Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(17):4795-4804. doi:10.7150/thno.26093 This issue Cite
Research Paper
Inhibition of Notch1 induces population and suppressive activity of regulatory T cell in inflammatory arthritis
Bo Youn Choi1*, Yuri Choi1*, Jong-Sung Park1*, Li-Jung Kang2, Seung Hyun Baek1, Jin Su Park1,3, Gahee Bahn1, Yoonsuk Cho1, Hark Kyun Kim1, Jihoon Han1, Jae Hoon Sul1, Sang-Ha Baik1,4, Dong Hoon Hyun5, Thiruma V. Arumugam1,4, Siyoung Yang2, Jeung-Whan Han1, Young Mo Kang6, Yong-Woo Cho7, Jae Hyung Park3,8,9 ![]() , Dong-Gyu Jo1,3,9
, Dong-Gyu Jo1,3,9 ![]()
1. School of Pharmacy, Sungkyunkwan University, Suwon, Korea
2. Department of Pharmacology, Ajou University School of Medicine, Suwon, Korea
3. Department of Health Science and Technology, Samsung Advanced Institute for Health Science and Technology, Sungkyunkwan University, Seoul, Korea
4. Department of Physiology, Yong Loo Lin School Medicine, National University of Singapore, Singapore
5. Department of Life Science, Ewha Womans University, Seoul, Korea.
6. Department of Internal Medicine (Rheumatology), Kyungpook National University School of Medicine, Daegu, Korea
7. Department of Chemical Engineering, Hanyang University, Ansan, Korea.
8. College of Engineering, Sungkyunkwan University, Suwon, Korea
9. Biomedical Institute for Convergence, Sungkyunkwan University, Suwon, Korea
*Equal contribution.n
Received 2018-3-15; Accepted 2018-7-27; Published 2018-9-9
Abstract

Inhibition of Notch signalling has shown anti-inflammatory properties in vivo and in vitro models of rheumatoid arthritis (RA). The objective of this study was to determine whether Notch1 might play a role in regulating T-regulatory cells (Tregs) in animal models of RA.
Methods: Collagen-induced arthritis (CIA) and collagen antibody-induced arthritis (CAIA) were induced in C57BL/6, Notch1 antisense transgenic (NAS) or DBA1/J mice. We examined whether pharmacological inhibitors of γ-secretase (an enzyme required for Notch1 activation) and antisense-mediated knockdown of Notch1 could attenuate the severity of inflammatory arthritis in CIA and CAIA mice. Proportions of CD4+CD25+Foxp3+ Treg cells were measured by flow cytometry. To assess the suppressive capacity of Treg toward responder cells, CFSE-based suppression assay of Treg was performed.
Results: γ-secretase inhibitors and antisense-mediated knockdown of Notch1 reduced the severity of inflammatory arthritis in both CIA and CAIA mice. Pharmacological and genetic inhibition of Notch1 signalling induced significant elevation of Treg cell population in CIA and CAIA mice. We also demonstrated that inhibition of Notch signalling suppressed the progression of inflammatory arthritis through modulating the expansion and suppressive function of regulatory T (Treg) cells.
Conclusion: Pharmacological and genetic inhibition of Notch1 signalling suppresses the progression of inflammatory arthritis through modulating the population and suppressive function of Treg cells in animal models of RA.
Keywords: rheumatoid arthritis, Notch1, Treg, CIA, CAIA, γ-secretase
Introduction
Rheumatoid arthritis (RA) is one of the most prevalent autoimmune diseases characterized by chronic inflammation of joints and progressive destruction of cartilage and bone [1, 2]. The cause of RA is obscure, although knowing its exact etiology makes it possible to applicate correct treatment [3, 4].
Notch signalling pathway is evolutionary conserved; it presents in most multicellular organisms. It regulates various cellular processes including development, differentiation, proliferation, adhesion, apoptosis, and communication [5, 6]. Notch1 signalling also plays pivotal roles in lipid metabolism, adaptive immune responses, and inflammatory disorders [7, 8]. Mammals have four Notch receptors (Notch 1-4) and five ligands (Delta-like[DLL]-1/-3/-4 and Jagged-1/-2). These are all type I transmembrane proteins [9, 10]. Binding of the extracellular domain of Notch receptor to Notch ligand on neighbouring cells initiates sequential two step proteolytic cleavage of Notch by extracellular ADAM (metalloprotease) protease and γ-secretase complex respectively, then releases Notch intracellular domain (NICD) [9]. NICD translocates into the nucleus, where it functions as a transcriptional activator and modulates its target genes such as Hes and p21 [5]. Recent studies have demonstrated that the expression and activation of Notch signalling can stimulate synoviocytes and accelerate production of pro-inflammatory cytokines in RA [11-15]. Moreover, Notch signalling is enhanced in RA patients and its expression pattern is altered in T helper (Th) cell [16].
Previously, we have demonstrated that inhibitors of γ-secretase, an enzyme required for Notch1 activation, can attenuate the severity of RA indices and suppress proinflammatory cytokines (TNF-α, IFN-γ, IL-6, IL-12, IL-17 and MCP-1), NF-κB, and MMP-3 in RA synoviocytes and in collagen-induced arthritis (CIA) mice [17, 18]. In addition, hyaluronan nanoparticles bearing a γ-secretase inhibitor effectively accumulated at the inflamed joint of CIA mice. They dramatically reduced the production of pro-inflammatory cytokines and collagen-specific auto-antibodies (IgG1 and IgG2a) in the sera of the CIA mice [18]. We have also shown that CIA progression is attenuated in Notch1 antisense transgenic (NAS) mice [11]. Consistently, down-regulation of Notch1 with Notch1-targeting siRNA delivery nanoparticles resulted in delayed bone erosion, cartilage damage, and progression of inflammation in CIA mice [17].
In the immune system, Notch signalling regulates T and B cell development, T cell activation, regulatory T (Treg) cell function and T helper (Th) cell differentiation [19-21]. Th cells from RA patients show highly expressed Notch receptors and increased activity of Notch signalling. Although the involvement of Notch signalling in RA has been suggested, the mechanism by which inhibiting Notch1 signalling could ameliorate the progression of inflammatory arthritis is currently unknown. Here we report that suppression of Notch1 signalling using antisense technology and low-molecular-weight γ-secretase inhibitors can ameliorate the disease process in animal models of RA by increasing the population and suppressive activity of Treg.
Methods
Mice
Mice overexpressing Notch1 antisense (NAS) construct under the control of a mouse mammary tumor virus long terminal repeat promoter were generated, backcrossed onto C57BL/6 background and characterized as previously described [22]. NAS and non-transgenic control C57BL/6 mice were kept under a 12 h light/12 h dark cycle with free access to food and water. Three-month-old male mice were used in experiments. This study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Sungkyunkwan University School of Medicine (SUSM). SUSM is an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International)-accredited facility that abides by the Institute of Laboratory Animal Resources (ILAR) guide.
γ-secretase inhibitors
We used very potent and selective γ-secretase inhibitors, including RO4929097 (2,2-dimethyl-N-((S)-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl)-N′-(2,2,3,3,3-pentafluoro-propyl)-malonamide) (Selleck) and L-685,458 ((5S)-(tert-Butoxy-carbonylamino)-6-phenyl-(4R)-hydroxy-(2R)-benzylhexanoyl)-L-leucy-L-phenylani-namide) (Tocris). RO4929097 is a cell-permeable dipeptide that can inhibit γ-secretase at an IC50 of 4 nM. L-685,458, an aspartyl protease transition state mimic, is a more potent and selective small molecule inhibitor of γ-secretase (IC50 = 17 nM) that displays more than 50-fold selectivity over a range of aspartyl, serine, and cysteine proteases.
Collagen-induced arthritis (CIA)
Collagen-induced arthritis (CIA) was induced in C57BL/6 or NAS mice as previously described [23]. Briefly, mice were injected intradermally at two sites at the base of the tail with 400 μg of chicken type II collagen (CII) (2 mg/mL, Chondrex, Redmond, WA, USA) emulsified in 200 μL of Complete Freund's Adjuvant (4 mg/mL, Chondrex) followed by a booster immunization 21 days later with CII emulsified in Incomplete Freund's Adjuvant (Chondrex). Mice were monitored for the induction of arthritis by clinical scoring or by measuring hind paw swelling using callipers. Paw scores included the following: 0 = normal, 1 = slight swelling and/or erythema, 2 = pronounced swelling, and 3 = ankylosis. These paw scores were summed for each mouse, giving a maximum possible score of 12 per mouse.
To induce CIA, DBA/1J mice were injected intradermally at the base of the tail with 200 μg of bovine type II collagen (CII) (2 mg/mL) emulsified in 100 μL of Complete Freund's Adjuvant (4 mg/mL), followed by a booster immunization 21 days later. Mice were injected intraperitoneally with 1 mg/kg RO4929097, 1 mg/kg L-685,458, or vehicle (10% DMSO) daily starting on the 32nd day of booster injection. DBA/1J mice were monitored for the induction of arthritis by clinical scoring. Paws were scored individually as follows: 0 = normal, 1 = mild swelling and erythema confined to the midfoot and ankle joint, 2 = mild swelling and erythema extending to the midfoot and ankle joint, 3 = moderate swelling and erythema extending from the metatarsal joints to the ankle, and 4 = severe swelling and erythema encompassing the foot, ankle and digits. These paw scores were summed for each mouse, giving a maximum possible score of 16 per mouse.
Synoviocyte culture
Human fibroblast-like synoviocytes (FLS) were cultured from RA patient tissue obtained during joint replacement surgery. Cells were subcultured at a 1:5 ratio. Passages between 5 and 8 were used in these experiments.
Quantitative real-time PCR
Total RNAs were prepared from FLS cells using RNA iso-plus reagent (TAKARA, 9109). Total RNA (1 μg) was used for cDNA synthesis with reverse transcriptase (TAKARA, RR036A). Synthesized cDNA was used as template for qPCR with SYBR premix EX taq II (Takara, RR820A) and gene-specific pair of primers (Table 1).
List of primers used in quantitative RT-PCR
| Gene | Primer sequences | Annealing temperature (℃) | PCR product size (bp) | |
|---|---|---|---|---|
| Forward | Reverse | |||
| Hey-1 | catacggcaggagggaaag | gcatctagtccttcaatgatgct | 60 | 200 |
| IL-6 | aattcggtacatcctcgacgg | ttggaaggttcaggttgttttct | 60 | 113 |
| IL-10 | gtgatgccccaagctgaga | cacggccttgctcttgtttt | 60 | 138 |
| IL-12 | tggagtgccaggaggacagt | tcttgggtgggtcaggtttg | 60 | 147 |
| GAPDH | caacaattcctggcgatacct | ggcatggactgtggtcatgag | 60 | 87 |
| TNF-α | cagagggcctgtacctcatc | ggaagacccctcccagatag | 55 | 219 |
| IL-17A | actacaaccgatccacctcac | actttgcctcccagatcacag | 55 | 84 |
| TGF-β | actactacgccaaggaggtcac | tgcttgaacttgtcatagatttcg | 55 | 73 |
| GAPDH | gacatcaagaaggtggtgaa | tgtcataccaggaaatgagc | 55 | 178 |
Collagen antibody-induced arthritis (CAIA)
C57BL/6 or NAS mice were injected intradermally with 2 mg Arthrogen-CIA 5-Clone monoclonal antibody Cocktail (αCII-IA) (Chondrex) on day 0 followed by 50 μg of lipopolysaccharide injection on day 3. On day 6, mice were administered 500 mg/kg adalimumab once and 1 mg/kg L-685,458 daily for 7 days. Mice were monitored for the induction of arthritis by clinical scoring as described above.
Histologic examination of joints and immunohistochemistry (IHC)
Knee joints were fixed in 10% neutral buffed formalin, decalcified using Decalcified Solution-Lite (Sigma, St. Louis, MO, USA) for 6 h, embedded in paraffin, and sectioned. Tissue sections were stained with H&E and synovitis and bone/cartilage destruction were evaluated with a five-point scale as follows: grade 0 (no sign of inflammation), grade 1 (mild inflammation with minimal hyperplasia of the synovial lining layer without cartilage destruction), and grades 2 through 4 (increasing degrees of inflammatory cell infiltration or cartilage and bone destruction).
For IHC, the tissue sections (5 μm) were deparaffined in xylene and rehydrated. After finishing heat-induced antigen retrieval, endogenous peroxidase was blocked with 0.3% H2O2 for 10 min and non-specific staining was prevented by soaking in 3% bovine serum albumin. Sections were incubated with primary antibody at 4 ℃ overnight. Primary antibodies were CD3 (Santacruz) and Foxp3 (Santacurz). Staining was done with SPlink HRP Mouse Detection (DAB) Kit (Life science division).
Bead-based cytokine assay
After 4 weeks of immunization, serum samples were obtained. Levels of TNF-α, INF-γ, IL-6, -12, -17 in serum were measured using a Milliplex Mouse Cytokine panel 1 (Millipore) and Luminex 100 system (Luminex) according to the manufacturer's instructions.
Analysis of T cell populations
For analysis of Treg population and effector T cell population, splenocytes were stained with CD4-FITC and CD25-PerCP-Cy5.5 antibodies (BD Bioscience, San Jose, CA, USA) or FITC anti-CD4 (BioLegend, San Diego, CA, USA), APC anti-CD44 (BioLegend), PB anti-CD62L (BioLegend). After fixation/permeabilization, cells were treated with Foxp3-PE antibody (BDbioscience). CD4 T, CD8 T, or Treg (CD4+CD25+Foxp3+) percentages were determined using a FACSCalibur (BD Bioscience) or guava easyCyteTM 8 (Millipore). Treg (CD4+Foxp3+) and effector T cells (CD44+CD62L-) populations were determined using FACS MACSQuant VYB flow cytometer.
Isolation of regulatory T cells
Treg cells were purified from splenocytes using CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec) and MACSTM (Miltenyi Biotec).
Suppression assay of regulatory T cells
Non-regulatory CD4+CD25- T responder (Tresp) cells were labeled with 1 μM of carboxy fluorescein diacetate succinimidyl ester (CFSE: eBioscience). Labeled Tresp cells (3 × 104) were mixed without or with primed CD4+CD25+ Treg cells (3 × 104) by plate-bound anti-CD3 (3 μg/mL) plus soluble anti-CD28 (5 μg/mL) mAb in the presence of anti-CD3 (3 μg/mL) plus irradiated (3000 rad) T cell-depleted splenocytes (3 × 104). CFSE signal of gated lymphocytes was analyzed by flow cytometry (guava easyCyteTM 8: Millipore). These experiments were run in quadruplicate. Results are expressed as relative inhibition of the percentage of CFSElow cells.
Statistical analysis
Prism 5.0 (Graphpad) was used for all statistical analyses. One-way analysis of variance (ANOVA) was used for multiple group analysis. Two-tailed unpaired t-test was used to compare data between two groups.
Results
γ-secretase inhibition mediates Treg induction and suppression of CIA propagation
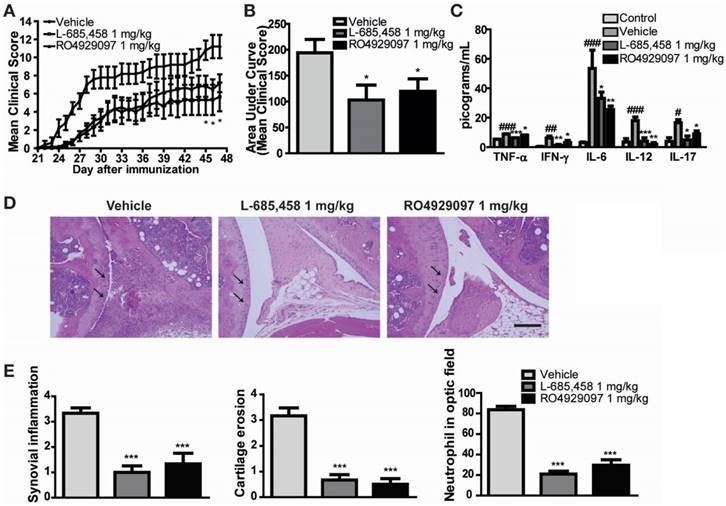
We first confirmed the protective effect of other γ-secretase inhibitors against CIA induction in DBA1/J mice. To avoid researcher bias, we concealed the identity of the treatment from the experimenter who scored the clinical condition of mice and histological sections for disease. We found that administering 1 mg/kg of two kinds of potent γ-secretase inhibitor (L-685,458 or RO4929097) starting at 21 days after primary immunization significantly improved clinical outcomes (Figure 1A-B) and serum levels of pro-inflammatory cytokines in CIA mice compared to vehicle-treated CIA controls (Figure 1C). Histologically, vehicle-treated CIA mice showed extensive synovitis, neutrophil infiltration, destruction of articular cartilage, and bone erosion. In contrast, joints of γ-secretase inhibitors-treated CIA mice showed significantly reduced synovial inflammation, cartilage erosion, and neutrophil infiltration (Figure 1D-E).
γ-secretase inhibitors ameliorate symptoms of CIA-induced RA. (A-B) Severity of arthritis was assessed using a visual arthritis scoring system in CIA mice treated with vehicle, L-685,458 or RO4929097. Values are mean ± SEM. *P < 0.05. (C) Bead-based cytokine analysis of serum using a Luminex 100 system at 47 days after primary immunization. Values are mean ± SEM. #P < 0.05, ##P < 0.01, ###P < 0.001 versus controls; *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle-treated controls. (D) Representative photomicrographs of H&E-stained tissue sections of knee joints of control mice and CIA mice treated with vehicle, L-685,458, or RO4929097. Original magnification, 200x. Scale bar: 100 μm. (E) Quantification of synovial inflammation, cartilage erosion, and neutrophil infiltration. Values are mean ± SEM. **P < 0.01, ***P < 0.001.
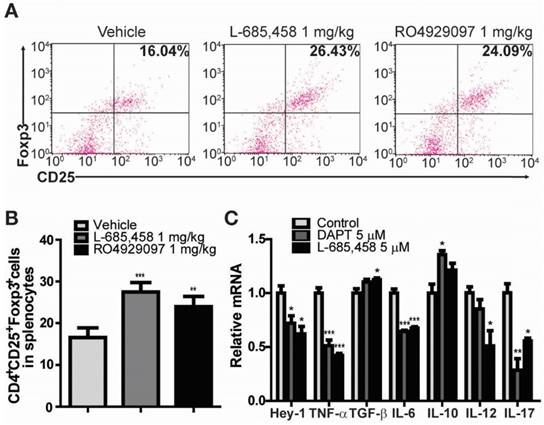
It is widely accepted that Treg cells defined by the expression of CD4, CD25, and transcription factor forkhead box P3 (Foxp3) play critical roles in the pathogenesis of several autoimmune diseases, including RA [24-28]. CD4+CD25+Foxp3+ Treg cells play a crucial role in preventing chronic inflammatory and autoimmune diseases by suppressing autoreactive T cells. Numerous studies in animal models of autoimmunity have shown that defects in Treg cells can contribute to the development of autoimmunity [24-26]. Therefore, we investigated the effect of γ-secretase inhibitors on the level of CD4+CD25+Foxp3+ Treg cell population in CIA mice. Treg cell population was significantly elevated in splenocytes isolated from either L-685,458- or RO4929097-treated CIA mice compared to that from vehicle-treated CIA mice (Figure 2A-B). Furthermore, treatment of fibroblast-like synoviocytes isolated from RA patients with γ-secretase inhibitors, DAPT or L-685,458, reduced mRNA expression levels of Hey-1 (a Notch1 target gene) and pro-inflammatory cytokines such as TNF-α, IL-6, IL-12, and IL-17, although expression levels of TGF-β and IL-10 known to induce Treg cells were increased by such treatment (Figure 2C). These results indicate that arthritis progression is protected by interruption of Notch signalling related to regulation of T cell population in CIA mice and synoviocytes from RA patients.
Administration of γ-secretase inhibitors induces Treg population in CIA-induced DBA1/J mice and cytokines are altered by γ-secretase inhibitors in human RA patients' synoviocytes. (A) Proportions of CD4+CD25+Foxp3+ cells among splenocytes of vehicle-, L-685,458-, or RO4929097-treated mice at 46 days after primary immunization evaluated by flow cytometry. Numbers indicate percentages of CD25+Foxp3+ cells among CD4+ cells. (B) Statistical analysis of CD4+CD25+Foxp3+ cells in splenocytes. Values are mean ± SEM. **P < 0.01. (C) Quantitative real-time PCR analyses of relative mRNA expression levels in RA patients' synoviocytes treated with 5 μM DAPT or 5 μM L-685,458 for 24 h. Value are mean ± SEM (n=3). *P < 0.05, **P < 0.01, ***P < 0.001 versus controls.
Treg population is increased in Notch1 antisense transgenic mice
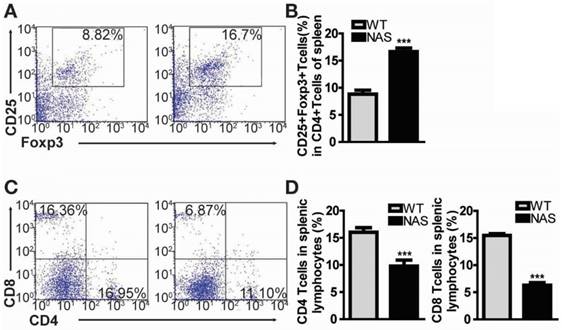
Encouraged by the effect of γ-secretase inhibitors on Treg population in our rodent model of RA, we examined mice transgenic for antisense Notch1 (NAS) [22] with CIA to determine whether a specific reduction of Notch1 signalling could increase Treg cell population accompanied by attenuated inflammatory arthritis. In our previous report, CIA progression was significantly reduced in NAS mice [11]. We also scrutinized the frequency of CD4+CD25+Foxp3+ Treg cells in WT CIA and NAS CIA mice. Treg cell population was significantly increased from 8.82% in WT CIA to 16.7% in NAS CIA mice (Figure 3A-B). Furthermore, both CD4+ and CD8+ T cells in NAS CIA mice were consistently decreased by 33% and 58%, respectively (Figure 3C-D) compared to those in WT CIA. The levels of these T cells showed similar patterns between WT and NAS mice before inducing CIA (data not shown). Since the CD4+ population contains all types of Th cells, these results imply that attenuated CIA progression of NAS mice seems to be dependent on increased population of Treg cells.
Treg population is increased in CIA-induced NAS mice. (A) Proportions of CD4+CD25+Foxp3+ cells among splenocytes of NAS CIA or WT CIA mice at 28 days after primary immunization evaluated by flow cytometry. Numbers indicate percentages of CD25+Foxp3+ cells among CD4+ cells. (B) Statistical analysis of CD4+CD25+Foxp3+ cells. Values are mean ± SEM. ***P < 0.001. (C) At 28 days after primary immunization, both CD4 and CD8 T cells from splenic lymphocytes of NAS or WT CIA mice were analysed by flow cytometry. (D) Statistical data shown as bar graphs. Values are mean ± SEM. ***P < 0.001.
Inhibition of Notch signalling ameliorates collagen antibody-induced arthritis (CAIA)
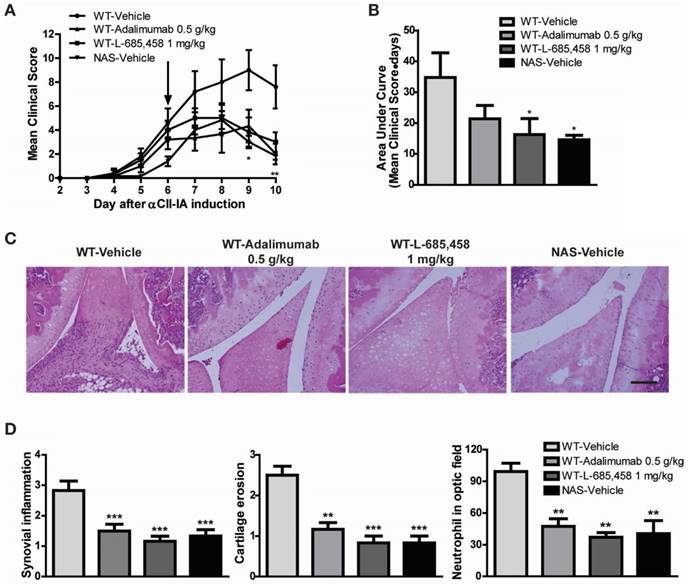
The CAIA model is useful for studying mechanisms of RA during the effector phase after CII-specific antibodies have been produced [29]. To determine whether Notch signalling might be engaged in the effector phase mediated by antibodies to CII, we injected a cocktail of several distinct high-affinity CII antibodies (αCII). After αCII injection, vehicle-treated C57BL/6 mice developed severe paw swelling and joint inflammation with cartilage erosion and neutrophil infiltration (Figure 4). In contrast, administration of L-685,458 or TNF-α-blocking antibody, adalimumab (HUMIRA), after arthritis onset (day 6) significantly suppressed CAIA compared to vehicle treatment (Figure 4A-B). Therapeutic treatment with L-685,458 or adalimumab also reduced synovial inflammation, cartilage erosion and neutrophil infiltration (Figure 4C-D). The efficacy of γ-secretase inhibitor (L-685,458) was comparable to that of the TNF-α blocker (adalimumab) in inhibiting CAIA (Figure 4). These data support that γ-secretase inhibitors can impair arthritis progression when they are administered at disease onset. They can also revert arthritis progression rapidly when they are given during established disease.
Therapeutic effect of Notch1 inhibition in CAIA. (A-B) Severity of arthritis was assessed using a visual arthritis scoring system in WT CAIA and NAS CAIA mice treated with vehicle, adalimumab, or L-685,458 (arrow). Values are mean ± SEM. *P < 0.05, **P < 0.01. (C) H&E-stained representative joint sections from the experiment shown in (A). Original magnification, 200x. Scale bar: 100 μm. (D) Quantification of synovial inflammation, cartilage erosion, and neutrophil infiltration. Values are mean ± SEM. **P < 0.01, ***P < 0.001.
Increased Treg population in NAS mice with CAIA
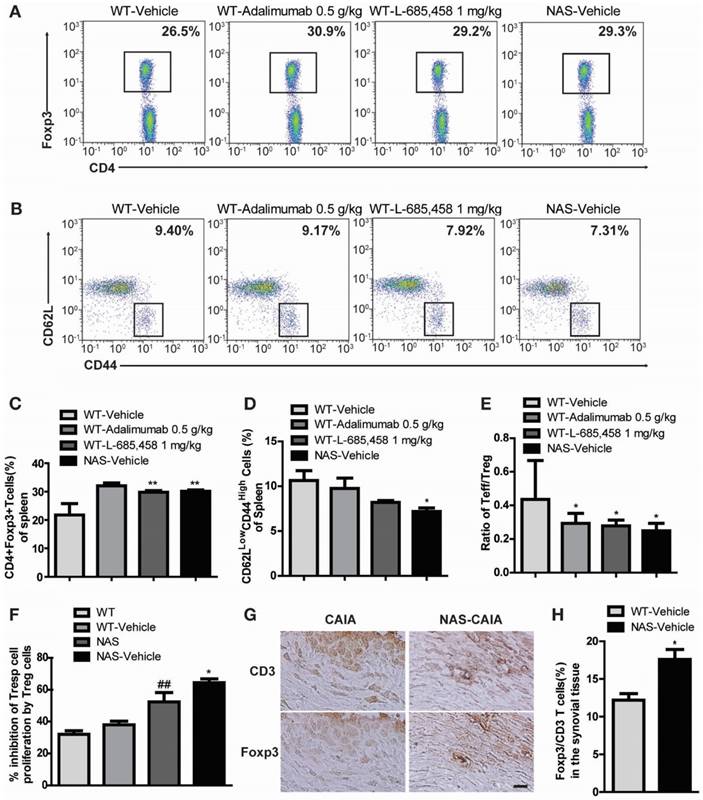
We also examined NAS mice with CAIA to determine whether a specific reduction of Notch signalling could attenuate the severity of inflammatory arthritis. Arthritis indices, paw thicknesses, synovitis, cartilage erosion, and neutrophil infiltration resulting from CAIA were significantly less in NAS mice (NAS-Vehicle) compared to those in non-transgenic mice (WT-Vehicle) (Figure 4). NAS mice showed very mild paw swelling with reduced number of infiltrating cells and less severe cartilage erosion in the CAIA model (Figure 4). Furthermore, Treg cell levels in the spleen were increased in L-685,458- and adalimumab-administrated WT CAIA (WT-Vehicle) or NAS CAIA (NAS-Vehicle) mice compared to those in vehicle-treated CAIA mice. To identify the Treg population, we analysed CD4+ and Foxp3+ populations in the same number of live cells because most Foxp3+ cells were CD25+ in the spleen and lymph nodes. As shown in Figure 3, the CD4+ population was greatly reduced by suppression of Notch signalling. On the other hand, inhibition of Notch by either pharmacological or genetic methods increased the Foxp3+ population in the CD4+ population (Figure 5A, C). Next, we assessed T effector cells known to maintain inflammation and consequently cause joint damage. Inhibition of Notch1 reduced the population of effector T (Teff) cells (CD62L-CD44+) in the spleen (Figure 5B, D). Ratios of Teff/Treg in spleens were decreased in L-685,458- and adalimumab-administrated WT CAIA or NAS CAIA mice compared to those in vehicle-treated CAIA mice (Figure 5E).
(A, C) Notch inhibition increases Treg population in the CAIA model. Proportions of CD4+Foxp3+ Treg cells among splenocytes of indicated mice at 9 days after αCII-IA induction were evaluated by flow cytometry. Numbers indicate percentages of CD4+Foxp3+ cells. Values are mean ± SEM. **P < 0.01. (B, D) Proportions of T effector cells among splenocytes. Numbers indicate percentages of CD62LlowCD44high cells. *P < 0.05 versus control. (E) Ratios between effector T cells and regulatory T cells (Teff/Treg). Values are mean ± SEM. *P < 0.05 versus control (WT/Vehicle). (F) Suppressive effects of Treg cells were anlayzed by CFSE-based suppression assay. Values are mean ± SEM. ##P < 0.01 versus WT mice; *P < 0.05 versus NAS mice. (G, H) Synovial tissues of CAIA were used for immunohistochemical staining analysis. Consecutive sections of synovial tissues from WT CAIA and NAS CAIA mice were DAB-stained for CD3 and Foxp3. Quantitative analysis is shown in the bar graph. Values are mean ± SEM. *P < 0.05. Scale bar: 25 μm.
Increased suppressive activity of Treg cells in NAS mice
To assess the suppressive capacity of Treg toward responder cells following increased Treg cell population, we performed a CFSE-based suppression assay of Treg cells. The suppressive activity of Treg cells was more intense in NAS and NAS CAIA mice compared to those in WT and WT CAIA mice (Figure 5F), indicating that blockade of Notch signalling resulted in increased population and suppressive activity of Treg cells. Furthermore, we evaluated the expression of Treg cells in the synovial tissue. Foxp3+ Treg cells in synovial tissues were significantly increased in NAS CAIA mice compared to WT CAIA mice (Figure 5G-H). These results suggest that pharmacological and genetic inhibition of Notch signalling can suppress the progression of inflammatory arthritis through modulating the population and suppressive function of Treg cells in animal models of RA.
Discussion
RA is a common human autoimmune disease characterized by chronic systemic inflammation and progressive bone destruction. It affects 1% of the population. Biologics, such as TNF blockade, have been proven to be effective in RA patients not responding to anti-rheumatic drugs including methotrexate, which is the first-line therapy for RA. However, still around 30% of patients treated with TNF blockades have no response. Despite numerous attempts to identify the molecular pathogenesis and etiology of RA, effective treatment without side effects is still lacking. In the present study, we demonstrated that Notch1 signalling was activated and played a crucial role in joint inflammation and Treg regulation in mouse models of RA, suggesting that Notch signalling might be potentially an important therapeutic target for arthritis.
Interference with Notch1 signalling using γ-secretase inhibitors protected mice against development of CIA, a model in which adaptive immune response is known to contribute to disease development similarly to human disease [18, 30]. Administration of γ-secretase inhibitors reverted CAIA progression rapidly when they were given during established disease. We found that interference with Notch1 expression reduced arthritis development as well as neutrophil accumulation in joints of both CIA and CAIA models. Innate immune cells are major players in initiation of CAIA while adaptive immune cells are also required for maintaining arthritis [31, 32].
Selective γ-secretase inhibitors are widely used to inhibit Notch signaling. However, they can affect substrates other than Notch. Therefore, we examined Notch1 antisense (NAS) transgenic mice that expressed 30% to 40% reduction of Notch1 protein [22]. We found that attenuation of Notch signalling reduced cartilage erosion and synovial inflammation in both CIA and CAIA models.
γ-secretase-mediated Notch pathway activation is a prerequisite for the activation of NF-κB and production of IFN-γ in lymphocytes, suggesting the importance of cross-talk between these signalling pathways in modulating the response of immune cells to stimulation [33, 34]. Previous studies have shown that Notch signalling can enhance T cell proliferation and cytokine production [19]. In fact, pro-inflammatory cytokines such as IFN-γ, TNF-α, IL-6, IL-12, and IL-17 play significant roles in the pathophysiology of autoimmune diseases including RA. Previous studies have reported elevated Notch activity in synovial membrane and synoviocytes from RA patients compared to that in control groups. In addition, activated Notch can promote synoviocyte proliferation induced by TNF-α in vitro [13, 35]. Increased NICD in inflamed joint can mediate hypoxia-induced angiogenesis related to the initiation and progression of RA [36]. In our previous study, we showed that Notch signalling not only stimulates synoviocytes in vivo but also accelerates the production of pro-inflammatory cytokines and immune responses influenced by up-regulated IgG1 and IgG2a [11]. When Notch signalling was blocked by genetic inhibition or pharmacological agents, levels of active NF-κB, ICAM-1, and pro-inflammatory cytokines were attenuated in the CIA model. Moreover, we proved that relative mRNA levels of Hey-1, and pro-inflammatory cytokines such as TNF-α, IL-6, IL-12, and IL-17 were reduced but levels of TGF-β and IL-10 were increased in human RA synoviocytes upon γ-secretase inhibitor treatment.
Although there have been many studies on the role of Notch signalling in the immune system, its precise role in autoimmune diseases remains elusive. Blockade of Notch signalling via γ-secretase inhibitor decreases TNF-α production and alters dendritic cells phenotype [37], and prevents experimental autoimmune encephalomyelitis (EAE) progression [38]. In contrast, overexpression of Jagged-1, a Notch ligand, can alleviate CIA while blockade of Jagged-1 via anti-Jagged-1 mAb exacerbates CIA [39]. In fact, one report has demonstrated that administration of anti-Jagged-1 mAb exacerbates clinical features of EAE while DLL-1 mAb alleviates disease severity [40]. These conflicting data not only indicate that Notch ligands can differentially regulate inflammation, but also suggest that an endogenous ligand other than Jagged-1 might mediate adverse effects of Notch signalling.
Approximately 75% of women with RA have improvement of symptoms during their pregnancy while their symptoms will worsen after delivery. It has been suggested that RA symptoms are alleviated due to increased Treg during pregnancy. Because a fetus is another autoantigen, Treg-deficient mice show increased abortion rates. However, pregnancy can induce Treg population to prevent maternal rejection [41, 42]. In a similar concept, induction of Treg cells expressing transcription factor Foxp3, a population of CD4+ T cells that possess potent immunosuppressive activities, is required to prevent autoimmunity [27]. However, the role of Notch1 signalling in Treg expansion and activity has been controversial. While a number of studies have shown that Notch signalling facilitates suppressor function and expansion of Treg cells [43-45], a few other studies have indicated a negative effect of Notch signalling on Treg cells in models of encephalomyelitis and autoimmune diabetes [20]. In the present study, we found that blockage of Notch signalling induced Treg expansion and suppressed autoimmune responses, and increased Treg cell numbers in synovial tissues of mouse models of RA. It has been recently reported that TNF-α blocker adalimumab can increase Treg population [46]. Indeed, the suppressor activity of NAS-derived Treg cells was significantly greater than that of WT-derived Treg cells. Therefore, it appears that Notch1 can exert diverse effects on Treg cells in a context-dependent manner. This discrepancy might be attributed to the use of different experimental systems and the intensity of Notch1 signalling.
The suppression of inflammatory arthritis by inhibition of Notch signalling in CIA and CAIA models might be due to dampening of T cell recruitment and decline of the effector function of T and B cells accompanied by increased Treg expansion and suppressor activity. We have previously shown that the blockade of Notch signalling via γ-secretase inhibition in immune cells of inflamed joints can alleviate symptoms of RA with therapeutic effects [11]. To avoid unwanted side effects of γ-secretase inhibitors, which block cleavage of Notch and substrates other than Notch in all cell types, treatments that specifically target activated lymphocytes are needed. Nonetheless, our study suggests that even global inhibition of Notch signalling can significantly alleviate clinical symptoms of RA as well as immune cell-mediated damage to joint tissues in animal models. Since there are no treatments for RA that can prevent damage to joint tissues, further studies on targeted Notch signalling inhibition at the site of inflammation in RA are warranted to provide a new therapeutic window.
Abbreviations
CAIA: collagen antibody-induced arthritis; CFSE: carboxyfluorescein diacetate succinimidyl ester; CIA: collagen-induced arthritis; CII: type II collagen, FLS: fibroblast-like synoviocytes; EAE: encephalomyelitis; IHC: immunohistochemistry; NAS: notch1 antisense; NICD: notch intracellular domain; RA: rheumatoid arthritis; Teff: effector T cell; Th: T helper cell; Treg: regulatory T cell; Tresp: T responder cell.
Acknowledgements
This research was supported by the National Research Foundation of Korea grants (NRF-2012R1A5A2A28671860, NRF-2018M3A9H1023767) funded by the Korean Government (MSIT), and by the Industrial Technology Innovation Program (10078392) of MOTIE.
Author contributions
B.Y.C., Y.C., J.-S.P., T.V.A., S.Y., Y.M.K., Y.W.C., J.H.P. and D.-G.J conceived the project and designed the experiments. J.-S.P., B.Y.C., Y.C., L.J.K., G.B., S.H.B., J.S.P., Y.C., H.K.K., J.H.S., J.H., D.H.H. and S.-H.B. conducted experiments and analyzed data. J.-S.P., B.Y.C., Y.C., J-W.H., J.H.P. and D.-G.J wrote the manuscript. All authors read and commented on the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429-42
2. Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397-440
3. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205-19
4. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388:2023-38
5. Fiuza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol. 2007;194:459-74
6. Bray S, Bernard F. Notch targets and their regulation. Curr Top Dev Biol. 2010;92:253-75
7. Cheng YL, Choi Y, Sobey CG, Arumugam TV, Jo DG. Emerging roles of the gamma-secretase-notch axis in inflammation. Pharmacol Ther. 2015;147:80-90
8. Song NJ, Yun UJ, Yang S, Wu C, Seo CR, Gwon AR. et al. Notch1 deficiency decreases hepatic lipid accumulation by induction of fatty acid oxidation. Sci Rep. 2016;6:19377
9. Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565-97
10. Woo HN, Park JS, Gwon AR, Arumugam TV, Jo DG. Alzheimer's disease and Notch signaling. Biochem Biophys Res Commun. 2009;390:1093-7
11. Park JS, Kim SH, Kim K, Jin CH, Choi KY, Jang J. et al. Inhibition of notch signalling ameliorates experimental inflammatory arthritis. Ann Rheum Dis. 2015;74:267-74
12. Khan IM, Palmer EA, Archer CW. Fibroblast growth factor-2 induced chondrocyte cluster formation in experimentally wounded articular cartilage is blocked by soluble Jagged-1. Osteoarthritis Cartilage. 2010;18:208-19
13. Nakazawa M, Ishii H, Aono H, Takai M, Honda T, Aratani S. et al. Role of Notch-1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum. 2001;44:1545-54
14. Ishii H, Nakazawa M, Yoshino S, Nakamura H, Nishioka K, Nakajima T. Expression of notch homologues in the synovium of rheumatoid arthritis and osteoarthritis patients. Rheumatol Int. 2001;21:10-4
15. Ando K, Kanazawa S, Tetsuka T, Ohta S, Jiang X, Tada T. et al. Induction of Notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene. 2003;22:7796-803
16. Jiao Z, Wang W, Guo M, Zhang T, Chen L, Wang Y. et al. Expression analysis of Notch-related molecules in peripheral blood T helper cells of patients with rheumatoid arthritis. Scand J Rheumatol. 2010;39:26-32
17. Kim MJ, Park JS, Lee SJ, Jang J, Park JS, Back SH. et al. Notch1 targeting siRNA delivery nanoparticles for rheumatoid arthritis therapy. J Control Release. 2015;216:140-8
18. Heo R, Park JS, Jang HJ, Kim SH, Shin JM, Suh YD. et al. Hyaluronan nanoparticles bearing gamma-secretase inhibitor: in vivo therapeutic effects on rheumatoid arthritis. J Control Release. 2014;192:295-300
19. Maillard I, Fang T, Pear WS. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu Rev Immunol. 2005;23:945-74
20. Bassil R, Zhu B, Lahoud Y, Riella LV, Yagita H, Elyaman W. et al. Notch ligand delta-like 4 blockade alleviates experimental autoimmune encephalomyelitis by promoting regulatory T cell development. J Immunol. 2011;187:2322-8
21. Tanigaki K, Honjo T. Regulation of lymphocyte development by Notch signaling. Nat Immunol. 2007;8:451-6
22. Cheng P, Zlobin A, Volgina V, Gottipati S, Osborne B, Simel EJ. et al. Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. J Immunol. 2001;167:4458-67
23. Inglis JJ, Simelyte E, McCann FE, Criado G, Williams RO. Protocol for the induction of arthritis in C57BL/6 mice. Nat Protoc. 2008;3:612-8
24. Esensten JH, Wofsy D, Bluestone JA. Regulatory T cells as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:560-5
25. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849-59
26. Valencia X, Lipsky PE. CD4+CD25+FoxP3+ regulatory T cells in autoimmune diseases. Nat Clin Pract Rheumatol. 2007;3:619-26
27. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775-87
28. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA. et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277-85
29. Khachigian LM. Collagen antibody-induced arthritis. Nat Protoc. 2006;1:2512-6
30. Trentham DE. Collagen arthritis as a relevant model for rheumatoid arthritis. Arthritis Rheum. 1982;25:911-6
31. Nandakumar KS, Backlund J, Vestberg M, Holmdahl R. Collagen type II (CII)-specific antibodies induce arthritis in the absence of T or B cells but the arthritis progression is enhanced by CII-reactive T cells. Arthritis Res Ther. 2004;6:R544-50
32. Skapenko A, Lipsky PE, Schulze-Koops H. T cell activation as starter and motor of rheumatic inflammation. Curr Top Microbiol Immunol. 2006;305:195-211
33. Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J. et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18:268-81
34. Shin HM, Minter LM, Cho OH, Gottipati S, Fauq AH, Golde TE. et al. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J. 2006;25:129-38
35. Jiao Z, Wang W, Ma J, Wang S, Su Z, Xu H. Notch signaling mediates TNF-alpha-induced IL-6 production in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Clin Dev Immunol. 2012;2012:350209
36. Gao W, Sweeney C, Connolly M, Kennedy A, Ng CT, McCormick J. et al. Notch-1 mediates hypoxia-induced angiogenesis in rheumatoid arthritis. Arthritis Rheum. 2012;64:2104-13
37. Perez-Cabezas B, Naranjo-Gomez M, Bastos-Amador P, Requena-Fernandez G, Pujol-Borrell R, Borras FE. Ligation of Notch receptors in human conventional and plasmacytoid dendritic cells differentially regulates cytokine and chemokine secretion and modulates Th cell polarization. J Immunol. 2011;186:7006-15
38. Minter LM, Turley DM, Das P, Shin HM, Joshi I, Lawlor RG. et al. Inhibitors of gamma-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat Immunol. 2005;6:680-8
39. Kijima M, Iwata A, Maekawa Y, Uehara H, Izumi K, Kitamura A. et al. Jagged1 suppresses collagen-induced arthritis by indirectly providing a negative signal in CD8+ T cells. J Immunol. 2009;182:3566-72
40. Elyaman W, Bradshaw EM, Wang Y, Oukka M, Kivisakk P, Chiba S. et al. JAGGED1 and delta1 differentially regulate the outcome of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5990-8
41. Munoz-Suano A, Kallikourdis M, Sarris M, Betz AG. Regulatory T cells protect from autoimmune arthritis during pregnancy. J Autoimmun. 2012;38:J103-8
42. Woidacki K, Meyer N, Schumacher A, Goldschmidt A, Maurer M, Zenclussen AC. Transfer of regulatory T cells into abortion-prone mice promotes the expansion of uterine mast cells and normalizes early pregnancy angiogenesis. Sci Rep. 2015;5:13938
43. Yvon ES, Vigouroux S, Rousseau RF, Biagi E, Amrolia P, Dotti G. et al. Overexpression of the Notch ligand, Jagged-1, induces alloantigen-specific human regulatory T cells. Blood. 2003;102:3815-21
44. Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A. et al. Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood. 2008;112:1813-21
45. Elyaman W, Bassil R, Bradshaw EM, Orent W, Lahoud Y, Zhu B. et al. Notch receptors and Smad3 signaling cooperate in the induction of interleukin-9-producing T cells. Immunity. 2012;36:623-34
46. Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J Exp Med. 2016;213:1241-53
Author contact
![]() Corresponding authors: Dong-Gyu Jo (email: jodgedu) or Jae Hyung Park (email: jhpark1edu)
Corresponding authors: Dong-Gyu Jo (email: jodgedu) or Jae Hyung Park (email: jhpark1edu)