis of the retinal tissues shows that phosphorylation of Akt, S6K1 and Erk1/2 were significantly increased in the proliferative retinal tissues of PDR patients (Figure 7B, P< 0.05). Collectively, the results show that Gαi1/3 upregulation in proliferative retinal tissues of PDR patients is correlated with Akt-mTOR and Erk1/2 over-activation, indicating a potential role of Gαi1/3 in the pathogenesis of PDR.
Gαi1 and Gαi3 upregulation in proliferative retinal tissues of PDR patients. The proliferative retinal membrane of six proliferative diabetic retinopathy (PDR) patients and retinas of three age-matched traumatic retinectomy patients were homogenized, and lysates were tested by Western blotting assay of the listed proteins (A-B) and Gαi/2/3 mRNAs (A, right panel). Expressions of the listed proteins were quantified using ImageJ software.*P< 0.05.
Discussion
In the pathogenesis of proliferative retinopathy, expression of VEGF and binding to the VEGFR2 receptor leads to receptor endocytosis and activation of multiple downstream signal pathways, including Erk, PI3K-Akt-mTOR, and PLCγ [34, 35]. Activation of these cascades promotes vascular cell proliferation, migration and vascularization [34, 35]. While bevacizumab and other VEGFR2 antibodies have displayed some success in treating proliferative retinopathy diseases, a large proportion of patients either develop resistance or do not respond well to VEGFR inhibition [34, 36]. A better understanding of the underlying mechanisms of VEGFR2 signaling is essential to develop novel and effective therapeutic strategies.
Our results provide compelling evidence that Gαi1 and Gαi3 are essential players in VEGFR2 signaling. Knockout or shRNA knockdown of Gαi1 and Gαi3 significantly inhibited VEGF-induced Akt-mTORC1 and Erk activation in MEFs. Re-expression of Gαi1 or Gαi3 restored VEGFR2 signaling in DKO MEFs. Using primary cultured HUVECs, shRNA-mediated knockdown of Gαi1 and/or Gαi3 also inhibited Akt-mTORC1 and Erk activation in response to VEGF. In contrast, ectopic overexpression of wild-type Gαi3 resulted in enhancement of VEGF-induced Akt-mTORC1 and Erk activation in the vascular cells. Activated VEGFR2 recruits Gab1, causing its phosphorylation and activation [23, 24, 37], and Gab1 recruitment of additional adaptor proteins [23, 24, 37]. Gαi1/3 knockout/knockdown largely attenuated VEGF-induced Gab1 activation in HUVECs and MEFs.
Mechanistically, we show that Gαi1 and Gαi3 are required for VEGF-induced VEGFR2 endocytosis. Following VEGF treatment, VEGFR2 is internalized within the clathrin-coated vesicles, an indispensable step for VEGF-induced signaling activation [9, 19]. In response to VEGF stimulation, we show that Gαi1 and Gαi3 co-immunoprecipitated with the VEGFR2 endocytosis complex (VEGFR2-Ephrin-B2-Dab2-PAR-3) in MEFs and HUVECs. Depletion, knockdown or mutation of Gαi1/3 disrupted the VEGF-induced VEGFR2 endocytosis complex formation. Further, Gαi1/3 silencing inhibited VEGF-induced VEGFR2 endocytosis, and VEGF-induced in vitro and in vivo angiogenesis as well as proliferation, migration, invasion and vessel like tube-formation of HUVECs. Hence, Gαi1 and Gαi3 are essential players in VEGF signaling possibly via facilitating VEGFR2 endocytosis.
Other studies have found that VEGFR2 can also be internalized through a clathrin-independent alternative route [38, 39]. VEGFR2 degradation persists even when the clathrin pathway is blocked. Macropinocytosis of VEGFR2 was also reported, and VEGF-induced VEGFR2 endocytosis was largely inhibited by EIPA, a commonly used inhibitor of macropinocytosis [38, 39]. Inhibition of macropinocytosis of VEGFR2 was also shown to inhibit VEGFR2 downstream signaling, including Akt and Erk cascades [38, 39]. We show that VEGF-induced VEGFR2 endocytosis and downstream signaling activation were blocked by Gαi1/3 knockout/knockdown. Thus, it is possible that Gαi1/3 could be required for both clathrin-dependent and -independent VEGFR2 endocytosis routes, to mediate downstream signaling transduction.
Importantly, Gαi1 and Gαi3 are upregulated in proliferative retinal tissues of PDR patients, and their upregulation is correlated with over-activation of Akt-S6K1 and Erk. In the animal models of OIR and alkali burn-induced neovascularization, Gαi1 and Gαi3 shRNA or knockout significantly inhibited angiogenesis in vivo. The present work suggests that inhibition of Gαi1 and Gαi3 in endothelial cells could provide a novel approach for the treatment of PDR and other proliferative retinal diseases.
Methods
Ethics
The study was approved by the Ethics Review Board of all authors' institutions.
Materials
Pertussis toxin (PTX) was purchased from Sigma (Shanghai, China). VEGF (-A), EGF and platelet-derived growth factor (PDGF)-BB were provided by Calbiochem (Shanghai, China). Puromycin was purchased from Sigma-Aldrich (Shanghai, China). The cell culture reagents were provided by Gibco BRL (Shanghai, China). The antibodies utilized in this study were provided by Cell Signaling Tech (Shanghai, China) and Santa Cruz Biotech (Santa Cruz, CA).
Cell Culture
Wild-type (WT), Gαi1 and Gαi3 double-knockout (DKO), Gαi1, Gαi2 or Gαi3 single-knockout (SKO) mouse embryonic fibroblasts (MEFs), as well as WT and Gab1 KO MEFs were described previously [11, 13, 14]. Culture of human umbilical vein endothelial cells (HUVECs) was described previously [27]. Cells were starved in 0.5% FBS medium overnight before any treatment.
Gαi1/3 shRNA in vitro
HUVECs were seeded onto six-well tissue culture plates (Corning, Shanghai, China) at 60% confluence. Lentivirus (20 μL virus per mL medium) containing shRNA of human Gαi1 (sc-105382-V) (Santa Cruz, CA) and/or human Gαi3 (sc-29325-V) (Santa Cruz, CA) was added. After 24 h, HUVECs with the target shRNA were selected by puromycin (1.0 μg/mL). The culture medium was replaced with fresh puromycin-containing culture medium every 2 days until resistant colonies were formed (7-8 days). The expression of Gαi1/3 in stable HUVECs was detected by Western blotting assay. Control HUVECs were infected with scramble non-sense shRNA lentiviral particles (Santa Cruz). The protocol of Gαi1 and Gαi3 shRNA in the WT MEFs was reported early [13, 14, 16].
Gαi1/3 adenovirus constructs
For adenovirus production, Gαi3 was amplified by RT-PCR. The full-length Gαi1/ Gαi3 cDNA was provided by Genepharm (Shanghai, China). The Gαi3(G202T) (“DN-Gαi3”) cDNA was also synthesized by Genepharm (Shanghai, China). The Gαi PCR fragment was sub-cloned into the BamHI/AgeI site of the of the pDC315 plasmid (with/out Flag tag) to produce pDC315-Gαi1/3. Using Lipofectamine 2000, HEK293 cells were transfected with pDC315-Gαi3 and pBHG lox ΔE1,3 Cre plasmid (from Genepharm, Shanghai, China) was used as the helper plasmid to generate the recombinant adenovirus, Ad-Gαi1/3, and the supernatant was harvested after one week. After viral amplification (3x), the supernatant was purified using an Adeno-X Virus Purification kit. To titer the virus, serially diluted adenovirus was used to transduce HEK293 cells. After one week, the labeled HEK293 cells were counted to calculate the viral titer (2.5×1010 pfu/mL). Ad-Gαi1/3 was added to cultured cells.
Tube formation assay
HUVECs (1×103 cells/well) were seeded onto 24-well tissue-culture plates, pre-coated with basement membrane matrix (BD Biosciences, Shanghai, China). Cells were then treated with VEGF for 24 h. Tube formation was photographed under an Olympus IX-73 microscope.
Generation of Gαi1/3 DKO mice
The generation of Gαi1/3DKO mice by the CRISPR-Cas9 method was described in detail in our previous study [16].
Oxygen-induced retinopathy (OIR) model
Briefly, newborn mice at P7 and their nursing mothers were exposed to 75% oxygen for 5 days, and then returned to room air for 5 days. The lentiviral GV493 Gαi3 shRNA, and the lentiviral GV493 murine Gαi1 shRNA were synthesized by Genepharma (Shanghai, China). Animals were anesthetized in 3% isoflurane in oxygen and injected intravitreally at P12 with Gαi1/3 shRNA lentivirus (1 μL volume per eye, injection within 20s) using a Hamilton syringe. Retinas were observed at P12/17. Mice were observed extremely carefully throughout the experimental period. The protocol was approved by the Soochow University Institutional Animal Care and Use Committee (IACUC) and Ethics Review Board (ERB).
Mice pups were fully anesthetized in 3% isoflurane in oxygen, then decapitated [40]. Eyes were enucleated and fixed in 4% paraformaldehyde solution for 1 h. Retinas were dissected and stained overnight at 4°C with FITC-conjugated lectin, isolectin B4 (Abcam, Shanghai, China, at 1:100). Retinas were whole mounted onto microscopy slides (Thermo Fisher Shanghai, China), which were then imaged at 10× using a Zeiss confocal microscope. Images were merged into a single file using the AxioVision software (Zeiss). Quantification of neovascularization was assessed using the SWIFT_NV methods as previously described [41].
Corneal alkali burn mouse model
As described previously [27], the mice ocular surface was rinsed with 20 mL of 0.9% saline solution for 1 min. Then, eyedrops of shRNA lentivirus (2 μL per eye) were instilled onto the alkali-burned eyes. Seven days after the alkali burn, the mice were euthanized and corneal neovascularization (“CoNV”) was observed.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR protocols using ABI Prism equipment and the SYBR Green PCR kit were performed using well-established methods, as previously described [14, 42]. Gαi1/2/3 mRNA expressions were quantified using the ΔΔCt protocol [14] and GAPDH as the internal control [42].
Additional assays
Western blotting assay and data quantification, co-immunoprecipitation (IP) assay, MTT cell proliferation assay, Transwell migration assay, Scratch wound healing assay, and BrdU incorporation ELISA assay were described in detail in our previous studies[11, 16, 43-46]. For all Western blotting assays, the same sets of lysate samples were run in sister gels to test different proteins. For each lane, the exact amount of protein lysates was loaded.
Plasma membrane fractionation
Plasma membranes were isolated as described in our previous studies [16].
Human retinas
A total of six (6) proliferative diabetic retinopathy (PDR) patients with lensectomy combined with vitrectomy surgery as well as three (3) age-matched traumatic retinectomy patients were enrolled. The anterior retinal hyperplastic membrane of PDR patients was stripped and immediately kept in lipid nitrogen for further analysis. The surgery-removed traumatic normal retinas were preserved as the control tissues. Written informed consent was obtained from each participant for clinical sample collection. The protocol was conducted according to the principles expressed in the Declaration of Helsinki and was approved by the Ethics Board of all authors' institutions.
Statistical analysis
All experiments were repeated at least three times; similar results were obtained in each case. The data presented in this study are expressed as mean ± standard deviation (SD). Comparison between any two groups was by two-tailed unpaired t-test for normally distributed data or nonparametric Mann-Whitney U-test for non-normally distributed data. Multiple group comparison was done by one-way analysis of variance (ANOVA) for data with a normal distribution. The Kruskal-Wallis test was used for data with non-normal distributions. Values of P< 0.05 were considered statistically significant.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was generously supported by grants from the National Natural Science Foundation of China (Grant No. 81302195, 31371139, 81571282, 81771457, 81700859, 81371055, 81570859, 81502162, 81670878), the Medical Science and Technology Development Project Fund of Nanjing (YKK16271, YKK15241, YKK16270), and the Natural Science Foundation of Jiangsu Province (BK20161568, BK20170060, BK20171065). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
All authors conceived the idea, designed the work, and contributed to acquisition of data.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17:611-25
2. Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A. et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163-77
3. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669-76
4. Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A. 1998;95:9349-54
5. Claesson-Welsh L. VEGF receptor signal transduction - A brief update. Vascul Pharmacol. 2016;86:14-7
6. Evans I. An overview of VEGF-mediated signal transduction. Methods Mol Biol. 2015;1332:91-120
7. Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169-83
8. Tae N, Lee S, Kim O, Park J, Na S, Lee JH. Syntenin promotes VEGF-induced VEGFR2 endocytosis and angiogenesis by increasing ephrin-B2 function in endothelial cells. Oncotarget. 2017;8:38886-901
9. Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T. et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465:487-91
10. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB. et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261-9
11. Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS. et al. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009;2:ra17
12. Liu YY, Chen MB, Cheng L, Zhang ZQ, Yu ZQ, Jiang Q. et al. microRNA-200a downregulation in human glioma leads to Galphai1 over-expression, Akt activation, and cell proliferation. Oncogene. 2018;37:2890-902
13. Zhang YM, Zhang ZQ, Liu YY, Zhou X, Shi XH, Jiang Q. et al. Requirement of Galphai1/3-Gab1 signaling complex for keratinocyte growth factor-induced PI3K-AKT-mTORC1 activation. J Invest Dermatol. 2015;135:181-91
14. Wang Z, Dela Cruz R, Ji F, Guo S, Zhang J, Wang Y. et al. G(i)alpha proteins exhibit functional differences in the activation of ERK1/2, Akt and mTORC1 by growth factors in normal and breast cancer cells. Cell Commun Signal. 2014;12:10
15. Cai S, Li Y, Bai JY, Zhang ZQ, Wang Y, Qiao YB. et al. Galphai3 nuclear translocation causes irradiation resistance in human glioma cells. Oncotarget. 2017;8:35061-8
16. Marshall J, Zhou XZ, Chen G, Yang SQ, Li Y, Wang Y. et al. Antidepression action of BDNF requires and is mimicked by Galphai1/3 expression in the hippocampus. Proc Natl Acad Sci U S A. 2018;115:E3549-E58
17. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960-76
18. Marty C, Ye RD. Heterotrimeric G protein signaling outside the realm of seven transmembrane domain receptors. Mol Pharmacol. 2010;78:12-8
19. Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, Drexler HC. et al. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol. 2013;15:249-60
20. Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483-6
21. Hubbard KB, Hepler JR. Cell signalling diversity of the Gqalpha family of heterotrimeric G proteins. Cell Signal. 2006;18:135-50
22. Barren B, Artemyev NO. Mechanisms of dominant negative G-protein alpha subunits. J Neurosci Res. 2007;85:3505-14
23. Dance M, Montagner A, Yart A, Masri B, Audigier Y, Perret B. et al. The adaptor protein Gab1 couples the stimulation of vascular endothelial growth factor receptor-2 to the activation of phosphoinositide 3-kinase. J Biol Chem. 2006;281:23285-95
24. Laramee M, Chabot C, Cloutier M, Stenne R, Holgado-Madruga M, Wong AJ. et al. The scaffolding adapter Gab1 mediates vascular endothelial growth factor signaling and is required for endothelial cell migration and capillary formation. J Biol Chem. 2007;282:7758-69
25. Ferrara N. VEGF and intraocular neovascularization: From discovery to therapy. Trans Vis Sci Techn. 2016;5:10
26. Amano S, Rohan R, Kuroki M, Tolentino M, Adamis AP. Requirement for vascular endothelial growth factor in wound- and inflammation-related corneal neovascularization. Invest Ophthalmol Vis Sci. 1998;39:18-22
27. Zhang XP, Li KR, Yu Q, Yao MD, Ge HM, Li XM. et al. Ginsenoside Rh2 inhibits vascular endothelial growth factor-induced corneal neovascularization. FASEB J. 2018:32 3782-91
28. Wang YN, Shan K, Yao MD, Yao J, Wang JJ, Li X. et al. Long noncoding RNA-GAS5: A novel regulator of hypertension-induced vascular remodeling. Hypertension. 2016;68:736-48
29. Liu C, Yao MD, Li CP, Shan K, Yang H, Wang JJ. et al. Silencing of circular RNA-ZNF609 ameliorates vascular endothelial dysfunction. Theranostics. 2017;7:2863-77
30. Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242-8
31. Stone J, Itin A, Alon T, Pe'er J, Gnessin H, Chan-Ling T. et al. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci. 1995;15:4738-47
32. Jardeleza MS, Miller JW. Review of anti-VEGF therapy in proliferative diabetic retinopathy. Semin Ophthalmol. 2009;24:87-92
33. Cheung N, Wong IY, Wong TY. Ocular anti-VEGF therapy for diabetic retinopathy: overview of clinical efficacy and evolving applications. Diabetes Care. 2014;37:900-5
34. Osaadon P, Fagan XJ, Lifshitz T, Levy J. A review of anti-VEGF agents for proliferative diabetic retinopathy. Eye (Lond). 2014;28:510-20
35. Abdallah W, Fawzi AA. Anti-VEGF therapy in proliferative diabetic retinopathy. Int Opthamol Clin. 2009;49:95-107
36. Bolinger MT, Antonetti DA. Moving past anti-VEGF: Novel therapies for treating diabetic retinopathy. Int J Mol Sci. 2016;17:E1498
37. Lu Y, Xiong Y, Huo Y, Han J, Yang X, Zhang R. et al. Grb-2-associated binder 1 (Gab1) regulates postnatal ischemic and VEGF-induced angiogenesis through the protein kinase A-endothelial NOS pathway. Proc Natl Acad Sci U S A. 2011;108:2957-62
38. Bruns AF, Herbert SP, Odell AF, Jopling HM, Hooper NM, Zachary IC. et al. Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic. 2010;11:161-74
39. Basagiannis D, Zografou S, Murphy C, Fotsis T, Morbidelli L, Ziche M. et al. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation through macropinocytosis. J Cell Sci. 2016;129:4091-104
40. Sitaras N, Rivera JC, Noueihed B, Bien-Aime M, Zaniolo K, Omri S. et al. Retinal neurons curb inflammation and enhance revascularization in ischemic retinopathies via proteinase-activated receptor-2. Am J Pathol. 2015;185:581-95
41. Stahl A, Connor KM, Sapieha P, Willett KL, Krah NM, Dennison RJ. et al. Computer-aided quantification of retinal neovascularization. Angiogenesis. 2009;12:297-301
42. Gong YQ, Huang W, Li KR, Liu YY, Cao GF, Cao C. et al. SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget. 2016;7:60123-32
43. Zuo Y, Lv Y, Qian X, Wang S, Chen Z, Jiang Q. et al. Inhibition of HHIP promoter methylation suppresses human gastric cancer cell proliferation and migration. Cell Physiol Biochem. 2018;45:1840-50
44. Chen MB, Liu YY, Xing ZY, Zhang ZQ, Jiang Q, Lu PH. et al. Itraconazole-Induced Inhibition on Human Esophageal Cancer Cell Growth Requires AMPK Activation. Mol Cancer Ther. 2018;17:1229-39
45. Cao C, Rioult-Pedotti MS, Migani P, Yu CJ, Tiwari R, Parang K. et al. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol. 2013;11:e1001478
46. Cao C, Sun Y, Healey S, Bi Z, Hu G, Wan S. et al. EGFR-mediated expression of aquaporin-3 is involved in human skin fibroblast migration. Biochem J. 2006;400:225-34
Author contact
![]() Corresponding authors: Drs. Jin Yao, Ke-ran Li and Qin Jiang, the Affiliated Eye Hospital, Nanjing Medical University. 138 Han-zhong Road, Nanjing City, China 210029. Tel:+86-025-86677699. Fax: +86-025-86677699. E-mail: dryaojincom (J. Y.), lkrnjmu7com (K. L) or Jqin710sina.com (Q. J.). Prof. Cong Cao, Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, 199 Ren-ai Road, Suzhou, Jiangsu 215123, China. Tel: +86-512-65883602, Fax: +86-512-65883602. E-mail: caocongedu.cn.
Corresponding authors: Drs. Jin Yao, Ke-ran Li and Qin Jiang, the Affiliated Eye Hospital, Nanjing Medical University. 138 Han-zhong Road, Nanjing City, China 210029. Tel:+86-025-86677699. Fax: +86-025-86677699. E-mail: dryaojincom (J. Y.), lkrnjmu7com (K. L) or Jqin710sina.com (Q. J.). Prof. Cong Cao, Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, 199 Ren-ai Road, Suzhou, Jiangsu 215123, China. Tel: +86-512-65883602, Fax: +86-512-65883602. E-mail: caocongedu.cn.