Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2019; 9(8):2158-2166. doi:10.7150/thno.30886 This issue Cite
Research Paper
NF-κB hijacking theranostic Pt(ll) complex in cancer therapy
Yingzhong Zhu1‡, Mingzhu Zhang1‡, Lei Luo3, Martin R Gill4, Cesare De Pace5, Giuseppe Battaglia5, Qiong Zhang1, Hongping Zhou1, Jieying Wu1, Yupeng Tian1 ![]() , Xiaohe Tian1,2,5
, Xiaohe Tian1,2,5 ![]()
1. Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei 230601, P. R. China.
2. School of Life Science. Anhui University, Hefei 230039, P. R. China.
3. College of Pharmaceutical Sciences, Southwest University, Chongqing 400716, China
4. CRUK/MRC Oxford Institute for Radiation Oncology, University of Oxford, Oxford OX3 7DQ, UK
5. Department of Chemistry, University College London, London, WC1H 0AJ, UK.
‡These authors contributed equally.
Received 2018-10-23; Accepted 2019-3-11; Published 2019-4-12
Abstract

Platinum complexes have been used for anti-cancer propose for decades, however, their high side effects resulting from damage to healthy cells cannot be neglected and prevent further clinical utilisation. Here, we designed a cyclometalated platinum (II) complex that can bind the endogenous nuclear factor-κB (NF-κB) protein. Employing detailed colocalization studies in co-culture cell line models, we show that by binding to NF-κB, the platinum (II) complex is capable of upregulated nuclear translocation specifically in cancer but not normal cells, thereby impairing cancer proliferation without disturbing healthy cells. In a murine tumour model, the platinum (II) complex prevents tumour growth to a greater extent than cisplatin and with considerably lower side-effects and kidney damage. Considering its weak damage to normal cells combined with high toxicity to cancer cells, this NF-κB-binding platinum complex is a potential anti-cancer candidate and acts to verify the strategy of hijacking endogenous trans-nuclear proteins to achieve cancer-cell specificity and enhance therapeutic indices.
Keywords: Theranostic, Platinum complex, Nuclear factor kappa B, Cancer therapy
Introduction
Platinum complexes have attracted sufficient attention as anti-tumour agents and thousands of platinum complexes have been synthesized and screened as potential anticancer candidates [1-3]. However, despite the fact a few platinum agents (e.g. cisplatin, carboplatin and oxalipatin) are extremely successful in clinical applications [4,5], 'fresh blood' in metallo-drug design is required to overcome drawbacks of these classical agents and broaden the range of treatable tumours [6,7]. As a significant defect, classical platinum drugs have a small therapeutic index and barely discriminate between cancer and healthy cells. Other factors that restricted their use are systemic toxicity and the almost inevitable drug resistance [8-11]. Researchers have found that functionalized nanoparticles could favour platinum complexes to deliver in place, as well as avoiding side-effect to certain degree [12-15]. Despite the efforts made in manufacturing artificial vectors for delivery, small and smart platinum molecular system (Mw < 1000) still remain advantageous, yet increasing the activity of platinum complexes in cancer cells whilst leaving normal cells unaffected would clearly be beneficial.
Endogenous trans-nuclear proteins represent one such potential target. There are several transcription factors periodically relocating between the cytosol and nucleus during inflammations including cancer [16-18]. Among them, nuclear factor-κB (NF-κB), a key mediator of inducible transcription in the immune systems, plays a critical role in immunology and cancer biology [19-23]. Five NF-κB subunits have been described: p50 (NF-κB1), p52 (NF-κB2), p55 (Rel A), p65 (Rel B), and c-Rel (Rel) [24]. It is known that in resting cells, NF-κB is inactive, residing predominantly in the cytosol where it is bound to IκB inhibitory proteins [19-22]. On the contrary, in cells infracted by inflammation and the genetic onset of cancer, NF-κB is activated through subsequent polyubiquitylation and proteasomal degradation, allowing NF-κB to translocate to the nucleus where it drives the expression of the genes associated with cell proliferation, angiogenesis, metastasis, tumor promotion, inflammation and suppression of apoptosis to affect/control the inflammation and cancer [25-28]. NF-κB intracellular trafficking therefore offers an elegant possibility to hijack it as an endogenous vector to deliver agents to the nucleus of cancer cells, discriminately and significantly impacting cancer cells whilst sparing healthy tissue.
In addition to cisplatin and its derivatives, the study of neutral platinum complexes with cyclometalated 2,6-diphenylpyridine ligand (C^N^C-H2) has been an area of considerable interest [29] and numerous pincer platinum complexes with anti-cancer properties have been described [12,30]. Notably, this includes work showing tridentate 2,6-diphenylpyridine square planar platinum complexes are able to inhibit NF-κB-dependent gene transcription [31]. Advantages of cyclometalated Pt complexes for bio-applications include the fact many are neutral molecules and so will not influence the extracellular/intracellular charge balance [32] while certain complexes emit from long-lived metal-to-ligand or ligand-to-ligand charge transfer states, providing a convenient method to track intracellular localization using fluorescence microscopy [33].
Here, we present the synthesis and biological activity of the cyclometalated Pt complex, [Pt(DMSO)(2,4,6-triphenylpyridine)]. This Pt complex consists of a planar cyclometalated tridentate ligand, 2,4,6-triphenylpyridine, where the two Pt-C bonds resulting a high persistent in extra-dilute solution and biotic environment than traditional Pt complexes with less heavy metal toxicity. The remaining coordination site is occupied by dimethyl sulfoxide, a ligand that could maintain the molecular stability in aqueous solution as well as be substituted by a stronger coordinating bio-species. We find that this Pt complex is able to bind NF-κB protein and shows strong co-localisation with NF-κB in cell co-staining experiments. Interestingly, in cancer cells, the platinum-bound active NF-κB protein complex was transported to the cell nucleus, which led to a high rate of apoptosis. In normal cells, however, the platinum complex remained in the cytosolic region of cells with minor cytotoxicity. The finding that an emissive Pt complex may distinguish normal cells and cancer cell by hijacking NF-κB protein not only exploits a new concept in the design of next-generation platinum drugs but also inspires the development of new therapeutics based on hijacking endogenous transcription factors.
Results and Discussion
Design and synthesis
We synthesized [Pt(DMSO)(2,4,6-triphenylpyridine)] (referred to as “Pt complex” hereafter) in a mild and straightforward method according to a mechanism described in the literature (Figure 1A-B and Scheme S1) [29]. After purification, the molecular structure of Pt complex was confirmed by 1H NMR, 13C NMR, 195Pt NMR, MALDI-TOF-MS spectra and crystal structural analysis (Figure S1-S3, Table S1-S2).
The molecular (A) and single-crystal (B, H atoms and solvent molecules were omitted for clarity) structure of Pt complex. (C) 2D, (D) 3D, (E) van der Waals interactions of Pt complex and inactive NF-κB protein by molecular modelling calculation.
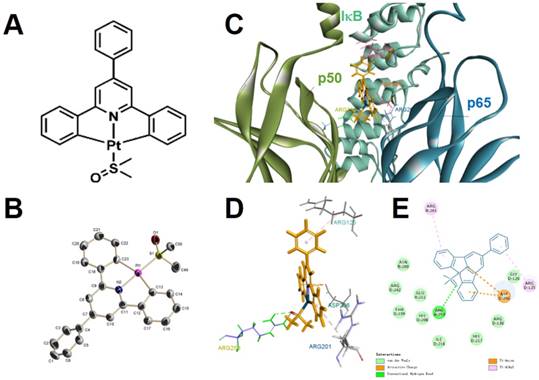
We conducted molecular modelling calculation to examine the binding ability between Pt complex and NF-κB protein by using Discovery Studio 4.1. For inactive NF-κB protein, the Pt complex inlays the cavity consisted by three polypeptides (p50, p60, iκB) and van der Waals interaction could be observed between Pt complex and inactive NF-κB protein with maximum binding energy -37.5294 Kcal/mol, (Figure 1C-D and Table S3). For active NF-κB protein, the primary interactions were ionic electrostatic action and hydrogen bonding which were generated between Pt complex and p50 polypeptides with maximum binding energy -30.5267 Kcal/mol, (Figure S4, Table S3). Considering the maximum binding energy, Pt complex was easier to separate from the active NF-κB protein than the inactive protein.
Photophysical properties
The absorption spectrum of Pt complex showed peak maxima at ~275 nm, ~350 nm and a broad absorption appearing at 375-550 nm (Figure S5). There is a small solvatochromic shift (~ 5 nm) upon changing the solvent from dichloromethane to acetonitrile. Due to non-radiative processes concerning low-lying d-d excited states and fast non-radiative decay rate, Pt complex was non-emissive in fluid solutions at room temperature. However, Pt complex displayed vibronically structured emission as a glassy solution in 2-MeTHF at 77K (Figure S6).
Visualizations of Pt complex in cells
According to the literature, certain cyclometalated Pt complexes display non-emission/weak emission in aqueous solutions, however, when combined with macromolecules, such as protein, DNA etc., distinct activation of emission or emission enhancement could be observed [34,35]. This is the result of hydrophobic interactions, which restrict the vibrations of platinum complexes, and the shielding of platinum complexes from O2 by the macromolecules.
We therefore attempted to evaluate the intracellular distribution of Pt complex in normal cells and cancer cells under confocal imaging. Firstly, five types of normal cells including HELF (Human embryo liver fibroblast), Astrocyte (mouse), bEND3 (mouse brain endothelium), 293T (Human embryonic kidney cells), and NIH 3T3 cells (mouse embryo fibroblast) were selected to incubate with 10 μM Pt complex for 30 min (Figure 2(A)). The micro-graphs along with a magnified region that indicated single cell clearly displayed the fluorescent signals with good photon-stability in cytosolic space but highly excluded from the nuclear region (Figure S7 and 2A). This is further confirmed via analysis of normalised fluorescence intensity, in which the cytosolic emission was much greater than that in the nucleus (Figure 2A, bar graphs). Strikingly, and in stark contrast to the results for normal cell lines, all nine evaluated cancer cell lines incubated with Pt complex (HepG2, HSS5787, Hela, MCF-7, H460, SHSY5Y, MDA-MB-231, A549, and U2O3 cells) displayed strong emission from the nucleus (Figure 2B). In contrast, the cytosolic fluorescence indicated relatively weaker internalisation in this cellular region (e.g. HepG2 and H460 cells). Along with the high stability of Pt complex in cell culture media (Figure S8), these results are consistent with the observed emission to be the result of the interaction of the Pt complex with a cellular protein.
Confocal imaging of normal cells (A) and cancer cells (B) incubated with Pt complex (10 μM, 1 h, λex = 405 nm, λem = 480-520 nm). The column picture in (A) and (B) was the emission intensity analysis (average per area) from the nuclear and cytosolic region, respectively. (C) The LPS inactive/active RAW264.7 cells treated with Pt complex (10 μM, 1 h). Scale bars = 10 μm, data point = 30 cells, Error bar SEM, p < 0.005.
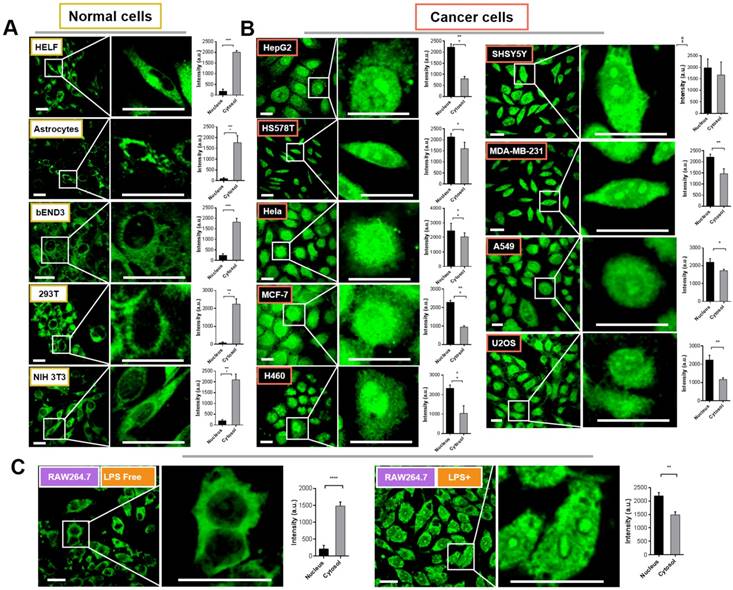
To further understand the differential subcellular distribution between normal and cancer cells, the murine macrophage cell line RAW264.7 cells were chosen as a healthy/inflammatory cell model. It is known that macrophages as an immunocyte are rich in NF-κB protein with an inactive form within the cytosol, yet by external stimulations (e.g. lipopolysaccharide, LPS), RAW264.7 cells undergo inflammation and the NF-κB protein is activated and shuttled between nucleus and cytosol [16]. As shown in Figure 2(C), the emission signal of Pt complex is principally within the cytosol in inactive RAW264.7 cells; consistent with previous results employing normal cells. Interestingly, when cells were stimulated with LPS, the Pt complex displayed clear fluorescent emission in the nucleus.
Transmission electron microscopy (TEM) and ICP-MS analysis
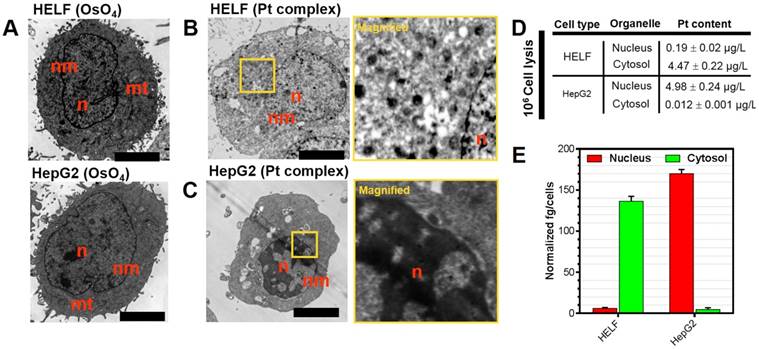
Platinum complexes with high electron density are ideal for scattering the electron beam used in transmission electron microscopy and this method may provide information on their intracellular distribution [36]. Accordingly, Pt complex-treated HELF and HepG2 cells (representatives for normal and cancer cells, respectively) were processed for TEM imaging. A classic membrane-localizing contrast agent osmium tetroxide (OsO4) was introduced as a control. It is shown in Figure 3A-C that HELF and HepG2 stained solely with OsO4 displayed clear contrast in subcellular membrane including mitochondria and nuclear membrane structures. Visualizing Pt complex-treated cells with no other contrast agent applied after TEM preparation did not provide any membrane signal, but significant accumulation in the nuclear region in cancer cells was apparent. In contrast, negligible signal in the nucleus of normal cells was observed. In a parallel experiment under similar treatment, inductively coupled plasma mass spectrometry (ICP-MS) was used to quantify the platinum content in isolated cytosol and nuclear fractions of Pt complex-treated cells (Figure 3(D)). ICP-MS indicated that platinum content was 0.19 ± 0.02 μg/L in the nucleus and 4.47 ± 0.22 μg/L in the cytosol for normal HELF cells. On the contrary, the content was 4.98 ± 0.24 μg/L in the nucleus and 0.012 ± 0.01 μg/L in the cytosol for cancer HepG2 cells. The normalized platinum substances in each cell also indicated the same distribution pattern (Figure 3E). However, compared with Pt complex, the ICP-MS data of cisplatin (Table S4) showed no obvious organelles distribution difference in normal and cancer cells.
(A) Untreated HELF or HepG2 cells stained with OsO4. (B,C) HELF or HepG2 cells treated with Pt complex (10 μM) for 30 min before processing for TEM. No other contrast stain was employed. (D) and (E) ICP-MS quantification of internalized Pt complex in nucleus and cytosol by HELF and HepG2 cells. Scale bars = 5 μm.
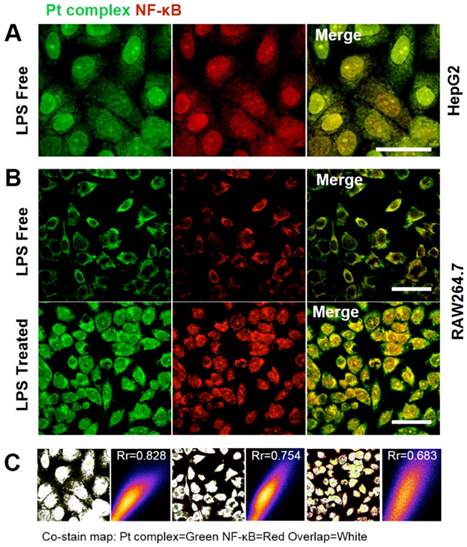
Co-localization with NF-κB protein by immunofluorescence method
The above experiments strongly suggested that the synthetic Pt complex displayed distinct intracellular distribution properties in normal compared to cancer cells and we speculated that this might be due to the hijacking of NF-κB protein by Pt complex. To explore this further, immunofluorescence was applied to determine cellular NF-κB localisation. Figure 4 shows the strong co-localization of Pt complex with NFκB (2nd ab: Cy3, λex = 550 nm, λem = 560-600 nm) in HepG2 cells, strongly implying that the Pt complex signal corresponds to NF-κB protein (Rr = 0.828). Importantly, LPS induced NF-κB protein translocation using the RAW264.7 inflammation cell line model confirmed that the transport of Pt complex in cells is NF-κB protein dependent, with positive Pearson coefficients of 0.754 and 0.683 for the LPS free and LPS treated group, respectively. In addition to these cellular experiments, protein electrophoresis analysis and fluorescence emission spectra using commercial purified NF-κB protein directly confirms cell-free binding interactions between platinum complex and NF-κB protein (Figure S9 and S10).
(A) Co-localization studies of Pt complex (10 μM, 30 min) with NF-κB protein in HepG2 cells. NF-κB protein localization determined by immunofluorescence and Cy3-labelled secondary antibodies. (B) Co-localization of Pt complex with NF-κB protein in LPS untreated/treated RAW264.7 cells. (C) The Pearson's correlation coefficient of Pt complex and Cy3 marked NF-κB protein. Scale bars = 20 μm.
Identification of normal and cancer cells in a metastases model
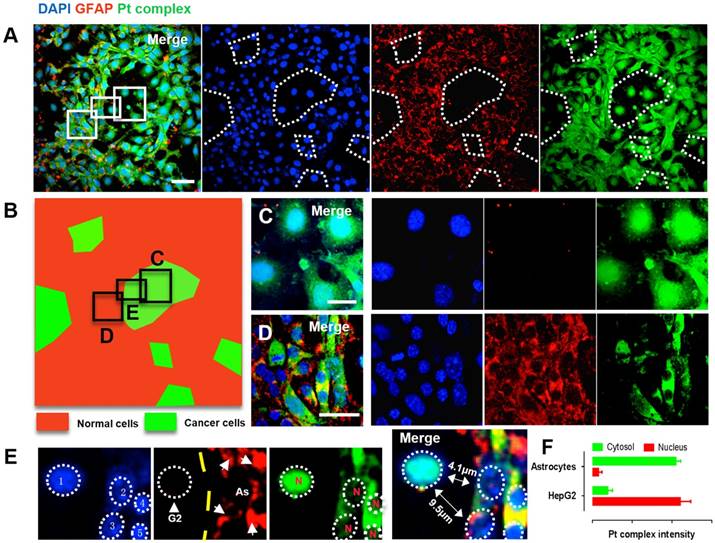
Brain metastases originating from liver, lung or other types of cancer represent a substantial threat to life [37,38]. Since the interaction between Pt complex and NF-κB protein was confirmed, we were therefore motivated to explore this concept in a multi-culture environment. Cancer liver HepG2 cells and normal parenchymal Astrocytes cells (cell number ratio 1:10) were co-cultured and distributed randomly in a single well plate to mimic a simple brain metastases model. In order to identify the difference between cancer cells and normal cells under confocal microscopy, the parenchymal astrocytes was immunofluorescently stained with GFAP (glial fibrillary acidic protein, 2nd ab: Cy5, λex = 633 nm, λem = 640-680 nm) while all cells were indicated by DAPI (4',6-diamidino-2-phenylindole). Using this method, two distinct populations was observed; positive GFAP staining showing normal astrocytes while regions of negative GFAP staining but with positive DAPI signal indicate the HepG2 population (Figure 5A). This was converted to a two-colour sketch map (Figure 5B Red: normal cells; Green: cancer cells), where magnified macrophages from red and green region clearly displayed normal cell and cancer cell morphology with differentiated Pt complex distribution pattern (Figure 5C and 5D). Notably, the cancer cells that could be distinguished displayed intense Pt complex signal that overlapped with DAPI originating from the nucleus (Figure 5E and 5F). This is consistent with previous results and support the concept that Pt complex can highjack NF-κB protein and accumulate within cancer cell nuclei, while remaining deactivated form within the cytosolic region in normal cells.
(A) Confocal imaging of co-cultured normal cells (HepG2 cells) and cancer cells (Astrocytes cells): DAPI (Blue), GFAP (Red), Pt complex (Green), and Merge channel. (B) Schematic representation of normal cells and cancer cells population distribution from captured micrograph. (C) GFAP negative cells represent cancer cells with intense nuclear uptake. (D) GFAP positive astrocytes displayed cytosolic uptake without nuclear staining. (E) A zoom in micrograph showed Pt complex different intracellular distribution in the co-culture model from 5 cells including 1 HepG2 cell and 4 astrocytes. (F) Pt complex fluorescence analysis at nuclear and cytosol from astrocytes and HepG2 cells. Scale bars = 20 μm. Abbreviations: G2: HepG2 cells; As: Astrocytes; N: nucleus.
The cytotoxicity of Pt complex towards cancer cells and normal cells
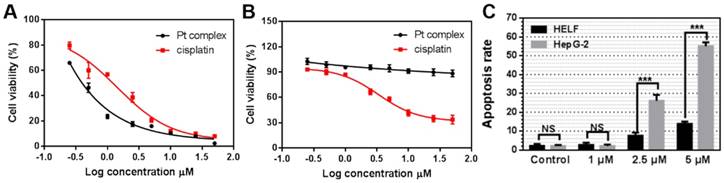
Considering the identified capability of Pt complex to target the nucleus of cancer - but not normal - cells, we measured its cytotoxicity by MTT method. HELF and HepG2 cells were chosen as normal and cancer cells, respectively. As shown in Figure 6A, B, Pt complex displayed high toxicity to HepG2 cells (IC50 = 0.28 μM) after 48 h incubation. Significantly reduced cytotoxicity towards normal HELF cells was observed, with negligible impact on cell viability observed even at high concentration (IC50 >50 μM). In comparison, cisplatin presented a nine-fold lower toxicity (IC50 = 2.56 μM, Figure 6A) to HepG2cancer cells and higher toxicity (IC50 = 3.36 μM, Figure 6B) to normal cells than Pt complex. To examine the mechanism of cell death, levels of apoptosis in cancer and normal cells treated with Pt complex was examined employing Annexin V staining and flow cytometry analysis. These results indicated substantially higher levels of early and late apoptotic cells in HepG2 cancer cells than normal HELF cells (Figure 6C, S11, 12), consistent with the death of cancer cells caused by Pt complex induced-apoptosis with low impact towards normal cells.
The Cytotoxicity of Pt complex (black line) and cisplatin (red line) (0.25 - 50 μM) against HepG2 (A) and HELF (B) cell lines (48 h incubation time). (C) The apoptosis rate of HELF and HepG2 cells cultured with Pt complex, as determined by AnnexinV/PI staining and flow cytometry analysis (24 h incubation time).
The UV-visible and emission spectra of Pt complex (Figure S13) were altered with the titration of DNA [39], indicating a form of reversible DNA binding. Meanwhile, comparing cells treated with or without DNA hydrolase, the emission intensity of Pt complex was stable (Figure S14). These results suggest that Pt complex can interact with DNA and operates by an altered anti-cancer pathway compared to cisplatin, which might be attributed to interference with intracellular NF-κB protein function or ligand-DNA interactions. However, determination of the precise mechanism of action would require further investigation. These results confirmed preferential activity of Pt complex towards cancer cells compared to normal cells and implied Pt complex might have lower side effects than cisplatin, thus offering the possibility to utilise the complex in vivo.
Anticancer ability in mice model and less side-effect towards kidney
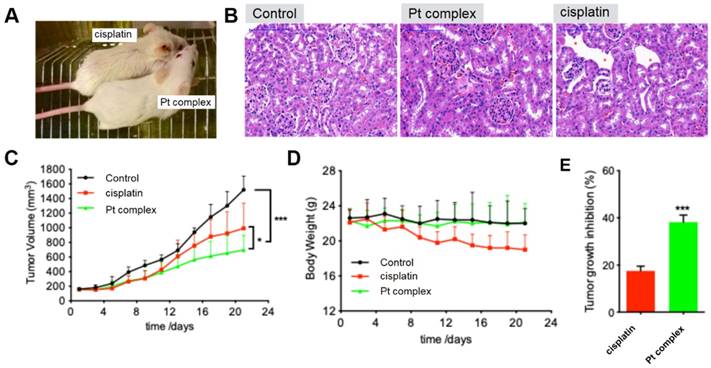
As indicated above, the Pt complex could selectively damage cancer cell in vitro. An untested cell, 4T1 cells, was chosen as cancer cell model to further confirm anti-cancer ability of Pt complex in vivo. With high cytotoxicity to 4T1 cells (Figure S15) and excellent penetration in 3D cancer cells tumour spheroids (Figure S16-17), studies with mouse xenograft models were performed. The mouse with solid tumour planted (n=6, 4T1 cells, flank injection) was treated with PBS, cisplatin, or Pt complex, respectively (3 weeks). Figure 7A directly presented that cisplatin treated mice showed significant side effects and gross changes in the skin including hair loss and messy pelage hair revealing considerable body damage; side effects that were not found in the Pt complex treatment group. Hematoxylineosin (HE) staining of kidney tissue sections of treated animals showed the region of cortex reins clearly presented that cisplatin had substantial damage to glomeruli kidney including decreasing glomerular volume and necrotic cells (Figure 7B). Conversely, the morphology of glomerular was complete with slight inflammation in Pt complex group. It is also encouraging to note that with extended treatment time (11 days), the growth of tumours in the Pt complex group were significantly inhibited and to a greater extent than animals treated with the clinical agent (Figure 7C and 7E). Furthermore, the body weight of the Pt complex group remained similar as for non-treated animal, while a decrease was observed as a result of cisplatin treatment (Figure 7D); findings consistent with reduced side-effects from Pt complex compared to cisplatin.
(A) Gross morphology of mice after intratumoural injection with Pt complex and cisplatin (3 mg/kg Pt drug at 1st, 3rd, and 5th day) at 11th day. (B) H&E staining of mice kidney treated with PBS, Pt complex, and cisplatin, the red-star indicated the damaged kidney region (C) The tumour volumes of mice model (murine breast cancer model, 4T1 cell) over 21 days (Injected doses: 20 μL for PBS, 3 mg/kg for cisplatin, 3 mg/kg for Pt complex. Injected on 1st, 3rd, and 5th day). (D) Mouse body weight over 21 days for animals treated as in (C). (E) Tumour growth inhibition rate with Pt complex or cisplatin treatment at 11 day for animals treated as in (C).
Conclusion
In summary, a novel Pt complex was designed and synthesized based on both theoretical and experimental methods, and cellular studies showed this complex to possess high anti-cancer activity with little effect on normal cells. The anti-cancer mechanism was that Pt complex could identify the cancer cells and normal cells by hijacking endogenous nuclear factor kappa B protein. More importantly, its high toxicity to cancer cells and low damage to healthy cells could also be applied in mouse model experiment. Due to its non-invasiveness against normal tissues, Pt complex could be used as a potential anti-cancer candidate with an improved therapeutic index and reduced safety concerns compared to cisplatin.
Supplementary Material
Supplementary methods, scheme, figures and tables.
Acknowledgements
This work was supported by a grant for the National Natural Science Foundation of China (21602003, 51432001, 51672002, 21871003 and 51772002), Anhui Provincial Natural Science Foundation of China (1708085MC68), and Anhui University Doctor Startup Fund (J01001962), Returnees innovation and entrepreneurship key support program, the Higher Education Revitalization Plan Talent Project (2013).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xiao H, Qi R, Li T. et al. Maximizing synergistic activity when combining RNAi and platinum-based anticancer agents. J Am Chem Soc. 2017;139:3033-3044
2. Thiabaud G, McCall R, He G. et al. Activation of Platinum (IV) Prodrugs By Motexafin Gadolinium as a Redox Mediator. Angew Chem Int Ed. 2016;128:12816-12821
3. Ritacco I, Mazzone G, Russo N. et al. Investigation of the Inertness to Hydrolysis of Platinum (IV) Prodrugs. Inorg Chem. 2016;55:1580-1586
4. Johnstone TC, Suntharalingam K, Lippard SJ. The next generation of platinum drugs: targeted Pt (II) agents, nanoparticle delivery, and Pt (IV) prodrugs. Chem Rev. 2016;116:3436-3486
5. Legin AA, Theiner S, Schintlmeister A. et al. Multi-scale imaging of anticancer platinum (IV) compounds in murine tumor and kidney. Chem Sci. 2016;7:3052-3061
6. Wang X, Wang X, Guo Z. Functionalization of platinum complexes for biomedical applications. Acc Chem Res. 2015;48:2622-2631
7. Wilson J. J, & Lippard, S. J. Synthetic methods for the preparation of platinum anticancer complexes. Chem Rev. 2013;114:4470-4495
8. Sancho-Martínez SM, Prieto-García L, Prieto M. et al. Subcellular targets of cisplatin cytotoxicity: an integrated view. Pharmacol Ther. 2012;136:35-55
9. Köberle B, Tomicic MT, Usanova S. et al. Cisplatin resistance: preclinical findings and clinical implications. BBA Rev Cancer. 2010. 1806:172-182
10. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573-584
11. Kuo WY, Hwu L, Wu CY. et al. STAT3/NF-κB-Regulated Lentiviral TK/GCV Suicide Gene Therapy for Cisplatin-Resistant Triple-Negative Breast Cancer. Theranostics. 2017;7:647-663
12. Tsai JLL, Zou T, Liu J. et al. Luminescent platinum (II) complexes with self-assembly and anti-cancer properties: hydrogel, pH dependent emission color and sustained-release properties under physiological conditions. Chem Sci. 2015;6:3823-3830
13. Zhu Z, Wang Z, Hao Y. et al. Glutathione boosting the cytotoxicity of a magnetic platinum (iv) nano-prodrug in tumor cells. Chem Sci. 2016;7:2864-2869
14. Dai Y, Xiao H, Liu J. et al. In vivo multimodality imaging and cancer therapy by near-infrared light-triggered trans-platinum pro-drug-conjugated upconverison nanoparticles. J Am Chem Soc. 2013;135:18920-18929
15. Miller MA, Zheng YR, Gadde S. et al. Tumor associated macrophages act as a slow-release reservoir of nano-therapeutic Pt (IV) pro-drug. Nat Commun. 2015;6:8692
16. Zambrano S, De Toma I, Piffer A. et al. NF-κB oscillations translate into functionally related patterns of gene expression. Elife. 2016;5:e09100
17. Purvis JE, Lahav G. Encoding and decoding cellular information through signaling dynamics. Cell. 2013;152:945-956
18. Deorukhkar A, Krishnan S. Targeting inflammatory pathways for tumor radiosensitization. Biochem Pharmacol. 2010;80:1904-1914
19. Qin X, Yan M, Wang X. et al. Cancer-associated Fibroblast-derived IL-6 Promotes Head and Neck Cancer Progression via the Osteopontin-NF-kappa B Signaling Pathway. Theranostics. 2018;8:921-940
20. Baud V, Karin M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33-40
21. Wang S, Liu Z, Wang L. et al. NF-[kappa] B signaling pathway, inflammation and colorectal cancer. Cell Mol Immunol. 2009;6:327
22. Wang H, Liang L, Dong Q. et al. Long noncoding RNA miR503HG, a prognostic indicator, inhibits tumor metastasis by regulating the HNRNPA2B1/NF-κB pathway in hepatocellular carcinoma. Theranostics. 2018;8:2814-2829
23. Cummins EP, Keogh CE, Crean D. et al. The role of HIF in immunity and inflammation. Mol Aspects Med. 2016;47:24-34
24. Ghosh S, Hayden MS. New regulators of NF-κB in inflammation. Nat Rev Immunol. 2008;8:837-848
25. Pasparakis M. Regulation of tissue homeostasis by NF-κB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778-788
26. Hayden MS, Ghosh S. NF-κB in immunobiology. Cell Res. 2011;21:223-244
27. Esatbeyoglu T, Huebbe P, Ernst I. et al. Curcumin—from molecule to biological function. Angew Chem Int Ed. 2012;51:5308-5332
28. Lee CH, Jeon YT, Kim SH. et al. NF-κB as a potential molecular target for cancer therapy. Biofactors. 2007;29:19-35
29. Kui SCF, Chui SSY, Che CM. et al. Structures, photoluminescence, and reversible vapoluminescence properties of neutral platinum (II) complexes containing extended π-conjugated cyclometalated ligands. J Am Chem Soc. 2006;128:8297-8309
30. Cutillas N, Yellol GS, de Haro C. et al. Anticancer cyclometalated complexes of platinum group metals and gold. Coordin Chem Rev. 2013;257:2784-2797
31. Liu J. Sun, RWY, Leung CH. et al. Inhibition of TNF-α stimulated nuclear factor-kappa B (NF-κB) activation by cyclometalated platinum (II) complexes. Chem Commun. 2012;48:230-232
32. Tian X, Zhu Y, Zhang M. et al. Mild acidic-enhanced mitochondrial-targeting by a neutral thiophene based terpyridine molecule with large two-photon action cross-section. Dyes Pigments. 2017;139:431-439
33. Yam VWW, Tang RPL, Wong KMC. et al. Syntheses, Electronic Absorption, Emission, and Ion-Binding Studies of Platinum (II) C^N^C and Terpyridyl Complexes Containing Crown Ether Pendants. Chem Eur J. 2002;8:4066-4076
34. Mauro M, Aliprandi A, Septiadi D. et al. When self-assembly meets biology: luminescent platinum complexes for imaging applications. Chem Soc Rev. 2014;43:4144-4166
35. Zou T, Lok CN, Fung YME. et al. Luminescent organoplatinum (II) complexes containing bis (N-heterocyclic carbene) ligands selectively target the endoplasmic reticulum and induce potent photo-toxicity. Chem Commun. 2013;49:5423-5425
36. Gill MR, Garcia-Lara J, Foster SJ. et al. A ruthenium (II) polypyridyl complex for direct imaging of DNA structure in living cells. Nat Chem. 2009;1:662-667
37. Chen EI, Hewel J, Krueger JS. et al. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007;67:1472-1486
38. Costa DB, Shaw AT, Ou SHI. et al. Clinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastases. J Clin Oncol. 2015;33:1881-1888
39. Ho YP, Au-Yeung SCF, To KKW. Platinum-based anticancer agents: Innovative design strategies and biological perspectives. Med Res Rev. 2003;23:633-655
Author contact
![]() Corresponding authors: yptianedu.cn; and xiaohe.tedu.cn
Corresponding authors: yptianedu.cn; and xiaohe.tedu.cn