Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(8):2167-2182. doi:10.7150/thno.30867 This issue Cite
Research Paper
GRP78-targeted ferritin nanocaged ultra-high dose of doxorubicin for hepatocellular carcinoma therapy
Bing Jiang1,2, Ruofei Zhang1,2, Jianlin Zhang1, Yaxin Hou1,2, Xuehui Chen1, Meng Zhou1, Xiuyun Tian3, Chunyi Hao3, Kelong Fan1 ![]() , Xiyun Yan1,2
, Xiyun Yan1,2 ![]()
1. Key Laboratory of Protein and Peptide Pharmaceuticals, CAS-University of Tokyo Joint Laboratory of Structural Virology and Immunology, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
2. College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China.
3. Key laboratory of Carcinogenesis and Translational Research, Department of Hepato-Pancreato-Biliary Surgery, Peking University Cancer Hospital & Institute, Beijing, 100142, China
Received 2018-10-22; Accepted 2018-12-29; Published 2019-4-12
Abstract

Hepatocellular carcinoma (HCC) remains one of the leading causes of cancer deaths, primarily due to its high incidence of recurrence and metastasis. Considerable efforts have therefore been undertaken to develop effective therapies; however, effective anti-HCC therapies rely on identification of suitable biomarkers, few of which are currently available for drug targeting.
Methods: GRP78 was identified as the membrane receptor of HCC-targeted peptide SP94 by immunoprecipitation and mass spectrum analysis. To develop an effective anti-HCC drug nanocarrier, we first displayed GRP78-targeted peptide SP94 onto the exterior surface of Pyrococcus furiosus ferritin Fn (HccFn) by genetic engineering approach, and then loaded doxorubicin (Dox) into the cavities of HccFn via urea-based disassembly/reassembly method, thereby constructing a drug nanocarrier called HccFn-Dox.
Results: We demonstrated that HccFn nanocage encapsulated ultra-high dose of Dox (up to 400 molecules Dox/protein nanocage). In vivo animal experiments showed that Dox encapsulated in HccFn-Dox was selectively delivered into HCC tumor cells, and effectively killed subcutaneous and lung metastatic HCC tumors. In addition, HccFn-Dox significantly reduced drug exposure to healthy organs and improved the maximum tolerated dose by six-fold compared with free Dox.
Conclusion: In conclusion, our findings clearly demonstrate that GRP78 is an effective biomarker for HCC therapy, and GRP78-targeted HccFn nanocage is effective in delivering anti-HCC drug without damage to healthy tissue.
Introduction
As one of the most common fatal malignancies, hepatocellular carcinoma (HCC) accounts for the second cause of cancer death worldwide [1]. Due to the high incidence of recurrence and metastasis of HCC, patients with HCC typically exhibit poor prognosis [2]. Clinical routine treatments for HCC include surgical resection, chemotherapy and radiation therapy, as well as other surgical approaches. However, relapse and metastasis of HCC commonly occur following surgical therapy [3]. Until now, only one chemotherapeutic drug, Sorafenib (Nexavar, a kinase inhibitor drug), was approved by FDA in 2008 as first-line treatment of HCC [4]. However, on average, this drug prolongs the overall survival of HCC patients for only two months [5]. Several other chemotherapeutic agents, including Dox, cisplatin, mitomycin, and epirubicin, have recently been evaluated for HCC treatment [6]. However, none of them exhibited clear clinical benefits due to their nonspecific biodistributions in vivo, which resulted in insufficient drug accumulation in tumors and severe side effects [6]. In comparison, targeted drug delivery overcomes these challenges by both its passive and active targeting properties. Thus, developing effective HCC targeted drug therapy strategies is in urgent demand.
The major barrier in developing HCC-targeted drug therapies remains the lack of appropriate tumor biomarkers that serve as specific entry target into the tumor cells to be attacked. Although several biomarkers for HCC tumors have so far been reported, such as folate receptor (FR), asialoglycoprotein receptor (ASGPR), transferrin receptor (TfR), epithelial growth factor (EGF), CD44, and integrin receptors [7], none of these receptor-targeted therapeutic strategies exhibit significant benefits in clinical trials until now [6].
One earlier study reported that a peptide called SP94 specifically binds to HCC cells [8], and was subsequently used as a promising ligand (of drug-carrier) for HCC-targeted therapy [9-11]. However, the tumor-specific receptor of SP94 expressed on HCC cells remains unknown. Considering the promising HCC therapeutic potential, it is worth identifying the receptor of SP94.
In this study, we first identified the receptor of SP94 as GRP78, a 78 kDa glucose-regulated protein on the cell membrane of HCC cells. GRP78 is a member of the heat-shock protein 70 (Hsp70) families [12], and has been identified earlier to be an endoplasmic reticulum (ER) chaperone that facilitates protein folding and trafficking [13]. More importantly, GRP78 serves as a marker for ER stress [14]. The expression of GRP78 typically increases and GRP78 tends to aggregate on the cell membrane of tumor cells in response to chronic stresses [14, 15], which facilitates evasion of the tumor from immune surveillance. Simultaneously, GRP78 overexpression increases its resistance to apoptosis and tolerance to chemotherapy [16-19]. Therefore, GRP78 overexpressed on the cell surface in tumor tissues typically indicate that this type of tumor is invasive and metastatic, representing a highly malignant state of tumor. Thus, patients with tumor cells positive for cell surface GRP78 generally exhibit shorter survival times, poorer treatment effects and higher recurrence rates [20-22].
Targeted drug delivery systems based on GRP78 targeting strategy for other malignant tumors have been described in other recent studies [23-26]; however, few studies investigated the specific functions or target properties of GRP78 in HCC tumor therapy.
Here, we set out to design a GRP78-targeted HccFn-Dox nanocage structure, in which SP94 was displayed onto the exterior surface of ferritin (from Pyrococcus furiosus) nanocage (HccFn), and the drug Dox was loaded into the cavities of HccFn. Due to the cell surface GRP78-based active targeting property and passive targeting property of enhanced permeability and retention (EPR) effects, together with the high loading efficiency of Dox, our HccFn-Dox nanocages were shown to be a very effective anti-HCC drug delivery system.
HccFn nanocage possesses three advantages over currently developed anti-HCC drug delivery systems. First, due to the surface display ability of ferritin nanocage, the binding affinity of HccFn nanocage to HCC cells was dramatically increased over 700-fold when compared with SP94 peptides. Second, our experimental data showed that ferritin nanocages from Pyrococcus furiosus possess ultra-high dose of Dox loading capacity, which is an unexpected superiority of Pyrococcus furiosus ferritin over ferritin from other species. Third, ferritin nanocages from human or mouse have been reported to interact with several membrane proteins [27, 28]. To avoid the targeting interference, as well as maintain the sole HCC targeting ability of SP94 peptide, we chose Pyrococcus furiosus ferritin nanocages without targeting ability as motif to construct HccFn nanocarriers.
Results
GRP78 is a receptor of SP94 peptide
To identify the receptor of SP94 which is specifically expressed on HCC [10], we incubated HepG2 cells with biotinylated SP94. Next, a chemical cross-linker DTSSP was added to stabilize the interaction of SP94 and its potential receptors. Following SDS-PAGE, Coomassie Blue staining identified a sharp band at ~72 kDa (Fig. 1B), which was then digested with trypsin. As listed in Fig. 1C, 11 tryptic peptides were identified as GRP78 fragments using searching algorithms provided by MASCOT software. The coverage of the identified peptides was 23.55% of the GRP78 sequence, together with a probability score of 34.01 (Fig. 1C), suggesting that the protein immunoprecipitated from the HepG2 cell surface was highly homologous to the GRP78 protein. Importantly, immunoprecipitation (IP) experiments using mouse anti-GRP78 mAbs generated positive results (Fig. 1D). Together, these assays strongly suggested that GRP78 is the membrane receptor recognized by the SP94 peptide.
SP94 peptide specifically binds to GRP78 in HepG2 cells. (A) The experimental procedure of identifying the membrane receptor of SP94 peptide. (B) SDS-PAGE analysis of the membrane proteins on HepG2 cells bound to biotin-SP94. The biotinylated FPWFPLPSPYGN peptide, which is not able to bind to HepG2 cells, was used as a negative control. Bands were identified by Coomassie Blue staining. (C) The peptide sequence of the target protein was deduced by mass spectrum analysis. (D) Protein immunoprecipitated from HepG2 cell surface by biotin-SP94 peptide was recognized by mouse anti-GRP78 mAbs and visualized with HRP-coupled anti-mouse IgG. (E) The binding activity of biotin-SP94 was reduced after GRP78 gene knockdown in HepG2 cells. (F) The binding activity of biotin-SP94 was enhanced when GRP78 was overexpressed in mouse 3T3-L1 cells.
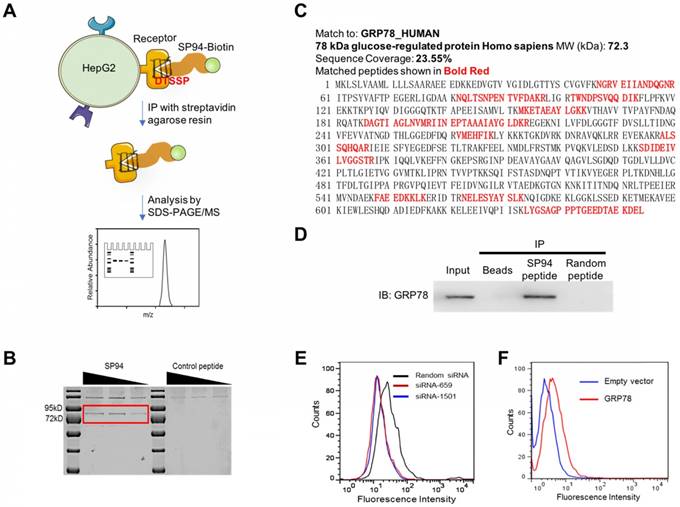
When GRP78 expression was silenced using GRP78-specific siRNA in HepG2 cells (Supplementary Fig. S1A), the binding activity of biotin-SP94 to GRP78 was decreased (Fig. 1E), whereas transient overexpression of GRP78 in 3T3 cells (Supplementary Fig. S1B) increased biotin-SP94 binding activity (Fig. 1F). These results further support that membrane GRP78 is the receptor of SP94 peptide.
A nanocaged drug carrier HccFn-Dox with high stability is developed
Having verified that SP94 specifically binds GRP78—a marker of HCC, we developed a novel nanocaged drug carrier HccFn-Dox nanocages (Fig.2A) suitable to specifically kill HCC tumor cells. First, SP94 was linked to the N-terminal of the ferritin subunit to be properly oriented and displayed on the exterior surface of the ferritin (Fn) nanocages by a genetic engineering approach (Fig. 2A), then Dox was loaded into the cavities of HccFn nanocages according to our previous report [29].
Construction and characterization of HccFn-Dox nanocages. (A) Schematic diagram of the construction of HccFn-Dox. (B) SEC analysis of HccFn-Dox. A specific absorption peak of Dox at 485 nm was detected in HccFn-Dox, but not in Fn and HccFn. (C) TEM imaging showed that HccFn-Dox possessed a well-defined morphology as a self-assembled spherical cage-like structure, similar to that of the control nanocages (Fn and HccFn). (D) DLS analysis revealed that HccFn-Dox exhibited uniform size distributions with an average radium of 8.7 nm in solution, which was close to those of 6.7, 8.6 nm for Fn and HccFn, respectively. (E) Native polyacrylamide gel electrophoresis (Native-PAGE) analysis of the HccFn-Dox. Upper panel, gel image with non-staining. Lower panel, gel image with Coomassie brilliant blue (CBB) R250 staining. (F) Stability test for Dox inside HccFn nanocages in 5% BSA and PBS buffer. No differences were found under the two conditions. (G) The release of Dox from HccFn-Dox nanocages at different pH conditions (pH 5.0 vs. pH 7.4). After 72 h, no Dox was released at pH 7.4, but a rapid release was detected at pH 5.0.
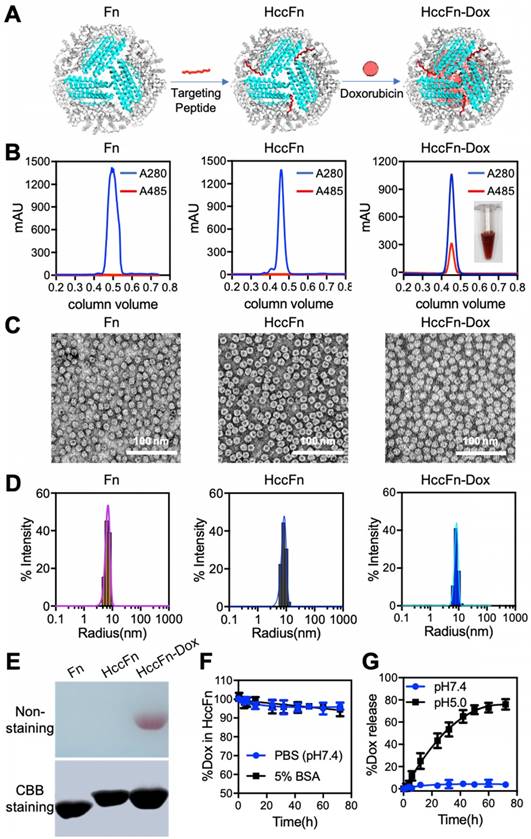
After these nanocages were purified from E. coli, we characterized their physical properties using size-exclusion chromatography (Fig. 2B) and Native-PAGE analysis (Fig. 2E). Two parameters were measured to ensure Dox was successfully loaded into our HccFn-Dox constructs. First, a specific absorption peak of Dox at 485 nm was detected in HccFn-Dox (Fig. 2B), but not in the control nanocages (Fn and HccFn). Second, a red (color of Dox) band on the gel was present in HccFn-Dox, not, however in Fn or HccFn (Fig. 2E). Importantly, the loading of Dox failed to affect the self-assembled spherical cage-like structure and monodisperse state of HccFn (Fig. 2C-D). As shown in Fig. 2C, our HccFn-Dox displayed a well-defined morphology, exhibiting a self-assembled spherical cage-like structure. Also, HccFn-Dox nanocages exhibited uniform size distributions, similar to those of the two controls. The average radius of HccFn-Dox, HccFn and Fn in solution were 8.7, 8.6, and 6.7 nm, respectively (Fig. 2D). Importantly, the amount of encapsulated Dox inside of an HccFn nanocage was determined to be 404 (Fig. S6). The Dox loading capability of HccFn was more than 10-fold higher than the average level of current reported ferritin based nanocarriers [29].
HccFn-Dox was stable under physiological conditions (pH=7.4), as indicated by lack of Dox release during a 72 h period of dialysis (Fig. 2F). However, at pH=5.0, Dox was rapidly released from the HccFn nanocages, and the portion of Dox remaining inside the cage decreased by 80% within 72 h (Fig.2G). Considering that the internal pH of lysosomes is approximately 5.0, HccFn nanocages most likely release their cargo once they are taken up by the lysosomal pathway downstream of the receptor internalization process.
HccFn-Dox nanocages specifically target HCC tumor both in vitro and in vivo
Following the successful construction of the drug nanocarrier HccFn-Dox (Fig.2A), we next tested whether HccFn-Dox specifically target HCC tumors, first in vitro, then in vivo.
HccFn were labeled with FITC and the binding of HccFn to HCC tumor cells was evaluated by confocal laser scanning microscopy (CLSM). As shown in Fig. 3A, HccFn specifically bound to the membrane of hepatocellular carcinoma cell line HepG2 in vitro, not however, the control Fn. The binding affinity of HccFn to HepG2 cells was calculated based on the flow cytometry (FACS) data. Results demonstrated that the binding affinity of HccFn to HepG2 cells was significantly higher (Kd ≤ 1 nM) than that of free SP94 peptide (Kd > 700 nM) (Fig. 3B). The cellular uptake of HccFn was reduced after GRP78 gene knockdown in HepG2 cells (Fig. S7), which further demonstrated that HccFn is taken up by the HepG2 cells via the interaction with GRP78.
HccFn-Dox nanocages specifically accumulated in HCC tumors both in vitro and in vivo. (A) After 30 min incubation of HepG2 cells with FITC-labeled HccFn and native Fn (control), FITC-HccFn was shown by CLSM (green signal) to accumulate in tumor cells. The nuclei were stained by DAPI (blue signal). Scale bar = 20 μm (B) The binding affinities of HccFn and free SP94 (as control) to HepG2 cells were calculated based on FACS data. (C) Confocal images of HccFn-Dox delivered Dox from lysosome to the nucleus in HepG2 tumor cells. HccFn-Dox were delivered into lysosome in 1h. Dox was detected in the nucleus in 24h. (Scale bar = 20 μm). (D) Following intravenous injection of Cy5.5-HccFn-Dox and Cy5.5-Fn-Dox (control) into HepG2 tumor xenografted mice, in vivo NIRF imaging (Left panel) showed that Cy5.5-HccFn-Dox accumulated specifically in the tumor area. The area containing the tumor cells is indicated by a white circle. Cy5.5 signals in the tumor area were quantitatively analyzed (Right panel, Mean±SD, n=3, ****p<0.0001, unpaired Student's t-test). (E) After different doses of Cy5.5-HccFn-Dox were intravenously injected into HepG2 tumor xenografted mice, Cy5.5-HccFn-Dox accumulated differently in HepG2 tumors (Left panel), which were further quantitatively analyzed (Mean±SD, n=3, ***p<0.001, ****p<0.0001, unpaired Student's t-test).
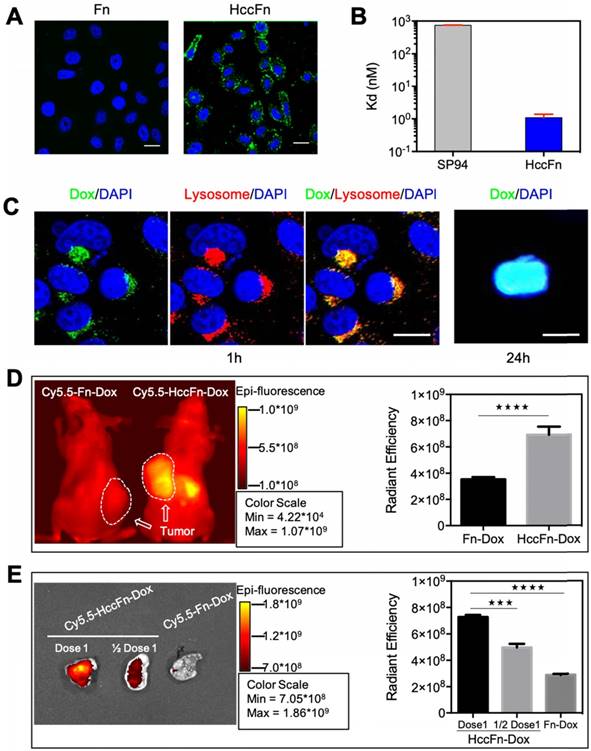
To test the hypothesis that the Dox release from HccFn nanocages occurs in endo/lysosomes, we incubated HepG2 cells with HccFn-Dox, followed by Lamp1 (a lysosomal marker) staining of live cells. After rapid binding of HccFn-Dox to the surface of HepG2 cells, the fluorescence signal of Dox merged with that of lysosome. Merging of these two signals occurred within 1h, and after 24 h, the majority of Dox was released from lysosome and transported into the cell nucleus. These results strongly suggest that HccFn-Dox nanocages are endocytosed into lysosomes whereupon Dox is released, before diffusing into the nucleus and inducing tumor cell apoptosis (Fig. 3C).
To show whether HccFn-Dox specifically target HCC tumors in vivo, Cy5.5-HccFn-Dox were intravenously (i.v.) injected into mice subcutaneously bearing HCC tumors in vivo. As shown in Fig. 3D, Cy5.5-HccFn-Dox nanocages specifically accumulated in HepG2 tumor cells, while accumulation of control nanocage Cy5.5-Fn-Dox in the tumor area was significantly lower (Fig. 3D; Fig. S2, S8). In addition to the tumor, liver and kidney, as the main organs for protein nanocages metabolism [29, 30], also exhibited HccFn-Dox and Fn-Dox accumulation (Fig. S2). Moreover, accumulation of Dox in the tumor area of HccFn-Dox treated mice was also significantly higher than that of Fn-Dox treated mice (Fig. S9).
Both our in vitro and in vivo experiments demonstrated that HccFn-Dox nanocages specifically target HCC tumors (Fig. 3A, B, D). Moreover, the accumulation of HccFn-Dox in tumor occurs to be dose-dependent (Fig. 3E; Fig. S8). Above all, these results indicate that HccFn-Dox nanocages specifically targeted HCC tumor.
HccFn-Dox nanocages deliver Dox effectively into HCC tumors, with low toxicity and long drug retention time
As our HccFn-Dox nanocages were found to specifically target HCC tumors, we next examined whether these nanocages effectively deliver the drug Dox into HCC tumors.
As shown in Fig. 4A, HccFn-Dox nanocages exhibited a longer drug retention time in plasma of mice than free Dox, suggesting that HccFn-Dox nanocages improved the levels of Dox retained in systemic circulation and facilitated time-dependent drug accumulation in tumors.
HccFn-Dox nanocages effectively delivered Dox into HCC tumors. (A) Dox concentration in plasma as a function of time post-injection of HccFn-Dox nanocages and free Dox into healthy BALB/c mice (n=3). (B) Alive tumor tissue became red after 4 hours of intravenous injection of HccFn-Dox. (C-E) Ex vivo imaging of Dox fluorescent signals in the major organs of mice after intravenous injection of HccFn-Dox. Heart, liver, spleen, lung, kidney, brain and tumor of the treated mice were collected at 1 h- (C), 4 h- (D), and 24 h-post injection (E), and these Dox fluorescent signals were quantitatively analyzed.
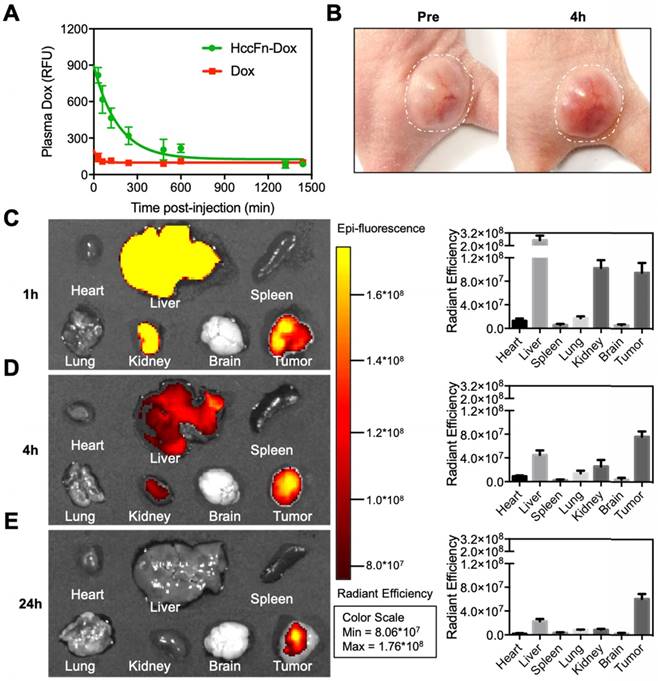
At 4h post-injection i.v. to mice bearing subcutaneous HepG2 tumors, the tumor tissue was gradually infiltrated by HccFn-Dox, as indicated by the color change (Fig. 4B). This result indicated that HccFn-Dox were successfully delivered into tumor cells.
Next, we studied the pharmacokinetic behavior of HccFn-Dox by tracking the fluorescence signal of Dox. As shown in Fig. 4C-E, at 1 h post-injection, Dox mainly accumulated in tumors, but not in other major organs except liver and kidney (Fig. 4C), suggesting that HccFn-Dox treatment will incur little toxic side effects on heart, spleen, lung or brain (further supported by the data from Fig. 4D, 4E). The amount of Dox accumulated in liver and kidney decreased rapidly at 4h post-injection and was almost completely eliminated at 24h post-injection, while the amount of Dox in tumors was kept relatively constant over the period studied (Fig. 4D, 4E). Together, these results indicated that HccFn-Dox represents as an effective drug nanocarrier characterized by high biosafety, a large portion of HccFn-Dox injected into mice was effectively delivered to tumors, and those not delivered to tumors were rapidly metabolized in liver and kidney. In contrast, tumor-targeting of HccFn-Dox nanocages is likely to be based on an active GRP78-mediated process. As a result, the drug cannot be easily eliminated from the body; instead, it is likely to be endocytosed into lysosomes and transported into cell nuclei, where it induces tumor cell apoptosis.
HccFn-Dox nanocages effectively kill HCC tumors
To test whether HccFn-Dox nanocages effectively kill HCC tumors, we determined the appropriate dosage of HccFn-Dox nanocages by first performing a tolerability study.
As shown in Fig. 5A, injection of HccFn-Dox at 20 mg/kg Dox equivalents to healthy BALB/c mice caused little visible effect since the body weight did not change. When HccFn-Dox nanocages were i.v. injected at 25 or 30 mg/kg Dox equivalents, the body weight gradually decreased before recovering to the levels of the control group. However, the recovery time following treatment of 30 mg/kg Dox equivalents was considerably longer than that for 25 mg/kg (Fig. 5A). In addition, blood chemistry and immune response parameters upon injection of HccFn-Dox at 25 mg/kg Dox equivalents failed to show significant differences compared to either HccFn- or PBS-injected mice (Table S1). Therefore, we chose HccFn-Dox at 25 mg/kg Dox equivalents for subsequent antitumor studies. In addition, we found that HccFn-Dox at 30 mg/kg Dox equivalents was the maximum dose tolerated by healthy mice, as their body weights still gradually recovered after the 11th day after injection (Fig. 5A). In comparison, the maximum tolerated dose for a single administration of free Dox was only 5 mg/kg [29]. This difference in drug tolerance clearly demonstrated that the encapsulation of Dox into HccFn nanocages significantly improves the tolerance of mice to Dox, thus enabling higher concentrations of Dox to be used in therapy.
HccFn-Dox nanocages effectively killed HCC tumor and exhibited less toxicity than Doxil. (A) Tolerability study of HccFn-Dox. HccFn-Dox at 20, 25, 30, or 40 mg/kg Dox equivalents were intravenously injected into healthy BALB/c mice (n=4 per group) on day 0, with PBS injection as control. Tumor growth (B), body weight change (C), survival time (D), and tumor cell morphology (E) for tumor-bearing mice upon injection of HccFn-Dox at 25 mg/kg Dox equivalents. HepG2 tumor cells were implanted subcutaneously into mice on day 0. Mice were intravenously administrated with HccFn-Dox (25 mg/kg Dox equivalents, n=6), and control substances such as Doxil (25 mg/kg Dox equivalents, n=6), Dox (5 mg/kg Dox equivalents, n=6), HccFn (60 mg/kg, n=6), or PBS (n=5) on day 9 and day 34. Black arrows indicated the administration time.
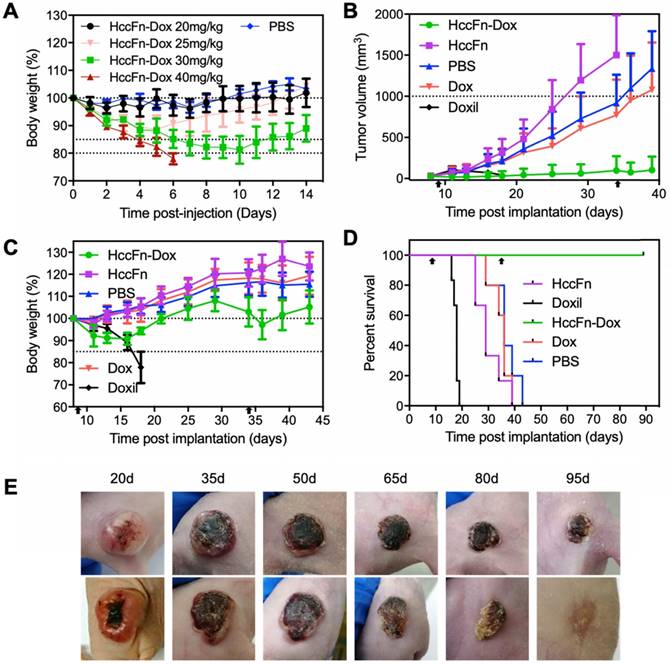
To test its therapeutic effects, we i.v. injected HccFn-Dox at 25 mg/kg Dox equivalents into mice subcutaneously bearing HCC tumors, with the first dose administrated at day 9 (when tumors reached ~50 mm3) and the second at day 34 post tumor implantation. We also administrated the same dose of non-targeted liposomal Dox (Doxil) (25 mg/kg Dox equivalents), Dox (5 mg/kg) as control. The tumor growth was significantly inhibited in HccFn-Dox administrated mice, while the tumors of the control groups grew rapidly and reached above 1,000 mm3 at day 36 (Fig. 5B). We also found complete tumor elimination from 4 of 6 HccFn-Dox-administrated mice, and all the HccFn-Dox administrated mice showed significant tumor necrosis (Fig. 5E). In addition, all HccFn-Dox administered mice survived for up to 90 d (Fig. 5D), with less than 10% weight loss after each drug application, as well as fast weight recovery (Fig. 5C). In contrast, the average survival times for mice injected with Doxil, free Dox, HccFn, or PBS were less than 36 days (Fig. 5D). Importantly, while application of Doxil effectively reduced the tumor growth rate, these mice died quickly (Fig. 5D), suggesting that at this dose, Doxil is highly toxic.
To further test the toxicity of HccFn-Dox in vivo, we evaluated multiple dose toxicity of HccFn-Dox. We injected HccFn-Dox intravenously at 5 mg/kg Dox equivalents into HCC tumors bearing mice, twice a week, 10 times in total. Multiple dosing i.v. administrations of HccFn-Dox also significantly suppressed the tumor growth (Fig. S5A). Importantly, HccFn-Dox administered mice showed minimal weight loss even after 10 doses of i.v. administrations of HccFn-Dox, while the mice failed to tolerate 5 doses of same concentration of free Dox or Doxil, indicating that HccFn-Dox exhibits excellent biosafety in mice (Fig. S5B). In two months after first administration, HccFn-Dox administered mice exhibited no marked weight loss and much longer survival time (Fig. S5C) than other groups (mice injected with Doxil, free Dox, HccFn, or PBS). This data indicated that HccFn-Dox is well tolerated in mice, showed low multiple dose toxicity.
Together, these results demonstrated the effective antitumor activity and less toxicity of HccFn-Dox, even compared to clinically approved Doxil.
HccFn-Dox nanocages suppress lung metastasis of HCC
Due to its high metastasis rate, HCC is commonly fatal [1]. In this study, we found that knockdown of GRP78 suppressed the migration of HCC cells (Fig.S3), indicating that GRP78 plays a role in the invasion and metastasis of HCC. Therefore, we hypothesized that GRP78 is suitable to be used as a therapeutic target for metastatic HCC.
As the lung is the most easily affected organ when extrahepatic metastases occur [31], we established a lung metastasis model of HCC to investigate the above hypothesis. HepG2-Luc cells (HepG2 cells stably transfected with a luciferase gene) were i.v. injected into nude mice, in which metastatic HCC tumor nodules were formed in lungs at 14-18 days post injection (Fig. 6A). After Cy5.5-HccFn-Dox was i.v. injected, it effectively accumulated in lung metastasis tissues (Fig. 6B), with a significantly higher fluorescent signal compared to that in the healthy lung (Fig. S4). Moreover, FITC-HccFn was shown to specifically recognize small HCC nodules in lung metastasis tissues from HCC bearing mice (Fig. 6C). Together, these results demonstrated that HccFn nanocages possess excellent targeting capability, effectively distinguishing metastatic lung HCC tumors from normal lung tissues.
HccFn-Dox nanocages inhibited HCC tumor lung metastases and growth in vivo. (A) Schematic diagram of the establishment of a mouse model of HCC lung metastasis. (B) In vivo fluorescent images of mice bearing metastatic lung HCC tumors (left) and healthy control mice (right) after intravenous injection of Cy5.5-HccFn-Dox for 4 h. (C) FITC-HccFn based fluorescence staining (left), and HE staining of paraffin-embedded lung slices from mice with lung metastasis tumors of HCC. (Scale bar = 200 μm). (D) Survival time for metastatic lung HCC tumor-bearing mice upon injection of HccFn-Dox at 25 mg/kg Dox equivalents. HepG2-Luc tumor cells were i.v. injected into mice on day 0. Mice were intravenously administrated with HccFn-Dox (25 mg/kg Dox equivalents, n=6), Dox (5 mg/kg Dox equivalents, n=6), or PBS (n=6) on day 14. (E) In vivo bioluminescence imaging (left) and statistical fluorescence analysis (right) of mice bearing metastatic HepG2-Luc lung tumors that were injected with HccFn-Dox at 25 mg/kg Dox equivalents. PBS was also injected into control mice (Mean ± SD, n=3. **p<0.01, unpaired Student's t-test). (F) HE staining of paraffin-embedded lung slices (left) and statistical analyses of the numbers of metastatic HCC tumor nodules (right) from HccFn-Dox administrated mice. Dox- and PBS-administrated ones were used as control. All mice were sacrificed, and lung tissues were collected at day 45 (Mean ± SD, n=4. ***p<0.001, unpaired Student's t-test). (Scale bar = 2 mm).
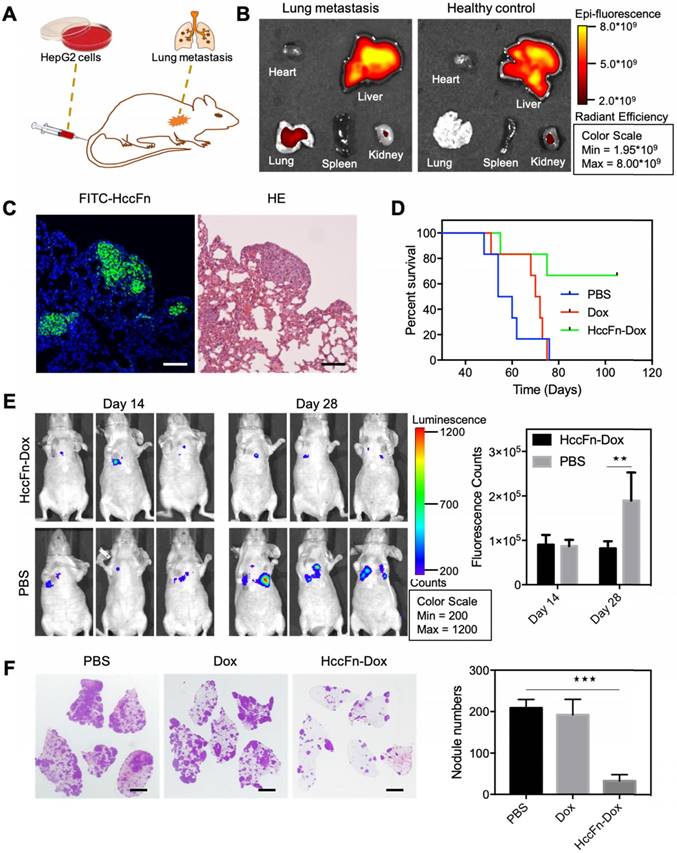
Two weeks after HccFn-Dox nanocages (25 mg/kg Dox equivalents) were i.v. injected into our mouse model, we found that the growth of metastasis lung HCC tumors was significantly inhibited (Fig. 6E, left). Statistical analysis of the fluorescence signal detected at day 28 in the metastatic HCC tumor nodules confirmed these results (Fig. 6E, right). At day 45, the number of metastatic HCC tumor nodules in the lung of HccFn-Dox treated group was significantly lower than that of control groups (Fig. 6F). In addition, 4 of 6 HccFn-Dox administered mice survived for up to 105 d (Fig. 6D). In contrast, all the mice injected with Dox or PBS died before day 76 (Fig. 6D).
Above all, our results demonstrated that GRP78 targeted HccFn-Dox nanocages significantly inhibited HCC tumor lung metastases and growth in vivo.
Discussion
In this study, we identified that cell surface GRP78 is a specific receptor of HCC targeted peptide SP94, and we developed a novel HCC-targeted drug nanocarrier, namely HccFn-Dox, which contains the ferritin nanocage with SP94 on its exterior surface and the drug Dox inside its cavity. The Pyrococcus furiosus ferritin nanocage we used in this study possesses ultra-high Dox loading capacity, is a more efficient nanodrug carrier than currently reported ferritin nanodrug carriers [29, 32]. We found that our GRP78-targeted HccFn-Dox drug delivery system significantly suppressed HCC tumor growth, and even eradicated tumors both in mice bearing subcutaneous and lung metastatic HCC tumor.
The research for HCC-targeted drugs lasts for decades due to lack of appropriate biomarkers, which is the major barrier in developing HCC-targeted drug therapy [1, 6]. Therefore, the identification of GRP78 as a biomarker for HCC represents an important progress in the field of HCC targeted therapy. The poor prognosis and high probability of HCC recurrence are largely due to the high rate of intrahepatic and extrahepatic metastases [33]. However, the underlying molecular mechanisms and biomarkers of HCC metastasis remain largely unclear. Searching the biomarkers and therapy targets for metastatic HCC will inevitably speed up the development of HCC metastasis therapy and thus prolong the overall survival time of HCC patients [34]. Here, we found that GRP78 promotes the invasion of HCC cells, and GRP78-targeted HccFn-Dox nanocages significantly suppress lung metastasis of HCC. Our results also demonstrated that cell surface GRP78 is an effective therapy target for primary and metastatic HCC tumors, thus confirming previous studies indicating that GRP78 represents a potential choice for HCC treatment [35-39]. These studies evaluated the anti-HCC efficiency of antibody-based GRP78 targeted nano-formulations by in vitro cell experiments. But none of them evaluated whether GRP78 is an effective therapy target for primary and metastatic HCC tumors in vivo.
An efficient HCC targeted nanomedicine greatly depends on the specific accumulation and high loading capacity of the potential drug carrier [7]. In this study, we used ferritin (Fn) nanocage as a motif to construct drug nanocarrier. Fn nanocages own a protein shell-cavity structure, whose cargo is loaded with high efficiency and which releases its drugs in a controlled manner. The protein shells of Fn nanocages are easily modified, either chemically or genetically, thus endowing these structures with a plethora of functionalities [32]. These features render Fn nanocage a powerful platform, with wide-ranging potential applications for clinical diagnostic and therapy.
Based on the self-assembly structure of ferritin, we believe that SP94 peptides are displayed on the 3-fold symmetry axes of the surface of ferritin in the form of 8×3 bundles. Moreover, based on our binding affinity data, these clustered SP94 peptides may be flexible for antigen binding in spatial structure.
HccFn-Dox nanocages possess excellent dual HCC-targeting ability. HccFn-Dox specifically recognizes HCC tumor cells through GRP78-based active targeting capability; meanwhile, the diameter of HccFn-Dox nanocage is approx. 17nm, the size makes HccFn-Dox nanocages easily passive penetrate tumor via EPR effects [40, 41]. Both active and passive HCC tumor targeting ability of HccFn-Dox nanocages resulted in a specific Dox accumulation at the tumor site.
Previously, Fn nanocages from human and horse were used to load Dox [42]. Here, we used Fn nanocages from Pyrococcus furiosus, as we found that they exhibit intrinsic higher Dox loading properties. We also performed experiments to compare the drug loading capacity of HccFn and Fn from Pyrococcus furiosus without SP94 fusion (Fig. S6). As we calculated, 404 Dox molecules were contained within the HccFn nanocage, and 413 Dox molecules were contained within the Pyrococcus furiosus ferritin nanocage. This result suggested that the Dox loading capacity is from the intrinsic nature of Pyrococcus furiosus ferritin. The reason for this difference might be that the inner surface structure of HccFn nanocages is more suitable for Dox binding than that of ferritin from other species.
Here, we showed that HccFn-Dox nanocages possess high drug-loading capability. This ability ensures a lower dosage of drug nanocarrier and thereafter less side effect. Thus, HccFn-Dox nanocages exhibited a better therapeutic effect than currently reported HCC-targeted nanomedicines. Most importantly, our toxicity analysis demonstrated that our HccFn-Dox exhibited excellent biosafety (Fig. S5, Table S1), even did not induce significant changes of immune cells in blood (Table S1). These unexpected results may be due to the highly conserved protein sequence and architecture between Pyrococcus furiosus ferritin and human ferritin [43]. Moreover, this study inspires us to rationally design protein cage nanocarriers according to the naturally evolved structure characteristics of Pyrococcus furiosus ferritin. Based on this work, we can conceive new ways to improve the Dox loading capacity of human ferritin-based nanocarrier and facilitate the translational research of protein cage nanocarriers.
Moreover, HccFn nanocarrier is expected to be a universal drug loading platform for tumor targeted delivery. As for HccFn nanocage, the diameter of its inner cavity is approx. 8 nm, is suitable for loading many small molecule chemotherapy drugs [32]. Previous, different types of small molecules have been successful encapsulated into ferritin nanocages [29, 44-47], indicating a potential universal drug loading ability of HccFn nanocage.
So far, there is no nanomedicine approved by FDA for HCC therapy [48], but meanwhile, there are two nanomedicines for HCC therapy are under clinical trials. One is liposomal Dox (ThermoDox), under phase III clinical trial; another is Lipid nanoparticles coated siRNA (DCR‑MYC), under phase I clinical trial. Compared with these passive targeted liposome or lipid nanocarriers, our HccFn nanocages possess GRP78-based active HCC targeting capability. In addition, HccFn nanocages are easily produced in E. coli. at a high yield (more than 50 mg/L). The drug loading of HccFn is both simple and economical. These two features make HccFn a more superior nanocarrier for HCC-targeted nanomedicine, for its easy to scale up in preparation processes.
In conclusion, we propose that GRP78-targeted HccFn-Dox nanocage is an ideal nanomedicine for HCC-targeted therapy. Also, the HccFn nanocarrier possesses a potential to be a universal chemotherapy drug nanocarrier for HCC therapy.
Materials and methods
The gene encoding peptide SP94 was constructed according to the amino acid sequence (SFSIIHTPILPL) provided by Albert et al. [8]. The gene sequence encoding Fn was obtained from NCBI (GenBank: DI342556.1 or LG067594.1), the amino acid sequence of Fn was also obtained from NCBI (NCBI Reference Sequence: WP_011011871.1). We constructed HccFn by linking the sequence of Pyrococcus furiosus Fn to the C terminus of the SP94 through a flexible amino acid sequence GGGSGGGGSGGGS. The gene encoding HccFn was amplified by overlap PCR from the DNA sequence of SP94 and Fn using two forward primers: (5' G GAA TTC CAT ATG AGC TTT AGC ATT ATT CAT ACC CCG 3'; with an NdeI restriction site) and (5' CAT GCA CTG AGG TTC TAC AAC TAC ATC TAC GAT C) and two reverse primers: (5' G ATC GTA GAT GTA GTT GTA GAA CCT CAG TGC ATG) and (5' CG GGA TCC TTA CTC TCC TCC CTG 3'; with a BamHI site). The PCR 600 bp product was inserted into the Escherichia coli (E. coli) expression vector pET-22b (+) plasmid (Novagen) with the NdeI and BamHI restriction sites. The recombinant plasmid HccFn-pET-22b (+) was subsequently transformed into E. coli DH5α (TransGen Biotech, Beijing, China), DNA sequencing was performed to select the positive colonies. An EasyPure Plasmid MiniPrep Kit (TransGen Biotech) was used to isolate the HccFn-pET-22b (+) plasmid.
To produce HccFn nanocages, the expression vector HccFn-pET-22b (+) was transformed into E. coli. Transetta (DE3) (TransGen Biotech). The transformed E. coli cells with HccFn-pET-22b (+) plasmids were cultured in LB medium with 100 mg/L ampicillin overnight. Then, HccFn protein nanocages were induced by adding 1 mM isopropyl-β-D-thiogalactoside (IPTG, Sigma), then the E. coli cells were cultured for 8h at 30°C. E. coli. cells were then harvested by centrifugation at 4,000 rpm for 15 min and the pellets were resuspended in Tris buffer (20 mM Tris, pH 8.0). Resuspended E. coli. cells were sonicated on ice and centrifuged at 12,000 rpm for 30 min. The supernatant was heat treated at 80°C for 15 min to denature and separate most E. coli. proteins. After centrifugated at 12,000 rpm for 30 min, the HccFn proteins were harvested from the supernatant and filtrated by 0.22 µm filter. Finally, HccFn protein was purified by size exclusion chromatography on a superdexTM 200pg column (GE Healthcare) followed by anion-exchange chromatography on Q-Sepharose Fast Flow (GE Healthcare). The concentration of HccFn was determined in triplicate by the BCA protein assay kit (Pierce) using bovine serum albumin as the standard. The typically yield of HccFn was 50 mg per 1 L patch.
Biophysical characterization of native Fn and HccFn nanocages
The prepared native Fn and HccFn nanocages were characterized using transmission electron microscope (TEM), and dynamic light scattering (DLS).
TEM. For TEM observation, the native Fn and HccFn nanocage samples (20μl, 0.1mg/mL) were embedded in Plasma Cleaner HPDC32G treated copper grid and stained with 1% uranyl acetate for 1 min, then imaged with a JEM-1400 80-kV TEM (JEOL, Japan).
DLS. The native Fn and HccFn protein samples (100μl, 0.25 mg/mL) were prepared in PBS buffer. DynaPro Titan (Wyatt Technology) was used to perform DLS analysis. The temperature was controlled at 25°C.
Labeling of HccFn nanocages
FITC-HccFn were prepared according to the protocol provided by Sigma-Aldrich. In brief, the FITC was dissolved in anhydrous DMSO at 1 mg/mL, then 50 μL of FITC solution was added to 2mg/ml HccFn solution in 1mL carbonate/bicarbonate buffer (100 mM carbonate, pH 9.0). The mixture was incubated at 4°C overnight. FITC-conjugated HccFn were concentrated by centrifugation at 12,000 g for 5 min, and buffer was exchanged with PBS in a Vivaspin-4 Centrifugal Concentrator (MWCO 100 kDa, Sartorius) by centrifugation at 10,000 g for 20 min. This procedure was repeated eight times. UV-Vis spectroscopy (Nanodrop 2000, ThermoFisher) was performed to determine the concentration of FITC in FITC-HccFn. Protein concentration of HccFn in FITC-HccFn was measured using a BCA protein assay kit (Pierce).
Preparation of Cy5.5-HccFn-Dox followed the identical protocol.
Cell line and Cell culture
The human hepatocellular carcinoma cell line HepG2 and mouse 3T3 cell line were purchased from American Type Culture Collection (ATCC). HepG2 cells were cultured in RPMI-1640 medium (Sigma-Aldrich) containing 10% fetal calf serum (Sigma-Aldrich), penicillin (100 U/mL, Sigma-Aldrich) and streptomycin (100 µg/mL, Sigma-Aldrich) at 37°C with 5% CO2. 3T3 cells were cultured in HDMEM medium (Sigma-Aldrich). Cells were cultured in 10 cm dishes (Corning) and passaged by trypsin-EDTA digestion every three days.
Cell-binding assays
The binding activities of native Fn and HccFn nanocages to HepG2 cells were measured on a FACSCalibur (Becton Dickinson) flow cytometry system (FACS) and confocal laser scanning microscope (CLSM).
The binding activities of Biotin-SP94 to 3T3 cells overexpressing GRP78 and GRP78 knockdown HepG2 cells were measured on a FACSCalibur (Becton Dickinson) flow cytometry system (FACS).
FACS. To perform the binding analysis, 100 µL detached HepG2 cell suspensions (2.5 x 106 cells/mL) were incubated with 0.1 nM-1 µM of Biotin-SP94 or FITC-HccFn for 45 min at 4°C in PBS containing 0.5% bovine serum albumin (BSA). Cells were washed by PBS for three times, then analyzed by a FACSCalibur flow cytometry system.
To confirm the binding activities of HccFn nanocages to GRP78, 100 µL detached control 3T3-L1 cell or GRP78 overexpressed 3T3-L1 cell suspensions (2.5 x 106 cells/mL) were incubated with 10 µg/ml of Biotin-SP94 for 45 min at 4°C in PBS containing 0.5% bovine serum albumin (BSA). 100 µL detached control HepG2 cell or GRP78 knockdowned HepG2 cell suspensions (2.5 x 106 cells/mL) were incubated with 10 µg/ml of Biotin-SP94 for 45 min at 4°C in PBS containing 0.5% bovine serum albumin (BSA). Cells were washed by PBS for three times, then analyzed by a FACSCalibur flow cytometry system.
CLSM. HepG2 cells were cultured in 35 mm confocal dishes (Corning). Once cells reached 90% confluency, the cells were washed two times with PBS, and then incubated in 5% normal goat serum for 45 min at 37°C for blocking. Cells were then incubated with 1 µg/ml of FITC-native Fn or FITC-HccFn for 1 h at 37°C in 5% normal goat serum. Cells were then washed three times with PBS and fixed in 4% cold formaldehyde in PBS for 10 min at room temperature. After washing with PBS, 4', 6-diamidino-2-phenylidole (DAPI, 1 µg/mL, Roche Applied Science) was used to stain the nuclei of cells for 10 min at room temperature. Finally, the confocal dishes were examined by a CLSM (Olympus FluoView FV-1000, Tokyo, Japan).
Construct the pEGFPN1 vector containing human GRP78
The human GRP78 gene was cloned from the human hepatocellular carcinoma cell line HepG2 cDNA library. Briefly, total RNA in HepG2 cells was extracted by 1ml Trizol, then the HepG2 cDNA library was obtained by RT-PCR. The gene encoding human GRP78 was amplified by PCR from the HepG2 cDNA library using a forward primer: (5' CCG CTC GAG ATG AAG CTC TCC CTG GTG 3', with an XhoI restriction site) and a reverse primer: (5' CG GGA TCC CAA CTC ATC TTT TTC TGC TGT ATC 3', with a BamHI restriction site). The PCR 1979 bp product was inserted into the plasmid vector pEGFPN1 (Novagen) with the XhoI and BamHI restriction sites.
The recombinant plasmid GRP78-pEGFPN1 was subsequently transformed into E. coli DH5α (TransGen Biotech, Beijing, China), DNA sequencing was performed to select the positive colonies. An EasyPure Plasmid MiniPrep Kit (TransGen Biotech) was used to isolate GRP78-pEGFPN1 plasmid.
Overexpression of human GRP78 in mouse 3T3 cells
3T3 cells were cultured in six-well culture dishes. The pEGFPN1 Mock vector and the vector containing human GRP78 were transfected into 3T3 cells using Lipo 2000 (Invitrogen). Briefly, changed the cell medium for Opti-MEM two hours before transfection. Mixed 2 μg plasmid vector and 4 μl Lipo 2000 for 20 min, then add the mixture to the dished dropwise. After cultured for 8h at 37°C with 5% CO2, changed Opti-MEM medium for HDMEM, then continue cultured for 36h. Finally, cells were harvested by 1 x SDS-PAGE sample buffer (GenStar).
Knockdown of GRP78 in human HepG2 cells
HepG2 cells were cultured in six-well culture dishes. The small interfering RNA (siRNA) (GRP78 siRNA-659, GRP78 siRNA-1501) and the Random siRNA were transfected into HepG2 cells using RNAiMax (Invitrogen). Briefly, changed the cell medium for Opti-MEM two hours before transfection. Mixed 20 pmol siRNA and 5 μl RNAiMax for 20 min, then add the mixture to the dished dropwise. After cultured for 48 h at 37°C with 5% CO2, cells were harvested by 1 x SDS-PAGE sample buffer (GenStar).
Immunoprecipitation and western blotting
The target protein of the SP94 peptide was identified by immunoprecipitation. HepG2 cells were cultured in 100mm cell culture dish (Corning). Once cells had grown to 90% confluency, cells were digested using PBS contianing 2% EDTA for 5 min at room temperature and washed two times with PBS. Cells were then incubated with 25 µg/ml of Biotin-labeled SP94 (Biotin-GGGSFSIIHTPILPL) or Biotin-labeled control peptide (Biotin-GGGFPWFPLPSPYGN) for 2 h at 4°C in PBS. The cells were washed two times by PBS, then incubated with 2 mM DTSSP (3, 3'-dithiobis [sulfosuccinimidylpropionate], Thermo Scientific) for 2 h at 4°C in PBS, before the reaction was stopped by adding 20 mM Tris-HCl. DTSSP is a chemical cross-linker, which was used to fix protein interactions in situ. Cells were lysed with RIPA Lysis Buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate) for 4 h at 4°C. Streptavidin Agarose Resin (Thermo Scientific) was added to the cell lysates and mixed thoroughly overnight at 4°C. Then the peptide-protein complexes were harvested by Centrifugation. Finally, the precipitated complexes were separated by 10% SDS-PAGE and Coomassie Blue-stained. The protein band was digested with trypsin, and the peptide fragments were identified by mass spectrum analysis.
Whether GRP78 is the binding receptor of SP94 peptide was also confirmed by immunoprecipitation. The precipitated complexes were separated by 10% SDS-PAGE and then transferred to a nitrocellulose membrane. After blocking with non-fat dry milk, the nitrocellulose membrane was incubated with a 1:1,000 dilution of mouse anti-GRP78 monoclonal antibody (mAbs, Rockland) overnight at 4°C. After washed with 0.1% Tween 20 in PBS three times, a 1:3,000 dilution of peroxidase linked goat anti-mouse antibody (GE Healthcare, UK) was incubated for 1 h at room temperature.
Western blotting was performed to confirm that human GRP78 was overexpressed in 3T3 cells and silenced in HepG2 cells. Lysates from the 3T3 cells transfected pEGFPN1 Mock vector and 3T3 cells transfected pEGFPN1 vector containing human GRP78 and lysates from the HepG2 cells transfected GRP78 siRNA-659, GRP78 siRNA-1501, random siRNA were analyzed by 10% SDS-PAGE and then transferred to a nitrocellulose membrane. After blocking with non-fat dry milk, the nitrocellulose membrane was incubated with a 1:1,000 dilution of mouse anti-GRP78 monoclonal antibody (mAbs, Rockland) and a 1:3,000 dilution of mouse anti-β-actin monoclonal antibody (mAbs, Sigma) overnight at 4°C, and developed with a 1:3,000 dilution of HRP-conjugated anti-mouse IgG.
Preparation of HccFn-Dox nanocages
Briefly, HccFn was dissolved in 8 M urea (Amredco) at 1 mg/mL and gently stirred for 30 min at room temperature to ensure fully denaturation and dissolution. Then Dox (Sangon Biotech) was added into the solution at 1 mg/mL. After incubation for 30 min at room temperature in the dark, the mixture was transferred into a dialysis bag (molecular weight cut-off 3 KDa, Thermofisher Scientific) and sequentially dialyzed against 6, 5, 4, 3, 2, 1, and 0 M urea buffer containing Dox at 1 mg/ml to refold HccFn protein shell. The solution was then dialyzed against 20mM PB buffer (pH 6.0) to remove free Dox. Finally, HccFn-Dox protein was purified by size exclusion chromatography on a SuperdexTM 200 10/300 GL column (GE Healthcare). The concentration of HccFn-Dox was determined in triplicates using a BCA protein assay kit (Pierce), with bovine serum albumin used as standard. The molar extinction coefficient of Dox was determined in PBS at 485 nm (ε = 1.00×104 M-1 cm-1) [49] and was used to determine Dox concentration in HccFn-Dox nanocages.
In vitro drug release analysis
HccFn-Dox nanocages in PBS buffer (600 μM Dox equivalents, 600 μL) were collected in the D-Tube Dialyzer (MWCO 6-8 KDa, Novagen), then dialyzed against either PBS buffer (pH 7.4) or acetate buffer (pH 5.0) at 37 °C away from light with gentle stirring. At different time points, the released free Dox in the dialysis buffer was determined by NanoDrop 2000 (Thermo Scientific) according to the absorbance peak value at 485 nm. HccFn-Dox was also dialyzed against normal mouse serum (ImmunoReagents, Inc.) for HccFn-Dox nanocage stability evaluation.
Pharmacokinetics analysis
HccFn-Dox (25 mg/kg body weight, Dox equivalents), or free Dox (25 mg/kg body weight) was injected into the tail vein of BALB/c mice (n = 5 for each group) to analyze the pharmacokinetics of HccFn-Dox. At different time points (30 min, 1 h, 2 h, 4 h, 8 h, 10 h, 22 h, 24 h) after injection, 10 μL blood was collected from the tail vein and mixed with 5 μL sodium citrate anticoagulant (DingGuo Biotec, Beijing). The blood sample mixture was centrifuged at 2000 rpm for 15 min to separate the supernatant. 10 μL supernatant was mixed with 490 μL of acidified isopropanol (0.75 M HCl in isopropanol) at -20°C overnight and away from light. After centrifugation at 15000 rpm for 10min and the supernatant was collected into a 96-well plate (Black Polystyrene, Costar, Corning). The concentration of Dox extracted from blood was determined by measuring the fluorescence at 485 nm excitation and 590 nm emission via Multiscan Spectrum EnSpire (Perkin Elmer, USA). The blood samples from untreated mice were used as control.
Animal experiments
All of the mice and their corresponding studies in this work were approved by the Chinese Academy of Sciences Institutional Animal Care and Use Committee (approval number: SYXK2017-33). For subcutaneous transplanted HepG2 tumor model therapeutic assessment, female BALB/c nude mice (Animal Center of the Chinese Academy of Medical Science) at 6-week-old were subcutaneous implanted with 1×106 HepG2 tumor cells in the right upper flank. When the size of tumors reached about 50 mm3, mice were divided into five groups randomly (n = 6 mice in each group) and i.v. administered a dose of free Dox (5 mg/kg), Doxil (25 mg/kg Dox equivalents), PBS, HccFn (60 mg/kg), HccFn-Dox (25 mg/kg Dox equivalents). During experimental period, the body weight and tumor volume of the mice in each group were measured every other day. Tumor volume was calculated by L × W2/2, where L represent the maximum diameter of tumor, while W represent the minimum diameter of tumor.
For HCC lung metastases mouse model therapeutic assessment, female BALB/c nude mice at 6-week-old were intravenous injected with 2×106 HepG2-Luc tumor cells. Two weeks later, mice were randomly assigned to three groups (n = 4 mice in each group) and intravenous administered HccFn-Dox nanocages (25 mg/kg Dox equivalents), free Dox (5 mg/kg) and PBS. As for survival time analysis, n = 6 mice in each group.
For in vivo tumor imaging, 1×106 HepG2 tumor cells were implanted into the right upper flank of 6-week old female BALB/c nude mice. When the size of tumors reached approx. 1.0 cm in diameter, mice were randomly divided into three groups (n = 3 mice in each group). Subsequently, a dose of Cy5.5-HccFn-Dox (2 nmol Cy5.5 equivalents), ½ dose of Cy5.5-HccFn-Dox (1 nmol Cy5.5 equivalents), and a dose of Cy5.5-Fn-Dox (2 nmol Cy5.5 equivalents) was injected into mice subcutaneously bearing HCC tumors. Tumors were imaged at 4 h post-injection by detecting the signal of Cy5.5 using IVIS Spectrum Imaging System (Xenogen). Then, mice were killed, tumors and the major tissues (heart, liver, spleen, lung, kidney) were collected to image using IVIS Spectrum Imaging System (Xenogen).
For biodistribution study, female BALB/c mice bearing HepG2 tumors were i.v. injected with HccFn-Dox (25 mg/kg Dox equivalents), then the signal of Dox was detected to quantify Dox distribution in each tissue. At 1, 4, and 24 h post-injection, mice were killed. Tumors and the major tissues (heart, liver, spleen, lung, kidney, brain) were collected, and Dox signals in each organ were measured using IVIS Spectrum Imaging System (Xenogen). Lastly, Dox concentrations in each tissue were quantified using IVIS Spectrum Imaging analysis software.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We thank Ms. Jingnan Liang for technical support in TEM imaging at the Core Facility of Equipment, Institute of Microbiology. We thank Dr. Xiang Ding and Dr. Fuquan Yang (Laboratory of Proteomics, Core Facility in the Institute of Biophysics) for their assistant with mass spectrum analysis. We thank Dr. Guizhi Shi (Institute of Biophysics, Chinese Academy of Sciences (CAS)) for assistance with pathological analysis. We thank Dr. Fanxia Meng and Dr. T. Juelich for linguistic assistance during the preparation of this manuscript.
This work was financially supported by the National Natural Science Foundation of China (No. 31871005, 31530026), Chinese Academy of Sciences under Grant No. YJKYYQ20180048, Young Elite Scientist Sponsorship Program by CAST (2015QNRC001), the Strategic Priority Research Program (No. XDB29040101), the Key Research Program of Frontier Sciences (No. QYZDY-SSW-SMC013), Chinese Academy of Sciences and National Key Research and Development Program of China (No. 2017YFA0205200).
Author contributions
K.F. and B.J. conceived and designed the experiments. B.J., R.Z., J.Z. and Y.H. synthesized and characterized the drug carrier. K.F., B.J., X.C. and M.Z. performed protein labeling and cell experiments. B.J., R.Z., X.T. and C.H. performed animal experiments. K.F., B.J. and X. Y. analyzed the data and wrote the paper. All authors discussed the results and commented on the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Waller Lisa P. Hepatocellular carcinoma: A comprehensive review. World J Hepatol. 2015;7:2648
2. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245-55
3. De Lope CR, Tremosini S, Forner A, Reig M, Bruix J. Management of HCC. J Hepatol. 2012;56:S75-S87
4. Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastro Hepat. 2010;7:448-58
5. Bellissimo F, Pinzone MR, Cacopardo B, Nunnari G. Diagnostic and therapeutic management of hepatocellular carcinoma. World J Gastro. 2015;21:12003-21
6. Lu J, Wang J, Ling D. Surface Engineering of Nanoparticles for Targeted Delivery to Hepatocellular Carcinoma. Small. 2017
7. Zhang X, Ng HLH, Lu A, Lin C, Zhou L, Lin G. et al. Drug delivery system targeting advanced hepatocellular carcinoma: Current and future. Nanomed Nanotechnol. 2016;12:853-69
8. Lo A, Lin CT, Wu HC. Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Mol Cancer Ther. 2008;7:579-89
9. Mohamed NK, Hamad MA, Hafez MZE, Wooley KL, Elsabahy M. Nanomedicine in Management of Hepatocellular Carcinoma: Challenges and Opportunities. Int J Cancer. 2016
10. Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA. et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10:389-97
11. Jin Y, Yang X, Tian J. Targeted polypyrrole nanoparticles for the identification and treatment of hepatocellular carcinoma. Nanoscale. 2018;10:9594-601
12. Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504-10
13. Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67:3496-9
14. Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell Surface Relocalization of the Endoplasmic Reticulum Chaperone and Unfolded Protein Response Regulator GRP78/BiP. J Biol Chem. 2010;285:15065-75
15. Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y. et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res. 2011;71:2848-57
16. Yao X, Liu H, Zhang X, Zhang L, Li X, Wang C. et al. Cell Surface GRP78 Accelerated Breast Cancer Cell Proliferation and Migration by Activating STAT3. PLoS One. 2015;10:e0125634
17. Zhang J, Jiang Y, Jia Z, Li Q, Gong W, Wang L. et al. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin Exp Metastas. 2006;23:401-10
18. Li Z, Zhang L, Zhao Y, Li H, Xiao H, Fu R. et al. Cell-surface GRP78 facilitates colorectal cancer cell migration and invasion. Int J Biochem Cell Biol. 2013;45:987-94
19. Yuan XP, Dong M, Li X, Zhou JP. GRP78 promotes the invasion of pancreatic cancer cells by FAK and JNK. Mol Cell Biochem. 2015;398:55-62
20. Wu CT, Wang WC, Chen MF, Su HY, Chen WY, Wu CH. et al. Glucose-regulated protein 78 mediates hormone-independent prostate cancer progression and metastasis through maspin and COX-2 expression. Tumor Biol. 2014;35:195-204
21. Daneshmand S, Quek ML, Lin E, Lee C, Cote RJ, Hawes D. et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum Pathol. 2007;38:1547-52
22. Zhuang L, Scolyer RA, Lee CS, McCarthy SW, Cooper WA, Zhang XD. et al. Expression of glucose-regulated stress protein GRP78 is related to progression of melanoma. Histopathology. 2009;54:462-70
23. Delie F, Petignat P, Cohen M. GRP78 Protein Expression in Ovarian Cancer Patients and Perspectives for a Drug-Targeting Approach. J Oncol. 2012;2012:468615
24. Delie F, Petignat P, Cohen M. GRP78-targeted nanotherapy against castrate-resistant prostate cancer cells expressing membrane GRP78. Target Oncol. 2013;8:225-30
25. Miao YR, Eckhardt BL, Cao Y, Pasqualini R, Argani P, Arap W. et al. Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin Cancer Res. 2013;19:2107-16
26. Zhao L, Li H, Shi Y, Wang G, Liu L, Su C. et al. Nanoparticles inhibit cancer cell invasion and enhance antitumor efficiency by targeted drug delivery via cell surface-related GRP78. Int J Nanomed. 2015;10:245-56
27. Li L, Fang CJ, Ryan JC, Niemi EC, Lebron JA, Bjorkman PJ. et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. P Natl Acad Sci U S A. 2010;107:3505-10
28. Chen TT, Li L, Chung DH, Allen CD, Torti SV, Torti FM. et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med. 2005;202:955-65
29. Liang MM, Fan KL, Zhou M, Duan DM, Zheng JY, Yang DL. et al. H-ferritin-nanocaged doxorubicin nanoparticles specifically target and kill tumors with a single-dose injection. P Natl Acad Sci USA. 2014;111:14900-5
30. Fan K, Jia X, Zhou M, Wang K, Conde J, He J. et al. Ferritin Nanocarrier Traverses the Blood Brain Barrier and Kills Glioma. Acs Nano. 2018
31. Uchino K, Tateishi R, Shiina S, Kanda M, Masuzaki R, Kondo Y. et al. Hepatocellular Carcinoma With Extrahepatic Metastasis Clinical Features and Prognostic Factors. Cancer-Am Cancer Soc. 2011;117:4475-83
32. Truffi M, Fiandra L, Sorrentino L, Monieri M, Corsi F, Mazzucchelli S. Ferritin nanocages: A biological platform for drug delivery, imaging and theranostics in cancer. Pharmacol Res. 2016;107:57-65
33. Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA. et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99-111
34. Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF. et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666-81
35. Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W. et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6:275-84
36. Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181-8
37. Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K. et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38:605-14
38. Jiang X, Kanda T, Nakamoto S, Miyamura T, Wu S, Yokosuka O. Involvement of androgen receptor and glucose-regulated protein 78 kDa in human hepatocarcinogenesis. Exp cell Res. 2014;323:326-36
39. Su RJ, Li Z, Li HD, Song HJ, Bao CF, Wei J. et al. Grp78 promotes the invasion of hepatocellular carcinoma. Bmc Cancer. 2010:10
40. Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS. et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7:383-8
41. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3-6
42. Belletti D, Pederzoli F, Forni F, Vandelli MA, Tosi G, Ruozi B. Protein cage nanostructure as drug delivery system: magnifying glass on apoferritin. Expert Opin Drug Deliv. 2017;14:825-40
43. Tatur J, Hagen WR, Matias PM. Crystal structure of the ferritin from the hyperthermophilic archaeal anaerobe Pyrococcus furiosus. J Biol Inorg Chem. 2007;12:615-30
44. Fan KL, Cao CQ, Pan YX, Lu D, Yang DL, Feng J. et al. Magnetoferritin nanoparticles for targeting and visualizing tumour tissues. Nat Nanotechnol. 2012;7:459-64
45. Yang Z, Wang X, Diao H, Zhang J, Li H, Sun H. et al. Encapsulation of platinum anticancer drugs by apoferritin. Chem Commun. 2007:3453-5
46. Li L, Munoz-Culla M, Carmona U, Lopez MP, Yang F, Trigueros C. et al. Ferritin-mediated siRNA delivery and gene silencing in human tumor and primary cells. Biomaterials. 2016;98:143-51
47. Hainfeld JF. Uranium-Loaded Apoferritin with Antibodies Attached - Molecular Design for Uranium Neutron-Capture Therapy. P Natl Acad Sci USA. 1992;89:11064-8
48. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2016
49. MacKay JA, Chen MN, McDaniel JR, Liu WG, Simnick AJ, Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat Mater. 2009;8:993-9
Author contact
![]() Corresponding authors: Kelong Fan, PhD. Email: fankelongac.cn; Xiyun Yan, MD, Email: yanxyac.cn.
Corresponding authors: Kelong Fan, PhD. Email: fankelongac.cn; Xiyun Yan, MD, Email: yanxyac.cn.