Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(17):4781-4794. doi:10.7150/thno.25189 This issue Cite
Research Paper
RGS4 deficit in prefrontal cortex contributes to the behaviors related to schizophrenia via system xc--mediated glutamatergic dysfunction in mice
Min-Wei Huang1,2,*, Yu-Jung Lin3,*, Chi-Wei Chang4, Fu-Ju Lei3, En-Peng Ho3, Ren-Shyan Liu4, Woei-Cherng Shyu5, Chia-Hung Hsieh3,6,7 ![]()
1. School of Medicine, China Medical University, Taichung, Taiwan
2. Department of Psychiatry, Chiayi Branch, Taichung Veterans General Hospital, Chiayi, Taiwan
3. Graduate Institute of Biomedical Science, China Medical University, Taichung, Taiwan
4. National PET/Cyclotron Center and Department of Nuclear Medicine, Taipei Veterans General Hospital, Taipei, Taiwan
5. Department of Neurology, Center for Translational Medicine, and Graduate Institute of Biomedical Science, China Medical University and Hospital, Taichung, Taiwan
6. Department of Medical Research, China Medical University Hospital, Taichung, Taiwan
7. Department of Biomedical Informatics, Asia University, Taichung, Taiwan
* these authors contributed equally to this work
Received 2018-1-28; Accepted 2018-4-9; Published 2018-9-9
Abstract

Rationale: Although molecular investigations of regulator of G-protein signaling 4 (RGS4) alterations in schizophrenia patients yielded partially inconsistent findings, the previous studies suggested that RGS4 is both a positional and functional candidate gene for schizophrenia and is significantly decreased in the prefrontal cortex. However, the exact role of RGS4 in the pathophysiology of schizophrenia is unclear. Moreover, a whole genome transcription profile study showed the possibility of RGS4-regulated expression of SLC7A11(xCT), a component of cysteine/glutamate transporter or system xc-. We hypothesized that system xc- is a therapeutic target of RGS4 deficit-mediated schizophrenia.
Methods: Pharmacological and genetic manipulation of RGS4 in organotypic brain slice cultures were used as an ex vivo model to investigate its role in system xc- and glutamatergic function. Lentiviral-based mouse models with RGS4 deficit in the prefrontal cortex and treatment with system xc- activator, N-acetyl cysteine (NAC), were utilized to observe their impacts on glutamatergic function and schizophrenic behaviors.
Results: Genetic and pharmacological inhibition of RGS4 resulted in a significant decrease in SLC7A11 (xCT) expression and hypofunction of system xc- and reduced glutamatergic function in organotypic brain slice cultures. However, NAC restored the dysregulation of RGS4-mediated functional deficits of glutamate. Moreover, knockdown of RGS4 specifically in the prefrontal cortex caused mice to exhibit behaviors related to schizophrenia such as increased stereotypy, impaired prepulse inhibition, deficits in social interactions, working memory, and nesting behavior, while enhancing sensitivity to the locomotor stimulatory effect of MK-801. These mice displayed glutamatergic dysfunction in the prefrontal cortex, which may have contributed to the behavioral deficits. RGS4 knockdown mice that received NAC treatment had improved glutamatergic dysfunction and schizophrenia behaviors.
Conclusion: Our results suggest that RGS4 deficit induces dysregulation and dysfunction of system xc-, which further results in functional deficits of the glutamatergic system and subsequently to schizophrenia-related behavioral phenotypes. Activation of system xc- offers a promising strategy to treat RGS4 deficit-mediated schizophrenia.
Keywords: regulator of G-protein signaling 4, system xc-, schizophrenia, glutamatergic transmission, N-acetyl cysteine
Introduction
Regulator of G-protein signaling 4 (RGS4) modulates several neurotransmitter systems implicated in schizophrenia, including the dopaminergic [1], serotonergic [2], and glutamatergic systems [3]. Additionally, a close proximity of the RGS4 locus to a chromosomal region associated with schizophrenia has been reported in linkage studies [4]. Moreover, a decrease in RGS4 expression in the prefrontal cortex or superior temporal gyrus of schizophrenia patients has been reported [5-8]. These findings implicate RGS4 as a susceptibility gene for schizophrenia. However, such changes could not be found in other related studies [9, 10]. Additionally, the behavioral studies in RGS4 knockout mice failed to show any differences in schizophrenia-like behavior [11]. This may be due to developmental adaptations and genetic compensation for RGS4 in the knockout mice, thereby masking the establishment of a clear phenotype. Although the mechanism of RGS4 involvement in the pathophysiology of schizophrenia remains poorly understood, a recent study demonstrated the effect of siRNA-mediated knockdown of RGS4 on the whole genome transcription profile. This indicated the possibility of RGS4-regulated expression of SLC7A11(xCT), a component of cysteine/glutamate transporter or system xc-, which is associated with glutamatergic transmission and antioxidant defense [12], dysfunction of which may contribute to the pathophysiology of schizophrenia.
The cystine-glutamate antiporter is a membrane-bound Na+-independent amino acid transporter, structurally composed of a heavy chain subunit common to all amino acid transporters, 4F2hc, and a light chain specific subunit, xCT [13, 14]. System xc- exchanges intracellular glutamate for extracellular cystine, thereby supporting intracellular glutathione (GSH) synthesis as well as non-vesicular glutamate release. Most previous studies have focused on its role in oxidative stress during cancer or other diseases [13]. However, results of recent studies using system xc--knockout mice indicated that the loss of system xc- does not induce oxidative stress but decreases extracellular glutamate, suggesting that system xc- is an important source of the extracellular glutamate that is involved in glutamatergic transmission [15]. Previous studies have indicated that system xc- represents a potential target in the treatment of schizophrenia [16]. A recent human study also suggested an association between system xc- dysregulation and schizophrenia [17].
The glutamate hypothesis of schizophrenia suggests that psychosis is associated with abnormal glutamate transmission and hypofunction of glutamatergic signaling via N-methyl-D-aspartate receptor (NMDAR) [18, 19]. This hypothesis was initially based on the effect of the psychotomimetic agent, phencyclidine (PCP), which is an antagonist of glutamate NMDA receptors [20]. Further clinical and laboratory studies supported the theory that various genetic and cellular susceptibility factors in schizophrenia may converge on glutamatergic dysfunction. Moreover, this hypothesis has provided novel therapeutic targets for schizophrenia that modulate glutamatergic transmission via a number of mechanisms including upstream and downstream mediators or effectors.
In this study, we hypothesized that RGS4 regulates system xc- function and its deficit contributes to disease progression in schizophrenia due to the hypofunction of glutamatergic transmission. We showed that RGS4 deficit induces dysregulation and dysfunction of system xc-, which further results in functional deficits of non-vesicular glutamate and NMDAR. Knockdown of RGS4 in the prefrontal cortex of mice displayed glutamatergic dysfunction and various behavioral phenotypes that correspond to the core symptoms of schizophrenia. Our findings suggest that RGS4 deficit in the prefrontal cortex contributes to the pathophysiology of schizophrenia via system xc- -mediated glutamatergic dysfunction. N-acetyl cysteine (NAC) is a promising potential pharmacotherapeutic candidate for the treatment of RGS4 deficit-mediated schizophrenia by restoring this mechanism.
Methods
Organotypic brain-slice cultures
Organotypic brain-slice cultures were prepared as previously described [21]. Briefly, the brains of adult C57BL/6 mice were removed and cut into coronal slices of 230 μm thickness using a Vibratome. Slices containing the cerebral cortex and hippocampus were placed onto Millicell-CM culture inserts (Millipore) in 6-well culture plates with culture medium.
Vector construction
The full-length mouse RGS4 (clone ID NM_009062.3, GeneScript) was amplified in a reaction with Platinum Taq DNA polymerase (Invitrogen) and was subcloned into pAS2.EYFP.puro (National RNAi core facility, Academia Sinica, Taiwan) at the NheI and EcoRI sites. The construct was confirmed by DNA sequencing. Lentiviral vectors carrying short hairpin RNA (shRNA)-targeting mouse RGS4 (5′-CCGGGAAGTCAAGAAATGGGCTGAACTCGAGTTCAGCCCATTTCTTGACTTCTTTTTG-3′) and scrambled shRNA (http://rnai.genmed.sinica.edu.tw/file/vector/C6-7/17.1.pLAS.Void.pdf) were provided by National RNAi core facility, Academia Sinica in Taiwan.
Lentivirus production and transduction
Lentiviral particles were generated by transiently cotransfecting 293T cells with the plasmids coding for RGS4, scrambled shRNA and shRNAs-targeting RGS4 in addition to plasmids encoding gag/pol and VSV-G envelope genes. Transfection was carried out with jetPEI reagent (Polyplus-Transfection). Organotypic brain-slices were infected with lentivirus in the presence of 8 μg/mL polybrene (Sigma-Aldrich). At 24 h post-infection, medium was removed and replaced with fresh growth medium containing puromycin (0.5 μg/mL) to select for infected cells 48 h post-infection.
Real-time quantitative polymerase chain reaction (Q-PCR)
Q-PCR analysis was performed as described previously [22]. The primers for quantitative analysis of RGS4, SLC7A11, and 18S rRNA (internal control) were: mouse RGS4 (F) 5′-CGGCTTCTTGCTTGAGGAGTG-3′ and (R) 5′-ATCCAGGTTCACATTCATGACT-3'; mouse SLC7A11 (F) 5′-CCTGGCATTTGGACGCTACAT-3′ and (R) 5′-TGAGAATTGCTGTGAGCTTGCA-3′; and mouse 18S rRNA (F) 5′-GTAACCCGTTGAACCCCATT-3′ and (R) 5′-CCATCCAATCGGTAGTAGCG-3′.
Western blot analysis
Tissues were lysed and extracts were prepared as previously described [22]. RGS4 and xCT proteins in brain slices were detected in 150 µg cell extract using monoclonal anti-RGS4 antibody (diluted 1:650; Santa Cruz BioTech; sc-6203) and anti-xCT antibody (diluted 1:600; Novus; NB300-317). Western blots were normalized using a monoclonal anti-β-actin antibody (diluted 1:10,000; Sigma-Aldrich; AC-74).
Cl--dependent [14C] L-cystine uptake
Cystine/glutamate antiporter activity was analyzed using Cl--dependent [14C] L-cystine uptake assay as previously described [23]. Briefly, brain slices were incubated with 0.8 µM [14C] L-cystine (Perkin Elmer) at 37 °C for 10 min. The uptake was terminated by rapidly rising tissue slices with ice-cold unlabeled uptake buffer. The tissue slices were then lysed by adding 0.8 mL 0.2 N NaOH containing 1% sodium dodecyl sulfate (SDS) and radioactivity was measured using Tri-Carb B2910TR liquid scintillation analyzer (Perkin Elmer).
In vitro extracellular glutamate release assay
Brain slices were incubated with 200 μL buffer solution containing 5.33 mM KCl, 26.19 mM NaHCO3, 117.24 mM NaCl, 1.01 mM NaH2PO4, 2.0 mM CaCl2, 5.56 mM D-glucose, and 100 μM cystine for 1 h at 37 °C. After incubation, the extracellular medium was collected for estimation of glutamate.
Glutamate estimation
Samples were diluted in 20 mM borate buffer at pH 9.0 and were derivatized for 1 min with N-tert-butyloxycarbonyl-L-cysteine and o-phthalaldehyde. Samples were then separated in a 5-mm C18 reverse-phase column (220 × 4.6 mm) Sheri-5 (Brownlee), and glutamate separation was monitored by fluorescence (excitation at 334 nm and emission at 433 nm) using an RF-10AXL fluorescence detector (Shimadzu).
[3H] MK-801 binding in organotypic brain-slices
After pretreatment under various conditions, brain slices were incubated with 10 nM [3H] MK-801 for 45 min, washed thrice in ice-cold phosphate-buffered saline (PBS), followed immediately by cell lysis in 1% SDS. Aliquots were taken for scintillation counting and protein assays. [3H] MK-801 binding radioactivity was normalized to total protein.
Mice and genotyping
Wild-type C57BL/6J mice were purchased from the Animal Facility of the National Science Counsel. xCT homozygous knockout (xCT-/-) mice with 129/Svj-C57BL/6J mixed genetic background kindly provided by Dr. Hideyo Sato. These mice were backcrossed with C57BL/6J mice. Genotyping of xCT homozygous knockout (xCT-/-) mice was carried out as previously described [24]. All animals were maintained on a 12-h light/dark cycle, with rodent chow and water ab libitum. All behavioral tests were carried out with male mice that were 10 weeks old at the start of testing. All animal experiments were conducted according to Institutional Guidelines of China Medical University after acquiring permission from the local Ethical Committee for Animal Experimentation.
Lentiviral microinjection
Bilaterally prefrontal cortical infusions were performed via a micro-injector with MAB 40 microdialysis pump fitted to the arm of the stereotaxic apparatus. Lentiviral vectors containing RGS4 or RGS4 shRNA were infused at a rate of 0.2 mL/min. Injectors were left in place for 2 min after completing the injection to ensure adequate diffusion from the injector tip. The microinjection sites were observed under a fluorescence microscope after frozen coronal sectioning (30 μm).
Open field test
Each subject was placed in an open field chamber (Coulbourn Instruments) and allowed to explore freely for 120 min. The locomotor activity including total distance traveled (in cm), vertical activity, time spent in the center, and the beam-break counts for stereotyped behaviors in the whole chamber as well as in the center area was monitored and analyzed by an automated system (the TruScan Activity System, Coulbourn Instruments).
Elevated plus-maze test
The elevated plus-maze consisted of four polycarbonate arms extending 30 cm in length, two of which had 16.5 cm-high walls. The plus maze was elevated 60 cm from the floor. Mice were placed in an open square dividing the four arms of the chamber. An overhead camera recorded the time spent on the open arms of the plus maze during each 5-min test session.
Pre-pulse inhibition tests
Pre-pulse inhibition (PPI) of the acoustic startle response was conducted using SR-LAB startle chambers (San Diego Instruments). Mice were placed into the plexiglas cylinder and apparatus for 10 min of habituation. Startle trials consisted of a 40 ms burst of 120 dB white-noise; pre-pulse trials consisted of a 20 ms pre-pulse stimulus that was 68, 71, or 77 dB above the white-noise background (64 dB), followed by 100 ms of the 120 dB startle stimulus. Non-stimulus or null trials consisted of the 64 dB white-noise background. PPI responses were calculated as a percentage score for each intensity of pre-pulse, where %PPI = [1 - (pre-pulse trials / startle-only trials)]*100.
Social interaction tests
A sociability assay was performed as previously described [25]. Briefly, a three-chamber apparatus was used to assess sociability and preference for social novelty. The test consisted of three subsequent 10-min phases: (i) Habituation: free exploration of the three chambers; (ii) Sociability: a stranger mouse was inserted into an enclosure in one of the side chambers; (iii) Social novelty: the by-now familiar mouse was left in place, and a different stranger mouse was inserted into an enclosure in the second side chamber. Interaction was quantified by counting how many times the mice sniffed the enclosure.
A delayed non-match to place task
A working memory task was performed as described [26]. Briefly, mice were food restricted and adapted to a T maze (two trials for 2 days for 5 min). The mice were given a forced run (the sample) to one arm and then, after a 5-s delay, given access to the original and opposite arm (the non-match) with an inter-trial interval of 40 s. Correct choices were reinforced with food reward at the end of the correct arm. After mice made at least 80% correct choices on two consecutive days, the testing phase began, where delays of 5, 15 or 30 s were randomly introduced between the sample run and choice run. Mice were given 4 trials of each delay on 3 days of testing for a total of 12 trials for each delay.
Nesting behavior test
Each mouse was placed alone in a cage with a piece of nesting material evenly spread on top of ordinary wood chips. After 1 h, photographs were taken of the floor of each cage to inspect whether there was a nest made of the cotton material. The number of scattered particles of the nestlets was counted for each cage by the NIH IMAGE program.
Sensitivity to MK-801
Mice were habituated to an open field for 1 h and were then intraperitoneally (i.p.) administered 0.6 mg/kg body weight MK-801 for 1 h.
Glutathione assay
Glutathione content in tissues was estimated using Glutathione Assay Kit (Sigma-Aldrich) according to the manufacturer's protocol.
Microdialysis
Mice were anesthetized with chloral hydrate (360 mg/kg, i.p.) and mounted on a stereotaxic frame. A cannula guide (0.65 mm outer diameter) was implanted in either the prefrontal cortex (relative to Bregma: angle 8°, A +1.94, L +0.73, V -3.28) and secured to the skull with an anchor screw and acrylic dental cement. On the next day, a microdialysis probe (CMA10, Carnegie Medicin, Stockholm, Sweden; membrane length: 1 mm) was inserted and connected to a microinfusion pump set to a speed of 1 μL/min and then perfused with Ringer's solution (147 mM NaCl, 4 mM KCl, and 2.3 mM CaCl2). Samples were collected every 30 min for the duration of the experiment. Probe positioning was histologically verified at the end of the experiments.
Radiochemistry
18F-labeled S-fluoroalkyl diarylguanidine-10 (18F-FSAG; radiochemical purity >95%) was synthesized via 18F‑fluorination of the protected precursor S-fluoroalkyl guanidine, followed by acidic hydrolysis, as previously described [27]. Briefly, the guanidines were radiolabeled by alkylation of the thiol precursor with [18F]-fluoromethyl tosylate. [18F]-Fluoromethyl tosylate was obtained with 20-26% radiochemical yield (non-decay-corrected, 90 min from end of bombardment) using the method reported by Neal et al. [28]. [18F]-fluoroalkylation of thiol precursor was carried out in acetonitrile in the presence of Cs2CO3 at 110 °C for 15 min. Following purification by reversed phase semi-preparative HPLC, the [18F]-guanidine was concentrated on a C18 Sep-Pak cartridge, eluted with ethanol (200 uL) and formulated for injection in either 0.9% w/w isotonic saline or phosphate buffered saline. The total synthesis time was 3-4 h from the end of bombardment.
Micro-positron emission tomography (PET) imaging
Each subject was injected with 37 MBq of 18F-FSAG. At 30 min after injection, mice were scanned on an animal SPECT/PET/CT device (Triumph Trimodality system; Gamma Medica) under isoflurane anesthesia. PET data was collected in list mode for 30 min followed by a CT examination for 3 min (Field of View (FOV) = 8.0 cm). The PET data was reconstructed using the ordered subsets expectation-maximization (OSEM) 3D algorithm (20 iterations). The CT raw files were reconstructed using Filter Back Projection (FBP). Images were obtained with a zoom factor of 2 in a 256 × 256 matrix. Calculations were corrected for radiation decay of 18F and the amount of injected dose, and the consistent color scale was applied to all PET images. All subsequent processing of the PET and CT images were performed in imaging software Amide 1.0.4. For quantitative data analysis, a region of interest (ROI) was placed on the virus injection sites of the prefrontal cortex. The average radioactivity was obtained from the average pixel value within the multiple ROI volumes. The counts in each ROI were converted to radioactivity per cubic centimeter of tissue (nCi/cc), assuming a tissue density of 1 g/mL, and were then normalized to percentage of injected dose per cubic centimeter of tissue (%ID/cc).
Biodistribution of 18F-FSAG
Animals received 118.4 MBq/kg of 18F-FSAG in 100 μL of PBS via lateral tail vein injection, and then were euthanized by CO2/O2 asphyxiation at 30 min after injection. After sacrifice, selected tissues of interest were then removed and weighed, and the radioactivity was measured using a γ-counter. The percentage injected dose per gram (%ID/g) was then calculated.
Statistical analysis
All data are reported as mean ± standard deviation (SD). Statistical analyses were performed with SPSS package (version 18.0) using unpaired Student's t test and ANOVA with Bonferroni's or Tukey's multiple comparison post hoc tests, where appropriate.
Results
RGS4 regulates xCT expression and the function of system xc - and NMDAR
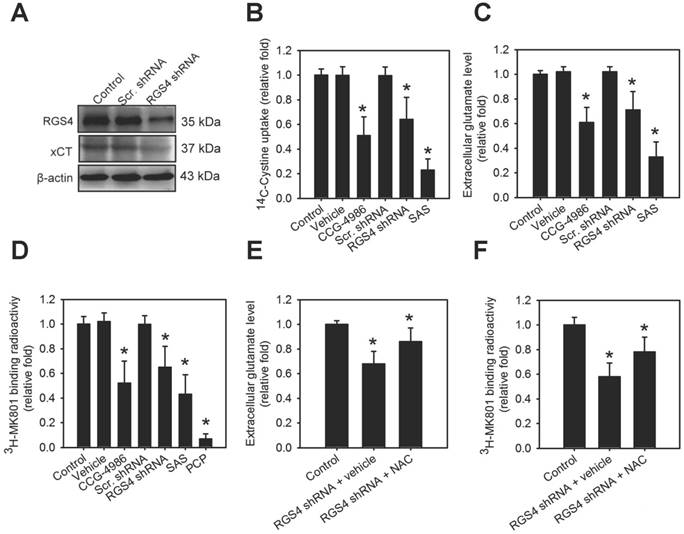
In an attempt to gain insight into the role of RGS4 on the modulation of system xc-, we utilized the organotypic brain slice culture from the adult mouse brain as a model to enable observation of temporal changes induced by various drugs in the living brain tissue [29]. RGS4 deficit in the brain was produced via genetic or pharmacological inhibition of RGS4. RGS4 knockdown in organotypic brain slice cultures via a lentiviral-based knockdown system significantly inhibited xCT expression (F(2, 6) = 30.05, p < 0.001, one-way ANOVA; Figure 1A and Figure S1A). Silencing RGS4 expression or pharmacological inhibition of RGS4 using CCG-4986 in organotypic brain slice cultures significantly attenuated tissue [14C] L-cystine radioactivity (F(5, 48) = 8.05, p < 0.0001, one-way ANOVA; Figure 1B) and extracellular glutamate content (F(5, 48) = 10.24, p < 0.0001, one-way ANOVA; Figure 1C). This indicated that RGS4 deficit triggers dysregulation and dysfunction of system xc-. Next, we further investigated whether RGS4 deficit-mediated dysfunction of system xc- affected downstream glutamatergic receptor function. This was done using a radioligand, [3H] MK-801, and observing the NMDAR function; [3H] MK-801 binds to the PCP site of NMDA ion channel when the channel is in the active and open state via NMDAR activation [30]. Pharmacological and genetic inhibition of RGS4 significantly inhibited the radioactive binding of [3H] MK-801 in brain slices (F(5, 48) = 6.36, p < 0.005, one-way ANOVA; Figure 1D), indicating that RGS4 deficit results in NMDAR hypofunction. Pretreatment of brain slice cultures with a system xc- inhibitor, sulfasalazine (SAS) [31], also decreased the radioactive binding of [3H] MK-801. Moreover, pretreatment of brain-slices with PCP, a non-competitive antagonist of the NMDAR, blocked the binding of [3H] MK-801, indicating the radioligand specificity for the activation of NMDAR. Importantly, the treatment with system xc- activator, NAC, improved RGS4 knockdown-mediated low extracellular glutamate (F(2, 24) = 26.83, p < 0.0001, one-way ANOVA; Figure 1E) and hypofunction of NMDAR (F(2, 24) = 12.45, p < 0.005, one-way ANOVA; Figure 1F), suggesting that system xc- is a critical mediator of RGS4 deficit-induced abnormal glutamatergic neurotransmission.
RGS4 deficit results in dysregulation of xCT and dysfunction of system xc- and N-methyl-D-aspartate receptor (NMDAR). (A) The protein levels of RGS4 and xCT in organotypic brain-slice cultures with or without RGS4 knockdown measured by western blot analysis. Organotypic brain-slices were infected with lentiviral vectors targeting RGS4 or scrambled shRNA sequences. Western blot analysis was conducted at 48 h after virus infection. N = 3 per group. Tissue [14C] L-cystine radioactivities (B), extracellular glutamate levels (C) and tissue [3H] MK801 radioactivities (D) in organotypic brain slices with or without the RGS4 inhibitor, CCG-4986 (30 μM), for 12 h, RGS4 knockdown for 48 h, sulfasalazine (SAS, 500 μM) for 12 h and phencyclidine (PCP, 10 μM) for 12 h. SAS and PCP treatments were used as positive controls for system xc- and NMDAR functional inhibition, respectively. Extracellular glutamate levels (E) and tissue [3H] MK801 radioactivities (F) in RGS4-knockdown brain slices treated with system xc- activator, N-acetyl-cysteine (NAC, 2.5 mM) for 24 h. N = 9 per group. *P < 0.0001 compared to controls, one-way ANOVA with Tukey's multiple comparison test.
System xc- is a critical effector for RGS4-regulated extracellular glutamate and NMDAR activation
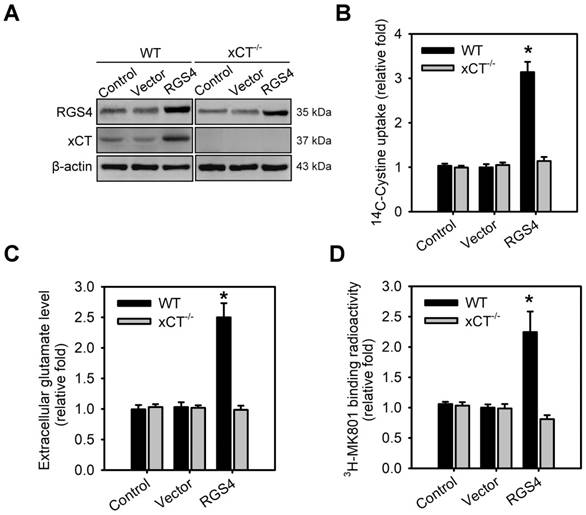
To validate system xc- as a critical effector involved in RGS4-mediated changes in extracellular glutamate and NMDAR, organotypic brain-slice cultures were prepared from the adult wild-type and xCT knockout (xCT-/-) mouse brains and transduced with RGS4-containing lentivirus. A significant effect was seen in the following parameters: genotype x viral transduction interaction in xCT expression (F 1,16 = 18.45, P < 0.001, two-way ANOVA), tissue [14C] L-cystine radioactivity (F1,52 = 38.23, P < 0.0001, two-way ANOVA), extracellular glutamate content (F 1,52 = 32.56, P < 0.0001, two-way ANOVA) and the radioactive binding of [3H] MK-801 (F 1,52 = 28.24, P < 0.0001, two-way ANOVA). RGS4 gain-of-function in wild-type brain slices largely increased xCT expression (Figure 2A and Figure S2B), tissue [14C] L-cystine radioactivity (Figure 2B), and extracellular glutamate content (Figure 2C). RGS4 overexpression in wild-type brain slices also subsequently promoted the radioactive binding of [3H] MK-801in brain slices (Figure 2D), indicating that RGS4 gain-of-function results in NMDAR hyperfunction. However, RGS4 overexpression in xCT-/- brain slices had no effect on xCT expression, tissue [14C] L-cystine radioactivity, extracellular glutamate levels and the radioactive binding of [3H] MK-801in brain slices (Figure 2A-D and Figure S1B), indicating that system xc- is a critical glutamate transporter involved in RGS4-mediated changes of extracellular glutamate and NMDAR activation.
System xc- is a critical effector for RGS4-regulated extracellular glutamate and NMDAR activation. (A) The protein levels of RGS4 and xCT in organotypic brain slice cultures with or without RGS4 overexpression measured by western blot analysis. Organotypic brain slices derived from wild-type (WT) or xCT knockout (xCT-/-) mice were infected with control lentiviral vectors or vectors overexpressing RGS4. Western blot analysis was conducted at 48 h after virus infection. N = 3 per group. Tissue [14C] L-cystine radioactivities (B), extracellular glutamate levels (n = 9) (C) and tissue [3H] MK801 radioactivities (D) in WT or xCT-/- organotypic brain slices with or without RGS4 overexpression. N = 9 per group. *P < 0.001 compared to controls, two-way ANOVA followed by Bonferroni test.
RGS4 deficit in the prefrontal cortex results in a decrease of non-vesicular glutamate and NMDAR activation
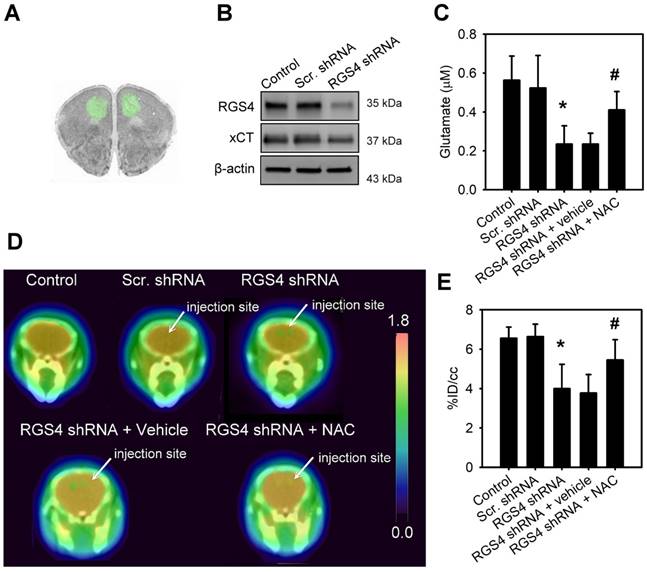
In order to mimic a decrease in RGS4 expression in the prefrontal cortex of schizophrenia patients, we used lentiviral shRNA vectors to target RGS4 expression in the mouse prefrontal cortex, in vivo. Injection of lentiviral vectors carrying the GFP reporter gene showed the infection area with an average radius of 1.84 ± 0.121 mm (Figure 3A). Western blotting analysis of infection area extracts at 21 days after virus injection showed that the prefrontal cortex with xCT knockdown had significantly lower levels of xCT compared to those expressing scramble shRNAs (F(2, 6) = 28.08, p < 0.001, one-way ANOVA; Figure 3B and Figure S1C). To examine the role of RGS4 in the maintenance of glutamate levels and NMDAR function, we knocked down RGS4 in the mouse prefrontal cortex. Microdialysis, 18F-labelled alkylthiophenyl guanidine (18F-FSAG) PET imaging, and ex vivo biodistribution studies were utilized to determine the extracellular glutamate levels and NMDAR activation in the prefrontal cortex of RGS4 knockdown mice and control mice. The extracellular glutamate levels (F(4, 35) = 21.67, p < 0.0001, one-way ANOVA) and radioactivity accumulation (F(4, 35) = 8.28, p < 0.0001, one-way ANOVA) in the prefrontal cortex were significantly decreased in the RGS4 knockdown mice compared with control mice (Figure 3C-E and Figure S2). Moreover, 1 week of NAC treatment restored the extracellular glutamate level and NMDAR activation in the prefrontal cortex of RGS4 knockdown mice compared to vehicle treatment (Figure 3C-E). On the other hand, no significant difference was observed in the glutathione content of the prefrontal cortex of any of the groups, indicating that RGS4 deficit in the prefrontal cortex does not alter the glutathione content (Figure S3). These results suggest that RGS4 deficit in the prefrontal cortex results in glutamate imbalance and hypofunction of NMDAR in vivo and the pharmacological activation of system xc- can restore it.
RGS4 deficit in prefrontal cortex results in a decrease of extracellular glutamate and NMDAR activation. (A) Microinjection sites and specific expressions of EGFP in the prefrontal cortex were observed by fluorescence microscopy. (B) The protein levels of RGS4 and xCT in prefrontal cortex infected with lentiviral vectors targeting RGS4 or scrambled shRNA sequences at 21 days after virus injection. The prefrontal cortical tissues of 3 mm in diameter around the injection site were punched out for western blot analysis. N = 3 per group. Microdialysis of extracellular glutamate content (n = 8) (C) and 18F-labelled alkylthiophenyl guanidine (18F-FSAG, a specific radioligand for PCP sites of the NMDAR) PET imaging (D) in the microinjection sites at 21 days after virus injection. Before microdialysis and PET imaging studies, mice were pre-treated with NAC (500 mg/kg/day) or vehicle for 1 week. (E) PET image-based quantification of 18F-FSAG accumulation in the injection areas of prefrontal cortex. N = 8 per group. *P < 0.0001 compared to control mice; # P < 0.01 compared to vehicle-treated mice, one-way ANOVA followed by Tukey's test.
RGS4 deficit in the prefrontal cortex increases stereotypy and reduces anxiety-like behavior
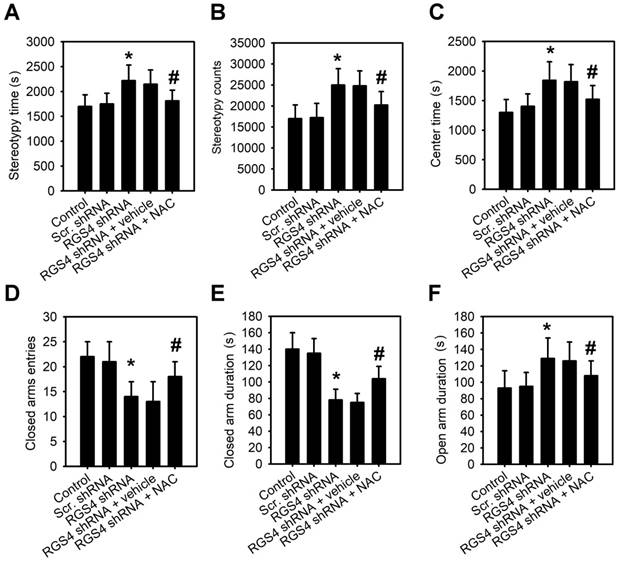
To observe the RGS4 deficit-induced behaviors, we first used the open field test to assess stereotypic behaviors and general locomotor activity. The time spent in stereotypic behavior (F(4, 55) = 12.45, p < 0.0001, one-way ANOVA) and stereotypic count (F(4, 55) = 18.24, p < 0.0001, one-way ANOVA) of the RGS4 knockdown mice were significantly higher than those of control mice (Figure 4A-B). No significant difference between groups was observed in the total distance traveled (F(4, 55) = 1.36, p = 0.259, one-way ANOVA) or resting time (F(4, 55) = 2.05, p = 0.099, one-way ANOVA; Figure S4A-B), indicating that RGS4 knockdown mice displayed no deficit in general motor activity. Additionally, the time spent in the center of the field was significantly longer in RGS4 knockdown mice than in control mice (F(4, 55) = 14.12, p < 0.0001, one-way ANOVA; Figure 4C), which may reflect low anxiety levels in the RGS4 knockdown mice. To confirm this phenotype of lesser anxiety-like behavior, we observed a fresh batch of RGS4 knockdown mice along with controls in an elevated plus-maze test. RGS4 knockdown mice showed a significantly lower closed-arm duration (F(4, 55) = 12.22, p < 0.0001, one-way ANOVA) and a lower ratio of closed-arm to total entries (F(4, 55) = 8.36, p < 0.0001, one-way ANOVA; Figure 4D-E). Moreover, the open-arm duration was significantly higher in RGS4 knockdown mice than in control mice (F(4, 55) = 10.52, p < 0.0001, one-way ANOVA; Figure 4F). The RGS4 deficit did not differ in terms of open-arm to total entries (F(4, 55) = 1.06, p = 0.385, one-way ANOVA) or distance traveled (F(4, 55) = 1.18, p = 0.329, one-way ANOVA; Figure S4C-D). Additionally, RGS4 knockdown mice showed significant improvement in stereotypic behavior and anxiety-like behavior after 1 week of NAC treatment, suggesting that these behaviors are mediated by system xc-.
RGS4 deficit in prefrontal cortex increases stereotypy and reduces anxiety-like behavior. Stereotypic behavior time (A), stereotypic behavior counts (B) and time spent in center (C) were significantly increased in RGS4 knockdown mice than in control mice. RGS4 knockdown mice logged significantly fewer entries (D) and durations (E) of visits to the closed arms. (F) RGS4 knockdown mice showed increased duration in the open arms. The open field test was used to assess stereotypic behaviors and general locomotor activity. The anxiety-like behaviors were observed by elevated plus-maze testing. All the behavior tests were carried out at 21 days after the virus injection. For the NAC treatment, mice were pre-treated with NAC (500 mg/kg/day) or vehicle for 1 week before behavior tests. N = 12 per group. *P < 0.05 compared to control mice; # P < 0.05 compared to vehicle-treated mice, one-way ANOVA followed by Tukey's test.
RGS4 deficit in the prefrontal cortex impairs pre-pulse inhibition and decreases social interaction
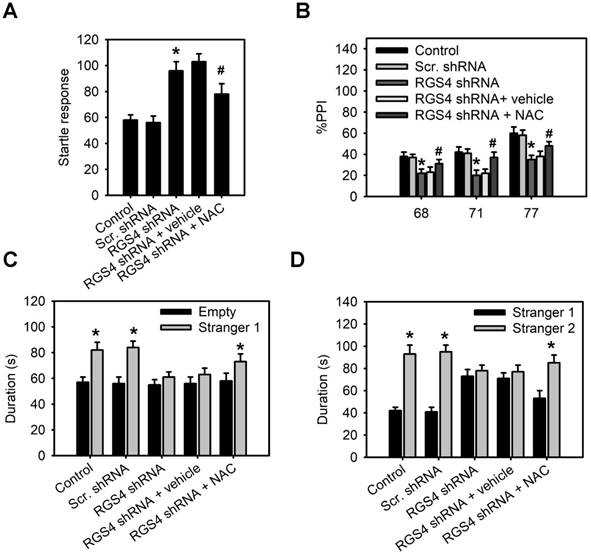
Schizophrenic patients display deficits in sensorimotor gating and social interaction [32, 33]. Therefore, we used the pre-pulse inhibition (PPI) of acoustic startle to determine whether RGS4 knockdown mice mimicked these abnormal phenotypes. Our results demonstrated that base startle reactivity in block 1 was significantly higher in RGS4 knockdown mice compared with control mice (F(4, 55) = 8.68, p < 0.0001, one-way ANOVA; Figure 5A). There was a significant effect of RGS4 knockdown and NAC treatment on the percent PPI (an index of sensorimotor gating) (F(4, 175) = 10.31, p < 0.0001, two-way ANOVA). Post-hoc comparisons revealed that PPI was significantly lower in RGS4 knockdown mice than in control mice, indicating impaired PPI in RGS4 knockdown mice (Figure 5B). We also analyzed social interactions in RGS4 knockdown mice using Crawley's sociability test and preference for social novelty test. A significant main effect of RGS4 knockdown and NAC treatment effect was found in Crawley's sociability test (F(4, 115) = 5.23, p < 0.001, two-way ANOVA) and preference for social novelty test (F(4, 115) = 6.04, p < 0.001, two-way ANOVA). Control mice spent significantly more time in the compartment with Stranger 1 compared to the compartment with an empty cup, indicating normal sociability, social motivation, and affiliation (Figure 5C). However, there were no significant differences between the time spent in the compartment with an empty cup as compared to that in the compartment containing Stranger 1 in RGS4 knockdown mice. Additionally, control mice recalled their previous contact with Stranger 1 and tended to spend more time with the newly encountered mouse (Stranger 2), indicating intact social memory and predilection for novel experience (Figure 5D). However, this function was decreased in RGS4 knockdown mice, indicating that they showed impaired sociability and decreased social motivation and preference for novelty. Furthermore, NAC treatment restored the deficits in PPI and decreased social interaction in RGS4 knockdown mice, indicating that these deficits were attributable to the hypofunction of system xc-.
RGS4 deficit in the prefrontal cortex impairs pre-pulse inhibition and decreases social interaction. (A) Base startle reactivity in block 1 was significantly higher in RGS4 knockdown mice compared with control mice (n = 12). *P < 0.001 compared to control mice, empty or Stranger 1; # P < 0.01 compared to vehicle-treated mice, one-way ANOVA followed by Tukey's test. (B) The percentage pre-pulse inhibition was significantly lower in RGS4 knockdown mice compared with control mice (n = 12). *P < 0.001 compared to control mice, empty or Stranger 1; # P < 0.05 compared to vehicle-treated mice, two-way ANOVA followed by Bonferroni test. RGS4 knockdown mice failed to demonstrate a preference for social proximity (C) and social novelty (D) by spending the same amount of time in both chambers. All the behavior tests were carried out at 21 days after virus injection. For the NAC treatment, mice were pre-treated with NAC (500 mg/kg/day) or vehicle for 1 week before behavior tests. N = 12 per group. *P < 0.001 compared to empty or Stranger 1; # P < 0.001 compared to vehicle-treated mice, two-way ANOVA followed by Bonferroni test.
RGS4 deficit in the prefrontal cortex impairs working memory and nesting behavior and decreases sensitivity to the locomotor stimulatory effect of MK-801
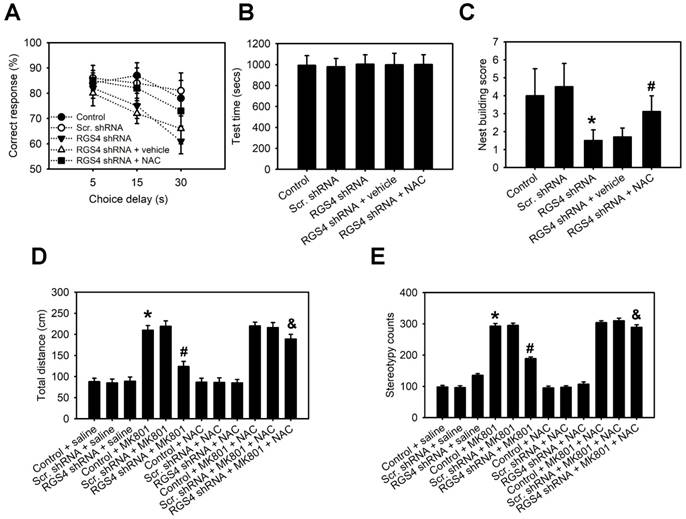
Impaired working memory and nesting behavior are behaviors related to schizophrenia in rodents. Therefore, we determined the role of RGS4 deficit in the prefrontal cortex in these behavioral tests. A delayed non-match-to-place task demonstrated that RGS4 knockdown mice learned the task and performed as well as control mice during the training. In the working memory test, however, RGS4 knockdown mice showed a consistent impairment compared with control mice (F(4, 175) = 8.18, p < 0.0001, two-way ANOVA; Figure 6A-B). Additionally, casual inspection of mice cages/housing showed poorly built nests of RGS4 knockdown mice. Control mice built fluffy nests using the wooden chips provided, at one corner of the cage floor, whereas RGS4 knockdown mice did not generally build distinguishable, discrete nests and tended to scatter pieces of nesting material across the cage floor. Images of the nests were captured and the number of scattered pieces of nesting material in each cage was counted. The number of pieces in the cages with RGS4 knockdown mice was significantly higher than those in the cages with control mice (F(4, 55) = 12.20, p < 0.0001, one-way ANOVA; Figure 6C). On the other hand, animal models of schizophrenia induced by NMDAR blockers such as PCP and MK-801, display an increase in both locomotion and stereotypic behaviors. Additionally, these behaviors are correlated with positive symptoms of schizophrenia [34]. To investigate the effects of NMDAR antagonism in the RGS4 knockdown mice, we assessed locomotion and stereotypic behaviors after MK-801 treatment. RGS4 knockdown mice showed dramatically decreased MK-801-mediated hyperlocomotion (F(11, 60) = 4.65, p < 0.0001, one-way ANOVA) and stereotypic behaviors (F(11, 60) = 5.46, p < 0.0001, one-way ANOVA) as compared to control mice, suggesting that dysfunction of RGS4 induces the hypofunction of NMDAR and further inhibits MK-801-mediated positive symptoms of schizophrenia (Figure 6D-E). Finally, we investigated whether oral administration of NAC could improve working memory, nesting behavior and sensitivity to the locomotor stimulatory effect of MK-801 in RGS4 knockdown mice. Indeed, 1-week supplementation of NAC in the drinking water significantly increased working memory, nesting capacities and sensitivity to the locomotor stimulatory effect of MK-801 in RGS4 knockdown mice.
RGS4 deficit in prefrontal cortex impairs working memory and nesting behavior and decreases sensitivity to the locomotor stimulatory effect of MK-801. (A) Working memory performance in a delayed non-match-to-place task. Vertical axis starts at 50% correct responses, which represents baseline response accuracy expected by chance. (B) Mean time to complete the working memory task measured in seconds. There were no differences in any of the groups, either in the number of days to reach the criterion or in the level of performance in the 3 days before testing. (C) Nest building activity was measured by complexity of the construction: 1 = no nest; 2 = saucer shaped nest; 3 = raised sides, and 4 = fully enclosed nest. Locomotor activity (D) and stereotypy (E) of untreated control or RGS4 knockdown mice and mice treated with 0.3 mg/kg MK-801, i.p. All the behavior tests were carried out at 21 days after virus injection. For the NAC treatment, mice were pre-treated with NAC (500 mg/kg/day) or vehicle for 1 week before behavior tests. N= 6-12 per group. *P < 0.001 compared to control mice; # P < 0.05 compared to vehicle-treated mice; P < 0.05 compared to MK801-treated mice, one-way ANOVA followed by followed by Tukey's test.
Discussion
Although there is inconsistent evidence for RGS4 alterations in the human schizophrenic brain, majority of the studies show evidence for a reproducible decrease of RGS4 expression in the human schizophrenic brain and in particular, in the prefrontal cortex [5-8]. However, there is no direct evidence that the actual reduction of RGS4 in the prefrontal cortex can induce schizophrenia-like behaviors in vivo. In the present study, we performed a comprehensive behavioral analysis of mice that received RGS4 knockdown specifically in the prefrontal cortex areas, and our results demonstrated increased stereotypy, low-anxiety phenotype, decreased social interaction, impaired attentional function, impaired working memory, impaired nesting behavior, and decreased sensitivity to the locomotor stimulatory effect of MK-801 in RGS4 knockdown mice compared to their control counterparts. Impairments in working memory [35], stereotypical behaviors [36], PPI [37], and social withdrawal [38] are prominent features of schizophrenia symptomatology. Decreased nesting behavior is characteristic of rodent models of schizophrenia [39-41] and could correspond to negative symptoms present in schizophrenic patients [42]. Therefore, the spectrum of abnormalities in mice with RGS4 deficit in the prefrontal cortex is strikingly similar to that observed in schizophrenia patients, suggesting that RGS4 deficit in the prefrontal cortex contributes to the pathophysiology of schizophrenia.
It has been demonstrated that RGS4 knockout mice had subtle sensorimotor deficits, but the other behavioral parameters did not support schizophrenia-like behaviors [11]. However, gene knockout mouse models carrying constitutive mutations were often associated with several disadvantages such as genetic compensation and developmental adaptations that could mask a clear phenotype. In contrast to RGS4 knockout strategies, the lentiviral strategies for RNA interference (RNAi)-mediated knockdown of RGS4 enable the suppression of RGS4 protein in a spatial and temporal manner. Therefore, we used this strategy to determine whether RGS4 deficit in the prefrontal cortex of adult brains can induce schizophrenia-like behaviors. Our results are inconsistent with previous findings derived from RGS4 knockout mice. It is possible that RGS4 knockdown in the prefrontal cortex of adult brains avoids the effects of genetic compensation and developmental adaptations and provides a clear phenotype induced by RGS4 deficit in specific brain areas.
RGS4 is a negative regulator of G-protein-coupled receptors and inhibits the signaling of group I metabotropic glutamate receptor (mGluR) subtype 1 and 5 in neuronal and non-neuronal cells [3]. Group I mGluRs are located on the postsynaptic membrane and modulate the ionotropic NMDARs involved in glutamatergic transmission [43]. Therefore, RGS4 deficits up-regulate the function of NMDAR and glutamatergic transmission. However, this inference does not support the general concept that NMDAR hypofunction is observed in the pathophysiology of schizophrenia. On the other hand, RGS4 is able to increase the function of NMDAR via blockage of the serotonin 5-HT1A-mediated downregulation of NMDAR function in pyramidal neurons of the prefrontal cortex [2]. RGS4 deficits have been shown to attenuate the effect of 5-HT1A on NMDAR function, suggesting the potential involvement of RGS4 in the pathophysiology of schizophrenia. Here, we provide a novel mechanism for RGS4-modulated glutamatergic transmission. We show that RGS4 promotes non-vesicular glutamate release and further increases the extracellular glutamate levels via upregulation of system xc-. Ex vivo and in vivo studies confirm that RGS4 knockdown results in a decrease in system xc- function via upregulation of xCT and subsequently reduces non-vesicular glutamate to activate the NMDAR. In contrast, RGS4 overexpression in wild-type brain slices but not in xCT-/- brain slices results in opposite effects, suggesting that system xc- is a critical effector for RGS4-regulated non-vesicular glutamate release and NMDAR activation. Additionally, we found that the RGS4 knockdown-mediated reduction of glutamate levels was more significant than the decreased xCT expression. This effect reflects the possibility of RGS4 being involved in the regulation of other glutamate transporters or glutamate synthesis. Further studies are necessary to confirm other mechanisms.
Although the detailed mechanism underlying the pathogenesis of schizophrenia remains unknown, altered regulation of fundamental mechanisms of oxidative stress may contribute to the pathogenesis of schizophrenia and related disorders [44]. Increasing evidence indicates that oxidative damage to lipids, proteins, and DNA is observed in schizophrenia [45-48]. Therefore, an imbalance in the generation and clearance of reactive oxygen species (ROS) and reactive nitrogen species (RNS) are hallmarks of schizophrenia. RGS4 deficit-mediated downregulation of system xc- may contribute to oxidative stress because system xc - provides intracellular cysteine for production of GSH and maintains plasma or cell redox balance [13]. GSH is a major antioxidant and a free-radical scavenger in the brain [49]. Recent studies have shown that individuals with psychiatric disorders such as bipolar disorder, schizophrenia, depression, and other psychiatric illnesses are deficient in GSH [50]. However, RGS4 deficits in mice did not affect GSH content in the prefrontal cortex, suggesting that alternative GSH synthesis pathways may exist in the brain. Therefore, redox dysregulation is not the main mechanism for RGS4 deficit-induced schizophrenic behaviors.
It has been proposed that reduced system xc- activity may lead to NMDAR hypofunction via decreased glutamate release or diminished NMDAR activation induced by the cysteine prodrug N-acetylcysteine. [51]. However, lack of direct evidence from in vivo data is a significant impediment to supporting this notion. In this study, a PET radiotracer was synthesized to visualize the PCP site of NMDA ion channel. Since this binding site is located within the channel, it is a unique target and is theoretically accessible only when the channel is in the active and "open" state, but not when it is in the inactive or "closed" state [52]. Therefore, this PET radiotracer can be used for non-invasive observation of NMDAR activation in vivo. The results derived from our previous study showed the radioligand specificity of our synthesized 18F-FSAG for the visualisation of NMDAR activation in vitro and in vivo using MK801 competition assay [53]. Here, our imaging data clearly showed that RGS4 deficit leads to NMDAR hypofunction in the prefrontal cortex, providing a better understanding of the complex nature of glutamate signaling in NMDAR-related disorders.
Notably, system xc- represents a novel and effective target in the treatment of schizophrenia and related disorders. Short-term administration of NAC has been shown to attenuate the psychotomimetic effects of phencyclidine [16]. Moreover, repeated NAC administration has been shown to significantly improve symptoms in patients with schizophrenia [54]. However, it is still unclear whether system xc-activity is altered in patients with schizophrenia. Clinical data derived from a small population of patients with schizophrenia showed significantly higher xCT protein expression in the dorsolateral prefrontal cortex [16]. Although this phenomenon can arise in response to chronic understimulation of system xc- as well as an increase in the expression of group II mGluR [55], this observation is not consistent with the notion that system xc- plays a role in the pathogenesis of schizophrenia. Therefore, it remains to be confirmed whether the progression of schizophrenia can trigger different alterations in the expression of system xc-. However, a recent study reported that patients with schizophrenia show downregulated expression of the two subunits of system xc-, SLC3A2 and SLC7A11, in white blood cells, suggesting that system xc- dysregulation may play a role in the pathogenesis of schizophrenia [17].
Although further clinical studies are required to confirm the impact of system xc- in the etiology and pathophysiology of schizophrenia, our results highlight system xc- as one of the critical downstream effectors of RGS4, the susceptibility gene for schizophrenia. RGS4 could modulate the expression and function of system xc- via transcriptional regulation. Decreased RGS4 expression is a common and specific feature of schizophrenia. Therefore, schizophrenia patients with RGS4 downregulation may display system xc- dysfunction and impaired glutamatergic transmission. System xc- can therefore be considered as a therapeutic target for improving symptoms of schizophrenia in these patients.
Abbreviations
%ID/g: percentage injected dose per gram; 18F-FSAG: 18F-labeled S-fluoroalkyl diarylguanidine-10; GSH: glutathione; i.p.: intraperitoneally; mGluR: group I metabotropic glutamate receptor; NAC: N-acetyl cysteine; NMDAR: N-methyl-D-aspartate receptor; PBS: phosphate-buffered saline; PCP: phencyclidine; PET: positron emission tomography; PPI: prepulse inhibition; Q-PCR: real-time quantitative polymerase chain reaction; RGS4: regulator of G-protein signaling 4; RNAi: RNA interference; RNS: reactive nitrogen species; ROS: reactive oxygen species; SAS: sulfasalazine; SDS: sodium dodecyl sulfate; shRNA: short hairpin RNA; xCT-/-: xCT homozygous knockout.
Acknowledgements
This work was supported by a grant from the National Science Council of the Republic of China (Grant No. 106-3114-8-039 -001 and 105-2628-B-039 -009 -MY3) and 2 grants from China Medical University Hospital (DMR-107-115 and DMR-106-124). We thank the National RNAi and Molecular Imaging Core Facilities, Academia Sinica, Taiwan, for technical support in gene knockdown and molecular imaging. We thank Dr. Hideyo Sato for providing the xCT-/- mice.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Taymans JM, Leysen JE, Langlois X. Striatal gene expression of RGS2 and RGS4 is specifically mediated by dopamine D1 and D2 receptors: clues for RGS2 and RGS4 functions. J Neurochem. 2003;84:1118-27
2. Gu Z, Jiang Q, Yan Z. RGS4 modulates serotonin signaling in prefrontal cortex and links to serotonin dysfunction in a rat model of schizophrenia. Mol Pharmacol. 2007;71:1030-9
3. Saugstad JA, Marino MJ, Folk JA, Hepler JR, Conn PJ. RGS4 inhibits signaling by group I metabotropic glutamate receptors. J Neurosci. 1998;18:905-13
4. Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS. Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science. 2000;288:678-82
5. Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001;6:293-301
6. Bowden NA, Scott RJ, Tooney PA. Altered expression of regulator of G-protein signalling 4 (RGS4) mRNA in the superior temporal gyrus in schizophrenia. Schizophr Res. 2007;89:165-8
7. Erdely HA, Tamminga CA, Roberts RC, Vogel MW. Regional alterations in RGS4 protein in schizophrenia. Synapse. 2006;59:472-9
8. Kimoto S, Glausier JR, Fish KN, Volk DW, Bazmi HH, Arion D. et al. Reciprocal alterations in regulator of G protein signaling 4 and microRNA16 in schizophrenia. Schizophr Bull. 2016;42:396-405
9. Rivero G, Gabilondo AM, Garcia-Sevilla JA, Callado LF, La Harpe R, Morentin B. et al. Brain RGS4 and RGS10 protein expression in schizophrenia and depression. Effect of drug treatment. Psychopharmacology (Berl). 2013;226:177-88
10. Lipska BK, Mitkus S, Caruso M, Hyde TM, Chen J, Vakkalanka R. et al. RGS4 mRNA expression in postmortem human cortex is associated with COMT Val158Met genotype and COMT enzyme activity. Hum Mol Genet. 2006;15:2804-12
11. Grillet N, Pattyn A, Contet C, Kieffer BL, Goridis C, Brunet JF. Generation and characterization of Rgs4 mutant mice. Mol Cell Biol. 2005;25:4221-8
12. Vrajova M, Pekova S, Horacek J, Hoschl C. The effects of siRNA-mediated RGS4 gene silencing on the whole genome transcription profile: implications for schizophrenia. Neuro Endocrinol Lett. 2011;32:246-52
13. Bridges RJ, Natale NR, Patel SA. System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20-34
14. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455-8
15. De Bundel D, Schallier A, Loyens E, Fernando R, Miyashita H, Van Liefferinge J. et al. Loss of system x(c)- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci. 2011;31:5792-803
16. Baker DA, Madayag A, Kristiansen LV, Meador-Woodruff JH, Haroutunian V, Raju I. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology. 2008;33:1760-72
17. Lin CH, Lin PP, Lin CY, Lin CH, Huang CH, Huang YJ. et al. Decreased mRNA expression for the two subunits of system x, SLC3A2 and SLC7A11, in WBC in patients with schizophrenia: Evidence in support of the hypo-glutamatergic hypothesis of schizophrenia. J Psychiatr Res. 2015;72:58-63
18. Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165-79
19. Javitt DC. Glutamatergic theories of schizophrenia. Isr J Psychiatry Relat Sci. 2010;47:4-16
20. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301-8
21. Tozuka Y, Fukuda S, Namba T, Seki T, Hisatsune T. GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron. 2005;47:803-15
22. Lin YJ, Shyu WC, Chang CW, Wang CC, Wu CP, Lee HT. et al. Tumor hypoxia regulates forkhead box C1 to promote lung cancer progression. Theranostics. 2017;7:1177-91
23. Soria FN, Perez-Samartin A, Martin A, Gona KB, Llop J, Szczupak B. et al. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J Clin Invest. 2014;124:3645-55
24. Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y. et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem. 2005;280:37423-9
25. Kaidanovich-Beilin O, Lipina T, Vukobradovic I, Roder J, Woodgett JR. Assessment of social interaction behaviors. J Vis Exp. 2011;48:2473
26. Aultman JM, Moghaddam B. Distinct contributions of glutamate and dopamine receptors to temporal aspects of rodent working memory using a clinically relevant task. Psychopharmacology (Berl). 2001;153:353-64
27. Robins EG, Zhao Y, Khan I, Wilson A, Luthra SK, Rstad E. Synthesis and in vitro evaluation of (18)F-labelled S-fluoroalkyl diarylguanidines: Novel high-affinity NMDA receptor antagonists for imaging with PET. Bioorg Med Chem Lett. 2010;20:1749-51
28. Neal TR, Apana S, Berridge MS. Improved synthesis of [18F]fluoromethyl tosylate, a convenient reagent for radiofluoromethylations. J Labelled Comp Radiopharm. 2005;48:557-68
29. Kim H, Kim E, Park M, Lee E, Namkoong K. Organotypic hippocampal slice culture from the adult mouse brain: a versatile tool for translational neuropsychopharmacology. Prog Neuropsychopharmacol Biol Psychiatry. 2013;41:36-43
30. Foster AC, Wong EH. The novel anticonvulsant MK-801 binds to the activated state of the N-methyl-D-aspartate receptor in rat brain. Br J Pharmacol. 1987;91:403-9
31. Shukla K, Thomas AG, Ferraris DV, Hin N, Sattler R, Alt J. et al. Inhibition of xc(-) transporter-mediated cystine uptake by sulfasalazine analogs. Bioorg Med Chem Lett. 2011;21:6184-7
32. Parwani A, Duncan EJ, Bartlett E, Madonick SH, Efferen TR, Rajan R. et al. Impaired prepulse inhibition of acoustic startle in schizophrenia. Biol Psychiatry. 2000;47:662-9
33. Couture SM, Penn DL, Roberts DL. The functional significance of social cognition in schizophrenia: a review. Schizophr Bull. 2006;32(Suppl 1):S44-63
34. Corbett R, Camacho F, Woods AT, Kerman LL, Fishkin RJ, Brooks K. et al. Antipsychotic agents antagonize non-competitive N-methyl-D-aspartate antagonist-induced behaviors. Psychopharmacology. 1995;120:67-74
35. Goldman-Rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6:348-57
36. Morrens M, Hulstijn W, Lewi PJ, De Hert M, Sabbe BG. Stereotypy in schizophrenia. Schizophr Res. 2006;84:397-404
37. Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry. 1990;47:181-8
38. Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261-76
39. Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H. et al. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci USA. 2003;100:8987-92
40. Koh HY, Kim D, Lee J, Lee S, Shin HS. Deficits in social behavior and sensorimotor gating in mice lacking phospholipase Cbeta1. Genes Brain Behav. 2008;7:120-8
41. Ouchi Y, Banno Y, Shimizu Y, Ando S, Hasegawa H, Adachi K. et al. Reduced adult hippocampal neurogenesis and working memory deficits in the Dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J Neurosci. 2013;33:9408-19
42. Powell CM, Miyakawa T. Schizophrenia-relevant behavioral testing in rodent models: a uniquely human disorder? Biol Psychiatry. 2006;59:1198-207
43. Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295-322
44. Ng F, Berk M, Dean O, Bush AI. Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int J Neuropsychopharmacol. 2008;11:851-76
45. Kim HK, Andreazza AC, Yeung PY, Isaacs-Trepanier C, Young LT. Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with bipolar disorder and schizophrenia. J Psychiatr Neurosci. 2014;39:130155
46. Che Y, Wang JF, Shao L, Young T. Oxidative damage to RNA but not DNA in the hippocampus of patients with major mental illness. J Psychiatr Neurosci. 2010;35:296-302
47. Mahadik SP, Mukherjee S. Free radical pathology and antioxidant defense in schizophrenia: a review. Schizophr Res. 1996;19:1-17
48. Reddy RD, Yao JK. Free radical pathology in schizophrenia: a review. Prostaglandins Leukot Essent Fatty Acids. 1996;55:33-43
49. Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649-71
50. Gawryluk JW, Wang JF, Andreazza AC, Shao L, Young LT. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol. 2011;14:123-30
51. Bridges R, Lutgen V, Lobner D, Baker DA. Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System xc-) to normal and pathological glutamatergic signaling. Pharmacol Rev. 2012;64:780-802
52. Poels EM, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA. et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry. 2014;19:20-9
53. Hsieh CH, Lin YJ, Chen WL, Huang YC, Chang CW, Cheng FC. et al. HIF-1alpha triggers long-lasting glutamate excitotoxicity via system xc(-) in cerebral ischaemia-reperfusion. J Pathol. 2017;241:337-49
54. Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I. et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64:361-8
55. Gupta DS, McCullumsmith RE, Beneyto M, Haroutunian V, Davis KL, Meador-Woodruff JH. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse. 2005;57:123-31
Author contact
![]() Corresponding author: Chia-Hung Hsieh, Ph.D. Mailing address: No. 91, Hsueh-Shih Road, Taichung 404, Taiwan. E-mail: chhsiehcmucmu.edu.tw; Tel: 886-4-22052121 extn. 7712; Fax: 886-4-22333641
Corresponding author: Chia-Hung Hsieh, Ph.D. Mailing address: No. 91, Hsueh-Shih Road, Taichung 404, Taiwan. E-mail: chhsiehcmucmu.edu.tw; Tel: 886-4-22052121 extn. 7712; Fax: 886-4-22333641