Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(21):5995-6007. doi:10.7150/thno.27706 This issue Cite
Research Paper
CDC20 regulates cardiac hypertrophy via targeting LC3-dependent autophagy
Yun-Peng Xie1, Song Lai2, Qiu-Yue Lin1, Xin Xie1, Jia-Wei Liao1, Hong-Xia Wang3, Cui Tian3, Hui-Hua Li1 ![]()
1. Department of Cardiology, Institute of Cardiovascular Diseases, First Affiliated Hospital of Dalian Medical University, Dalian 116011, China;
2. Department of Cardiology, Peking University Third Hospital and Key Laboratory of Cardiovascular Molecular Biology and Regulatory Peptides, Ministry of Health, Key Laboratory of Molecular Cardiovascular Sciences, Ministry of Education and Beijing Key Laboratory of Cardiovasicular Receptors Research, Beijing 100191, China;
3. Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China.
Received 2018-6-6; Accepted 2018-10-8; Published 2018-11-15
Abstract

Rationale: Sustained cardiac hypertrophy often leads to heart failure (HF). Understanding the regulation of cardiomyocyte growth is crucial for the treatment of adverse ventricular remodeling and HF. Cell division cycle 20 (CDC20) is an anaphase-promoting complex activator that is essential for cell division and tumorigenesis, but the role of CDC20 in cardiac hypertrophy is unknown. We aimed to test whether CDC20 participates in the regulation of pathological cardiac hypertrophy and investigate the underlying mechanism in vitro and in vivo.
Methods: Male C57BL/6 mice were administered a recombinant adeno-associated virus serotype 9 (rAAV9) vector expressing CDC20 or a siRNA targeting CDC20 and their respective controls by tail intravenous injection.
Results: Microarray analysis showed that CDC20 was significantly upregulated in the heart after angiotensin II infusion. Knockdown of CDC20 in cardiomyocytes and in the heart reduced cardiac hypertrophy upon agonist stimulation or transverse aortic constriction (TAC). Conversely, enforced expression of CDC20 in cardiomyocytes and in the heart aggravated the hypertrophic response. Furthermore, we found that CDC20 directly targeted LC3, a key regulator of autophagy, and promoted LC3 ubiquitination and degradation by the proteasome, which inhibited autophagy leading to hypertrophy. Moreover, knockdown of LC3 or inhibition of autophagy attenuated Ang II-induced cardiomyocyte hypertrophy after deletion of CDC20 in vitro.
Conclusions: Our study reveals a novel cardiac hypertrophy regulatory mechanism that involves CDC20, LC3 and autophagy, and suggests that CDC20 could be a new therapeutic target for patients with hypertrophic heart diseases.
Keywords: CDC20, transverse aortic constriction, cardiac hypertrophy, LC3, ROS
Introduction
Cardiac hypertrophy is an adaptive response to physiological and pathological overload at an early stage. However, sustained pathological overload induces maladaptation and cardiac remodeling, thereby leading to heart failure (HF) and sudden death [1]. In response to pressure or volume stress, intracellular hypertrophic signaling pathways become activated to reuse embryonic transcription factors and increase the synthesis of various proteins [2]. One key process in cardiac hypertrophy is the adaptation of protein turnover, which is the balance between protein synthesis and degradation [3]. A main mechanism controlling protein degradation is the ubiquitin-proteasome system (UPS), which is known to be important for many cellular processes, including the regulation of cell apoptosis, signal transduction and transcriptional activity [4].
Cell division cycle 20 homologue (CDC20, also called Fizzy) is an activator of the anaphase-promoting complex (APC) and belongs to the E3 ubiquitin ligase complex [5]. Mounting evidence has revealed that CDC20 plays a critical role in cell apoptosis and tumorigenesis via targeting different signaling mediators [6, 7]. Importantly, CDC20 is an essential developmental gene whose disruption in mice causes embryonic lethality via aberrant mitotic arrest [8]. However, the functional roles of CDC20 and the molecular targets of CDC20 during cardiac hypertrophy remain to be identified.
Autophagy is an evolutionarily conserved process that is used by the cell to degrade cytoplasmic contents for quality control, survival during temporary energy crises, and catabolism and recycling [9]. Central to this process is the formation of autophagosomes, which is controlled by a set of autophagy genes, such as ATG5, ATG6/beclin 1, ATG7, and ATG8/LC3. Increasing evidence has revealed an important pathogenic role of altered activity of the autophagosome-lysosome pathway in cardiac hypertrophy and HF. Normal levels of autophagy protect cells from environmental stimuli, but excessive autophagy or insufficient autophagy may lead to disease. Cardiomyocyte autophagy plays an important role in maintaining myocardial function. Studies have found that autophagy is attenuated by siRNA knockdown of the autophagy-related gene Atg7 in neonatal rat cardiomyocytes, and cardiomyocytes exhibit significantly hypertrophy. Similar results have been obtained in in vivo experiments. After inducing autophagy by Atg7-RNAi in neonatal rats, the rat heart showed obvious characteristics of cardiac hypertrophy [10]. MIF deficiency exacerbates pressure overload-induced cardiac hypertrophy and contractile anomalies, probably through an AMPK-mTOR-autophagy-dependent mechanism [11].
In this study, we aimed to test whether CDC20 is able to regulate cardiac hypertrophy via targeting autophagy. Our results showed that CDC20 is markedly upregulated in cardiomyocytes in response to hypertrophic stimulation. Knockdown of CDC20 significantly attenuates agonist- and pressure overload-induced cardiac hypertrophy, but the overexpression of CDC20 aggravates this effect in vitro and in vivo. Furthermore, we found that autophagy flux can be regulated by CDC20 through targeting LC3 ubiquitination and proteasomal degradation. Thus, our results revealed a novel role of CDC20 in regulating cardiac hypertrophy and suggest that inhibition of CDC20 may represent a potential therapeutic target for treating hypertrophic diseases.
Methods
Antibodies and reagents
Antibodies: LC3B (Sigma; L7532), ATG5 (Cell Signalling; 12994), AKT (Cell Signalling; 9272), p-AKT (Cell Signalling; 4060S), ERK1/2(Cell Signalling; 9102), p-ERK1/2 (Cell Signalling; 9101), Beclin-1 (Cell Signalling; 3738), horseradish peroxidase-linked anti-mouse or anti-rabbit IgG (Cell Signalling; 7074S). GAPDH (Proteintech; 10494), β-actin (SantaCruz; sc-47778), ATG7 (Santa Cruz sc-33211), EGFR (abcam; ab52894). Primers including ANP, BNP, α-MHC, LC3 were purchased from Sangon Biotech (Shanghai, China). The protein A/G plus-agarose were purchased from Selleck Chemicals (Houston, TX, USA), Ang II (Sigma; F3165), wheat germ agglutinin (WGA) and pentobarbital was purchased from Sigma-Aldrich (St Louis, MO). Collagenase II, DMEM/F-12, fetal bovine serum (FBS) penicillin and streptomycin were purchased from HyClone Laboratories Inc. (Logan, UT). TRizol and lipofectamine 2000 were obtained from Invitrogen (Carlsbad, CA). All other chemicals frequently used in our laboratory were purchased from either Sigma-Aldrich or BD Pharmingen (San Jose, CA).
Experimental animals
C57BL/6 male mice (8-10 weeks old) and Sprague-Dawley (SD) rats (1-2 days old) that were raised in a pathogen-free animal house were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All procedures were approved by the Animal Care and Use committee of Dalian Medical University. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH; Publication No. 85-23, revised 1996).
A HF mouse model was established by transverse aortic constriction (TAC) [12]. Briefly, male wild-type (WT) mice (8 weeks old) were anesthetized with isoflurane. The left side of the chest was then opened. A 7.0 nylon suture ligature was tied against a 27-gauge needle at the transverse aorta to produce a 65-70% constriction after removal of the needle. Doppler analysis was performed to confirm that adequate aortic constriction was induced. The sham operation was performed with a similar procedure without ligation. All surgeries and subsequent analyses were conducted in a blinded manner.
rAAV9 vector delivery
Recombinant adeno-associated virus serotype 9 (rAAV9) expressing an siRNA targeting CDC20 (rAAV9/siCDC20), full-length CDC20 cDNA (AAV9/CDC20) and a scramble control (rAAV9/ZsGreen) were generated by Biowit Technologies Co., Ltd. (Shenzhen, China), according to the manufacturer's protocol. The sequence of rAAV9-shRNA-CDC20 was GCC TAG TGG TCC TGG AGA AAG. We tested the tail injection efficiency of wild-type mice at two different doses (1×109 or 1×1011 vg) and found that rAAV9 injection at a dose of 1x1011 vg for 2 weeks resulted in over 90% of the myocardial area being ZsGreen-positive.
Echocardiography
Echocardiography was performed with a Vevo 2100 High-Resolution Imaging System (VisualSonics Inc.) in mice that were sedated with 1-1.5% isoflurane and placed on a heating pad to maintain body temperature.
Cell culture and isolation of cardiac cells
Rat neonatal cardiac myocytes (NRCMs) were isolated from 24-hour-old SD rats. All operations were performed under a sterile hood. After the hearts were removed, they were cut to approximately 1 mm3 and digested with 0.04% trypsin and 0.07% type II collagenase. Then, the dispersed cells were incubated on 100 mm culture dishes for 90 minutes at 37°C with 5% CO2. The cardiomyocytes were resuspended, collected and transferred to laminin-coated (10 µg/ml) 6-well plates or 24-well plates. The cardiomyocytes were cultured in DMEM/F12 with 10% fetal bovine serum (FBS) for 16-20 hours, and the medium was then replaced with serum-free DMEM/F12 for 12 hours before the experiments.
Adenoviruses and cell infection
Adenovirus expressing CDC20 (Ad-CDC20/Ad-siCDC20 with or without GFP) or GFP (or empty vector) alone was used to infect cell cultures according to the manufacturer's protocol (Hanbio, China). NRCMs were infected with Ad-CDC20/Ad-siCDC20 or Ad-GFP at a multiplicity of infection (MOI) of 50 and then treated with Ang II (100 nM, Sigma) or phenylephrine (PE) (10 µM, Sigma) for 24 hours. To determine the efficiency of CDC20 gene infection, immunofluorescence microscopy and Western blot analyses were performed 24 hours after Ad-CDC20/Ad-siCDC20 or Ad-GFP infection.
RNA isolation and real-time PCR
Total RNA was isolated from NRCMs or heart tissue using TRIzol (Invitrogen). cDNA was used for PCR amplification with gene-specific primers for atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC). GAPDH was used as the endogenous control. The value relative to the control sample is given by 2-ΔΔCT. qPCR was performed with an iCycler IQ system (Bio- Rad, USA).
Immunoprecipitation assay
Immunoprecipitation was performed with the Protein A/G Magnetic Beads system (Selleck Chemicals, Houston, TX, USA) according to the manufacturer's protocols. Briefly, cardiomyocytes or heart tissues were lysed with washing buffer (50 mM Tris, 150 mM NaCl, 0.1%-0.5% NP-40, pH 7.5) containing Protease Inhibitor Cocktail Tablets (04693132001, Roche). The primary antibody (Ab) was covalently immobilized on protein A/G agarose using magnetic beads. The Ab (typically 2-4 μg) was added to 50 μl of binding buffer (50 mM Tris, 150 mM NaCl, 0.1%-0.5% detergent: Triton X-100 or NP-40, pH 7.5) to dilute the Ab to the appropriate concentration. The Ab and the protein sample mixture were added to the protein A/G magnetic beads. The tube was rotated for 1 hour at room temperature or 4 hours at 4°C to allow the antigen (Ag) to bind to the protein A/G magnetic beads-Ab complex. The samples were washed, 10-50 μl of elution buffer (0.1 M -0.2 M glycine, 0.1%-0.5% NP-40, pH 1.5) was added to the tube with magnetic beads-Ab-Ag complex, and the supernatant was collected. The final solution was used for denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or subjected to further analysis.
List of primers
| Rat ANF | Forward | 5'-CTT CTC CAT CAC CAA GGG CTT-3' | Mouse ANF | Forward | 5'-CAC AGA TCT GAT GGA TTT CAA GA-3' |
| Reverse | 5'-GGA TTT GCT CCA ATA TGG CCT-3' | Reverse | 5'-CCT CAT CTT CTA CCG GCA TC-3' | ||
| Rat BNP | Forward | 5'-TGA TTC TGC TCC TGC TTT TC-3' | Mouse BNP | Forward | 5'-GAA GGT GCT GTC CCA GAT GA-3' |
| Reverse | 5'-GTG GAT TGT TCT GGA GAC TG-3' | Reverse | 5'-CCA GCA GCT GCA TCT TGA AT-3' | ||
| Rat GAPDH | Forward | 5'-GGC AAG TTC AAT GGC ACA GT-3' | Mouse GAPDH | Forward | 5'-GGT TGT CTC CTG CGA CTT CA-3' |
| Reverse | 5'-TGG TGA AGA CGC CAG TAG ACT C-3' | Reverse | 5'-GGT GGT CCA GGG TTT CTT ACT C-3' | ||
| Rat CDC20 | Forward | 5'-GTC AAG GCT GTT GCA TGG TG-3' | Mouse CDC20 | Forward | 5'-GCC CAC CAA AAA GGA GCA TC-3' |
| Reverse | 5'-GAT GGA GCA CAC CTG GGA AT-3' | Reverse | 5'-ATT CTG AGG TTT GCC GCT GA-3' | ||
| Rat LC3 | Forward | 5'-GAG TGG AAG ATG TCC GGC TC-3' | |||
| Reverse | 5'-CCA GGA GGA AGA AGG CTT GG-3' |
In vivo ubiquitylation assay
The in vivo ubiquitination assay was performed following the previously described protocol. Protein lysates from cultured cardiomyocytes were precipitated and detected by Western blot analysis using appropriate antibodies (including LC3 and ubiquitin).
In vitro ubiquitylation assay
Immunocomplexes were added to a ubiquitination reaction (final volume of 30 μl) containing 10× E3 Ligase Reaction Buffer, 60 ng of E1, 600 ng of E2, 1 μg of purified GFP-CDC20, 1 μg of purified LC3, and 10 μg of ubiquitin (following the process described by Ju¨rgen Behrens et al. [13]). The reactions were incubated at 30°C for 2 hours unless otherwise indicated, terminated by boiling for 5 minutes with SDS loading buffer, and resolved by SDS-PAGE, followed by immunoblotting with appropriate antibodies.
Histopathology
The hearts were fixed in a 4% paraformaldehyde solution for 24 hours, embedded in paraffin and sectioned (5 μm). Hematoxylin and eosin (H&E) and Masson's trichrome staining were performed on the heart sections using standard procedures. The positive areas were analyzed by Image Pro Plus 3.0 (Nikon).
Wheat germ agglutinin (WGA)
Sections were stained with 50 μg/ml WGA for 60 minutes to evaluate the cardiomyocyte cross-sectional area, and the cell area was calculated by measuring 15-20 cells per slide.
Reactive oxygen species (ROS)
The ROS generated in the cells were detected by using dihydroethidium (DHE). DHE is capable of staining superoxide anion (O2·-). DHE (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in DMSO to generate the stock solution. After the cells were treated, they were washed twice with ice-cold phosphate-buffered saline (PBS), treated with DHE (10-5 M), and incubated for 30 minutes at 37°C. After probe treatment, the cells were washed at least twice with ice-cold PBS. The positive areas were analyzed by Image Pro Plus 3.0 (Nikon).
Pulse-chase analysis
Cardiomyocytes were infected with adenovirus expressing GFP alone, CDC20, siRNA-control or siRNA-CDC20 for 24 hours. Protein lysates were prepared at the indicated time points after the addition of cycloheximide (CHX, 20 μM) or CHX (20 μM) plus MG132 (10 μM). The levels of endogenous LC3 protein were detected by Western blot analysis with an anti-LC3 Ab, and the results were quantified and normalized to β-actin.
Western blot analysis
Total proteins were extracted from snap-frozen heart tissue or cultured cells by using radioimmunoprecipitation assay (RIPA) buffer (PMSF:RIPA=1:100, Solarbio Science Technology Co.). The protein lysates were separated by 8-12% SDS-PAGE and transferred to polyvinylidenedifluoride (PVDF) membranes by the Bio-Rad Western blotting system. The membranes were incubated with the appropriate antibodies at 4°C overnight and then with horseradish peroxidase-conjugated secondary antibodies (1:2500) for 1 hour at room temperature. All blots were developed by using a chemiluminescence system, and signal intensities were analyzed with a Gel-pro 4.5 Analyzer (Media Cybernetics, USA).
Statistical analysis
All results are expressed as the mean ± SD. An unpaired 2-tailed t-test was used to compare the 2 groups. Comparisons between the control and TAC groups were performed using one-way ANOVA with the Newman-Keuls multiple comparison test from GraphPad Prism 7 (GraphPad Prism Software). For the other comparisons, we determined the significance of differences between the means of the groups by one-way ANOVA. Differences were considered statistically significant at P < 0.05.
Results
Upregulation of CDC20 in cardiomyocytes by hypertrophic stimuli
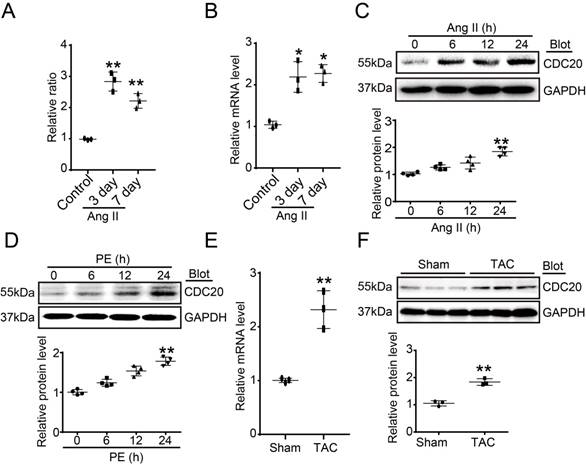
To identify which ubiquitin E3 ligase regulates cardiac hypertrophy, a microarray analysis was performed in Ang II-infused hearts. CDC20 was found to be significantly upregulated at different time points after Ang II infusion (Figure 1A). Ang II-induced upregulation of CDC20 in the same samples was confirmed by qPCR analysis (Figure 1B). Moreover, the CDC20 protein level was increased in neonatal rat cardiomyocytes (NRCMs) in a time-dependent manner after Ang II (100 nM) or PE (100 μM) treatment (Figure 1C-1D). However, CDC20 expression was not altered in Ang II- or PE-treated cardiac fibroblasts (Figure S1A-S1B). Increased expression of CDC20 at both the mRNA and protein levels was further confirmed in the hearts after TAC (Figure 1E-1G). Together, these results indicate that CDC20 expression is upregulated in cardiomyocytes in response to hypertrophic stimuli and may play a role in cardiac hypertrophy.
Upregulation of CDC20 in cardiomyocytes by hypertrophic stimuli. (A) Microarray analysis of CDC20 gene expression in the angiotension II-infused mouse heart at days 3 and 7 (n=3). (B) qPCR analysis of CDC20 expression in the angiotension II-infused mouse heart at days 3 and 7 (n=3). (C and D) CDC20 protein levels in neonatal rat cardiomyocytes (NRCMs) treated with phenylephrine (PE, 100 μM) or angiotensin II (Ang II, 100 nM) at different time points (n=4). (E) The mRNA expression of CDC20 in the heart after 4 weeks of transverse aortic constriction (TAC) (n=3). (F) The protein expression of CDC20 in the heart after 4 weeks of TAC (n=3).
CDC20 regulates cardiomyocyte hypertrophy in vitro
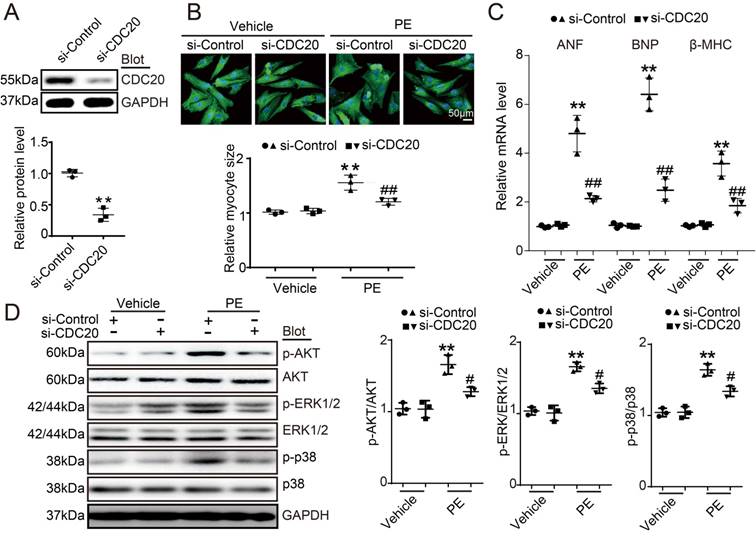
To investigate the role of CDC20 in the heart during pathological stress, NRCMs were infected with a CDC20-knockdown recombinant adenovirus (Ad-siCDC20) or a scramble Ad-siRNA. The expression of CDC20 was significantly reduced by 2.2-fold after Ad-siCDC20 infection compared with that of the Ad-siRNA-control (Figure 2A). Moreover, Ad-siCDC20 knockdown in cardiomyocytes inhibited PE-induced cardiomyocyte hypertrophy, as evidenced by reduced cell surface areas and reduced mRNA expression of ANP, BNP, and β-MHC compared with that of the scramble Ad-siRNA group (Figure 2B-2C). Accordingly, PE-induced activation of hypertrophic signals, such as AKT, ERK1/2 and p38, was also inhibited in Ad-siCDC20-infected cells after PE treatment (Figure 2D). In contrast, the enforced expression of CDC20 by infection with an adenovirus overexpressing CDC20 (Ad-CDC20) resulted in an augmentation of hypertrophic responses, including cell surface area, expression of ANP, BNP and β-MHC, and activation of AKT, ERK1/2 and p38 signaling, compared with those of the Ad-GFP control, both at baseline and after Ang II treatment (Figure S2A-2D). These data suggest that CDC20 exerts a prohypertrophic effect.
The downregulation of CDC20 inhibited the phenylephrine (PE)-induced hypertrophic response in cultured cardiomyocytes. (A) Immunoblotting analysis of CDC20 in neonatal rat cardiomyocytes infected with adenovirus siCDC20 or siControl (n=3). (B) Representative images of double immunostaining (red for α-actinin, blue for DAPI) of NRCMs after 24 hours of PE (100 nM) (left). Scale bar: 50 μm. Quantification of the myocyte surface area (n=3, 150 cells counted per experiment; right). (C) qPCR analysis of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC) mRNA levels in NRCMs treated as described in (B) (n=3). (D) Immunoblot analysis of AKT, ERK1/2, p38 and GAPDH in NRCMs treated as described in (B) (n=3); * P < 0.05, ** P < 0.01 vs. control; # P < 0.05, ## P < 0.01 vs. Ad-siRNA-control plus PE.
CDC20 influences hypertrophy in vivo
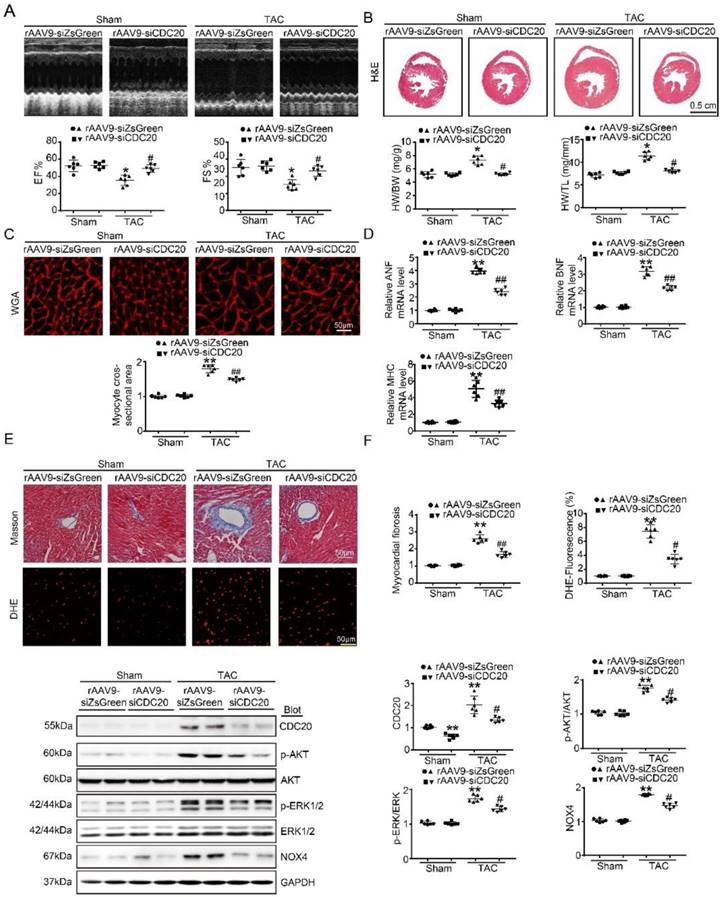
To better understand the function of CDC20 in the heart in vivo, we generated a cardiotropic recombinant adeno-associated virus (rAAV9) serotype containing a siRNA that mediates cardiac-specific knockdown of CDC20 (rAAV9-siCDC20) or a scrambled sequence (rAAV9-siZsGreen) because deletion of the CDC20 gene resulted in embryonic lethality in mice [8]. After two weeks of rAAV9-siCDC20 injection at 1×1011 vg, we observed more than 95% transfection efficiency and reduced CDC20 expression by 2-fold in the heart compared with the rAAV9-siZsGreen control, but no changes in the lung or liver were observed (Figure S3A-3B). Subsequently, the mice were subjected to TAC or sham operation for an additional 5 weeks. Knockdown of CDC20 in mice by rAAV9-siCDC20 injection significantly ameliorated left ventricular (LV) contractile function, as reflected by the LV ejection fraction (EF%) and fractional shortening (FS%), compared with that of the rAAV9-siZsGreen group after the TAC operation (Figure 3A). Concomitantly, rAAV9-siCDC20-injected mice exhibited a reduced hypertrophic response, as evidenced by decreased LV wall thickness, heart weight/body weight (HW/BW) and heart weight/tibia length (HW/TL) ratios, cross-sectional areas of myocytes and mRNA levels of ANP, BNP and β-MHC compared with those of the rAAV9-siZsGreen group (Figure 3B-D). We also analyzed cardiac fibrosis and ROS levels and observed that TAC-induced increases in fibrotic area and DHE density were reduced in rAAV9-siCDC20-injected mice (Figure 3E). In addition, rAAV9-siCDC20 injection reduced the activation of AKT, ERK1/2 and NOX4 compared with that of the rAAV9-siZsGreen group after the TAC operation (Figure 3F).
The knockdown of CDC20 suppresses hypertrophy in vivo. (A) Wild-type (WT) mice injected with rAAV9-siZsGreen or rAAV9-siCDC20 were subjected to sham operation or TAC for 5 weeks. Echocardiographic assessment of ejection fraction (EF%) and fractional shortening (FS%) (n=6). (B) H&E staining of heart sections (upper). Scale bar 0.5 cm. The ratios of heart weight to tibia length (HW/TL) and body weight (HW/BW) (lower) (n=6). (C) Heart cross-sections were stained with TRITC-labeled wheat germ agglutinin (WGA, upper panel). Scale bar: 10 μm. Quantification of the relative myocyte cross-sectional area (n=6, 200 cells counted per heart; lower panel). (D) qPCR analysis of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC) mRNA levels in the hearts of rAAV9-siZsGreen- and rAAV9-siCDC20-treated mice following sham operation or TAC (n=6). (E) Representative images of Masson's Trichrome (upper) and DHE staining (middle) of ventricular sections. Scale bar: 50 μm. Quantification of the relative fibrotic area and ROS fluorescence intensity (n=6, lower). (F) Immunoblotting analysis of p-AKT, AKT, p-ERK1/2, ERK1/2, NOX4 and GAPDH in heart samples (n=6). * P < 0.05, ** P < 0.01 vs. sham; # P < 0.05, ## P < 0.01 vs. the rAAV9-siZsGreen plus TAC group.
To further test whether the overexpression of CDC20 enhances hypertrophic remodeling in vivo, WT mice received rAAV9-CDC20 or rAAV9-ZsGreen alone. After 2 weeks of rAAV9-CDC20 injection at 1×1011 vg, we observed more than 95% transfection efficiency and increased CDC20 expression by 3.4-fold in LV tissues compared with those of the rAAV9-ZsGreen control, but no changes in the lung or the liver were observed (Figure S3C-3D). The mice were then subjected to TAC for 3 weeks. Compared with the injection of rAAV9-ZsGreen, the injection of rAAV9-CDC20 markedly enhanced mouse death post-TAC operation (Figure S4A). TAC-induced HF, as reflected by decreased LV EF% and FS%, was aggravated in rAAV9-CDC20-injected mice compared with that in the rAAV9-ZsGreen group (Figure S4B). Moreover, the TAC operation significantly increased the hypertrophic response (HW/BW and HW/TL ratios, cross-sectional areas of myocytes, ROS production, and expression of ANP, BNP and β-MHC) in rAAV9-ZsGreen-injected animals, and these effects were further accelerated in rAAV9-CDC20-injected mice (Figure S4C-F). In contrast to the findings from rAAV9-siCDC20-injected mice, the overexpression of CDC20 further enhanced the activation of AKT, ERK1/2 and NOX4 compared with that of the rAAV9-ZsGreen control after TAC stress (Figure S4G). Thus, these data indicate that CDC20 exerts a prohypertrophic function in the animal model.
CDC20 regulates autophagic flux
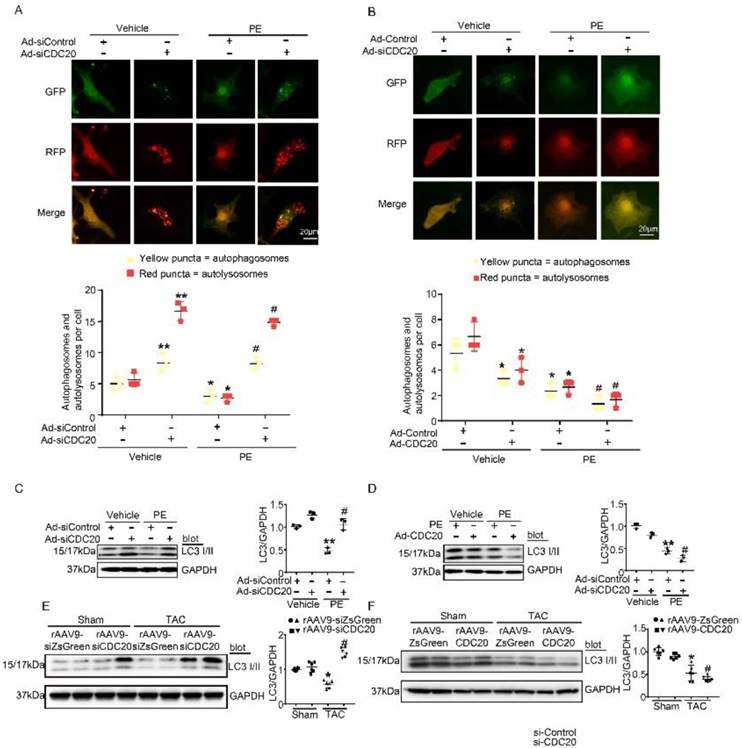
Autophagy activation has been proved to play a critical role in cardiac hypertrophy and HF [9], so we tested whether CDC20 influences autophagic flux in vitro in cardiomyocytes using mRFP-GFP-LC3. In this experiment, red dots represent autolysosomes, while yellow dots represent autophagosomes. Following fusion with lysosomes, GFP is quenched in the acidic environment, while RFP continues to emit fluorescence, marking the autolysosomal compartment. Using this method, autophagosome-autolysosome numbers can readily be calculated [13]. Primary cardiomyocytes were infected with Ad-siCDC20 (without GFP) or the Ad-siRNA-control together with Ad-mRFP-GFP-LC3 for 24 hours and then treated with PE for an additional 48 hours. Compared with the siRNA-control, Ad-siCDC20 infection markedly enhanced the numbers of both GFP and RFP dots, both at baseline and after PE treatment, and red dots (autolysosome formation) were more abundant than yellow dots (autophagosomes), suggesting that the deletion of CDC20 promoted autophagic flux (Figure 4A). In contrast, the overexpression of CDC20 by infection with Ad-CDC20 reduced autophagic flux, as indicated by decreased numbers of both GFP and RFP dots compared with the Ad-control at baseline and after PE treatment (Figure 4B).
CDC20 regulates autophagic flux and LC3 stability. (A) Immunofluorescence showing the autophagic flux after treatment with PE and/or si-CDC20. Scale bar: 40 μm (n=3). (B) Immunofluorescence showing the autophagic flux after treatment with PE and/or CDC20 (n=3). (C) Representative Western blot showing the expression of LC3 I/II after transfection with siCDC20 and treatment with phosphate-buffered saline or PE for 72 hours (n=3). (D) Representative Western blot showing the expression of LC3 I/II after transfection with CDC20 and treatment with phosphate-buffered saline or PE for 72 hours (n=3). (E) Representative Western blot showing the expression of LC3 I/II in heart samples from rAAV9-siZsGreen- and rAAV9-siCDC20-treated mice following sham operation or TAC. * P < 0.05 vs. sham; # P < 0.05 vs. the rAAV9-siZsGreen plus TAC group (n=6). (F) Representative Western blot showing the expression of LC3 I/II and GAPDH in heart samples from rAAV9-ZsGreen- and rAAV9-CDC20-treated mice following sham operation or TAC (n=6). * P < 0.05, ** P < 0.01; # P < 0.05.
CDC20 regulates LC3 stability
Since LC3 is a key regulator of autophagy activation, we next assessed whether CDC20 regulated LC3 protein levels in vitro. Knockdown of CDC20 by siRNA in cardiomyocytes significantly increased LC3 I and II protein levels compared with those of the siRNA-control under basal conditions and after 48 hours of PE treatment (Figure 4C), but LC3 I and II protein levels were reduced by overexpression of CDC20 compared to those of the Ad-control under basal conditions and after 48 hours of PE treatment (Figure 4D). Furthermore, we examined the effect of CDC20 on LC3 stability in the heart in vivo. rAAV9-siCDC20-injected mice showed increased protein levels of LC3 I and II compared with the those of the rAAV9-siZsGreen group after the TAC operation (Figure 4E). In contrast, overexpression of CDC20 in mice by rAAV9-CDC20 injection further aggravated the reduction in LC3 I and II levels compared with those of the rAAV9- ZsGreen control after TAC stress (Figure 4F). Overall, these results suggest that CDC20 regulates the activation of autophagy via reducing LC3 stability.
CDC20 interacts with LC3 and promotes LC3 ubiquitination and degradation by the proteasome
To determine whether CDC20 influences LC3 stability through their mutual interaction, we performed coimmunoprecipitation (Co-IP) assays in primary cardiomyocytes. The LC3 protein was efficiently precipitated by the anti-CDC20 antibody but not by an IgG control, and this interaction between CDC20 and LC3 was enhanced by PE treatment (Figure 5A). To identify the sequence in CDC20 that interacts with LC3, we tested full-length Myc-tagged CDC20 and three Myc-tagged CDC20 deletion mutants for their ability to bind LC3 expressed in 293T cells. Co-IP assays with an anti-Myc antibody revealed that the sequences containing amino acids 478-499 of the IR domain were required for binding to LC3 (Figure 5B). Thus, these findings indicate that CDC20 associates with LC3 through the IR domain-containing amino acids 477-499 (Figure 5C).
CDC20 interacts with LC3 and directly promotes LC3 ubiquitination and degradation by the proteasome. (A) Western blot showing the expression of LC3 I/II by co-IP. (B) Western blot showing the expression of CDC20 mutant proteins (tagged with Myc) and LC3 I/II. (C) Schematic diagram depicting the CDC20 domain structure and summary of in vitro binding experiments. (D and E) Lysates harvested from NRCMs after infection with siRNA-control and siRNA-CDC20 or infection with Ad-GFP and Ad-CDC20 and pretreatment with MG132 for 6 hours were immunoprecipitated with anti-LC3. The ubiquitin-conjugated LC3 was detected by Western blotting with anti-ubiquitin (Ub) and anti-CDC20 antibodies (upper panel). Input showing the expression of the corresponding proteins in whole-cell lysates (lower panel). (F) Ubiquitin-conjugated LC3 was detected by Western blotting with anti-ubiquitin (Ub) (left). Input showing the expression of the corresponding proteins in whole-cell lysates (right). (G) CDC20 ubiquitinated LC3 in vitro. GFP-tagged CDC20 was tested for E3 ubiquitin ligase activity in the presence and absence of GST-E1, His6-E2, GFP-CDC20 (full-length 1-499 aa), GFP-CDC20 (168-499 aa) and GFP-CDC20 (1-476 aa). The protein blot was analyzed using an anti-ubiquitin antibody. (H and I) NRCMs were infected with adenovirus Ad-GFP and Ad-CDC20 or with siRNA-control and siRNA-CDC20 and then treated with cycloheximide (CHX, 10 μM) and/or MG132 (10 μM) and harvested at the indicated time points. Representative immunoblotting analyses of LC3 protein levels for each group.
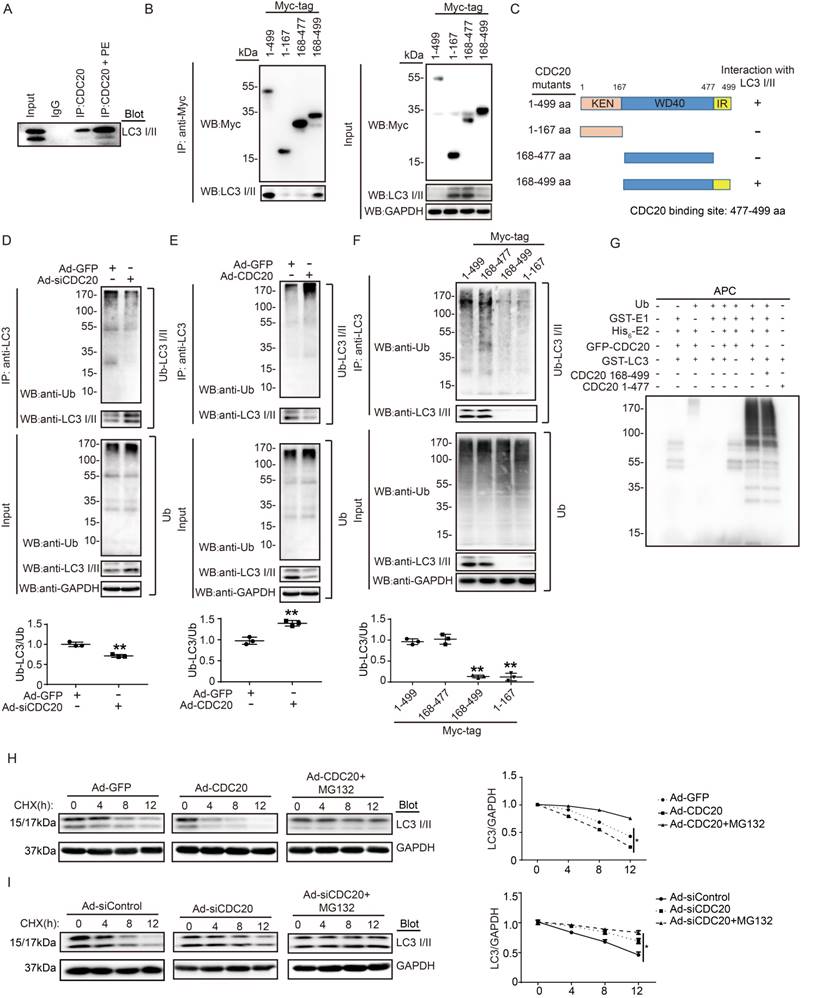
To evaluate whether CDC20 reduces LC3 stability through ubiquitination and proteasomal degradation, primary cardiomyocytes were infected with si-CDC20 or si-control adenovirus for 24 hours. Immunoprecipitation (IP) was performed with an anti-LC3 antibody. Immunoblotting assays showed that the knockdown of CDC20 significantly inhibited LC3 ubiquitination but increased the protein level of LC3 compared with that of the siRNA control (Figure 5D). Conversely, the overexpression of Ad-CDC20 enhanced LC3 ubiquitination but decreased LC3 protein levels compared with those of the Ad-GFP control (Figure 5E). To test whether the IR domain is involved in mediating LC3 ubiquitination, 293T cells were transfected with a full-length Myc-tagged CDC20 and different CDC20 deletion mutants. Immunoblotting with an anti-Ub Ab showed that deletion of amino acids 477-499 within CDC20 abolished LC3 ubiquitination (Figure 5F), suggesting that sequences within the amino acids 477-499 mediated LC3 ubiquitination.
To further determine the effect of CDC20 on LC3 ubiquitination, we performed an in vitro ubiquitination assay with immunopurified GFP-LC3, ubiquitin (Ub), APC, recombinant GST-E1, His6-E2, GFP-CDC20 (full-length 1-499 aa), GFP-CDC20 (168-499 aa) and GFP-CDC20 (1-477 aa). We found that polyubiquitylation of LC3 proceeded efficiently in the presence of CDC20 (1-499 aa), CDC20 (168-499 aa) and other components of the reaction but was abolished by the deletion of UBA1, CDC20 (478-499 aa) or any other component of the reaction (Figure 5G). These results demonstrate that CDC20 directly ubiquitinated LC3.
To test whether ubiquitinated LC3 is degraded by the proteasome, we performed a pulse-chase assay in primary cardiomyocytes with cycloheximide (CHX, an inhibitor of protein synthesis) and MG132 (a proteasome inhibitor). Overexpression of CDC20 by infection with Ad-CDC20 resulted in a shortened half-life of LC3 I and II compared with that of the Ad-GFP control, and this reduction was reversed by the addition of MG132 (Figure 5H). In contrast, the knockdown of CDC20 by Ad-siCDC20 increased the half-life of LC3 protein I and II compared with that of the Ad-siControl, and the degradation of LC3 I and II was completely blocked in Ad-siCDC20-infected cells (Figure 5I). The knockdown of endogenous CDC20 in NRCMs did not affect the expression of LC3 at the transcriptional level (Figure S3). Collectively, these results indicated that CDC20 regulated LC3 protein levels in a proteasome-dependent manner.
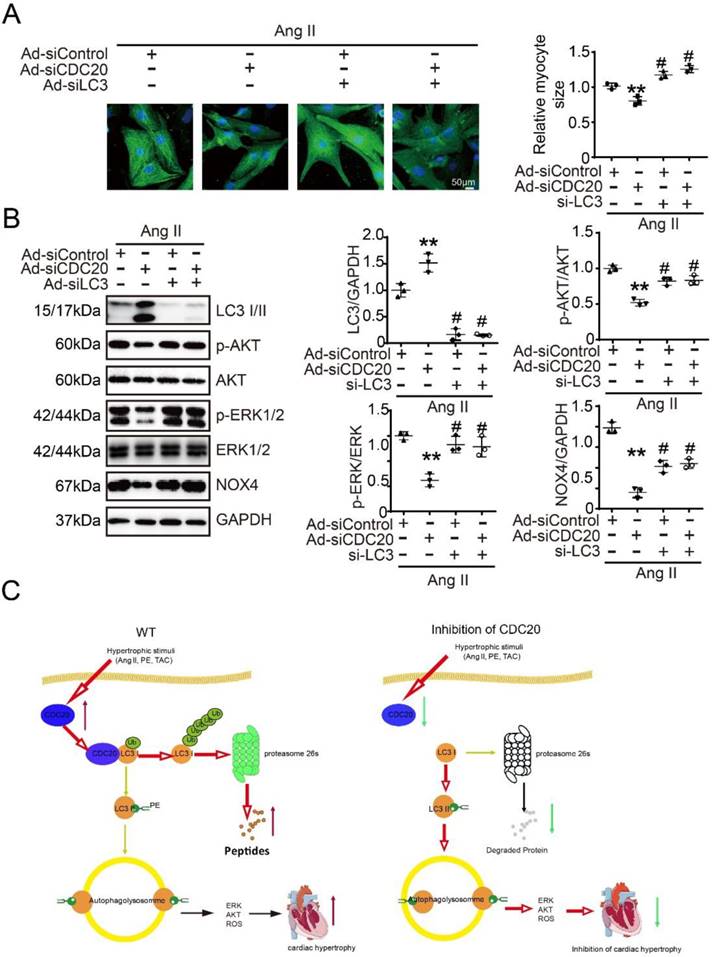
Knockdown of LC3 reversed the reduction of cardiomyocyte size after the deletion of CDC20 in vitro
Next, we tested whether CDC20 regulates cardiomyocyte hypertrophy through LC3-mediated autophagy. Immunostaining showed that the knockdown of CDC20 by siRNA resulted in significant reductions of cardiomyocyte size (Figure 6A) and the activation of AKT, ERK1/2 and NOX4 compared with that of the siControl after Ang II treatment (Figure 6B, lane 2 versus 1), which was markedly reversed by siRNA-LC3 infection (Figure 6B, lane 3 versus 2) and was fully restored by a combination of siRNA-CDC20 and siRNA-LC3 infection (Figure 6B, lane 4 versus 3). Similarly, the effect of CDC20 knockdown on the reduction of cardiomyocyte size (Figure S6A) and the inactivation of AKT, ERK1/2 and NOX4 was also confirmed in siRNA-CDC20-infected cells in the presence of chloroquine (CQ), an autophagy inhibitor (Figure S6B). Thus, these results indicate that CDC20 knockdown inhibits cardiomyocyte hypertrophy via the LC3-mediated activation of autophagy.
Knockdown of LC3 reversed the reduction of cardiomyocyte size after deletion of CDC20 in vitro. (A) Representative images of double immunostaining (green for α-actinin) of NRCMs transfected with siRNA-CDC20 or siRNA-control with or without Ad-siLC3 treatment after 24 hours of Ang II stimulation (left). Scale bar 50 μm. Quantification of the myocyte surface area (right) (n=3). (B) Representative Western blot analyses of p-AKT, AKT, p-ERK1/2, ERK1/2, NOX4, LC3 I/II and GAPDH in NRCMs infected with siRNA-control or siRNA-CDC20 with or without Ad-siLC3 treatment after 24 hours of Ang II stimulation (n=3). C, A functional link between CDC20 and LC3-dependent autophagy in hypertrophy. ** P < 0.01 vs. saline; # P < 0.05, ## P < 0.01 vs. saline
Discussion
Our study identified CDC20 as a prohypertrophic regulator. The knockdown of CDC20 in cardiomyocytes inhibited the hypertrophic response after agonist or TAC treatment. In contrast, enforced expression of CDC20 in cardiomyocytes aggravated the hypertrophic response in vitro and in vivo. Furthermore, we identified LC3, a key regulator of autophagy, as a CDC20 target. CDC20 is able to directly bind to and promote LC3 ubiquitination and proteasomal degradation, thereby resulting in autophagy inhibition and hypertrophy. These data suggest that there is a functional link between CDC20 and LC3-dependent autophagy in hypertrophy. Our findings are summarized in Figure 6C.
Cardiac hypertrophy and consequent HF are major health challenges around the world [15, 16]. Over the past decade, growing evidence has indicated the causative contribution of the UPS system and autophagy to hypertrophy and HF [12, 17]. The UPS is a selective proteolytic system in which the conjugation of ubiquitin to substrates induces degradation by the proteasome. During this process, E3 ligases, as key enzymes, specifically recognize protein substrates for ubiquitination and play an important role in regulating cardiac hypertrophy [18, 19]. CDC20 plays important roles in cell cycle progression by activating the APC E3 ubiquitin ligase, which initiates chromatid separation and entrance into anaphase [20, 21]. CDC20-APC was found to ubiquitinate multiple substrates, including Nek2A, cyclin A, securin, cyclin B1, Bim, and Mcl-1, and target them for degradation by the proteasome [6, 7, 20, 22]. Mounting evidence indicates that CDC20 expression is remarkably elevated in higher grades of cancers, and CDC20 has been linked to poor prognosis in multiple cancers [23, 24]. Interestingly, several studies have revealed unexpected nonmitotic functions for CDC20-APC in the development of the mammalian brain and glioblastoma stem-like cells [25]. However, little is known about the functional roles of CDC20 in regulating cardiac hypertrophy. Here, our results showed that CDC20 was upregulated in cardiomyocytes in response to hypertrophic stimuli (Figure 1). The knockdown of CDC20 in cardiomyocytes inhibited cardiac hypertrophy after hypertrophic stress, but the overexpression of CDC20 aggravated this response (Figure 2-4), demonstrating that CDC20 exerts a prohypertrophic effect in both cultured cardiomyocytes and mice.
Autophagy is a conserved intracellular catabolic process that is responsible for the degradation of long-lived proteins and organelles by the lysosome [26]. Recent works on autophagy changed our understanding of the regulation of cardiac hypertrophy. Notably, a specific set of autophagy-related genes, such as ATG5, ATG6/beclin1, and ATG7, are essential for autophagy [27]. Recent studies indicated that autophagy is essential for the development of cardiac hypertrophy [27-30]. Among autophagy-related proteins, LC3 (microtubule-associated protein 1A and 1B light-chain 3; MAP-LC3, MAP1-LC3/MAP1A1B-LC3) is a unique ubiquitin-like modifier that is localized on isolated membranes and plays a critical role in the formation of autophagosomes. During autophagy, cytosolic LC3 (LC3-I) is activated by ATG7 (an E1-like enzyme), transferred to ATG3 (an E2-like enzyme), and ultimately conjugated to phospholipids to form the LC3-phospholipid conjugate (LC3-II). LC3-II is localized to autophagosomes. During the fusion of autophagosomes with lysosomes, LC3-II on the cytosolic face of autophagosomes is delipidated by ATG4B to form LC3-I, and LC3-II in the autolysosomal lumen is degraded by lysosomal hydrolases. Thus, LC3-II is a promising autophagosomal marker, and the lysosomal turnover of LC3-II is a marker for autophagic activity. In this study, we first monitored the effect of CDC20 on autophagic flux using the RFP-GFP-LC3 double fluorescence system and found that CDC20 indeed regulated autophagic activity in cardiomyocytes (Figure 5). CDC20 could interact with LC3 directly and promote LC3 ubiquitination and degradation by the proteasome, thereby leading to the inhibition of autophagy (Figures 5 and 6). Importantly, we knocked down LC3 or inhibited autophagy to study its role in cardiomyocyte hypertrophy and demonstrated that LC3 is required for Ang II-induced hypertrophy after CDC20 deletion in vitro (Figure 6C). Thus, we identified LC3 as a new target of CDC20 in hypertrophy.
In conclusion, our present study identified CDC20 as a novel regulator of pathological cardiac hypertrophy and dysfunction. The knockdown of CDC20 improved hypertrophic remodeling, but the overexpression of CDC20 aggravated this response in cellular and animal models. Our results further revealed that CDC20 directly binds to LC3 and promotes LC3 ubiquitination and degradation by the proteasome, resulting in the inhibition of autophagy. However, the knockdown of LC3 or the inhibition of autophagy reversed the antihypertrophic effect of CDC20 deletion, indicating that the knockdown of CDC20 inhibits hypertrophy by stabilizing LC3. Thus, our data provide a novel mechanistic insight into the likely links among CDC20, LC3-dependent autophagy and hypertrophy and reveal a potential therapeutic target for treating hypertrophic diseases.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors gratefully acknowledge Mrs. Huimin Gu for her help in pathology.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81330003, 81600197, and 81630009), the Dalian High-Level Talents Innovation and Entrepreneurship Projects (2015R019), and the Chang Jiang Scholar Program (T2011160).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Diwan A, Dorn GN. Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda). 2007;22:56-64
2. Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407:221-6
3. Cui Z, Scruggs SB, Gilda JE, Ping P, Gomes AV. Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J Mol Cell Cardiol. 2014;71:32-42
4. Zhang J, Wan L, Dai X, Sun Y, Wei W. Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochim Biophys Acta. 2014;1845:277-93
5. Hadjihannas MV, Bernkopf DB, Bruckner M, Behrens J. Cell cycle control of Wnt/beta-catenin signalling by conductin/axin2 through CDC20. Embo Rep. 2012;13:347-54
6. Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. Embo J. 2010;29:2407-20
7. Wan L, Tan M, Yang J, Inuzuka H, Dai X, Wu T. et al. APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev Cell. 2014;29:377-91
8. Li M, York JP, Zhang P. Loss of Cdc20 causes a securin-dependent metaphase arrest in two-cell mouse embryos. Mol Cell Biol. 2007;27:3481-8
9. Wang X, Cui T. Autophagy modulation: a potential therapeutic approach in cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2017;313:H304-19
10. McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T. et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050-5
11. Xihui X, Yinan H, Sreejayan N, Richard B, Jun R. Macrophage Migration Inhibitory Factor Deletion Exacerbates Pressure Overload-Induced Cardiac Hypertrophy through Mitigating Autophagy. Hypertension. 2013;63:490-9
12. Hui Li, Kedar V, Zhang C, McDonough H, Arya R, Da W. et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex, J. Clin. Invest. 2004;114:1058-1071
13. Hadjihannas MV, Bernkopf DB, Bruckner M, Behrens J. Cell cycle control of Wnt/beta-catenin signalling by conductin/axin2 through CDC20. Embo Rep. 2012;13:347-54
14. Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 2009;452:1-12
15. Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007-18
16. Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol Ther. 2016;161:52-66
17. Barac YD, Emrich F, Krutzwakd-Josefson E, Schrepfer S, Sampaio LC, Willerson JT. et al. The ubiquitin-proteasome system: A potential therapeutic target for heart failure. J Heart Lung Transplant. 2017;36:708-14
18. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and Autophagy in the Heart. Circ Res. 2016;118:1563-76
19. Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ. et al. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211-23
20. Kravtsova-Ivantsiv Y, Ciechanover A. Ubiquitination and degradation of proteins. Methods Mol Biol. 2011;753:335-57
21. Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379-93
22. Chang DZ, Ma Y, Ji B, Liu Y, Hwu P, Abbruzzese JL. et al. Increased CDC20 expression is associated with pancreatic ductal adenocarcinoma differentiation and progression. J Hematol Oncol. 2012;5:15
23. Mao DD, Gujar AD, Mahlokozera T, Chen I, Pan Y, Luo J. et al. A CDC20-APC/SOX2 Signaling Axis Regulates Human Glioblastoma Stem-like Cells. Cell Rep. 2015;11:1809-21
24. Moura IM, Delgado ML, Silva PM, Lopes CA, Do AJ, Monteiro LS. et al. High CDC20 expression is associated with poor prognosis in oral squamous cell carcinoma. J Oral Pathol Med. 2014;43:225-31
25. Yang Y, Kim AH, Yamada T, Wu B, Bilimoria PM, Ikeuchi Y. et al. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science. 2009;326:575-8
26. Berardi DE, Campodonico PB, Diaz BM, Urtreger AJ, Todaro LB. Autophagy: friend or foe in breast cancer development, progression, and treatment. Int J Breast Cancer. 2011;2011:595092
27. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M. et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619-24
28. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T. et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428-38
29. Tanida I, Ueno T, Uchiyama Y. A super-ecliptic, pHluorin-mKate2, tandem fluorescent protein-tagged human LC3 for the monitoring of mammalian autophagy. Plos One. 2014;9:e110600
30. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM. et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284-97
Author contact
![]() Corresponding author: Hui-Hua Li, E-mail: hhli1935com. Department of Cardiology, Institute of Cardiovascular Diseases, First Affiliated Hospital of Dalian Medical University, Dalian 116011, China. Fax: 8641183635963
Corresponding author: Hui-Hua Li, E-mail: hhli1935com. Department of Cardiology, Institute of Cardiovascular Diseases, First Affiliated Hospital of Dalian Medical University, Dalian 116011, China. Fax: 8641183635963