Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(4):1154-1169. doi:10.7150/thno.29146 This issue Cite
Research Paper
Activated platelets in the tumor microenvironment for targeting of antibody-drug conjugates to tumors and metastases
May Lin Yap1,2, James D McFadyen1,3,4, Xiaowei Wang1,3, Melanie Ziegler1, Yung-Chih Chen1, Abbey Willcox1,3,4, Cameron J Nowell5, Andrew M Scott6,7, Erica K Sloan5, P Mark Hogarth2,8,9, Geoffrey A Pietersz1,2,8,9,10*, Karlheinz Peter1,3,9* ![]()
1. Baker Heart and Diabetes Institute, Melbourne, 3004, Australia
2. Department of Clinical Pathology, The University of Melbourne, Melbourne, 3010, Australia
3. Department of Medicine, Monash University, Melbourne, 3800, Australia
4. Department of Hematology, The Alfred Hospital, Melbourne, 3004, Australia
5. Drug Discovery Biology Theme, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, Victoria 3052, Australia
6. Olivia Newton-John Cancer Research Institute, and School of Cancer Medicine, La Trobe University, Melbourne, Victoria, Australia
7. Department of Molecular Imaging and Therapy, Austin Health, and University of Melbourne, Melbourne, Victoria, Australia.
8. Burnet Institute, Melbourne, 3004, Australia
9. Department of Immunology, Monash University, Melbourne, 3800, Australia
10. College of Health and Biomedicine, Victoria University, Melbourne, 3021, Australia
*equally contributing senior authors
Received 2018-8-10; Accepted 2019-1-12; Published 2019-2-7
Abstract

Rationale: Platelets are increasingly recognized as mediators of tumor growth and metastasis. Hypothesizing that activated platelets in the tumor microenvironment provide a targeting epitope for tumor-directed chemotherapy, we developed an antibody-drug conjugate (ADC), comprised of a single-chain antibody (scFv) against the platelet integrin GPIIb/IIIa (scFvGPIIb/IIIa) linked to the potent chemotherapeutic microtubule inhibitor, monomethyl auristatin E (MMAE).
Methods: We developed an ADC comprised of three components: 1) A scFv which specifically binds to the high affinity, activated integrin GPIIb/IIIa on activated platelets. 2) A highly potent microtubule inhibitor, monomethyl auristatin E. 3) A drug activation/release mechanism using a linker cleavable by cathepsin B, which we demonstrate to be abundant in the tumor microenvironment. The scFvGPIIb/IIIa-MMAE was first conjugated with Cyanine7 for in vivo imaging. The therapeutic efficacy of the scFvGPIIb/IIIa-MMAE was then tested in a mouse metastasis model of triple negative breast cancer.
Results: In vitro studies confirmed that this ADC specifically binds to activated GPIIb/IIIa, and cathepsin B-mediated drug release/activation resulted in tumor cytotoxicity. In vivo fluorescence imaging demonstrated that the newly generated ADC localized to primary tumors and metastases in a mouse xenograft model of triple negative breast cancer, a difficult to treat tumor for which a selective tumor-targeting therapy remains to be clinically established. Importantly, we demonstrated that the scFvGPIIb/IIIa-MMAE displays marked efficacy as an anti-cancer agent, reducing tumor growth and preventing metastatic disease, without any discernible toxic effects.
Conclusion: Here, we demonstrate the utility of a novel ADC that targets a potent cytotoxic drug to activated platelets and specifically releases the cytotoxic agent within the confines of the tumor. This unique targeting mechanism, specific to the tumor microenvironment, holds promise as a novel therapeutic approach for the treatment of a broad range of primary tumors and metastatic disease, particularly for tumors that lack specific molecular epitopes for drug targeting.
Keywords: Cancer, Activated platelets, GPIIb/IIIa, Antibody-drug conjugate
Introduction
The mainstay of treatment for many cancers remains systemic chemotherapy. However, this approach is associated with inevitable 'off target' effects of chemotherapy, resulting in systemic toxicity due to the limited selectivity for tumor cells. Recently, antibody-directed chemotherapy has offered a very promising approach to overcome these inherent limitations of systemic chemotherapy by leveraging the targeting capability of antibodies towards tumor-specific antigens or proteins for the delivery of chemotherapy [1]. Consequently, antibody-drug conjugates (ADC) provide the means to deliver high local concentrations of chemotherapy specifically to the tumor whilst avoiding systemic toxicity [2]. The efficacy of ADCs has been demonstrated by the success of trastuzumab emtansine for HER-2 positive breast cancer and brentuximab vedotin in anaplastic large cell lymphoma and Hodgkin lymphoma [3,4], and holds promise in malignancies with limited prognosis such as glioblastoma [5].
The efficacy of ADCs is highly contingent upon the expression and abundance of a specific target epitope on the cancer cells. However, this represents a major limitation for the use of ADCs since in several cancer types, such as triple negative breast cancer, specific molecular targets are lacking or are yet to be determined [6,7]. Here, we develop a unique strategy to overcome this limitation by targeting a specific component of the tumor microenvironment, as opposed to a molecular epitope unique to a specific cancer cell. There is growing evidence demonstrating an intimate relationship between platelets and cancer, and the abundance of activated platelets in the microenvironment of a range of tumors [8-10] including ovarian cancer [11], colorectal cancer [12] and oesophageal cancer [13]. In this regard, platelets express an abundant and specific surface epitope, the integrin glycoprotein (GP)IIb/IIIa (αIIbß3; CD41/CD61). Importantly, on circulating platelets this integrin exhibits an 'unactivated' or low affinity conformation. In contrast, upon platelet activation, GPIIb/IIIa undergoes a confirmation change such that it adopts a high affinity conformation for ligand binding [14]. Indeed, we recently provided proof of concept that the high affinity conformation of GPIIb/IIIa on activated platelets can serve as a molecular targeting epitope for the tumor microenvironment in several types of cancers in mice and humans [15]. Therefore, targeting activated platelets in the tumor microenvironment, using a highly abundant and specific epitope, represents a novel approach for the targeted delivery of tumor directed chemotherapy.
Here, we describe the generation of a unique ADC, scFvGPIIb/IIIa-MMAE, that targets platelets in the microenvironment of tumors as a novel approach for cancer treatment. This ADC is based on three major components: 1) The targeting scFv (scFvGPIIb/IIIa), which selectively binds to an epitope that is cryptic on the low-affinity GPIIb/IIIa on circulating platelets but becomes exposed on the high affinity GPIIb/IIIa, expressed on activated platelets [16,17]. 2) A synthetic microtubule inhibitor, monomethyl auristatin E (MMAE), which is an ideal drug for targeting by utilizing its high cytotoxic potency and obviating its otherwise dominant non-specific cell toxicity. 3) A cancer-specific, Valine-citrulline (Val-Cit) linker, utilizing the abundance of cathepsin B in the tumor microenvironment for cleavage and release of the active MMAE, thereby locally releasing cytotoxic effects [18,19].
To demonstrate the functionality of our novel anti-cancer ADC, with its unique activated platelet targeting, we first confirmed the efficacy of scFvGPIIb/IIIa-MMAE for tumor killing in human cell lines of triple negative breast cancer, colorectal cancer, fibrosarcoma and prostate cancer. Next, we used a triple negative breast cancer metastasis model for proof of concept in vivo studies. Treatment of mice with scFvGPIIb/IIIa-MMAE resulted in significant regression of primary tumors and prevented metastasis without systemic side effects. Together, these findings indicate the generation of a promising ADC and establishes a unique concept that holds promise as a novel, potentially broadly applicable anti-cancer therapy, which is of relevance for both difficult to treat tumors and those with limited specific target epitopes.
Materials and Methods
Study Approval
All animal studies were conducted in strict accordance with protocols approved by the Alfred Medical Research Education Precinct Animal Ethics Committee and the Monash University Animal Ethics Committee.
Generation of targeting scFv-LPETG and coupling enzyme sortase A
The generation of the scFvGPIIb/IIIa and a control, non-binding, scFv (scFvmut) has been described previously [20]. Using polymerase chain reaction, an LPETG-tag (sortase A recognition sequence), a V5-tag and a His-tag were introduced to the C-terminal end of the scFv [21]. The entire scFv was then subcloned into a pSectag 2A vector (Invitrogen, Carlsbad, CA, United States) for expression in human embryonic kidney (HEK) cells (Invitrogen, Carlsbad, CA, United States) [22]. Sortase A was used to induce an enzymatic reaction for the conjugation of the scFv, carrying an LPETG sequence to MMAE which was produced incorporating a triple glycine sequence. Sortase A, a transpeptidase cloned from Staphylococcus aureus was produced and purified as previously described [23]. All proteins (scFvs and sortase A) contain a 6x His-tag, which was used for purification with nickel-based affinity chromatography (Invitrogen, Carlsbad, CA, United States).
Conjugation of scFv with MMAE and Cy7
MMAE, carrying a Val-Cit linker and a triple glycine sequence (GGG-Val-Cit-PAB-MMAE) was synthesized by Levena Biopharma (San Diego, CA, United States). The scFvGPIIb/IIIa and scFvmut (each constructed with a LPETG-tag) were linked to GGG-Val-Cit-PAB-MMAE using a sortase A enzyme-based protocol to produce scFvGPIIb/IIIa-MMAE and scFvmut-MMAE, as described previously [21]. Excess scFv which contains a His-tag was removed using metal affinity chromatography (Invitrogen, Carlsbad, CA, United States) and excess MMAE was removed using a 10 kDa spin column. For imaging studies, Cy7 was incorporated into the conjugate by incubating scFvGPIIb/IIIa-MMAE and scFvmut-MMAE with 2x excess Cy7 via amine labeling (AAT Bioquest, Sunnyvale, CA, United States). Excess free dye was removed by dialysis in PBS. The purified scFv-Cy7-MMAE was analyzed by SDS-PAGE gel and the protein and near-infrared signal from the band of interest was confirmed using the Odyssey Imager (Licor Biosciences, Lincoln, NE, United States). Additionally, Western blot was performed with rabbit anti-MMAE antibody (Levena Biopharma, San Diego, CA, United States), detected with an HRP-anti-rabbit antibody (Cell Signaling, Danvers, MA, United States) to confirm conjugation of MMAE to the scFv.
Preparation of platelet rich plasma and flow cytometry
Blood was collected from healthy volunteers in citrate and centrifuged at 180 g for 10 minutes. The platelet rich plasma was then collected, stored at 37°C and used within two hours. For flow cytometry, platelet rich plasma was diluted 1:20 in Tyrode's buffer. To induce platelet activation, ADP was added at a final concentration of 20 μM for 5 minutes before adding the scFv. Binding was determined by anti-V5-FITC (ThermoFisher Scientific, Waltham, MA, United States) or rabbit anti-MMAE antibody (Levena Biopharma, San Diego, CA, United States) and detected with an anti-rabbit mAb coupled to AF647 (Invitrogen, Carlsbad, CA, United States). Flow cytometry was performed using a FACS Fortessa scanner (BD Biosciences, Franklin Lakes, NJ, United States). Results were analyzed using the Flowlogic software.
To determine the ability of cancer cells to activate platelets, platelet rich plasma was incubated with the cancer cell lines MDA-MB-231, HT29, HT1080 and PC3 for 6 hours at 37°C. As a positive control with the same experimental setting, ADP-activated platelet rich plasma was incubated with the cancer cells for 6 hours at 37°C. The cancer cell and platelet (resting/ADP-activated platelets) mixtures were then stained with an anti-CD41-PE monoclonal antibody (BD Biosciences, Franklin Lakes, NJ, United States) and scFvGPIIb/IIIa or scFvmut binding was detected by an anti-V5-FITC monoclonal antibody (Abcam, Cambridge, United Kingdom). Flow cytometry and analysis was performed as described above.
Cancer cell lines
A metastatic variant of the MDA-MB-231 triple-negative breast adenocarcinoma cell line (a kind gift from Dr Zhou Ou, Fudan University Shanghai Cancer Center, China) was transduced with a lentiviral vector containing codon-optimized firefly luciferase-mCherry under the control of the ubiquitin-C promoter [24,25] and was cultured in DMEM medium + Glutamax (Life Technologies, Carlsbad, CA, United States), supplemented with 10% (v/v) FBS (Invitrogen, Carlsbad, CA, United States), at 37°C in a 5% CO2 humidified atmosphere. Human cancer cell lines HT29, HT1080 and PC3 were cultured in RPMI media (Life Technologies, Carlsbad, CA, United States) supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin at 37°C in a 5% CO2 humidified atmosphere. Cell identity was confirmed by karyotyping.
Cytotoxicity assays
The cytotoxic activity of GGG-Val-Cit-PAB-MMAE and scFv-MMAE were assessed in a cytotoxicity assay in the presence of cathepsin B. To induce cathepsin B cleavage, MMAE, GGG-Val-Cit-PAB-MMAE or scFv-MMAE were incubated with 0.01 units of pre-activated cathepsin B (Sigma Aldrich, St Louis, MO, United States) for 4 hours at 37°C in 25 mM acetate buffer pH 4.8. MDA-MB-231, HT29, HT1080 and PC3 cells were seeded on a 96 well plate at 6000 cells/well overnight. The next day, cells were treated with MMAE, GGG-Val-Cit-PAB-MMAE or scFv-MMAE in the presence or absence of cathepsin B and incubated for 72 hours. Metabolism of the yellow tetrazolium salt (XTT) was determined according to the manufacturer's instructions (Sigma Aldrich, St Louis, MO, United States). Percentage killing relative to untreated control cultures was calculated using the following formula: 100 - [(test value/untreated value) x100]. Each assay was repeated in triplicate.
For platelet assays, 100 µL of platelet rich plasma (containing approximately 2x107 platelets) was activated with 20 µM ADP. Activated platelets were incubated with scFv-MMAE for 30 minutes. To remove unbound scFv-MMAE, the platelet-scFv-MMAE mixture was centrifuged at 2000 g for 2 minutes, the supernatant was removed, and platelets were resuspended in PBS. As a control, unwashed platelet-scFv-MMAE mixture was used. Either washed or unwashed (control) platelet-scFv-MMAE mixture was added to MDA-MB-231, HT29, HT1080 and PC3 cells in the absence of exogenous cathepsin B and incubated for 72 hours before determination of XTT metabolism as described above.
Immunohistochemical analysis of cathepsin B in tumors
Tumors were fixed in formalin solution (Sigma Aldrich, St Louis, MO, United States) for 24 hours, paraffin embedded, and microtome sectioned (Leica, Wetzlar, Germany) to 5 µm - 30 µm onto a glass slide. Sections were deparaffinized and underwent antigen retrieval with 0.01 M Citric Acid in 90°C for 20 minutes. Tumor sections were stained overnight with an anti-cathepsin B antibody (Abcam, Cambridge, United Kingdom) and detected with an Alexa Fluor 488 labeled anti-mouse antibody (Life Technologies, Carlsbad, CA, United States) and cell surface membrane-reactive anti-sodium/potassium ATPase antibody (Abcam, Cambridge, United Kingdom), counterstained with Hoechst® (ThermoFisher Scientific, Waltham, MA, United States) and visualized using the A1r Plus Confocal Microscope (Nikon, Shinagawa, Tokyo, Japan), 20x water objective.
Breast cancer metastasis model
To establish metastasis, 2 × 105 MDA-MB-231 cells in 20 μL PBS were injected into the fourth left mammary fat pad of 5-6 weeks old anaesthetized BALB/C athymic nude mice. Primary tumor growth was measured by caliper and tumor volume was determined using the formula (length × width2)/2. Additionally, primary tumor growth was measured via bioluminescence using the IVIS Lumina II (Perkin Elmer, Waltham, MA, United States) imaging system by measuring luciferase activity after a tail-vein injection of 150 mg/kg D-luciferin (ThermoFisher Scientific, Waltham, MA, United States). Metastasis development was monitored via bioluminescence using IVIS Lumina II by measuring luciferase activity in the chest region, distant from the primary tumor, for a longer time point (60 sec) as previously described [25]. Metastasis development was quantified using the Living Image software v4.5.1 (Perkin Elmer, Waltham, MA, United States) by quantifying photon/s in a region of interest around the chest bioluminescence signal.
For therapy studies, mice underwent bioluminescence imaging on day 3 and day 7 post tumor inoculation to confirm tumor growth. Mice with tumors were then randomly assigned to three groups and treated with either scFvGPIIb/IIIa-MMAE, scFvmut-MMAE or left untreated. Treatment was initiated 7 days following tumor inoculation, via intravenous injection of 6 mg/kg body weight of either scFvGPIIb/IIIa-MMAE or scFvmut-MMAE followed by three additional treatments every fourth day. Primary tumor size and metastasis development was monitored twice a week as described above.
In vivo fluorescence imaging of scFvGPIIb/IIIa-Cy7-MMAE
Animals were injected intravenously with 20 µg of scFvGPIIb/IIIa-Cy7-MMAE or scFvmut-Cy7-MMAE. Fluorescence imaging of the ADC in mice was performed 24 hours later using the IVIS Lumina using the following settings (Filter Passband = Excitation 710-760 nm, Emission 810-875 nm). Following imaging, mice were sacrificed, organs perfused to remove circulating blood and reimaged.
Flow cytometry and ex vivo fluorescence imaging of scFvGPIIb/IIIa-GFP and GP1bβ
BALB/C nude mice with MDA-MB-231 mammary tumor, or non-tumor bearing mice were injected intravenously with 20 µg of scFvGPIIb/IIIa-GFP or scFvmut-GFP and 20 µL of DyLight 649 anti-GP1bβ (Emfret Analytics, Eibelstadt, Bayern, Germany). After 24 hours, animals were sacrificed, and tumor, spleen and femurs were extracted.
Tumor samples were fixed in formalin (Sigma Aldrich, St Louis, MO, United States) for 24 hours, paraffin embedded, and microtome sectioned (Leica, Wetzlar, Germany) to 20 µm onto a glass slide. Sections were deparaffinized, stained with Hoechst® and visualized using the A1r Plus Confocal Microscope using a 60x oil objective.
For flow cytometry, spleen sections were teased apart, disaggregated and filtered through a 100 µm filter into single cell suspensions. Bone marrow cells were extracted from the femurs and filtered through a 100 µm filter into single cell suspensions. Spleen and bone marrow cells were then suspended in 1 mL red cell lysis buffer (0.155 M NH4Cl, 0.01 M KHCO3, 0.01 mM EDTA) for 10 min and neutralized with wash buffer (PBS, 2mM EDTA, 0.1% BSA). Cells were stained with mouse CD41-PE (eBioscience, Waltham, MA, United States) and flow cytometry was performed using the Fortessa scanner. Results were analyzed using the Flowlogic software.
Mouse blood collection and toxicity measurements
Mouse blood was collected via submandibular bleeds into EDTA-coated microtainer collection tubes (BD Biosciences, Franklin Lakes, NJ, United States) and blood counts were performed using the XS-1000i hematologic analyzer (Sysmex Corporation, Kobe, Hyogo, Japan) to determine white blood cells (WBC) and platelets counts. For liver and kidney function tests, 500 µL of mouse blood was collected in citrate. Plasma was collected following centrifugation of blood at 1000 g for 10 minutes and alkaline phosphatase (ALP), alanine aminotransferase (ALT) and urea levels were measured using the Synchron LX20PRO System (Beckman Coulter Diagnostics, High Wycombe, United Kingdom) by Monash Pathology.
Statistical analyses
All data are reported as mean ± SEM of at least 3 independent assays unless otherwise noted. Statistical analyses were performed using unpaired Student's T tests for comparison of two groups. Statistical analyses were performed using one-way ANOVA, with Dunnett's multiple comparisons test for analysis of metastasis volume and two-way ANOVA, with Tukey's multiple comparisons test for analysis of primary tumor growth volume. A P value of < 0.05 was considered significant.
Results
ScFvGPIIb/IIIa-Cy7-MMAE binds to activated platelets
To allow the unique targeting of a potent anti-mitotic agent to activated platelets, we developed a novel antibody-drug conjugate combining several advanced biotechnological tools and methods. We utilized a unique scFvGPIIb/IIIa, which specifically targets activated platelets, combined with a sortase A based site-directed biological conjugation method to produce a novel ADC, scFvGPIIb/IIIa-MMAE incorporating the potent anti-mitotic agent (MMAE). In order to facilitate its conjugation to the scFv and enzymatic release, a MMAE linker was designed with a triglycine (GGG) sequence, a cathepsin B cleavable peptide (Val-Cit) and a para-aminobenzylalcohol (PABA) self-immolative spacer (Figure 1). Each scFv has a sortase A recognition sequence, LPETG, at the C-terminus used for a site-directed, enzymatic coupling to the glycine sequence of GGG-Val-Cit-PAB-MMAE. To enable in vivo detection, the scFv-MMAE was labeled with a Cy7 dye by NHS labeling to yield, scFvGPIIb/IIIa-Cy7-MMAE (Figure 1). Analysis of this final construct using SDS page electrophoresis and visualized using the Odyssey reader confirmed successful Cy7 labeling of the scFv which has a molecular weight of 34 kDa (Figure 2A). The scFvGPIIb/IIIa carries a hexahistidine tag downstream of the LPETG sequence, which is cleaved following sortase A reaction, resulting in a final scFvGPIIb/IIIa-Cy7-MMAE product with reduced molecular weight compared to the uncleaved scFvGPIIb/IIIa. MMAE conjugation to the scFv was analyzed via Western blotting using an anti-MMAE antibody and confirmed by the presence of a band at ~33 kDa (Figure 2B). Additionally, flow cytometry experiments demonstrated that specific binding of the scFvGPIIb/IIIa-Cy7-MMAE construct to activated platelets was maintained post conjugation (Figure 2C). The scFvGPIIb/IIIa used in this study binds to only the activated form of platelet-specific GPIIb/IIIa, importantly both in human, as well as in mouse platelets. As a negative control, we used an scFvmut, which we have previously developed with a point mutation, such that it acts as a non-binding control (Figure 2D) [16].
Structure of GGG-Val-Cit-PAB-MMAE and labeling strategy for the generation of scFvGPIIb/IIIa-Cy7-MMAE and cathepsin B drug release. (A) scFvGPIIb/IIIa was conjugated to GGG-Val-Cit-PAB-MMAE using an enzymatic sortase A reaction, (B) followed by Cy7 conjugation, via NHS labelling to produce scFvGPIIb/IIIa-Cy7-MMAE. (C) In vivo, cathepsin B cleaves the scFvGPIIb/IIIa-Cy7-MMAE at the MMAE Val-Cit linker, releasing the potent MMAE for tumor killing.
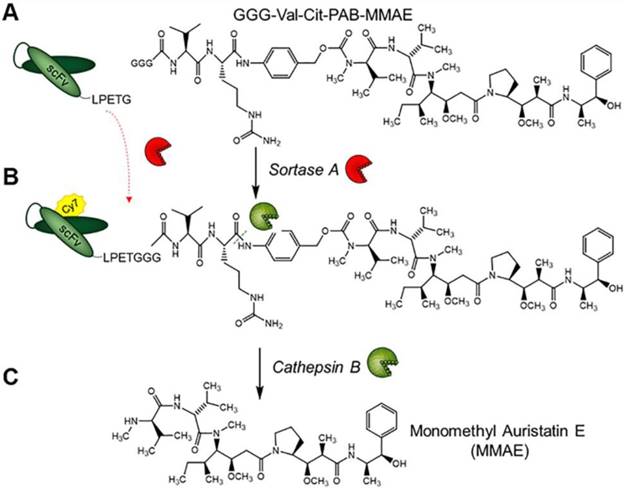
scFvGPIIb/IIIa-Cy7-MMAE characterization and specific binding to activated platelets. (A) Coomassie (red) and near infrared (green) imaging of scFvGPIIb/IIIa -Cy7-MMAE (I), unmodified scFvGPIIb/IIIa (II), scFvmut-Cy7-MMAE (III) and unmodified scFvmut (IV). Yellow color indicates the overlap between Coomassie in red and near infrared in green. (B) Western blot of scFvGPIIb/IIIa-Cy7-MMAE (I), unmodified scFvGPIIb/IIIa (II), scFvmut-Cy7-MMAE (III) and unmodified scFvmut (IV). (C) Flow cytometry profile showing comparable binding of scFvGPIIb/IIIa (red) and scFvGPIIb/IIIa-Cy7-MMAE (blue) to activated platelets. The scFvGPIIb/IIIa contains a V5 tag, which allows using an anti-V5-FITC secondary antibody for detection. The anti-V5-FITC antibody alone was used as a negative control (grey) (all n=3). (D) Flow cytometry profile of scFvGPIIb/IIIa-Cy7-MMAE and scFvmut-Cy7-MMAE binding to ADP-activated platelets (red), resting platelets (black), and secondary anti-V5 antibody (grey filled) only. Data expressed as mean ± SEM.
Cathepsin B cleaves scFvGPIIb/IIIa-MMAE and scFvmut-MMAE to its potent and cytotoxic form
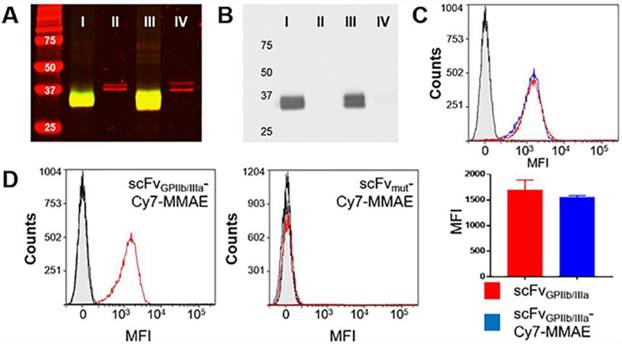
Next, we assessed the cytotoxic activity of MMAE on tumor cells in culture after the drug was released from the ADC by exogenous cathepsin B and confirmed equal dose-dependent killing of the triple-negative breast adenocarcinoma cell line MDA-MB-231 by scFvGPIIb/IIIa-MMAE and scFvmut-MMAE. On MDA-MB-231 cells, the IC50 of scFvGPIIb/IIIa-MMAE was 1.98x10-10 M while the IC50 of the scFvmut-MMAE was 1.78x10-10 M (Figure 3A). These studies confirmed that our approach of utilizing a sortase A conjugation of MMAE to the scFv allowed for a controlled and equal coupling of MMAE to the scFv and the equivalent cathepsin B-mediated release of cytotoxic MMAE by scFvGPIIb/IIIa and scFvmut.
Conjugation of MMAE to scFvGPIIb/IIIa and scFvmut produces an active and potent MMAE in the presence of cathepsin B present in tumor cells and the tumor microenvironment. (A) Cytotoxicity assay of triple negative breast adenocarcinoma MDA-MB-231 cells, cultured in MMAE, GGG-Val-Cit PAB-MMAE and scFvGPIIb/IIIa-MMAE, with (+C) or without (-C) cathepsin B (all n=3). Data expressed as mean ± SEM. (B) Cytotoxicity assay of MDA-MB-231 cells that were cultured in scFvGPIIb/IIIa-MMAE versus scFvmut-MMAE in the presence of cathepsin B (all n=3). Data expressed as mean ± SEM. (C) Cytotoxicity assay of MDA-MB-231 cells, which were cultured in activated platelets that had been pre-incubated with scFvGPIIb/IIIa-MMAE (■) or scFvmut-MMAE (■), and undergone washing to remove unbound scFv (wash) in the absence of exogenous cathepsin B. As a control, MDA-MB-231 cells were cultured in activated platelets that had been pre-incubated with scFvGPIIb/IIIa-MMAE (▼) or scFvmut-MMAE (▼), which did not undergo a washing step to remove unbound scFv (all n=3). Data expressed as mean ± SEM. (D) MDA-MB-231 tumor sections were stained with an anti-cathepsin B antibody (green) and anti-sodium/potassium ATPase antibody (red) and counterstained with Hoechst® nucleic acid stain (blue). Imaging at 20x, demonstrating the abundance of cathepsin B (green) within the tumor microenvironment (left panel). Secondary antibody staining of the sections showed no fluorescence signal (right panel). (E) Further 4x magnification of the anti-cathepsin B antibody stained MDA-MB-231 tumor sections, demonstrating the abundance of cathepsin B (green) within the tumor. White arrow indicates pericellular localization of Cathepsin B.
The incorporation of a cathepsin B cleavable linker enables the activation of an inactive prodrug to an active cytotoxic form by utilizing the abundance of cathepsin B in the tumor microenvironment [26]. Prior to animal studies, in vitro studies were performed to demonstrate that the scFvGPIIb/IIIa-MMAE is released and maintains cytotoxicity upon exposure to cathepsin B. MDA-MB-231 cells were incubated with either MMAE, GGG-Val-Cit-PAB-MMAE or scFvGPIIb/IIIa-MMAE in the absence and presence of cathepsin B. The IC50 killing efficacy of GGG-Val-Cit-PAB-MMAE on MDA-MB-231 tumor cells was 1.86x10-8 M and the addition of cathepsin B increased the cellular killing efficacy by approximately 40-fold to 4.70x10-10 M (Figure 3B), which is similar to the IC50 of the unmodified MMAE. The IC50 killing efficacy of scFvGPIIb/IIIa-MMAE was 2.21x10-8 M and the addition of cathepsin B enhanced the killing efficacy to 1.96x10-10 M (Figure 3B). We confirmed an increased cell killing by the GGG-Val-Cit-PAB-MMAE and scFv-MMAE in the presence of cathepsin B, indicating release of MMAE. We also observed a lower level (~10-8 M) of non-specific killing of cancer cells with scFv-MMAE and GGG-Val-Cit-PAB-MMAE in the absence of exogenous Cathepsin B. We expect this to be due to the secretion of small amounts of cathepsin B from the cancer cells, or release of other cathepsins and proteases that can also cleave the Val-Cit linker, as previously reported [27-29]. These findings confirm the cytotoxic potency and the efficacy of the cathepsin B dependent release and activation of the potent MMAE from its inactive form in the newly created scFvGPIIb/IIIa-MMAE.
To investigate if the therapeutic efficacy of scFvGPIIb/IIIa-MMAE is based on its specificity for activated platelets, the scFvGPIIb/IIIa-MMAE or scFvmut-MMAE was incubated with ADP-stimulated platelets in vitro. Platelets were subsequently washed to remove unbound scFvGPIIb/IIIa-MMAE or scFvmut-MMAE and then added to MDA-MB-231 cells (Figure 3C). Consistent with the specific binding of the targeted ADC to activated GPIIb/IIIa, incubation of scFvGPIIb/IIIa-MMAE with activated platelets resulted in increased MDA-MB-231 cell killing, with an IC50 of 5.9x10-9 M. In contrast, activated platelets incubated with scFvmut-MMAE demonstrated no cellular killing. As a control to confirm the equal coupling of MMAE to the scFvGPIIb/IIIa and scFvmut, a mixture of activated platelets and either scFvGPIIb/IIIa-MMAE or scFvmut-MMAE, which did not undergo washing, were added to MDA-MB-231 tumor cells (Figure 3C). No significant differences in killing were observed between the scFvGPIIb/IIIa-MMAE and scFvmut-MMAE. The IC50 killing efficacy of scFvGPIIb/IIIa-MMAE on MDA-MB-231 was 1.27x10-8 M and the IC50 killing efficacy of scFvmut-MMAE on MDA-MB-231 was 1.38x10-8 M, thereby confirming equal coupling efficacy of MMAE to both, scFvGPIIb/IIIa and scFvmut.
To examine whether our novel ADC displays efficacy against different tumor types, these experiments were repeated using a human colorectal cell line (HT29), a human fibrosarcoma cell line (HT1080) and a human prostate cancer cell line (PC3). We first demonstrated that MDA-MB-231, HT29, HT1080 and PC3 tumor cells cultured in GGG-Val-Cit PAB-MMAE and scFvGPIIb/IIIa-MMAE, in the presence of cathepsin B were able to induce cellular killing, albeit at slightly varying concentrations (Supplementary Figure 1A). Next, HT29, HT1080 and PC3 cell lines were cultured in ADP-activated platelets mixed with scFvGPIIb/IIIa-MMAE and scFvmut-MMAE (Supplementary Figure 1B). These studies confirmed that the scFvGPIIb/IIIa-MMAE bound to activated platelets and induced cell killing of the HT29, HT1080 and PC3 cell lines, thus supporting the notion that this platelet-targeted ADC is active across a broad range of tumor cell types.
In this study, we introduce a novel approach of utilizing Val-Cit-PAB-MMAE, a cathepsin B dependent prodrug, conjugated to a scFv that targets activated platelets as a component of the tumor microenvironment. Previous studies have indicated the abundance of secreted or cell surface-associated cathepsin B within tumor cells [30,31]. To confirm the suitability of the platelet-targeting approach for therapeutic application, we investigated the presence and localization of cathepsin B within the tumor via immunofluorescence imaging of tumor xenograft sections stained with a cathepsin B antibody. Immunostaining confirmed the presence of cathepsin B within MDA-MB-231 tumor cells and the tumor microenvironment (Figure 3D). To ascertain the cellular localization of cathepsin B in tumor cells, a magnified image was acquired (Figure 3E). This confirms the intracellular and pericellular location (indicated by white arrows) of cathepsin B within tumor cells.
Cancer cells induce platelet activation and can be imaged using scFvGPIIb/IIIa-Cy7-MMAE
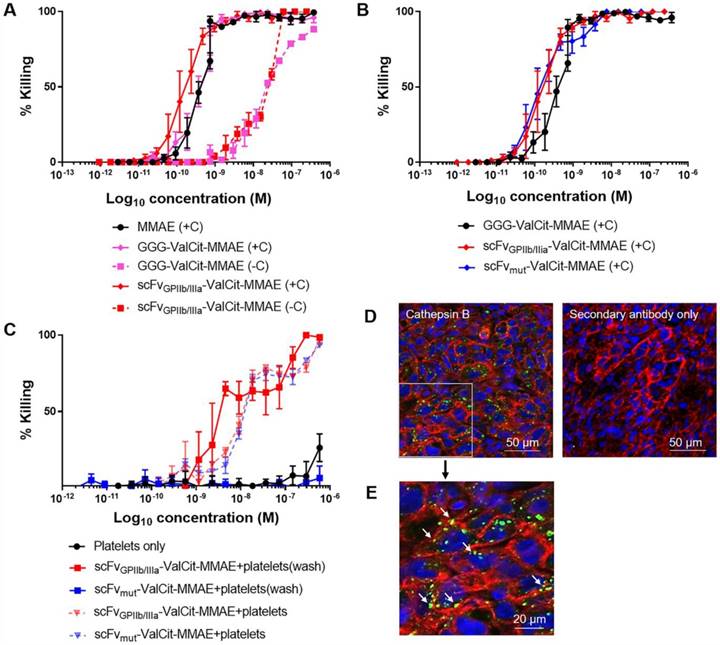
The ability of cancer cells to activate platelets and the functionality of the scFvGPIIb/IIIa-MMAE to target activated platelets in the tumor microenvironment was first analyzed in vitro via fluorescence imaging. MDA-MB-231, HT29, HT1080 and PC3 cells were incubated with washed human platelets and stained with anti-CD41 antibody as a platelet marker and scFvGPIIb/IIIa, as a platelet activation marker. Strikingly, flow cytometry analysis of the platelet population (as defined by CD41 expression) revealed that incubation of platelets with MDA-MB-231, HT29, HT1080 or PC3 tumor cells resulted in activation of the entire platelet population, to a similar extent as ADP-activated platelets, thus confirming the ability of scFvGPIIb/IIIa to detect tumor cell-induced platelet activation (Figure 4A and 4B, red line). In contrast, scFvmut, as negative control, showed no binding to platelets incubated with cancer cells (Figure 4B, blue line). Furthermore, gating on the cancer cell population confirmed that platelets bind directly to cancer cells, to varying degrees, depending on the cell line (Figure 4A and 4C). Indeed, the percentage of MDA-MB-231, HT29, HT1080 and PC3 cells with adherent platelets were 10%, 15%, 44% and 11%, respectively (Figure 4C). To further illustrate the platelet-cancer cell interactions, we performed immunofluorescence imaging, demonstrating that platelets can bind directly to cancer cells and that cancer cells induce platelet activation, which can be detected by binding of the activation-specific scFvGPIIb/IIIa (Figure 4D).
Tumor cells induce platelet activation. (A) Washed platelet rich plasma was added to MDA-MB-231, HT29, HT1080 and PC3 tumor cells and stained with a PE-conjugated anti-CD41 antibody and scFvGPIIb/IIIa or scFvmut, detected with an anti-V5-FITC antibody. Flow cytometry dot plots represent the gating strategy differentiating regions of cancer cells and platelets. The cancer cell region was further gated to select for CD41-positive cancer cells (CD41+ve). (B) Analysis of the CD41-positive region demonstrated that 100% of platelets were activated upon incubation with cancer cells for 6 hours, determined by positive scFvGPIIb/IIIa binding (red), which was equivalent to scFvGPIIb/IIIa binding to ADP-activated platelets (green). As a negative control, platelets incubated with cancer cells were also stained with scFvmut (blue) and displayed negative binding (all n=3). (C) Analysis of the region of cancer cells positive for CD41 (red), confirming that platelets can bind directly to cancer cells (all n=3). (D) Fluorescence imaging (20x) demonstrated that MDA-MB-231 cells bind directly to platelets (red) and induces platelet activation as shown by positive scFvGPIIb/IIIa staining (green) and negative scFvmut staining.
scFvGPIIb/IIIa-Cy7-MMAE localizes to activated platelets in the tumor microenvironment and sites of metastasis
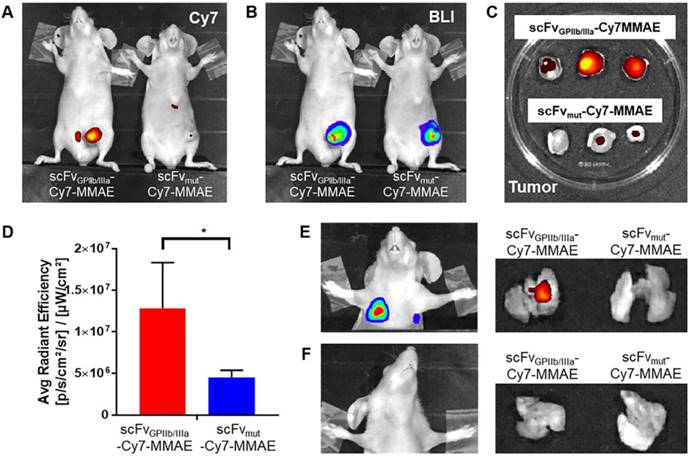
For in vivo characterization, scFvGPIIb/IIIa-MMAE and scFvmut-MMAE were conjugated to Cy7 and injected intravenously to mice with MDA-MB-231 primary tumor that had already metastasized. Fluorescence imaging of mice performed 24 hours post injection of scFv-Cy7-MMAE demonstrated enrichment of scFvGPIIb/IIIa-Cy7-MMAE at the primary tumor region, with no tumor localization of the scFvmut-Cy7-MMAE (Figure 5A). As the MDA-MB-231 cells had been previously transduced with a lentiviral vector to express luciferase, bioluminescence imaging was used to detect areas of primary tumor as shown in Figure 5B. Concordant with in vivo imaging, ex vivo analyses showed significant enrichment of scFvGPIIb/IIIa-Cy7-MMAE but not scFvmut-Cy7-MMAE in the primary tumors (Figure 5C and 5D). Bioluminescence imaging of lung and lymph node metastases was performed by covering the primary tumor site and imaging with a longer exposure time (60 sec) (Figure 5E and 5F). We further demonstrated that the scFvGPIIb/IIIa-Cy7-MMAE localized in the lungs of MDA-MB-231 tumor-bearing mice with lung metastases (Figure 5E), but not in mice without lung metastases (Figure 5F), confirming that scFvGPIIb/IIIa-Cy7-MMAE localization was specific to activated platelets within tumors. Again, no tumor localization of the scFvmut-Cy7-MMAE was observed in the lungs in the presence or absence of metastases (Figure 5E and 5F).
Activated platelets are present in the tumor microenvironment and can be detected and imaged using the scFvGPIIb/IIIa. (A) In vivo fluorescence imaging of MDA-MB-231 tumor-bearing mice, injected with scFvGPIIb/IIIa-Cy7-MMAE and scFvmut-Cy7-MMAE. (B) In vivo bioluminescence imaging of MDA-MB-231 tumor-bearing mice confirming primary tumor localization in the mammary gland. (C) Ex vivo biodistribution of the primary tumor of MDA-MB-231 tumor-bearing mice injected with scFvGPIIb/IIIa-Cy7-MMAE and scFvmut-Cy7-MMAE. (D) Quantification of fluorescence signal of MDA-MB-231 tumor-bearing mice, injected with scFvGPIIb/IIIa-Cy7-MMAE and scFvmut-Cy7-MMAE (all n=3). Data expressed as mean ± SEM. *P<0.05 analyzed by unpaired Student's T tests. (E) Representative bioluminescence in vivo lung imaging of MDA-MB-231 tumor-bearing mice demonstrated the presence of lung metastases (left panel) and representative ex vivo fluorescence imaging of the lung (right panel) demonstrating positive scFvGPIIb/IIIa-Cy7-MMAE signal but negative scFvmut-Cy7-MMAE. (F) Representative in vivo lung imaging of MDA-MB-231 tumor-bearing mice with no lung metastasis (left panel) and representative ex vivo fluorescence imaging, demonstrating negative scFvGPIIb/IIIa-Cy7-MMAE and scFvmut-Cy7-MMAE signal (right panel). BLI-Bioluminescence.
scFvGPIIb/IIIa localizes to activated platelets in the tumor microenvironment but not to resting platelets in the spleen and bone marrow
To further confirm the selectivity of the targeted ADC for platelets in the tumor microenvironment in vivo, MDA-MB-231 tumor-bearing mice were injected with scFvGPIIb/IIIa-GFP or scFvmut-GFP and a Dylight 649 anti-GP1bβ antibody. This in vivo approach for immunofluorescence was used as the activated form of GPIIb/IIIa undergoes a conformational change and antigen masking upon tissue fixation and is not recognized by the scFvGPIIb/IIIa. After 24 hours, tumors were extracted, sectioned and imaged using fluorescence microscopy to demonstrate the localization of platelets, detected by GP1bβ staining co-localizing with activated platelets, as indicated by scFvGPIIb/IIIa-GFP in the tumor microenvironment (Figure 6A). In contrast, we saw no GFP signal in mice injected with scFvmut-GFP (Figure 6B), indicating the specificity of the scFvGPIIb/IIIa targeting to activated platelets.
Activated platelets are present in the tumor microenvironment but absent in the spleen and bone marrow. (A, B) Immunofluorescence imaging of tumor sections of MDA-MB-231 tumor-bearing mice injected with DyLight 649 anti-GP1bβ (red) and scFvGPIIb/IIIa-GFP or scFvmut-GFP (green), counterstained with Hoechst® (blue). (A) Immunofluorescence imaging (20x) demonstrated the abundance of platelets within the tumor microenvironment (red), and the specificity of the platelet binding of scFvGPIIb/IIIa to tumor-associated platelets in vivo (green). (B) In contrast, the scFvmut does not bind platelets in vivo. (C, D) Flow cytometry of the spleen and bone marrow of BALB/C nude mice injected with PBS (control) or DyLight 649 anti-GP1bβ and scFvGPIIb/IIIa-GFP or scFvmut-GFP. (C) Flow cytometry of spleen cells, which were co-stained for CD41 and gated on the CD41-positive region, demonstrated the presence of platelets in the spleen via DyLight 649 anti-GP1bβ staining, but the absence of activated platelets, demonstrated by the absence of scFvGPIIb/IIIa-GFP staining (n=3). (D) Flow cytometry of bone marrow cells from the femur, co-stained for CD41 and gated on the CD41-positive region, demonstrates the presence of platelets in the bone marrow via DyLight 649 anti-GP1bβ staining, but the absence of activated platelets, demonstrated by the absence of scFvGPIIb/IIIa-GFP staining (n=3).
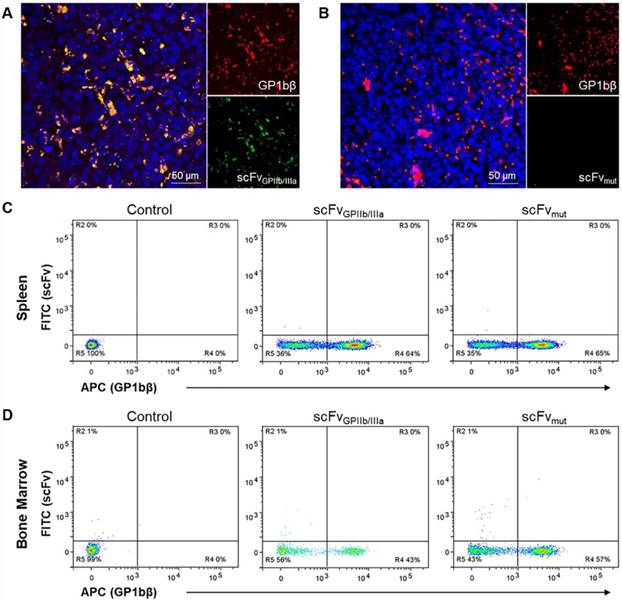
To demonstrate the specificity of the scFvGPIIb/IIIa to activated platelets in the tumor microenvironment, we next investigated whether the scFv localized to the spleen and bone marrow, which are two abundant sources of non-activated platelets. BALB/C nude mice were injected with scFvGPIIb/IIIa-GFP or scFvmut-GFP and the Dylight 649 anti-GP1bβ antibody and the femurs and spleen were extracted after 24 hours and stained for CD41. Single cell analysis via flow cytometry of the spleen and bone marrow, gated on CD41-positive cells displayed binding of the GP1bβ antibody in the spleen and bone marrow but no scFvGPIIb/IIIa binding to these regions (Figure 6C and 6D), thus confirming the specificity of scFvGPIIb/IIIa for activated platelets in vivo.
Treatment with scFvGPIIb/IIIa-Cy7-MMAE reduces tumor growth and metastasis
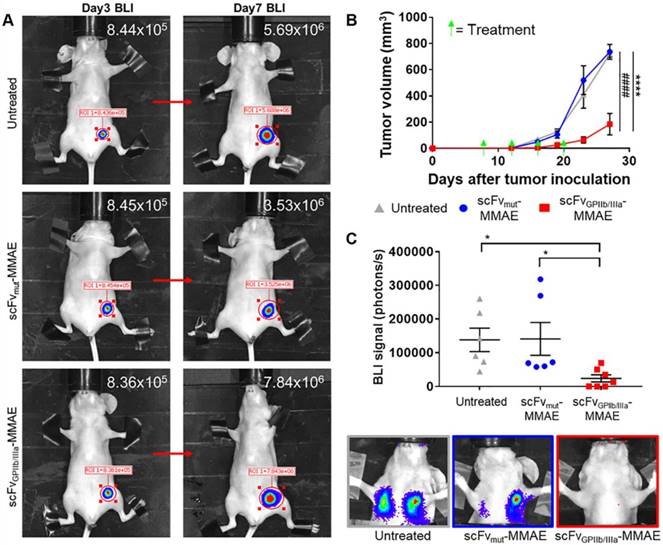
To investigate the in vivo effect of the scFvGPIIb/IIIa-MMAE therapeutic, MDA-MB-231 metastatic tumor-bearing mice were imaged for bioluminescence signal at day 3 post MDA-MB-231 inoculation, and again at day 7 to confirm the presence of growing tumors (Figure 7A). Mice with established tumors were then divided into three groups and treated with scFvGPIIb/IIIa-MMAE or scFvmut-MMAE, or left untreated, and the effect of therapy on tumor growth and metastasis formation was assessed. Strikingly, treatment of tumor-bearing mice with scFvGPIIb/IIIa-MMAE at a dose of 6 mg/kg (administered every 4 days) resulted in a marked reduction in the primary tumor size by over 70% as compared to mice treated with scFvmut-MMAE or mice in the untreated group (Figure 7B). Accordingly, a significant reduction in the development of lung and lymph node metastases on day 23, as measured by total bioluminescence, was seen in scFvGPIIb/IIIa-MMAE treated mice compared to non-targeted and vehicle controls (Figure 7C). Significantly, treatment with scFvGPIIb/IIIa-MMAE prevented the development of metastasis in 40% of mice (3 out of 7 mice), as indicated by bioluminescence imaging, while all mice in the untreated group or treated with scFvmut-MMAE developed metastases. Importantly, no therapeutic effects were observed with scFvmut-MMAE, thus highlighting the impressive ability of our targeting approach to concentrate therapeutic doses of MMAE specifically within the tumor and to prevent metastases. Consistent with our data demonstrating the high degree of specificity of scFvGPIIb/IIIa-MMAE for activated platelets, we did not observe any off-target side effects in treated mice. Mice were healthy during the course of the treatment, did not lose weight (Supplementary Figure 2A), and hematological parameters, liver function and renal function were within normal ranges (Supplementary Figure 2B). Taken together, these findings demonstrate the potential of therapeutic targeting of activated platelets as a strategy for the delivery of chemotherapy to maximize potency and potentially minimize systemic side effects.
scFvGPIIb/IIIa-MMAE treatment inhibits tumor growth and metastasis development in a murine model of triple negative breast cancer. (A) Bioluminescence imaging and signal quantification of MDA-MB-231 tumor-bearing mice on day 3 and the same mouse imaged again at day 7 post tumor inoculation (indicated by a red arrow), which were then randomly assigned to scFvGPIIb/IIIa-MMAE, scFvmut-MMAE or untreated groups. (B) Primary tumor volume of MDA-MB-231 tumor-bearing mice treated with 6 mg/kg of scFvGPIIb/IIIa-MMAE (■) (n=7), scFvmut-MMAE (●) (n=6) or untreated (▲) (n=6). Data expressed as mean ± SEM. ****P<0.0001 between untreated and scFvGPIIb/IIIa-MMAE and ####P<0.0001 between scFvmut-MMAE and scFvGPIIb/IIIa-MMAE analyzed by two-way ANOVA with Tukey's multiple comparison's test. (C) Metastasis burden of MDA-MB-231 tumor-bearing mice treated with 6 mg/kg of scFvGPIIb/IIIa-MMAE (■) (n=7), scFvmut-MMAE (●) (n=6) or untreated (▲) (n=6), measured via bioluminescence imaging, 60 sec exposure, and representative images of lung and lymph node metastasis from each group. Data expressed as mean ± SEM. *P<0.05 analyzed by one-way ANOVA with Dunnett's multiple comparison's test. BLI-Bioluminescence.
Discussion
In this study we demonstrate the utility of targeting activated platelets within the tumor microenvironment as a novel strategy for the treatment of primary tumors and prevention of metastatic disease. This approach is based on the development of a unique ADC, which targets tumor-associated platelets using the activated GPIIb/IIIa as an epitope for localized delivery of MMAE, a potent synthetic anti-mitotic agent. Importantly, this novel approach allows for the controlled-release of MMAE, via tumor-derived cathepsin B, sparing the undesirable systemic side effects commonly seen with traditional chemotherapy.
The efficacy of an ADC is highly contingent upon the presence and abundance of a specific molecular target on tumor cells. This is a major limitation for the use of ADCs, as many cancer types do not express a clinically validated antigenic molecular target. In this regard, a particular advantage of our approach is the possible ubiquitous nature of activated platelets in a broad range of human tumors, including breast [32], colorectal [33], lung [34], ovarian [11] and esophageal [13] tumors, indicating that our platelet-targeting strategy holds promise for a large range of tumors.
Additionally, we show the ability to target the tumor stroma as a novel approach to deliver a pro-drug for the killing of tumor cells. Valine-citrulline (Val-Cit), a cathepsin B cleavable linker requires the presence of cathepsin B for cleavage and release of free MMAE, the active anti-cancer agent. The protease cathepsin B is tightly regulated in healthy tissues. However, it is well established that cathepsin B is overexpressed in tumor cells [30,31]. Moreover, tumor cells, as well as cells in the tumor microenvironment, such as stromal fibroblasts and macrophages, can secrete cathepsin B, resulting in the accumulation of cathepsin B within the tumor microenvironment [31,35-37]. There remains some debate regarding the role of cathepsin B in regulating tumor biology given it has been demonstrated to facilitate tumor progression and metastasis [38], and promote tumor apoptosis [39,40]. Nevertheless, it is well established that cathepsin B is highly expressed in the tumor microenvironment [30,31,38]. Therefore, incorporating a cathepsin B cleavable linker, which allows for the specific release of the potent cytotoxic MMAE at the tumor site, represents a major advantage of our ADC design.
Therapeutic approaches using ADCs, such as MMAE are commonly used to target antigens expressed on cancer cells. The binding of the ADC to the tumor cell is typically followed by internalization of the ADC and release of the cytotoxic drug [38,41]. However, studies by Dal Corso et al, have also demonstrated the functionality of ADCs with cathepsin B-cleavable linkers, which are coupled to an antibody with a specificity to non-internalizing targets in the tumor microenvironment [42,43]. Our study further illustrates that tumor-specific cytotoxicity can be achieved by targeting a platelet surface epitope using scFvGPIIb/IIIa-MMAE, conjugated via a Val-Cit linker. A similarly elegant and successful targeting approach for MMAE to the tumor microenvironment was recently reported by Li F et al., who used the antigen-unspecific Fc-FcγR interaction to deliver a MMAE-conjugated ADC to tumor-associated macrophages, also resulting in the release of MMAE in the tumor environment [44]. Our approach demonstrates an alternative mechanism for targeting the tumor microenvironment as the scFv does not possess an Fc region. Nevertheless, both selective deliveries of MMAE to the tumor microenvironment represent novel promising anti-cancer approaches.
Previous studies have confirmed that cathepsin B can be transported to the extracellular surface (pericellular expression), leading to its secretion into the microenvironment as well as being associated with the cell surface [45,46]. This concept has recently been further supported by Kramer L et al., who demonstrated that tumors could be imaged using a cell impermeable Cathepsin B-specific DARPin labelled with Cy5.5, thus highlighting the abundance of membrane associated/secreted cathepsin B in the tumor microenvironment in vivo [47]. Furthermore, the secreted cathepsin B is made up of the “active” Cathepsin B (in contrast to the precursor form of cathepsin B, known as pro-cathepsin B) [48]. This “active” cathepsin B is required for cleavage of the Val-Cit linker, resulting in the release of the active and highly potent cytotoxic drug into the extracellular space [49,50]. Based on these findings, we postulate that scFvGPIIb/IIIa-MMAE is cleaved and activated by cathepsin B in the extracellular space. An alternative possibility is that the uncleaved ADC could be taken up by tumor cells, and the cytotoxic MMAE released by intracellular cathepsin B. The exact tumor cell killing mechanism remains to be investigated in future detailed studies.
Whilst many ADCs have utilized whole antibodies, we chose to use a scFv as an antibody format, since it offers several significant advantages. The small size allows quick and efficient penetration of the scFv based ADC to the tumor environment, whilst ensuring quick clearance of the scFv from non-affected tissues. The recombinant nature of production offers flexibility with respect to modification of the scFv and the incorporation of additional tags, such as the LPETG sequence which we have used in the sortase A conjugation method [45]. Furthermore, the scFvGPIIb/IIIa possesses two additional advantages for clinical translation. Firstly, the scFvGPIIb/IIIa was developed from a human scFv library, reducing the risk of antigenicity. Second, the scFvGPIIb/IIIa is cross reactive between mice and human GPIIb/IIIa, and therefore binds with similar affinity to activated platelets from both species [16].
Our data suggests that this newly designed ADC has the capability to target and efficiently kill a broad range of tumor cells in vitro. Therefore, future studies will be aimed at investigating the efficacy of scFvGPIIb/IIIa-MMAE across different tumor types in vivo. In addition, whilst tumors can display or acquire resistance mechanisms to MMAE, a major advantage of our scFv platform is the ability to conjugate the scFv with different cytotoxic drugs, thus providing versatility in both the range of tumors that can be targeted, and the cytotoxic payload to be delivered. Whilst our preliminary safety studies demonstrated no off-target toxicity associated with the scFvGPIIb/IIIa-MMAE, it is important to note that mice are approximately 3 to 6 times less sensitive to the toxic effects of MMAE compared to humans [46]. Thus, studies to examine for signs of systemic toxicity in other species, which share a similar MTD to humans, such as rats and cynomolgus monkeys, will be important for clinical translation. In general, further systematic toxicity studies would need to be undertaken prior to any potential clinical study.
Cancer associated thrombosis is one of the major causes of mortality in cancer patients. It is therefore common for cancer patients to be treated with anti-thrombotics [47]. As such, another important aspect for clinical translation relates to the question of whether the concurrent use of anti-thrombotic drugs impacts on the therapeutic efficacy of our ADC. Indeed, the use of GPIIb/IIIa inhibitors during anti-cancer therapy might reduce the effective binding of the GPIIb/IIIa targeting ADC. However, GPIIb/IIIa inhibitor therapy is only used, albeit infrequently, in the acute setting of percutaneous coronary intervention [14]. In addition, GPIIb/IIIa inhibitors are associated with a high bleeding risk and would frequently be contraindicated in patients with active cancer [48]. Nevertheless, cancer patients might receive treatment with aspirin or P2Y12 inhibitors. Although platelet function is partially inhibited with these drugs, they do not fully prevent the conformational change of the GPIIb/IIIa receptor [49], which exposes the targeting epitope of our ADC. Furthermore, given the multiple mechanisms by which tumors can activate platelets, and the high abundance of GPIIb/IIIa receptor on platelets, the ADC targeting epitope should still be present in high abundance following aspirin or P2Y12 therapy. Interestingly, anticoagulants such as heparin, have been demonstrated to increase cathepsin B activity [50], which could potentiate the effect of this ADC therapy. Overall, we expect that the described activated platelet targeting therapeutic approach will maintain efficacy in the presence of anti-thrombotic drugs. However, the issue of concomitant anti-thrombotic therapies requires further investigation during the clinical development of scFvGPIIb/IIIa-MMAE.
Moreover, the ability of the scFvGPIIb/IIIa-MMAE to act as an imaging agent to detect activated platelets within tumors is an advantage in the context of concurrent treatment modalities that could affect platelet activation, and to identify various tumor types with abundant platelets as a predictor of treatment response. Therefore, as part of developing a personalized medicine approach, patients could first be imaged with a specific imaging version of scFvGPIIb/IIIa, e.g. which is coupled to a PET/MRI contrast agent, allowing the sensitive detection of tumors and metastases. Upon confirming the presence of activated platelets within tumors, patients could then be treated with scFvGPIIb/IIIa conjugated to the cytotoxic drug. This personalized theranostic approach is a particularly attractive capability of our activated-platelet targeting strategy.
In conclusion, we introduce a novel approach for the safe and effective delivery of anti-cancer therapy, within the tumor microenvironment, by targeting activated platelets. We designed and successfully tested a novel antibody-drug conjugate incorporating a scFv targeting activated GPIIb/IIIa and an additional selective drug activation/release mechanism via cathepsin B, for the delivery of the cytotoxic agent, monomethyl auristatin E specifically to the tumor site. Addressing a major clinical need, we provide proof of concept of the therapeutic efficacy of our novel ADC in treating both primary tumor and preventing metastasis using a murine model of the difficult to treat triple negative breast cancer. Moreover, we have previously established the ability of the activated platelet-specific scFv for imaging a broad range of tumors [15] which raises the prospect of an integrated approach for cancer diagnosis and therapy, by utilizing molecular imaging as a means to detect tumor-associated platelets, and therefore predict those likely to respond to platelet-targeted therapy. Together, these findings demonstrate the broad potential of our unique activated platelet-targeting approach as a novel theranostic anti-cancer strategy, especially for cancers that do not express specific molecular epitopes suitable for therapeutic targeting.
Abbreviations
ADC: antibody-drug conjugate; ADP: adenosine diphosphate; AF: Alexa Fluor; ALP: alkaline phosphatase; ALT: alanine aminotransferase; BLI: Bioluminescence; Cy7: Cyanine 7; DARPin: Designed ankyrin repeat protein; GPIIb/IIIa: Glycoprotein IIb/IIIa; IC50: half maximal inhibitory concentration; IVIS: In vivo imaging system; MMAE: monomethyl auristatin E; Mut: Mutant; NHS: N-hydroxysuccinimide; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; scFv: single-chain antibodies; Val-Cit: Valine-citrulline; HRP: horseradish peroxidase; WBC: white blood cells and XTT: tetrazolium salt.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Dr Alexander Ziegler and Dr Adam Walker from the Monash Institute of Pharmaceutical Sciences and Joy Yao from the Baker Heart and Diabetes Institute for their technical support. We would like to acknowledge Iśka Carmichael from the Monash Micro Imaging team and Michael Daskalakis from Monash Pathology for their technical support. We would like to thank Tobias Ziegler for his assistance with the schematic drawing.
Author contributions
MLY, JDM, XW, MZ and AW designed and performed the experiments. MLY, JDM, XW, MZ, YCC, AW, CJN, EKS and GAP analyzed the data. MLY, JDM, XW, MZ, PMH, EKS, GAP and KP wrote the manuscript. AMS, EKS, PMH, GAP and KP supervised the project. GAP and KP developed the ADC and conceived the project.
Funding
This work was funded by the National Health and Medical Research Council (NHMRC project grant: 1108670) of Australia and in part by the Victorian Government Operational Infrastructure Support Program. In addition, MLY is supported by the Research Training Program Scholarship, JDM is supported by the Sylvia and Charles Viertel Foundation Clinical Investigator Award, XW is supported by the National Heart Foundation of Future Leader Fellowship with the Paul Korner Innovation Award, YCC is supported by the National Heart Foundation Postdoctoral Fellowship, AMS is supported by a NHMRC Senior Practitioner Fellowship, EKS is supported by NHMRC 1147498. PMH is supported by the Rebecca Cooper Foundation and NHMRC project grants and KP is supported by a NHMRC Principal Research Fellowship.
Competing Interests
KP is an inventor on patents describing activated platelet-targeting recombinant antibodies. All other authors have declared that no competing interest exists.
References
1. Parakh S, Parslow AC, Gan HK, Scott AM. Antibody-mediated delivery of therapeutics for cancer therapy. Expert Opin Drug Deliv. 2016;13(3):401-19
2. Adair JR, Howard PW, Hartley JA, Williams DG, Chester KA. Antibody-drug conjugates - a perfect synergy. Expert Opin Biol Ther. 2012;12(9):1191-206
3. Amiri-Kordestani L, Blumenthal GM, Xu QC, Zhang L, Tang SW, Ha L. et al. FDA Approval: Ado-Trastuzumab Emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin Cancer Res. 2014;20(17):4436-41
4. Dada R, Zekri J, Saadi RA. Brentuximab vedotin in pretreated Hodgkin lymphoma patients: a systematic review and meta-analysis. Expert Opin Biol Ther. 2016;16(6):739-45
5. Gan HK, Bent M van den, Lassman AB, Reardon DA, Scott AM. Antibody-drug conjugates in glioblastoma therapy: the right drugs to the right cells. Nat Rev Clin Oncol. 2017;14(11):695
6. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938-48
7. Palma G, Frasci G, Chirico A, Esposito E, Siani C, Saturnino C. et al. Triple negative breast cancer: looking for the missing link between biology and treatments. Oncotarget. 2015;6(29):26560-74
8. Yan M, Jurasz P. The role of platelets in the tumor microenvironment: From solid tumors to leukemia. Biochim Biophys Acta. 2016;1863(3):392-400
9. Lonsdorf AS, Krämer BF, Fahrleitner M, Schönberger T, Gnerlich S, Ring S. et al. Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 integrin mediates interaction of melanoma cells with platelets a connection to hematogenous metastasis. J Biol Chem. 2012;287(3):2168-78
10. Elaskalani O, Berndt MC, Falasca M, Metharom P. Targeting platelets for the treatment of cancer. Cancers. 2017;9(7):94
11. Stone RL, Nick AM, McNeish IA, Balkwill F, Han HD, Bottsford-Miller J. et al. Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med. 2012;366(7):610-8
12. Garcia-Albeniz X, Chan AT. Aspirin for the prevention of colorectal cancer. Best Pract Res Clin Gastroenterol. 2011;25(0):461-72
13. Shimada H, Oohira G, Okazumi S, Matsubara H, Nabeya Y, Hayashi H. et al. Thrombocytosis associated with poor prognosis in patients with esophageal carcinoma. J Am Coll Surg. 2004;198(5):737-41
14. Armstrong PC, Peter K. GPIIb/IIIa inhibitors: from bench to bedside and back to bench again. Thromb Haemost. 2012;107(5):808-14
15. Yap ML, McFadyen JD, Wang X, Zia NA, Hohmann JD, Ziegler M. et al. Targeting activated platelets: A unique and potentially universal approach for cancer imaging. Theranostics. 2017;7(10):2565-74
16. Schwarz M, Meade G, Stoll P, Ylanne J, Bassler N, Chen YC. et al. Conformation-specific blockade of the integrin GPIIb/IIIa a novel antiplatelet strategy that selectively targets activated platelets. Circ Res. 2006;99(1):25-33
17. Schwarz M, Röttgen P, Takada Y, Le Gall F, Knackmuss S, Bassler N. et al. Single-chain antibodies for the conformation-specific blockade of activated platelet integrin alphaIIbbeta3 designed by subtractive selection from naive human phage libraries. FASEB J. 2004;18(14):1704-6
18. Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF. et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21(7):778-84
19. Sanderson RJ, Hering MA, James SF, Sun MMC, Doronina SO, Siadak AW. et al. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin Cancer Res. 2005;11(2 Pt 1):843-52
20. Wang X, Hagemeyer CE, Hohmann JD, Leitner E, Armstrong PC, Jia F. et al. Novel single-chain antibody-targeted microbubbles for molecular ultrasound imaging of thrombosis validation of a unique noninvasive method for rapid and sensitive detection of thrombi and monitoring of success or failure of thrombolysis in mice. Circulation. 2012;125(25):3117-26
21. Ta HT, Prabhu S, Leitner E, Jia F, von Elverfeldt D, Jackson KE. et al. Enzymatic single-chain antibody tagging: a universal approach to targeted molecular imaging and cell homing in cardiovascular disease. Circ Res. 2011;109(4):365-73
22. Hohmann JD, Wang X, Krajewski S, Selan C, Haller CA, Straub A. et al. Delayed targeting of CD39 to activated platelet GPIIb/IIIa via a single-chain antibody: breaking the link between antithrombotic potency and bleeding? Blood. 2013;121(16):3067-75
23. Wang X, Gkanatsas Y, Palasubramaniam J, Hohmann JD, Chen YC, Lim B. et al. Thrombus-targeted theranostic microbubbles: A new technology towards concurrent rapid ultrasound diagnosis and bleeding-free fibrinolytic treatment of thrombosis. Theranostics. 2016;6(5):726-38
24. Li D-Q, Hou Y-F, Wu J, Chen Y, Lu J-S, Di G-H. et al. Gene expression profile analysis of an isogenic tumour metastasis model reveals a functional role for oncogene AF1Q in breast cancer metastasis. Eur J Cancer. 2006;42(18):3274-86
25. Le CP, Nowell CJ, Kim-Fuchs C, Botteri E, Hiller JG, Ismail H. et al. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat Commun. 2016;7:10634
26. Schmid B, Chung D-E, Warnecke A, Fichtner I, Kratz F. Albumin-binding prodrugs of camptothecin and doxorubicin with an Ala-Leu-Ala-Leu-linker that are cleaved by cathepsin B: synthesis and antitumor efficacy. Bioconjug Chem. 2007;18(3):702-16
27. Tan G-J, Peng Z-K, Lu J-P, Tang F-Q. Cathepsins mediate tumor metastasis. World J Biol Chem. 2013;4(4):91-101
28. Sudhan DR, Siemann DW. Cathepsin L targeting in cancer treatment. Pharmacol Ther. 2015;155:105-16
29. Caculitan NG, Dela Cruz Chuh J, Ma Y, Zhang D, Kozak KR, Liu Y. et al. Cathepsin B is dispensable for cellular processing of cathepsin B-cleavable antibody-drug conjugates. Cancer Res. 2017;77(24):7027-37
30. Frosch BA, Berquin I, Emmert-Buck MR, Moin K, Sloane BF. Molecular regulation, membrane association and secretion of tumor cathepsin B. APMIS. 1999;107(1):28-37
31. Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 2003;22(2-3):271-86
32. Lal I, Dittus K, Holmes CE. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. 2013;15:207
33. Li N, Yu Z, Zhang X, Liu T, Sun Y, Wang R. et al. Elevated mean platelet volume predicts poor prognosis in colorectal cancer. Sci Rep. 2017;7(1):10261
34. Ji Y, Sheng L, Du X, Qiu G, Su D. Elevated platelet count is a strong predictor of poor prognosis in stage I non-small cell lung cancer patients. Platelets. 2015;26(2):138-42
35. Edgington LE, Verdoes M, Bogyo M. Functional imaging of proteases: recent advances in the design and application of substrate- and activity- based probes. Curr Opin Chem Biol. 2011;15(6):798-805
36. Yhee JY, Kim SA, Koo H, Son S, Ryu JH, Youn I-C. et al. Optical imaging of cancer-related proteases using near-infrared fluorescence matrix metalloproteinase-sensitive and cathepsin B-sensitive probes. Theranostics. 2012;2(2):179-89
37. Erdel M, Trefz G, Spiess E, Habermaas S, Spring H, Lah T. et al. Localization of cathepsin B in two human lung cancer cell lines. J Histochem Cytochem. 1990;38(9):1313-21
38. Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets. 2013;17(3):281-91
39. Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U. et al. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 2008;283(27):19140-50
40. de Castro M a. G, Bunt G, Wouters FS. Cathepsin B launches an apoptotic exit effort upon cell death-associated disruption of lysosomes. Cell Death Discov. 2016;2:16012
41. Ruan H, Hao S, Young P, Zhang H. Targeting cathepsin B for cancer therapies. Horiz Cancer Res. 2015;56:23-40
42. Dal Corso A, Gébleux R, Murer P, Soltermann A, Neri D. A non-internalizing antibody-drug conjugate based on an anthracycline payload displays potent therapeutic activity in vivo. J Control Release. 2017;264:211-8
43. Dal Corso A, Cazzamalli S, Gébleux R, Mattarella M, Neri D. Protease-cleavable linkers modulate the anticancer activity of noninternalizing antibody-drug conjugates. Bioconjugate Chem. 2017;28(7):1826-33
44. Li F, Ulrich M, Jonas M, Stone IJ, Linares G, Zhang X. et al. Tumor-associated macrophages can contribute to antitumor activity through FcγR-mediated processing of antibody-drug conjugates. Mol Cancer Ther. 2017;16(7):1347-54
45. Mai J, Finley RL, Waisman DM, Sloane BF. Human procathepsin B interacts with the annexin II tetramer on the surface of tumor cells. J Biol Chem. 2000;275(17):12806-12
46. Sloane BF, Rozhin J, Johnson K, Taylor H, Crissman JD, Honn KV. Cathepsin B: association with plasma membrane in metastatic tumors. Proc Natl Acad Sci USA. 1986;83(8):2483-7
47. Kramer L, Renko M, Završnik J, Turk D, Seeger MA, Vasiljeva O. et al. Non-invasive in vivo imaging of tumour-associated cathepsin B by a highly selective inhibitory DARPin. Theranostics. 2017;7(11):2806-21
48. Aggarwal N, Sloane BF. Cathepsin B: multiple roles in cancer. Proteomics Clin Appl. 2014;8(5-6):427-37
49. Wang Q, Zhong Y-J, Yuan J-P, Shao L-H, Zhang J, Tang L. et al. Targeting therapy of hepatocellular carcinoma with doxorubicin prodrug PDOX increases anti-metastatic effect and reduces toxicity: a preclinical study. J Transl Med. 2013;11:192
50. Shao L-H, Liu S-P, Hou J-X, Zhang Y-H, Peng C-W, Zhong Y-J. et al. Cathepsin B cleavable novel prodrug Ac-Phe-Lys-PABC-ADM enhances efficacy at reduced toxicity in treating gastric cancer peritoneal carcinomatosis: an experimental study. Cancer. 2012;118(11):2986-96
51. Pietersz GA, Wang X, Yap ML, Lim B, Peter K. Therapeutic targeting in nanomedicine: the future lies in recombinant antibodies. Nanomedicine. 2017;12(15):1873-89
52. Mirsalis JC, Schindler-Horvat J, Hill JR, Tomaszewski JE, Donohue SJ, Tyson CA. Toxicity of dolastatin 10 in mice, rats and dogs and its clinical relevance. Cancer Chemother Pharmacol. 1999;44(5):395-402
53. Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122(10):1712-23
54. McFadyen JD, Schaff M, Peter K. Current and future antiplatelet therapies: emphasis on preserving haemostasis. Nat Rev Cardiol. 2018;15(3):181-91
55. Wijeyeratne YD, Heptinstall S. Anti-platelet therapy: ADP receptor antagonists. Br J Clin Pharmacol. 2011;72(4):647-57
56. Almeida PC, Nantes IL, Chagas JR, Rizzi CCA, Faljoni-Alario A, Carmona E. et al. Cathepsin B activity regulation heparin-like glycosaminoglycans protect human cathepsin B from alkaline pH-induced inactivation. J Biol Chem. 2001;276(2):944-51
Author contact
![]() Corresponding author: Karlheinz.Peteredu.au
Corresponding author: Karlheinz.Peteredu.au