Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusions
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(21):6008-6024. doi:10.7150/thno.24365 This issue Cite
Research Paper
Nanoparticles targeting extra domain B of fibronectin-specific to the atherosclerotic lesion types III, IV, and V-enhance plaque detection and cargo delivery
Mikyung Yu1, Carleena A. Ortega1, Kevin Si1, Roberto Molinaro6, Frederick J. Schoen2, Renata F. C. Leitao1,3, Xiaoding Xu1, Morteza Mahmoudi1, Suyeon Ahn1, Jerry Liu1, Phei Er Saw1,4, In-Hyun Lee1,4, Mirna M. B. Brayner1,5, Azita Lotfi1, Jinjun Shi1, Peter Libby6, Sangyong Jon4 ![]() , Omid C. Farokhzad1,7
, Omid C. Farokhzad1,7 ![]()
1. Center for Nanomedicine and Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA
2. Department of Pathology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115
3. Department of Morphology, School of Medicine, Federal University of Ceará, Fortaleza, CE, Brazil
4. KAIST Institute for the BioCentury, Department of Biological Sciences, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea
5. School of Medicine, Federal University of Ceará, Sobral, CE, Brazil
6. Department of Cardiovascular Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115
7. King Abdulaziz University, Jeddah 21589, Saudi Arabia
Received 2017-12-14; Accepted 2018-8-22; Published 2018-11-15
Abstract

Extra domain B of fibronectin (FN-EDB) is upregulated in the extracellular matrix during tissue remodeling and has been postulated as a potential biomarker for atherosclerosis, yet no systematic test for FN-EDB in plaques has been reported. We hypothesized that FN-EDB expression would intensify in advanced plaques. Furthermore, engineering of FN-EDB-targeted nanoparticles (NPs) could enable imaging/diagnosis and local delivery of payloads to plaques.
Methods: The amount of FN-EDB in human atherosclerotic and normal arteries (ages: 40 to 85 years) was assessed by histological staining and quantification using an FN-EDB-specific aptide (APTFN-EDB). FN-EDB-specific NPs that could serve as MRI beacons were constructed by immobilizing APTFN-EDB on the NP surface containing DTPA[Gd]. MRI visualized APTFN-EDB-[Gd]NPs administered to atherosclerotic apolipoprotein E-deficient mice in the brachiocephalic arteries. Analysis of the ascending-to-descending thoracic aortas and the aortic roots of the mice permitted quantitation of Gd, FN-EDB, and APTFN-EDB-[Gd]NPs. Cyanine, a model small molecule drug, was used to study the biodistribution and pharmacokinetics of APTFN-EDB-NPs to evaluate their utility for drug delivery.
Results: Atherosclerotic tissues had significantly greater FN-EDB-positive areas than normal arteries (P < 0.001). This signal pertained particularly to Type III (P < 0.01), IV (P < 0.01), and V lesions (P < 0.001) rather than Type I and II lesions (AHA classification). FN-EDB expression was positively correlated with macrophage accumulation and neoangiogenesis. Quantitative analysis of T1-weighted images of atherosclerotic mice revealed substantial APTFN-EDB-[Gd]NPs accumulation in plaques compared to control NPs, conventional MRI contrast agent (Gd-DTPA) or accumulation in wild-type C57BL/6J mice. Additionally, the APTFN-EDB-NPs significantly prolonged the blood-circulation time (t1/2: ~ 6 h) of a model drug and increased its accumulation in plaques (6.9-fold higher accumulation vs. free drug).
Conclusions: Our findings demonstrate augmented FN-EDB expression in Type III, IV, and V atheromata and that APTFN-EDB-NPs could serve as a platform for identifying and/or delivering agents locally to a subset of atherosclerotic plaques.
Keywords: atherosclerosis, aptides, extra domain B of fibronectin, magnetic resonance imaging, nanoparticles
Introduction
Atherosclerotic cardiovascular disease remains the leading cause of death worldwide despite wide and successful use of cholesterol-lowering drugs [1]. One of the current challenges in the prevention of cardiovascular disease is identifying subclinical plaque burdens and timely treatment before clinical events occur [2]. Recent evidence suggests that so-called 'vulnerable plaques', characterized by large lipid pools, many macrophages, inflammation, and thin fibrous caps, rarely rupture to cause clinical events—only ~5% of thin-capped vulnerable plaques provoked acute coronary syndrome over 3.4 years [3]. Since atherosclerosis is a systemic, multifocal disease that develops concurrently at different sites in the arterial tree [4], a single patient generally carries diverse lesion types. Additionally, the clinically significant complications of atherosclerotic plaques (particularly plaque rupture, erosion, and thrombosis) depend more on the cellular and molecular content and state of activation within plaques than the degree of stenosis [5]. For example, superficial erosions that cause sudden cardiac death are often barely calcified, less inflamed than ruptured plaques, and exhibit intact elastic laminas and a well-developed media with contractile smooth muscle cells without distinct morphological features. Moreover, the eroded plaques will likely increase as a proportion of the thrombotic complications of atherosclerosis given the widespread use of lipid-lowering drugs such as statins [6,7]. Such observations strongly suggest the necessity of developing new approaches for plaque identification and treatment for better individual management.
2-deoxy-2-[18F]fluoro-d-glucose positron emission tomography has been most widely used to detect inflamed plaques [8,9]. To better define atherosclerotic plaque burden and individual risk, researchers have actively explored various targeted diagnostic approaches, such as imaging with an elastin-specific magnetic resonance imaging (MRI) contrast agent or with 18F-labeled mannose (2-deoxy-2-[18F]fluoro-D-mannose), or detection of plaque microcalcification with 18F-sodium fluoride [10-12]. However, advanced imaging technologies and preventive strategies often do not ensure effective therapy, because the correlation between increased uptake of imaging agents and future adverse events has yet to be validated [13,14]. In this regard, there is a pressing need for an integrative approach that both enables biologically based plaque detection with high specificity and directs local drug delivery to plaque subtypes.
Nanotechnology provides innovative tools for developing precision diagnostics and targeted therapeutics [14-17]. Recently, it has been reported that companion MRI nanoparticles (NPs) can predict therapeutic nanomedicine efficacy by identifying individuals with higher enhanced permeability and retention (EPR), and thus greater likelihood of NP accumulation [18]. Although initial applications of nanomedicine have largely focused on cancer [19], the field has undergone remarkable growth beyond oncology, including a few recent clinical studies of atherosclerosis [20,21]. Intravenously administered liposomal nanoparticles encapsulating prednisolone showed significantly improved pharmacokinetic profile and increased localization in macrophages within human atherosclerotic plaques; however, accumulation of the nanomedicine in atherosclerotic lesions did not elicit an anti-inflammatory therapeutic effect, presumably because prednisolone might not be ideal for atherosclerotic disease treatment [22]. NPs that target atherosclerotic plaques—including those specific to αvβ3 integrin, vascular cell adhesion molecule 1, collagen, macrophages, and fibrin—have been investigated in animals with advanced atherosclerosis [23-25], but efficient targeting often requires a heavily impaired endothelium [23]. Moreover, the poor correlation between experimental atherosclerotic lesions observed in mouse models (i.e., hypercholesterolemic apolipoprotein E-deficient (apoE-KO) mice fed a western diet (WD)) and actual complex lesions found in humans is one of the reasons hindering clinical translation of nanomedicines.
Here we propose an extra domain B of fibronectin (FN-EDB)-targeting platform whose application might not only improve the triaging of patients into preventive therapy categories based on further lesion-specific imaging but also guide therapeutic regimens. FN-EDB is a splice variant of FN, one of the best-characterized models of alternative splicing [26]. It consists of 91 amino acids and has an identical sequence in mice and humans, but adult FN generally lacks the FN-EDB domain [27]. Instead, it can become inserted into FN during active tissue remodeling, such as that which occurs in tumor angiogenesis and wound healing, as well as embryogenesis [28]. FN-EDB was originally identified as a tumor-specific biomarker, and several antibodies against it have been developed for use in imaging and therapy applications; of these, single-chain Fv (scFv) L19 antibody-conjugated IL-2 and tumor necrosis factor-α are currently undergoing clinical trials for cancer treatment [28,29]. In addition, studies have evaluated fluorescent or radiolabeled scFv L19 derivatives in detection of atherosclerotic plaques in mice [30-32]. Although FN-EDB holds potential for selectively targeting progressive plaques that exhibit arterial remodeling, its correlation with atherosclerosis grade has not been systemically investigated, nor has a nanotechnological approach to atherosclerosis based on FN-EDB targeting been considered.
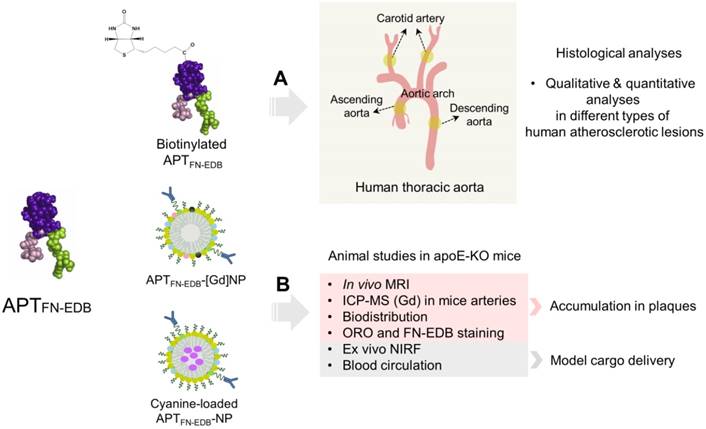
We recently developed a platform technology, termed 'aptides', which enables screening and identification of target-specific, high-affinity peptides [33]. As aptides are chemically synthesized and, thus, can easily introduce functional groups for site-specific conjugation, they are suitable candidates as targeting ligands for drug delivery applications. In particular, because aptides have little immunogenicity and do not have redundant Fc regions, Fc receptor binding-mediated side effects or immunostimulation can be minimized, suggesting superior clinical safety versus antibody-based ligands [34-37]. Recently, we showed that the FN-EDB-specific aptide, APTFN-EDB, replaced anti-FN-EDB antibodies (e.g., L19, BC-1) and it was capable of delivering anticancer drugs, such as docetaxel and SN38, to FN-EDB-expressing tumors [38,39]. In the present study, we examine for the first time the feasibility of using APTFN-EDB to detect FN-EDB expression in different types of human atherosclerotic lesions via immunohistochemistry (IHC) and evaluate the correlation of FN-EDB expression with macrophage accumulation, neovascularization, and plaque hypoxia (Figure 1A). In addition, we have designed an FN-EDB-specific nanoplatform functionalized with APTFN-EDB capable of plaque identification by MRI in apoE-KO mice, ultimately achieving efficient delivery of a model cargo to atherosclerotic plaques (Figure 1B).
Schematic of study design. (A) APTFN-EDB (left) was functionalized with biotin for histological analyses in different types of atherosclerotic lesions of human carotid arteries and ascending-to-descending aortas. (B) APTFN-EDB (left) was immobilized on the surface of NPs (APTFN-EDB-NPs) for animal studies. APTFN-EDB-NPs were further modified by incorporating DTPA(Gd) or cyanine in the shell or core of NPs, respectively, and intravenously injected into advanced atherosclerotic mice through tail vein to demonstrate plaque identification, accumulation, and model cargo delivery.
Results and Discussion
FN-EDB expression is more extensive in advanced atherosclerotic lesions than in early-stage atherosclerosis
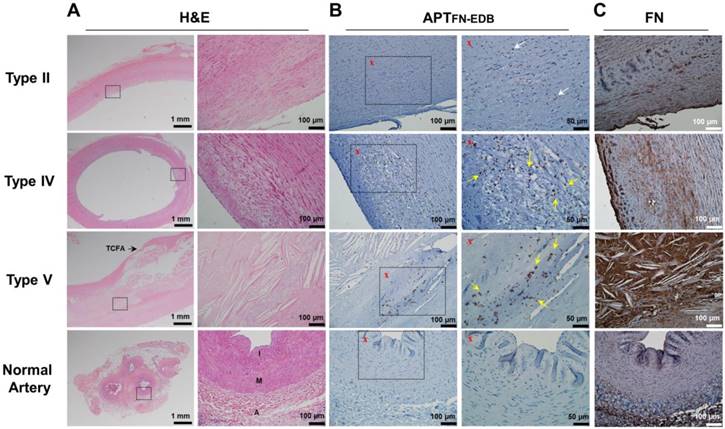
To investigate FN-EDB expression in different types of human atherosclerotic lesions and normal arteries, we analyzed sections of aortas and carotid arteries with atherosclerosis and normal arteries without atherosclerosis, classified by American Heart Association (AHA) criteria (Figure 2A and Figure S1A) [40]. After incubation with biotin-labeled APTFN-EDB and visualization using avidin-biotin horseradish peroxidase, staining of APTFN-EDB revealed more extensive FN-EDB expression in intermediate and advanced plaques than in early-stage atherosclerosis (Figure 2B and Figure S1B). In addition, the advanced necrotic core, which typically includes abundant free cholesterol clefts underlying the thin-cap fibroatheroma (TCFA) as seen in AHA lesion type V, exhibited intensive FN-EDB expression. Conversely, normal arteries showed little or essentially no FN-EDB expression. We further found that a biotin-labeled control scrambled aptide (APTSCR) could not bind to FN-EDB in atherosclerotic lesions, and could not discriminate between atherosclerotic lesions and normal arteries (Figure S2). We also examined the expression level of the dominant FN form in AHA lesion types I-VI and normal arteries using anti-FN antibody. Unlike the clear association of FN-EDB expression with the presence of advanced plaques, the expression of usual FN in these plaques did not differ significantly from that found in normal arteries (Figure 2C and Figure S1C).
FN-EDB expression is intensified in advanced human atherosclerotic lesions. Representative photomicrographs of type II, type IV, and type V atherosclerotic lesions and normal arteries stained with H&E, APTFN-EDB (for FN-EDB), or FN. (A) Left: Low-magnification images of H&E-stained sections. Scale bar = 1 mm. Right: Magnified views corresponding to the black rectangles in adjacent H&E images. Scale bars = 100 μm. (B) Left: Low-magnification images of APTFN-EDB staining results. Scale bar = 100 μm. Right: Magnified views corresponding to the black rectangles in adjacent images labeled with an “x”. Scale bar = 50 μm. White arrows, FN-EDB-positive areas; yellow arrows, FN-EDB expression around progressive atherosclerotic lesions. (C) FN staining results using anti-FN antibody. Scale bar = 100 μm. A: adventitia; I: intima; M: media; TCFA: thin-cap fibroatheroma.
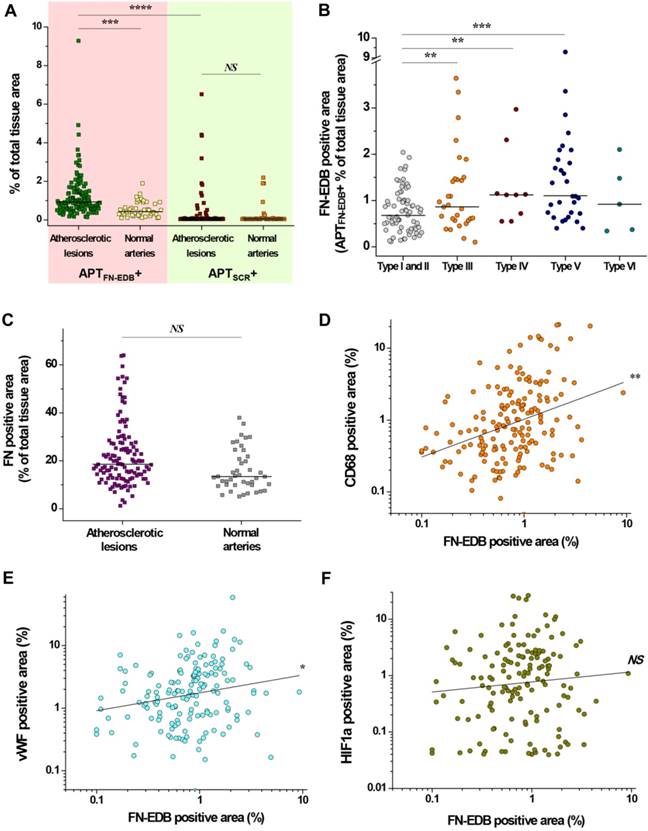
Quantitative analyses of FN-EDB- or FN-positive areas in atherosclerotic lesions and normal arteries affirmed the plaque-detection capability of APTFN-EDB (Figure 3). Sections of atherosclerotic aortic and carotid arteries showed significantly higher FN-EDB than APTSCR staining (P < 0.00001) or compared to normal artery sections (P < 0.0005) (Figure 3A). By contrast, staining with APTSCR showed non-specific binding in a few atherosclerotic lesions and normal arteries (Figure 3A), but the level was not considered significant based on the sample size. FN-EDB expression was notably more intense in AHA type III (P < 0.005), IV (P < 0.005), and V (P < 0.0005) lesions than in type I and II lesions (Figure 3B). Although FN-positive staining trended higher in atherosclerotic aortic and carotid artery sections compared with normal arteries, this difference did not reach statistical significance (Figure 3C). Conformational changes in FN after the insertion of EDB enable particular intermolecular interactions with extracellular matrix (ECM) proteins, suggesting a specific role for FN-EDB in the regulation of ECM assembly and matrix-dependent cell growth in establishing structural integration during tissue remodeling [41]. In addition, alternative splicing of FN is promoted in the arterial endothelium by recruited macrophages under disturbed-flow conditions and has been identified as an important protective mechanism against hemorrhagic rupture of the vessel wall [42]. While the mechanisms that underlie alternative splicing of FN-EDB during expansive arterial remodeling remain poorly understood, FN-EDB expression in particular types of advanced plaques in our study suggests how patients with a variety of lesion types in varying proportions could be effectively approached for early diagnosis and treatment to improve prognosis [43].
FN-EDB expression pertains particularly to Type III, IV, and V lesions, and is positively correlated with macrophage accumulation and neoangiogenesis. (A) The percentage of FN-EDB-positive areas stained with APTFN-EDB was assessed in sections of aortas and carotid arteries with atherosclerosis (n = 147) and normal arteries without atherosclerosis (n = 43) and compared with the corresponding areas stained with APTSCR (negative control). (B) FN-EDB-positive areas in different types of human atherosclerotic tissue sections (type I, n = 17; type II, n = 54; type III, n = 30; type IV, n = 9; type V, n = 32; type VI, n = 5). (C) The percentage of FN-positive areas was assessed using anti-FN antibody in sections of aortas and carotid arteries with atherosclerosis (n = 121) and normal arteries without atherosclerosis (n = 43). (D-F) Correlation of FN-EDB with CD68, vWF, and HIF1α. The percentages of FN-EDB-positive areas in human atherosclerotic lesions and normal arteries were related to the percentages of (D) CD68-, (E) vWF-, or (F) HIF1α-positive areas of the corresponding tissue sections (correlation between FN-EDB and CD68, n = 174; correlation between FN-EDB and vWF, n = 158; correlation between FN-EDB and HIF1α, n = 148). Data are for individual tissue sections, with means shown as horizontal lines. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.00001. NS, not significant.
FN-EDB expression positively correlates with macrophage accumulation and neovascularization
We next sought to determine whether FN-EDB expression in different types of atherosclerotic lesions correlates with macrophage accumulation, neovascularization, or plaque hypoxia—putative markers of advanced plaques [44-46]. We first stained for CD68, von Willebrand factor (vWF), and hypoxia-inducible factor 1a (HIF1α) in consecutive sections of AHA type I-VI atherosclerotic lesions used for FN-EDB expression analysis (Figure S3). Although type I lesions contained a few macrophages, type II-VI lesions had modest to extensive macrophage content. Staining for vWF revealed features of adventitial-derived vasa vasorum and plaque neovascularization in most types of lesions. Similarly, we found HIF1α-positive areas, indicating hypoxia, in all lesion types.
Quantitative assessments of CD68-, vWF-, and HIF1α-positive areas provide insight into the levels of inflammation, neovascularization, and plaque hypoxia in different types of atherosclerotic lesions (Figure S4). CD68 staining indicated extensive macrophage accumulation in type IV (P < 0.01, vs. type I and II; P < 0.05, vs. type III), type V (P < 0.001, vs. type I and II; P < 0.01, vs. type III), and type VI (P < 0.001, vs. type I and II) lesions (Figure S4A). The adventitial-derived vasa vasorum and plaque neovascularization were more intense in type V (P < 0.01) than in type I or II lesions (Figure S4B). These findings indicate that FN-EDB expression in atherosclerotic lesions correlated positively with macrophage accumulation (P < 0.005) and neovascularization (P < 0.05) (Figure 3D-E), supporting the frequent association of FN-EDB expression with unstable advanced atherosclerotic lesions. In particular, FN-EDB expression was significantly higher in type III lesions compared to early-stage lesions, which were not recognized by other putative markers (Figure 3B and Figure S4). On the other hand, HIF1α-positive areas identified in different types of lesions revealed no significant differences in plaque hypoxia (Figure S4C), and the index of plaque hypoxia did not correlate with FN-EDB expression (Figure 3F). These observations suggest that low O2 varies in different lesions and that upregulation of HIF-1α is not directly associated with the physiological function of FN-EDB in plaques.
NP conjugation with APTFN-EDB to construct plaque-specific MRI probes
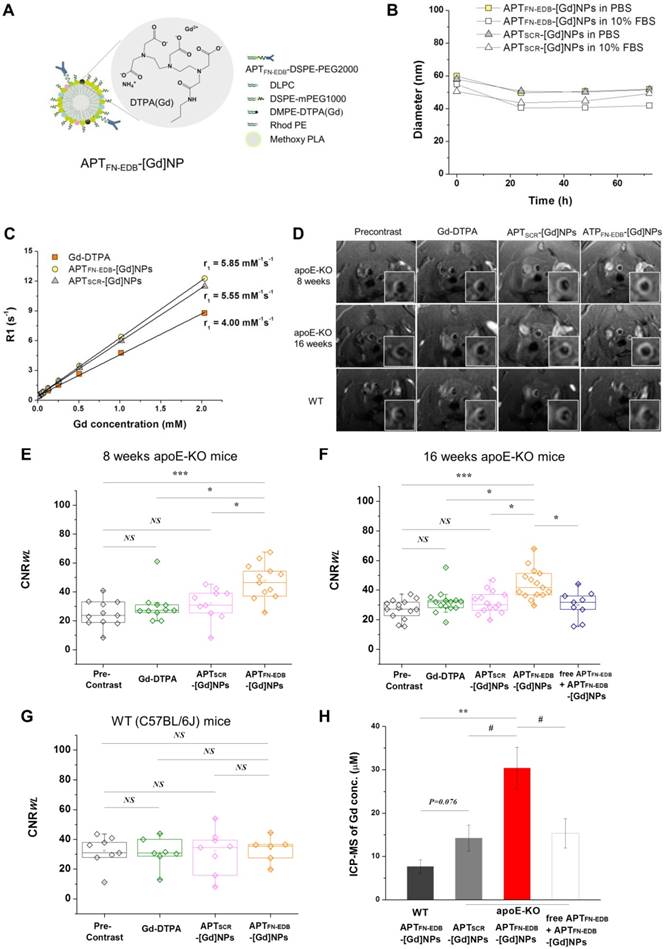
The utility of APTFN-EDB for assessing the correlation of FN-EDB expression with plaque stage prompted us to develop an FN-EDB-targeting nanoplatform to probe plaques in vivo. Previously, we reported that biorecognition of lipid-polymer hybrid NPs was maximized when the length of the polyethylene glycol (PEG) coating molecules on the lipid-polymer NPs was properly shortened relative to the ligand linker [47]. To this end, we fabricated FN-EDB-targeting nanoparticles (APTFN-EDB-NPs) in which APTFN-EDB, used as a targeting ligand, was attached to the surface of NPs consisting of biodegradable poly(D,L-lactide) (PLA) as the NP core and a PEGylated phospholipid layer (DSPE-PEG1000/DLPC/APTFN-EDB-DSPE-PEG2000) as a shell surrounding the PLA core. Unlike antibody conjugation, which exhibits batch-to-batch variation and loss of affinity after conjugation, APTFN-EDB enables fine-tuning of the surface engineering parameters of NPs such as ligand density variation. We further fine-tuned the density of APTFN-EDB on FN-EDB-targeting NPs to optimize FN-EDB targeting efficacy. To do this, we varied the molar ratios of the aptide-attached PEGylated phospholipid (APTFN-EDB-DSPE-PEG2000) from 1% to 5%, yielding APTFN-EDB-NPs with a mean hydrodynamic size range of 53 ± 0.5 ~ 67 ± 1.8 nm (PDI: 0.2) (Figure S5). The surface charges of APTFN-EDB-NPs were 1.86 ± 1.1, -3.43 ± 1.8, and -5.17 ± 2.3 mV for aptide densities of 1%, 2.5%, and 5%, respectively. APTSCR-NPs with a mean hydrodynamic size of 52 ± 1.4 nm (PDI: 0.2) and surface charge of 0.6 ± 1.1 mV were also prepared as a control.
Further experiments assessed the ability of APTFN-EDB-NPs to detect atherosclerotic lesions in human tissue specimens. Confocal microscopy-based optical sectioning revealed that APTFN-EDB-NPs at aptide densities of 1% or 2.5% showed a notable ability to detect atherosclerotic lesions (Figure S6). Contrary to our expectations, APTFN-EDB-NPs with an aptide density of 5% exhibited diminished colloidal stability, resulting in much less binding to atherosclerotic lesions. On the other hand, control APTSCR-NPs showed little binding to atherosclerotic lesions, and all NPs exhibited negligible binding to normal arteries. Based on these findings, we considered an aptide density of 1-2.5% for APTFN-EDB-NPs optimal for use in in vivo studies.
We next examined whether APTFN-EDB-NPs could detect atherosclerotic plaques in vivo. To detect atherosclerotic plaques by MRI, considered an important emerging non-invasive modality that does not utilize ionizing radiation [48], we incorporated a gadolinium (Gd)-based MRI contrast agent with a phospholipid tail (DMPE-DPTA[Gd]) into APTFN-EDB-NPs and APTSCR-NPs (Figure 4A). The resulting MRI contrast agent-incorporated NPs, APTFN-EDB-[Gd]NPs and APTSCR-[Gd]NPs, had a diameter of ~40 ± 0.8 ~ 60 ± 1.3 nm (PDI: 0.2) and exhibited high colloidal stability in both phosphate-buffered saline (PBS; pH 7.4) and serum-containing medium (10% fetal bovine serum [FBS]) for 72 h (Figure 4B). Spin-lattice relaxation time (T1)-weighted MR images were acquired for APTFN-EDB-[Gd]NPs or APTSCR-[Gd]NPs at Gd concentrations between 0 and 2 mM, yielding longitudinal relaxivity values (r1) of 5.85 ± 0.02 and 5.55 ± 0.02 mM-1s-1, respectively (Figure 4C), values that were higher than that of the conventional MRI contrast agent, Gd-DTPA (4.00 ± 0.28 mM-1s-1) .
Atherosclerotic plaque detection ability of APTFN-EDB-[Gd]NPs is superior to that of Gd-DTPA or non-targeting NPs in vivo. (A) Schematic of APTFN-EDB-[Gd]NP. (B-C) Characterization of APTFN-EDB-[Gd]NPs. (B) In vitro stabilities of APTFN-EDB-[Gd]NPs and APTSCR-[Gd]NPs in PBS and 10% FBS, respectively, at 37 °C. (C) The relaxivity (r1) of APTFN-EDB-[Gd]NPs at 7.0 T. The r1 values for Gd-DTPA and APTSCR-[Gd]NPs are also presented for comparison. (D-H) In vivo MRI of APTFN-EDB-[Gd]NPs in apoE-KO mice on a WD for 8 or 16 wk, or in WT mice (C57BL/6J) on a normal chow diet, compared with that of Gd-DTPA or APTSCR-[Gd]NPs in the corresponding animal models. (D) Representative cross-sectional views of brachiocephalic arteries obtained before and after injection of Gd-DTPA, APTSCR-[Gd]NPs, or APTFN-EDB-[Gd]NPs in each group. (E) CNRWL values in apoE-KO mice on an 8-wk WD before (Precontrast, n = 11) and after administration of Gd-DTPA (n = 11), APTSCR-[Gd]NPs (n = 10), or APTFN-EDB-[Gd]NPs (n = 12). (F) CNRWL values in apoE-KO mice on a 16-wk WD before (Precontrast, n = 14) and after administration of Gd-DTPA (n = 14), APTSCR-[Gd]NPs (n = 14), or APTFN-EDB-[Gd]NPs (n = 15). Free APTFN-EDB was injected through the tail vein 2 h prior to the injection of APTFN-EDB-[Gd]NPs (n = 9). (G) CNRWL values in WT mice before (Precontrast, n = 8) and after administration of Gd-DTPA (n = 7), APTSCR-[Gd]NPs (n = 8), or APTFN-EDB-[Gd]NPs (n = 6). Data are for individual mice. The lower and upper ends of boxes represent the 25th and 75th percentiles, respectively, and the line across the box indicates the median. (H) ICP-MS assessment of arteries collected from WT mice and apoE-KO mice after administration of APTFN-EDB-[Gd]NPs, APTSCR-[Gd]NPs, or free APTFN-EDB + APTFN-EDB-[Gd]NPs. #P<0.05, *P<0.005, **P<0.001, ***P<0.0001. NS, not significant.
APTFN-EDB-[Gd]NPs detect in vivo atherosclerotic plaques in hypercholesterolemic apoE-deficient mice
Although experimental atherosclerosis imperfectly recapitulates the various plaque stages found in human atherosclerotic lesions, it nonetheless has utility for in vivo studies. Accordingly, we examined the plaque-detection capability of APTFN-EDB-[Gd]NPs after intravenous (i.v.) injection in hypercholesterolemic apoE-KO mice that consumed a WD for 8 or 16 wk (Figure 4D-F). The brachiocephalic vessel wall, which has served to determine the predilection for developing progressive atherosclerotic lesions, was visualized by MRI. MRI signal intensities of the aortic wall-to-lumen were measured to determine the contrast-to-noise ratio in those lesions (CNRWL). A conventional MRI contrast agent (Gd-DTPA) was used as a standard nonspecific control to obtain the same MR images [10], but there was no apparent change in MR images between precontrast and intravenous injection of Gd-DTPA (0.2 mmol/kg) (Figure 4D-F).
Next, MR images were acquired with APTFN-EDB-[Gd]NPs or APTSCR-[Gd]NPs (50 μmol Gd/kg) after i.v. injection into atherosclerotic mice. Similar to the findings for the Gd-DTPA-treated group, APTSCR-[Gd]NPs showed little contrast enhancement in the atherosclerotic vessel wall (Figure 4D-F). However, strong contrast enhancement was evident in the brachiocephalic arteries of apoE-KO mice injected with APTFN-EDB-[Gd]NPs (Figure 4D-F). Notably, treatment with APTFN-EDB-[Gd]NPs caused a considerable increase in plaque CNRWL in apoE-KO mice on an 8-wk WD (P < 0.0001, vs. precontrast; P < 0.005, vs. Gd-DTPA; P < 0.005, vs. APTSCR-[Gd]NPs) and in apoE-KO mice on a 16-wk WD (P < 0.0001, vs. precontrast; P < 0.005, vs. Gd-DTPA; P < 0.005, vs. APTSCR-[Gd]NPs), indicating unambiguous detection of established atherosclerotic lesions by APTFN-EDB-[Gd]NPs. To normalize the MRI contrast enhancement of the brachiocephalic arteries for each muscle, we examined the normalized enhancement ratio (NER) (Table 1). Consistent with the previous results, the APTFN-EDB-[Gd]NP-treated group demonstrated significant aortic MRI signal enhancement, with an average NER % of 42.9% in apoE-KO mice on an 8-wk WD (*P < 0.001, vs. Gd-DTPA; P < 0.05, vs. APTSCR-[Gd]NPs) and 23.6 % in apoE-KO mice on an 16-wk WD (†P < 0.05, vs. Gd-DTPA; P < 0.05, vs. APTSCR-[Gd]NPs). Our findings revealed a similar MR contrast enhancement trend in apoE-KO mice injected with APTFN-EDB-[Gd]NPs on 8- or 16-wk WD, demonstrating its ability to reliably identify plaques. A competition experiment, performed by injecting an excess of free APTFN-EDB (100 mg/kg) 2 h prior to the injection of APTFN-EDB-[Gd]NPs, showed a marked decrease in CNRWL values compared with those obtained using APTFN-EDB-[Gd]NPs (P < 0.005, vs. APTFN-EDB-[Gd]NPs) (Figure 4F). These results suggest that specific binding of APTFN-EDB to FN-EDB mediates the MRI detection of atherosclerotic lesions by APTFN-EDB-[Gd]NPs. Furthermore, wild-type (WT) mice (C57BL/6J) used as a control showed no statistically meaningful accumulation of APTFN-EDB-[Gd]NPs or APTSCR-[Gd]NPs in brachiocephalic arteries (Figure 4D, G). Direct comparison of CNRWL values of ApoE-KO and WT mice treated with APTFN-EDB-[Gd]-NPs (Figure 4F-G) additionally demonstrates remarkable differences in plaque accumulation (P<0.05) (Figure S7).
Summary of in vivo MRI results showing NER % for each group.
| Average NER % | |
|---|---|
| (8 weeks apoE-KO) | |
| Gd-DTPA | 2.3 % |
| APTSCR-[Gd]NPs | 19.0 % |
| APTFN-EDB-[Gd]NPs | 42.9 %* |
| (16 weeks apoE-KO) | |
| Gd-DTPA | 7.7 % |
| APTSCR-[Gd]NPs | 6.7 % |
| APTFN-EDB-[Gd]NPs | 23.6 %# |
| (WT-C57BL6) | |
| Gd-DTPA | -0.3 % |
| APTSCR-[Gd]NPs | 9.3 % |
| APTFN-EDB-[Gd]NPs | 8.8 % |
NER %: normalized enhancement ratio (percent change). MRI contrast enhancement of the brachiocephalic arteries was normalized for each muscle. *P < 0.001 vs. Gd-DTPA and P < 0.05 vs. APTSCR-[Gd]NPs; # P < 0.05 vs. Gd-DTPA and P < 0.05 vs. APTSCR-[Gd]NPs.
In vivo plaque-detection capability of APTFN-EDB-[Gd]NPs is related to FN-EDB expression in the plaque
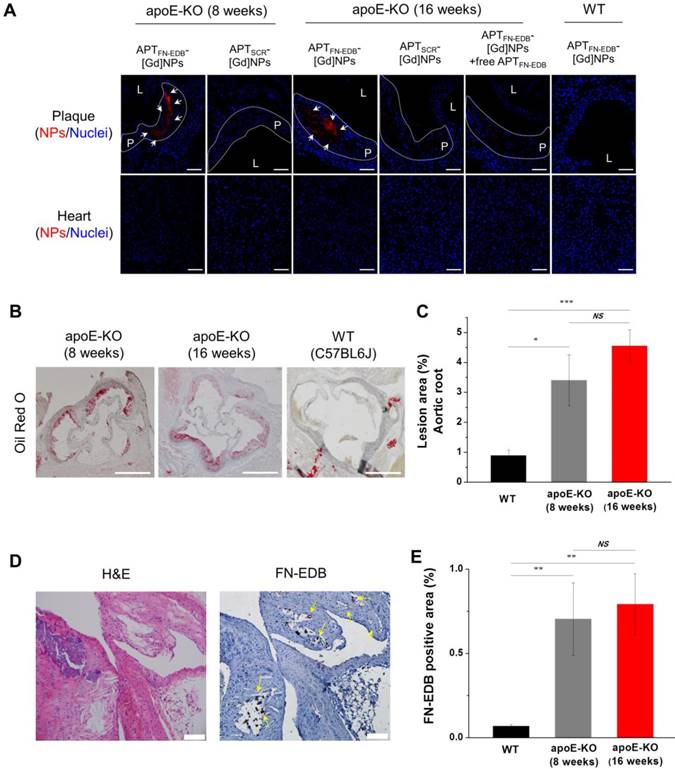
Twenty-four hours after in vivo MRI, the aorta and other organs were harvested for further analysis. We first quantified the amounts of Gd in the collected ascending-to-descending thoracic aortas of mice treated with NPs by ICP-MS (Figure 4H). We found that the CNRWL values obtained in MRI were in good agreement with the amount of Gd in the thoracic aortas of the apoE-KO mice on WD and WT mice treated with the NPs. The Gd concentration in the aortas from apoE-KO mice treated with APTFN-EDB-[Gd]NPs was approximately 2-fold higher than those treated with APTSCR-[Gd]NPs or an excess of free APTFN-EDB prior to injection of APTFN-EDB-[Gd]NPs (P < 0.05). On the other hand, WT mice treated with APTFN-EDB-[Gd]NPs showed diminished accumulation of Gd in aortas (P < 0.001), suggesting that APTFN-EDB decorated on the surface of NPs results in substantial accumulation of the NPs in the atherosclerotic lesions. Second, we found that the targeted accumulation of APTFN-EDB-[Gd]NPs in atherosclerotic lesions was more evident in confocal microscopic analyses (Figure 5A). Strong fluorescence signals from the rhodamines of NPs were observed in the atherosclerotic plaques of apoE-KO mice (both 8- and 16-wk WD) treated with APTFN-EDB-[Gd]NPs, whereas relatively little or no fluorescence was observed in plaques treated with APTSCR-[Gd]NPs or the competition regimen. Nakashiro et al. reported that non-targeting poly(lactic-co-glycolic-acid) NPs were found in circulating monocytes and aortic macrophages [49], but immunostaining of the atherosclerotic plaques of apoE-KO mice injected with Cy5.5-labeled APTFN-EDB-NPs revealed that accumulation of APTFN-EDB-NPs in the plaque was substantially co-localized with FN-EDB expression (Figure S8, arrows) rather than with endothelial cell or macrophage expression, demonstrating that the NP accumulation in plaque was mainly attributed to FN-EDB targeting. In addition to being distributed in plaques, APTFN-EDB-[Gd]NPs also localized in other organs including liver, but barely accumulated in heart (Figure S9 and Figure 5A). Third, we further quantified the gross lesion areas of aortic roots in apoE-KO and WT mice by Oil red O (ORO) staining (Figure 5B-C). Quantitative analysis revealed that atherosclerotic lesion areas (%) in apoE-KO mice on 8- and 16-wk WD were significantly larger than those in WT mice. We also found that the size of lesions was similar in apoE-KO mice between 8- and 16-wk WD groups, supporting the in vivo plaque-detection capability of APTFN-EDB-[Gd]NPs. Fourth, we assessed FN-EDB expression in plaques from untreated apoE-KO mice. These plaques showed the presence of cholesterol crystal clefts, a sign of progressive atherosclerotic lesions, and substantial FN-EDB expression around the lesions (Figure 5D-E). In contrast, WT mice exhibited no FN-EDB expression in the corresponding region. Together, the considerable MRI contrast enhancement, concentration of Gd, fluorescence intensity, and histological staining found in the atherosclerotic aortas of mice treated with APTFN-EDB-[Gd]NPs indicate the aptide-targeting, ligand-mediated, specific accumulation of NPs in atherosclerotic plaques. Dysfunctional endothelia, including those with large gaps and microvascular permeability derived from the vasa vasorum around the plaques, allow NPs to extravasate from the bloodstream [14,50] and remain locally owing to specific binding to FN-EDB expressed within the plaques.
APTFN-EDB-[Gd]NPs accumulate within atherosclerotic plaques through FN-EDB targeting. (A) Representative confocal microscopy images of DAPI-stained aortic sinus and heart sections showing NPs (red) and nuclei (blue). Atherosclerotic plaques of apoE-KO mice are outlined (white dotted line). The apoE-KO mice (fed a WD for 8 or 16 wk) injected with APTFN-EDB-[Gd]NPs exhibited substantial plaque accumulation (arrows), whereas such accumulation was not observed in corresponding sections from WT mice or mice injected with APTSCR-[Gd]NPs or pre-injected with free APTFN-EDB. Scale bar = 60 µm. L and P indicate lumen and plaque, respectively. (B-E) Characterization of plaques in apoE-KO mice, and comparison with normal arteries of WT mice. (B) Representative images of Oil red O-stained atherosclerotic lesions in the aortic root and corresponding regions of WT mice. Scale bar = 500 µm. (C) Quantitative analysis shows percentages of lesion areas in WT (n = 8) and apoE-KO mice (n = 10 per group). (D) Expression of FN-EDB in atherosclerotic plaques was investigated in aortic arches of untreated apoE-KO mice by immunohistochemical staining using biotinylated APTFN-EDB. Scale bar = 100 µm. Arrows indicate FN-EDB-positive areas. (E) Quantitative analysis shows percentages of FN-EDB-positive areas in WT (n = 7) and apoE-KO mice (n = 4 for 8 wk of WD and n = 7 for 16 wk of WD). *P<0.05, **P<0.01, ***P<0.001. NS, not significant.
APTFN-EDB-NP-mediated delivery enhances the circulation half-life and plaque accumulation of a model drug
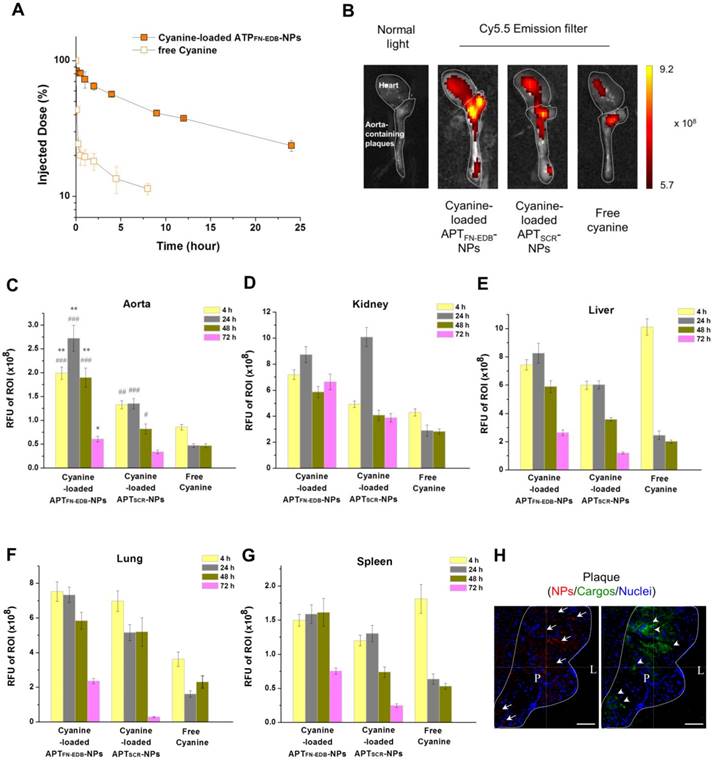
Finally, we examined the suitability of APTFN-EDB-NPs as a drug carrier for targeted delivery to atherosclerotic plaques. As a model small molecule drug, the fluorescent compound cyanine (MW: 716.31 Da) was loaded into APTFN-EDB-NPs. Circulation in the blood and the biodistribution of cargo-loaded APTFN-EDB-NPs were evaluated in healthy BALB/c mice and apoE-KO mice. Upon i.v. injection, the parent cyanine exhibited rapid clearance from the blood, with a half-life (t1/2) of ~50 min, whereas cyanine-loaded APTFN-EDB-NPs showed a significantly extended blood circulation time, with a t1/2 of ~6 h (Figure 6A). Our previous pharmacokinetics studies indicated that the half-life of therapeutics encapsulated in non-targeted nanoparticles was not significantly different from that of therapeutics encapsulated in targeted nanoparticles when the nanoparticle surfaces were composed of the same components except for a targeting ligand [51]. Rather, surface coating results in remarkable differences in half-life [52]. For example, while siRNAs encapsulated in DSPE-PEG5K- and DSPE-PEG3K-coated NPs indicated similar t1/2 values of 8.1 h and 7.1 h, respectively, those encapsulated in ceramide-PEG5K-coated NPs exhibited significantly shorter t1/2 (~30 min). Since the surface-coating materials used for APTSCR-NPs are DSPE-PEG1000, ligand-conjugated DSPE-PEG2000, and DLPC, with the same molar ratios used for APTFN-EDB-NPs, we would expect that the t1/2 of cyanine encapsulated in APTSCR-NPs (as a model drug) would be similar to that of cyanine encapsulated in APTFN-EDB-NPs. In addition to significantly improving its pharmacokinetic characteristics, cyanine-loaded APTFN-EDB-NPs also showed substantial cyanine accumulation in plaque-containing aortas by near-infrared (NIR) fluorescence imaging, exhibiting 2.2-fold and 6.9-fold higher intensity than the same cargo-loaded APTSCR-NPs and free cyanine, respectively, at 24 h post injection (**P < 0.0005, vs. APTSCR-NPs; ###P < 0.00001, vs. free cyanine) (Figure 6B-C). These results support the ability of APTFN-EDB-NPs to detect plaques and enable targeted drug delivery to the disease lesion. In particular, the cyanine intensity from the APTFN-EDB-NPs in aortas gradually decreased to 21% over 72 h (Figure 6C) despite the stable profile of the NPs shown under physiological conditions (Figure 4B); thus, therapeutic strategies will need to consider an injection schedule of every 3 or 4 days. Confocal microscopy analyses of the aortic roots further revealed that the cyanine cargo was distributed around areas within the atherosclerotic plaque occupied by APTFN-EDB-NPs (Figure 6H). We also observed higher cargo accumulation in mice treated with cargo-loaded APTSCR-NPs than in those treated with free cargo, which was not found in the MRI results within the time frame we studied. Non-specifically accumulated NIR fluorochrome (cyanine) in plaque areas after delivery through the microvasculature may be detected with better sensitivity compared to detection under MRI [53,54]. A biodistribution study showed relatively higher uptake of the cargo in the liver, lung, and kidney compared with the spleen at earlier time points (Figure 6D-G). Cargo in the liver and lung were substantially cleared within ~72 h, whereas it continuously remained in the kidney, suggesting renal excretion as a major clearance route.
APTFN-EDB-NPs prolong circulation time of a model drug and enhance drug accumulation in plaques. (A) Blood circulation profile of cyanine cargo-loaded APTFN-EDB-NPs and free cyanine after intravenous administration. Cargo in blood samples collected at each time point was detected using near infrared fluorescence measurement (n = 3 per time point for each group). (B) Ex vivo fluorescence images of representative aortas obtained from apoE-KO mice and injected with cyanine-loaded APTFN-EDB-NPs, APTSCR-NPs, or free cyanine. Images were obtained 4 h post injection. Heart and aorta containing plaques in each group are outlined (white dotted line). Intensiometric scale units: (photons/s)/(μW/cm2). (C-G) In vivo accumulation of cyanine cargo-loaded APTFN-EDB-NPs, APTSCR-NPs, and free cyanine in aorta, kidney, liver, lung, and spleen (n = 3 per time point for each group). *P<0.005, **P<0.0005, versus corresponding time points for cyanine-loaded APTSCR-NPs; #P<0.01, ##P<0.001, ###P<0.00001, versus corresponding time points for free cyanine groups. (H) Cross-sectional view of plaque region (outlined with white dotted line) in apoE-KO mice injected with cyanine-loaded APTFN-EDB-NPs shows NPs (red, arrow), cyanine (green, arrow head), and nuclei (blue). Scale bar = 60 μm. L and P indicate lumen and plaque, respectively.
Although determining the therapeutic response to drug-loaded APTFN-EDB-NPs was not among our study objectives, the superior ability of our FN-EDB-targeting platform technology to identify atherosclerotic lesions as well as its pharmacokinetic advantages drove successful payload delivery to plaques, suggesting an important novel means for identifying and locally treating early stages of atherosclerosis. Polymeric and lipid-based nanomedicines, as primary classes of commercialized NPs, have shown promise for delivering various therapeutics and have reached clinical-stage evaluation [19]. Additionally, those results suggest effective strategies for delivery of emerging therapeutic candidates, including RNA therapeutics (siRNA, mRNA, miRNA, etc.) and CRISPR (clustered, regularly interspaced, short palindromic repeats)/CRISPR-associated protein 9 (Cas9) systems [55-58].
Conclusions
We have for the first time i) identified FN-EDB intensified expression in AHA human plaque types III, IV, and V lesions; ii) developed a novel FN-EDB-specific NP platform capable of plaque identification; ultimately achieving iii) efficient payload delivery to atherosclerotic plaques. Our findings suggest that our FN-EDB-targeting platform and its unique features contributed by nanotechnology offers a comprehensive strategy for personalized medicine, in which diagnosis could guide therapeutic procedures and estimate local therapy. Since mouse and human FN-EDB domains are identical homologues [27], we anticipate that translatability of this study will be high. We envision that a platform consisting of a polymeric core and an FN-EDB-specific lipid surface, targeting particular phenotypes of progressive atherosclerotic lesions, in tandem with the nanotechnological approach described here, may offer exciting possibilities for better stratification of patients at risk and guiding optimal treatments using appropriate therapeutic agents.
Methods
Materials
Poly(D,L-lactide) with terminal ester groups (PLA-OCH3, inherent viscosity 0.26-0.54 dL/g in chloroform), was purchased from Durect Lactel Absorbable Polymers (Pelham, AL, USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-1000] (DSPE-mPEG1000), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000-maleimide), 1,2-dimyristoyl-snglycero-3-phosphoethanolamine-N-diethylenetriaminepentaacetic acid (gadolinium salt) (DMPE-DTPA (Gd)), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (18:1 Liss Rhod PE), 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(Cyanine 5.5) (18:1 Cy5.5 PE) were obtained from Avanti Polar Lipids (Alabaster, AL, USA). Cy5.5-NHS monoester was purchased from GE Healthcare Life Sciences (Pittsburgh, PA, USA). Dimethyl sulfoxide (DMSO), dimethylformamide (DMF), chloroform, methanol, benzyl alcohol, and ethyl acetate were purchased from Sigma-Aldrich (St. Louis, MO, USA). The FN-EDB-targeting bipodal peptides, APTFN-EDB (SSSPIQGSWTWENGK(Cys)WTWKGIIRLEQ) and biotin-labeled APTFN-EDB (SSSPIQGSWTWENGK(biotin)WTWKGIIRLEQ; >95% purity) and corresponding scrambled peptide controls, APTSCR (HASDRNGSWTWENGK(Cys)WTWKGLHEQSD) and biotin-labeled APTSCR (HASDRNGSWTWENGK(biotin)WTWKGLHEQSD; >95% purity) were purchased from AnyGen Co., Ltd. (Gwangju, South Korea). NP sizes and ζ-potentials were obtained by quasielectric laser light scattering using a ZetaPALS dynamic light-scattering (DLS) detector (15 mW laser, incident beam ¼ 676 nm; Brookhaven Instruments (Holtsville, NY, USA). Matrix-assisted laser desorption/ionization time of flight (MALDI TOF) mass spectrometry was performed on a Bruker Dalton system.
Histological analyses
Human atherosclerotic aortas and carotid arteries were obtained at autopsy from patients with atherosclerosis (n = 30, male and female, age 40-85 y), following a post-mortem interval shorter than 24 h. As a control group, normal (non-atherosclerotic) arteries were similarly obtained from patients (n = 10, male and female, age 40-85 y). The autopsies were performed at Brigham and Women's Hospital (BWH) following typical procedures; specimens for study were acquired according to an approved Institutional Review Board protocol. Collected tissues (2-6 pieces per patient) were immediately fixed in formalin fixative and stored in 70% ethanol solution until use; fixed tissue samples were subsequently cut into serial 5-μm paraffin cross-sections for further analysis. Additional paraffin cross-sections of human atherosclerotic arteries (n = 22) and normal arteries (n = 4) were obtained from Origene (Rockville, MD, USA). Tissues were classified by an experienced cardiovascular pathologist (F.J.S.) into atherosclerotic lesion type according to criteria described in an American Heart Association (AHA) consensus document [40]. For immunohistological characterization, all available sections were treated with an antigen retrieval solution, peroxidase blocking solution, and avidin/biotin blocking solution (DAKO, CA, USA). The sections were then incubated with biotin-labeled APTFN-EDB or APTSCR (50 μg/mL in PBS) for 1 h, treated with VECTASTAIN ABC kit (Vector labs, CA, USA) for 30 min, visualized with 3,3'-diaminobenzidine chromogen substrate (DAKO), and counterstained with Mayer's hematoxylin. For antibody staining, consecutive sections were incubated with primary antibodies against CD68 (1:500 dilution; Abcam, MA, USA), HIF1α (1:800 dilution; Abcam), vWF (1:20 dilution; Abcam), or fibronectin (1:200 dilution; Abcam). All stained cross-sections were digitally imaged with an MVX10 MacroView Dissecting Scope (with a 1.25× lens) or an Olympus BX63 automated microscope (with 20× and 40× lenses). Fiji software was used for quantitative analysis of immunopositive areas. Five hot spots—regions of strong expression—on each stained cross-section were identified under the BX63 microscope, photographed at a magnification of 200× (2,070 × 1,548 pixels), and expressed as the average percentage of total tissue area obtained per image.
Serial cross-sections of the aortic root area, aortic arches, and other major organs of WT and apoE-KO mice were cut to a thickness of 5 μm. Lesion areas at the aortic root were visualized by fat staining (Oil red O), photographed with an Olympus BX63 automated microscope at a magnification of 400× (2,070 × 1,548 pixels), and quantitatively analyzed. Aortic arch sections of untreated apoE-KO mice fed a WD were stained with biotin-labeled APTFN-EDB and analyzed as described for human plaque studies.
Synthesis and purification of APTFN-EDB-DSPE-PEG2000 and APTSCR-DSPE-PEG2000 conjugates
DSPE-PEG2000-maleimide (50 mg, 0.017 mmol), dissolved in chloroform, was added to cysteine-inserted APTFN-EDB (50 mg, 0.016 mmol) or APTSCR (50 mg, 0.016 mmol), previously dissolved in dry DMF. The reaction was stirred at room temperature for 24 h, and the final products, APTFN-EDB-DSPE-PEG2000 and APTSCR-DSPE-PEG2000 conjugates, were purified by high-performance liquid chromatography (HPLC) on a TMS-250 C1 column (4.6 × 75 mm) at a flow rate of 0.3 mL/min using a gradient of 20% A (0.05% TFA, 100% water)/80% B (0.043% TFA, 80% ACN) to 100% B for 60 min after running initially at 20% A/80% B for 15 min (yield: 70~80%). Purified conjugates were characterized by MALDI TOF mass spectrometry, yielding values of 6189.9 Da (calculated, 6117.5 Da) for the APTFN-EDB-DSPE-PEG2000 conjugate and 6142.3 Da (calculated, 6155.4 Da) for the APTSCR-DSPE-PEG2000 conjugate.
Synthesis and characterization of APTFN-EDB-NPs
PLA-OCH3 was first dissolved in a mixture of ethyl acetate and benzyl alcohol (40 mg/mL). For optimization of ligand density, different DSPE-mPEG1000:DLPC:APTFN-EDB-DSPE-PEG2000 molar ratios (69.5:29.5:1, 69:28.5:2.5, and 67.5:27.5:5) were then prepared by dissolving separately in a mixture of ethyl acetate and benzyl alcohol (40 mg/mL) at 70% of the PLA weight, after which 18:1 Liss Rhod PE (0.5 wt% of total polymer/lipid mixture) was added to the solution. The PLA/lipid mixture in a co-solvent of ethyl acetate and benzyl alcohol was then added drop-wise into an aqueous solution (molecular biology-grade water; Hyclone) with gentle stirring and then sonicated with a probe. NPs were then allowed to self-assemble overnight with continuous stirring while the organic phase was allowed to evaporate. The remaining organic solvent and free molecules were removed by washing the NP solution three times using an Amicon Ultra-15 centrifugal filter (EMD Millipore, Darmstadt, Germany) with a molecular weight cut-off of 100 kDa, and then resuspended in either water or PBS to obtain the final desired concentration. NPs were used immediately or stored at 4 °C. APTSCR-NPs were synthesized using the same procedure. Hydrodynamic size and surface charge of NPs were measured in PBS by DLS using a ZetaPALS system (Brookhaven). Human atherosclerotic and normal artery tissues were rehydrated, heated in 10 mM sodium citrate for antigen retrieval, treated with 0.1% sudan black solution (in 70% ethanol), and stained with APTFN-EDB-NPs or APTSCR-NPs for determination of the optimal NP ligand density. Tissue sections were mounted in Vectorshield DAPI-containing mounting medium (Vectorlabs) and analyzed using an Olympus FV1000 confocal microscope.
Synthesis and characterization of APTFN-EDB-[Gd]NPs
PLA-OCH3 was first dissolved in a mixture of ethyl acetate and benzyl alcohol (40 mg/mL). DSPE-mPEG1000, DLPC, DMPE-DTPA-Gd (pre-dissolved in chloroform:methanol, 9:1 v/v), and APTFN-EDB-DSPE-PEG2000 (70:9:20:1, molar ratio) were then dissolved separately in a mixture of ethyl acetate and benzyl alcohol (40 mg/mL) at 70% of the PLA weight, after which 18:1 Liss Rhod PE (0.5 wt% of total polymer/lipid mixture) was added to the mixture, and samples were processed as described for APTFN-EDB-NPs in the previous section. APTSCR-[Gd]NPs were synthesized following the same procedure. Gd concentration was analyzed using inductively coupled plasma mass spectrometry (ICP-MS). The relaxivities of NPs were analyzed using an MRI phantom study. NPs at different concentrations (0-2 mM Gd) were dispersed in PBS in 1.5 mL microcentrifuge tubes, which were scanned on a 7.0 T MRI system (Bruker Instruments, Germany). T1 relaxation time was measured using the following parameters: TR = 50, 100, 150, 250, 400, 750, 1,500, 3,000, 5,000, and 10,000 ms; TE = 10.7 ms; flip angle = 180°; field-of-view = 50 mm × 80 mm; slice thickness = 2 mm. The stability of NPs in PBS and in 10% FBS was tested at 37 °C with shaking (100 rpm) following various incubation times (0 to 72 h), and changes in the hydrodynamic size of NPs were measured by DLS.
Animals and diet
Male apoE-KO mice (B6.129P2-Apoetm1Unc) 5-6 weeks old, purchased from Jackson laboratory, were fed a Western-type diet (WD) containing 20.3% fat, 22.9% protein, 45.7% carbohydrate, and 0.2% cholesterol for 8 or 16 wk (Research Diets Inc., NJ, USA). At each end point, mice were scanned by T1-weighted MRI and analyzed. WT mice (C57BL/6J, 20-wk-old, male) fed a normal chow diet were used as controls. All animal procedures followed a protocol approved by a standing Institutional Animal Care and Use Committee at Harvard Medical School.
In vivo MRI
Brachiocephalic arteries from mice were visualized with a 7.0 T MRI system (Bruker). Mice were anesthetized with a 4% isoflurane/O2 mixture and maintained with a 1.5% isoflurane/O2 mixture delivered through a nose cone. A respiratory sensor connected to a monitoring system was placed on the back to monitor the rate and depth of respiration. A constant body temperature of 37 °C was maintained using a thermocouple heating system. The brachiocephalic arteries were identified in an axial section on a localizing sequence. T1-weighted MRI was performed using a black blood IntraGate FLASH sequence with a navigator echo and retrospective gating. Imaging parameters were as follows: hermite-shaped RF pulse; repetition time/echo time, 20/2.112 ms; 10 cardiac frames; field of view, 12.8 × 12.8 mm2; matrix, 160 × 160; slice thickness, 0.6 mm; spatial resolution, 0.08 mm per pixel; number of repetitions, 200.
Plaque targeting of NPs was investigated by first subjecting apoE-KO mice to a baseline MRI scan for precontrast images, followed by injection of a 50 μmol Gd/kg dose of either APTFN-EDB-[Gd]NPs or APTSCR-[Gd]NPs via the tail vein. The in vivo specificity of APTFN-EDB-[Gd]NPs was confirmed by pre-injecting mice with 100 mg/kg free APTFN-EDB (SSSPIQGSWTWENGKWTWKGIIRLEQ; >95% purity) via the tail vein 2 h prior to injection of APTFN-EDB-[Gd]NPs. Gd-DTPA (Magnevist; Bayer Schering AG, Germany), a standard, nonspecific MRI contrast agent, was intravenously injected as a control. WT mice were injected with equivalent doses of APTFN-EDB-[Gd]NPs, APTSCR-[Gd]NPs, or Gd-DTPA. Postcontrast MRI of APTFN-EDB-[Gd]NPs and APTSCR-[Gd]NPs was performed 4-5 h post injection, and that of Gd-DTPA was performed 30 min post injection. Black blood images with the lowest signal intensity at the brachiocephalic arteries were used for quantitative analysis of MRI results. Signal intensities of regions of interest (ROIs) were measured on the aortic wall with four points in four quadrants of the brachiocephalic artery on each slice. Signal intensities of the aortic lumen and muscle were also measured in each slice. The standard deviation of noise (SDnoise) was recorded in tracheal air for each slice. The contrast-to-noise of aortic wall-to-lumen (CNRWL) was determined by the formula CNRWL = [(W1 + W2 + W3 + W4)/4 - L]/SDnoise, where Wn is the signal intensity value of the aortic wall in quadrant n, and L is the signal intensity of the lumen. The normalized enhancement ratio (NER) and normalized percent change (NER %) were used to measure the normalized signal intensity (SI) enhancement of the aortic wall after the administration of Gd-DTPA, APTSCR-[Gd]NPs, or APTFN-EDB-[Gd]NPs. NER was calculated by the following formula.
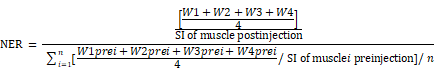
NER was normalized as NER % = (NER-1) × 100.
Twenty-four hours post injection; animals were sacrificed and exsanguinated by perfusion of heparinized saline (10 mL) through the heart. Aortas (from ascending aorta to descending thoracic aorta) from mice injected with APTFN-EDB-[Gd]NPs or APTSCR-[Gd]NPs were isolated and fixed for ICP-MS analysis. Aortic roots containing the aortic sinus and other major organs (liver, heart, lung, spleen, and kidney) from all groups were frozen in OCT for sectioning. Immunostaining of frozen cross sections was performed using a rat monoclonal anti-CD31 (endothelial cells staining, BD Pharmingen), a mouse anti-FN-EDB (Abcam), and a rabbit anti-Mac2 (macrophage staining, Abcam) at concentrations of 1:50, 1:200, and 1:50, respectively. Cy5.5-labeled APTFN-EDB NPs were prepared using a similar synthesis as that described for APTFN-EDB NPs using 18:1 Cy5.5 PE for co-localization study. Aortic roots of apoE-KO mice injected with Cy5.5-labeled APTFN-EDB NPs were harvested 24 h post-injection for immunofluorescence study. In situ images were captured digitally using a BX61WI microscope coupled with a Fluoview FV1000 confocal unit (Olympus) equipped with an Olympus DP72 camera and running Fluoview 10-ASW Software (Olympus) for confocal microscopy.
Confocal laser-scanning fluorescence microscopy
NPs injected into mice were visualized in each tissue section with an Olympus FV1000 confocal microscope. Frozen sections were washed with PBS, fixed in a 4% paraformaldehyde solution, and washed three times with PBS. All sections were mounted with DAPI-containing Vectashield, covered with cover slips (protected from light), and kept at -20 °C until laser-scanning confocal fluorescence microscopy imaging was performed. Images were acquired within 48 h using a 20×/0.75 NA objective lens.
In vivo biodistribution studies
Cyanine-loaded APTFN-EDB-NPs were prepared by adding 1.7 mg of Cy5.5-NHS monoester (10 wt% of total feed materials, 80 mg/mL in DMSO), as a model cargo, to the PLA/lipid solution. Cyanine-loaded APTFN-EDB-NPs and APTSCR-NPs were prepared as described above for other NPs. ApoE-KO mice fed a WD were administered cyanine-loaded APTFN-EDB-NPs, cyanine-loaded APTSCR-NPs, or free cyanine via tail vein injection. At each time point (4, 24, 48, and 72 h; n = 3 per time point) mice were sacrificed, and their aortas, livers, spleens, lungs, and kidneys were collected and imaged with an IVIS Lumina system (Xenogen, USA) using 605 nm excitation and Cy5.5 emission filters. Photon counts were used to quantify the fluorescence intensity in each tissue at each time point. Images were processed with Living Image 4.5.2 integrated software (Xenogen). Aortic roots from mice injected with cargo-loaded NPs were obtained 4 h post injection and frozen in OCT for sectioning.
Pharmacokinetics
Male BALB/c mice 5-6 weeks old (n = 3 per group) were intravenously injected with either cyanine-loaded APTFN-EDB-NPs or free cyanine through the tail vein. At different time points, blood samples were collected retro-orbitally and the fluorescence intensity of each sample was measured on a microplate reader (Infinite 200 PRO; TECAN, Männedorf, Switzerland) using an NIR filter. Different amounts of NPs or free cyanine were added to blood obtained from untreated mice and their fluorescence intensities were measured to generate standard curves.
Statistical analyses
Values are expressed as mean ± SD for size, zeta-potential, and relaxivity results, and mean ± SEM for the others. Statistical significance of differences was determined using a one-way analysis of variance (ANOVA) with a post hoc Tukey's test. Correlations between FN-EDB and CD68, vWF, or HIF1α were determined based on apparent fits obtained by linear analysis using Pearson correlation. P-values < 0.05 were considered statistically significant.
Abbreviations
apoE-KO: apolipoprotein E-deficient; APTFN-EDB: FN-EDB-specific aptide; APTFN-EDB-NPs: FN-EDB-targeting NPs; APTFN-EDB-[Gd]NPs: APTFN-EDB-NPs incorporating DTPA(Gd); APTSCR: scrambled aptide; CNRWL: MRI signal intensities in the aortic wall-to-lumen; ECM: extracellular matrix; EPR: enhanced permeability and retention; FBS: fetal bovine serum; FN-EDB: extra domain B of fibronectin; Gd: gadolinium; HIF1α: hypoxia-inducible factor 1α; IHC: immunohistochemistry; MRI: magnetic resonance imaging; NER: normalized enhancement ratio; NIR: near-infrared; NPs: nanoparticles; ORO: Oil red O; PEG: polyethylene glycol; PLA: poly(D,L-lactide); scFv: single-chain Fv; TCFA: thin-cap fibroatheroma; vWF: von Willebrand factor; WD: western diet; WT: wild-type.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the NHLBI Program of Excellence in Nanotechnology (PEN) contract HHSN268201000045C from the National Heart, Lung, and Blood Institute (O.C.F.); the National Institute of Health (NIH) HL127464 (O.C.F.); the Global Research Laboratory (GRL) (NRF-2012K1A1A2045436) (S.J. and O.C.F.) and Basic Science Research Program (NRF-2018R1A3B1052661) (S.J.) through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT; the David Koch-Prostate Cancer Foundation Award in Nanotherapeutics (O.C.F.); and the American Heart Association (AHA) 18CSA34080399 (O.C.F., P.L., and M.Y.). P.L. was supported by the Heart, Lung, and Blood Institute (HL080472) and the RRM Charitable Fund.
Competing Interests
O.C.F. declares financial interests in Selecta Biosciences, Tarveda Therapeutics, Placon Therapeutics, and Seer Biosciences.
References
1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R. et al. Heart disease and stroke statistics-2017 update: a report from the American heart association. Circulation. 2017;135:e146-e603
2. Shah PK. Screening asymptomatic subjects for subclinical atherosclerosis: can we, does it matter, and should we? J Am Coll Cardiol. 2010;56:98-105
3. Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS. et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364:226-35
4. Tarkin JM, Joshi FR, Rudd JH. PET imaging of inflammation in atherosclerosis. Nat Rev Cardiol. 2014;11:443-57
5. Moreno PR, Bernardi VH, López-Cuéllar J, Murcia AM, Palacios IF, Gold HK. et al. Macrophages, smooth muscle cells, and tissue factor in unstable angina. Implications for cell-medicated thrombogenicity in acute coronary syndromes. Circulation. 1996;94:3090-7
6. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852-66
7. Crea F, Libby P. Acute coronary syndromes: the way forward from mechanisms to precision treatment. Circulation. 2017;136:1155-66
8. Rogers IS, Nasir K, Figueroa AL, Cury RC, Hoffmann U, Vermylen DA. et al. Feasibility of FDG imaging of the coronary arteries: comparison between acute coronary syndrome and stable angina. JACC Cardiovasc Imaging. 2010;3:388-97
9. Rudd JH, Narula J, Strauss HW, Virmani R, Machac J, Klimas M. et al. Imaging atherosclerotic plaque inflammation by fluorodeoxyglucose with positron emission tomography: ready for prime time? J Am Coll Cardiol. 2010;55:2527-35
10. Makowski MR, Wiethoff AJ, Blume U, Cuello F, Warley A, Jansen CH. et al. Assessment of atherosclerotic plaque burden with an elastin-specific magnetic resonance contrast agent. Nat Med. 2011;17:383-8
11. Tahara N, Mukherjee J, de Haas HJ, Petrov AD, Tawakol A, Haider N. et al. 2-deoxy-2-[18F]fluoro-D-mannose positron emission tomography imaging in atherosclerosis. Nat Med. 2014;20:215-9
12. Irkle A, Vesey AT, Lewis DY, Skepper JN, Bird JL, Dweck MR. et al. Identifying active vascular microcalcification by (18)F-sodium fluoride positron emission tomography. Nat Commun. 2015;6:7495
13. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36-46
14. Duivenvoorden R, Tang J, Cormode DP, Mieszawska AJ, Izquierdo-Garcia D, Ozcan C. et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065
15. Lee JH, Huh YM, Jun YW, Seo JW, Jang JT, Song HT. et al. Artificially engineered magnetic nanoparticles for ultra-sensitive molecular imaging. Nat Med. 2007;13:95-9
16. Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater. 2014;13:125-38
17. Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16:1035-41
18. Miller MA, Gadde S, Pfirschke C, Engblom C, Sprachman MM, Kohler RH. et al. Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci Transl Med. 2015;7:314ra183
19. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
20. Silencing inflammatory activity by injecting nanocort in patients at risk for atherosclerotic disease (SILENCE). May 21, 2012. https://clinicaltrials.gov/ct2/show/NCT01601106
21. A proof of concept study to determine the local delivery and efficacy of nanocort (DELIVER). July 23, 2012. https://clinicaltrials.gov/ct2/show/NCT01647685
22. Van der Valk FM, van Wijk DF, Lobatto ME, Verberne HJ, Storm G, Willems MC. et al. Prednisolone-containing liposomes accumulate in human atherosclerotic macrophages upon intravenous administration. Nanomedicine. 2015;11:1039-46
23. Lobatto ME, Fuster V, Fayad ZA, Mulder WJ. Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat Rev Drug Discov. 2011;10:835-52
24. Kheirolomoom A, Kim CW, Seo JW, Kumar S, Son DJ, Gagnon MK. et al. Multifunctional nanoparticles facilitate molecular targeting and miRNA delivery to inhibit atherosclerosis in apoE(-/-) mice. ACS Nano. 2015;9:8885-97
25. Beldman TJ, Senders ML, Alaarg A, Pérez-Medina C, Tang J, Zhao Y. et al. Hyaluronan nanoparticles selectively target plaque-associated macrophages and improve plaque stability in atherosclerosis. ACS Nano. 2017;11:5785-99
26. White ES, Baralle FE, Muro AF. New insights into form and function of fibronectin splice variants. J Pathol. 2008;216:1-14
27. Zardi L, Carnemolla B, Siri A, Petersen TE, Paolella G, Sebastio G. et al. Transformed human cells produce a new fibronectin isoform by preferential alternative splicing of a previously unobserved exon. EMBO J. 1987;6:2337-42
28. Kaspar M, Zardi L, Neri D. Fibronectin as target for tumor therapy. Int J Cancer. 2006;118:1331-9
29. A phase I/II dose escalation study of the tumor-targeting human L19-IL2 monoclonal antibody-cytokine fusion protein in combination with dacarbazine for patients with metastatic melanoma. October 31, 2016. https://clinicaltrials.gov/ct2/show/NCT02076646
30. Matter CM, Schuler PK, Alessi P, Meier P, Ricci R, Zhang D. et al. Molecular imaging of atherosclerotic plaques using a human antibody against the extra-domain B of fibronectin. Circ Res. 2004;95:1225-33
31. Dietrich T, Perlitz C, Licha K, Stawowy P, Atrott K, Tachezy M. et al. ED-B fibronectin (ED-B) can be targeted using a novel single chain antibody conjugate and is associated with macrophage accumulation in atherosclerotic lesions. Basic Res Cardiol. 2007;102:298-307
32. Dietrich T, Berndorff D, Heinrich T, Hucko T, Stepina E, Hauff P. et al. Targeted ED-B fibronectin SPECT in vivo imaging in experimental atherosclerosis. Q. J. Nucl. Med. Mol. Imaging. 2015;59:228-37
33. Kim S, Kim D, Jung HH, Lee IH, Kim JI, Suh JY. et al. Bio-inspired design and potential biomedical applications of a novel class of high-affinity peptides. Angew Chem Int Ed Engl. 2012;51:1890-4
34. Xiang D, Zheng C, Zhou SF, Qiao S, Tran PH, Pu C. et al. Superior performance of aptamer in tumor penetration over antibody: implication of aptamer-based theranostics in solid tumors. Theranostics. 2015;5:1083-97
35. Eyetech Study Group. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina. 2002;22:143-52
36. Ireson CR, Kelland LR. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006;5:2957-62
37. Lao YH, Phua KK, Leong KW. Aptamer nanomedicine for cancer therapeutics: barriers and potential for translation. ACS Nano. 2015;9:2235-54
38. Kim H, Lee Y, Kang S, Choi M, Lee S, Kim S. et al. Self-assembled nanoparticles comprising aptide-SN38 conjugates for use in targeted cancer therapy. Nanotechnology. 2016;27:48LT01
39. Kim H, Lee Y, Lee IH, Kim S, Kim D, Saw PE. et al. Synthesis and therapeutic evaluation of an aptide-docetaxel conjugate targeting tumor-associated fibronectin. J Control Release. 2014;178:118-24
40. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr. et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, American Heart Association. Circulation. 1995;92:1355-74
41. Bencharit S, Cui CB, Siddiqui A, Howard-Williams EL, Sondek J, Zuobi-Hasona K. et al. Structural insights into fibronectin type III domain-mediated signaling. J Mol Biol. 2007;367:303-9
42. Murphy PA, Hynes RO. Alternative splicing of endothelial fibronectin is induced by disturbed hemodynamics and protects against hemorrhage of the vessel wall. Arterioscler Thromb Vasc Biol. 2014;34:2042-50
43. Adams A, Bojara W, Schunk K. Early diagnosis and treatment of coronary heart disease in symptomatic subjects with advanced vascular atherosclerosis of the carotid artery (type III and IV b findings using ultrasound). Cardiol Res. 2017;8:7-12
44. Osborn EA, Jaffer FA. Imaging inflammation and neovascularization in atherosclerosis: clinical and translational molecular and structural imaging targets. Curr Opin Cardiol. 2015;30:671-80
45. Sadat U, Jaffer FA, van Zandvoort MA, Nicholls SJ, Ribatti D, Gillard JH. Inflammation and neovascularization intertwined in atherosclerosis: imaging of structural and molecular imaging targets. Circulation. 2014;130:786-94
46. Mateo J, Izquierdo-Garcia D, Badimon JJ, Fayad ZA, Fuster V. Noninvasive assessment of hypoxia in rabbit advanced atherosclerosis using 18F-fluoromisonidazole positron emission tomographic imaging. Circ Cardiovasc Imaging. 2014;7:312-20
47. Yu M, Amengual J, Menon A, Kamaly N, Zhou F, Xu X. et al. Targeted nanotherapeutics encapsulating liver x receptor agonist GW3965 enhance antiatherogenic effects without adverse effects on hepatic lipid metabolism in ldlr-/- mice. Adv Healthc Mater. 2017;6:1700313
48. Lindsay AC, Choudhury RP. Form to function: current and future roles for atherosclerosis imaging in drug development. Nat Rev Drug Discov. 2008;7:517-29
49. Nakashiro S, Matoba T, Umezu R, Koga J, Tokutome M, Katsuki S. et al. Pioglitazone-incorporated nanoparticle prevent plaque destabilization and rupture by monocyte/macrophage differentiation in apoe-/- mice. Arterioscler Thromb Vasc Biol. 2016;36:491-500
50. Taqueti VR, Di Carli MF, Jerosch-Herold M, Sukhova GK, Murthy VL, Folco EJ. et al. Increased microvascularization and vessel permeability associate with active inflammation in human atheromata. Circ Cardiovasc Imaging. 2014;7:920-9
51. Xu X, Wu J, Liu Y, Saw PE, Tao W, Yu M. et al. Multifunctional envelope-type siRNA delivery nanoparticle platform for prostate cancer therapy. ACS Nano. 2017;11:2618-27
52. Zhu X, Xu Y, Solis LM, Tao W, Wang L, Behrens C. et al. Long-circulating siRNA nanoparticles for prohibitin 1-targeted non-small cell lung cancer treatment. Proc Natl Acad Sci U S A. 2015;112:7779-84
53. Mulder WJ, Jaffer FA, Fayad ZA, Nahrendorf M. Imaging and nanomedicine in inflammatory atherosclerosis. Sci Transl Med. 2014;6:239sr1
54. Han Z, Wu X, Roelle S, Chen C, Schiemann WP, Lu ZR. Targeted gadofullerene for sensitive magnetic resonance imaging and risk-stratification of breast cancer. Nat Commun. 2017;8:692
55. Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U. et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638-46
56. Ramaswamy S, Tonnu N, Tachikawa K, Limphong P, Vega JB, Karmali PP. et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci. U S A. 2017;114:E1941-50
57. Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q. et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34:328-33
58. Xu X, Saw PE, Tao W, Li Y, Ji X, Yu M. et al. Tumor microenvironment-responsive multistaged nanoplatform for systemic RNAi and cancer therapy. Nano Lett. 2017;17:4427-35
Author contact
![]() Corresponding authors: syjonac.kr (S.J.), Email: ofarokhzadharvard.edu (O.C.F.).
Corresponding authors: syjonac.kr (S.J.), Email: ofarokhzadharvard.edu (O.C.F.).