Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(21):5801-5813. doi:10.7150/thno.29380 This issue Cite
Research Paper
miR-200c/141 Regulates Breast Cancer Stem Cell Heterogeneity via Targeting HIPK1/β-Catenin Axis
Bingjie Liu1,2*, Ruikai Du3*, Lei Zhou1,2, Jiahui Xu1, Song Chen4,5, Ji Chen6, Xiaoli Yang1, Dong-xu Liu7, Zhi-ming Shao1,8, Lixing Zhang1, Zuoren Yu9, Ni Xie10 ![]() , Jun-lin Guan11
, Jun-lin Guan11 ![]() , Suling Liu1
, Suling Liu1 ![]()
1. Shanghai Cancer Center & Institutes of Biomedical Sciences, Cancer Institute, Key Laboratory of Breast Cancer in Shanghai, Key Laboratory of Medical Epigenetics and Metabolism, Innovation Center for Cell Signaling Network, Shanghai Medical College, Fudan University, Shanghai 200032, China;
2. School of Life Science, University of Science & Technology of China, Hefei, Anhui 230027, China;
3. State Key Laboratory of Space Medicine Fundamentals and Application, China Astronaut Research and Training Center, Beijing, China;
4. Institute of Medicinal Biotechnology, Jiangsu College of Nursing, Huaian, China
5. Department of Oncology, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
6. Department of Pathology, Suizhou Affiliated Hospital to Hubei University of Medicine, Suizhou, Hubei, 441300, China
7. The Centre for Biomedical and Chemical Sciences, School of Science, Faculty of Health and Environmental Sciences, Auckland University of Technology, Auckland 1010, New Zealand.
8. Department of Oncology, Department of Breast Surgery, Shanghai Medical College, Fudan University, Shanghai 200032, China;
9. Key Laboratory of Arrhythmias of the Ministry of Education of China, Research Center for Translational Medicine, Translational Medical Center for Stem Cell Therapy, East Hospital, Tongji University School of Medicine, 150 Jimo Road, Shanghai 200120, China;
10. Institute of translation medicine, Shenzhen Second People's Hospital, The first Affiliated Hospital of Shenzhen University Health Science Center
11. Department of Cancer Biology, University of Cincinnati College of Medicine, Cincinnati, OH 45267
*These authors contributed equally to this article.
Received 2018-8-21; Accepted 2018-10-9; Published 2018-11-10
Abstract

Increasing evidence demonstrates the existence of two inter-convertible states of breast cancer stem cells (BCSCs) with distinct behaviors in proliferation and mobility, and the BCSC heterogeneity is accurately regulated by sophisticated mechanisms including microRNAs. The microRNA-200 family including miR-200c/141 cluster was reported to affect cancer cell invasion and metastasis by regulating epithelial to mesenchymal transition (EMT). However, the effect of miR-200 family on BCSC heterogeneity is uncertain. Thus, we investigated whether the miR-200c/141 cluster had different effects on breast tumor growth and metastasis by switching the two states of BCSC.
Methods: The spontaneous mammary tumor mouse model with miR-200c/141 conditional knockout was utilized for analyzing the role of miR-200c/141 cluster in vivo. The effect of miR-200c/141 cluster on BCSCs was performed by CD24/CD29 staining and ALDEFLUOR assay. miR-200c/141 target expression and EMT-related marker expression were verified in tumor sections, primary cells and breast cancer cell lines by qRT-PCR or western blotting. Statistical analysis was determined using two-way ANOVA and Student's t-test. All values were presented as the mean ± s.e.m.
Results: The deletion of miR-200c/141 cluster regulated BCSC heterogeneity and promoted the EMT-like BCSC generation, which resulted in increased tumor metastasis and inhibited tumor growth by directly upregulating the target gene homeodomain-interacting protein kinase 1 (HIPK1) and sequential β-catenin activation.
Conclusions: Our results indicated that miR-200c/141 played biphasic roles in breast tumor progression via affecting the BCSC heterogeneity, suggesting targeting BCSC heterogeneity to simultaneously restrict breast cancer initiation and metastasis could be a promising therapeutic strategy for breast cancer.
Keywords: miR-200c/141, breast cancer, cancer stem cell, heterogeneity, HIPK1
Introduction
Cancer stem cells (CSCs) were first described in acute myeloid leukemia by John Dick [1]as a rare cell population possessing self-renewal capacity and causing relapse by giving rise to new tumors [2]. The concept of CSC was subsequently expanded to solid tumors by the successful identification of such rare cell population in breast cancer by Clarke and Wicha[3], followed by several studies in other solid tumors. More and more studies indicate that clarifyying the characteristics of BCSCs is crucial to developing effective treatments for breast cancer. Tumor heterogeneity is a well-recognized characteristic in the bulk tumor population (e.g. existence of both CSCs and non-CSCs) that is important in our understanding of the complexity of tumor biology and for developing effective tumor treatment[4]. Recent research also showed that the heterogeneity or plasticity occurred in CSCs[5]. Shipitsin, M et al observed different gene expression patterns between CSCs isolated from metastases and their primary tumors, which generated the hypothesis for two types of CSCs - stationary CSCs and mobile CSCs[6]. In recent years, increasing evidence demonstrates the existence of two inter-convertible states of breast cancer stem cells (BCSCs) with epithelial-like (ALDH+ BCSCs) and mesenchymal-like (CD24-CD44+ BCSCs) properties which have distinct behaviors in proliferation and mobility [7-9]. But the underlying molecular mechanisms governing the CSC plasticity or heterogeneity are still poorly understood.
microRNAs have potent effects on diverse cellular functions[10] and play an essential role in the regulation of BCSC self-renewal and differentiation [11-13]. The microRNA-200 (miR-200) family consists of five members that are located in two gene clusters (miR-200c/141 and miR200a/b/429) and is implicated in various biological and disease processes[14, 15] such as tumor cell chemosensitivity and apoptosis[16, 17]. Gibbons et al showed that forced expression of miR-200 inhibits tumor cells to undergo Epithelial-mesenchymal transformation (EMT), invade and metastasize [18]. The EMT hypothesis suggest that overexpression of miR-200 could result in a decrease in the metastatic potential of cancer cells, and silencing zinc finger E box-binding homeobox (Zeb) protein expression through their 3′ untranslated regions, giving rise to increased E-cadherin expression and the acquisition of epithelial characteristics[14]. But the effect of miR-200 on cancer cell proliferation is controversial. Jun Yu et al suggested that high levels of miR-200c expression correlated with low invasion ability would stimulate cell proliferation in pancreatic cancer[19]. However, Lei Liu et al found that miR-200c suppressed cell invasion, migration and proliferation in bladder cancer through targeting Bmi-1 and E2F3[20]. Such apparently contradicting observations raise the interesting possibility that miR-200c may differentially regulate the proliferation and invasion property of certain cancer cells.
Our previous study showed that the expressions of miR-200c and miR-141, very closely located next to each other in the same cluster, were significantly increased in the epithelial-like ALDH+ BCSCs [7, 21]. Studies by other groups also showed that miR-200c/ 141 play an important role in mesenchymal-epithelial-transition (MET)[22, 23] in cultured cells or xenograft models. Since there is a good correlation between epithelial-mesenchymal-transition (EMT) and CSC characteristics [24], these results suggest that miR-200c/141 cluster could affect BCSC heterogeneity and plasticity. Nevertheless, there have been very few studies to examine directly such potential functions and mechanisms using in vivo models for miRNAs, and none for miR-200c/141 cluster. In this study, we utilized a miR-200c/141 conditional knockout mouse model that bearing spontaneous mammary tumor and identified a critical role of miR-200c/141 in regulating BCSC heterogeneity. This effect is yielded by its direct targeting of homeodomain-interacting protein kinase 1 (HIPK1), HIPK1 induces the EMT of breast cancer cells by activating Wnt/β-catenin signaling pathway. Our results show that miR-200c/ 141-HIPK1-Wnt/β-catenin axis mediates breast tumor cell proliferation and invasion by modulation of EMT and inter-conversion between the epithelial and mesenchymal states of BCSCs.
Materials and Methods
Mice
C57BL/6 miR-141/200cflox/flox mice (Jackson Laboratory, USA) were backcrossed to the FVB background for six generations. Obtained FVB miR-141/ 200cflox/flox mice were crossed to MMTV-Cre transgenic mice (provided by Dr. Yi Arial Zeng) to generate mice with mammary specific deficient miR-141 and miR-200c (miR-141/200c-/-) then bred to MMTV-PyMT mice (provided by Yi Arial Zeng). miR-141/ 200cflox/flox; PyMT mice were used as wild type controls in MMTV-Cre; miR-141/200cflox/flox; PyMT experiments. All mice were maintained in a specific pathogen-free facility and animal experimentation was conducted in accordance with institutional guidelines.
Tumorigenesis and metastasis studies
Littermate controls were used in all experiments when possible. For spontaneous tumorigenesis studies, female mice carrying the specific oncogenes were examined weekly for mammary tumors. Tumors were considered for initiation when they reached 2-3mm and tumors were measured weekly by calipers for calculation of tumor volumes (length × width2/2). Mice were sacrificed when the diameter of bearing tumor reached 15mm. For each tumor, one part was embedded in paraffin for histological analysis and the rest was digested with collagenase/hyaluronidase (Stem Cell Technologies, USA) for flow cytometry. Lung nodules were counted after sectioning and staining of the lungs.
Immunohistochemistry and immunofluorescence
The human breast cancer tissues used in the study were obtained from Fudan University Shanghai Cancer Center (Shanghai, China). The slices of paraffin-embedded tissues were dewaxed and rehydrated in xylene and graded alcohol solutions. Anti-HIPK1 (1:50, Abcam, USA), anti-Ki67 (1:100, Abcam, USA), anti-E-cadherin (1:100, Proteintech, USA) and anti-Vimentin (1:100, CST, USA) were used as primary antibodies. Goat anti-mouse/rabbit IgG conjugated with HRP and DAB Kit (DAB-0031, Maxim, China) were used for immunohistochemistry staining. For immunofluorescent, both of goat anti-mouse IgG AlexaFluor-488 and goat anti-rabbit IgG AlexaFluor-679 (1:200, Life Technologies, USA) were used and nuclei was co-stained with DAPI (Life Technologies, USA).
Cell culture
Breast cancer cell lines MCF-7, BT474 and T47D were purchased from ATCC and SUM149 were obtained from Asterand. All cell lines were cultured according to the recommends from ATCC or Asterand. Primary cells used in the study were derived from digested tumor tissues and cultured with EpiCult™-B Mouse Medium Kit (#05610, STEMCELL, USA) under directions.
Interference of gene expression
Sequence-specific miRNA inhibitor (RIBOBIO, China) was used to inhibit endogenous miR-200c or miR-141 by combining with mature miRNA. Transfection experiments were carried out using Lipofectamine 3000 Reagent (Invitrogen, USA). Lentiviral system was used for establishment of stable cell lines with HIPK1 knockdown or overexpression.
MTT assay
Cells were seeded in 96-well plates one thousand per well and cultured for 3, 5, or 7 days. Each group was performed triplicate. For each well 20µl MTT (5mg/ml, Biosharp, China) was added and plates were incubated at 37°C for 3 hours. After removing the supernatant, 100 µl DMSO per well was added and kept shaking for 10 minutes. The optical density (OD) value was measured at 490 nm with microplate reader (Elx800, BioTek, USA).
Colony formation assays
Cells were plated in a 6-well plate as 1000 cells per well. When visible colonies formed, remove media and fix cells with 4% paraformaldehyde. Cells were stained with 0.5% crystal violet and colonies were counted under microscope.
Invasion assay
Transwell chambers (#3422, Corning, USA) pre-coated with matrigel (354234, Corning, USA) were placed in 24-well plate at 37°C for 3-4 hours. Then 4 X104 cells were plated on chambers without serum and medium containing 10% fetal bovine serum offered in the bottom well. After 36 hours, chambers were fixed (methyl alcohol: glacial acetic acid=3:1) and stained with 0.1% crystal violet, then invaded cells were photographed for statistical analysis.
Flow cytometry
For the ALDEFLUOR assay (StemCell, USA), dissociated cells were suspended in assay buffer containing ALDEFLUOR substrate and incubated with or without aldehyde dehydrogenase inhibitor DEAB. A CD24/CD44 or CD24/CD29 assay was performed with anti-CD24 (1:20, 561647, BD), anti-CD44 (1:100, 560532, BD), anti-CD24 (1:40, 101814, BioLegend, USA) and anti-CD29 (1:80, 102226, BioLegend). For analysis of tumor cells from spontaneous breast cancer mice, anti-mouse-lineage antibodies were used for gating: H2Kd (1:100, 116607, Biolegend), CD45 (1:50, 555483, BD), CD31 (1:50, 555446, BD), CD140b (1:50, 558821, BD), and CD235a (1:50, 555570, BD). The CytoFLEX instrument (Beckman Coulter, USA) was used for data acquisition and analysis were performed in CytExpert software.
RNA extraction and quantitative real-time PCR
Total RNA were extracted by TRIzol Reagent (Takara, China) and extracted RNA was measured on Nanodrop (Thermo Fisher Scientific, USA). cDNA was prepared from 1 µg RNA using ReverTra Ace qPCR RT Kit (TOYOBO, China). QRT-PCR was performed using SYBR Green PCR Kit (Vazyme Biotech, China) with 7300 Real-Time PCR system (Applied Biosystems, USA). 5sRNA or GAPDH were used as a reference gene for miRNA or mRNA, respectively. All primers used are shown in Table S1.
Western blotting
Cells were lysed in RIPA buffer (Beyotime, China) and protein concentration was measured by BCA Protein Assay Kit (Pierce, USA). Protein sample mixed with loading buffer were separated by SDS-PAGE and subsequently transferred onto PVDF membranes (Millipore, USA). Membrane was blocked in 5% de-fat milk and incubated with primary antibody and HRP-conjugated secondary antibody sequentially. Following antibodies were used in this study: anti-HIPK1 (ab152109, Abcam), anti-E-cadherin (20874-1-AP, Proteintech), anti-Vimentin (5741s, CST), anti-snail (3879, CST), anti-Actin (HC201, TransGen, China), goat anti-mouse IgG-HRP (sc-2005, Santa Cruz, USA) and goat anti-rabbit IgG-HRP (sc-2004, Santa Cruz). ImageQuant LAS 4000 mini imaging system (GE, Fairfield, USA) and Western HRP Substrate (WBLUF0500, Millipore) were used in chemiluminescent detection.
Luciferase reporter assay
293T cells were transfected with miR-200c or miR-141 overexpression plasmid (pTRIPZ vector) were seeded in 96-well plate and were transfected with 200 ng luciferase reporter plasmids containing sequence of HIPK1 3'UTR wild type or the corresponding deletion. 24 hours after transfection, luciferase activity was measured according to the instructions of luciferase assay kit (E2920, Promega, USA).
Patients and clinical samples
The human breast cancer tissue used in this study, obtained from the Fudan Universitay Shanghai Cancer Center (Shanghai, China), was comprised of 25 pairs of breast cancer tissues and their corresponding adjacent normal tissues (Table S2). The study was approved by ethical committee of Fudan University.
Statistical analysis
All values are presented as the mean ± s.e.m. and at least three repeated individual experiments were performed for each group, except where otherwise indicated. Statistical analysis was determined using two-way ANOVA and Student's t-test with GraphPad Prism 6 (GraphPad Software, USA). A p-value < 0.05 was considered statistically significant.
Results
Deletion of miR-200c/141 cluster inhibits mammary tumor formation but increases lung metastasis in vivo
To investigate the role of miR-200c/141 in regulating BCSC heterogeneity in vivo, we conditionally deleted the miR-200c/141 cluster in mammary gland by intercrossing miR-200c/141fl/fl mice with MMTV-PyMT and MMTV-Cre transgenic mice using a similar strategy as described previously [25] and in Figure 1A. Genotyping by PCR analysis of genomic DNA identified cohorts of offsprings containing miR-200c/141fl/fl, MMTV-PyMT and MMTV-Cre alleles (designated as cKO mice) and miR-200c/141fl/fl and MMTV-PyMT alleles (designated as Control mice, WT) (Figure S1A). QRT-PCR analysis of mammary glands showed significantly decreased expression of miR-200c and miR-141 in cKO mice (Figure S1B, S1C).
Deletion of miR-200c/141 cluster inhibits mammary tumor formation. (A) Schematic view of cross-breeding among MMTV-PyMT, MMTV-Cre transgenic mice and mice carrying loxP sites flanking the miR-200c/141fl/fl, and the generation of spontaneous breast tumor mice with mammary-specific deletion of miR-200c/141 cluster (miR-200c/141-/-; PyMT). (B) Kinetics of mammary tumor onset in MMTV-PyMT female mice of indicated genotypes. (log-rank test; WT: n=40,cKO:n=41) *p < 0.05. (C) The percentage of tumor-free mammary glands at indicated ages and indicated genotypes of mice. (D) Total tumor burden in WT and cKO cohorts evaluated at 15 weeks in the same mice as in (C). *p < 0.05. (E) The representative images for Ki67 IHC staining in tumor sections and quantification of Ki67 positive cells per tumor section in the same cohort of mice from (C). ***p < 0.001
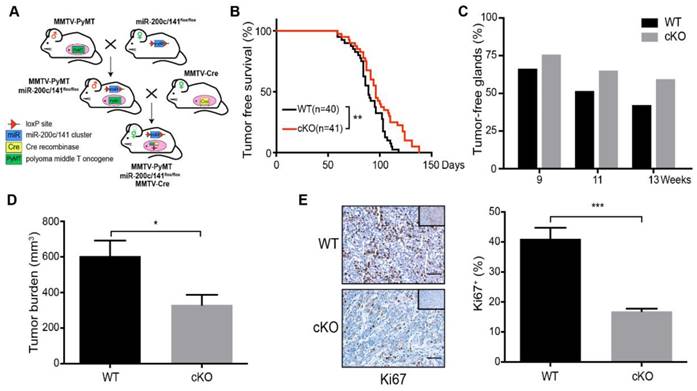
Relative to control (WT) mice, cKO mice presented a delay in mammary tumor development (Figure 1B). The inhibition effect of miR-200c/141 cluster deletion on tumor formation was also supported by a greater number of tumor-free mammary glands (Figure 1C) and reduced tumor burden (Figure 1D) in cKO mice. We next stained the tumor sections for a cell proliferation marker Ki67 and found a significantly decreased number of Ki67+ cells in breast tumors of cKO mice (Figure 1E). Surprisingly, in contrast to the reduced tumor development, histological analysis showed significantly increased lung metastatic nodules in cKO mice (Figures 2A and 2B). However, decreased proliferating (i.e. Ki67+) cells were also detected in metastatic nodules in cKO mice, (Figure 2C). Together, these results suggest that miR-200c/141 may confer a growth advantage during tumorigenesis at both the primary sites and metastasis sites but inhibit the tumor metastasis.
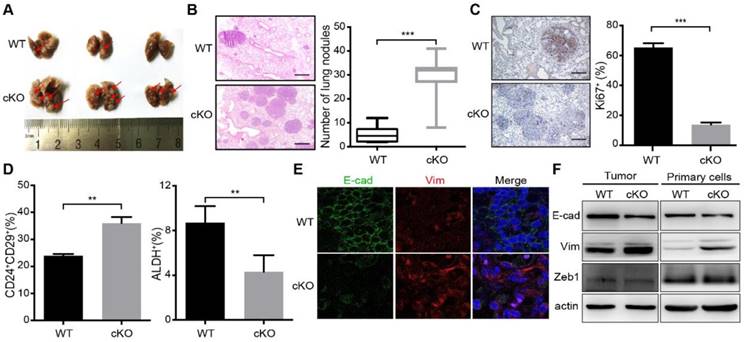
Deletion of miR-200c/141 cluster increases mammary tumor lung metastasis and induces EMT. (A) Images of lungs from the same cohort of mice as shown in Figure 1B. Red arrows represent metastatic nodules in lungs. (B) HE staining of lung sections from (A) and the numbers of metastatic lesions per lung section were counted. ***p < 0.001. (C) The representative images for Ki67 IHC staining in lung sections from WT or cKO mice and quantification of Ki67 positive cells. ***p < 0.001. (D) FACS was performed to detect the effect of miR-200c/141 cluster on breast cancer stem cells (BCSCs) with CD24/CD29 and ALDEFLUOR assay. (E) E-cadherin (E-cad) and Vimentin (Vim) expression was verified by Immuno- fluorescence staining in tumor sections with indicated genotypes. (F) E-cadherin (E-cad), Vimentin (Vim) and Zeb1 protein immunoblotting in freshly lytic tumors and primary cells from the mice with indicated genotypes.
miR-200c/141 mediates mammary tumor progression in a biphasic manner via regulating BCSC plasticity
Previous research demonstrated that BCSCs exist in alternative epithelial-like and mesenchymal-like states. The mesenchymal-like state characterized as CD24-CD44+ is associated with expression of mesenchymal markers, relative quiescence, and high invasive capacity, whereas the epithelial-like state characterized as ALDH+ is associated with expression of epithelial markers, establishment of cell polarity, and extensive proliferation[7, 8]. More recent study showed two distinct subpopulations of BCSCs with CD24+CD29+ markers for the mesenchymal population and ALDH+ for the epithelial BCSCs [8, 26]. To examine the effect of miR-200c/141 cluster deletion on BCSCs directly, tumors from cKO and WT mice were minced and digested into single cell suspensions for BCSC analysis by flow cytometry using these two sets of BCSC markers. Intriguingly, the CD24+CD29+ mesenchymal subset was significantly increased, but the ALDH+ epithelial subset was decreased in cKO mice (Figures 2D and S2) suggesting that miR-200c/141 could regulate BCSC heterogeneity in PyMT mouse model of breast cancer. Considering the role of miR-200c/141 cluster in mesenchymal to epithelial transition (MET), the epithelial marker (E-cadherin) and mesenchymal marker (Vimentin) were detected by immunofluorescence on tumor sections and immunoblotting analysis on mouse primary tumors and tumor cells isolated and cultured from primary tumors (Primary cells). We found that miR-200c/141 deletion decreased E-cadherin and increased Vimentin expression (Figure 2E and 2F), suggesting that increased EMT in bulk tumors may be driven by the elevated levels of mesenchymal BCSCs, which could contribute to increased lung metastases in cKO mice.
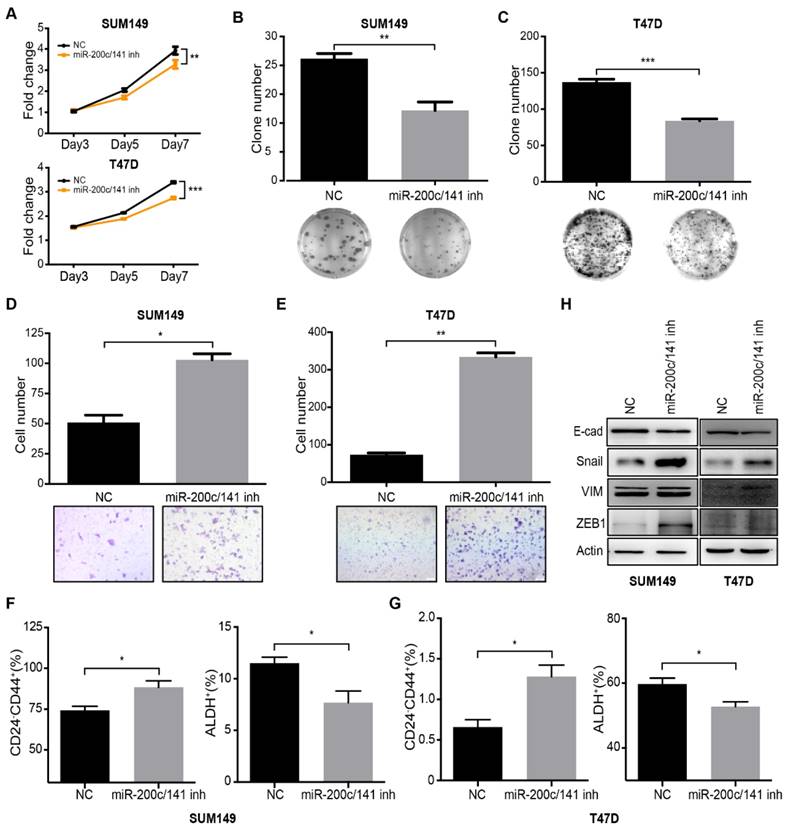
To evaluate a potential role of miR-200c/141 in human BCSCs and further investigate the involved mechanisms, we treated human breast cancer cell lines SUM149 (Basal subtype) and T47D (Luminal subtype) with specific miR-200c/141 inhibitors or scramble controls and examined the effects on the cell functions. MTT and colony formation assays showed that miR-200c/141 antagonists inhibited the cell proliferation of both cell lines (Figure 3A-3C). Interestingly, we found that miR-200c/141 inhibitor treatment increased invasive capacity of both cell lines (Figure 3D and 3E). These results markedly resembled the phenotypes of reduced tumorigenesis and increased metastasis upon miR-200c/141 deletion that we observed in the PyMT mouse model in vivo (see Figures 1 and 2). Then we examined the BCSC status by flow cytometry using CD24-CD44+ markers to detect mesenchymal-like human BCSC as described previously [7]. Similar to our findings in PyMT mouse models, inhibition of miR-200c/141 increased mesenchymal-like CD24-CD44+ BCSC population but decreased epithelial-like ALDH+ BCSC population (Figures 3F, 3G and S3). Furthermore, attenuated E-cadherin and elevated Vimentin and Snail were found after miR-200c/141 inhibition, suggesting an EMT occurred in these cells (Figure 3H). All those results were also confirmed in another human cancer cell line BT474 (Her2 subtype) and primary cultured cells isolated from PyMT mouse tumors (Figure S4). All these results suggest that miR-200c/141 could regulate BCSC heterogeneity via inhibiting EMT in human breast cancers through similar mechanisms as in mouse models.
Inhibition of miR-200c and miR-141 results in reduced cell proliferation and increased cell invasion in vitro by regulating BCSC plasticity. SUM149 and T47D cells were treated with 200nM miR-200c and miR-141 inhibitors (miR200c- 141inh) or negative control inhibitor (NC) for 48h. (A) Treated SUM149 or T47D cells were accessed for proliferation ability which was performed with MTT assay as described in methods. **p<0.01. (B-C) The plate colony formation assay was carried out with 500 treated SUM149 (B) or T47D(C) colonies were counted in the whole field for statistics. **p<0.01. (D-E) The invasive ability of treated SUM149 (D) or T47D (E) cells was investigated with the transwell assay according to the manufacturer's protocol. Quantitative analysis was accessed for the total invasive cells from three independent experiments. *p<0.05, **p<0.01. (F-G) Flow cytometry analysis of BCSCs was carried out for the CD24/CD44 and ALDEFLUOR assay in treated SUM149 (F) and T47D (G) cells. (H) After being treated for 48 hours, cells were harvested to detect the protein expression levels of E-cadherin (E-cad), Snail, Vimentin (VIM) and ZEB1 by Western Blotting.
HIPK1 is a direct target of miR-200c/141 and correlated with breast cancer malignancy
To study the mechanisms of miR-200c and miR-141 in controlling breast cancer development and progression, we explored downstream gene targets common for these two miRNAs. Analyses of two databases miRBase and targetScan for candidate targets identified 10 overlapping genes for miR-200c and miR-141 (Figure 4A). We then examined mRNA expression of these 10 genes in mammary tumors and isolated primary cells from cKO and WT mice, and found a greatest increase for HIPK1 and YWHAG in mammary tumor tissues from cKO mice (Figure 4B). The proteins levels for these two proteins were then analyzed by Western blotting. We found a more significant increase for HIPK1 than YWHAG in cKO mice compared to those from WT mice (Figure 4C). Similar increases in the expression of HIPK1 were found upon inhibition of miR-200c/141 in SUM149 and T47D cells (Figure 4D and 4E). To further assess the possibility of HIPK1 as a direct repression target by miR-200c/141, we cloned the human HIPK1 3'UTR into a luciferase reporter plasmid. The HIPK1 3'UTR contains two predicted binding sites for miR-200c/141: one for miR-200c and another for miR-141 (Figure 4F). Indeed, mutation of each of the binding site released repression by miR-200c and miR-141, respectively (Figure 4G), suggesting that HIPK1 is a direct downstream target of miR-200c/141 through these sites [2].
HIPK1 is a direct and functional target of miR-200c and miR-141, and is correlated with breast cancer malignancy. (A) Schematic diagram of genes targeted by both of miR-200c and miR-141 were analyzed based on miRBase and targetScan databases. (B) Candidate target genes were confirmed with qRT-PCR assay in both tumor and primary cells from miR-200c/141 cKO mice. (C) The protein expression levels of HIPK1 and YWHAG were detected by Western Blotting. (D) mRNA expression levels of HIPK1 and YWHAG were confirmed with qRT-PCR assay in SUM149 treated with miR200c and miR141 inhibitors or negative control. **p<0.01. (E) Protein expression levels of HIPK1 and YWHAG were confirmed by Western Blotting in SUM149 and T47D cells treated with indicated inhibitors. (F) Predicted miR-200c and miR-141 binding region sequence in 3'UTR of HIPK1. (G) The potential miR-200c and miR-141 binding regions in 3'UTR of HIPK1 were confirmed with luciferase reporter assay. The relative luciferase activity is defined as the value of activity of luciferase gene folded over internal control. **p<0.01. (H) The Kaplan-Meier survival curve of breast cancer patients (log-rank test, ***p < 0.001), data from TCGA. (I) The HIPK1 protein level in clinical breast tumor tissues and paratumor tissues was shown by immunohistochemistry (IHC) (Brown: HIPK1). Bar, 200um. (J) Quantification of HIPK1-positive cells ratio in IHC staining sections of breast cancer patient tissues from (I). ***p<0.001
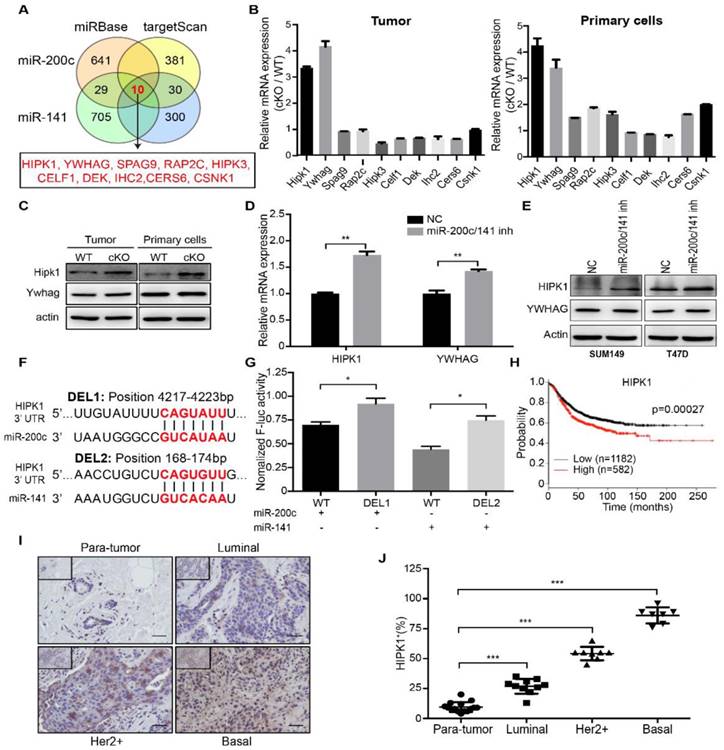
We next examined the functional role of HIPK1 in breast cancer. A Kaplan-Meier survival curves between HIPK1 mRNA expression and breast cancer patients was carried out based on TCGA database, and it showed that patients with low HIPK1 expression had a longer overall survival (Figure 4H). At the same time, paired breast tumors with different subtypes (Luminal, Her2+, Basal) and their adjacent normal tissues (paratumor) from same patients were analyzed and quantified for HIPK1 expression by IHC (Figure 4I and 4J). HIPK1 was up-regulated in tumors significantly and when the samples were classified into paratumor, Luminal, Her2 and Basal, HIPK1 expression was increased with the malignant level. These results indicated the oncogenic role of HIPK1 in breast cancer.
HIPK1 knockdown rescues the effects of miR-200c/141 inhibition on breast cancer cells
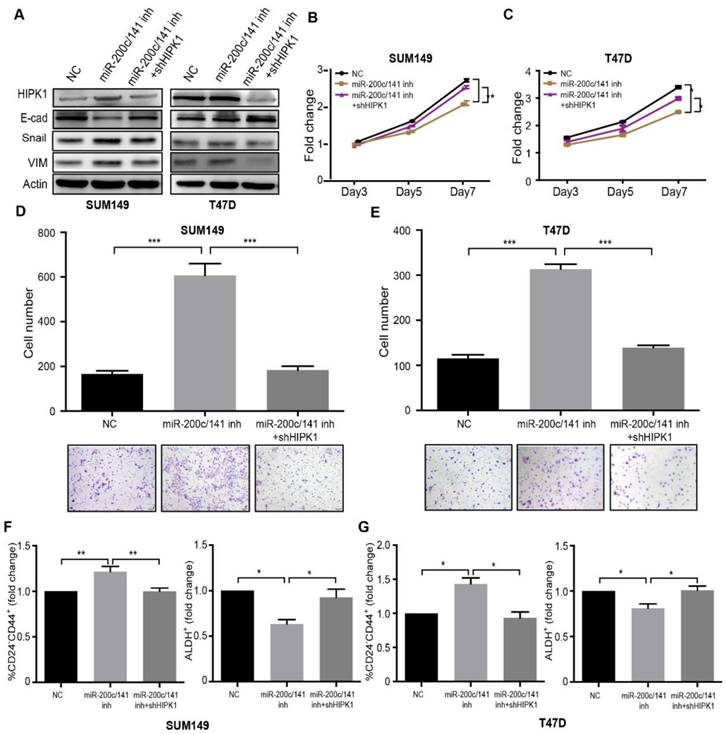
We had identified HIPK1 as a clinically relevant direct target of miR-200c/141. In order to investigate whether HIPK1 served as a major downstream effector of miR-200c/141, we established HIPK1 knockdown SUM149 cell line via a specific shRNA lentiviral infection. Epithelial/mesenchymal markers, E-cadherin, Vimentin and Snail were detected by western blotting (Figure 5A). The results showed that the EMT induced by the miR-200c/141 inhibition could be attenuated by the HIPK1 knockdown. The in vitro assays revealed an increased cell proliferation and reduced invasion in rescued group compared to miR-200c/141 inhibitor treatment alone (Figure 5B-5E). Furthermore, the HIPK1 knockdown abrogated the effects of the miR-200c/141 antagonist on both CD24-CD44+ and ALDH+ BCSC population (Figures 5F, 5G and S5). These results confirmed that HIPK1 served as a functional direct target of miR-200c/141 in regulating cancer cell proliferation, cell invasion, and BCSCs heterogeneity regulation.
HIPK1 knockdown rescues the effects of miR-200c/141 inhibition on breast cancer cells. SUM149 and T47D cells were transfected with HIPK1-shRNA lentivirus and then treated with miR-200c/141 inhibitors. (A) Protein expression level of HIPK1, E-Cadherin (E-cad), Snail and Vimentin (VIM) were detected by Western Blotting. Samples were harvested after cells had been treated for 48 hours. (B-C) MTT assay was performed with treated SUM149 (B) and T47D (C) cells for cell proliferation ability. *p<0.05, **p<0.01. (D-E) The invasive ability was investigated with treated SUM149 (D) or T47D (E) cells by transwell assay. Quantitative analysis of the total invasive cells from three independent experiments. ***p<0.001 (F-G) The heterogeneity of BCSCs was analyzed by Flow cytometry with two sets of markers (CD24-CD44+ and ALDH+) in treated SUM149 (F) and T47D (G) cells.
In addition, to examine whether HIPK1 can mimic the effect of miR-200c/141 cluster deletion on breast cancer cell functions and BCSCs, we overexpressed HIPK in SUM149 cells (Figure S6A) and examined the effects on the cell functions. MTT and colony formation assays showed that HIPK1 overexpression inhibited the cell proliferation of both cell lines (Figures S6B, S6C). Similar to the effects of miR-200c/141 cluster deletion, we found that HIPK1 overexpression increased invasive capacity of SUM149 cells (Figure S6D). Furthermore, attenuated E-cadherin and elevated Vimentin were also found after HIPK1 overexpression, suggesting an EMT occurred in these cells (Figure S6E). HIPK1 overexpression increased mesenchymal-like CD24-CD44+ BCSC population but decreased epithelial-like ALDH+ BCSC population (Figure S6F). These results markedly resembled the effects of miR-200c/141 inhibition on breast cancer cell functions that we observed above (Figures 3, S3 and S4).
Beta-catenin (β-catenin) was activated to regulate the BCSC heterogeneity and cancer progression by both the deletion of miR-200c/141 cluster and the HIPK1 overexpression
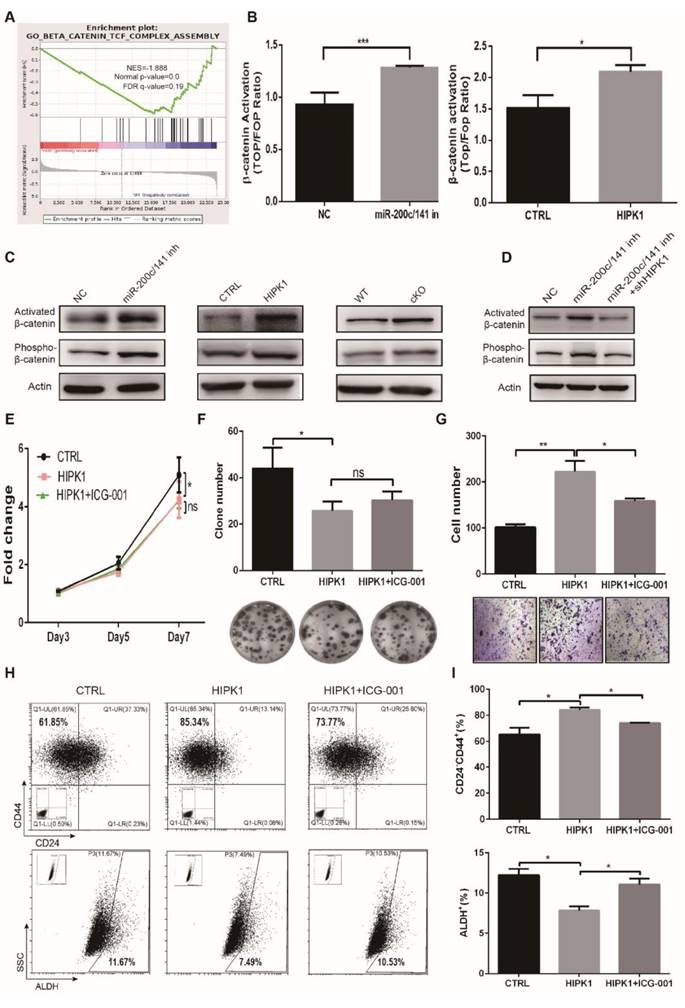
In order to further elucidate the downstream signaling pathway regulation by the deletion of the miR-200c/141 cluster, we performed the RNA-Seq assay in primary tumor cells from cKO and WT PyMT mice. Gene Set Enrichment Analysis (GSEA) indicated that the downregulated genes in the cKO PyMT primary tumor cells were negatively correlated to the Wnt/β-catenin pathway which exerts a critical role in cell self-renewal and differentiation (Figure 6A) and HIPKs were also reported to directly interact with β-catenin [27, 28]. Then, we verified the β-catenin transcription activity was increased in both miR-200c/141- inhibited and HIPK1-overexpressed cells by TOP/FOP luciferase assay (Figure 6B). Furthermore, we found that the active form of β-catenin was upregulated consistently in both miR-200c/141- inhibited and HIPK1-overexpressed cells as well as in the tumors from miR-200c/141 knockout mice (Figure 6C), and the HIPK1 knockdown diminished the activation of β-catenin by miR-200c/141 inhibition (Figure 6D). In addition, we confirmed the corresponding response of the β-catenin downstream effector c-Myc in both miR-200c/141-inhibited and HIPK1-overexpressed cells (Figure S7A, S7B) and proved that HIPK1 phosphorylated β-catenin protein at Ser552 in vitro (Figure S8). According to the findings above, we treated HIPK1-overexpressing SUM149 cells with Wnt/β-catenin pathway inhibitor ICG-001 resulting in slightly rescued cell proliferation but significantly diminished invasion from HIPK1 overexpression (Figures 6E-6G). There might be some other suspicious signals mediating the effects of HIPK1 on cell proliferation, such as p21 which was significantly upregulated in miR-200c/141 knocking-down or HIPK1-overexpressing cells (data not shown). In addition, increase of CD24-CD44+ and decrease of ALDH+ BCSC population in HIPK1-overexpressing group were partially inhibited after β-catenin inhibition (Figures 6H, 6I). These results demonstrated that HIPK1/β-catenin axis was crucial in regulating BCSC heterogeneity.
miR-200c/141 cluster regulates breast cancer metastasis and BCSCS states through β-catenin activity. (A) The downregulated genes from primary tumors of cKO PyMT mice compared to WT PyMT mice were applied to Gene Set Enrichment Analysis (GSEA). (B) The transcriptional activity of β-catenin was measured in both miR-200c/141-inhibited and HIPK1-overexpressed cells by TOP/FOP luciferase assay. (C) The activated β-catenin (non-phosphorylation at ser33/37/thr41 sites) and Phosphorylation of β-catenin in ser552 site in both miR-200c/141-inhibited and HIPK1- overexpressed cells as well as in the tumors from miR-200c/141 knockout mice were measured by Western Blotting. (D) Rescue experiment was performed in miR-200c/141 and HIPK1 double knockdown cell lines by Western Blotting. (E-G) The cell proliferation ability was measured for HIPK1-overexpressing SUM149 cells in the absence or presence of ICG-001 (10uM, 24h) by the MTT assay (E) and plate colony formation assay (F) and the cell invasive ability was tested by transwell assay (G). Quantitative analysis of the cells was done from three independent experiments. ICG- 001: Wnt/β-catenin pathway inhibitor. (H-I) Flow cytometry analysis of BCSCs was carried out for the CD24/CD44 analysis and ALDEFLUOR assay in SUM149 cells.
Discussion
Developing understandings of CSCs make it a consensus that targeting CSCs could be a better therapeutic approach to cure cancer. The fundamental problem is that ascertain the mechanism of regulating CSC heterogeneity. We previously identified two distinctive BCSC populations with mesenchymal-like or epithelial-like characters which show different tumor proliferation or invasion properties [7]. Chen et al [29] found CD133+/ALDHhigh cells appear to be a tumor initiating subpopulation, whereas CD133-/ ALDHlow cells harbor more chemoresistance and invasion property in hepatoma. Therefore, unveiling the potential mechanisms regulating BCSC transformation cross distinctive states is necessary to understand the plasticity of CSCs. In this study, we describe that two states of BCSC populations could mutually transform in EMT process mediated by miR-200c/141-HIPK1/β-catenin axis.
For exploring the mechanisms of BCSC transformation, we chose miR-200c/141 cluster which we found is highly expressed in the epithelial state BCSCs (ALDH+) and the MMTV-PyMT transgenic mice as entry points. Delayed tumor development and increased lung metastasis were observed in miR-200c/141 cKO mice. However, Fischer KR et al reported miR-200 overexpression did not impair the ability of MMTV-PyMT tumor cells to form distant lung metastases [30]. Considering MMTV-PyMT mouse model is a widely used autochthonous mammary tumor model which develops luminal adenocarcinoma [31] and the majority cells from primary tumors in MMTV-PyMT mouse are epithelial type [30], conditional knockout of miR-200/141 cluster may present more remarkable mesenchymal phenotype responsible for the lung metastasis compared with miR-200/141-expressing mouse model. And human breast cancer cell lines including T47D, BT474 and SUM149 which covered luminal, Her2 and basal subtypes of human breast cancer were used for experimental assays in vitro for addition. The similar phenotype gained a consistent conclusion that inhibition of miR-200c/141 induced cells more invasive but is anti-proliferative, along with enriched mesenchymal BCSCs and reduced ALDH+ population, which is very stimulating and intrigues us to further explore the molecular mechanisms.
Previous studies have demonstrated that miR-200c/141 cluster is involved in cancer progression by promoting MET. As we know, EMT is a process in which epithelial cells lose their cell polarity and cell-cell adhesion, and gain migratory and invasive mesenchymal properties, and MET is the reverse process. Both of the two processes are critical for development of many tissues and organs[32]. EMT has been shown to occur in the initiation of metastasis and drug resistance across different cancer types [33-35] and EMT process was reported to induce CD24-CD44+ BCSCs [36], but less is known about the effects on ALDH+ BSCSs. Our current results showed EMT could indeed increase invasion but decrease the tumor initiating and proliferation via differentially modulating BCSC population.
The classical targets of miR-200 are the E-cadherin transcriptional repressors ZEB1 and ZEB2 [22] in which there is no miR-141 binding site at 3'UTR region. However, miR-200c and miR-141 are located in the same cluster regulated with one promoter[37] and our study showed that inhibiting any of those two miRNAs had similar effects on tested breast cancer cell function (data not shown), so we focused on the common predicted target genes for both. With the analysis of online miRNA-binding predicting tools and subsequent experimental confirmation, we provided evidence for the functional role of miR-200c/141 in regulating BCSC heterogeneity by directly repressing HIPK1, a homeodomain-interacting protein kinase.
HIPK1 is observed highly expressed in several cancers, but its function appears to be complicated as a tumor repressor or an oncogene in a context-dependent manner [38-40]. Although high expression of HIPK1 is identified in invasive breast cancer cells, the mechanism is not certain [41]. Here, we identified HIPK1 as a direct functional target of miR-200c/141 in regulating BCSC heterogeneity and tumor progression. In addition, we authenticated β-catenin was activated by both miR-200c/141 inhibition and ectopic expression of HIPK1 that played a critical role in the regulation of breast cancer.
Taken together, our study supports the finding of distinct BCSC states harboring different properties and reveals a miR-200c/141- HIPK1-dependent mechanism in regulating BCSC heterogeneity and breast cancer progression. This effect is subsequently mediated by deregulation of Wnt/β-catenin pathway. Combination of targeting BCSC heterogeneity especially the mesenchymal-like population and the traditional chemotherapies to simultaneously restrict breast cancer initiation and metastasis may be a promising therapeutic strategy for breast cancer.
Abbreviations
BCSCs: Breast cancer stem cells; EMT: Epithelial to mesenchymal transition; HIPK1: Homeodomain-interacting protein kinase 1; CSCs: Cancer stem cells; ALDH: Aldehyde dehydrogenase; Zeb: zinc finger E box-binding homeobox; qRT-PCR: Quantitative real-time PCR; GSEA: Gene Set Enrichment Analysis.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Thanks to Dr. Stephen Ethier for generously providing the breast cancer cell line SUM149.
Funding
This work was supported by the National Key Research and Development Program of China [Stem Cell and Translational Research 2016YFA0101202], NSFC grant [81530075], the MOST grant [2015CB553800], NSFC grants [81472741, 81772799, 81572877, 81773155, 31800708], Fudan-SIMM Joint Research Fund [FU-SIMM 20172007], Fudan University Research Foundation grant [IDH 1340042], Research Foundation of the Fudan University Shanghai Cancer Center grant [YJRC1603], and Science & Technology Program of Jiangsu Province [BK20151287], the Natural Science Foundation of Guangdong (2016A030313029, 2017A030313668), Sanming Project of Medicine in Shenzhen (SZSM201612031 ), Shenzhen Municipal Government of China (GJHZ20160301163138685, JCYJ20170817171808368, JCYJ20170818085657917).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645
2. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727-38
3. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8
4. Easwaran H, Tsai H-C, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716-27
5. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16:41
6. Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J. et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259-73
7. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y. et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2014;2:78-91
8. Yeo SK, Wen J, Chen S, Guan JL. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfbeta/Smad Signaling. Cancer Res. 2016;76:3397-410
9. Liu M, Liu Y, Deng L, Wang D, He X, Zhou L. et al. Transcriptional profiles of different states of cancer stem cells in triple-negative breast cancer. Mol Cancer. 2018;17:65
10. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
11. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C. et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109-23
12. Yu F, Deng H, Yao H, Liu Q, Su F, Song E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene. 2010;29:4194-204
13. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D. et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592-603
14. Dykxhoorn DM. MicroRNAs and metastasis: little RNAs go a long way. Cancer Res. 2010;70:6401-6
15. Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C. et al. Tumour angiogenesis regulation by the miR-200 family. Nat Commun. 2013;4:2427
16. Feng B, Wang R, Chen LB. Review of miR-200b and cancer chemosensitivity. Biomed Pharmacother. 2012;66:397-402
17. Li Y, VandenBoom TG 2nd, Kong D, Wang Z, Ali S, Philip PA. et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704-12
18. Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ. et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140-51
19. Yu J, Ohuchida K, Mizumoto K, Sato N, Kayashima T, Fujita H. et al. MicroRNA, hsa-miR-200c, is an independent prognostic factor in pancreatic cancer and its upregulation inhibits pancreatic cancer invasion but increases cell proliferation. Mol Cancer. 2010;9:169
20. Liu L, Qiu M, Tan G, Liang Z, Qin Y, Chen L. et al. miR-200c inhibits invasion, migration and proliferation of bladder cancer cells through down-regulation of BMI-1 and E2F3. J Transl Med. 2014;12:305
21. Deng L, Shang L, Bai S, Chen J, He X, Martin-Trevino R. et al. MicroRNA100 inhibits self-renewal of breast cancer stem-like cells and breast tumor development. Cancer Res. 2014;74:6648-60
22. Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910-4
23. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G. et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593-601
24. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
25. Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S. et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009;69:466-74
26. Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ. et al. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13:141-52
27. Marinaro C, Pannese M, Weinandy F, Sessa A, Bergamaschi A, Taketo MM. et al. Wnt signaling has opposing roles in the developing and the adult brain that are modulated by Hipk1. Cereb Cortex. 2012;22:2415-27
28. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192-205
29. Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol. 2011;55:838-45
30. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
31. Wan L, Lu X, Yuan S, Wei Y, Guo F, Shen M. et al. MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell. 2014;26:92-105
32. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
33. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559-64
34. Ye X, Weinberg RA. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015;25:675-86
35. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741-51
36. Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA. et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:235-52
37. Vrba L, Jensen TJ, Garbe JC, Heimark RL, Cress AE, Dickinson S. et al. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS One. 2010;5:e8697
38. Lee D, Park SJ, Sung KS, Park J, Lee SB, Park SY. et al. Mdm2 associates with Ras effector NORE1 to induce the degradation of oncoprotein HIPK1. EMBO Rep. 2012;13:163-9
39. Kondo S, Lu Y, Debbas M, Lin AW, Sarosi I, Itie A. et al. Characterization of cells and gene-targeted mice deficient for the p53-binding kinase homeodomain-interacting protein kinase 1 (HIPK1). Proc Natl Acad Sci U S A. 2003;100:5431-6
40. Rey C, Soubeyran I, Mahouche I, Pedeboscq S, Bessede A, Ichas F. et al. HIPK1 drives p53 activation to limit colorectal cancer cell growth. Cell Cycle. 2013;12:1879-91
41. Park BW, Park S, Koo JS, Kim SI, Park JM, Cho JH. et al. Homeodomain-interacting protein kinase 1 (HIPK1) expression in breast cancer tissues. Jpn J Clin Oncol. 2012;42:1138-45
Author contact
![]() Corresponding authors: Suling Liu, Key Laboratory of Breast Cancer in Shanghai, Cancer Institute, Fudan University Shanghai Cancer Center, Shanghai 200032, China; Tel/Fax: 086-21-34771023. Email: sulingedu.cn OR Jun-Lin Guan, Department of Cancer Biology, University of Cincinnati College of Medicine, Cincinnati, OH 45267; Tel: 1-513-558-5323; Fax: 1-513-558-5061; Email: guanjledu OR Ni Xie, Institute of translation medicine, Shenzhen Second People's Hospital, The first Affiliated Hospital of Shenzhen University Health Science Center, Shenzhen 518035, China; Tel: +86-13501580802; Fax: +86-755-83003435; Email: xn100edu.cn
Corresponding authors: Suling Liu, Key Laboratory of Breast Cancer in Shanghai, Cancer Institute, Fudan University Shanghai Cancer Center, Shanghai 200032, China; Tel/Fax: 086-21-34771023. Email: sulingedu.cn OR Jun-Lin Guan, Department of Cancer Biology, University of Cincinnati College of Medicine, Cincinnati, OH 45267; Tel: 1-513-558-5323; Fax: 1-513-558-5061; Email: guanjledu OR Ni Xie, Institute of translation medicine, Shenzhen Second People's Hospital, The first Affiliated Hospital of Shenzhen University Health Science Center, Shenzhen 518035, China; Tel: +86-13501580802; Fax: +86-755-83003435; Email: xn100edu.cn